
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper–oxygen adducts: new trends in characterization and properties towards C–H activation
Jonathan
De Tovar
 a,
Rébecca
Leblay
b,
Yongxing
Wang
b,
Laurianne
Wojcik
c,
Aurore
Thibon-Pourret
a,
Marius
Réglier
*b,
A. Jalila
Simaan
*b,
Nicolas
Le Poul
*c and
Catherine
Belle
*a
a,
Rébecca
Leblay
b,
Yongxing
Wang
b,
Laurianne
Wojcik
c,
Aurore
Thibon-Pourret
a,
Marius
Réglier
*b,
A. Jalila
Simaan
*b,
Nicolas
Le Poul
*c and
Catherine
Belle
*a
aUniversité Grenoble-Alpes, CNRS, Département de Chimie Moléculaire, Grenoble, France. E-mail: catherine.belle@univ-grenoble-alpes.fr
bAix Marseille Univ, CNRS, Centrale Marseille, iSm2, Institut des Sciences Moléculaires de Marseille, Marseille, France. E-mail: marius.reglier@univ-amu.fr; jalila.simaan@univ-amu.fr
cUniversité de Brest, Laboratoire de Chimie, Electrochimie Moléculaires et Chimie Analytique, Brest, France. E-mail: lepoul@univ-brest.fr
First published on 13th May 2024
Abstract
This review summarizes the latest discoveries in the field of C–H activation by copper monoxygenases and more particularly by their bioinspired systems. This work first describes the recent background on copper-containing enzymes along with additional interpretations about the nature of the active copper–oxygen intermediates. It then focuses on relevant examples of bioinorganic synthetic copper–oxygen intermediates according to their nuclearity (mono to polynuclear). This includes a detailed description of the spectroscopic features of these adducts as well as their reactivity towards the oxidation of recalcitrant Csp3–H bonds. The last part is devoted to the significant expansion of heterogeneous catalytic systems based on copper–oxygen cores (i.e. within zeolite frameworks).
 From left to right: Yongxing Wang, A. Jalila Simaan, Nicolas Le Poul, Catherine Belle, Marius Réglier, Laurianne Wojick, Rébecca Leblay, Aurore Thibon-Pourret and Jonathan De Tovar. | Organized as a French collaborative consortium (from Grenoble, Marseille and Brest), the authors of this review carry out research on the structure–function relationships of copper–oxygen species for the activation of C–H bonds in alkanes. Two French ANR grants, the COMEBAC project (ANR-13-BSO7-0018) and COSACH project (ANR-22-CE07-0032), supported their research. Based on new catalysts inspired by the structure and functioning of the copper-containing monooxygenase pMMO, the COMEBAC project aimed to design and study a mixed-valent bis(μ-oxo)CuIICuIII species that was proposed to be active in the oxidation of alkanes. The subsequent COSACH project continues the objective of COMEBAC but in extending its investigations towards mononuclear and dinuclear copper–oxygen entities. The architecture of these new catalysts is based on recent experimental and theoretical research on bioinspired models of copper-containing monoxygenases (pMMO, LPMO and PHM). COMEBAC and COSACH cover different areas of chemistry, including the synthesis/characterization of new ligands/complexes, generation and identification of transient copper–oxygen species using for example cryo-spectroelectrochemical approaches, theoretical calculations and studies of reactivity for the oxidation of substrates. |
1. Introduction
Copper–oxygen species (mono to multi-copper, derived from O2 or O-atom donors represented by Cu/O) have been widely investigated since the 1980s due to their ubiquitous involvement in various biological processes such as oxidation and oxygenation reactions. Given their ability to act on various substrates as different as methane and phenols (pMMO vs. Ty, Scheme 1) copper-containing monooxygenases have drawn particular attention1 for the development of synthetic catalysts for efficient C–H bond activation. The approach, which consists of going back and forth between enzymes and chemical models, has allowed us to better understand the structure/reactivity of Cu/O species in enzymes as well as the O-atom transfer (OAT) mechanisms. This has been particularly well illustrated by studies of type III copper-containing proteins (hemocyanin, catechol oxidase and tyrosinase) which now constitute an important part of several bioinorganic chemistry textbooks.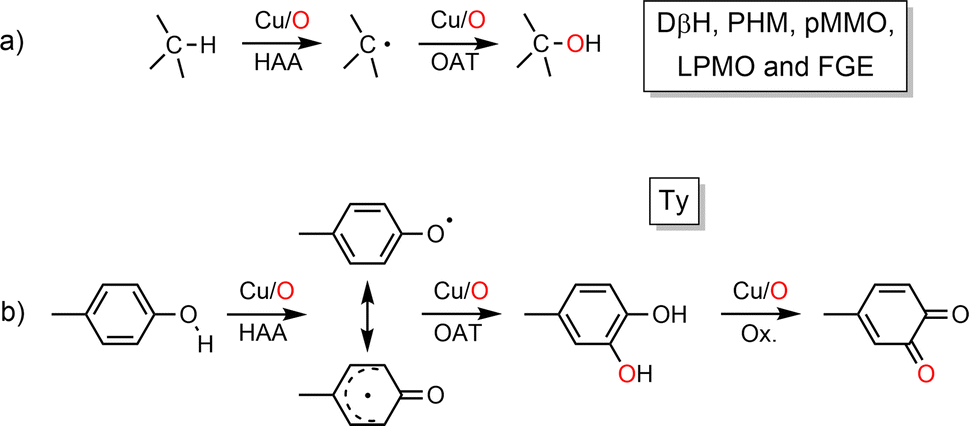 | ||
| Scheme 1 Reaction type catalyzed by copper-containing monooxygenases (a) DβH (Dopamine β-monooxygenase); PHM (Peptidylglycine α-Hydroxylating Monooxygenase); pMMO (particulate Methane Monooxygenase); LPMO (Lytic Polysaccharide Monooxygenase); FGE (Formylglycine-Generating Enzyme) and (b) Ty (Tyrosinase). | ||
Over the past decade, some aspects of the structure and reactivity of copper–oxygen models have been reported by Stack,2,3 Itoh,4 Tolman,5 Karlin,6 and Garcia-Bosch.7 This review covers the literature from the year 2017 to early 2024, focusing on recent advances on the structures and reactivities of the characterized copper–oxygen species possibly involved in C–H bond activation (Scheme 1a), as models for copper containing monooxygenases. Many articles in the literature report the oxidation of catechol to quinone or non-catalytic oxidation of phenol to catechol and subsequently to quinone by bioinspired complexes modelling tyrosinase's activity (Scheme 1b). The majority of these articles fall outside the scope of this review. Only those which aimed at highlighting the reactivity of Cu/O entities towards X–H bonds are discussed herein. Beyond the inventory of research work accomplished in the field of structure/reactivity of copper–oxygen species, our objective is to highlight the trends towards the challenging goal of efficient C–H bond activation. Mechanisms of C–H bond activation by reactive Cu/O intermediates (mono to polynuclear) are discussed with particular emphasis on the latest understandings and key catalytic systems (including heterogeneous catalysts). Charts 1 and 2 in this review offer a comprehensive list of the main ligands discussed here, along with their respective abbreviations, towards the formation and stabilization of both monodentate and polydentate Cu/O species.
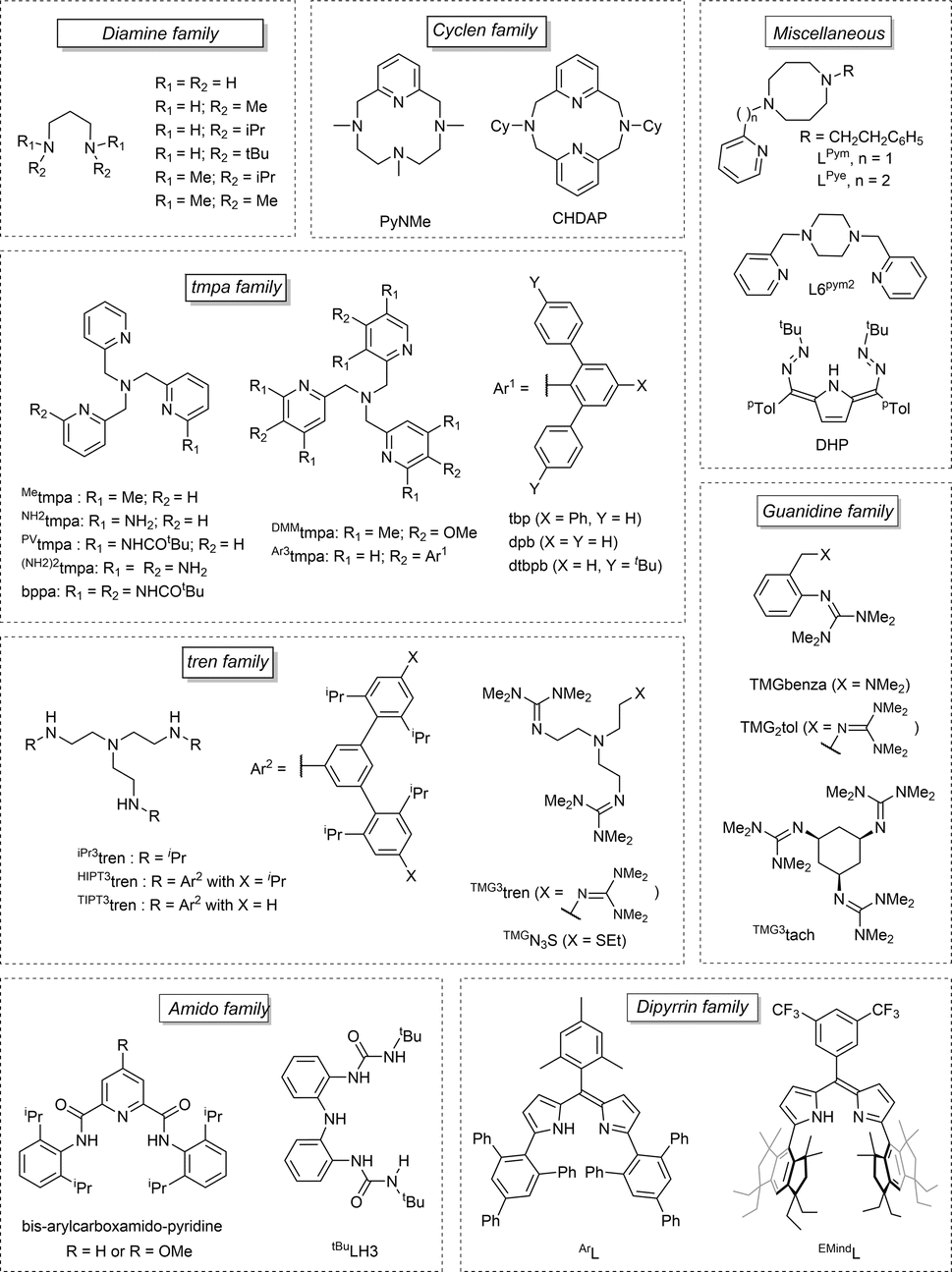 | ||
| Chart 1 Representation of the monodentate ligands reported in this review. | ||
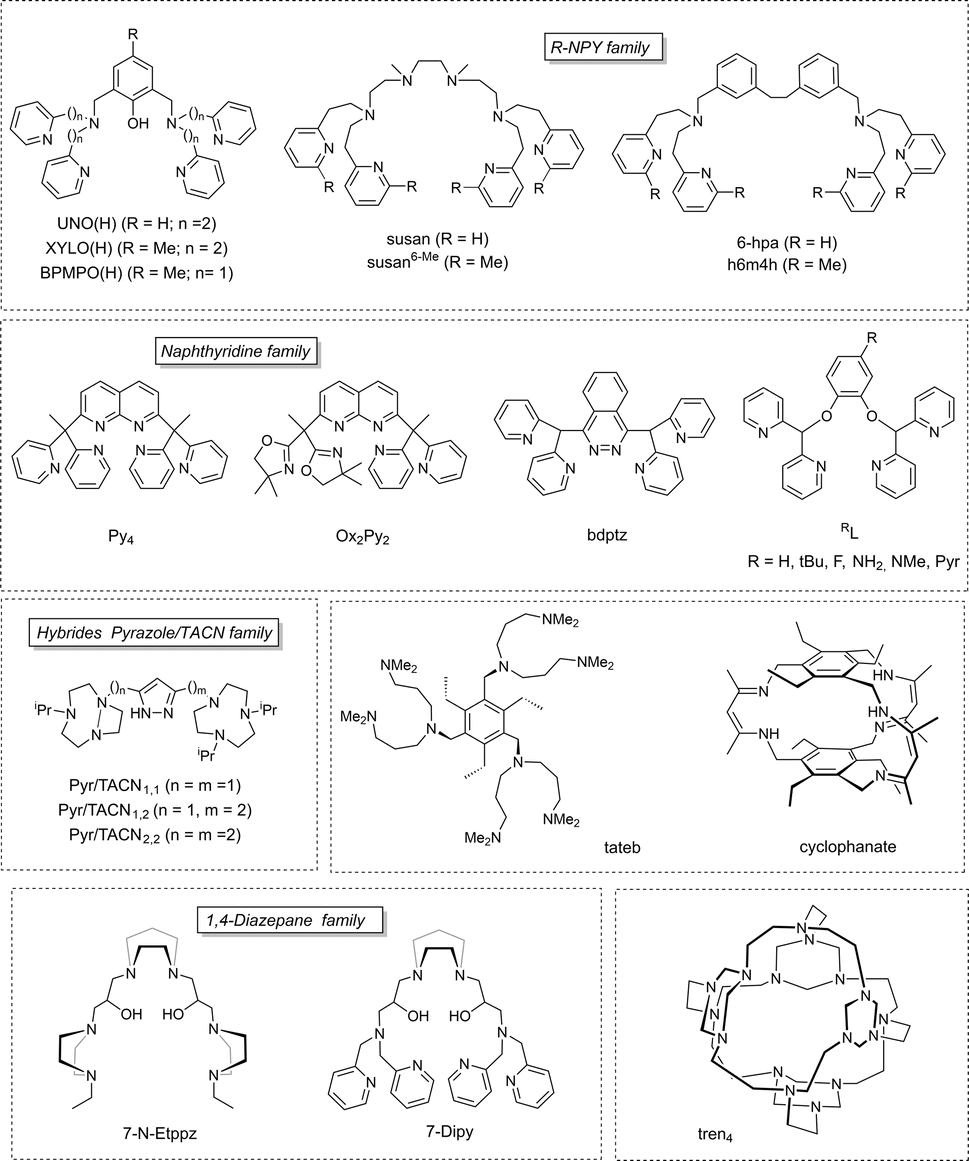 | ||
| Chart 2 Representation of the polydentate ligands reported in this review. | ||
2 General context
Recent years have seen the evolution of our knowledge on some copper-containing monooxygenases. Indeed, without mentioning the particulate Methane Monooxygenase (pMMO), which has not yet revealed all its secrets, or Lytic Polysaccharide Monooxygenase (LPMO), the activity of which is still debated (monooxygenase vs. peroxygenase), others like Peptidylglycine α-Hydroxylating Monooxygenase (PHM) and Dopamine β-monooxygenase (DβH), could well go from uncoupled to coupled binuclear (see Section 2.1).2 Moreover, the emerging advances could allow polyethylene deconstruction with hexamerins.8 The first section of this review is an opportunity to update new knowledge related to copper-containing monooxygenases.Another development observed in recent years concerns the thermodynamic and kinetic evaluation of reactions linked to proton-coupled electron transfer (PCET) processes. These are important advances for a better understanding of the intrinsic properties of the catalysts. Therefore, we have devoted a second section to these thermodynamic aspects.
2.1. Copper-containing monooxygenases
Among the copper-containing monooxygenases described so far, tyrosinases (Tys) occupy an emblematic place due to the important role played by chemical models in deciphering their oxy-state structure and their reactivity. Tyrosinases do not catalyze the oxidation of phenols through the activation of a C–H bond but more precisely via an SRN1 mechanism (vide infra). However, as the mechanism of this transformation involves Cu/O species common to the other monooxygenases, we start this section with tyrosinase. Two recent reviews from Tuczek9 and Mukherjee10 deal with this close link. Tys are present in prokaryotes and eukaryotes (plants, arthropods, fungi and mammals) where they are involved in tissues pigmentation, wound healing, radiation protection and primary immune response. Tys belong to the commonly called binuclear coupled enzymes due to their active site composed of two copper ions coordinated to three histidines each and spaced from each other by 3.1–4.2 Å depending on the redox and/or ligation state. The Ty-catalyzed oxygenation of phenolic compounds consists of two consecutive reactions, the ortho-hydroxylation of phenol to catechol (cresolase or monophenolase activity, EC 1.14.18.1) and its subsequent oxidation into ortho-quinone (catecholase or diphenolase activity, EC 1.10.3.1) (Fig. 1). Beyond many applications in the medical and biotechnological fields,11,12 recent studies on tyrosinases from Solomon's group have revisited the mechanism of the O-atom transfer that was previously considered as an aromatic electrophilic substitution (SEAr) on a phenolate anion bound to the μ-η2:η2 peroxido dicopper(II) core (Fig. 1b).13,14 On the basis of new kinetic studies and QM/MM calculations, the group of Solomon concluded that the most likely mechanism would be a radical-nucleophilic aromatic substitution (SRN1) initiated by a H-bonding of the present phenol (no deprotonation) and then proton-coupled electron transfer (PCET) to the μ-η2:η2 peroxido dicopper(II) core of oxy-Ty (Fig. 1c).15–18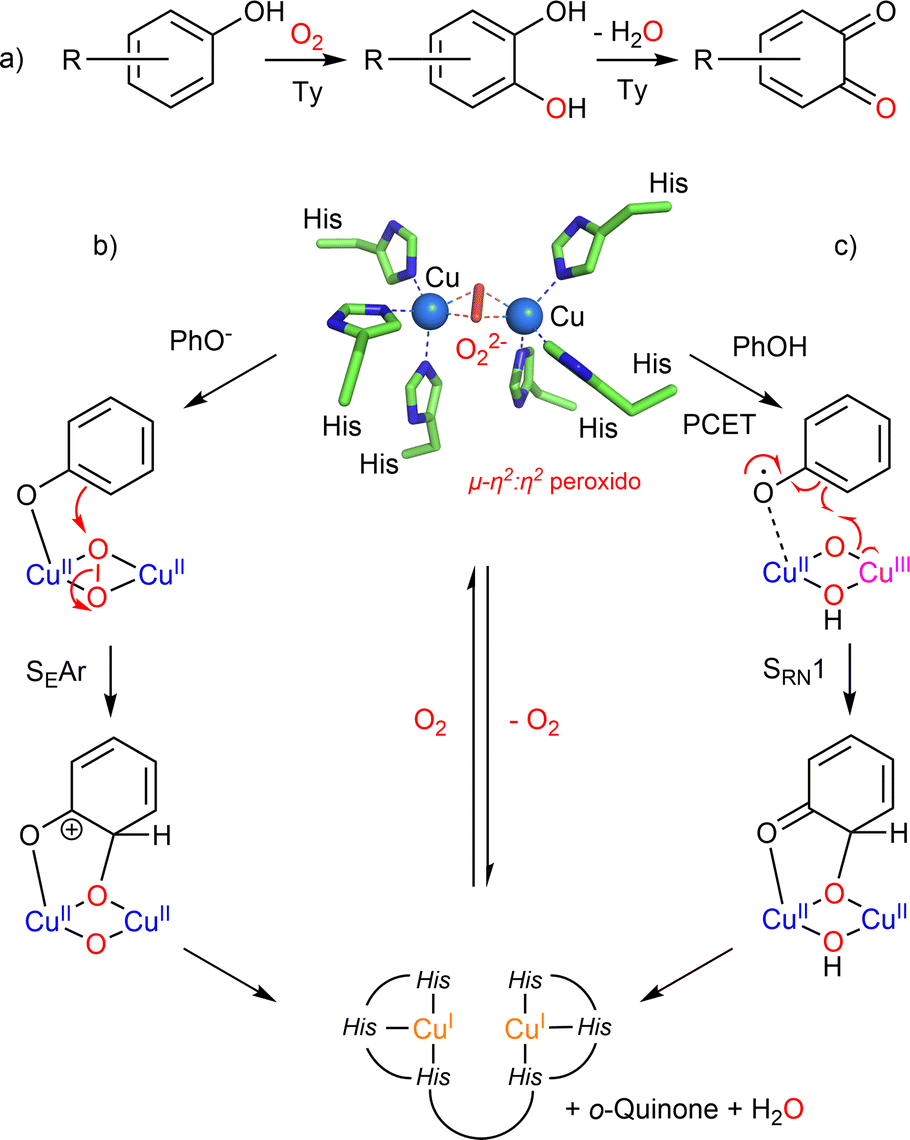 | ||
| Fig. 1 Type III copper-containing proteins, (a) reactions catalyzed by Ty and oxy-Ty 3D-structure of the active site (PDB ID: 1wx4); (b) mechanism involving an electrophilic aromatic substitution (SEAr) and (c) mechanism involving a radical-nucleophilic aromatic substitution (SRN1). | ||
Two other copper-containing monooxygenases have recently gained renewed interest. Dopamine β-monooxygenase (DβH, EC 1.14.17.1, Fig. 2b) and Peptidylglycine α-Hydroxylating Monooxygenase (PHM, EC 1.14.17.3, Fig. 2a) were until now considered as uncoupled binuclear copper-containing monooxygenases with two copper ions coordinated one to three histidines (CuH) and the second to two histidines and one methionine (CuM). CuH and CuM, which are spaced from each other by approximately 11 Å, were proposed to possess distinct functions: CuM would be involved in the hydroxylation process through a copper-superoxido species while CuH would act as an electronic relay (Fig. 2d).19 In this representation, the copper-superoxido species would abstract an H-atom (Hydrogen Atom Abstraction, HAA) from the substrate which would be followed by a rebound event (Scheme 2a). This proposal, which was based on initial X-ray data (Fig. 2c and d), spectroscopic and kinetic studies on the reduced and oxy-PHM, was recently revisited and could become obsolete according to the recent X-ray 3D structure of the human DβH (Fig. 2e and f).20
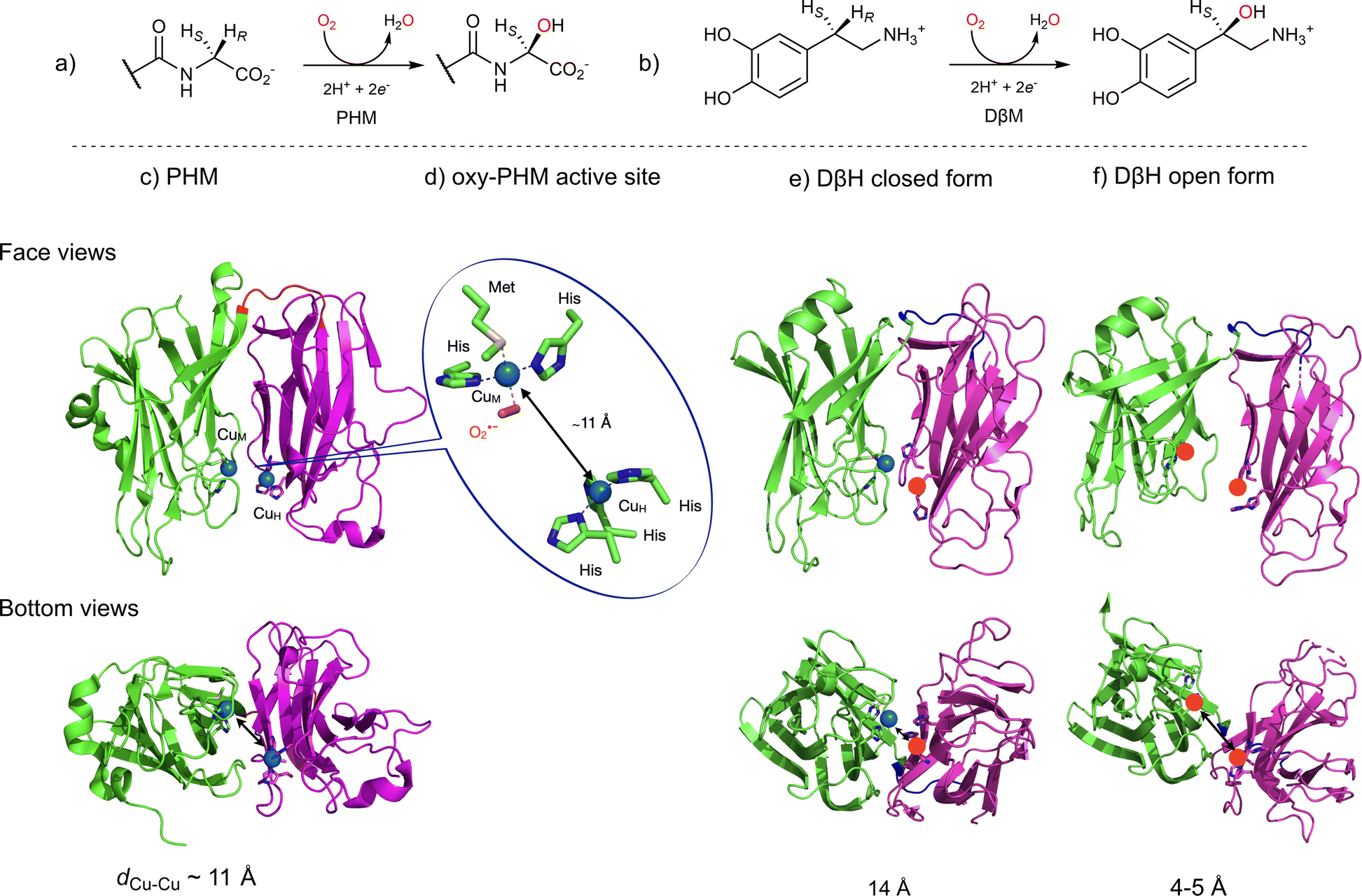 | ||
| Fig. 2 Uncoupled binuclear copper-containing monooxygenases, PHM and DβH. (a) and (b) reactions catalyzed by PHM and DβH, respectively; (c) X-ray structure of PHM from Rattus norvegicus showing the two domains (CuM domain in green and CuH domain in magenta) connected by a loop (in blue) in which each domain binds a single copper atom spaced from each other by approximately 11 Å and (d) structure of the CuM-superoxido intermediate in PHM (PDB ID 1opm); (e) and (f) X-ray structure of the human DβM (PDB ID 4zel) showing the open (e) and the closed (f) forms. Only the CuM domain of the closed form contains a copper atom, the other copper ions (red sphere) are manually placed. | ||
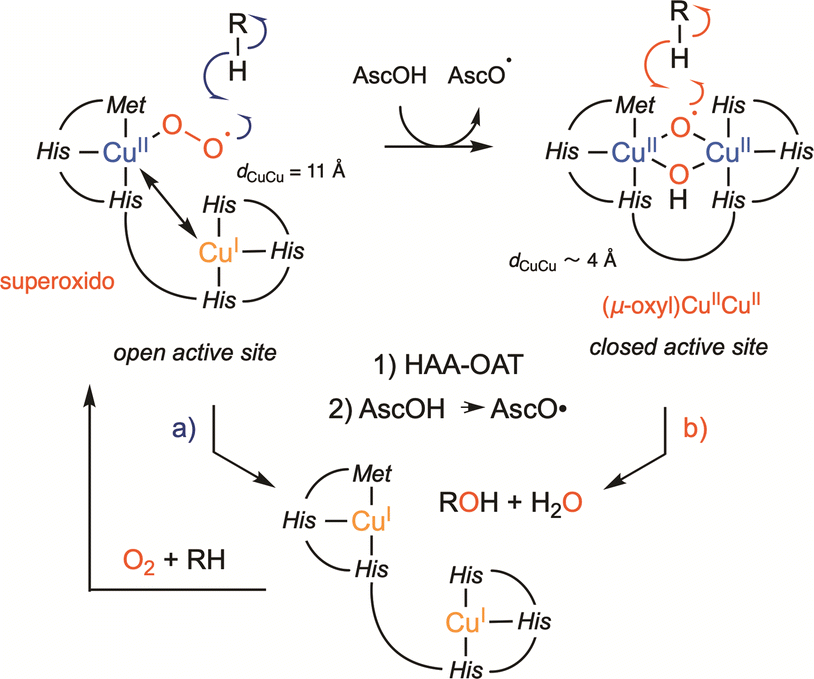 | ||
| Scheme 2 Recently revisited mechanism for PHM and DβH showing the mechanisms involving an HAA by (a) CuM-superoxido intermediate in an open dicopper active site and (b) μ-oxyl dicopper intermediate in closed dicopper active site. Abbreviation: AscOH for ascorbic acid. | ||
Indeed, the DβH X-ray structure revealed that the enzyme can adopt two different conformations: (i) an open conformation with two distant copper ions, resembling the previously reported PHM structure (Fig. 2e) and (ii) a closed conformation, in which the two copper sites are only 4 to 5 Å apart, in what might be a coupled binuclear copper site (Fig. 2f).
Could PHM and DβH leave the uncoupled binuclear family for the coupled binuclear ones? This question is relevant since an alternative mechanism corroborated by QM/MM calculations,21 involving a dinuclear copper intermediate formed via an open to closed conformational transition managed to better account for the experimental spectroscopic data collected over decades on PHM and the DβH (Scheme 2b).22–25 On one hand, it is interesting to note that this proposal was already tentatively suggested in 2002, though without sufficient support from the scientific community at the time.26 On the other hand, if confirmed, this new finding illustrates once more the important role of chemical models in these studies. This takes us back few decades to Réglier’s proposals when: (i) the first DβH functional model involving the occurrence of a μ-η2:η2 peroxido dicopper(II) species was reported27,28 and (ii) based on the substrate binding ligand approach29,30 the occurrence of a dicopper mixed-valent species such as (μ-oxido)CuIICuIII ↔ (μ-oxyl)CuIICuII was proposed to explain the O-atom transfer to the ligand.31
The membrane-bound Methane Monooxygenase (pMMO, EC 1.14.18.3) continues to be in the headlines of discussions about the nature, localization and nuclearity of the active site, as well as on the mechanism. Methane monooxygenases (MMOs) are crucial enzymes in bacterial methane metabolism since they catalyze the conversion of methane to methanol (CH4 + O2 + 2e− + 2H+ → CH3OH + H2O). They represent a potential route towards the use and mitigation of methane emissions. MMOs exist in two forms, a cytoplasmic soluble form (sMMO) and a membrane-bound enzyme form (pMMO). These MMOs are completely unrelated, with different architectures, metal cofactors (iron for sMMO and copper for pMMO), and mechanisms.32 While the mechanism of sMMO is well described and agreed upon, that is not the case for pMMO. To date, crystal structures of pMMOs from five species have been determined.33,34 These structures show that pMMOs are heterotrimers in which each monomer is composed of three subunits, PmoA (α), PmoB (β), and PmoC (γ). The heterotrimer (αβγ)3 adopts a cylindrical shape composed of twelve transmembrane helices almost entirely embedded in the lipid bilayer (Fig. 3a).
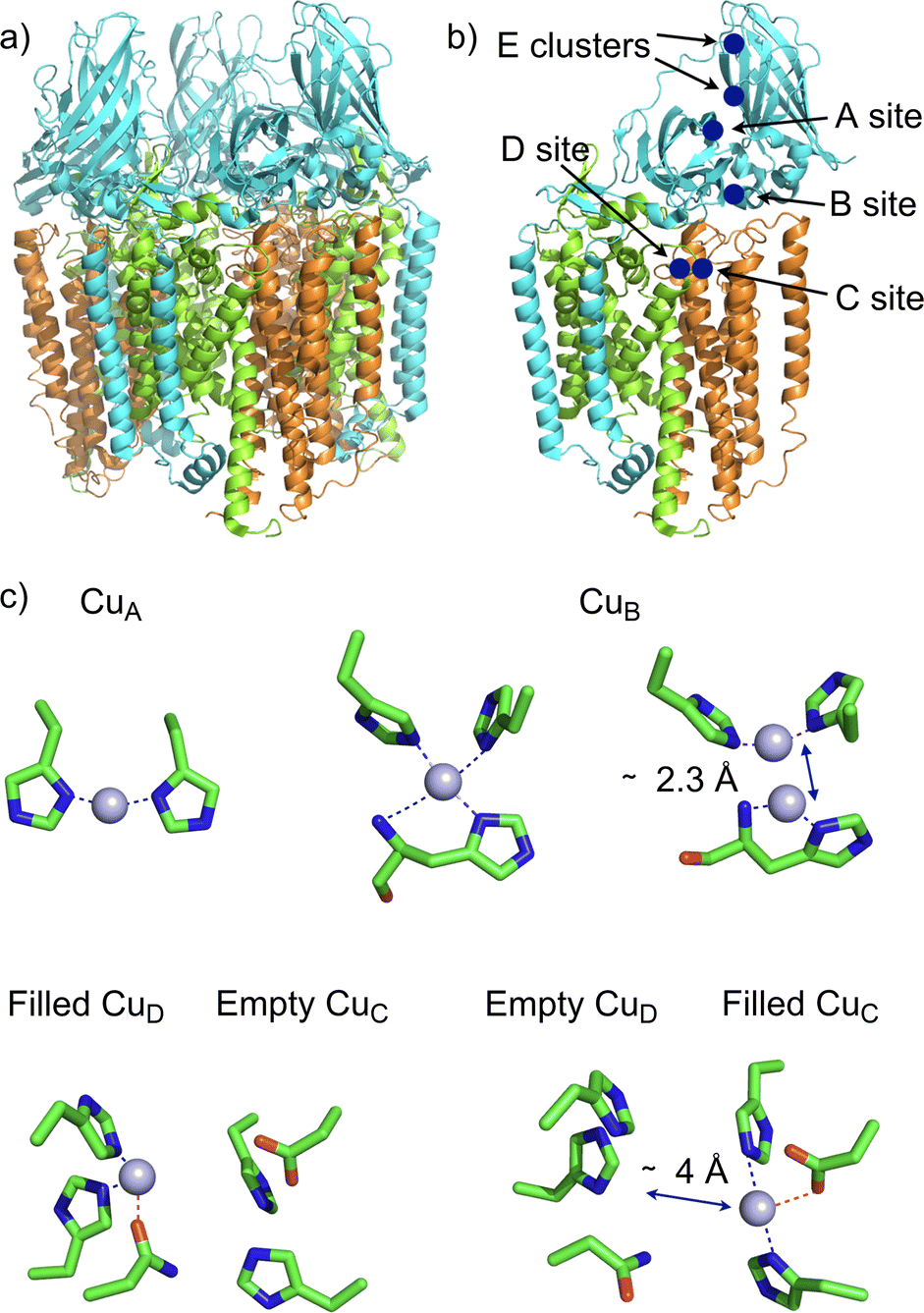 | ||
| Fig. 3 X-ray structure of M. capsulatus (Bath) pMMO (PDB ID 3rgb). (a) Heterotrimer (αβγ)3 adopting a cylindrical shape composed of twelve transmembrane helices; (b) 3D-view of one subunit αβγ showing the different location of copper ions, PmoA (α) in green, PmoB (β) in blue and PmoC (γ) in orange; (c) 3D-views of the identified copper center by cryo-EM. The CuB center displays a histidine brace similar to the one reported on LPMO (see Fig. 4). | ||
Despite these structures, the identity of the copper sites remains unclear. According to several spectroscopic and biochemical analyses there would be 12–14 copper ions found within each functional pMMO. Only two sites (CuB and CuC) have been clearly identified by X-ray crystallography in all pMMO sources (Fig. 3). The nuclearity of the CuB site was the subject of a long debate. In the original crystal structure of the pMMO of M. capsulatus (Bath), this site could be modeled with one or two copper ions. However, thanks to spectroscopic35–37 and computational38 studies, the debate has evolved in favor of a mononuclear site. In contrast, the CuC site located within the transmembrane domain of the PmoC subunit has been identified as mononuclear featuring two histidines and one aspartate as ligands. Biochemical studies of pMMO activity suggest that the CuC site would likely be the catalytic site for O2 activation and CH4 oxidation.34 A recent publication by Shaik attempts to address this question through a QM/MM study of dioxygen activation at the CuC center in the presence of the physiological reductant duroquinol (DQH2).39 This simulation has converged towards the occurrence of the CuCII-oxyl species which would be responsible for the C–H activation. These results reveal the role that phenols could play in the activation of O2 at the monocopper center.
However, two recent cryo-electron microscopy (cryo-EM) structures of the pMMO have re-opened the debate (Fig. 3). The first structure published by Chan regards pMMO from M. capsulatus (Bath) in the copper(I) oxidation state. This structure reveals a dinuclear copper B site, and more surprising unveils additional copper clusters at the PmoA/PmoC interface within the membrane (D site) and in the water-exposed C-terminal subdomain of the PmoB (E site).40 The other structures have been published by Rosenzweig of the pMMOs M. capsulatus (Bath), M. alcaliphilum and Methylocystis sp. strain Rockwell in the copper(II) oxidation state and embedded in the lipid bilayer closer to the in vivo environment.41 These new structures reveal a mononuclear copper A site (only for M. capsulatus and M. alcaliphilum), a mononuclear copper B site and the presence of copper in C or D sites but never together in both sites. The different nuclearities observed for the copper B site (mono vs. binuclear) could be linked to the oxidation state of copper ions, mono for copper(II) vs. dinuclear for copper(I). This observation is perhaps not surprising, since copper tridentate complexes bearing the same ligand were shown to crystallize in dimeric and monomeric forms in the copper(I) and copper(II) oxidation states, respectively.42 More interesting is the proximity of the copper C and D sites which are only approximately 4 Å apart from each other. Recent QM/MM calculations43 suggest that the CuDI site would be more reactive than the CuCI site in dioxygen activation for the generation of a CuII-oxyl species. Furthermore, these simulations which tend to demonstrate that the natural reductant ubiquinol (CoQH2) prefers to bind to the CuDI site rather than to the CuCI site could support the fact that the active site of pMMO is CuD rather than CuC.43 Even if the structures did not reveal the presence of copper ions in the two sites, given the short distance between them, one may raise the question of the physiological relevance of a possible binuclear CuC–CuD site for methane oxidation (Fig. 3c).
Lytic polysaccharide monooxygenases (LPMOs, EC 1.14.99.54) are copper-containing enzymes that comprise a large enzyme super-family. LPMOs hydroxylate polysaccharide substrates at either the C1 or the C4 position followed by the subsequent formation of the aldonic acids (C1 hydroxylation) or 4-keto sugars (C4 hydroxylation) at oxidized chain ends (Fig. 4a).44 All LPMOs display a mononuclear copper active site that is located on the binding surface of the enzyme (Fig. 4b). Coordinated with two histidine residues, the copper ion exhibits a distinctive feature: it is linked to the N-amino-terminal histidine through its NH2-terminal end and with the imidazole side chain, a coordination motif that has been named “histidine brace”.45 Since the discovery in the early 2010s of the first LPMO, these enzymes have continuously attracted interest and stimulated the curiosity of the scientific community.44–47 One major reason for interest in LPMOs is their key role in the deconstruction of recalcitrant polysaccharides (cellulose, chitin, etc.), a role of paramount commercial importance to produce advanced biofuels or bio-based chemicals.48 Another matter of interest is that LPMOs were the first “real” mononuclear copper-containing monooxygenases easily accessible by genetic expression.
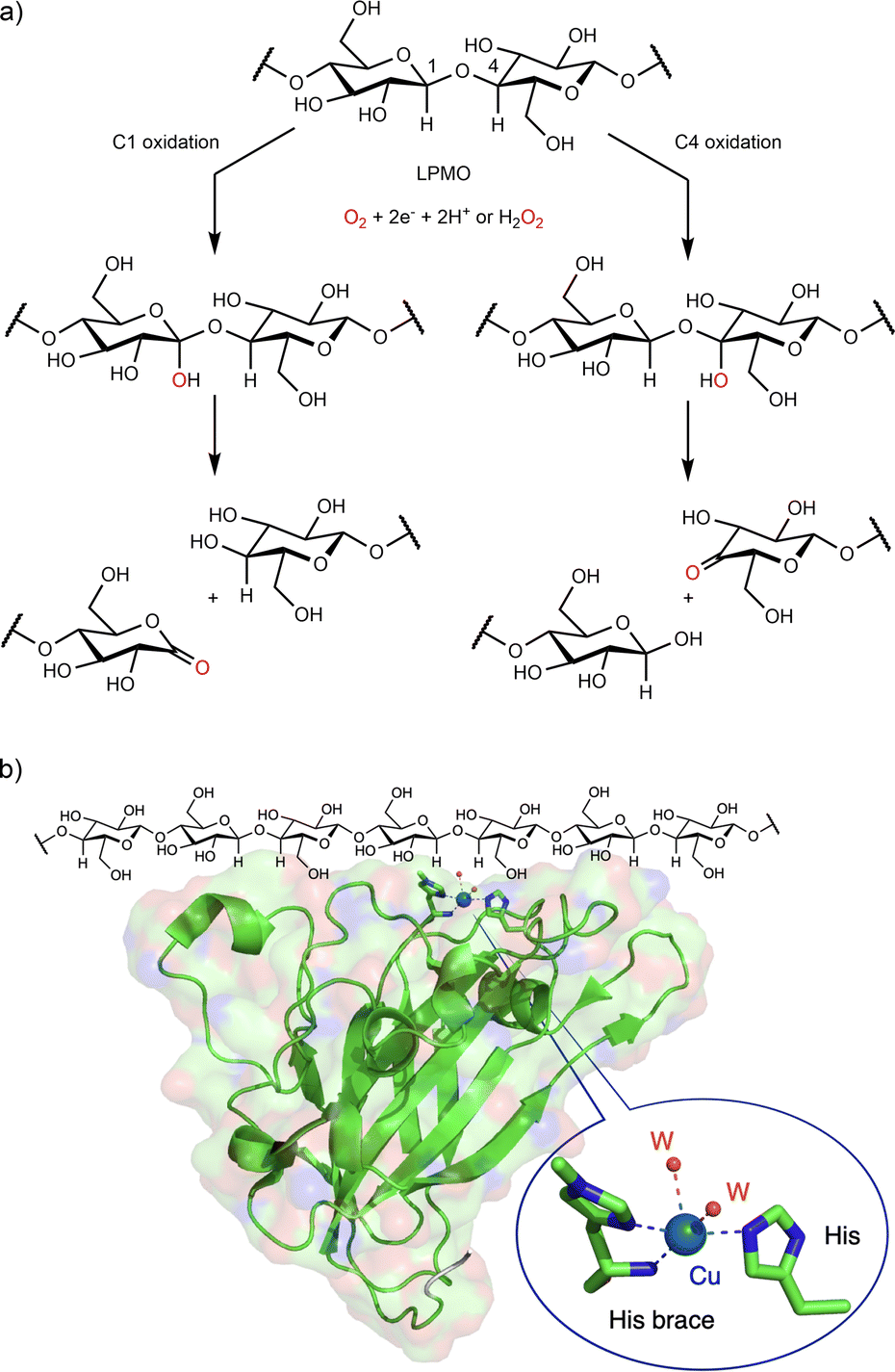 | ||
| Fig. 4 Lytic polysaccharide monooxygenase (LPMO). (a) Reactions catalyzed by LPMO; (b) LPMO X-ray structure from Thermoascus aurantiacus (PDB ID 2yet) showing the copper active site at the binding surface of the LPMO. | ||
LMPOs are active in complex media, with heterogeneous substrates (cellulose, chitine, etc.) in the presence of numerous reducing polyphenolic compounds (degradation of lignin), and in the presence of both dioxygen and hydrogen peroxide. Therefore, the mechanism associated with their activity is difficult to study. Although LPMOs were considered at first as monooxygenases (with dioxygen as the co-substrate), recent investigations have suggested that they could instead be considered as peroxygenase (with hydrogen peroxide as the co-substrate).49 The exact nature of this oxygenated co-substrate (dioxygen or hydrogen peroxide) is still debated50–53 as well as the nature of the copper–oxygen species involved in polysaccharide hydroxylation.54,55 Recently, Schröder et al. resolved a new cryo-trapped structure with an activated dioxygen intermediate in a mixture of superoxido and hydroperoxido states.56 Several alternative active species have been proposed to be formed within LPMOs' catalytic mechanism, including CuII-oxyl, or even the hydroxyl radical57,58 without having been spectroscopically characterized or observed. Scheme 3 summarizes the possible intermediates and mechanisms for LPMO catalyzed hydroxylation.
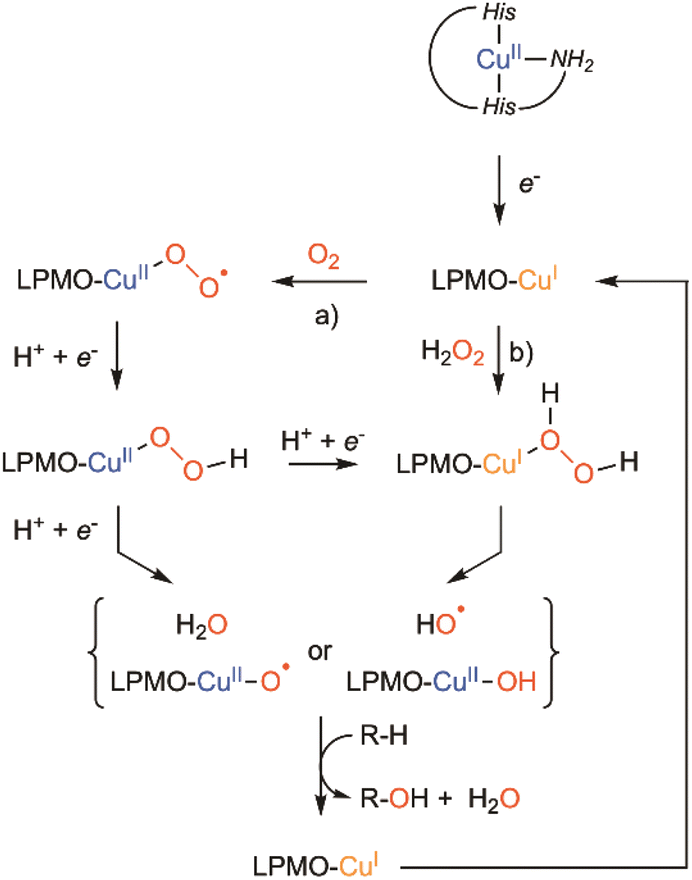 | ||
| Scheme 3 Putative mechanisms to explain LPMO catalyzed hydroxylation: (a) the monooxygenase pathway and (b) peroxygenase pathway. | ||
The Formylglycine-Generating Enzyme (FGE, EC 1.8.3.7), which catalyzes the post-translational activation of type I sulfatases (cysteine residues in formylglycine, Fig. 5a) in eukaryotes and aerobic microbes, was discovered at the end of the 1990s.59 Recently, it drew renewed interest not only because of its potential in protein engineering,60 but also because of its unique active site which differentiates it from other copper-containing monooxygenases (Fig. 5).61 Indeed, while copper ions bind mainly to histidines with tetrahedral or square-planar coordination spheres in the classical copper-containing monooxygenases (Ty, DβH, PHM, pMMO and LPMO), a single copper(I) binds two cysteines in a linear coordination configuration in FGE. Thanks to several techniques (stopped-flow absorbance spectroscopy,62 X-ray,63 QM/MM64), a mechanism has been proposed in which a non-coordinated dioxygen complex was suggested as a precatalytic complex. The dioxygen would not be coordinated to the copper(I) but rather bound in a hydrophobic pocket juxtaposed to the metal. Thus, the binding and activation of dioxygen by FGE would be a stepwise process conversely to many other metal-containing oxygenases. The mechanism would then involve HAA by a copper superoxido intermediate followed by the formation of an alkyl-peroxido intermediate precursor of the aldehyde. However, the mechanism is far from being fully elucidated and investigation using small copper model complexes could be highly beneficial.
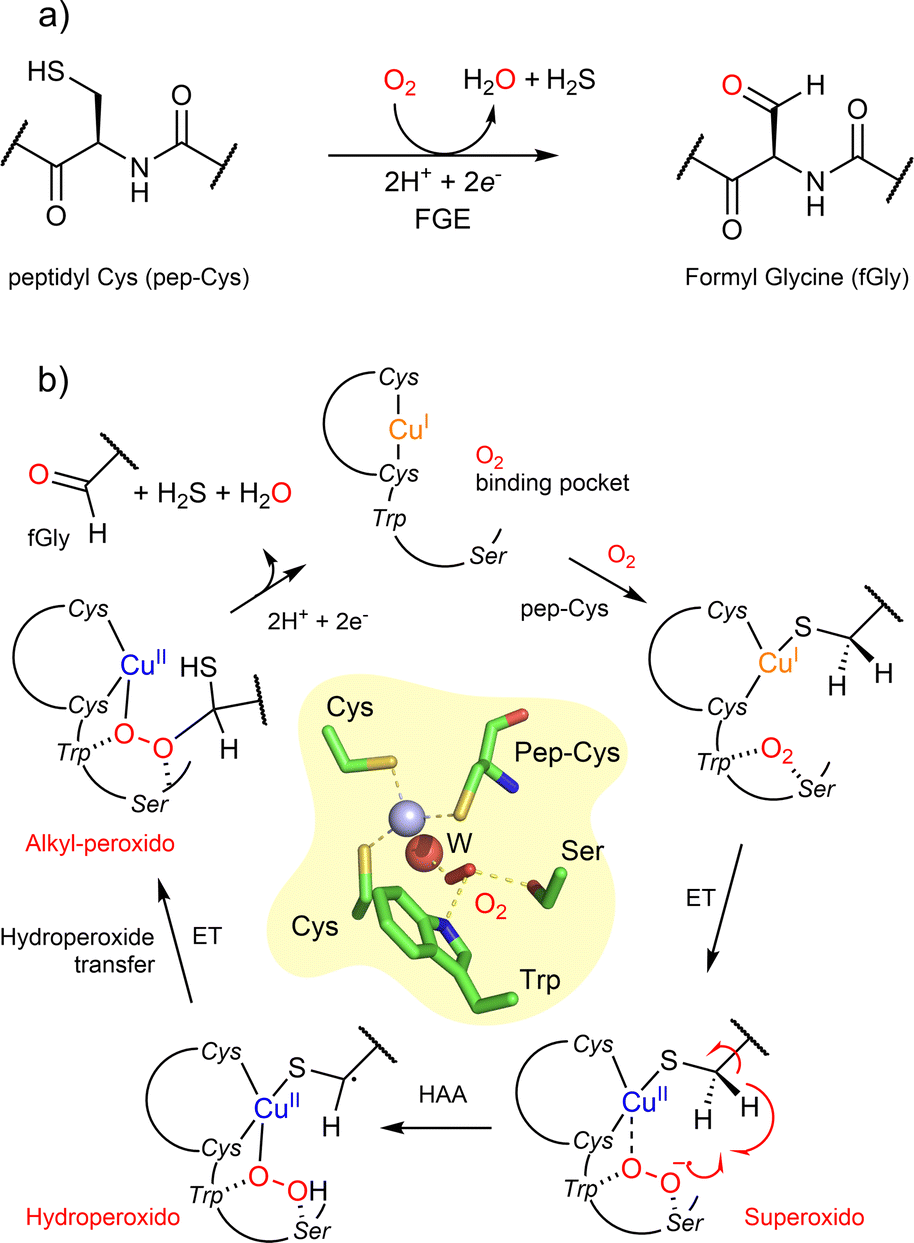 | ||
| Fig. 5 Formylglycine-Generating Enzyme (FGE). (a) Reaction catalyzed by FGE; (b) the proposed mechanism and within the center the 3D view of the FGE active site from Thermomonospora curvata (PDB ID 6xtr) showing the copper center linked to two cysteines and the peptide substrate as well as the hydrophobic pocket consisting of a tryptophan and a serine where the dioxygen binds before to be reduced to superoxide. | ||
In 2023, it was reported that the buccal secretion of the lepidopteran Galleria mellonella larvae was capable of oxidizing and depolymerizing polyethylene at room temperature in the presence of a copper salt. The cryo-EM analysis and 3D-reconstructions revealed that the lepidopteran buccal secretion is mainly composed of four hexamerins (Demetra, Cibeles, Ceres and Cora) belonging to the type III copper-containing protein family.8 However, despite close homologies with Hc and Ty, the type III canonical active site present in such lepidopteran hexamerins does not have the capacity to bind copper ions. Surprisingly, it consists of a combination of aromatic/hydrophobic and charged residues instead of histidine residues present in Hc/Ty. However, X-ray fluorescence spectroscopy at the Cu-absorption edge indicated the presence of 6 copper ions per subunit in lepidopteran hexamerins and the 3D reconstructions revealed the presence of several different metal ion binding sites. The diverse location of metal sites, sometimes present at subunit interfaces, could have a structural function, but it cannot be excluded that the PE-modifying activity of these proteins is assisted by any of these metals. The results are still quite vague, and the occurrence of copper ions needs to be confirmed. However, this work must receive our full attention considering that plastic waste management is a pressing ecological, societal, and economical challenge.
In this constantly evolving context where knowledge of biological systems is refined over the years, the design of model complexes of the different active sites is necessary to contribute to this progress. From these studies, a clear emphasis on the chemistry of dioxygen and/or hydrogen peroxide at mono, bi or trinuclear copper centers en route to produce CuII-oxyl or (μ-oxido)CuIICuIII ↔ (μ-oxyl)CuIICuII active species is more relevant than ever. This review focuses on this subject.
2.2. Thermodynamic and kinetic aspects of C–H bond abstraction
Evaluation of the intrinsic mechanisms occurring in HAA with the afore-mentioned enzymes is obviously extremely difficult due to the transient character of the copper–oxygen adducts generated along the catalytic cycle. Nevertheless, synthetic models of copper enzymes, although generally less effective than their natural counterparts, have been extensively studied since they afford better understanding of oxygen activation through a fine characterization of the intermediates, mostly by using low-temperature conditions. In recent years, a common strategy has been targeted to evaluate reaction thermodynamics and kinetics related to Proton-Coupled Electron Transfer (PCET) processes to obtain better insights into the intrinsic properties of the catalysts. In particular, the bond dissociation free energy (BDFE) of the O–H bond of hydrogenated copper–oxygen adducts is considered as a strong indicator of the oxidizing HAA power of the catalyst, although it provides no direct information on the stepwise/concerted pathways for H-atom transfer and the related kinetics.65BDFEs for solution-phase PCET processes are usually determined by using a square scheme involving two parallel ET and PT processes, where XH denotes the hydrogenated copper–oxygen catalyst (Scheme 4).66,67
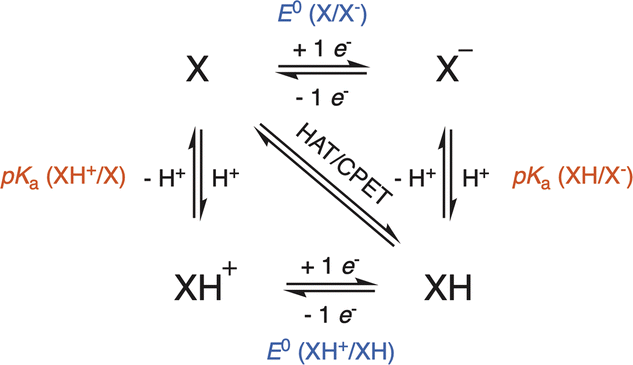 | ||
| Scheme 4 Square-scheme mechanism for the HAA reaction: concerted (HAT/CPET) vs. stepwise (ET/PT) pathways. X is the reactive copper–oxygen species and XH stands for the hydrogenated copper–oxygen species. | ||
According to this scheme, the BDFE of the X–H bond can be obtained by considering stepwise electron transfer (ET) and proton transfer (PT) processes. Indeed, independent determination of pKa and one-electron reduction standard potential (E0 in V) values related to ET–PT or PT–ET pathways yields BDFE according to eqn (1):66,67
BDFE = 23.06![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) E0 + 1.37pKa + CG E0 + 1.37pKa + CG | (1) |
Given the high uncertainty on CG, Mayer and co-workers have recently proposed another general expression for BDFE (see eqn (2)) that includes the potential of hydrogenation E0 (V vs. H2) formally related to the PCET conversion of X into XH involving H2, as well as the free enthalpy of formation of H˙:66,68
| BDFE = 23.06E0 (V vs. H2) + ΔG0f(H˙) | (2) |
According to Mayer, this alternative approach is of interest since (i) E0 (V vs. H2) is easily accessible from open-circuit potential (OCP) measurements, and (ii) ΔG0f(H˙) is almost constant (52.2 +/− 0.6 kcal mol−1) for a wide range of commonly used solvents, in contrast to CG. As a consequence, the solution-phase measured values of E0 (V vs. H2) are considered as equivalent to BDFE ones. In practice, OCP measurements require the presence in solution of known concentrations of X, XH, acid/base buffer and electrolyte species.66 Although it was shown to be fully reliable for stable X/XH systems, it is more difficult to use with highly unstable X/XH species such as those encountered with copper–oxygen adducts.
Beyond thermochemical considerations for PCET processes, supplementary information can be obtained by different kinetic analyses and/or computational studies. Various experimental approaches are possible to decipher the concerted or stepwise character of the studied reaction. According to the general definition adopted in recent years by chemists working in the field of PCET reactions, two classes of concerted HAA processes are usually considered, namely CPET (also coined as cPCET) and HAT. The difference between concerted proton–electron transfer (CPET) and H-atom transfer (HAT) relies on the locus of the electron and proton transfer, i.e. on the same orbital (HAT) or on different orbitals (CPET).67 For these two mechanisms, high-energy intermediates are precluded in strong contrast to elementary ET and PT stepwise pathways. Interestingly, in-between situations which were referred to as concerted asynchronous HAA reactions have been recently proposed on the basis of computational studies.69–72 Hence, a basic asynchronous mechanism may develop a PT-like character in the transition-state whereas an oxidatively asynchronous process would feature an ET-like character.
The rate constant k of an HAA reaction is classically determined experimentally from the UV-Vis monitoring of either the catalyst or substrate spectroscopic signal throughout the reaction.65,73,74 For instance, the determination of k for various concentrations of hydrogenated and deuterated substrates allows the determination of a Kinetic Isotope Effect (KIE) value, suggesting either a stepwise (ET–PT/PT–ET) process if the KIE is close to 1, or a concerted (HAT/CPET) reaction if KIE > 1. Other approaches like Hammett plots (lnk vs. Hammett coefficients of substrates), Marcus plots (lnk vs. E0(substrate)) and Bell–Evans–Polanyi plots (Gibbs free energy of activation ΔG* vs. BDFE(substrate)) have been also used to obtain mechanistic information.73 Hence, the rate constant for a concerted PCET process is expected to increase as the BDFE of the substrate decreases, as classically found for KIE (H/D) analysis. In contrast, a stepwise PCET featuring a PT rate-determining step shows an increase of k with the BDFE of the substrate. In addition to these experimental approaches, recent computational studies have proposed to differentiate between HAT and CPET processes on the basis of the variation of the dipolar moment of the catalyst–substrate adduct bond throughout the HAA reaction.72
Overall, quantification of thermodynamics and kinetics of HAA reactions involving copper–oxygen species has been extensively standardized over the last few years, allowing direct comparison between the different families of copper catalysts. With the support of computational studies, such approach gives more insights into the mechanisms and helps in the rationalization of the oxidative properties in relation to the structure of the active species.
3 Mononuclear copper–oxygen species
3.1. Cu/O2 entities: copper-superoxido or copper-peroxido adducts
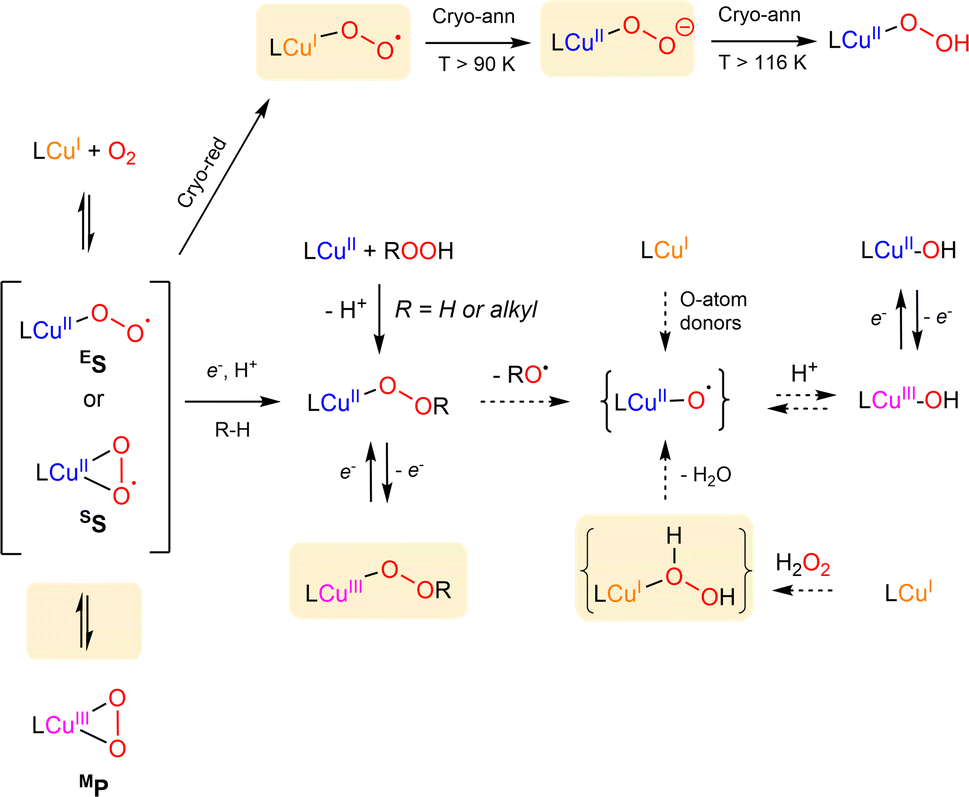 | ||
| Scheme 5 Summary of known formation and interconversion pathways of mononuclear copper–oxygen entities. Recently isolated species or pathways are highlighted in orange. Dotted arrows correspond to postulated conversions pathways. The nomenclature used is derived from Stack et al.2 | ||
In 2017, only four Cu/O2 entities had been structurally characterized.78–81 Two additional structures were since reported by the group of Betley82,83 using sterically encumbered dipyrrin ligands (ArL or EMindL, with Ar = 5-mesityl-1,9-(2,4,6-Ph3-C6H2) and EMind = 1,1,7,7-tetraethyl-1,2,3,5,6,7-hexahydro-3,3,5,5-tetramethyl-s-indacene, Chart 1). In both cases, the dioxygen derived ligand is bound in the η2 mode. In particular, high-quality single crystals were obtained by exposing single crystals of the (EMindL)Cu(N2) precursor to air. The reaction proceeded in the solid state, reflecting that replacement of N2 by O2 induces minimal changes in lattice parameters (Fig. 6a).83 The molecular structure of (EMindL)Cu(O2) revealed a square-planar geometry around the copper center with relatively short Cu–O bonds of 1.824 and 1.834 Å, together with an O–O distance of 1.379 Å. The resonance Raman spectrum of the intermediate showed the presence of a ν(O–O) vibration at 1003 cm−1. These structural and spectroscopic data, combined with DFT calculations, led to the assignment to a predominant SS description of the two intermediates.
 | ||
| Fig. 6 Selected crystallographic structures of recently described mononuclear copper–oxygen adducts. a) η2-superoxido based on the EMindL ligand.83 Hydrogen atoms were omitted for clarity; b) The anion [(L)Cu(OOCm)]− based on the bis(arylcarboxamido)-pyridine ligand.85 Hydrogen atoms and the Et4N+ counter ion were omitted for clarity; c) η1-Hydroperoxido complex in an artificial enzyme based on a streptavidin scaffold: [(biot-et-dpea)CuII(OOH)(OH2)]+⊂Sav;86 d) High-valent [(L)CuOH] species based on the bis(arylcarboxamido)-pyridine ligand (W as deprotonated water ligand).87 | ||
Although no crystallographic structures could be obtained, the groups of Karlin and Hoffman have recently reported on new types of copper–oxygen intermediates using tmpa and tren-derived ligands (L = PVtmpa, DMMtmpa, TMG3tren, Chart 1).84 The well-known ES [(L)CuII(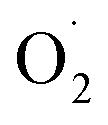 )]+ species were generated and cryo-reduced by γ-irradiation at 77 K using a 60Co source. This allows the generation of energetic mobile electrons through solvent ionization processes. All primary cryo-reduced species display similar axial Q-band and X-band EPR spectra with g∥ = 2.52 and g⊥ ≈ 1.95. Detailed spectroscopic analysis led to the proposal that [(L)CuI(η1-
)]+ species were generated and cryo-reduced by γ-irradiation at 77 K using a 60Co source. This allows the generation of energetic mobile electrons through solvent ionization processes. All primary cryo-reduced species display similar axial Q-band and X-band EPR spectra with g∥ = 2.52 and g⊥ ≈ 1.95. Detailed spectroscopic analysis led to the proposal that [(L)CuI(η1-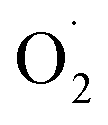 )] species are formed upon reduction of the copper ions, which are trapped in the geometry of the parent complexes (Scheme 5).
)] species are formed upon reduction of the copper ions, which are trapped in the geometry of the parent complexes (Scheme 5).
To the best of our knowledge, this corresponds to the first isolation of such species in the literature. Warming of the frozen solution of [(DMMtmpa)CuI(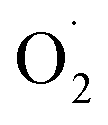 )] at 90 K for 5 minutes (a temperature below the MeTHF solvent melting point) led to the change of the EPR signature that was assigned to the formation of the [(DMMtmpa)CuII(η1-O2−)] intermediate resulting from internal electron transfer between the copper center and the superoxido ligand (Scheme 5). Although already crystallographically detected in metalloenzymes (e.g. LPMO88), such η1-Cu(II)-peroxido species was never evidenced in model complexes. Finally, warming at a temperature above the solvent melting point allowed protonation of the terminal peroxido ligand to provide the η1-hydroperoxido [(DMMtmpa)CuII(η1 –OOH)]+ intermediate.
)] at 90 K for 5 minutes (a temperature below the MeTHF solvent melting point) led to the change of the EPR signature that was assigned to the formation of the [(DMMtmpa)CuII(η1-O2−)] intermediate resulting from internal electron transfer between the copper center and the superoxido ligand (Scheme 5). Although already crystallographically detected in metalloenzymes (e.g. LPMO88), such η1-Cu(II)-peroxido species was never evidenced in model complexes. Finally, warming at a temperature above the solvent melting point allowed protonation of the terminal peroxido ligand to provide the η1-hydroperoxido [(DMMtmpa)CuII(η1 –OOH)]+ intermediate.
As previously mentioned, the dioxygen-derived ligand of the primary Cu/O2 adducts is found either in η1 or η2 coordination modes. Yet, using the bulky TIPT3tren ligand (Chart 1), the group of Itoh observed the coexistence of the two binding modes with one single ligand-system.89 Indeed, oxygenation of the Cu(I) precursor at low temperature led to the detection of an ES species together with another intermediate, assigned to the MP adduct based on UV-Vis, Raman, NMR and DFT calculations.89
Finally, using a redox active ligand (DHP = (2,5-bis((2-t-butylhydrazono)(p-tolyl)methyl)-pyrrole, Chart 1), the group of Anderson prepared a diamagnetic CuII–L˙ complex.90 Upon oxygenation at low temperature, an ES intermediate was characterized arising from electron transfer between the ligand and dioxygen.
ES species have been shown to display promising HAA reactivity towards O–H and C–H bonds, leading to the corresponding Cu–OOH species and radical products.5,19,75–77 The group of England prepared bulky tmpa derived ligands (bearing aryl substituents) and successfully stabilized [(Ar3tmpa)CuII(η1- )]+ species that are stable up to −20 °C.92 This allowed extensive reactivity studies using a broad range of substrates bearing O–H, N–H, or C–H bonds. As already observed for other complexes, HAA from C–H bonds proved to be more challenging than O–H and N–H bonds and the intermediates were found to react only with weak C–H bonds (ca. 73 kcal mol−1).
)]+ species that are stable up to −20 °C.92 This allowed extensive reactivity studies using a broad range of substrates bearing O–H, N–H, or C–H bonds. As already observed for other complexes, HAA from C–H bonds proved to be more challenging than O–H and N–H bonds and the intermediates were found to react only with weak C–H bonds (ca. 73 kcal mol−1).
It has been reported that introduction of thioether ligation to the copper, modelling the coordination sphere found in PHM and DβH enzymes, enhances the oxidizing power of η1-Cu(II)-superoxido adducts.93 Although several complexes were prepared with thioether-containing ligands, it has to be noted that the occurrence of thioether ligation to copper was only recently experimentally attested to using EXAFS on the previously reported [(TMGN3S)CuII( )]+ species.94,95
)]+ species.94,95
Second coordination sphere interactions are also known to modulate the reactivity of metal–oxygen species.96 In order to probe the effect of second coordination sphere H-bonding effects on the reactivity of η1 cupric-superoxido species, the group of Karlin has prepared a series of tmpa-based ligands with varying H-bonding ability.97,98 Increasing H-bonding to the superoxido moiety by changing the nature and the number of H-bonding substituents on the tmpa ligand was shown to both decrease the amount of binuclear entities formed to the profit of ES species and increase the electrophilic reactivity of the intermediate towards HAA (Fig. 7). Accordingly, the bis-pivilamido substituted ligand provided the most reactive superoxido intermediate that was able to react with O–H BDEs up to 91.5 kcal mol−1 and C–H BDEs up to 79.7 kcal mol−1, emphasizing the importance of second-coordination sphere elements to modulate the reactivity of copper–oxygen species, as already reported for other reactive intermediates. Very recently, Borovik et al. demonstrated that intramolecular H-bonds, introduced by external ligands, could influence the structural and magnetic properties of the mononuclear complex.99
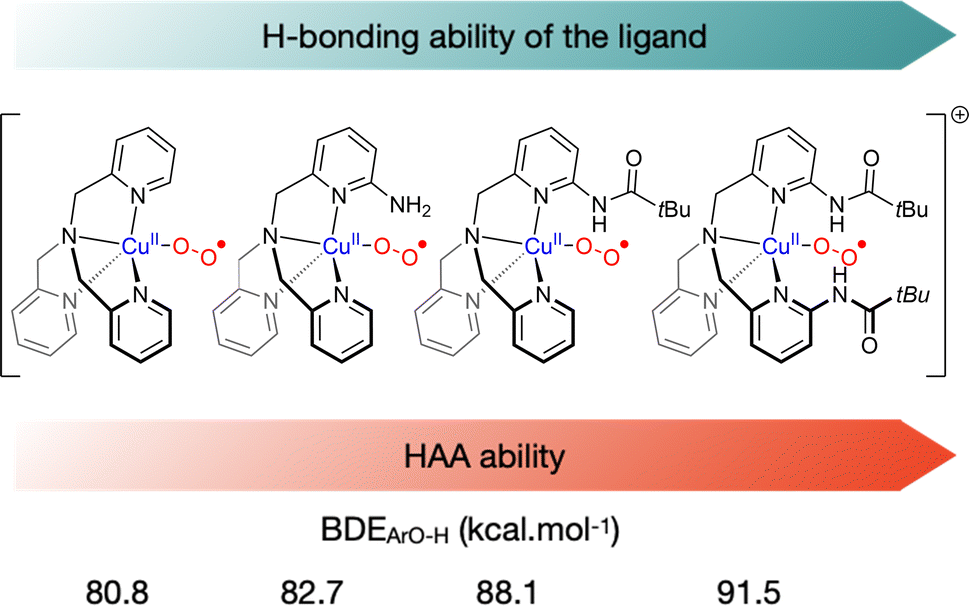 | ||
| Fig. 7 Representation of selected tmpa-based ligands prepared with different H-bonding substituents to modulate their HAA reactivity.97,98 Highest BDEOH from phenolic substrates that react with the superoxido intermediate. | ||
The nucleophilic reactivity of the ES species based on the bis(arylcarboxamido)-pyridine anionic ligand [N,N′-bis(2,6-diisoproylphenyl)-2,6-pyridinedicarboxamido]100 towards the aldehyde deformylation reaction has been reported in the literature.101 The group of Tolman has recently re-investigated the mechanism of this reaction.102 The preparation of the intermediate was improved by the use of a cryptand (Kryptofix® 222) when reacting the Cu(II) precursor with KO2. This allowed additional mechanistic investigations suggesting HAA rather than nucleophilic attack processes.
Following this first study, the reaction of this anionic [(L)CuII(η1- )]− with para-substituted phenols XArOH was investigated at low temperature.103 Interestingly, kinetic data suggested a change in mechanism across the series depending on the nature of the substituents of the phenols. In the case of phenols with low acidities (X = OMe, NMe2) a CPET/HAT mechanism was proposed. The reaction rates were significantly lower than that obtained with the high-valent [(L)CuIII(OH)] core containing the same ligand.65 The O–H BDE value for the [(L)CuIIOOH]− species was estimated to be ∼3 kcal mol−1 lower than that for [(L)CuII(OH2)]2−, consistent with the higher PCET/HAT rates of [(L)CuIII(OH)] species as compared to [(L)CuII(η1-
)]− with para-substituted phenols XArOH was investigated at low temperature.103 Interestingly, kinetic data suggested a change in mechanism across the series depending on the nature of the substituents of the phenols. In the case of phenols with low acidities (X = OMe, NMe2) a CPET/HAT mechanism was proposed. The reaction rates were significantly lower than that obtained with the high-valent [(L)CuIII(OH)] core containing the same ligand.65 The O–H BDE value for the [(L)CuIIOOH]− species was estimated to be ∼3 kcal mol−1 lower than that for [(L)CuII(OH2)]2−, consistent with the higher PCET/HAT rates of [(L)CuIII(OH)] species as compared to [(L)CuII(η1- )]− (Scheme 6). In the case of electron-deficient and acidic phenols (X = NO2, CF3, Cl), the results were consistent with a rate-determining proton transfer (PT) from the phenol to the [(L)CuII(
)]− (Scheme 6). In the case of electron-deficient and acidic phenols (X = NO2, CF3, Cl), the results were consistent with a rate-determining proton transfer (PT) from the phenol to the [(L)CuII( )]− species to yield the formally CuIII–OOH adduct. The enhanced basicity of [(L)CuII(
)]− species to yield the formally CuIII–OOH adduct. The enhanced basicity of [(L)CuII( )]− as compared to [(L)CuIII(OH)]+ (pKa ∼19 vs. 12 respectively) likely favors PT from acidic phenols while for the least acidic ones a CPET/HAT mechanism is followed. Finally, using the ligand Lpym (Chart 1), the group of Itoh has observed an unprecedented reactivity of the corresponding SE intermediate towards aldehydes and ketones, leading to C–C bond formation with acetone (the reaction solvent), and to the corresponding β-hydroxy-ketones (aldols).104 Nucleophilic reactivity was supported by detailed kinetic studies and DFT calculations. A Cu-alkylperoxido species was transiently detected and characterized by rR spectroscopy, which is in agreement with nucleophilic addition of the Cu(II)-superoxido species on the carbonyl carbon of the substrate.
)]− as compared to [(L)CuIII(OH)]+ (pKa ∼19 vs. 12 respectively) likely favors PT from acidic phenols while for the least acidic ones a CPET/HAT mechanism is followed. Finally, using the ligand Lpym (Chart 1), the group of Itoh has observed an unprecedented reactivity of the corresponding SE intermediate towards aldehydes and ketones, leading to C–C bond formation with acetone (the reaction solvent), and to the corresponding β-hydroxy-ketones (aldols).104 Nucleophilic reactivity was supported by detailed kinetic studies and DFT calculations. A Cu-alkylperoxido species was transiently detected and characterized by rR spectroscopy, which is in agreement with nucleophilic addition of the Cu(II)-superoxido species on the carbonyl carbon of the substrate.
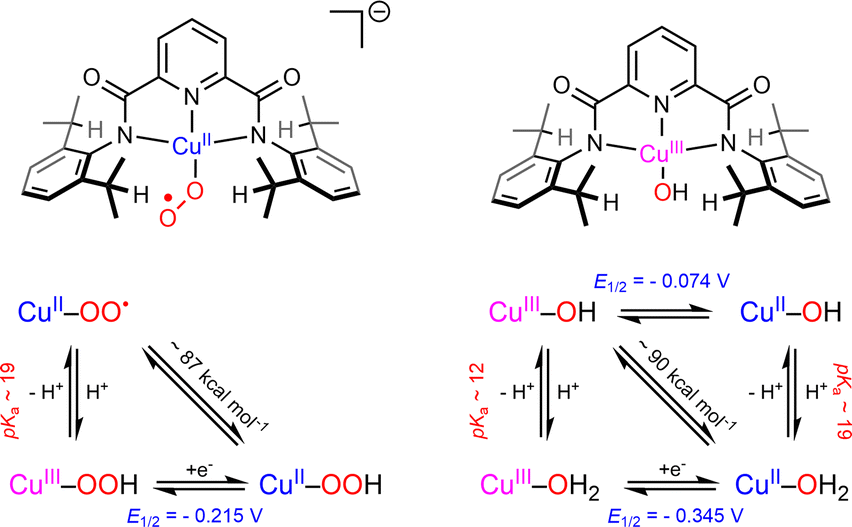 | ||
Scheme 6 Comparison of the thermodynamic properties of [(L)Cu(II)(η1- )]− and [(L)CuIII(OH)] using the same bis(arylcarboxamido)-pyridine ligand. Adapted from ref. 103. )]− and [(L)CuIII(OH)] using the same bis(arylcarboxamido)-pyridine ligand. Adapted from ref. 103. | ||
The reactivity of η2-Cu/O2 species for HAA is less documented. The group of Betley has studied the reactivity of two η2-Cu/O2 based on dipyrrin ligands.82,83 In the case of the ArL ligand, although thermally stable at room temperature, the intermediate reacted with 1,4-cyclohexadiene and was able to perform HAA and acid/base chemistry towards a range of phenolic substrates as well as O-atom transfer to triphenylphosphine. Although the mechanistic features remain unclear, it was proposed that an end-on ES could be formed prior to reaction with substrates. The intermediate [(EMindL)CuII(η2-O2)] reacted with a stoichiometric amount of 1,2-diphenylhydrazine to provide azoarene and H2O2 as well as the initial [(EMindL)CuII(η2-O2)] species. When the experiments were conducted under an inert atmosphere, a new diamagnetic species was detected and assigned to [(EMindL)CuI(H2O2)] (Scheme 7). The same species could also be produced upon reaction of the initial [(EMindL)CuI(N2)] species with dry H2O2 ((Ph3PO)2(H2O2))105 supporting the assignment as a Cu(I)–(H2O2) species. Such intermediate has been proposed to be pivotal in the LPMO catalytic cycle both in monooxygenase and peroxygenase pathways.55 Further understanding of its structural and reactivity properties would therefore be of high interest.
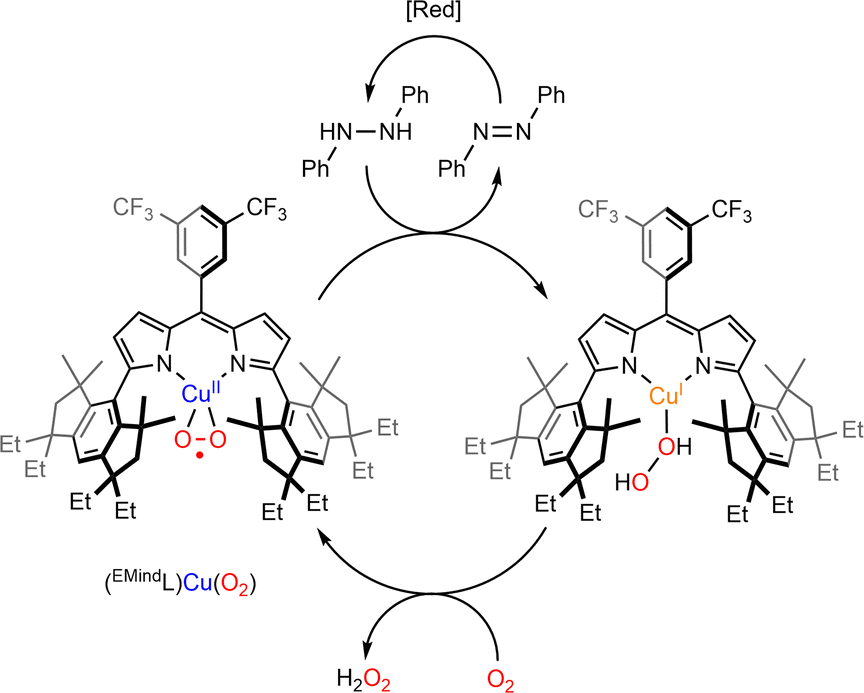 | ||
| Scheme 7 Reaction of [(EMindL)CuII(O2)] with 1,2-diphenylhydrazine to produce azoarene and [(EMindL)CuII(H2O2)]. Adapted from ref. 83. | ||
3.2. CuOOR entities: copper-hydroperoxido or alkylperoxido adducts
 )]+ precursor on a substrate to generate the corresponding [CuII(η1-OOH)]+ species (many examples were reported using TEMPOH as a hydrogen atom donor) or (ii) a “peroxide-shunt” approach, i.e. treatment of a Cu(II) precursor (or Cu(I)) by H2O2 or ROOH (in the presence or absence of a base) yielding the corresponding [CuII(η1-OOR)]+ species. Several new examples were recently prepared following these strategies.85,92,94,107–111 It has to be noted that a novel route involving cryoreduction of a superoxide [CuII(η1-
)]+ precursor on a substrate to generate the corresponding [CuII(η1-OOH)]+ species (many examples were reported using TEMPOH as a hydrogen atom donor) or (ii) a “peroxide-shunt” approach, i.e. treatment of a Cu(II) precursor (or Cu(I)) by H2O2 or ROOH (in the presence or absence of a base) yielding the corresponding [CuII(η1-OOR)]+ species. Several new examples were recently prepared following these strategies.85,92,94,107–111 It has to be noted that a novel route involving cryoreduction of a superoxide [CuII(η1- )]+ precursor followed by annealing has recently been described by the group of Karlin (Scheme 5).84 This route has also allowed the isolation of unprecedented CuII-η1-peroxido species.
)]+ precursor followed by annealing has recently been described by the group of Karlin (Scheme 5).84 This route has also allowed the isolation of unprecedented CuII-η1-peroxido species.
Copper(II)-hydro(alkyl)peroxido intermediates that have been isolated in model systems all display an η1 coordination mode. They are characterized by intense UV-Vis features at 350 nm assigned to peroxide-to-copper LMCT as well as EPR signatures characteristic of copper(II) species. Two characteristic vibration modes are generally detected in their rRaman spectra: ν(O–O) ∼850 cm−1 and ν(C–O) ∼550 cm−1. In 2017, only two [CuII(η1-OOR)]+ species (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H or cumyl, Cm) had been structurally characterized.112,113 Three additional structures were recently reported.85–87
H or cumyl, Cm) had been structurally characterized.112,113 Three additional structures were recently reported.85–87
The group of Tolman has used the dianionic bis(arylcarboxamido)-pyridine ligand (Chart 1) and reacted the [(L)CuIIOH]− precursor with excess ROOH (with R = tBu or Cm).12 The corresponding [(L)CuII(OOR)](Et4N) complexes were successfully crystallized (Fig. 6b for R = Cm).87 These correspond to the only additional examples of copper-alkylperoxide species structurally characterized after the first structure reported in 1993. The intermediates display O–O bond distances of 1.416 and 1.468 Å for R = tBu and Cm respectively, which agree well with that of the already reported Cu-OOCm intermediate (1.460 Å).112 Interestingly, cyclic voltammetry measurements of the two intermediates revealed quasi-reversible processes at E1/2 = −150 and −200 mV vs. Fc+/Fc, respectively. Chemical oxidations led to the isolation of one-electron oxidized species, formally Cu(III)-alkylperoxido species. These intermediates are EPR silent and display intense absorptions in the visible region (500–700 nm). Resonance Raman experiments revealed a ν(O–O) = 831 cm−1 sensitive to isotopic labeling with 18O. The exact electronic description (either CuIII-alkylperoxido, or antiferromagnetically coupled singlet ground-state CuII-ligand radical or ferromagnetically coupled triplet CuII-ligand radical) could not be unambiguously confirmed using DFT calculations. These formally CuIII-alkylperoxido species did not show any reactivity towards weak O–H or C–H bonds. They were however found to react with 4-dimethylamino-phenol (NMe2ArOH) yielding a Cu(II)-phenoxyl radical product, via possible PCET or ET/PT pathways.
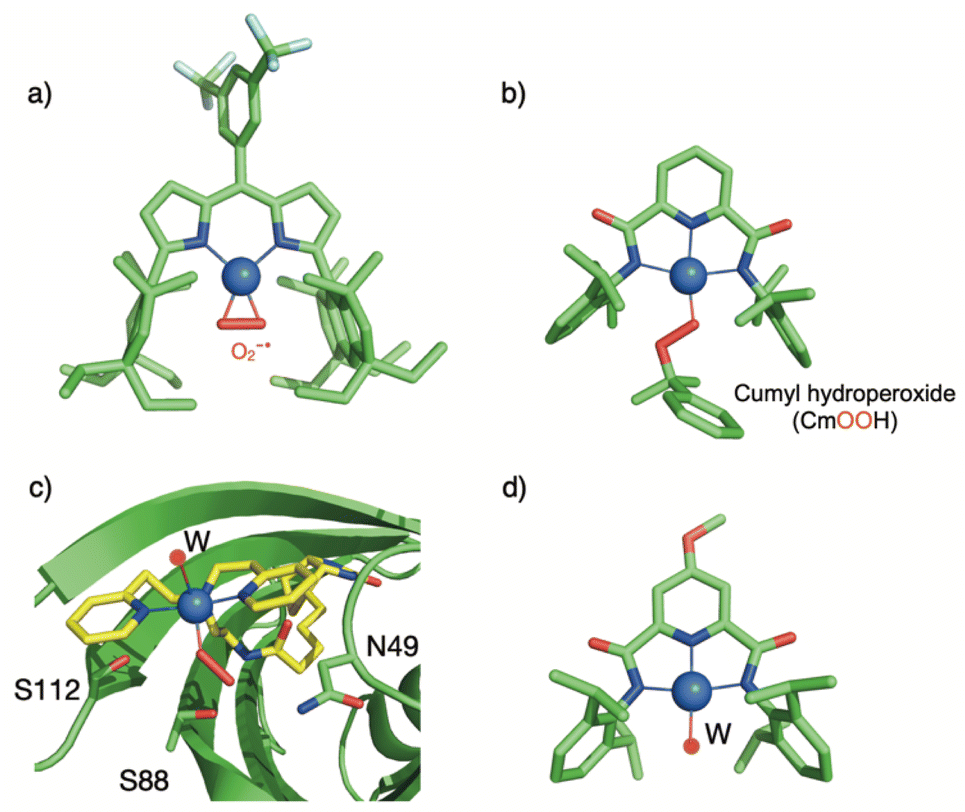
Using the biotin-streptavidin strategy used to develop artificial metalloenzymes,114 the group of Borovik have anchored a biotinylated dpea (dipyridine ethylamine) ligand into a wild-type (WT) streptavidin (Sav) scaffold ([biot-et-dpea) CuII(H2O)2]2+⊂Sav (with an ethyl spacer between biotin and dpea).115 Addition of hydrogen peroxide to a buffered solution of the Cu(II) artificial enzyme led to the formation of a [(biot-et-dpea)CuII(OOH)(OH2)]+⊂Sav intermediate, persistent for one day at room temperature.86 The intermediate was also generated by incubating crystals of the Cu(II) precursor with hydrogen peroxide, which allowed the obtention of crystallographic data at 1.62 Å resolution (Fig. 6c). The copper is found in a 5-coordinated distorted trigonal bipyramidal geometry with 3 nitrogen atoms from the dpea ligand, a water molecule and an end-on hydroperoxido ligand. The Cu–O distance is found to be equal to 1.94 Å and the O–O distance is equal to 1.52 Å. The structure reveals that the streptavidin scaffold provides a H-bonding network to both the distal and the proximal O-atoms of the hydroperoxido ligand, H-bonded to asparagine residue N49 or to a water molecule stabilized by an S112 residue respectively. Remarkably, this structure represents the second crystallographic data obtained on Cu(II)–OOH species. Mutations of serine (S112) or asparagine (N89) into alanine were performed to evaluate the effect of H-bonding disruption on the stability of the intermediate. In the case of N89A, no drastic difference in stability was noted. In contrast, mutation of S122 into alanine resulted into drastic loss of stability of the intermediate. Although no crystal structure of the CuII–OOH species could be obtained, the structure of the ([biot-et-dpea) CuII(H2O)2]2+⊂Sav S112A precursor indicated that the occupancy of the targeted water molecule was reduced by 50%. In addition, the Cu complex was no longer located into a single position but over two positions, consistent with the importance of H-bonding interactions to govern the position of the complex within the Sav cavity. The reactivity of the different copper-hydroperoxido species confined within the native and mutant sav scaffolds was evaluated using 4-chlorobenzylamine as substrate. The copper-hydroperoxido species confined within the native and N49A Sav scaffolds were not active. In contrast, ([biot-et-dpea) CuII(H2O)(OOH)]2+⊂Sav S112A was found to react with the substrate. These results nicely corroborate the known effects of H-bond interactions to either the proximal or distal atoms of the OOH ligand on the properties of Cu(II)–OOH species.116–118
In most cases, the Cu-OOR intermediates are found in square-pyramidal or bipyramidal geometries. Interestingly, the groups of Itoh and Comba have isolated a Cu-OOCm intermediate using a rigid and bulky tridentate ligand, TMG3tach (Chart 1), that imposes an unusual tetrahedral geometry to the copper.110 The intermediate was prepared by reacting the methoxide precursor [(TMG3tach)CuII(OMe)]+ with excess CmOOH. Its spectral properties were highly similar to those of alkylperoxido intermediates with different geometries.
Finally, although many Cu-OOR(H) species have been isolated and characterized, very few Cu-acylperoxido species have been studied.119–121 Using a ligand based on a rigid 6-membered cyclic diamine with two CH2Py side arms (L6pym2, Chart 1), the group of Itoh has recently isolated a Cu(II)-acylperoxido intermediate upon reaction of Cu(II) complexes with mCPBA.122 The purple complex [(L6pym2)CuII(OOCOAr)]+ was found to be stable at 30 °C. Using similar ligands but with 7- or 8-membered rings, the adducts were unstable and converted to the corresponding m-chlorobenzoic acid (m-CBA) adducts.
Using the ligand CHDAP (N,N′-dicyclohexyl-2,11-diaza[3,3](2,6)pyridinophane) the group of Cho prepared two [(CHDAP)CuII(OOR)]+ intermediates (with R = C(CH3)2Ph and tBu) that were characterized using UV-Vis, EPR, resonance Raman and mass spectrometry.107 Self-decay of the intermediates at 25 °C followed first-order kinetics and was shown to involve O–O bond homolysis. The stability of the intermediates was not affected by the addition of thioanisole or cyclohexadiene, consistent with the fact that they are poorly/not reactive in electrophilic C–H bond activation. In contrast, reaction with aldehydes and benzoyl chlorides was observed and a mechanism involving nucleophilic attack of the alkylperoxido ligand on the carbonyl groups was proposed. A few years later, the same group showed that a copper-hydroperoxido intermediate, [(iPr3tren)CuII(OOH)]+, was also capable of nucleophilic reactivity with acyl chlorides and with aldehydes for deformylation reactions.108 In the case of aldehyde deformylation, two routes were discussed that involve a nucleophilic attack of either the proximal or the distal oxygen from the intermediate. These two studies highlight new properties of CuOOR species that will need to be further explored and understood. Aromatic hydroxylation by Cu–OOH species was also reported by the groups of Mayilmurugan and Ghosh.129,130
In the catalytic cycle of LPMO, it has been suggested that the Cu(I)–(H2O2) intermediate might be involved (Scheme 3).55 Although already suggested as possible intermediates in oxidation reactions,131,132 such species have only been recently detected in a model complex (Scheme 2).83
Recently, the groups of Karlin and Solomon have provided experimental evidence that such a peroxygenase pathway can be efficient in bioinspired model chemistry.133 The complex [(TMG3tren)CuI]+ was treated with a sub-stoichiometric amount of dry H2O2 yielding the hydroxylated TMG3tren-OH ligand. Interestingly, no Cu(II) was detected at the end of the reaction suggesting a Cu(I) redox state of the complex. When hydroxyl radical scavengers were added, ligand hydroxylation was cancelled supporting the involvement of hydroxyl radicals and Fenton-like pathways. In the presence of a slight excess (3 equivalents) of H2O2, overoxidation occurred and the alkoxide complex [(TMG3tren-O)CuII]+ resulting from ligand hydroxylation was isolated. Overall, experimental results pointed towards a “peroxygenase-like” mechanism with the involvement of controlled Fenton pathways for the hydroxylation of adequately positioned C–H bonds (Scheme 8).
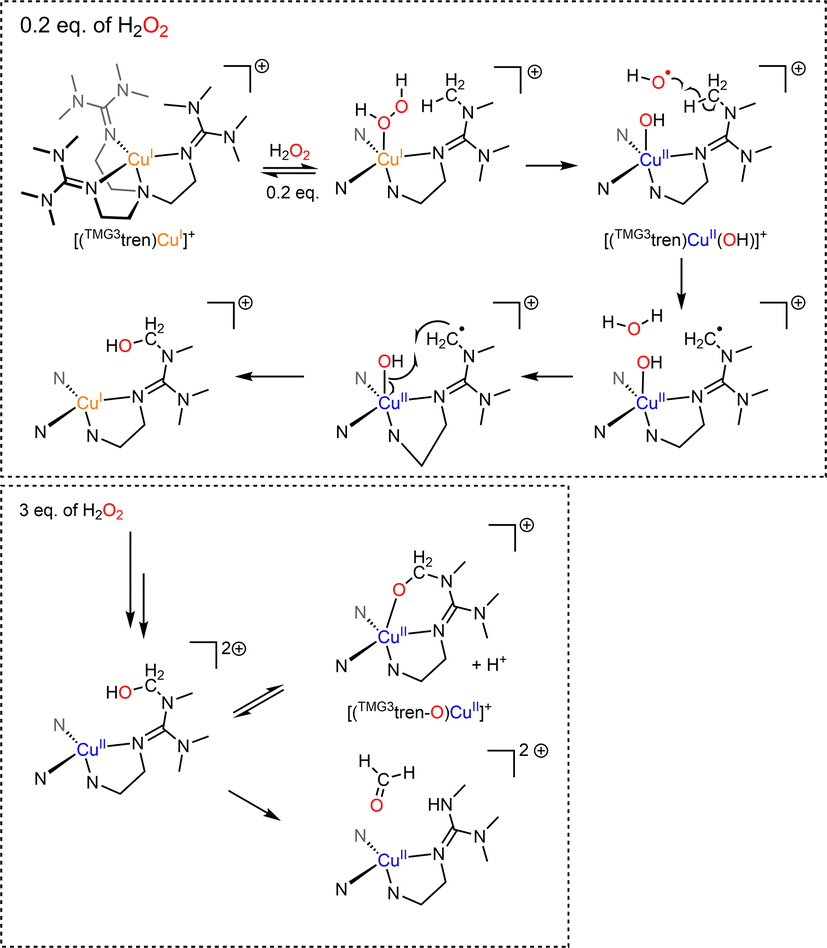 | ||
| Scheme 8 Reaction of [(TMG3tren)CuI]+ with varying amounts of H2O2. Adapted from ref. 133. | ||
Similar conclusions were obtained by the group Garcia-Bosch using bppa, a tmpa based ligand (noted LH2 by the authors) that possesses two H-bond donors on the pyridine ligands.124 The authors have investigated the reactivity of mononuclear Cu complexes with O2 and H2O2. [(LH2)CuI]+ was reacted with dioxygen in acetonitrile at −35 °C and, in the presence of TEMPOH, full conversion to [(LH2)CuII(OOH)]+ species was achieved. Addition of triflic acid led to the release of H2O2 together with [(LH2)CuII]+. This reactivity was discussed as relevant for the production of hydrogen peroxide from dioxygen by LPMO. Upon reaction of [(LH2)CuI]+ with H2O2, [(LH2)CuII(OH)]+ was produced which then evolved towards [(LH2)CuII(OOH)]+ when excess H2O2 was used. Hydroxyl radical formation was also evidenced. The chemistry in play here seems therefore highly similar to the one described above133 and could be of relevance for LPMO peroxygenase pathway.
3.3. Copper-oxyl and high-valent copper oxygen species
High-valent Cu cores have been proposed as a key species in the catalytic cycle of Cu-containing monooxygenases and in synthetic oxidation catalysts. Increasing number of formally Cu(III) complexes, mainly organometallic ones, are described in the literature, although the existence of Cu(III) and the exact redox state of the metal center is questioned.72,87,134–143 Most of these species are beyond the scope of this review which focuses on copper–oxygen species for C–H bond activation. Two main species will therefore be discussed here: [CuO]+ and the protonated [CuOH]2+ cores.[CuO]+ cores are often postulated in the catalytic cycles of copper-containing monooxygenases and synthetic model complexes for the activation of strong C–H bonds such as those of polysaccharides or methane.5,55,140,144 Calculations support the electronic description as triplet ground state Cu(II)-oxyl species ([CuIIO˙]+). Based on the biradical character as well as the predicted weak Cu–O bond, they are expected to be highly reactive, probably explaining the lack of experimental evidence gathered on this reactive species. However, beyond numerous computational studies, the only data on the reactivity of such high-valent species came from gas-phase reactions, confirming their ability to perform C–H abstraction on alkanes, including methane.144–147 Attempts to isolate [CuO]+ cores in solution upon reaction of a Cu(I) precursor with oxo transfer reagents such as pyridinium N-oxides or PhIO (Scheme 5) remained unsuccessful (with the exception of one ESI-MS data set).148 Following this strategy, the group of Karlin has recently reacted several Cu(I) complexes with para-substituted N,N-dimethylaniline N-oxides (DMAO) as O-atom donors. The reported results pointed towards the involvement of reactive copper-oxyl species, able to hydroxylate the strong C–H bonds (∼90 kcal mol−1) of the N,N-dimethylaniline product generated after O-atom transfer reactions to the Cu(I) precursors.98
The suggestions that [CuOH]2+ cores could be of biological relevance came from the work of the group of Tolman.5,140 Using electron donating bis(arylcarboxamido) ligands, [CuOH]2+ species were generated upon one-electron oxidation of the corresponding Cu(II) precursors and a Cu(III)–OH formulation was suggested based on spectroscopic data and DFT calculations. These intermediates were shown to react with strong C–H bonds (up to 99 kcal mol−1 for cyclohexane).65 This strong C–H bond activation ability was correlated with the strong basicity of the Cu(II)–OH precursors compensating the weak oxidizing power of the [CuOH]2+ core leading to a strong O–H BDE of the Cu(II)–(OH2) species and thus to a strong thermodynamic driving force to attack energetic C–H bonds (Scheme 6).140,149 The group of Cramer computationally rationalized the effect of various factors on the reactivity of such high valent [CuOH]2+ cores, including ligand modifications, counter ions, van der Waals interactions and quantum-mechanical tunneling effects.150 Based on their oxidative power, it has been anticipated that such species could be involved in the catalytic cycle of LPMO, possibly stabilized by deprotonation of the terminal primary amine of the histidine brace motif.151 It has to be noted that deprotonation of the terminal amine of the histidine brace has been suggested to occur at high pH based on EPR and UV-Vis data.152 Interestingly, high valent [CuOH]2+ cores were recently postulated in the course of H2O2 splitting by a second-coordination sphere mutant of a fungal LPMO based on DFT calculations.153
Following previously reported work, the group of Tolman has successfully stabilized [(L)Cu(OR)] cores (with RH or CH2CF3) and obtained X-ray structural data using a bis(arylcarboxamido)-pyridine ligand bearing a methoxy substituent on the pyridine moiety (Fig. 6d).87 The two neutral intermediates were found in square-planar geometries with metal–ligand bond distances significantly shortened (average of 0.102 Å) as compared to that found in the Cu(II) precursors, in agreement with previously predicted contractions using EXAFS analyses and DFT calculations. Resonance Raman spectra of [(L)Cu(OR)] complexes displayed a ν(Cu–OR) vibration around 635 cm−1 similar to the ones already reported for known high-valent Cu–OH species using similar ligands. Finally, NMR spectra were consistent with diamagnetic S = 0 ground-states supporting the description as CuIII–OH cores, in agreement with previous proposals.140 This study represents the first structural characterization of a [CuOH]2+ core as well as the first isolation of a CuIII-alkoxide analog.
The groups of Tolman and Cramer have also reported on a complete mechanistic investigation of the C–H and O–H bond abstraction mechanism of three different high valent species using the bis(arylcarboxamido)-pyridine ligand:72 [(L)CuIII(OH)], [(L)CuIII(OOCm)]103 and [(L)CuIII(OCOAr)] a formally Cu(III)-carboxylate species (Ar = meta-chlorophenyl) prepared for the first time in this study. It was observed that the most reactive species among the three was the Cu(III)–OH species. Then the carboxylate complex was found to be more reactive than the alkylperoxido one (mostly unreactive). Detailed DFT calculations indicated that O–H or C–H bond abstraction proceeds with different synchronicity of the CPET/HAT depending on the nature of the intermediate and on the substrate. Although C–H bond abstraction from dihydroanthracene (DHA) by [(L)Cu(OH)] follows a HAT mechanism, O–H bond abstraction from phenol by the same intermediate is best described as a concerted “separated” cPCET with proton and electron transfers involving different acceptor centers (the O and the Cu atoms respectively). In the case of [(L)Cu(OCOAr)], both reactions were found to proceed by oxidative asynchronous cPCET, probably driven by the greater oxidizing power of the copper(III) species and the lower basicity of the carboxylate ligand.
It is worth mentioning that several additional [Cu-Y]+ cores have been isolated and studied using bis(arylcarboxamido)-pyridine ligands (Y = SR, halide, CN, NO2) providing interesting tools to investigate the electronic structures and reactivity of high-valent copper cores.141–143,154,155 In particular, using halides, the group of Zhang has stabilized high-valent [Cu-X]+ species.142 Calculations have suggested significant halide radical characters for all the complexes which are therefore best described as [(L)CuII(X˙)], making these species analogs to copper-oxyl ones. The [(L)CuX] species were able to perform HAA from DHA. In addition, upon heating of [(L)CuX] species in the presence of azobis(isobutyronitrile) (AIBN) as a carbon-centered radical precursor, formation of 2-haloisobutyronitriles was observed demonstrating the ability of these high-valent species to perform radical capture and halogenation. [(L)CuF] was then employed for the direct fluorination of a variety of alkyl substrates with allylic and benzylic C–H bonds as well as α-C–H bonds of ethers in a 2:1 Cu:substrate stoichiometry. These findings were further developed to establish electrocatalytic C–H fluorination of various substrates.155
Finally, the groups of Lancaster and Garcia-Bosch have studied a copper-hydroxide complex based on tridentate redox active ligand (Scheme 9).73 The starting [(tBuL)CuII-OH]2− species was shown to undergo two reversible ligand-centered oxidation processes. The three Cu–OH cores did not show any reactivity with C–H bonds but oxidized phenols featuring relatively weak O–H bonds.
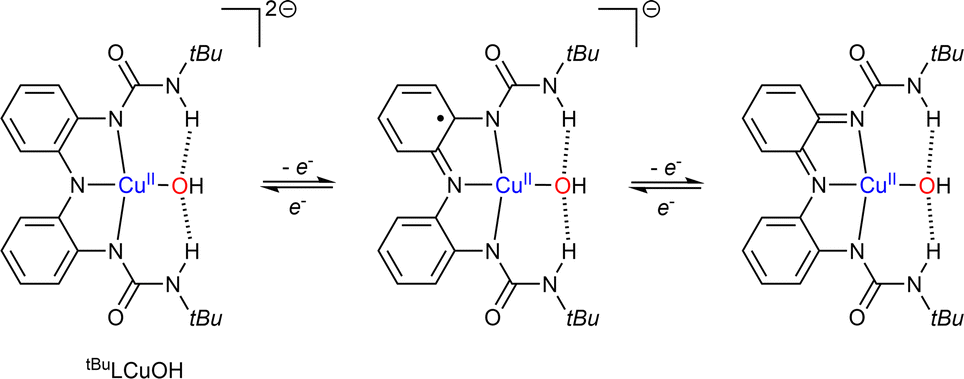 | ||
| Scheme 9 Characterized oxidation states reached via reduction/oxidation of tBuLCuOH. Adapted from ref. 73. | ||
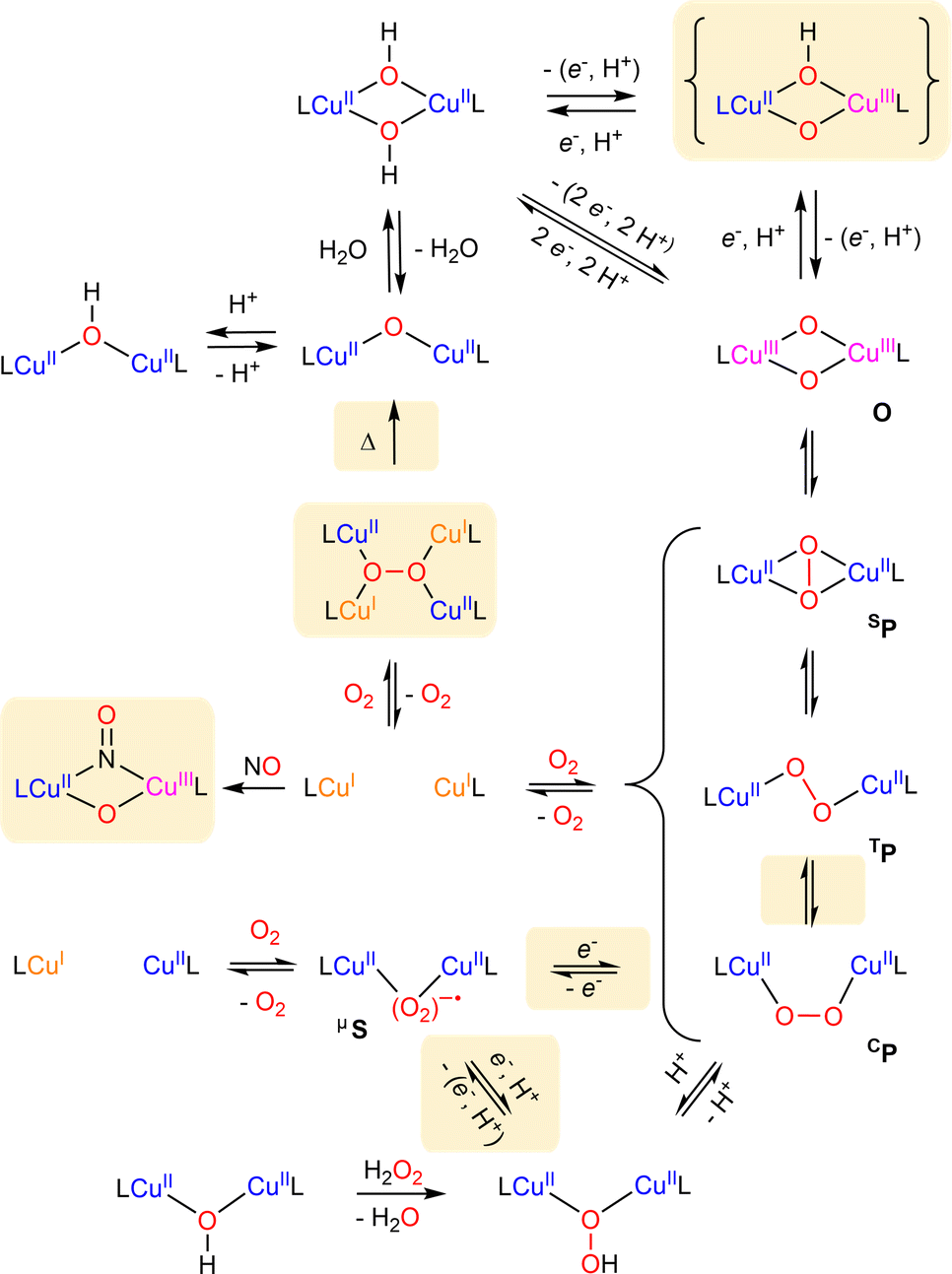 | ||
| Scheme 10 Summary of known formation and interconversion pathways of dinuclear copper–oxygen entities. Recently described species and pathways are highlighted. | ||
4 Dinuclear copper–oxygen species
Since the first structurally characterized trans-peroxido (TP) and μ-η2:η2-peroxido (SP) dicopper(II) species (Scheme 10) by Karlin156 and Kitajima157 in the late eighties, numerous studies related to the characterization and reactivity of dicopper-oxygen adducts in biological and synthetic systems have been reported. Reviews related to dinuclear systems for the 2014–2017 period1,3,5,6,158 have remarkably highlighted various equilibria between isomeric copper–oxygen species, demonstrating how this could impact their oxidative properties. In the following section, we present recent advances in the field of dinuclear Cu2/O2 adducts particularly on isomer interconversion and C–H bond activation.4.1 Peroxido dicopper(II) core
As shown in Scheme 10, peroxido dicopper(II) complexes can be prepared from the oxygenation of either mononuclear or dinuclear copper(I) complexes. As previously reported in many reviews,3,5,6,158,159 various types of dicopper(II) peroxides differing by their configuration can be obtained through subtle variation of controllable parameters (ligand, solvent, temperature…), as described in this section.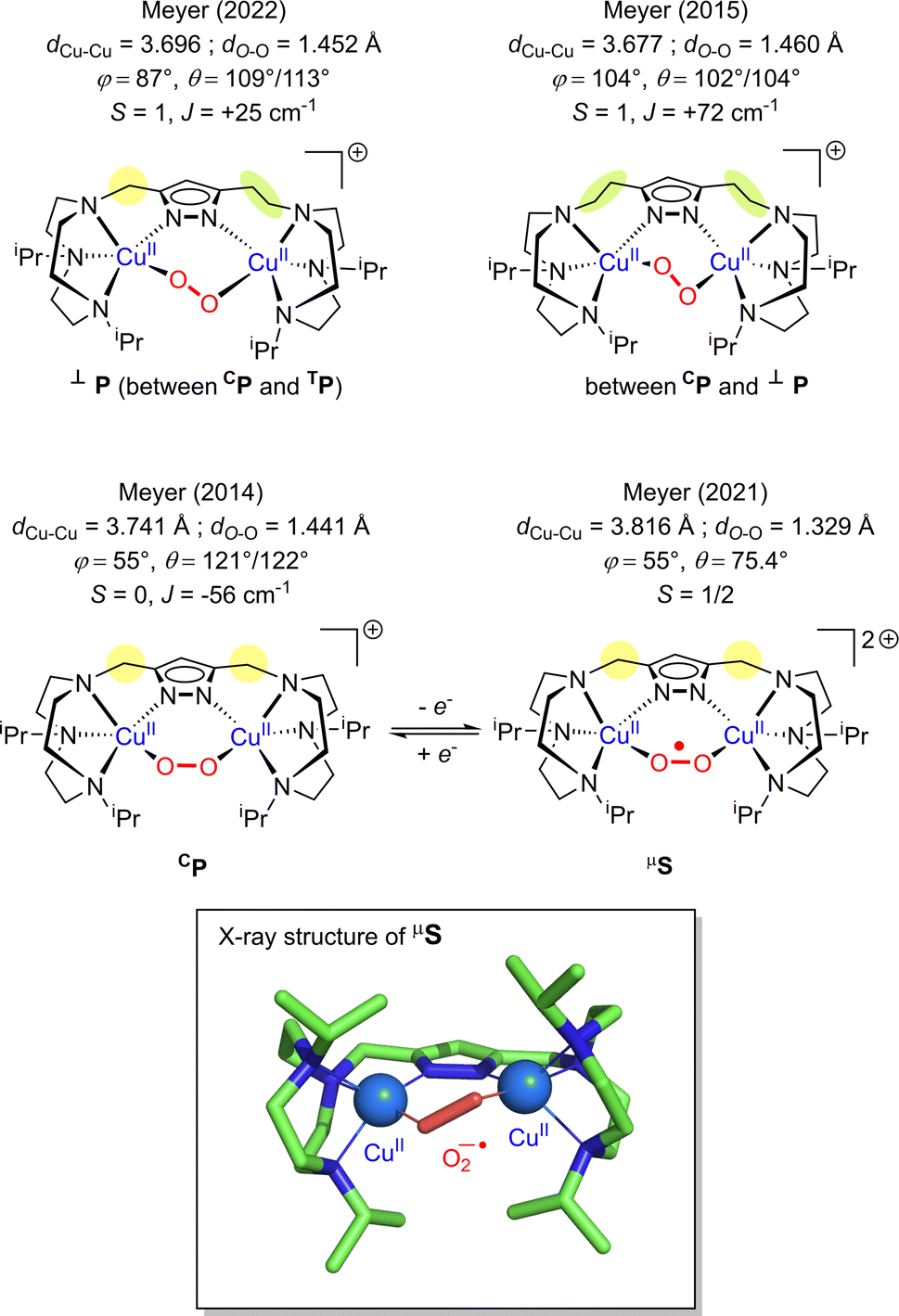 | ||
| Fig. 8 Summary of the structurally characterized dicopper(II) peroxido species with pyrazolato/TACN based ligands from the Meyer group.160–162,167φ represents the torsion angle Cu–O–O–Cu and θ the Cu–O–O angle. | ||
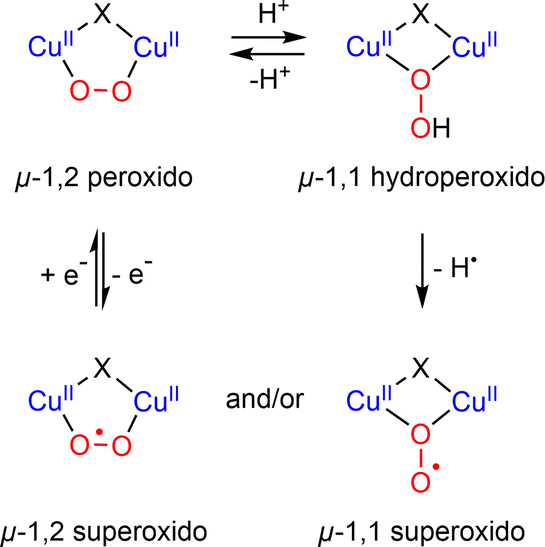 | ||
| Scheme 11 Dinuclear CuII peroxido/superoxido species interconversions. | ||
In 2019, Karlin reported the generation and characterization of two different superoxido dicopper species by chemical oxidation from a μ-1,2 peroxido complex based on the XYLO(H) dinucleating ligand (XYLO(H) = 2,6-bis{[(bis(2-pyridylethyl)amino]methyl}phenol)} (Chart 2 and Table 1).168 A mixture of μ-1,2 and μ-1,1 superoxido species was detected by UV-Vis and rR spectroscopies exhibiting similar behavior to other superoxido derivatives previously described. In 2021, by adopting the ligand used for the previous structural characterization of a dinuclear copper(II) cis peroxido,162 Meyer reported the X-ray structures of both peroxido and superoxido species bearing the same pyrazolato/TACN2,2 ligand167 (Chart 2 and Fig. 8). Redox investigation revealed one electron process characterized by a small reorganization energy. This was in accordance with other superoxido/peroxido interconversions reported in solution, reflecting that the redox process occurs at the peroxido-unit.169–171 Very recently Karlin and co-workers have completed the studies using a phenolato bridged ligand (BPMPO(H), Chart 2 and Table 1) analogous to XYLO(H).172 A peroxido-dicopper(II) complex was characterized and shown to undergo reversible oxidation to a superoxido-dicopper(II) species. The various Cu2II( ) complexes (in the nature of the dinucleating ligand) were compared and discussed with respect to physical properties and reactivity.172 Based on spectroscopic, titration and electrochemical data, thermodynamic parameters (E1/2, pKa) were obtained, allowing a comparative analysis (Table 1). For each species described (peroxido, superoxido and hydroperoxido) despite various bridging ligands and various experimental conditions (temperature and solvent), the UV-Vis absorptions for each form are in a similar energy range. Similarly, O–O stretching frequencies obtained from rR (ν(O–O)) are between 798 and 815 cm−1 for all peroxido species (Table 1). The rR data on superoxido species range from 1070 cm−1 (Cu2II-
) complexes (in the nature of the dinucleating ligand) were compared and discussed with respect to physical properties and reactivity.172 Based on spectroscopic, titration and electrochemical data, thermodynamic parameters (E1/2, pKa) were obtained, allowing a comparative analysis (Table 1). For each species described (peroxido, superoxido and hydroperoxido) despite various bridging ligands and various experimental conditions (temperature and solvent), the UV-Vis absorptions for each form are in a similar energy range. Similarly, O–O stretching frequencies obtained from rR (ν(O–O)) are between 798 and 815 cm−1 for all peroxido species (Table 1). The rR data on superoxido species range from 1070 cm−1 (Cu2II- based on the Pyr-TACN2,2 ligand) to 1143 cm−1 (Cu2II-
based on the Pyr-TACN2,2 ligand) to 1143 cm−1 (Cu2II- based on the XYLO− ligand). The decrease of the O–O stretching frequencies correlates with the weaker O–O bonds. In more detail, from various phenolato (Karlin group)168,172 and pyrazolato (Meyer group)161,167,171 bridging ligands, the particular binucleating ligand design was shown to considerably influence/control the reduction potential of the superoxido-dicopper(II) complex, the OO–H pKa and BDFE (see part 2.2, Table 1 and Scheme 4). The phenoxido-bridged complexes appeared to be more basic compared to the pyrazolato/TACN2,2 based complex (Pyr/TACN2,2 ligand, Chart 2 and Table 1). They were also demonstrated to be good candidates for HAA of inert C–H bonds (BDFE > 80 kcal mol−1). EPR analysis for [Cu2II(Pyr/TACN2,2)(
based on the XYLO− ligand). The decrease of the O–O stretching frequencies correlates with the weaker O–O bonds. In more detail, from various phenolato (Karlin group)168,172 and pyrazolato (Meyer group)161,167,171 bridging ligands, the particular binucleating ligand design was shown to considerably influence/control the reduction potential of the superoxido-dicopper(II) complex, the OO–H pKa and BDFE (see part 2.2, Table 1 and Scheme 4). The phenoxido-bridged complexes appeared to be more basic compared to the pyrazolato/TACN2,2 based complex (Pyr/TACN2,2 ligand, Chart 2 and Table 1). They were also demonstrated to be good candidates for HAA of inert C–H bonds (BDFE > 80 kcal mol−1). EPR analysis for [Cu2II(Pyr/TACN2,2)(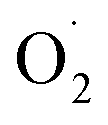 )]2+ and [Cu2II(BPMPO)(
)]2+ and [Cu2II(BPMPO)(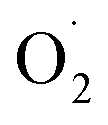 )]2+ displayed simple mono-CuII (S = ½) type axial spectra, thus indicating that the unpaired electron at the superoxide moiety was antiferromagnetically coupled to one of the CuII centers, leaving the remaining spin on the detected second CuII ion. In contrast [Cu2II(XYLO)(
)]2+ displayed simple mono-CuII (S = ½) type axial spectra, thus indicating that the unpaired electron at the superoxide moiety was antiferromagnetically coupled to one of the CuII centers, leaving the remaining spin on the detected second CuII ion. In contrast [Cu2II(XYLO)( )]2+ and its analog [Cu2II(UNO)(
)]2+ and its analog [Cu2II(UNO)(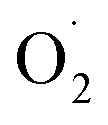 )]2+ displayed at g = ∼2.0 a typical organic radical single line spectra, in accordance to a localized unpaired electron on the
)]2+ displayed at g = ∼2.0 a typical organic radical single line spectra, in accordance to a localized unpaired electron on the 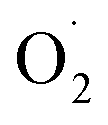 moiety associated with antiferromagnetic coupling occurring between the two copper(II) ions of the complexes.169 This different behaviors pointed out the influence of chelate ring size on electronic-bonding properties.
moiety associated with antiferromagnetic coupling occurring between the two copper(II) ions of the complexes.169 This different behaviors pointed out the influence of chelate ring size on electronic-bonding properties.
| Ligands (references) | Pyr/TACN2,2 (ref. 162, 167 and 171) | XYLO(H) (ref. 168) | BPMPO(H) (ref. 172) |
|---|---|---|---|
| Conditions of the studies | CH3CN, −20 °C | MeTHF, −125 °C | MeTHF, −90 °C |
| Dicopper( II ) peroxido | |||
| rR, ν(O–O) cm−1 | 798 | 803 | 815 |
| UV-Vis | 527; ε = 5000 | 523; ε = 7150 | 536; ε = 8500 |
| λ max (nm); ε (M−1 cm−1) | 648; ε = 3900 | 420 | |
| 620 | |||
|
|||
| Dicopper( II ) superoxido | |||
| rR (ν(O–O) cm−1 | 1070 | 1143 | 1086 |
| UV-Vis, λmax (nm); ε (M−1 cm−1) | 440; ε = 11500 |
402; ε = 18750 |
405; ε = 15100 |
| 480; ε = 4380 | |||
| E 1/2 vs. Fc+/0 (O2˙−/O22−) | −0.59 V | −0.53 V | −0.44 V |
|
|||
| Dicopper( II ) hydroperoxido | |||
| UV-Vis | 416; ε = 5700 | 398; ε = 10725 |
391; ε = 5800 |
| λ max (nm); ε (M−1 cm−1) | 630 | ||
| OO–H pKA | 22.2 | 24 | 22.3 |
| OO–H BDFE kcal mol−1 | 69.4 | 80.7 | 80.3 |
These significant advances owe a lot to the recent development of cryogenic spectroscopies and analysis for unstable species identification173 (i.e. low-temperature spectroelectrochemistry reviewed in 2021 by Le Poul174) associated with theoretical modelling at low temperatures. Since a lot of reactive intermediates have been characterized by cryo-ESI-MS (cold-spray ionization-mass spectrometry), the association of such ESI together with tandem MS could be of interest in the field of copper as reported for iron oxido intermediates.175
In another example, DeBeer used Valence-to-Core (VtC) X-ray emission spectroscopy (XES)182 as a sensitive probe to discriminate between SP and O cores through O–O bond detection.183 More generally, such synchrotron-based X-ray spectroscopy approaches have been more frequently used in these last few years for the characterization of dicopper(II)-oxygen adducts, with the support of theoretical calculations, and provide their electronic structure description.138
4.2 Peroxido species as an active intermediate and reactivity
Iron, manganese or cobalt complexes are the most predominant dinuclear systems reported in the literature for C–H hydroxylation.184 They display multiple oxidation or spin states and can generate binuclear peroxido, superoxido or oxido reactive intermediates in the presence of dioxygen. Although dinuclear copper complexes share similar features, few studies have exploited these behaviors to activate substrates.Nevertheless, in the last few years, significant advancements were made in the use of low molecular weight dicopper complexes capable of activating inert C–H bond in a direct manner through bioinspired approaches. Such developments are presented in the following parts.
In some cases, such side-one (SP) species exhibited no phenol ortho-hydroxylation (tyrosinase like-activity) as reported by Company for dinuclear PyNMe-based peroxides (PyNMe = 3,6,9-trimethyl-3,6,9-triaza-1(2,6)-pyridinacyclodecaphane, Chart 1).195 The presence of the SP species was ascertained by a unique highly intense UV-Vis signature at 353 nm analogously to other SP compounds. The steric hindrance, rigidity and the saturated coordination imposed by the tetradentate PyNMe ligand prevented phenolic substrate binding and explained that no phenol ortho-hydroxylation could be detected. In another example, the room-temperature stable and inert SP complex based on an hexapyridine ligand196 (h6m4h = 1,2-bis[2-(bis(6-methyl-2-pyridyl)methyl)-6-pyridyl]ethane ligand, Chart 2) was investigated by Kodera and Le Poul.197 The two-electron reduction of the SP complex resulted in the O–O bond cleavage, but no C–H or O–H bond activation of exogeneous substrates was detected. This result was attributed to the negative standard potential and weak basicity of the bis-reduced species, precluding any HAA process due to a low BDFE.
Dicopper(III) bis(μ-oxido) adducts (O) resulting from the conversion of sP species by O–O bond breakage have also been suggested to be active species.198,199 Since the pioneering work of Itoh,200–202 various examples of such reactivity have been described and reviewed.203,204 Karlin, Solomon and Garcia-Bosch74 converted a non-active SP in an active O species via two possible processes following coordination of either: (i) an external substrate (sodium phenolate) that was further ortho-hydroxylated (tyrosinase like-activity) or (ii) a Lewis acid (one equivalent of Sc(CF3SO3)3 or two equivalents of B(C6F5)3 or DMF·CF3SO3H) in the ligand scaffold able to enhance the C–H bond activation ability. More recently, the formation of the reactive O species after addition of two equivalents of a phenolic substrate was explored by Stack with diamine ligands (Chart 1).205 The reactivity studies were extended to hydroxylation associated with the dehalogenation of 2-halophenolate substrates.
An attractive one-pot approach has been recently developed by Göttlich and Schindler,206 involving a dicopper(II)–O2 adduct in combination with a photochemical reducing agent. Reaction of 2 equivalents of the LCuI complex (L = tmpa ligand) with O2 yielded an active dinuclear intermediate (O or SP). In toluene solution, selective catalytic oxidation of toluene into benzaldehyde at room temperature was achieved, without formation of benzyl alcohol or over-oxidized products (Scheme 12). Regeneration of the LCuI active species was performed by using a ruthenium bipyridine photosensitizer [Ru(bpy)3](PF6)2 in the presence of triethyl amine (TEA, sacrificial electron donor) and irradiation at 475 nm. Under these conditions, the tmpa complex achieved a toluene conversion of 42%. Briefly, the proposed mechanism involved a dinuclear intermediate leading to the benzyl radical  through a H-atom abstraction from toluene. Reaction of
through a H-atom abstraction from toluene. Reaction of  with O2 yielded the benzylperoxido radical (PhCH2OO˙) after additional hydrogen abstraction on toluene to the corresponding reactive hydroperoxide PhCH2OOH species which evolves to benzaldehyde and water.
with O2 yielded the benzylperoxido radical (PhCH2OO˙) after additional hydrogen abstraction on toluene to the corresponding reactive hydroperoxide PhCH2OOH species which evolves to benzaldehyde and water.
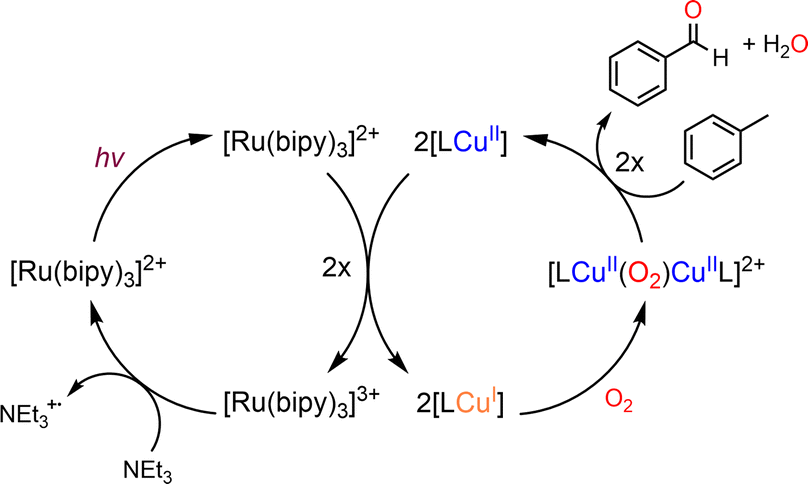
4.3 Dicopper-μ-oxido species as active intermediate
Dicopper(II)-μ-oxido complexes are accessible from multiple routes, including the reaction of Cu(I) complexes (mono or dinuclear) with O2, PhIO and even with NO.207 Already, Limberg mentioned in 2014 that “complexes featuring CuII–O–CuII units do not have a developed chemistry”.207 Since then, studies have remained focused on the spectroscopic features and reactivity of such entities. Dicopper(II)-μ-oxido species have been proposed as active intermediates in homogeneous catalytic activation of inert alkanes (such methane) as well as in heterogeneous catalysts (see Section 6 below).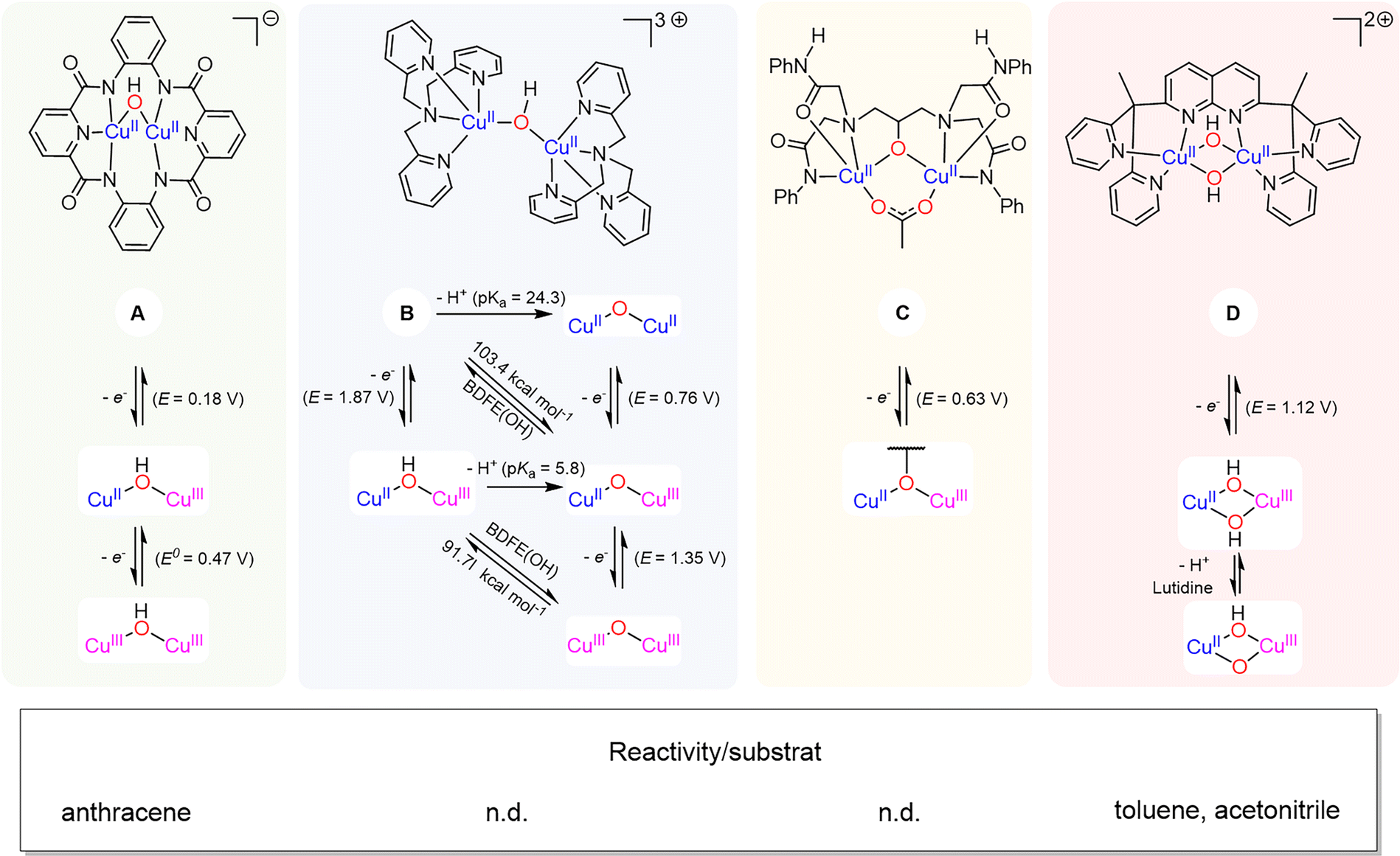 | ||
| Fig. 9 (Top) Starting materials for the preparation of reported dicopper high valent species. (Down) Selected experimental properties (A (wet DMF),209 B215 and D216 (CH3CN) and C210 (DMF)), E in V vs. Fc+/Fc. | ||
Although no structural report has yet been reported for such species, given their high instability, they remain of high interest since μ-oxido bridged CuIICuIII cores have been postulated as putative intermediates for C–H bond activation.215,217,218 More to this point, De Tovar recently evidenced the tunability of their electrochemical properties through the formation of intimate ion pairs with surrounding anions by non-covalent interactions, highlighting the importance of second sphere effects when designing such [Cu2II(μ-OH)2]2+ species.219 Interestingly and counter-intuitively, CuIICuIII(μ-O)x intermediates could be more efficient for strong C–H bond activation than their corresponding CuIIICuIII(μ-O)x counterparts. In 2013, Yoshizawa and Shiota postulated that HAA activation could preferably occur after protonation and reduction of an O species, leading to a mixed-valent intermediate.217 The catalytic oxidative ability of such an intermediate was experimentally demonstrated by Belle using a naphthyridine-based spacer [Py4CuIICuII(μ-OH)2]2+ complex (Py4 = 2,7-bis(1,1-dipyridylethyl)-1,8-naphthyridine ligand, Chart 2 and Fig. 9, structure D). Mono-electrochemical oxidation of the complex [LCuIICuII(μ-OH)2]2+ in acetonitrile at room temperature resulted in the transient generation of a mixed-valent LCuIICuIII species,213 that could perform catalytic HAA from strong Csp3–H bonds (toluene and CH3CN with BDFE = 87.0 and 94.2 kcal mol−1, respectively) in the presence of a relatively weak base (lutidine, pKa = 14.16).216 On the other hand, Thibon-Pourret reported on an intramolecular ligand C–H bond activation using a monooxidized dicopper(II) bis(μ-OH) complex with the Ox2Py2 ligand (Chart 2) bearing a bis-oxazoline arm in place of a bis-pyridine arm on a Py4 ligand (Fig. 9-structure D).220
Finally, in 2020, Kiebers-Emmons215 rationalized these observations through experimental determination of thermodynamic parameters (E1/2, pKa, BDFEs) for tmpa-based complexes (Fig. 9, structure B). BDFEs(OH) for the HAA reactions LCuII-OH-CuIIL → LCuII-O-CuIIIL and LCuII-OH-CuIIIL → LCuIII-O-CuIIIL of 103.4 and 91.7 kcal mol−1 (Fig. 9), respectively indicated that the higher oxidation CuIII–O–CuIII state had a lower HAA ability than the mixed valent CuII–O–CuIII state. It was suggested that geometry (specifically the Cu–O–Cu angle), solvation, magnetic, and mixed valency contributions in dicopper complexes participate in the tuning of the BDFE(OH) contrarily to mononuclear species. Altogether, the results demonstrated that such high-valent dicopper(II,III) species could be key reactive intermediates in copper-based oxidative processes.
H, Chart 2) prepared from RLCuICuI and an excess of NO (Fig. 10),221 or alternatively from RLCuICuII and NO2. This complex was able to perform oxygen atom transfer (OAT), HAA, and C–H hydroxylation of various substrates such as phosphines, cyclohexadienes, and isochroman, yielding phosphine oxide, benzene, and 1-isochromanone, respectively. The oxidizing entity displayed a spin-switching behavior between S = 1/2 and S = 3/2 which was shown to be temperature-dependent. Modifications on the ligand (R groups introduced in position 4 of the catechol spacer (Chart 2)) induced an unsymmetrical environment for the dicopper center [RLCu2(μ-O)(μ-NO)]2+ and allowed spin state control of the intermediate.222 The S = 1/2 state appeared more reactive for HAA than the S = 3/2 state. Best activities were obtained with RF and NH2, despite possessing opposing electron-donating/withdrawing properties.
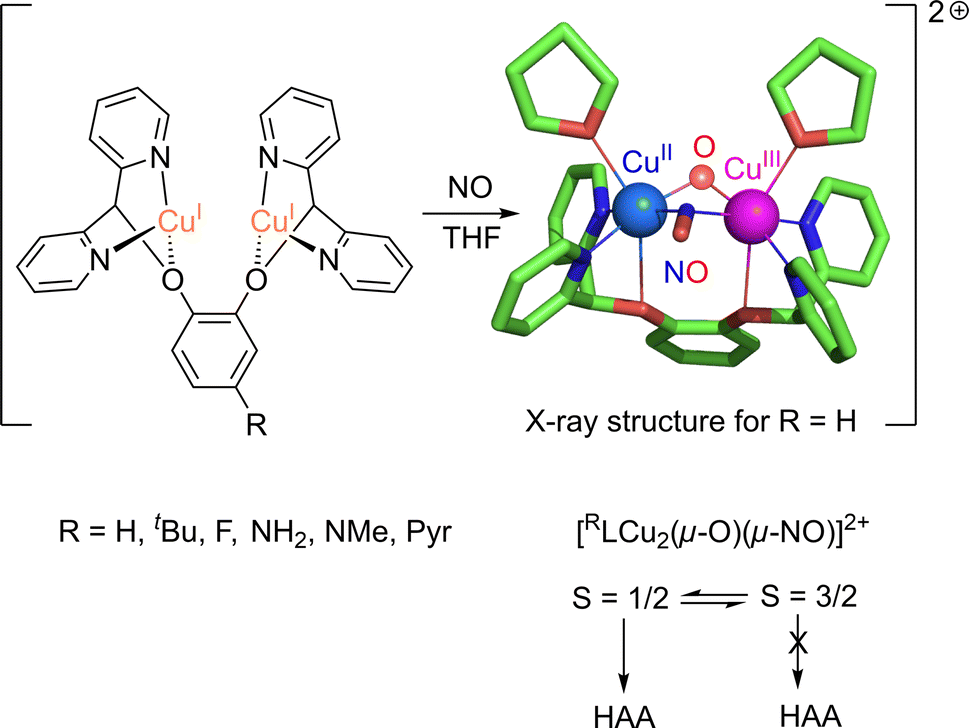
4.4 Dicopper-hydroperoxido as active intermediates
Catalytic hydroxylation of benzene to phenol considering CuII(μ-OH)CuII species and H2O2 or Cu2I and O2 has been mechanistically investigated by Kodera223 and very recently by Glaser224 starting from dinuclear species [Cu2II(μ-OH)(6-hpa)]3+ or [Cu2II(μ-OH)(susan)]3+ respectively (6-hpa and susan ligands are depicted in Chart 2). In both cases, remarkably high activities compared to phenol oxidation in the ortho position (tyrosinase like activity) were observed (see Section 4.2.1). The active catalytic species were examined and both a Fenton-like radical mechanism or dicopper-hydroperoxido based mechanism were shown to be feasible depending of the catalytic conditions (temperature, excess amount of H2O2…). Subsequently, Kodera reported that the same dinuclear [Cu2II(μ-OH)(6-hpa)]3+ species placed in the presence of H2O2 was able to oxidize methane (using a high-pressure reactor) to methanol (and formaldehyde as the minor product) with TON of 43 and 7.4 respectively.225 The copper-oxyl and peroxyl radicals species detected by cryo-ESI MS were proposed to be the active species for benzene oxidation.2235 Polynuclear copper–oxygen species
5.1 Trinuclear copper–oxygen adducts and C–H bond activation
So far, very few trinuclear copper complexes have been designed to activate dioxygen and perform oxidative reaction of hydrogenated substrates. Amongst reported works, Chan and co-workers have been particularly prone to develop synthetic tricopper models of the D active site of particulate methane monooxygenases (pMMOs) (see Section 2.1). Over several studies, they designed tricopper diazepane-based complexes capable of mediating methane oxidation in the presence of hydrogen peroxide in acetonitrile, but with a low efficiency under homogeneous conditions. According to DFT calculations226 and EXAFS experiments,227 the active species was proposed to be a mixed-valent adduct [CuIICuII(μ-O)2CuIII(L)]1+ (where L denotes a trinucleating ligand, for example L = 7-N-Etppz, Chart 2) generated from the reaction of tris-copper(I) complexes with O2 (see Scheme 13). The presence of a third copper center was suggested to increase the reactivity of the putative intermediate towards methane through a combination of different effects such as (i) a favored planar conformation and lengthening of the CuII–CuII distance, (ii) an increased Lewis acidity and (iii) an overall singlet state of the “oxene” cluster. Interestingly, recent work by Nandi et al. has investigated the role of acetonitrile in the methane oxidation process mediated by the tricopper piperazine complexes from the group of Chan.228 In particular, they showed that methanol could be produced from acetonitrile in the absence of methane, but in the presence of a copper complex and hydrogen peroxide.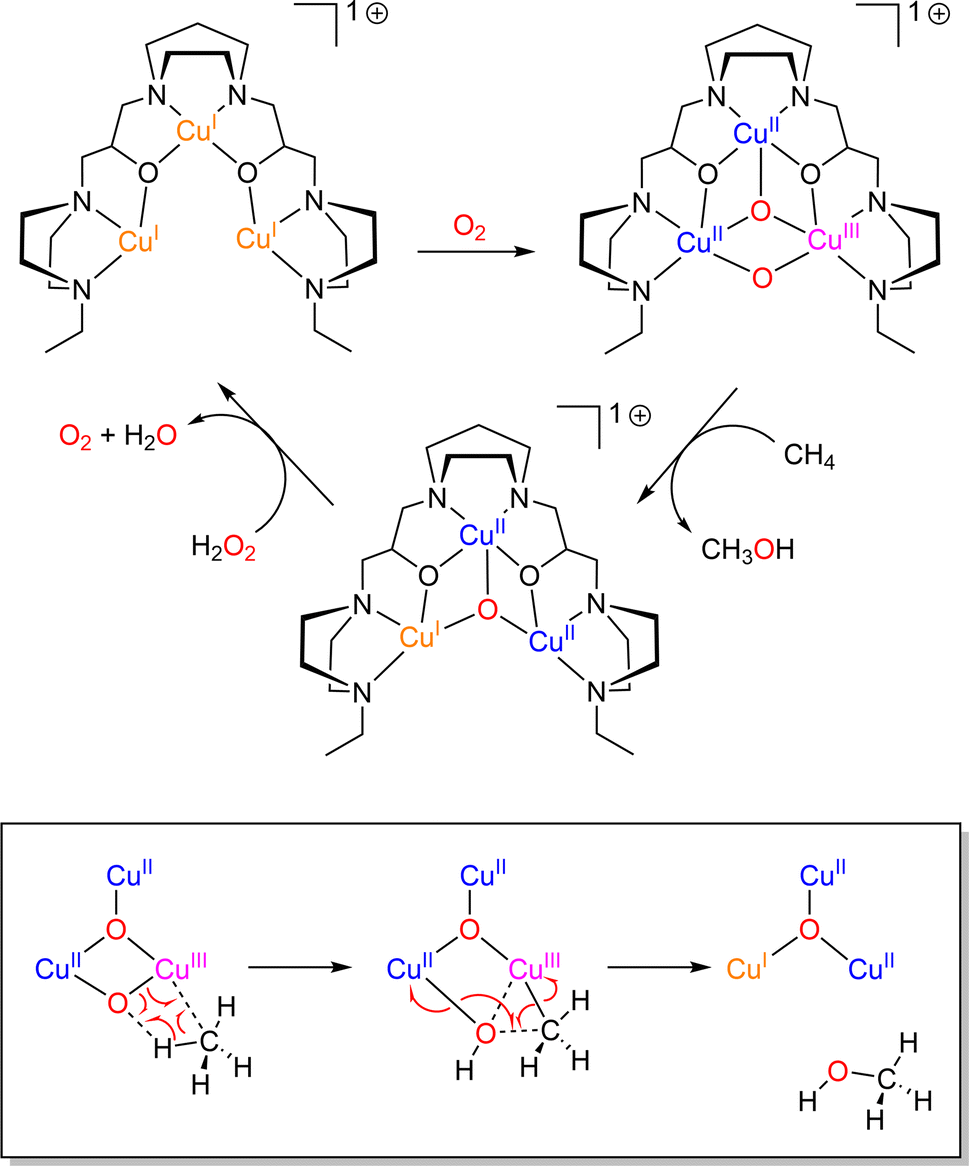 | ||
| Scheme 13 Top: Catalytic cycle for the oxidation of CH4 mediated by the [CuICuICuI(7-N-Etppz)]1+ complex and O2 in the presence of hydrogen peroxide. Bottom: Proposed intermediates for methane oxidation. Adapted from ref. 227 and 229. | ||
Significant progress in methane-to-methanol conversion was more recently obtained by Chan's group by carrying out catalysis under heterogeneous conditions.229 Experimental results demonstrated that grafting of the copper catalysts onto mesoporous silica nanoparticles significantly improved methane oxidation relative to bulk studies. One main reason for this enhancement was proposed to originate from the over-solubility of methane in the confined spaces of nanoporous materials, hence increasing the turnover frequency by raising methane concentration and avoiding the competitive direct oxidation of hydrogen peroxide by the active copper–oxygen species.
In a further paper, Chan reported the catalytic oxidation of ethane and propane using the same strategy as that used with methane.230 Indeed, the [Cu3II(7-N-Etppz)]4+ complex (see Scheme 13) was immobilized on anionic-based mesoporous silica nanoparticles (TP-MSN, 70 nm size, 2 nm pore size) and in situ reduced into a tris-copper(I) species by sodium ascorbate at room temperature. In the presence of O2 and H2O2, the grafted complex mediated the oxidation of ethane into ethanol with a 7-fold higher efficiency (21% conversion) compared to the same reaction under homogeneous conditions. Oxidation of propane led to the formation of both isopropanol and acetone. Notably, kinetic studies for a mixture of gases demonstrated that the catalyst was more selective for propane than for ethane or methane conversions into alcohols (30:22:1, distribution, respectively).
Catalytic methane oxidation by the tricopper-(7-N-Etppz) complex was also investigated by DFT calculations.231 Two mechanisms were considered for CH4 oxidation by the reactive [Cu3(μ-O)2(7-N-Etppz)]1+ complex: (i) a concerted electrophilic oxene insertion and (ii) a nucleophilic hydrogen-atom abstraction (HAA) followed by a radical rebound process. Lower activation energies were obtained for the latter, hence suggesting the occurrence of a two-step mechanism. Calculations revealed that C–O recombination was the rate-determining step in the radical rebound mechanism, hence predicting a low H/D Kinetic Isotopic Effect (KIE).
In 2020, Chan investigated the activation of the tricopper(I) complex [CuICuICuI(7-Dipy)]1+ (for ligand 7-Dipy see Chart 2) by H2O2 by using rapid-freeze–quench and cryo-ESI MS.232 The complex was activated by H2O2 rather than O2 to offer better chances in terms of kinetics to observe and characterize copper–oxygen adducts. Through an optimized methodology, the targeted [CuIICuII(μ-O)2CuIII(7-Dipy)]1+ species was detected together with other copper oxygen intermediates. A deactivation pathway involving this reactive species and a tris-Cu(I) bis-protonated complex was proposed to explain the detection of [CuIICuIII(μ-O)2CuIII(7-Dipy)]2+ by high resolution mass spectrometry (Scheme 14).
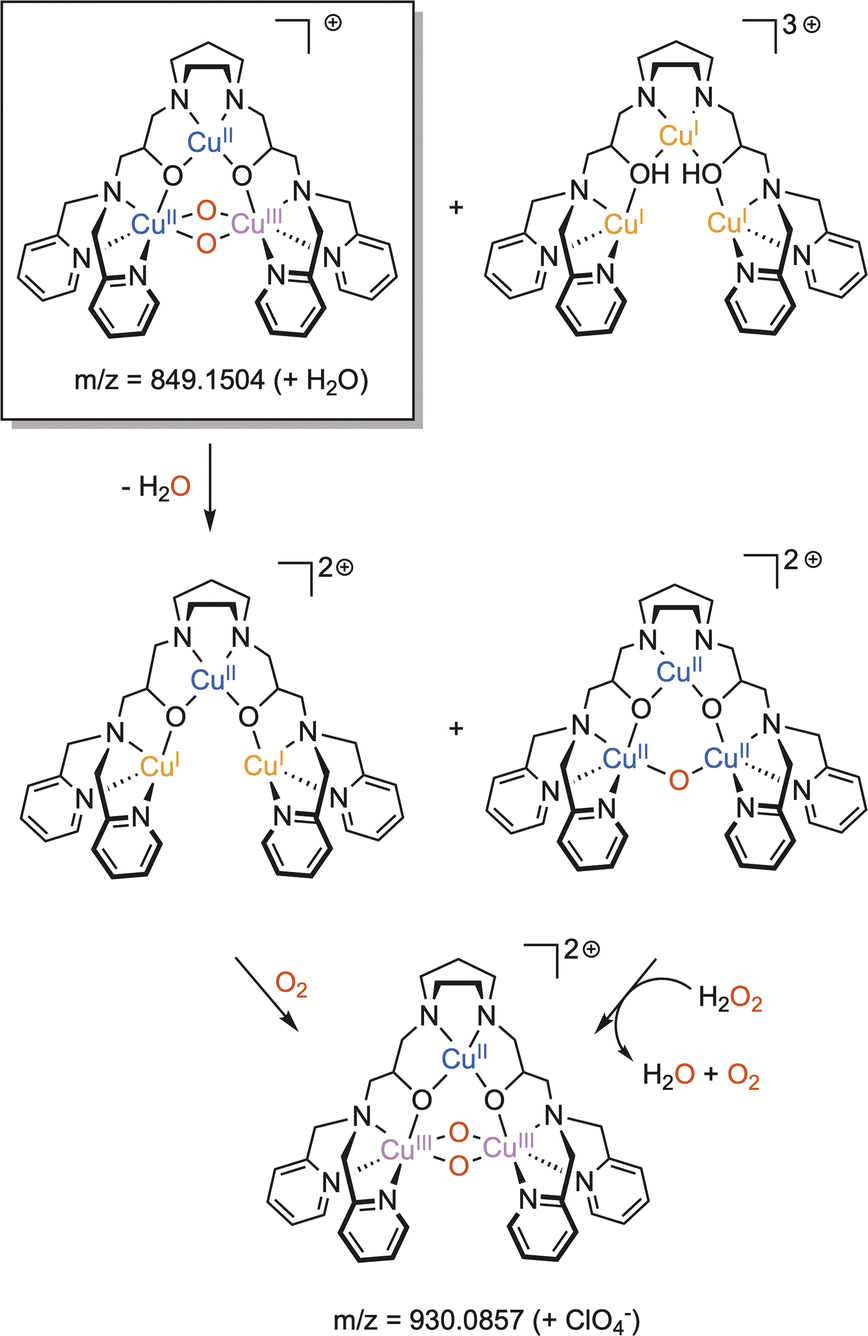 | ||
| Scheme 14 Deactivation pathway proposed from mass-spectrometry experiments with the tricopper (7-Dipy) complex. Adapted from ref. 232. | ||
In a more recent paper, the same group reported the development of electrocatalytic devices to perform methane-to-methanol conversion in aqueous media.233 As a first step, the tricopper(II)-oxido (7-N-Etppz) catalyst was immobilized in mesoporous silica nanoparticles. The modified particles were then dispersed in water and deposited onto screen-printed carbon electrodes through a drop casting method, yielding a surface concentration of 0.17 nmol cm−2. The electrocatalytic oxidation of methane was carried out by electrochemical reduction below −0.50 V vs. Ag/AgCl in potassium perchlorate (pH = 6.5). Production of methanol with high turnover number (TON = approx. 2400 after 3 h of electrolysis at Eapp = −0.9 V) was evidenced by gas chromatography from bulk electrolysis under air. However, the catalytic device suffered from the competitive oxygen reduction reaction (ORR), as well as copper leaching over time. To overcome this, sodium ascorbate and copper(II) chloride were added to the solution, showing significant improvements for the recyclability. Comparative studies with pMMO's modified electrodes emphasized the interest of using such synthetic catalysts instead of proteins in terms of efficiency over mass of catalyst.
The group of Doerrer has recently reported the generation of tricopper [Cu3O2] cores using various monocopper(I) complexes bearing fluorinated alkoxide ligands, the main objective being to perform efficient C–H bond activation. The utilization of perfluoropinacolate bidentate ligands, such as pinF, afforded the formation of bis(μ3-oxido) triscopper(II,II,III) adducts.234 These species were characterized by UV/Vis spectroscopy at −80 °C, cryo-ESI MS, and EPR spectroscopy. Reactivity studies demonstrated that the trinuclear adduct was able to catalytically oxidize p-hydroquinone into benzoquinone but with a limited efficiency. It also displayed OAT (Oxygen Atom Transfer) properties towards triphenylphosphines. A further step was obtained later using a fluorinated O-donor monodentate ligand (OC4F9).235 UV-Vis kinetic analysis suggested the transient formation of bis-μ-oxido intermediates that rapidly converted into bis-(μ3-O)CuIICuIICuIII species. The latter were characterized by resonance Raman spectroscopy (the related [Cu3O2] core associated with stretching at 679 cm−1 shifted to 718 cm−1 after isotopic labeling with 18O) and UV-Vis (λmax = 307, 520 and 602 nm) spectroscopy, as well as mass spectrometry. The (OC4F9) copper–oxygen trimer was shown to catalytically oxidize hydroquinone. Furthermore, it was also able to stoichiometrically convert 2,4-di-tert-butylphenolate into 3,5-di-tert-butyl-1,2-catechol. Such property that was not observed with the bidentate pinF analogue was ascribed to the better flexibility of the monodentate ligand. Aiming at reproducing multicopper oxidase activity, Murray recently reported the C–H bond activation of diverse organic substrates (toluene, dihydroanthracene…) starting from a tricopper complex bearing a cyclophanate ligand (Chart 2).236 Low-temperature UV-Vis stopped-flow experiments highlighted the reaction of the tricopper complex with O2, leading to the generation of trinuclear copper–oxygen adducts (λmax = 490 and 630 nm) with a similar spectroscopic signature to that of Chan's complex. Computational studies suggested that oxidation of exogeneous substrates may occur through a concerted HAA reaction from a reactive (μ-oxyl)(μ3-oxido)tricopper(II) intermediate, followed by radical rebound and OAT. Competitive intramolecular ligand oxidation was invoked to explain the low yield obtained for substrate oxidation (<1%).
5.2. Trinuclear copper complexes: related reactivities
The four-electron reduction of O2 to water mediated by a trinuclear copper complex has been poorly investigated in the last 5 years despite its technological significance in fuel cell applications, and its relevance to the biological process in multicopper oxidases (laccase, bilirubin oxidase).Ray reported in 2018 a comparative study of the 4e−, 4H+ reduction of dioxygen mediated by either monocopper(I) or tricopper(I) complexes bearing identical amine-based coordination spheres.237 Low-temperature UV-Vis spectroscopic measurements, supported by X-ray absorption near-edge spectroscopy (XANES), were employed to monitor the reaction of the trinuclear complex [Cu3I(L)]3+ (L = tateb = 1,3,5-tris-3,3-iminobis(N,N-dimethylpropylamine)-2,4,6 triethylbenzene, Chart 2) with O2 in the absence of reductant and proton sources. A green species assigned to a [CuICuII(O22−)CuII(L)]3+ intermediate was hence detected at −80 °C. Increasing temperature to −60 °C revealed its conversion into a brownish [CuIICuII(O23−)CuII(L)]2+ species. In the presence of both reductant and proton sources at −20 °C, the tricopper(I) complex produced water as the sole product. Kinetic studies revealed that O2 binding was the rate-determining step (RDS), resulting from the high instability of the trinuclear copper–oxygen adducts which underwent a very fast PCET step. In contrast, a different mechanism was found for its mononuclear analogue, generating a bis-μ-oxido Cu2III intermediate which ran the PCET reaction in a slower manner.
A supramolecular tricopper(II) complex bearing four assembled tren units was developed by Zhang as a catalytic model of multicopper oxidases for oxygen reduction into water (Fig. 11).238 The ligand was specifically designed to provide protons in the secondary sphere of the copper ions and to prevent any disassembling of the catalyst structure through the PCET reactions. Successive reduction/protonation of [Cu3II(μ-O)3(LH)]5+ (L = tren4, Chart 2) led to the generation of a tricopper(I) (μ-OH2)2 species (Fig. 11). The different redox and protonated states were assigned thanks to voltammetric and spectroscopic characterizations. Interestingly, these reactions were shown to occur within a narrow potential range (170 mV). This effect was ascribed to favor proton relay by the ligand scaffold, as occurring in multicopper oxidases.
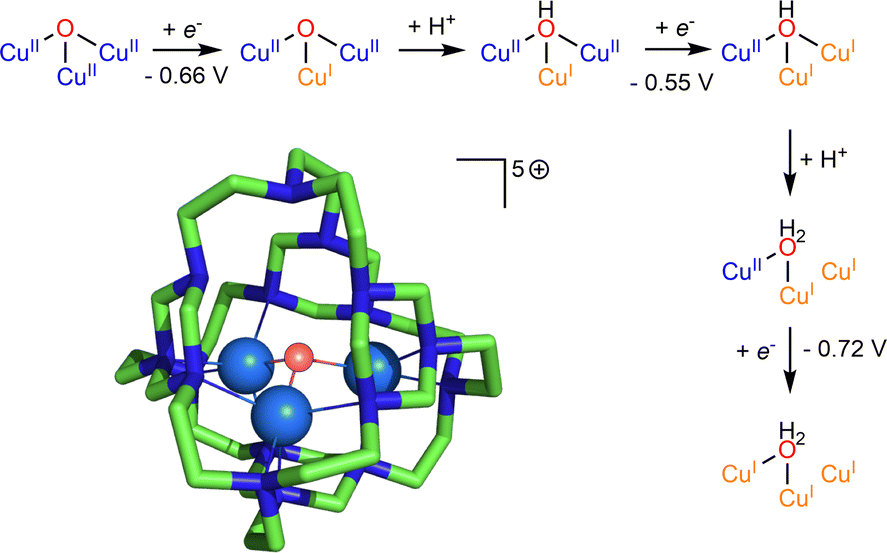 | ||
| Fig. 11 Stepwise proton-coupled reduction of the [Cu3II(μ-O)3(LH)]5+ complex (L = tren4) studied by Zhang as a model of multicopper oxidases for the reduction of dioxygen into water. Adapted from ref. 238. | ||
In 2018, Chan et al. reported on the efficient electrocatalytic reduction of dioxygen into water thanks to heterogeneous tricopper catalysts.239 Four different copper derivatives were investigated to account for the ligand topology and the effect of the number of copper centers on the catalytic properties was investigated. All of them were immobilized onto screen-printed carbon electrodes through a dropcasting approach. Amongst the complexes, [Cu3I(7-N-Etppz-CH2OH)]+ exhibited the best performances with a 680 mV vs. RHE overpotential value at pH = 7, a turnover frequency of 12 s−1, and the production of 27% H2O2. Kinetic studies demonstrated that the rate-determining step was not dioxygen binding to the catalyst but merely the O–O bond cleavage through the third electron transfer to the copper cluster. Further comparative studies between dinuclear and trinuclear analogues demonstrated the key role played by the third copper center in terms of overpotential.
6 Heterogenized copper–oxygen species
6.1 Materials framework for the immobilization of copper–oxygen species
Heterogenized copper–oxygen species typically refer to Cu-containing compounds where Cu ions/clusters are incorporated into a heterogeneous support or matrix. These heterogenized species play a crucial role in catalysis due to copper's ability to undergo redox reactions and participate in O2 activation and C–H bond activation, particularly in methane oxidation. This represents a crucial process with significant environmental implications. The development of selective oxidation heterogenized systems has thus received great attention over the last few years becoming a hot approach due to its advantages arising from its straight-forward implementation into industrial devices particularly focusing on methane-to-methanol (MTM) transformation.240,241 Within this context, understanding the nature of the true active species in such heterogenized systems has become a target towards improved performances. In the literature, there are several heterogenized systems covering the use of zeolites as coordinating materials for Cu–O species immobilization (Fig. 12) and their further MTM conversion utilization.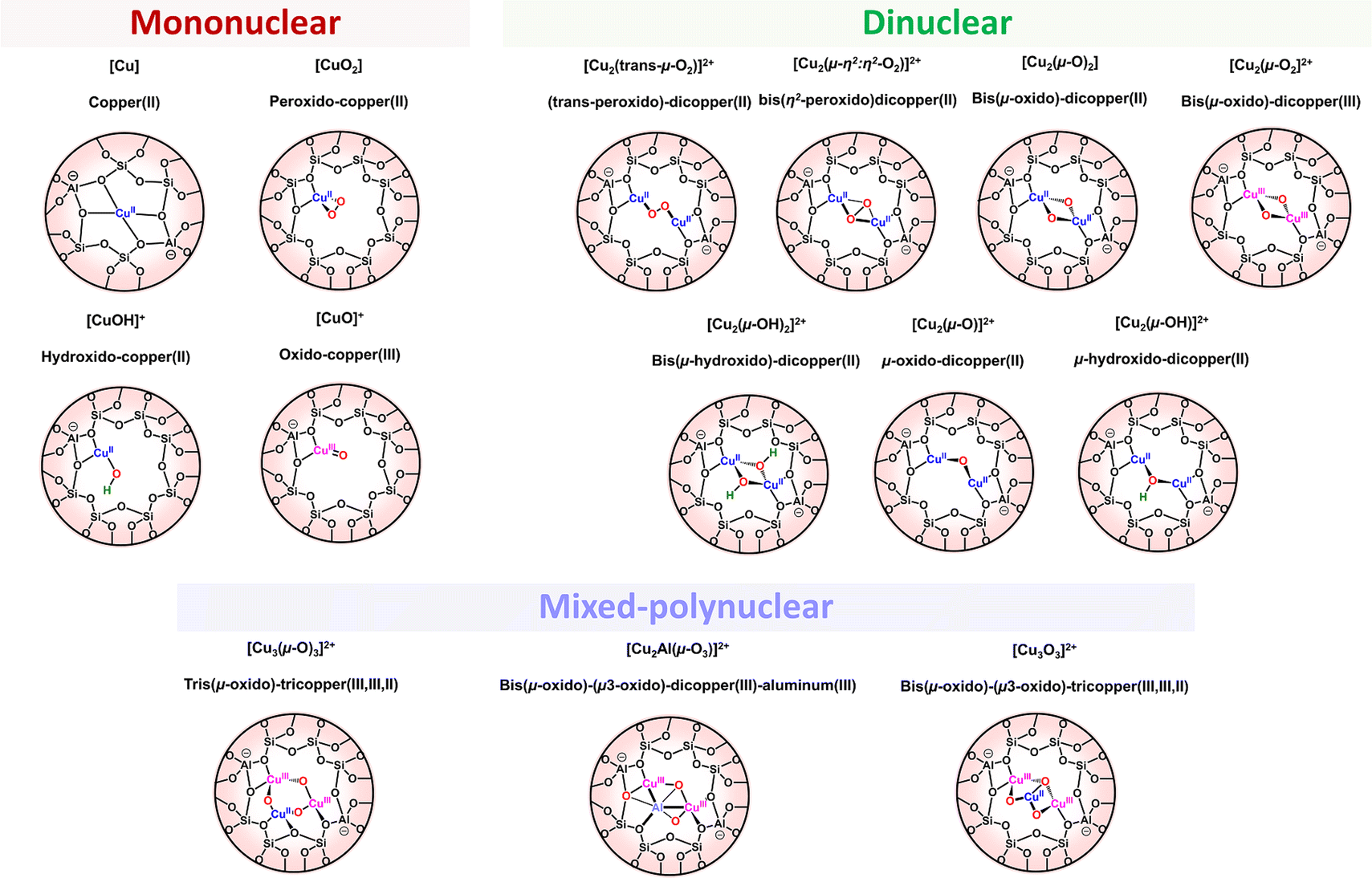 | ||
| Fig. 12 Selected zeolite-based Cu–O catalyst. | ||
Zeolites are traditionally referred to as a family of open-framework aluminosilicate-based materials exhibiting distributed micropores at molecular dimensions. From here, depending on how aluminosilicate tetrahedra are connected, a different zeolite framework type can be assigned. This classification follows a three-letter code by the International Zeolite Association. Within this context, different zeolite frameworks have been preferentially studied for MTM transformation such as beta (BEA), faujasite (FAU), mordenite (MOR), mordenite framework inverted (MFI), chabazite (CHA) and omega (MAZ) (Fig. 13) among the identified 235 distinct zeolite framework types. However, despite the efforts for the characterization of such heterogenized systems, the nature of the true active species is still a topic of debate since it depends on many factors ranging from the catalytic conditions used for the MTM conversion, the nature of such material lattices, the copper incorporation method and the signal treatment for the identification and mechanism elucidation of active species.242 Additionally, many techniques and analyses have been applied for this purpose, ranging from rR spectroscopy, isotope labelling experiments and DFT calculations and machine learning.243,244 The aim of this section is to present the approach dealing with the parameters mentioned above towards a better understanding covering the reusability of the zeolite-containing copper-heterogenized systems.
 | ||
| Fig. 13 Selected zeolite framework types. Adopted from ref. 240. | ||
6.2 Heterogenization of copper–oxygen species in zeolites
Cu–O species immobilized in zeolite micropores have been shown to be efficient catalysts for the selective oxidation of methane, among other substrates. Despite the lack of exact structure characterization of these Cu–O species, different structures have been proposed to exist within the Cu-exchange zeolites (Fig. 12).This phenomenon is due to the relative distance between the monomeric [CuOH]+ species within the zeolite lattice (Fig. 14). These monomeric species (Fig. 14b) facilitate the formation of molecular methanol rather than the methoxy species [Cu2(μ-OCH3)]+ formed by copper dimers.
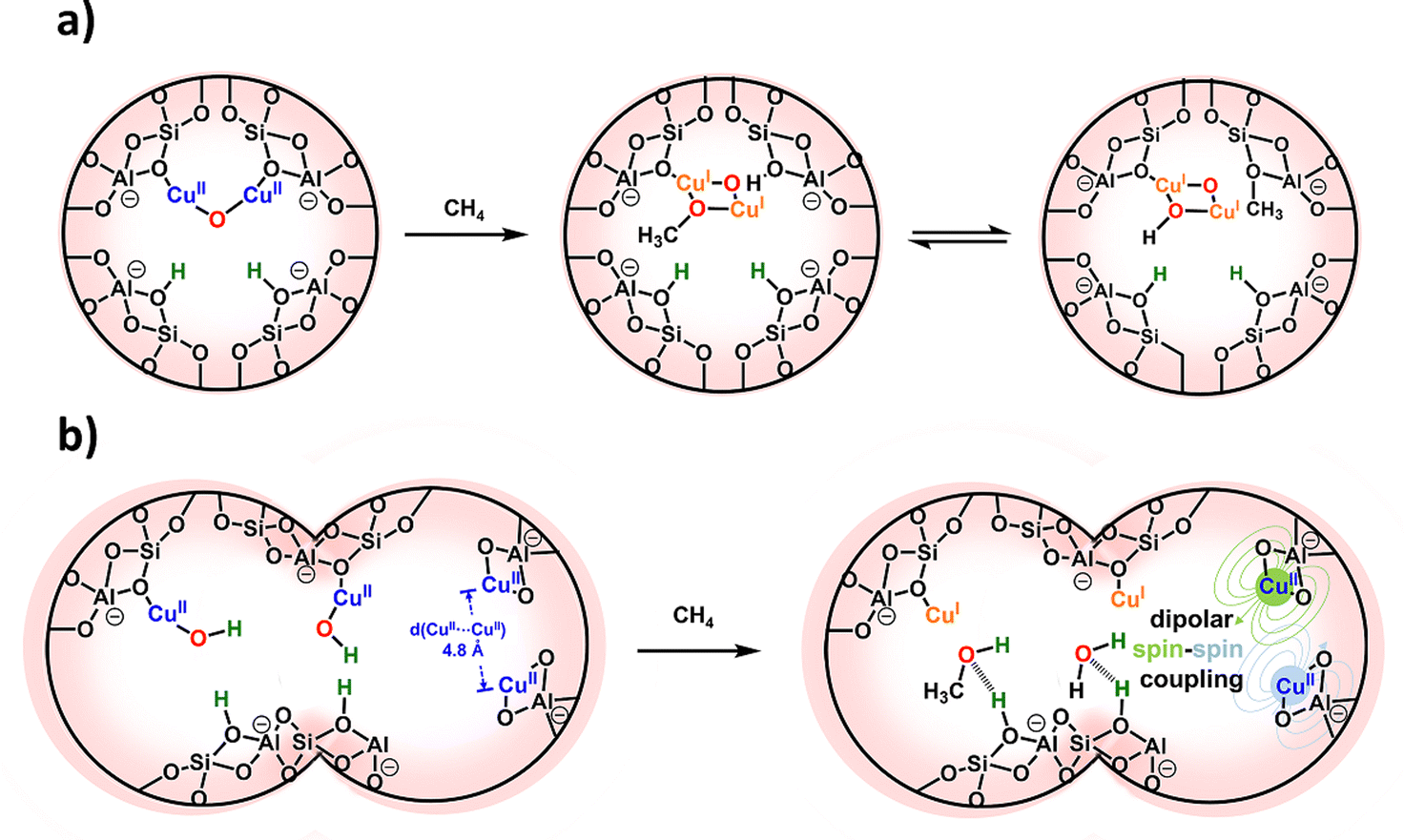 | ||
| Fig. 14 Suggested reaction pathways of Cu–O species in Cu-MOR and their transformations during the reaction with CH4. (a) Pathway involving [Cu2(μ-O)]2+ species. (b) Pathway involving [CuOH]+ species, highlighting the inactivity of monomeric Cu species exhibiting dipolar spin–spin coupling at a separation distance of 4.8 Å. | ||
However, if these monomeric species are at a distance of ca. 4.8 Å, a significant dipolar spin–spin coupling between two copper sites makes them inactive (Fig. 14b). Recently, Lercher246 studied Cu-MOR catalysts finding that the most active Cu site was a [Cu2Al(μ-O3)]2+ cluster (Fig. 12) being able even to form an ethyl methyl ether when replacing H2O by ethanol in the product extraction step (see Section 6.2.2). Additionally,247 pretreated MFI with large cations (K+, Cs+) tunes the formation of the Cu–O species, generating a new [Cu2(μ-O)]2+ site, responsible for methanol production at low copper loadings. In general, the formation of Cu-based nanoparticles or aggregates hinders the ability of Cu-zeolites to activate methane. However, depending on their size, location and nature, they are able to perform such activation.248,249
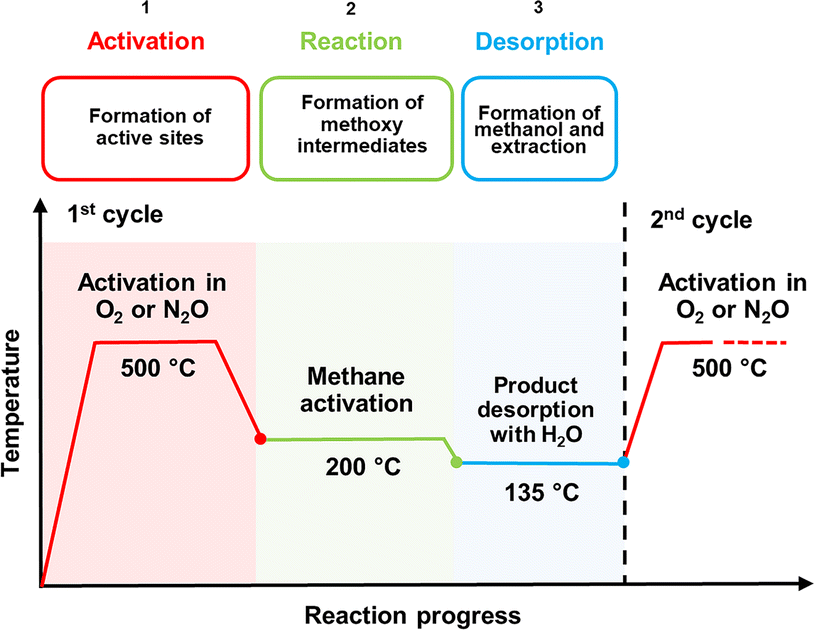 | ||
| Fig. 15 Typical experimental protocol of the catalytic conversion of methane to methanol over Cu-exchanged zeolites. Adapted from ref. 246. | ||
(i) Catalyst activation and treatment. The activation temperature plays a pivotal role in both the formation and nature of the copper sites prior to methane activation. In general, the oxidation of Cu-heterogenized materials requires temperatures higher than 300 °C in order to increase the population of active species. However, depending on the intrinsic kinetics of the Cu site types and the distribution of such types within a given material, this activation temperature has an impact on the overall methanol formation as well as on the selectivity.250 Additionally, although O2 as oxidizing gas has been largely used for such catalyst activation, other gases like N2O have also been employed showing a higher proportion of [Cu2O]2+ species.251 Furthermore, both [Cu2(trans-μ-O2)]2+ and [Cu2(μ-O)]2+ species can be generated through a [Cu2(μ-η2:η2-O2)]2+ intermediate.252 Additionally, although [Cu2(μ-O)]2+ species are postulated to be one type of active species under these conditions, their formation ratio can be tuned by applying a self-reduction treatment, usually performed at T > 500 K under He, CO or mixtures of both gases.252,253 However, the formation of Cu–O sites can be drastically affected by the presence of O2 or water impurities.
(ii) Methane activation. After the catalyst activation, CH4 can react with this latter in a stoichiometric way at 120 °C to 200 °C. However, the reactivity of the catalyst is sensitive to H2O due to the intrinsic instability of the species.
(iii) Methanol extraction. Methanol extraction is usually performed by desorption of the methoxy or adsorbed methanol species with a steam flow at lower temperatures allowing the catalytic species to be regenerated.254,255 Recently, Cu-MOR catalysts have256 been found to form both  and H˙ radicals under non-thermal dielectric barrier discharge (DBD) plasma conditions facilitating methanol desorption.257 The authors also noticed that longer reaction times lead to the reduction of the [Cu2(μ-O)]2+ species into [Cu]+ and [CuOH]+ as well as the formation of a carbon deposit. Nevertheless, the [Cu2(μ-O)]2+ species can be regenerated rapidly while eliminating the carbon deposit under O2.
and H˙ radicals under non-thermal dielectric barrier discharge (DBD) plasma conditions facilitating methanol desorption.257 The authors also noticed that longer reaction times lead to the reduction of the [Cu2(μ-O)]2+ species into [Cu]+ and [CuOH]+ as well as the formation of a carbon deposit. Nevertheless, the [Cu2(μ-O)]2+ species can be regenerated rapidly while eliminating the carbon deposit under O2.
6.3 Parameters affecting the catalytic activity in copper-exchanged zeolites
The reactivity of the catalytic system seems to be notably influenced by the specific type of zeolite. Despite the need to individually consider and discuss these materials, only a few factors have been identified as affecting the overall performance of the catalysts. This variation is attributed to the distinct nature of the copper sites within the zeolite framework, dependent on factors such as the Si/Al ratio, loading and distribution, pore size, lattice conformation and 2nd sphere effects.The decrease of products by partial oxidation at higher reaction temperatures is associated with their further transformation via three main pathways. First is their further oxidation by active Cu–O sites resulting in the formation of surface [CuCO]+ and CO2. The second direction implies the decomposition of methoxy species to CO above 630 K. The last pathway is C–C coupling into light saturated hydrocarbons, such as ethane and propane. Additionally, the previously formed methanol can be converted into hydrocarbons because of the presence of acid sites in Cu-CHA. More to this point, the lattice of Cu-MOR allows the [Cu3(μ-O3)]2+ species to be formed as an intermediate active species able to activate and promote the oxidation of two CH4 molecules (Fig. 16).259 Then, both zeolite topology and zeolite lattice constraints are found to play a significant role as they strengthen the Cu⋯Cu interaction and consequently weaken the μ–O bond destabilizing the [Cu2(μ-O)x]2+ species for CH4 activation.260
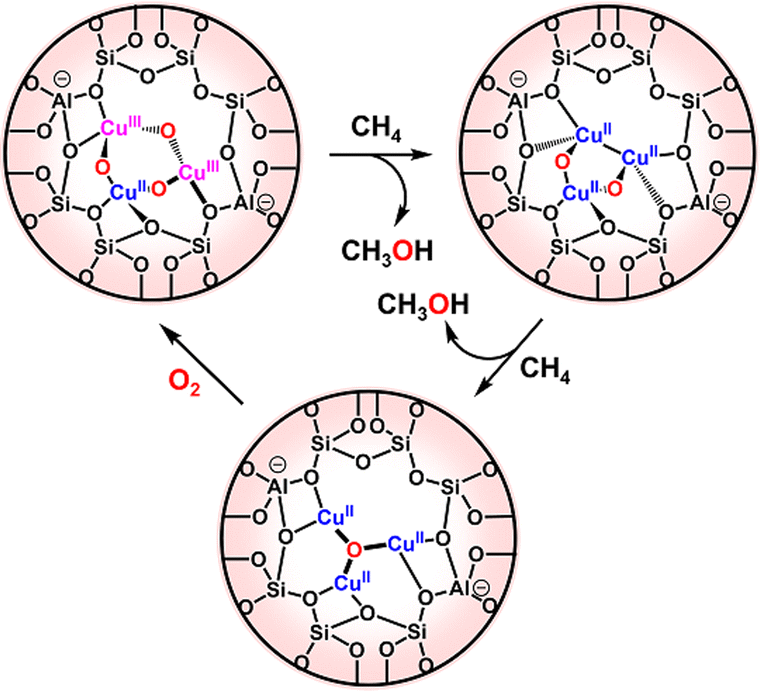 | ||
| Fig. 16 Formation of a [Cu3(μ3-O)]2+ species and two CH3OH molecules from the reaction of CH4 with [Cu3(μ-O)3]2+ in MOR zeolite. Adapted from ref. 252. | ||
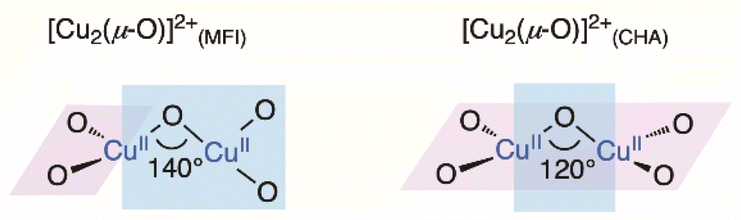 | ||
| Fig. 17 [Cu2(μ-O)]2+ in MFI (left) and CHA (right). | ||
These phenomena where the active site is found to be located in a constricted region of the zeolite lattice are denominated as 2nd sphere effects. Here, close van der Waals contact between the substrate and the zeolite lattice in the vicinity of the active site is increased leading thus to higher activities. Recently, the authors found that the superior activity of such [Cu2(μ-O)]2+ species within the CHA framework is tightly related to their inner ferromagnetic character due to the π bonding ligation with their oxido bridges rather than their Cu–O–Cu angles as previously observed for homogeneous complexes.278 Within this context, when bending the Cu–O–Cu angle, each CuII nucleus binds with a different oxido p-orbital, lowering overlap and leading to orthogonal magnetic orbitals on the bridge. Also, Stepanov279 studied the reactivity of [Cu3(μ-O)3]2+ oxido clusters on Cu-ZSM-5 after methane activation for both benzene and CO methylation finding that methoxy species at [Cu3(μ-O)3]2+ oxido clusters easily interact with benzene yielding toluene.
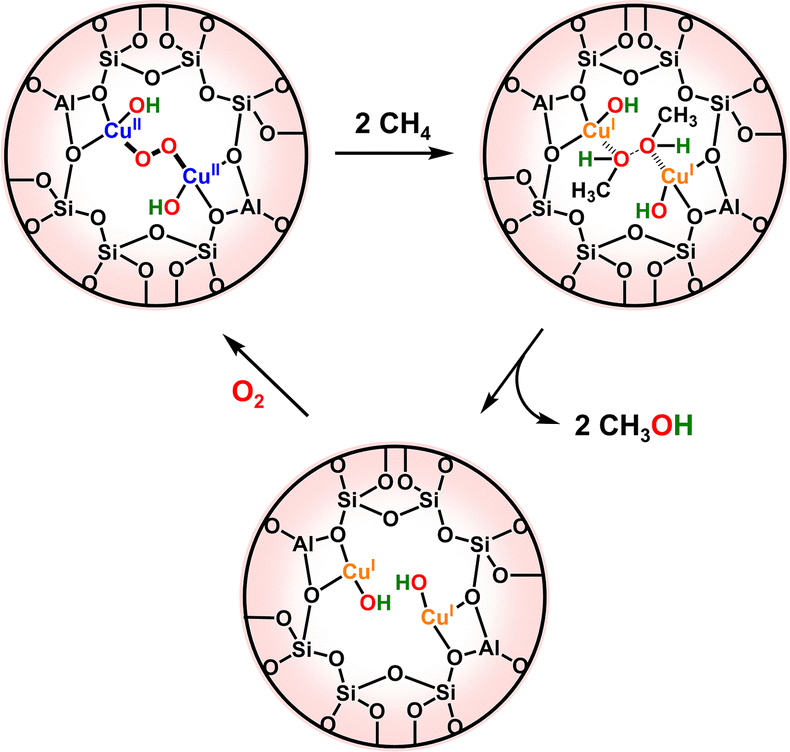 | ||
| Fig. 18 Proposed catalytic cycle for the CH4/O2/H2O reaction over Cu zeolites. | ||
6.4 Other supports
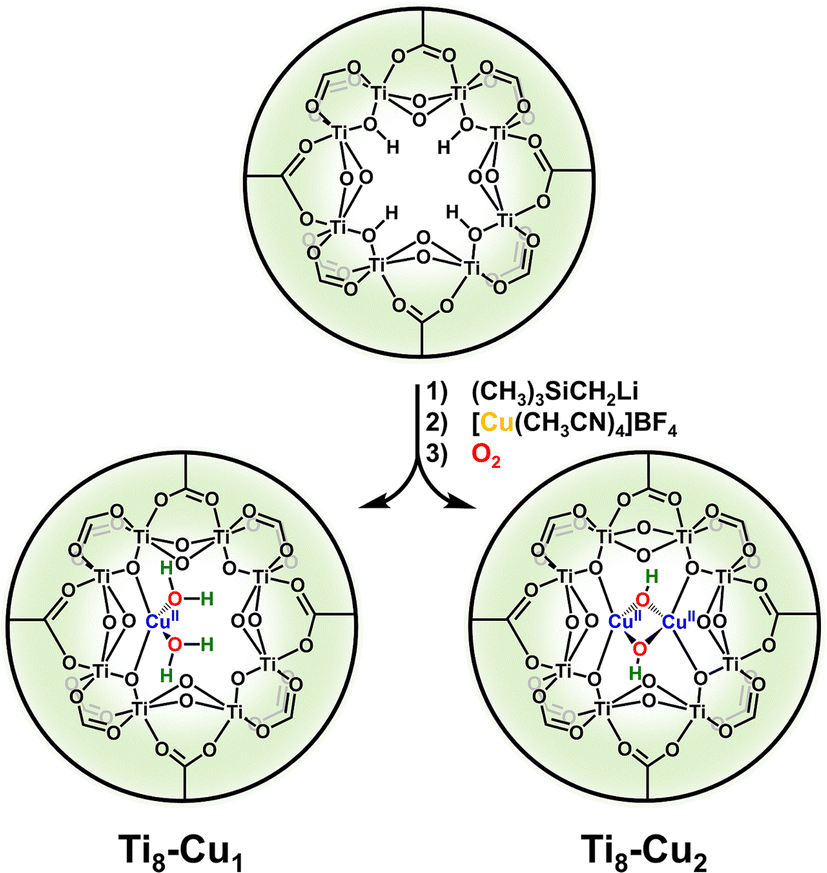 | ||
| Fig. 19 Incorporation of copper centers and subsequent treatment with O2 affording Ti8–Cu1 and Ti8–Cu2. | ||
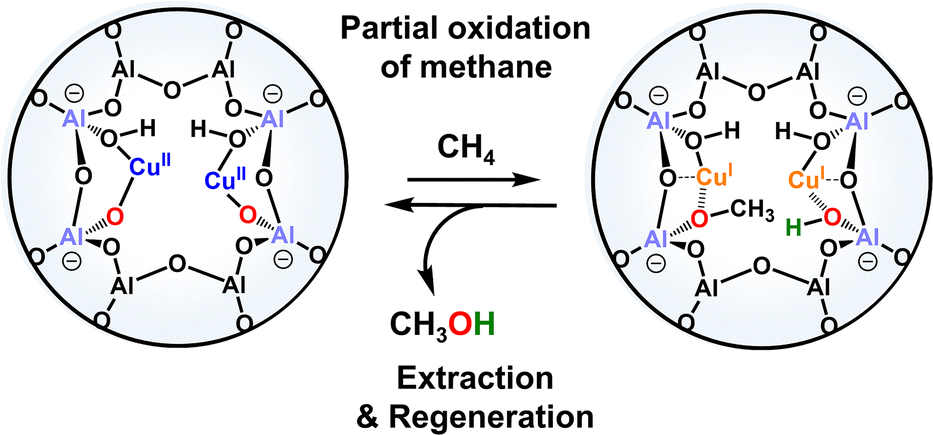 | ||
| Fig. 20 Strategy used to generate monomeric [Cu]2+ sites in a pure alumina environment and the subsequent reaction with CH4 to form hydroxyl and methoxy species. | ||
6.5 New strategies
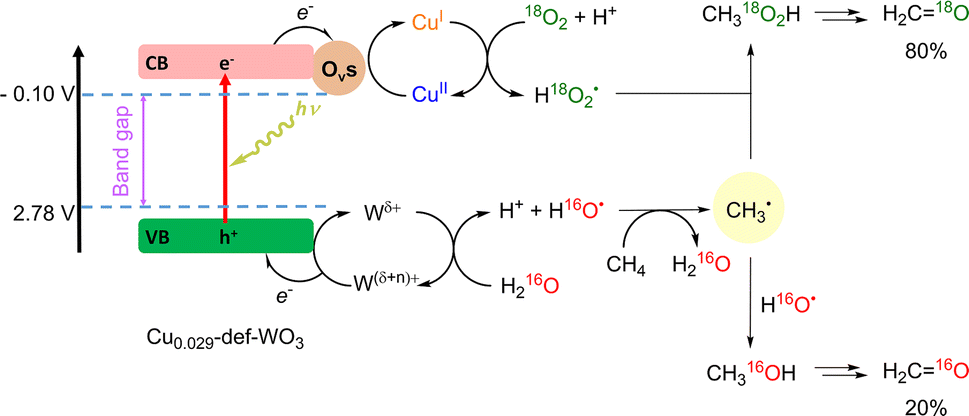 | ||
| Fig. 21 Proposed mechanism for charge transfer steps during selective CH4 oxidation over Cu0.029-def-WO3. | ||
7 Concluding remarks and perspectives
Copper–oxygen species constitute a distinctive and important class of oxidation catalysts within natural systems. Although significant advancements have been achieved over the past few years in terms of fundamental and applied research still significant gaps persist in our understanding of their efficiency. This is particularly highlighted by the ongoing reconsideration of some established mechanisms in light of recent breakthroughs. The typical examples are the recent debates about the nuclearity of the reactive copper–oxygen species (mono or di) in DβH, as well as the re-evaluation of an LPMO enzyme as peroxidases rather than monooxygenases.In the bio-inspired chemistry field, strong efforts have been particularly made during these last few years to develop synthetic copper–oxygen species able to perform efficient oxygenation of recalcitrant C–H bonds, methane being the targeted substrate.
Noteworthily, recent developments have included the exploration of copper cluster complexes incorporated within heterogeneous systems, such as zeolites. These catalysts were shown to advantageously afford the generation of highly oxidative mono- and polynuclear active sites which are hardly accessible in solution. Aside from the promising results obtained with heterogeneous systems, homogeneous copper–oxygen species still remain relevant due to valuable insights on the description of catalytic intermediates. Recent technological breakthroughs have strongly contributed to the characterization of new types of copper–oxygen transient species, such as dinuclear mixed-valent systems. In particular, in situ spectroscopic cryogenic techniques and synchrotron-based methods have played pivotal roles in the field in defining structure/function correlations and reaction coordinates in catalysis. Additionally, cryo-electron microscopy (cryo-EM) introduces innovative tools for elucidating the three-dimensional structure of proteins and has played a role in reopening the debate on pMMO. Altogether, these approaches have allowed easier determination of the thermodynamic properties (BDFEs) of copper–oxygen species related to their oxidative HAA power. In addition, significant progress in the field of computational studies has obviously demonstrated that these theoretical approaches have become crucial tools to improve our in-depth understanding and assist in the analysis of ambiguous experimental data.
Future work in the field of copper–oxygen species will have to consider all these new aspects/findings in order to design efficient catalysts for the activation of inert C–H bonds. The development of coupled techniques (spectroscopy, electrochemistry, spectrometry) for the in situ and time-resolved characterization of transient species will be of help to elucidate more deeply the different mechanisms.298 Alternatively, novel approaches based on photoinduced copper catalysis and radical chemistry may be applied to activate substrates under mild conditions, as recently reported.299
Author contributions
J. Simaan, M. Réglier, J. De Tovar, N. Le Poul and C. Belle contributed equally to writing the original draft and to reviewing and editing the manuscript. A. Thibon-Pourret, L. Wojcik, R. Leblay, Y. Wang contributed to writing, reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the French Agence Nationale de la Recherche (ANR-13-BSO7-0018 and ANR-22-CE07-0032). This work has been partially supported by CBH-EUR-GS (ANR-17-EURE-0003) and Labex ARCANE (ANR-11-LABX-0003-01) programs, in the framework of which this work was carried out. Region-Sud PACA and CSC are acknowledged for the PhD fellowships of R. L. and Y. W. respectively.References
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Copper Active Sites in Biology, Chem. Rev., 2014, 114(7), 3659–3853, DOI:10.1021/cr400327t.
- L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Structure and Spectroscopy of Copper−Dioxygen Complexes, Chem. Rev., 2004, 104(2), 1013–1046, DOI:10.1021/cr020632z.
- W. Keown, J. B. Gary and T. D. P. Stack, High-Valent Copper in Biomimetic and Biological Oxidations, J. Biol. Inorg. Chem., 2017, 22(2–3), 289–305, DOI:10.1007/s00775-016-1420-5.
- S. Itoh, Developing Mononuclear Copper–Active-Oxygen Complexes Relevant to Reactive Intermediates of Biological Oxidation Reactions, Acc. Chem. Res., 2015, 48(7), 2066–2074, DOI:10.1021/acs.accounts.5b00140.
- C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Copper–Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity, Chem. Rev., 2017, 117(3), 2059–2107, DOI:10.1021/acs.chemrev.6b00636.
- D. A. Quist, D. E. Diaz, J. J. Liu and K. D. Karlin, Activation of Dioxygen by Copper Metalloproteins and Insights from Model Complexes, JBIC, J. Biol. Inorg. Chem., 2017, 22(2–3), 253–288, DOI:10.1007/s00775-016-1415-2.
- R. Trammell, K. Rajabimoghadam and I. Garcia-Bosch, Copper-Promoted Functionalization of Organic Molecules: From Biologically Relevant Cu/O2 Model Systems to Organometallic Transformations, Chem. Rev., 2019, 119(4), 2954–3031, DOI:10.1021/acs.chemrev.8b00368.
- M. Spínola-Amilibia, R. Illanes-Vicioso, E. Ruiz-López, P. Colomer-Vidal, F. Rodriguez-Ventura, R. Peces Pérez, C. F. Arias, T. Torroba, M. Solà, E. Arias-Palomo and F. Bertocchini, Plastic Degradation by Insect Hexamerins: Near-Atomic Resolution Structures of the Polyethylene-Degrading Proteins from the Wax Worm Saliva, Sci. Adv., 2023, 9(38), eadi6813, DOI:10.1126/sciadv.adi6813.
- A. Koch, T. A. Engesser, R. Jurgeleit and F. Tuczek, Monooxygenation of phenols by small-molecule models of tyrosinase: correlations between structure and catalytic activity, in Copper Bioinorganic Chemistry, World Scientific, 2023, pp. 123–152, DOI:10.1142/9789811269493_0004.
- P. Gupta and R. Mukherjee, Modeling tyrosinase activity using m-Xylyl-based ligands: ring hydroxylation, reactivity, and theoretical investigation, in Copper Bioinorganic Chemistry, World Scientific, 2023, pp. 81–122, DOI:10.1142/9789811269493_0003.
- A. Bijelic, A. Rompel and C. Belle, Tyrosinases: enzymes, models and related applications, in Series on Chemistry, Energy and the Environment, World Scientific, 2019, pp. 155–183, DOI:10.1142/9789813274440_0007.
- C. Faure, A. du Moulinet d'Hardemare, H. Jamet, C. Belle, E. Bergantino, L. Bubacco, M. Benfatto, J. A. Simaan and M. Réglier, Transition state analogue molecules as mechanistic tools and inhibitors for tyrosinase, in Copper Bioinorganic Chemistry, From Health to Bioinspired Catalysis, World Scientific, 2023 Search PubMed.
- N. Fujieda, K. Umakoshi, Y. Ochi, Y. Nishikawa, S. Yanagisawa, M. Kubo, G. Kurisu and S. Itoh, Copper–Oxygen Dynamics in the Tyrosinase Mechanism, Angew. Chem., Int. Ed., 2020, 59(32), 13385–13390, DOI:10.1002/anie.202004733.
- M. Rolff, J. Schottenheim, H. Decker and F. Tuczek, Copper–O2 Reactivity of Tyrosinase Models towards External Monophenolic Substrates: Molecular Mechanism and Comparison with the Enzyme, Chem. Soc. Rev., 2011, 40(7), 4077–4098, 10.1039/C0CS00202J.
- I. Kipouros, A. Stańczak, J. W. Ginsbach, P. C. Andrikopoulos, L. Rulíšek and E. I. Solomon, Elucidation of the Tyrosinase/O2/Monophenol Ternary Intermediate That Dictates the Monooxygenation Mechanism in Melanin Biosynthesis, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(33), e2205619119, DOI:10.1073/pnas.2205619119.
- I. Kipouros and E. I. Solomon, New Mechanistic Insights into Coupled Binuclear Copper Monooxygenases from the Recent Elucidation of the Ternary Intermediate of Tyrosinase, FEBS Lett., 2023, 597(1), 65–78, DOI:10.1002/1873-3468.14503.
- I. Kipouros, A. Stańczak, M. Culka, E. Andris, T. R. Machonkin, L. Rulíšek and E. I. Solomon, Evidence for H-Bonding Interactions to the μ-η2:η2 -Peroxide of Oxy-Tyrosinase That Activate Its Coupled Binuclear Copper Site, Chem. Commun., 2022, 58(24), 3913–3916, 10.1039/D2CC00750A.
- I. Kipouros, A. Stańczak, E. M. Dunietz, J. W. Ginsbach, M. Srnec, L. Rulíšek and E. I. Solomon, Experimental Evidence and Mechanistic Description of the Phenolic H-Transfer to the Cu2O2 Active Site of Oxy-Tyrosinase, J. Am. Chem. Soc., 2023, 145(42), 22866–22870, DOI:10.1021/jacs.3c07450.
- A. L. Concia, A. Munzone, M.-C. Kafentzi, A. Kochem, M. Réglier, C. Decroos and A. Jalila Simaan, Modeling the mononuclear copper monooxygenase active site, in Series on Chemistry, Energy and the Environment, World Scientific, 2019, pp. 185–263, DOI:10.1142/9789813274440_0008.
- T. V. Vendelboe, P. Harris, Y. Zhao, T. S. Walter, K. Harlos, K. El Omari and H. E. M. Christensen, The Crystal Structure of Human Dopamine β-Hydroxylase at 2.9 Å Resolution, Sci. Adv., 2016, 2(4), e1500980, DOI:10.1126/sciadv.1500980.
- P. Wu, F. Fan, J. Song, W. Peng, J. Liu, C. Li, Z. Cao and B. Wang, Theory Demonstrated a “Coupled” Mechanism for O2 Activation and Substrate Hydroxylation by Binuclear Copper Monooxygenases, J. Am. Chem. Soc., 2019, 141(50), 19776–19789, DOI:10.1021/jacs.9b09172.
- E. F. Welch, K. W. Rush, R. J. Arias and N. J. Blackburn, Pre-Steady-State Reactivity of Peptidylglycine Monooxygenase Implicates Ascorbate in Substrate Triggering of the Active Conformer, Biochemistry, 2022, 61(8), 665–677, DOI:10.1021/acs.biochem.2c00080.
- E. F. Welch, K. W. Rush, R. J. Arias and N. J. Blackburn, Copper Monooxygenase Reactivity: Do Consensus Mechanisms Accurately Reflect Experimental Observations?, J. Inorg. Biochem., 2022, 231, 111780, DOI:10.1016/j.jinorgbio.2022.111780.
- R. J. Arias, E. F. Welch and N. J. Blackburn, New Structures Reveal Flexible Dynamics between the Subdomains of Peptidylglycine Monooxygenase. Implications for an Open to Closed Mechanism, Protein Sci., 2023, 32(4), e4615, DOI:10.1002/pro.4615.
- K. W. Rush, K. A. S. Eastman, E. F. Welch, V. Bandarian and N. J. Blackburn, Capturing the Binuclear Copper State of Peptidylglycine Monooxygenase Using a Peptidyl-Homocysteine Lure, J. Am. Chem. Soc., 2024, 146(8), 5074–5080, DOI:10.1021/jacs.3c14705.
- I. Blain, P. Slama, M. Giorgi, T. Tron and M. Réglier, Copper-Containing Monooxygenases: Enzymatic and Biomimetic Studies of the O-Atom Transfer Catalysis, Rev. Mol. Biotechnol., 2002, 90(2), 95–112, DOI:10.1016/S1389-0352(01)00068-X.
- S. Itoh, T. Kondo, M. Komatsu, Y. Ohshiro, C. Li, N. Kanehisa, Y. Kai and S. Fukuzumi, Functional Model of Dopamine β.-Hydroxylase. Quantitative Ligand Hydroxylation at the Benzylic Position of a Copper Complex by Dioxygen, J. Am. Chem. Soc., 1995, 117(16), 4714–4715, DOI:10.1021/ja00121a033.
- E. A. Lewis and W. B. Tolman, Reactivity of Dioxygen−Copper Systems, Chem. Rev., 2004, 104(2), 1047–1076, DOI:10.1021/cr020633r.
- I. Blain, P. Bruno, M. Giorgi, E. Lojou, D. Lexa and M. Réglier, Copper Complexes as Functional Models for Dopamine β-Hydroxylase – Stereospecific Oxygen Atom Transfer, Eur. J. Inorg. Chem., 1998, 1998(9), 1297–1304, DOI:10.1002/(SICI)1099-0682(199809)1998:9<1297::AID-EJIC1297>3.0.CO;2-M.
- I. Blain, M. Giorgi, I. De Riggi and M. Réglier, Copper Complexes as Functional Models for Dopamine β-Hydroxylase – Mechanistic Study of Oxygen Atom Transfer from Cu/O Species to Benzylic C−H Bonds, Eur. J. Inorg. Chem., 2001, 2001(1), 205–211, DOI:10.1002/1099-0682(20011)2001:1<205::AID-EJIC205>3.0.CO;2-U.
- I. Blain, M. Giorgi, I. De Riggi and M. Réglier, Substrate-Binding Ligand Approach in Chemical Modeling of Copper-Containing Monooxygenases, Intramolecular Stereoselective Oxygen Atom Insertion into a Non-Activated C–H Bond, Eur. J. Inorg. Chem., 2000, 2000(2), 393–398, DOI:10.1002/(SICI)1099-0682(200002)2000:2<393::AID-EJIC393>3.0.CO;2-2.
- C. W. Koo and A. C. Rosenzweig, Biochemistry of Aerobic Biological Methane Oxidation, Chem. Soc. Rev., 2021, 50(5), 3424–3436, 10.1039/D0CS01291B.
- S. I. Chan, W.-H. Chang, S.-H. Huang, H.-H. Lin and S. S.-F. Yu, Catalytic Machinery of Methane Oxidation in Particulate Methane Monooxygenase (pMMO), J. Inorg. Biochem., 2021, 225, 111602, DOI:10.1016/j.jinorgbio.2021.111602.
- F. J. Tucci and A. C. Rosenzweig, Direct Methane Oxidation by Copper- and Iron-Dependent Methane Monooxygenases, Chem. Rev., 2024, 124(3), 1288–1320, DOI:10.1021/acs.chemrev.3c00727.
- M. O. Ross, F. MacMillan, J. Wang, A. Nisthal, T. J. Lawton, B. D. Olafson, S. L. Mayo, A. C. Rosenzweig and B. M. Hoffman, Particulate Methane Monooxygenase Contains Only Mononuclear Copper Centers, Science, 2019, 364(6440), 566–570, DOI:10.1126/science.aav2572.
- G. E. Cutsail, M. O. Ross, A. C. Rosenzweig and S. DeBeer, Towards a Unified Understanding of the Copper Sites in Particulate Methane Monooxygenase: An X-Ray Absorption Spectroscopic Investigation, Chem. Sci., 2021, 12(17), 6194–6209, 10.1039/D1SC00676B.
- R. J. Jodts, M. O. Ross, C. W. Koo, P. E. Doan, A. C. Rosenzweig and B. M. Hoffman, Coordination of the Copper Centers in Particulate Methane Monooxygenase: Comparison between Methanotrophs and Characterization of the CuC Site by EPR and ENDOR Spectroscopies, J. Am. Chem. Soc., 2021, 143(37), 15358–15368, DOI:10.1021/jacs.1c07018.
- L. Cao, O. Caldararu, A. C. Rosenzweig and U. Ryde, Quantum Refinement Does Not Support Dinuclear Copper Sites in Crystal Structures of Particulate Methane Monooxygenase, Angew. Chem., Int. Ed., 2018, 57(1), 162–166, DOI:10.1002/anie.201708977.
- W. Peng, X. Qu, S. Shaik and B. Wang, Deciphering the Oxygen Activation Mechanism at the CuC Site of Particulate Methane Monooxygenase, Nat. Catal., 2021, 4(4), 266–273, DOI:10.1038/s41929-021-00591-4.
- W.-H. Chang, H.-H. Lin, I.-K. Tsai, S.-H. Huang, S.-C. Chung, I.-P. Tu, S. S.-F. Yu and S. I. Chan, Copper Centers in the Cryo-EM Structure of Particulate Methane Monooxygenase Reveal the Catalytic Machinery of Methane Oxidation, J. Am. Chem. Soc., 2021, 143(26), 9922–9932, DOI:10.1021/jacs.1c04082.
- C. W. Koo, F. J. Tucci, Y. He and A. C. Rosenzweig, Recovery of Particulate Methane Monooxygenase Structure and Activity in a Lipid Bilayer, Science, 2022, 375(6586), 1287–1291, DOI:10.1126/science.abm3282.
- A. L. Concia, M. R. Beccia, M. Orio, F. T. Ferre, M. Scarpellini, F. Biaso, B. Guigliarelli, M. Réglier and A. J. Simaan, Copper Complexes as Bioinspired Models for Lytic Polysaccharide Monooxygenases, Inorg. Chem., 2017, 56(3), 1023–1026, DOI:10.1021/acs.inorgchem.6b02165.
- W. Peng, Z. Wang, Q. Zhang, S. Yan and B. Wang, Unraveling the Valence State and Reactivity of Copper Centers in Membrane-Bound Particulate Methane Monooxygenase, J. Am. Chem. Soc., 2023, 145(46), 25304–25317, DOI:10.1021/jacs.3c08834.
- A. Munzone, V. G. H. Eijsink, J.-G. Berrin and B. Bissaro, Expanding the Catalytic Landscape of Metalloenzymes with Lytic Polysaccharide Monooxygenases, Nat. Rev. Chem, 2024, 8(2), 106–119, DOI:10.1038/s41570-023-00565-z.
- P. H. Walton, G. J. Davies, D. E. Diaz and J. P. Franco-Cairo, The Histidine Brace: Nature's Copper Alternative to Haem?, FEBS Lett., 2023, 597(4), 485–494, DOI:10.1002/1873-3468.14579.
- J. A. Hangasky, T. C. Detomasi and M. A. Marletta, Glycosidic Bond Hydroxylation by Polysaccharide Monooxygenases, Trends Chem., 2019, 1(2), 198–209, DOI:10.1016/j.trechm.2019.01.007.
- K. K. Meier, S. M. Jones, T. Kaper, H. Hansson, M. J. Koetsier, S. Karkehabadi, E. I. Solomon, M. Sandgren and B. Kelemen, Oxygen Activation by Cu LPMOs in Recalcitrant Carbohydrate Polysaccharide Conversion to Monomer Sugars, Chem. Rev., 2018, 118(5), 2593–2635, DOI:10.1021/acs.chemrev.7b00421.
- Lytic polysaccharide monooxygenases in special issue of biotechnology for biofuels, ed. J. G. Berrin and P. Walton, 2019.
- B. Bissaro, Å. K. Røhr, G. Müller, P. Chylenski, M. Skaugen, Z. Forsberg, S. J. Horn, G. Vaaje-Kolstad and V. G. H. Eijsink, Oxidative Cleavage of Polysaccharides by Monocopper Enzymes Depends on H2O2, Nat. Chem. Biol., 2017, 13(10), 1123–1128, DOI:10.1038/nchembio.2470.
- B. Bissaro and V. G. H. Eijsink, Lytic Polysaccharide Monooxygenases: Enzymes for Controlled and Site-Specific Fenton-like Chemistry, Essays Biochem., 2023, 67(3), 575–584, DOI:10.1042/EBC20220250.
- J. A. Hangasky, A. T. Iavarone and M. A. Marletta, Reactivity of O2 versus H2O2 with Polysaccharide Monooxygenases, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(19), 4915–4920, DOI:10.1073/pnas.1801153115.
- T. M. Hedison, E. Breslmayr, M. Shanmugam, K. Karnpakdee, D. J. Heyes, A. P. Green, R. Ludwig, N. S. Scrutton and D. Kracher, Insights into the H2O2 -driven Catalytic Mechanism of Fungal Lytic Polysaccharide Monooxygenases, FEBS J., 2021, 288(13), 4115–4128, DOI:10.1111/febs.15704.
- R. Kont, B. Bissaro, V. G. H. Eijsink and P. Väljamäe, Kinetic Insights into the Peroxygenase Activity of Cellulose-Active Lytic Polysaccharide Monooxygenases (LPMOs), Nat. Commun., 2020, 11(1), 5786, DOI:10.1038/s41467-020-19561-8.
- B. Wang, E. M. Johnston, P. Li, S. Shaik, G. J. Davies, P. H. Walton and C. Rovira, QM/MM Studies into the H 2 O 2 -Dependent Activity of Lytic Polysaccharide Monooxygenases: Evidence for the Formation of a Caged Hydroxyl Radical Intermediate, ACS Catal., 2018, 8(2), 1346–1351, DOI:10.1021/acscatal.7b03888.
- B. Wang, Z. Wang, G. J. Davies, P. H. Walton and C. Rovira, Activation of O 2 and H 2 O 2 by Lytic Polysaccharide Monooxygenases, ACS Catal., 2020, 10(21), 12760–12769, DOI:10.1021/acscatal.0c02914.
- G. C. Schröder, W. B. O'Dell, S. P. Webb, P. K. Agarwal and F. Meilleur, Capture of Activated Dioxygen Intermediates at the Copper-Active Site of a Lytic Polysaccharide Monooxygenase, Chem. Sci., 2022, 13(45), 13303–13320, 10.1039/D2SC05031E.
- B. Wang, X. Zhang, W. Fang, C. Rovira and S. Shaik, How Do Metalloproteins Tame the Fenton Reaction and Utilize •OH Radicals in Constructive Manners?, Acc. Chem. Res., 2022, 55(16), 2280–2290, DOI:10.1021/acs.accounts.2c00304.
- S. M. Jones, W. J. Transue, K. K. Meier, B. Kelemen and E. I. Solomon, Kinetic Analysis of Amino Acid Radicals Formed in H2O2-Driven CuI LPMO Reoxidation Implicates Dominant Homolytic Reactivity, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(22), 11916–11922, DOI:10.1073/pnas.1922499117.
- T. Dierks, B. Schmidt and K. Von Figura, Conversion of Cysteine to Formylglycine: A Protein Modification in the Endoplasmic Reticulum, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(22), 11963–11968, DOI:10.1073/pnas.94.22.11963.
- M. J. Appel and C. R. Bertozzi, Formylglycine, a Post-Translationally Generated Residue with Unique Catalytic Capabilities and Biotechnology Applications, ACS Chem. Biol., 2015, 10(1), 72–84, DOI:10.1021/cb500897w.
- D. A. Miarzlou, F. Leisinger, D. Joss, D. Häussinger and F. P. Seebeck, Structure of Formylglycine-Generating Enzyme in Complex with Copper and a Substrate Reveals an Acidic Pocket for Binding and Activation of Molecular Oxygen, Chem. Sci., 2019, 10(29), 7049–7058, 10.1039/C9SC01723B.
- M. J. Appel, K. K. Meier, J. Lafrance-Vanasse, H. Lim, C.-L. Tsai, B. Hedman, K. O. Hodgson, J. A. Tainer, E. I. Solomon and C. R. Bertozzi, Formylglycine-Generating Enzyme Binds Substrate Directly at a Mononuclear Cu(I) Center to Initiate O 2 Activation, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(12), 5370–5375, DOI:10.1073/pnas.1818274116.
- F. Leisinger, D. A. Miarzlou and F. P. Seebeck, Non-Coordinative Binding of O2 at the Active Center of a Copper-Dependent Enzyme, Angew. Chem., Int. Ed., 2021, 60(11), 6154–6159, DOI:10.1002/anie.202014981.
- Y. Wu, C. Zhao, Y. Su, S. Shaik and W. Lai, Mechanistic Insight into Peptidyl-Cysteine Oxidation by the Copper-Dependent Formylglycine-Generating Enzyme, Angew. Chem., 2023, 135(7), e202212053, DOI:10.1002/ange.202212053.
- D. Dhar and W. B. Tolman, Hydrogen Atom Abstraction from Hydrocarbons by a Copper(III)-Hydroxide Complex, J. Am. Chem. Soc., 2015, 137(3), 1322–1329, DOI:10.1021/ja512014z.
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications, Chem. Rev., 2022, 122(1), 1–49, DOI:10.1021/acs.chemrev.1c00521.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications, Chem. Rev., 2010, 110(12), 6961–7001, DOI:10.1021/cr100085k.
- C. F. Wise, R. G. Agarwal and J. M. Mayer, Determining Proton-Coupled Standard Potentials and X–H Bond Dissociation Free Energies in Nonaqueous Solvents Using Open-Circuit Potential Measurements, J. Am. Chem. Soc., 2020, 142(24), 10681–10691, DOI:10.1021/jacs.0c01032.
- D. Usharani, D. C. Lacy, A. S. Borovik and S. Shaik, Dichotomous Hydrogen Atom Transfer vs Proton-Coupled Electron Transfer During Activation of X–H Bonds (X = C, N, O) by Nonheme Iron–Oxo Complexes of Variable Basicity, J. Am. Chem. Soc., 2013, 135(45), 17090–17104, DOI:10.1021/ja408073m.
- D. Bím, M. Maldonado-Domínguez, L. Rulíšek and M. Srnec, Beyond the Classical Thermodynamic Contributions to Hydrogen Atom Abstraction Reactivity, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(44), E10287–E10294, DOI:10.1073/pnas.1806399115.
- C. E. Elwell, M. Mandal, C. J. Bouchey, L. Que, C. J. Cramer and W. B. Tolman, Carboxylate Structural Effects on the Properties and Proton-Coupled Electron Transfer Reactivity of [CuO2CR]2+ Cores, Inorg. Chem., 2019, 58(23), 15872–15879, DOI:10.1021/acs.inorgchem.9b02293.
- M. Mandal, C. E. Elwell, C. J. Bouchey, T. J. Zerk, W. B. Tolman and C. J. Cramer, Mechanisms for Hydrogen-Atom Abstraction by Mononuclear Copper(III) Cores: Hydrogen-Atom Transfer or Concerted Proton-Coupled Electron Transfer?, J. Am. Chem. Soc., 2019, 141(43), 17236–17244, DOI:10.1021/jacs.9b08109.
- T. Wu, S. N. MacMillan, K. Rajabimoghadam, M. A. Siegler, K. M. Lancaster and I. Garcia-Bosch, Structure, Spectroscopy, and Reactivity of a Mononuclear Copper Hydroxide Complex in Three Molecular Oxidation States, J. Am. Chem. Soc., 2020, 142(28), 12265–12276, DOI:10.1021/jacs.0c03867.
- I. Garcia-Bosch, R. E. Cowley, D. E. Díaz, R. L. Peterson, E. I. Solomon and K. D. Karlin, Substrate and Lewis Acid Coordination Promote O–O Bond Cleavage of an Unreactive L2CuII2(O22–) Species to Form L2CuIII2(O)2 Cores with Enhanced Oxidative Reactivity, J. Am. Chem. Soc., 2017, 139(8), 3186–3195, DOI:10.1021/jacs.6b12990.
- B. Kim and K. D. Karlin, Ligand–Copper(I) Primary O2-Adducts: Design, Characterization, and Biological Significance of Cupric–Superoxides, Acc. Chem. Res., 2023, 56(16), 2197–2212, DOI:10.1021/acs.accounts.3c00297.
- S. Itoh, T. Abe, Y. Morimoto and H. Sugimoto, 2-(2-Pyridyl)Ethylamine (Pye) Ligands in Copper(I)-Dioxygen Chemistry, Inorg. Chim. Acta, 2018, 481, 38–46, DOI:10.1016/j.ica.2017.09.017.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Structure and Reactivity of the First-Row d-Block Metal-Superoxo Complexes, Dalton Trans., 2019, 48(26), 9469–9489, 10.1039/C9DT01402K.
- K. Fujisawa, M. Tanaka, Y. Moro-oka and N. Kitajima, A Monomeric Side-On Superoxocopper(II) Complex: Cu(O2)(HB(3-tBu-5-iPrpz)3), J. Am. Chem. Soc., 1994, 116(26), 12079–12080, DOI:10.1021/ja00105a069.
- N. W. Aboelella, E. A. Lewis, A. M. Reynolds, W. W. Brennessel, C. J. Cramer and W. B. Tolman, Snapshots of Dioxygen Activation by Copper: The Structure of a 1:1 Cu/O2 Adduct and Its Use in Syntheses of Asymmetric Bis(μ-Oxo) Complexes, J. Am. Chem. Soc., 2002, 124(36), 10660–10661, DOI:10.1021/ja027164v.
- A. M. Reynolds, B. F. Gherman, C. J. Cramer and W. B. Tolman, Characterization of a 1:1 Cu−O2 Adduct Supported by an Anilido Imine Ligand, Inorg. Chem., 2005, 44(20), 6989–6997, DOI:10.1021/ic050280p.
- C. Würtele, E. Gaoutchenova, K. Harms, M. C. Holthausen, J. Sundermeyer and S. Schindler, Crystallographic Characterization of a Synthetic 1:1 End-On Copper Dioxygen Adduct Complex, Angew. Chem., Int. Ed., 2006, 45(23), 3867–3869, DOI:10.1002/anie.200600351.
- D. A. Iovan, A. T. Wrobel, A. A. McClelland, A. B. Scharf, G. A. Edouard and T. A. Betley, Reactivity of a Stable Copper–Dioxygen Complex, Chem. Commun., 2017, 53(74), 10306–10309, 10.1039/C7CC05014C.
- K. M. Carsch, A. Iliescu, R. D. McGillicuddy, J. A. Mason and T. A. Betley, Reversible Scavenging of Dioxygen from Air by a Copper Complex, J. Am. Chem. Soc., 2021, 143(43), 18346–18352, DOI:10.1021/jacs.1c10254.
- R. Davydov, A. E. Herzog, R. J. Jodts, K. D. Karlin and B. M. Hoffman, End-On Copper(I) Superoxo and Cu(II) Peroxo and Hydroperoxo Complexes Generated by Cryoreduction/Annealing and Characterized by EPR/ENDOR Spectroscopy, J. Am. Chem. Soc., 2022, 144(1), 377–389, DOI:10.1021/jacs.1c10252.
- B. D. Neisen, N. L. Gagnon, D. Dhar, A. D. Spaeth and W. B. Tolman, Formally Copper(III)–Alkylperoxo Complexes as Models of Possible Intermediates in Monooxygenase Enzymes, J. Am. Chem. Soc., 2017, 139(30), 10220–10223, DOI:10.1021/jacs.7b05754.
- S. I. Mann, T. Heinisch, T. R. Ward and A. S. Borovik, Peroxide Activation Regulated by Hydrogen Bonds within Artificial Cu Proteins, J. Am. Chem. Soc., 2017, 139(48), 17289–17292, DOI:10.1021/jacs.7b10452.
- V. M. Krishnan, D. Y. Shopov, C. J. Bouchey, W. D. Bailey, R. Parveen, B. Vlaisavljevich and W. B. Tolman, Structural Characterization of the [CuOR]2+ Core, J. Am. Chem. Soc., 2021, 143(9), 3295–3299, DOI:10.1021/jacs.0c13470.
- J.-P. Bacik, S. Mekasha, Z. Forsberg, A. Y. Kovalevsky, G. Vaaje-Kolstad, V. G. H. Eijsink, J. C. Nix, L. Coates, M. J. Cuneo, C. J. Unkefer and J. C.-H. Chen, Neutron and Atomic Resolution X-Ray Structures of a Lytic Polysaccharide Monooxygenase Reveal Copper-Mediated Dioxygen Binding and Evidence for N-Terminal Deprotonation, Biochemistry, 2017, 56(20), 2529–2532, DOI:10.1021/acs.biochem.7b00019.
- S. Paria, Y. Morimoto, T. Ohta, S. Okabe, H. Sugimoto, T. Ogura and S. Itoh, Copper(I)–Dioxygen Reactivity in the Isolated Cavity of a Nanoscale Molecular Architecture, Eur. J. Inorg. Chem., 2018, 2018(19), 1976–1983, DOI:10.1002/ejic.201800029.
- M. E. Czaikowski, A. J. McNeece, J.-N. Boyn, K. A. Jesse, S. W. Anferov, A. S. Filatov, D. A. Mazziotti and J. S. Anderson, Generation and Aerobic Oxidative Catalysis of a Cu(II) Superoxo Complex Supported by a Redox-Active Ligand, J. Am. Chem. Soc., 2022, 144(34), 15569–15580, DOI:10.1021/jacs.2c04630.
- D. A. Quist, M. A. Ehudin and K. D. Karlin, Unprecedented Direct Cupric-Superoxo Conversion to a Bis-μ-Oxo Dicopper(III) Complex and Resulting Oxidative Activity, Inorg. Chim. Acta, 2019, 485, 155–161, DOI:10.1016/j.ica.2018.10.015.
- S. Y. Quek, S. Debnath, S. Laxmi, M. Van Gastel, T. Krämer and J. England, Sterically Stabilized End-On Superoxocopper(II) Complexes and Mechanistic Insights into Their Reactivity with O–H, N–H, and C–H Substrates, J. Am. Chem. Soc., 2021, 143(47), 19731–19747, DOI:10.1021/jacs.1c07837.
- S. Kim, J. Y. Lee, R. E. Cowley, J. W. Ginsbach, M. A. Siegler, E. I. Solomon and K. D. Karlin, A N3S(thioether)-Ligated CuII -Superoxo with Enhanced Reactivity, J. Am. Chem. Soc., 2015, 137(8), 2796–2799, DOI:10.1021/ja511504n.
- M. Bhadra, W. J. Transue, H. Lim, R. E. Cowley, J. Y. C. Lee, M. A. Siegler, P. Josephs, G. Henkel, M. Lerch, S. Schindler, A. Neuba, K. O. Hodgson, B. Hedman, E. I. Solomon and K. D. Karlin, A Thioether-Ligated Cupric Superoxide Model with Hydrogen Atom Abstraction Reactivity, J. Am. Chem. Soc., 2021, 143(10), 3707–3713, DOI:10.1021/jacs.1c00260.
- T. Hoppe, P. Josephs, N. Kempf, C. Wölper, S. Schindler, A. Neuba and G. Henkel, An Approach to Model the Active Site of Peptidglycine-α-hydroxylating Monooxygenase (PHM), Z. Anorg. Allg. Chem., 2013, 639(8–9), 1504–1511, DOI:10.1002/zaac.201300066.
- R. L. Shook and A. S. Borovik, Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation, Inorg. Chem., 2010, 49(8), 3646–3660, DOI:10.1021/ic901550k.
- M. Bhadra, J. Y. C. Lee, R. E. Cowley, S. Kim, M. A. Siegler, E. I. Solomon and K. D. Karlin, Intramolecular Hydrogen Bonding Enhances Stability and Reactivity of Mononuclear Cupric Superoxide Complexes, J. Am. Chem. Soc., 2018, 140(29), 9042–9045, DOI:10.1021/jacs.8b04671.
- D. E. Diaz, D. A. Quist, A. E. Herzog, A. W. Schaefer, I. Kipouros, M. Bhadra, E. I. Solomon and K. D. Karlin, Impact of Intramolecular Hydrogen Bonding on the Reactivity of Cupric Superoxide Complexes with O−H and C−H Substrates, Angew. Chem., Int. Ed., 2019, 58(49), 17572–17576, DOI:10.1002/anie.201908471.
- D. L. Ross, A. J. Jasniewski, J. W. Ziller, E. L. Bominaar, M. P. Hendrich and A. S. Borovik, Modulation of the Bonding between Copper and a Redox-Active Ligand by Hydrogen Bonds and Its Effect on Electronic Coupling and Spin States, J. Am. Chem. Soc., 2024, 146(1), 500–513, DOI:10.1021/jacs.3c09983.
- P. J. Donoghue, A. K. Gupta, D. W. Boyce, C. J. Cramer and W. B. Tolman, An Anionic, Tetragonal Copper(II) Superoxide Complex, J. Am. Chem. Soc., 2010, 132(45), 15869–15871, DOI:10.1021/ja106244k.
- P. Pirovano, A. M. Magherusan, C. McGlynn, A. Ure, A. Lynes and A. R. McDonald, Nucleophilic Reactivity of a Copper(II)–Superoxide Complex, Angew. Chem., Int. Ed., 2014, 53(23), 5946–5950, DOI:10.1002/anie.201311152.
- W. D. Bailey, N. L. Gagnon, C. E. Elwell, A. C. Cramblitt, C. J. Bouchey and W. B. Tolman, Revisiting the Synthesis and Nucleophilic Reactivity of an Anionic Copper Superoxide Complex, Inorg. Chem., 2019, 58(8), 4706–4711, DOI:10.1021/acs.inorgchem.9b00090.
- W. D. Bailey, D. Dhar, A. C. Cramblitt and W. B. Tolman, Mechanistic Dichotomy in Proton-Coupled Electron-Transfer Reactions of Phenols with a Copper Superoxide Complex, J. Am. Chem. Soc., 2019, 141(13), 5470–5480, DOI:10.1021/jacs.9b00466.
- T. Abe, Y. Hori, Y. Shiota, T. Ohta, Y. Morimoto, H. Sugimoto, T. Ogura, K. Yoshizawa and S. Itoh, Cupric-Superoxide Complex That Induces a Catalytic Aldol Reaction-Type C–C Bond Formation, Commun. Chem., 2019, 2(1), 12, DOI:10.1038/s42004-019-0115-6.
- K. J. Kadassery, S. K. Dey, A. F. Cannella, R. Surendhran and D. C. Lacy, Photochemical Water-Splitting with Organomanganese Complexes, Inorg. Chem., 2017, 56(16), 9954–9965, DOI:10.1021/acs.inorgchem.7b01483.
- D. Jeong, J. Selverstone Valentine and J. Cho, Bio-Inspired Mononuclear Nonheme Metal Peroxo Complexes: Synthesis, Structures and Mechanistic Studies toward Understanding Enzymatic Reactions, Coord. Chem. Rev., 2023, 480, 215021, DOI:10.1016/j.ccr.2023.215021.
- B. Kim, D. Jeong and J. Cho, Nucleophilic Reactivity of Copper(ii)–Alkylperoxo Complexes, Chem. Commun., 2017, 53(67), 9328–9331, 10.1039/C7CC03965D.
- B. Kim, D. Jeong, T. Ohta and J. Cho, Nucleophilic Reactivity of a Copper(II)-Hydroperoxo Complex, Commun. Chem., 2019, 2(1), 81, DOI:10.1038/s42004-019-0187-3.
- T. Abe, Y. Morimoto, K. Mieda, H. Sugimoto, N. Fujieda, T. Ogura and S. Itoh, Geometric Effects on O-O Bond Scission of Copper(II)-Alkylperoxide Complexes, J. Inorg. Biochem., 2017, 177, 375–383, DOI:10.1016/j.jinorgbio.2017.08.016.
- I. Shimizu, Y. Morimoto, G. Velmurugan, T. Gupta, S. Paria, T. Ohta, H. Sugimoto, T. Ogura, P. Comba and S. Itoh, Characterization and Reactivity of a Tetrahedral Copper(II) Alkylperoxido Complex, Chem.–Eur. J., 2019, 25(47), 11157–11165, DOI:10.1002/chem.201902669.
- S. Paria, T. Ohta, Y. Morimoto, H. Sugimoto, T. Ogura and S. Itoh, Structure and Reactivity of Copper Complexes Supported by a Bulky Tripodal N4 Ligand: Copper(I)/Dioxygen Reactivity and Formation of a Hydroperoxide Copper(II) Complex, Z. Anorg. Allg. Chem., 2018, 644(14), 780–789, DOI:10.1002/zaac.201800083.
- N. Kitajima, T. Katayama, K. Fujisawa, Y. Iwata and Y. Morooka, Synthesis, Molecular Structure, and Reactivity of (Alkylperoxo)Copper(II) Complex, J. Am. Chem. Soc., 1993, 115(17), 7872–7873, DOI:10.1021/ja00070a041.
- A. Wada, M. Harata, K. Hasegawa, K. Jitsukawa, H. Masuda, M. Mukai, T. Kitagawa and H. Einaga, Structural and Spectroscopic Characterization of a Mononuclear Hydroperoxo–Copper(II) Complex with Tripodal Pyridylamine Ligands, Angew. Chem., Int. Ed., 1998, 37(6), 798–799, DOI:10.1002/(SICI)1521-3773(19980403)37:6<798::AID-ANIE798>3.0.CO;2-3.
- A. D. Liang, J. Serrano-Plana, R. L. Peterson and T. R. Ward, Artificial Metalloenzymes Based on the Biotin–Streptavidin Technology: Enzymatic Cascades and Directed Evolution, Acc. Chem. Res., 2019, 52(3), 585–595, DOI:10.1021/acs.accounts.8b00618.
- S. I. Mann, T. Heinisch, A. C. Weitz, M. P. Hendrich, T. R. Ward and A. S. Borovik, Modular Artificial Cupredoxins, J. Am. Chem. Soc., 2016, 138(29), 9073–9076, DOI:10.1021/jacs.6b05428.
- S. Yamaguchi, S. Nagatomo, T. Kitagawa, Y. Funahashi, T. Ozawa, K. Jitsukawa and H. Masuda, Copper Hydroperoxo Species Activated by Hydrogen-Bonding Interaction with Its Distal Oxygen, Inorg. Chem., 2003, 42(22), 6968–6970, DOI:10.1021/ic035080x.
- S. Yamaguchi, A. Kumagai, S. Nagatomo, T. Kitagawa, Y. Funahashi, T. Ozawa, K. Jitsukawa and H. Masuda, Synthesis, Characterization, and Thermal Stability of New Mononuclear Hydrogenperoxocopper(II) Complexes with N3O-Type Tripodal Ligands Bearing Hydrogen-Bonding Interaction Sites, Bull. Chem. Soc. Jpn., 2005, 78(1), 116–124, DOI:10.1246/bcsj.78.116.
- S. Yamaguchi, A. Wada, S. Nagatomo, T. Kitagawa, K. Jitsukawa and H. Masuda, Thermal Stability of Mononuclear Hydroperoxocopper(II) Species. Effects of Hydrogen Bonding and Hydrophobic Field, Chem. Lett., 2004, 33(12), 1556–1557, DOI:10.1246/cl.2004.1556.
- I. Sanyal, P. Ghosh and K. D. Karlin, Mononuclear Copper(II)-Acylperoxo Complexes, Inorg. Chem., 1995, 34(11), 3050–3056, DOI:10.1021/ic00115a035.
- P. Ghosh, Z. Tyeklar, K. D. Karlin, R. R. Jacobson and J. Zubieta, Dioxygen-Copper Reactivity: X-Ray Structure and Characterization of an (Acylperoxo)Dicopper Complex, J. Am. Chem. Soc., 1987, 109(22), 6889–6891, DOI:10.1021/ja00256a073.
- N. Kitajima, K. Fujisawa and Y. Moro-oka, Formation and Characterization of a Mononuclear (Acylperoxo)Copper(II) Complex, Inorg. Chem., 1990, 29(3), 357–358, DOI:10.1021/ic00328a001.
- Y. Morimoto, M. Kawai, A. Nakanishi, H. Sugimoto and S. Itoh, Controlling the Reactivity of Copper(II) Acylperoxide Complexes, Inorg. Chem., 2021, 60(12), 8554–8565, DOI:10.1021/acs.inorgchem.1c00475.
- P. Specht, A. Petrillo, J. Becker and S. Schindler, Aerobic C−H Hydroxylation by Copper Imine Complexes: The Clip-and-Cleave Concept – Versatility and Limits, Eur. J. Inorg. Chem., 2021, 2021(20), 1961–1970, DOI:10.1002/ejic.202100185.
- K. Sagar, M. Kim, T. Wu, S. Zhang, E. L. Bominaar, M. A. Siegler, M. Hendrich and I. Garcia-Bosch, Mimicking the Reactivity of LPMOs with a Mononuclear Cu Complex, Eur. J. Inorg. Chem., 2024, e202300774, DOI:10.1002/ejic.202300774.
- I. Castillo, A. P. Torres-Flores, D. F. Abad-Aguilar, A. Berlanga-Vázquez, M. Orio and D. Martínez-Otero, Cellulose Depolymerization with LPMO-inspired Cu Complexes, ChemCatChem, 2021, 13(22), 4700–4704, DOI:10.1002/cctc.202101169.
- H. Oh, W.-M. Ching, J. Kim, W.-Z. Lee and S. Hong, Hydrogen Bond-Enabled Heterolytic and Homolytic Peroxide Activation within Nonheme Copper(II)-Alkylperoxo Complexes, Inorg. Chem., 2019, 58(19), 12964–12974, DOI:10.1021/acs.inorgchem.9b01898.
- T. Tano, M. Z. Ertem, S. Yamaguchi, A. Kunishita, H. Sugimoto, N. Fujieda, T. Ogura, C. J. Cramer and S. Itoh, Reactivity of Copper(II)-Alkylperoxo Complexes, Dalton Trans., 2011, 40(40), 10326, 10.1039/c1dt10656b.
- Y. Lee, B. Kim, S. Kim, E. W. H. Ng, S. Ariyasu, O. Shoji, S. Yoon, H. Hirao and J. Cho, Influence of Solvents on Catalytic C–H Bond Oxidation by a Copper(II)–Alkylperoxo Complex, ACS Catal., 2024, 3524–3532, DOI:10.1021/acscatal.3c05643.
- S. Muthuramalingam, K. Anandababu, M. Velusamy and R. Mayilmurugan, Benzene Hydroxylation by Bioinspired Copper(II) Complexes: Coordination Geometry versus Reactivity, Inorg. Chem., 2020, 59(9), 5918–5928, DOI:10.1021/acs.inorgchem.9b03676.
- S. Kumari, S. Muthuramalingam, A. K. Dhara, U. P. Singh, R. Mayilmurugan and K. Ghosh, Cu(I) Complexes Obtained via Spontaneous Reduction of Cu(II) Complexes Supported by Designed Bidentate Ligands: Bioinspired Cu(I) Based Catalysts for Aromatic Hydroxylation, Dalton Trans., 2020, 49(39), 13829–13839, 10.1039/D0DT02413A.
- W. Ghattas, M. Giorgi, Y. Mekmouche, T. Tanaka, A. Rockenbauer, M. Réglier, Y. Hitomi and A. J. Simaan, Identification of a Copper(I) Intermediate in the Conversion of 1-Aminocyclopropane Carboxylic Acid (ACC) into Ethylene by Cu(II)−ACC Complexes and Hydrogen Peroxide, Inorg. Chem., 2008, 47(11), 4627–4638, DOI:10.1021/ic702303g.
- S. Góger, J. S. Pap, D. Bogáth, A. J. Simaan, G. Speier, M. Giorgi and J. Kaizer, Copper Catalyzed Oxidation of Amino Acids, Polyhedron, 2014, 73, 37–44, DOI:10.1016/j.poly.2014.02.007.
- B. Kim, M. T. Brueggemeyer, W. J. Transue, Y. Park, J. Cho, M. A. Siegler, E. I. Solomon and K. D. Karlin, Fenton-like Chemistry by a Copper(I) Complex and H2O2 Relevant to Enzyme Peroxygenase C–H Hydroxylation, J. Am. Chem. Soc., 2023, 145(21), 11735–11744, DOI:10.1021/jacs.3c02273.
- Q. Zhang, S. Tong and M.-X. Wang, Unraveling the Chemistry of High Valent Arylcopper Compounds and Their Roles in Copper-Catalyzed Arene C–H Bond Transformations Using Synthetic Macrocycles, Acc. Chem. Res., 2022, 55(19), 2796–2810, DOI:10.1021/acs.accounts.2c00316.
- M. Bera, S. Kaur, K. Keshari, A. Santra, D. Moonshiram and S. Paria, Structural and Spectroscopic Characterization of Copper(III) Complexes and Subsequent One-Electron Oxidation Reaction and Reactivity Studies, Inorg. Chem., 2023, 62(14), 5387–5399, DOI:10.1021/acs.inorgchem.2c04168.
- R. Eerlapally, S. Gupta, A. Awasthi, R. Kumar and A. Draksharapu, Spectroscopic Characterization and the Reactivity of a High Valent (L)Cu(iii) Species Supported by a Proline-Based Pseudopeptide, Dalton Trans., 2023, 52(25), 8645–8653, 10.1039/D3DT00697B.
- Y. Liu, S. G. Resch, I. Klawitter, G. E. Cutsail, S. Demeshko, S. Dechert, F. E. Kühn, S. DeBeer and F. Meyer, An Adaptable N-Heterocyclic Carbene Macrocycle Hosting Copper in Three Oxidation States, Angew. Chem., Int. Ed., 2020, 59(14), 5696–5705, DOI:10.1002/anie.201912745.
- B. L. Geoghegan, Y. Liu, S. Peredkov, S. Dechert, F. Meyer, S. DeBeer and G. E. I. Cutsail, Combining Valence-to-Core X-Ray Emission and Cu K-Edge X-Ray Absorption Spectroscopies to Experimentally Assess Oxidation State in Organometallic Cu(I)/(II)/(III) Complexes, J. Am. Chem. Soc., 2022, 144(6), 2520–2534, DOI:10.1021/jacs.1c09505.
- I. M. DiMucci, J. T. Lukens, S. Chatterjee, K. M. Carsch, C. J. Titus, S. J. Lee, D. Nordlund, T. A. Betley, S. N. MacMillan and K. M. Lancaster, The Myth of d8 Copper(III), J. Am. Chem. Soc., 2019, 141(46), 18508–18520, DOI:10.1021/jacs.9b09016.
- N. Gagnon and W. B. Tolman, [CuO] + and [CuOH] 2+ Complexes: Intermediates in Oxidation Catalysis?, Acc. Chem. Res., 2015, 48(7), 2126–2131, DOI:10.1021/acs.accounts.5b00169.
- J. K. Bower, M. S. Reese, I. M. Mazin, L. M. Zarnitsa, A. D. Cypcar, C. E. Moore, A. Yu. Sokolov and S. Zhang, C(Sp3)–H Cyanation by a Formal Copper(iii) Cyanide Complex, Chem. Sci., 2023, 14(5), 1301–1307, 10.1039/D2SC06573H.
- J. K. Bower, A. D. Cypcar, B. Henriquez, S. C. E. Stieber and S. Zhang, C(Sp3)–H Fluorination with a Copper(II)/(III) Redox Couple, J. Am. Chem. Soc., 2020, 142(18), 8514–8521, DOI:10.1021/jacs.0c02583.
- C. J. Bouchey and W. B. Tolman, Involvement of a Formally Copper(III) Nitrite Complex in Proton-Coupled Electron Transfer and Nitration of Phenols, Inorg. Chem., 2022, 61(5), 2662–2668, DOI:10.1021/acs.inorgchem.1c03790.
- N. Dietl, M. Schlangen and H. Schwarz, Thermal Hydrogen-Atom Transfer from Methane: The Role of Radicals and Spin States in Oxo-Cluster Chemistry, Angew. Chem., Int. Ed., 2012, 51(23), 5544–5555, DOI:10.1002/anie.201108363.
- N. Dietl, C. van der Linde, M. Schlangen, M. K. Beyer and H. Schwarz, Diatomic [CuO]+ and Its Role in the Spin-Selective Hydrogen- and Oxygen-Atom Transfers in the Thermal Activation of Methane, Angew. Chem., Int. Ed., 2011, 50(21), 4966–4969, DOI:10.1002/anie.201100606.
- G. Yassaghi, E. Andris and J. Roithová, Reactivity of Copper(III)–Oxo Complexes in the Gas Phase, ChemPhysChem, 2017, 18(16), 2217–2224, DOI:10.1002/cphc.201700490.
- N. R. M. De Kler and J. Roithová, Copper Arylnitrene Intermediates: Formation, Structure and Reactivity, Chem. Commun., 2020, 56(84), 12721–12724, 10.1039/D0CC05198E.
- D. Maiti, A. A. Narducci Sarjeant and K. D. Karlin, Copper−Hydroperoxo-Mediated N-Debenzylation Chemistry Mimicking Aspects of Copper Monooxygenases, Inorg. Chem., 2008, 47(19), 8736–8747, DOI:10.1021/ic800617m.
- D. Dhar, G. M. Yee, A. D. Spaeth, D. W. Boyce, H. Zhang, B. Dereli, C. J. Cramer and W. B. Tolman, Perturbing the Copper(III)–Hydroxide Unit through Ligand Structural Variation, J. Am. Chem. Soc., 2016, 138(1), 356–368, DOI:10.1021/jacs.5b10985.
- B. Dereli, M. R. Momeni and C. J. Cramer, Density Functional Modeling of Ligand Effects on Electronic Structure and C–H Bond Activation Activity of Copper(III) Hydroxide Compounds, Inorg. Chem., 2018, 57(16), 9807–9813, DOI:10.1021/acs.inorgchem.8b01530.
- L. Ciano, G. J. Davies, W. B. Tolman and P. H. Walton, Bracing Copper for the Catalytic Oxidation of C–H Bonds, Nat. Catal., 2018, 1(8), 571–577, DOI:10.1038/s41929-018-0110-9.
- P. J. Lindley, A. Parkin, G. J. Davies and P. H. Walton, Mapping the Protonation States of the Histidine Brace in an AA10 Lytic Polysaccharide Monooxygenase Using CW-EPR Spectroscopy and DFT Calculations, Faraday Discuss., 2022, 234, 336–348, 10.1039/D1FD00068C.
- K. R. Hall, C. Joseph, I. Ayuso-Fernández, A. Tamhankar, L. Rieder, R. Skaali, O. Golten, F. Neese, Å. K. Røhr, S. A. V. Jannuzzi, S. DeBeer, V. G. H. Eijsink and M. Sørlie, A Conserved Second Sphere Residue Tunes Copper Site Reactivity in Lytic Polysaccharide Monooxygenases, J. Am. Chem. Soc., 2023, 145(34), 18888–18903, DOI:10.1021/jacs.3c05342.
- W. Wu, J. T. De Hont, R. Parveen, B. Vlaisavljevich and W. B. Tolman, Sulfur-Containing Analogues of the Reactive [CuOH]2+ Core, Inorg. Chem., 2021, 60(7), 5217–5223, DOI:10.1021/acs.inorgchem.1c00216.
- H. Hintz, J. Bower, J. Tang, M. LaLama, C. Sevov and S. Zhang, Copper-Catalyzed Electrochemical C–H Fluorination, Chem Catal., 2023, 3(1), 100491, DOI:10.1016/j.checat.2022.100491.
- R. R. Jacobson, Z. Tyeklar, A. Farooq, K. D. Karlin, S. Liu and J. Zubieta, A Copper-Oxygen (Cu2-O2) Complex. Crystal Structure and Characterization of a Reversible Dioxygen Binding System, J. Am. Chem. Soc., 1988, 110(11), 3690–3692, DOI:10.1021/ja00219a071.
- N. Kitajima, K. Fujisawa, Y. Morooka and K. Toriumi, Μ-N2:N2-Peroxo Binuclear Copper Complex, [Cu(HB(3,5-(Me2CH)2pz)3)]2(O2), J. Am. Chem. Soc., 1989, 111(24), 8975–8976, DOI:10.1021/ja00206a062.
- C. Citek, S. Herres-Pawlis and T. D. P. Stack, Low Temperature Syntheses and Reactivity of Cu2O2 Active-Site Models, Acc. Chem. Res., 2015, 48(8), 2424–2433, DOI:10.1021/acs.accounts.5b00220.
- E. I. Solomon, Dioxygen Binding, Activation, and Reduction to H2O by Cu Enzymes, Inorg. Chem., 2016, 55(13), 6364–6375, DOI:10.1021/acs.inorgchem.6b01034.
- N. Kindermann, E. Bill, S. Dechert, S. Demeshko, E. J. Reijerse and F. Meyer, A Ferromagnetically Coupled (S=1) Peroxodicopper(II) Complex, Angew. Chem., Int. Ed., 2015, 54(6), 1738–1743, DOI:10.1002/anie.201409709.
- T. Lohmiller, C.-J. Spyra, S. Dechert, S. Demeshko, E. Bill, A. Schnegg and F. Meyer, Antisymmetric Spin Exchange in a μ-1,2-Peroxodicopper(II) Complex with an Orthogonal Cu–O–O–Cu Arrangement and S = 1 Spin Ground State Characterized by THz-EPR, JACS Au, 2022, 2(5), 1134–1143, DOI:10.1021/jacsau.2c00139.
- K. E. Dalle, T. Gruene, S. Dechert, S. Demeshko and F. Meyer, Weakly Coupled Biologically Relevant CuII2(μ-ŋ1:ŋ1-O2) Cis-Peroxo Adduct That Binds Side-On to Additional Metal Ions, J. Am. Chem. Soc., 2014, 136(20), 7428–7434, DOI:10.1021/ja5025047.
- J. Nehrkorn, J. Telser, K. Holldack, S. Stoll and A. Schnegg, Simulating Frequency-Domain Electron Paramagnetic Resonance: Bridging the Gap between Experiment and Magnetic Parameters for High-Spin Transition-Metal Ion Complexes, J. Phys. Chem. B, 2015, 119(43), 13816–13824, DOI:10.1021/acs.jpcb.5b04156.
- J. Nehrkorn, K. Holldack, R. Bittl and A. Schnegg, Recent Progress in Synchrotron-Based Frequency-Domain Fourier-Transform THz-EPR, J. Magn. Reson., 2017, 280, 10–19, DOI:10.1016/j.jmr.2017.04.001.
- N. P. Vargo, J. B. Harland, B. W. Musselman, N. Lehnert, M. Z. Ertem and J. R. Robinson, Calcium-Ion Binding Mediates the Reversible Interconversion of Cis and Trans Peroxido Dicopper Cores, Angew. Chem., Int. Ed., 2021, 60(36), 19836–19842, DOI:10.1002/anie.202105421.
- A. Brinkmeier, K. E. Dalle, L. D'Amore, R. A. Schulz, S. Dechert, S. Demeshko, M. Swart and F. Meyer, Modulation of a μ-1,2-Peroxo Dicopper(II) Intermediate by Strong Interaction with Alkali Metal Ions, J. Am. Chem. Soc., 2021, 143(42), 17751–17760, DOI:10.1021/jacs.1c08645.
- A. Brinkmeier, R. A. Schulz, M. Buchhorn, C.-J. Spyra, S. Dechert, S. Demeshko, V. Krewald and F. Meyer, Structurally Characterized μ-1,2-Peroxo/Superoxo Dicopper(II) Pair, J. Am. Chem. Soc., 2021, 143(27), 10361–10366, DOI:10.1021/jacs.1c04316.
- D. A. Quist, M. A. Ehudin, A. W. Schaefer, G. L. Schneider, E. I. Solomon and K. D. Karlin, Ligand Identity-Induced Generation of Enhanced Oxidative Hydrogen Atom Transfer Reactivity for a CuII2 (O2•–) Complex Driven by Formation of a CuII2(−OOH) Compound with a Strong O–H Bond, J. Am. Chem. Soc., 2019, 141(32), 12682–12696, DOI:10.1021/jacs.9b05277.
- R. Cao, C. Saracini, J. W. Ginsbach, M. T. Kieber-Emmons, M. A. Siegler, E. I. Solomon, S. Fukuzumi and K. D. Karlin, Peroxo and Superoxo Moieties Bound to Copper Ion: Electron-Transfer Equilibrium with a Small Reorganization Energy, J. Am. Chem. Soc., 2016, 138(22), 7055–7066, DOI:10.1021/jacs.6b02404.
- I. López, R. Cao, D. A. Quist, K. D. Karlin and N. Le Poul, Direct Determination of Electron-Transfer Properties of Dicopper-Bound Reduced Dioxygen Species by a Cryo-Spectroelectrochemical Approach, Chem.–Eur. J., 2017, 23(72), 18314–18319, DOI:10.1002/chem.201705066.
- N. Kindermann, C.-J. Günes, S. Dechert and F. Meyer, Hydrogen Atom Abstraction Thermodynamics of a μ-1,2-Superoxo Dicopper(II) Complex, J. Am. Chem. Soc., 2017, 139(29), 9831–9834, DOI:10.1021/jacs.7b05722.
- P. K. Hota, A. Jose, S. Panda, E. M. Dunietz, A. E. Herzog, L. Wojcik, N. Le Poul, C. Belle, E. I. Solomon and K. D. Karlin, Coordination Variations within Binuclear Copper Dioxygen-Derived (Hydro)Peroxo and Superoxo Species; Influences upon Thermodynamic and Electronic Properties, J. Am. Chem. Soc., 2024, 146(19), 13066–13082, DOI:10.1021/jacs.3c14422.
- I. López and N. Le Poul, Low-Temperature Electrochemistry and Spectroelectrochemistry for Coordination Compounds, Coord. Chem. Rev., 2021, 436, 213823, DOI:10.1016/j.ccr.2021.213823.
- I. López and N. Le Poul, Theoretical Aspects of Electrochemistry at Low Temperature, J. Electroanal. Chem., 2021, 887, 115160, DOI:10.1016/j.jelechem.2021.115160.
- K. Bleher, P. Comba, J. H. Gross and T. Josephy, ESI and Tandem MS for Mechanistic Studies with High-Valent Transition Metal Species, Dalton Trans., 2022, 51(22), 8625–8639, 10.1039/D2DT00809B.
- S. Mahapatra, J. A. Halfen, E. C. Wilkinson, G. Pan, C. J. Cramer, L. Que Jr. and W. B. Tolman, A New Intermediate in Copper Dioxygen Chemistry: Breaking the O-O Bond To Form a {Cu2(μ-O)2}2+ Core, J. Am. Chem. Soc., 1995, 117(34), 8865–8866, DOI:10.1021/ja00139a026.
- J. A. Halfen, S. Mahapatra, E. C. Wilkinson, S. Kaderli, V. G. Young, L. Que, A. D. Zuberbühler and W. B. Tolman, Reversible Cleavage and Formation of the Dioxygen O-O Bond Within a Dicopper Complex, Science, 1996, 271(5254), 1397–1400, DOI:10.1126/science.271.5254.1397.
- M. T. Kieber-Emmons, J. W. Ginsbach, P. K. Wick, H. R. Lucas, M. E. Helton, B. Lucchese, M. Suzuki, A. D. Zuberbühler, K. D. Karlin and E. I. Solomon, Observation of a CuII2(μ-1,2-peroxo)/CuIII2(μ-oxo)2 Equilibrium and Its Implications for Copper–Dioxygen Reactivity, Angew. Chem., Int. Ed., 2014, 53(19), 4935–4939, DOI:10.1002/anie.201402166.
- V. E. Goswami, A. Walli, M. Förster, S. Dechert, S. Demeshko, M. C. Holthausen and F. Meyer, Acid/Base Triggered Interconversion of μ-η2:η2 -Peroxido and Bis(μ-Oxido) Dicopper Intermediates Capped by Proton-Responsive Ligands, Chem. Sci., 2017, 8(4), 3031–3037, 10.1039/C6SC04820J.
- H.-C. Liang, M. J. Henson, L. Q. Hatcher, M. A. Vance, C. X. Zhang, D. Lahti, S. Kaderli, R. D. Sommer, A. L. Rheingold, A. D. Zuberbühler, E. I. Solomon and K. D. Karlin, Solvent Effects on the Conversion of Dicopper(II) μ-ŋ2:ŋ2-Peroxo to Bis-μ-Oxo Dicopper(III) Complexes: Direct Probing of the Solvent Interaction, Inorg. Chem., 2004, 43(14), 4115–4117, DOI:10.1021/ic0498283.
- A. Singha, A. Rana and A. Dey, Nitrogen Hybridization Controls Peroxo-Oxo Equilibrium in Ethylenediamine Bound Binuclear [Cu2O2] Complexes, Inorg. Chim. Acta, 2019, 487, 63–69, DOI:10.1016/j.ica.2018.11.026.
- P. Zimmermann, S. Peredkov, P. M. Abdala, S. DeBeer, M. Tromp, C. Müller and J. A. van Bokhoven, Modern X-Ray Spectroscopy: XAS and XES in the Laboratory, Coord. Chem. Rev., 2020, 423, 213466, DOI:10.1016/j.ccr.2020.213466.
- G. E. Cutsail III, N. L. Gagnon, A. D. Spaeth, W. B. Tolman and S. DeBeer, Valence-to-Core X-Ray Emission Spectroscopy as a Probe of O−O Bond Activation in Cu2O2 Complexes, Angew Chem. Int. Ed. Engl., 2019, 58(27), 9114–9119, DOI:10.1002/anie.201903749.
- S. Fukuzumi, T. Kojima, Y.-M. Lee and W. Nam, High-Valent Metal-Oxo Complexes Generated in Catalytic Oxidation Reactions Using Water as an Oxygen Source, Coord. Chem. Rev., 2017, 333, 44–56, DOI:10.1016/j.ccr.2016.09.018.
- T. Matsumoto, K. Ohkubo, K. Honda, A. Yazawa, H. Furutachi, S. Fujinami, S. Fukuzumi and M. Suzuki, Aliphatic C−H Bond Activation Initiated by a (μ-η2:η2-Peroxo)Dicopper(II) Complex in Comparison with Cumylperoxyl Radical, J. Am. Chem. Soc., 2009, 131(26), 9258–9267, DOI:10.1021/ja809822c.
- M. Réglier, C. Jorand and B. Waegell, Binuclear Copper Complex Model of Tyrosinase, J. Chem. Soc. Chem. Commun., 1990, 24, 1752–1755, 10.1039/C39900001752.
- R. Schneider, T. A. Engesser, J. N. Hamann, C. Näther and F. Tuczek, Dinuclear Copper(I) Complexes Supported by Bis-Tridentate N -Donor-Ligands: Turning-On Tyrosinase Activity, Eur. J. Inorg. Chem., 2022, 2022(35), e202200509, DOI:10.1002/ejic.202200509.
- B. Herzigkeit, B. M. Flöser, N. E. Meißner, T. A. Engesser and F. C. Tuczek, Coordinate. Catalyze. Using CuAAC Click Ligands in Small-Molecule Model Chemistry of Tyrosinase, ChemCatChem, 2018, 10(23), 5402–5405, DOI:10.1002/cctc.201801606.
- A. Koch, T. A. Engesser, C. Näther and F. Tuczek, Oligodentate Aminotriazole Ligands for CuAAC and Copper-Mediated Monooxygenation of Phenols: Influence of Denticity, Chain Length and N-Alkylation on Catalytic Activity, ChemCatChem, 2023, e202301316, DOI:10.1002/cctc.202301316.
- M. Paul, M. Teubner, B. Grimm-Lebsanft, C. Golchert, Y. Meiners, L. Senft, K. Keisers, P. Liebhäuser, T. Rösener, F. Biebl, S. Buchenau, M. Naumova, V. Murzin, R. Krug, A. Hoffmann, J. Pietruszka, I. Ivanović-Burmazović, M. Rübhausen and S. Herres-Pawlis, Exceptional Substrate Diversity in Oxygenation Reactions Catalyzed by a Bis(μ-Oxo) Copper Complex, Chem.–Eur. J., 2020, 26(34), 7556–7562, DOI:10.1002/chem.202000664.
- M. Paul, A. Hoffmann and S. Herres-Pawlis, Room Temperature Stable Multitalent: Highly Reactive and Versatile Copper Guanidine Complexes in Oxygenation Reactions, J. Biol. Inorg. Chem., 2021, 26(2–3), 249–263, DOI:10.1007/s00775-021-01849-9.
- L. Laurini, S. M. Conte, K. Hüser, P. R. F. Cordero, H. M. Núñez Ponce, S. Zimmer, L. Lauterbach and S. Herres-Pawlis, From Phenols to Antimicrobial Phenazines: Tyrosinase-like Catalytic Activity of a Bisguanidine Based Bis(μ-oxido) Complex, Eur. J. Inorg. Chem., 2024, e202300700, DOI:10.1002/ejic.202300700.
- E. L. Presti, E. Monzani, L. Santagostini and L. Casella, Building Biomimetic Model Compounds of Dinuclear and Trinuclear Copper Clusters for Stereoselective Oxidations, Inorg. Chim. Acta, 2018, 481, 47–55, DOI:10.1016/j.ica.2017.09.045.
- E. L. Presti, M. L. Perrone, L. Santagostini, L. Casella and E. Monzani, A Stereoselective Tyrosinase Model Compound Derived from an M-Xylyl-l-Histidine Ligand, Inorg. Chem., 2019, 58(11), 7335–7344, DOI:10.1021/acs.inorgchem.9b00473.
- C. Magallón, J. Serrano-Plana, S. Roldán-Gómez, X. Ribas, M. Costas and A. Company, Inorg. Chim. Acta, 2018, 481, 166–170, DOI:10.1016/j.ica.2017.08.061.
- M. Kodera, K. Katayama, Y. Tachi, K. Kano, S. Hirota, S. Fujinami and M. Suzuki, Crystal Structure and Reversible O2-Binding of a Room Temperature Stable μ-ŋ2:ŋ2-Peroxodicopper(II) Complex of a Sterically Hindered Hexapyridine Dinucleating Ligand, J. Am. Chem. Soc., 1999, 121(47), 11006–11007, DOI:10.1021/ja992295q.
- I. López, A. G. Porras-Gutiérrez, B. Douziech, L. Wojcik, Y. Le Mest, M. Kodera and N. Le Poul, O–O Bond Cleavage via Electrochemical Reduction of a Side-on Peroxo Dicopper Model of Hemocyanin, Chem. Commun., 2018, 54(39), 4931–4934, 10.1039/C8CC01959B.
- L. M. Mirica, M. Vance, D. J. Rudd, B. Hedman, K. O. Hodgson, E. I. Solomon and T. D. P. Stack, Tyrosinase Reactivity in a Model Complex: An Alternative Hydroxylation Mechanism, Science, 2005, 308(5730), 1890–1892, DOI:10.1126/science.1112081.
- A. Company, S. Palavicini, I. Garcia-Bosch, R. Mas-Ballesté, L. Que, E. V. Rybak-Akimova, L. Casella, X. Ribas and M. Costas, Tyrosinase-Like Reactivity in a CuIII2(μ-O)2 Species, Chem.–Eur. J., 2008, 14(12), 3535–3538, DOI:10.1002/chem.200800229.
- S. Itoh, H. Nakao, L. M. Berreau, T. Kondo, M. Komatsu and S. Fukuzumi, Mechanistic Studies of Aliphatic Ligand Hydroxylation of a Copper Complex by Dioxygen: A Model Reaction for Copper Monooxygenases, J. Am. Chem. Soc., 1998, 120(12), 2890–2899, DOI:10.1021/ja972809q.
- M. Taki, S. Itoh and S. Fukuzumi, C−H Bond Activation of External Substrates with a Bis(μ-Oxo)Dicopper(III) Complex, J. Am. Chem. Soc., 2001, 123(25), 6203–6204, DOI:10.1021/ja015721s.
- S. Itoh, M. Taki, H. Nakao, P. L. Holland, W. B. Tolman, L. Que Jr. and S. Fukuzumi, Aliphatic Hydroxylation by a Bis(μ-Oxo)Dicopper(III) Complex, Angew. Chem., Int. Ed., 2000, 39(2), 398–400, DOI:10.1002/(SICI)1521-3773(20000117)39:2<398::AID-ANIE398>3.0.CO;2-2.
- J. N. Hamann, B. Herzigkeit, R. Jurgeleit and F. Tuczek, Small-Molecule Models of Tyrosinase: From Ligand Hydroxylation to Catalytic Monooxygenation of External Substrates, Coord. Chem. Rev., 2017, 334, 54–66, DOI:10.1016/j.ccr.2016.07.009.
- L. Marais, H. C. M. Vosloo and A. J. Swarts, Homogeneous Oxidative Transformations Mediated by Copper Catalyst Systems, Coord. Chem. Rev., 2021, 440, 213958, DOI:10.1016/j.ccr.2021.213958.
- P. Kang, B.-L. Lin, T. A. G. Large, J. Ainsworth, E. C. Wasinger and T. D. P. Stack, Phenolate-Bonded Bis(μ-Oxido)-Bis-Copper(III) Intermediates: Hydroxylation and Dehalogenation Reactivities, Faraday Discuss., 2022, 234, 86–108, 10.1039/D1FD00071C.
- C. Noß, R. Göttlich and S. Schindler, Photochemically Mediated Toluene Oxidation through a Copper Complex, Chem.–Eur. J., 2023, 29(46), e202301142, DOI:10.1002/chem.202301142.
- P. Haack and C. Limberg, Molecular CuII-O-CuII Complexes: Still Waters Run Deep, Angew. Chem., Int. Ed., 2014, 53(17), 4282–4293, DOI:10.1002/anie.201309505.
- R. Jurgeleit, B. Grimm-Lebsanft, B. M. Flöser, M. Teubner, S. Buchenau, L. Senft, J. Hoffmann, M. Naumova, C. Näther, I. Ivanović-Burmazović, M. Rübhausen and F. Tuczek, Catalytic Oxygenation of Hydrocarbons by Mono-μ-oxo Dicopper(II) Species Resulting from O−O Cleavage of Tetranuclear CuI/CuII Peroxo Complexes, Angew. Chem., Int. Ed., 2021, 60(25), 14154–14162, DOI:10.1002/anie.202101035.
- M. R. Halvagar, P. V. Solntsev, H. Lim, B. Hedman, K. O. Hodgson, E. I. Solomon, C. J. Cramer and W. B. Tolman, Hydroxo-Bridged Dicopper(II,III) and -(III,III) Complexes: Models for Putative Intermediates in Oxidation Catalysis, J. Am. Chem. Soc., 2014, 136(20), 7269–7272, DOI:10.1021/ja503629r.
- A. Kochem, F. Gennarini, M. Yemloul, M. Orio, N. Le Poul, E. Rivière, M. Giorgi, B. Faure, Y. Le Mest, M. Réglier and A. J. Simaan, Characterization of a Dinuclear Copper(II) Complex and Its Fleeting Mixed-Valent Copper(II)/Copper(III) Counterpart, ChemPlusChem, 2017, 82(4), 615–624, DOI:10.1002/cplu.201600636.
- F. Gennarini, R. David, I. López, Y. Le Mest, M. Réglier, C. Belle, A. Thibon-Pourret, H. Jamet and N. Le Poul, Influence of Asymmetry on the Redox Properties of Phenoxo- and Hydroxo-Bridged Dicopper Complexes: Spectroelectrochemical and Theoretical Studies, Inorg. Chem., 2017, 56, 7707–7719, DOI:10.1021/acs.inorgchem.7b00338.
- A. Thibon-Pourret, F. Gennarini, R. David, J. A. Isaac, I. Lopez, G. Gellon, F. Molton, L. Wojcik, C. Philouze, D. Flot, Y. Le Mest, M. Réglier, N. Le Poul, H. Jamet and C. Belle, Effect of Monoelectronic Oxidation of an Unsymmetrical Phenoxido-Hydroxido Bridged Dicopper(II) Complex, Inorg. Chem., 2018, 57, 12364–12375, DOI:10.1021/acs.inorgchem.8b02127.
- J. A. Isaac, F. Gennarini, I. Lopez, A. Thibon-Pourret, R. David, G. Gellon, B. Gennaro, C. Philouze, F. Meyer, S. Demeshko, Y. Le Mest, M. Réglier, H. Jamet, N. Le Poul and C. Belle, Room-Temperature Characterization of a Mixed-Valent μ-Hydroxodicopper(II,III) Complex, Inorg. Chem., 2016, 55, 8263–8266, DOI:10.1021/acs.inorgchem.6b01504.
- N. G. Connelly and W. E. Geiger, Chemical Redox Agents for Organometallic Chemistry, Chem. Rev., 1996, 96(2), 877–910, DOI:10.1021/cr940053x.
- P. E. VanNatta, D. A. Ramirez, A. R. Velarde, G. Ali and M. T. Kieber-Emmons, Exceptionally High O–H Bond Dissociation Free Energy of a Dicopper(II) μ-Hydroxo Complex and Insights into the Geometric and Electronic Structure Origins Thereof, J. Am. Chem. Soc., 2020, 142(38), 16292–16312, DOI:10.1021/jacs.0c06425.
- J. A. Isaac, A. Thibon-Pourret, A. Durand, C. Philouze, N. Le Poul and C. Belle, High-Valence CuIICuIII Species in Action: Demonstration of Aliphatic C–H Bond Activation at Room Temperature, Chem. Commun., 2019, 55, 12711–12714, 10.1039/C9CC04422A.
- Y. Shiota, G. Juhász and K. Yoshizawa, Role of Tyrosine Residue in Methane Activation at the Dicopper Site of Particulate Methane Monooxygenase: A Density Functional Theory Study, Inorg. Chem., 2013, 52(14), 7907–7917, DOI:10.1021/ic400417d.
- Y. Shiota and K. Yoshizawa, Comparison of the Reactivity of Bis(μ-Oxo)CuIICuIII and CuIIICuIII Species to Methane, Inorg. Chem., 2009, 48(3), 838–845, DOI:10.1021/ic8003933.
- J. De Tovar, C. Philouze, A. Thibon-Pourret and C. Belle, Insights into Non-Covalent Interactions in Dicopper(ii,ii) Complexes Bearing a Naphthyridine Scaffold: Anion-Dictated Electrochemistry, Chem. Commun., 2024, 60, 2228–2231, 10.1039/D3CC06264C.
- J. A. Isaac, G. Gellon, F. Molton, C. Philouze, N. Le Poul, C. Belle and A. Thibon-Pourret, Symmetrical and Unsymmetrical Dicopper Complexes Based on Bis-Oxazoline Units: Synthesis, Spectroscopic Properties and Reactivity, Inorganics, 2023, 11(8), 332, DOI:10.3390/inorganics11080332.
- W. Tao, J. K. Bower, C. E. Moore and S. Zhang, Dicopper μ-Oxo, μ-Nitrosyl Complex from the Activation of NO or Nitrite at a Dicopper Center, J. Am. Chem. Soc., 2019, 141(26), 10159–10164, DOI:10.1021/jacs.9b03635.
- S. Carter, W. Tao, R. Majumder, A. Yu. Sokolov and S. Zhang, Two-State Hydrogen Atom Transfer Reactivity of Unsymmetric [Cu2(O)(NO)]2+ Complexes, J. Am. Chem. Soc., 2023, 145(32), 17779–17785, DOI:10.1021/jacs.3c04510.
- T. Tsuji, A. A. Zaoputra, Y. Hitomi, K. Mieda, T. Ogura, Y. Shiota, K. Yoshizawa, H. Sato and M. Kodera, Specific Enhancement of Catalytic Activity by a Dicopper Core: Selective Hydroxylation of Benzene to Phenol with Hydrogen Peroxide, Angew. Chem., Int. Ed., 2017, 56(27), 7779–7782, DOI:10.1002/anie.201702291.
- L. Siebe, C. Butenuth, A. Stammler, H. Bögge, S. Walleck and T. Glaser, Generation and Reactivity of μ-1,2-Peroxo CuIICuII and Bis-μ-Oxo CuIIICuIII Species and Catalytic Hydroxylation of Benzene to Phenol with Hydrogen Peroxide, Inorg. Chem., 2024, 63(5), 2627–2639, DOI:10.1021/acs.inorgchem.3c03914.
- H. Takahashi, K. Wada, K. Tanaka, K. Fujikawa, Y. Hitomi, T. Endo and M. Kodera, Alkane Oxidation with H2O2 Catalyzed by Dicopper Complex with 6-Hpa Ligand: Mechanistic Insights as Key Features for Methane Oxidation, Bull. Chem. Soc. Jpn., 2022, 95(8), 1148–1155, DOI:10.1246/bcsj.20220138.
- P. P.-Y. Chen and S. I. Chan, Theoretical Modeling of the Hydroxylation of Methane as Mediated by the Particulate Methane Monooxygenase, J. Inorg. Biochem., 2006, 100(4), 801–809, DOI:10.1016/j.jinorgbio.2005.12.014.
- S. Maji, J. C. M. Lee, Y. Lu, C. Chen, M. Hung, P. P. Y. Chen, S. S. F. Yu and S. I. Chan, Dioxygen Activation of a Trinuclear CuICuICuI Cluster Capable of Mediating Facile Oxidation of Organic Substrates: Competition between O-Atom Transfer and Abortive Intercomplex Reduction, Chem.–Eur. J., 2012, 18(13), 3955–3968, DOI:10.1002/chem.201103075.
- E. Moharreri, T. Jafari, D. Rathnayake, H. Khanna, C.-H. Kuo, S. L. Suib and P. Nandi, Role of Catalytic Nitrile Decomposition in Tricopper Complex Mediated Direct Partial Oxidation of Methane to Methanol, Sci. Rep., 2021, 11(1), 19175, DOI:10.1038/s41598-021-98721-2.
- C.-C. Liu, C.-Y. Mou, S. S.-F. Yu and S. I. Chan, Heterogeneous Formulation of the Tricopper Complex for Efficient Catalytic Conversion of Methane into Methanol at Ambient Temperature and Pressure, Energy Environ. Sci., 2016, 9(4), 1361–1374, 10.1039/C5EE03372A.
- C.-C. Liu, D. Janmanchi, D.-R. Wen, J.-N. Oung, C.-Y. Mou, S. S.-F. Yu and S. I. Chan, Catalytic Oxidation of Light Alkanes Mediated at Room Temperature by a Tricopper Cluster Complex Immobilized in Mesoporous Silica Nanoparticles, ACS Sustainable Chem. Eng., 2018, 6(4), 5431–5440, DOI:10.1021/acssuschemeng.8b00270.
- C. Yeh, S. S. F. Yu, S. I. Chan and J. Jiang, Quantum Chemical Studies of Methane Oxidation to Methanol on a Biomimetic Tricopper Complex: Mechanistic Insights, ChemistrySelect, 2018, 3(18), 5113–5122, DOI:10.1002/slct.201800550.
- Y. Chen, C. Wu, P. Sung, S. I. Chan and P. P. Chen, Turnover of a Methane Oxidation Tricopper Cluster Catalyst: Implications for the Mechanism of the Particulate Methane Monooxygenase (pMMO), ChemCatChem, 2020, 12(11), 3088–3096, DOI:10.1002/cctc.202000322.
- Y.-F. Tsai, T. Natarajan, Z.-H. Lin, I.-K. Tsai, D. Janmanchi, S. I. Chan and S. S.-F. Yu, Voltage-Gated Electrocatalysis of Efficient and Selective Methane Oxidation by Tricopper Clusters under Ambient Conditions, J. Am. Chem. Soc., 2022, 144(22), 9695–9706, DOI:10.1021/jacs.2c01169.
- S. F. Hannigan, A. I. Arnoff, S. E. Neville, J. S. Lum, J. A. Golen, A. L. Rheingold, N. Orth, I. Ivanović-Burmazović, P. Liebhäuser, T. Rösener, J. Stanek, A. Hoffmann, S. Herres-Pawlis and L. H. Doerrer, On the Way to a Trisanionic {Cu3O2} Core for Oxidase Catalysis: Evidence of an Asymmetric Trinuclear Precursor Stabilized by Perfluoropinacolate Ligands, Chem.–Eur. J., 2017, 23(34), 8212–8224, DOI:10.1002/chem.201605926.
- S. E. N. Brazeau, E. E. Norwine, S. F. Hannigan, N. Orth, I. Ivanović-Burmazović, D. Rukser, F. Biebl, B. Grimm-Lebsanft, G. Praedel, M. Teubner, M. Rübhausen, P. Liebhäuser, T. Rösener, J. Stanek, A. Hoffmann, S. Herres-Pawlis and L. H. Doerrer, Dual Oxidase/Oxygenase Reactivity and Resonance Raman Spectra of {Cu3O2} Moiety with Perfluoro- t -Butoxide Ligands, Dalton Trans., 2019, 48(20), 6899–6909, 10.1039/C9DT00516A.
- B. J. Cook, G. N. Di Francesco, M. T. Kieber-Emmons and L. J. Murray, A Tricopper(I) Complex Competent for O Atom Transfer, C–H Bond Activation, and Multiple O2 Activation Steps, Inorg. Chem., 2018, 57(18), 11361–11368, DOI:10.1021/acs.inorgchem.8b00921.
- X. Engelmann, E. R. Farquhar, J. England and K. Ray, Four-Electron Reduction of Dioxygen to Water by a Trinuclear Copper Complex, Inorg. Chim. Acta, 2018, 481, 159–165, DOI:10.1016/j.ica.2017.11.014.
- W. Zhang, C. E. Moore and S. Zhang, Multiple Proton-Coupled Electron Transfers at a Tricopper Cluster: Modeling the Reductive Regeneration Process in Multicopper Oxidases, J. Am. Chem. Soc., 2022, 144(4), 1709–1717, DOI:10.1021/jacs.1c10948.
- N. Thiyagarajan, D. Janmanchi, Y. Tsai, W. H. Wanna, R. Ramu, S. I. Chan, J. Zen and S. S. F. Yu, A Carbon Electrode Functionalized by a Tricopper Cluster Complex: Overcoming Overpotential and Production of Hydrogen Peroxide in the Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2018, 57(14), 3612–3616, DOI:10.1002/anie.201712226.
- M. A. Newton, A. J. Knorpp, V. L. Sushkevich, D. Palagin and J. A. van Bokhoven, Active Sites and Mechanisms in the Direct Conversion of Methane to Methanol Using Cu in Zeolitic Hosts: A Critical Examination, Chem. Soc. Rev., 2020, 49(5), 1449–1486, 10.1039/C7CS00709D.
- D. Kiani, S. Sourav, Y. Tang, J. Baltrusaitis and I. E. Wachs, Methane Activation by ZSM-5-Supported Transition Metal Centers, Chem. Soc. Rev., 2021, 50(2), 1251–1268, 10.1039/D0CS01016B.
- J. W. A. Fischer, F. Buttignol, A. Brenig, D. Klose, D. Ferri, V. Sushkevich, J. A. Van Bokhoven and G. Jeschke, Design Principles of Operando Ultraviolet-Visible and Electron Paramagnetic Resonance Spectroscopy Setups for Active Site Characterization in Ion-Exchanged Zeolites, Catal. Today, 2024, 429, 114503, DOI:10.1016/j.cattod.2023.114503.
- A. Wijerathne, A. Sawyer, R. Daya and C. Paolucci, Competition between Mononuclear and Binuclear Copper Sites across Different Zeolite Topologies, JACS Au, 2024, 4(1), 197–215, DOI:10.1021/jacsau.3c00632.
- M. Gallego, A. Corma and M. Boronat, An Alternative Catalytic Cycle for Selective Methane Oxidation to Methanol with Cu Clusters in Zeolites, Phys. Chem. Chem. Phys., 2024, 26(7), 5914–5921, 10.1039/D3CP05802F.
- V. L. Sushkevich, M. Artsiusheuski, D. Klose, G. Jeschke and J. A. Bokhoven, Identification of Kinetic and Spectroscopic Signatures of Copper Sites for Direct Oxidation of Methane to Methanol, Angew. Chem., Int. Ed., 2021, 60(29), 15944–15953, DOI:10.1002/anie.202101628.
- I. Lee, M.-S. Lee, L. Tao, T. Ikuno, R. Khare, A. Jentys, T. Huthwelker, C. N. Borca, A. Kalinko, O. Y. Gutiérrez, N. Govind, J. L. Fulton, J. Z. Hu, V.-A. Glezakou, R. Rousseau, M. Sanchez-Sanchez and J. A. Lercher, Activity of Cu–Al–Oxo Extra-Framework Clusters for Selective Methane Oxidation on Cu-Exchanged Zeolites, JACS Au, 2021, 1(9), 1412–1421, DOI:10.1021/jacsau.1c00196.
- D. Plessers, A. J. Heyer, H. M. Rhoda, M. L. Bols, E. I. Solomon, R. A. Schoonheydt and B. F. Sels, Tuning Copper Active Site Composition in Cu-MOR through Co-Cation Modification for Methane Activation, ACS Catal., 2023, 13(3), 1906–1915, DOI:10.1021/acscatal.2c05271.
- A. J. Knorpp, A. B. Pinar, C. Baerlocher, L. B. McCusker, N. Casati, M. A. Newton, S. Checchia, J. Meyet, D. Palagin and J. A. van Bokhoven, Paired Copper Monomers in Zeolite Omega: The Active Site for Methane-to-Methanol Conversion, Angew. Chem., Int. Ed., 2021, 60(11), 5854–5858, DOI:10.1002/anie.202014030.
- K. Kvande, S. Prodinger, F. Schlimpen, P. Beato, P. Pale, S. Chassaing and S. Svelle, Copper-Zeolites Prepared by Solid-State Ion Exchange - Characterization and Evaluation for the Direct Conversion of Methane to Methanol, Top. Catal., 2023, 66(17), 1406–1417, DOI:10.1007/s11244-022-01763-7.
- O. Suleiman, D. Panthi, O. Adeyiga and S. O. Odoh, Methane C–H Activation by [Cu2O]2+ and [Cu3O3]2+ in Copper-Exchanged Zeolites: Computational Analysis of Redox Chemistry and X-Ray Absorption Spectroscopy, Inorg. Chem., 2021, 60(9), 6218–6227, DOI:10.1021/acs.inorgchem.0c03693.
- H. M. Rhoda, D. Plessers, A. J. Heyer, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Spectroscopic Definition of a Highly Reactive Site in Cu-CHA for Selective Methane Oxidation: Tuning a Mono-μ-Oxo Dicopper(II) Active Site for Reactivity, J. Am. Chem. Soc., 2021, 143(19), 7531–7540, DOI:10.1021/jacs.1c02835.
- B. Ipek, M. J. Wulfers, H. Kim, F. Göltl, I. Hermans, J. P. Smith, K. S. Booksh, C. M. Brown and R. F. Lobo, Formation of [Cu2O2]2+ and [Cu2O]2+ toward C–H Bond Activation in Cu-SSZ-13 and Cu-SSZ-39, ACS Catal., 2017, 7(7), 4291–4303, DOI:10.1021/acscatal.6b03005.
- D. T. Bregante, L. N. Wilcox, C. Liu, C. Paolucci, R. Gounder and D. W. Flaherty, Dioxygen Activation Kinetics over Distinct Cu Site Types in Cu-Chabazite Zeolites, ACS Catal., 2021, 11(19), 11873–11884, DOI:10.1021/acscatal.1c03471.
- J. Wieser, A. J. Knorpp, D. C. Stoian, P. Rzepka, M. A. Newton and J. A. van Bokhoven, Assessing the Productivity of the Direct Conversion of Methane-to-Methanol over Copper-Exchanged Zeolite Omega (MAZ) via Oxygen Looping, Angew. Chem., Int. Ed., 2023, 62(40), e202305140, DOI:10.1002/anie.202305140.
- T. Pereira, P. Caroline, J. V. R. Vieira, C. H. F. Da Cunha, S. C. M. Mizuno, Y. O. Carvalho, T. Faheina, M. Picinini, A. L. Blanco, A. C. M. Tello, E. A. Urquieta-Gonzalez, A. Lopez-Castillo, A. M. De Lima, J. B. O. D. Santos and J. M. C. Bueno, Conversion of Methane to Methanol over Cu-MAZ (Zeolite Omega): An Oxygen-Free Process Using H2O and CO2 as Oxidants, Appl. Catal., B, 2024, 342, 123370, DOI:10.1016/j.apcatb.2023.123370.
- M. A. Artsiusheuski, R. Verel, J. A. van Bokhoven and V. L. Sushkevich, Mechanism of Hydrocarbon Formation in Methane and Methanol Conversion over Copper-Containing Mordenite, ACS Catal., 2023, 13(9), 5864–5875, DOI:10.1021/acscatal.2c06312.
- Y. Tang, Y. Cui, G. Ren, K. Ma, X. Ma, C. Dai and C. Song, One-Step Synthesis of Methanol and Hydrogen from Methane and Water Using Non-Thermal Plasma and Cu-Mordenite Catalyst, Fuel Process. Technol., 2023, 244, 107722, DOI:10.1016/j.fuproc.2023.107722.
- P. Xiao, Y. Wang, K. Nakamura, Y. Lu, J. N. Kondo, H. Gies and T. Yokoi, C -Axis-Oriented Sheet-like Cu/AEI Zeolite Contributes to Continuous Direct Oxidation of Methane to Methanol, Catal. Sci. Technol., 2023, 13(20), 5831–5841, 10.1039/D3CY00584D.
- M. H. Mahyuddin, T. Tanaka, A. Staykov, Y. Shiota and K. Yoshizawa, Dioxygen Activation on Cu-MOR Zeolite: Theoretical Insights into the Formation of Cu2O and Cu3O3 Active Species, Inorg. Chem., 2018, 57(16), 10146–10152, DOI:10.1021/acs.inorgchem.8b01329.
- M. H. Mahyuddin, A. G. Saputro, R. P. P. Sukanli, F. Fathurrahman, J. Rizkiana, A. Nuruddin and H. K. Dipojono, Molecular Insight into the Role of Zeolite Lattice Constraints on Methane Activation over the Cu–O–Cu Active Site, Phys. Chem. Chem. Phys., 2022, 24(7), 4196–4203, 10.1039/D1CP05371J.
- L. Tao, I. Lee, R. Khare, A. Jentys, J. L. Fulton, M. Sanchez-Sanchez and J. A. Lercher, Speciation of Cu-Oxo Clusters in Ferrierite for Selective Oxidation of Methane to Methanol, Chem. Mater., 2022, 34(10), 4355–4363, DOI:10.1021/acs.chemmater.1c04249.
- B. E. R. Snyder, P. Vanelderen, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Second-Sphere Effects on Methane Hydroxylation in Cu-Zeolites, J. Am. Chem. Soc., 2018, 140(29), 9236–9243, DOI:10.1021/jacs.8b05320.
- H. Lv, X. Liu, Y. Hao and Y. Yi, Coupling of Dielectric Barrier Discharge and Cu–S-1 Catalyst for Direct Oxidation of Methane to Methanol, Plasma Chem. Plasma Process., 2023, 43(6), 1963–1978, DOI:10.1007/s11090-023-10333-y.
- X. Tang, J. Ye, L. Guo, T. Pu, L. Cheng, X.-M. Cao, Y. Guo, L. Wang, Y. Guo, W. Zhan and S. Dai, Atomic Insights into the Cu Species Supported on Zeolite for Direct Oxidation of Methane to Methanol via Low-Damage HAADF-STEM, Adv. Mater., 2023, 35(25), 2208504, DOI:10.1002/adma.202208504.
- K. Kvande, S. Prodinger, B. G. Solemsli, S. Bordiga, E. Borfecchia, U. Olsbye, P. Beato and S. Svelle, Cu-Loaded Zeolites Enable the Selective Activation of Ethane to Ethylene at Low Temperatures and Pressure, Chem. Commun., 2023, 59(40), 6052–6055, 10.1039/D3CC00948C.
- K. Nakamura, P. Xiao, R. Osuga, Y. Wang, S. Yasuda, T. Matsumoto, J. N. Kondo, M. Yabushita, A. Muramatsu, H. Gies and T. Yokoi, Impacts of Framework Al Distribution and Acidic Properties of Cu-Exchanged CHA-Type Zeolite on Catalytic Conversion of Methane into Methanol Followed by Lower Hydrocarbons, Catal. Sci. Technol., 2023, 13(9), 2648–2651, 10.1039/D3CY00127J.
- Y. Tsuchimura, H. Yoshida, M. Machida, S. Nishimura, K. Takahashi and J. Ohyama, Investigation of the Active-Site Structure of Cu-CHA Catalysts for the Direct Oxidation of Methane to Methanol Using In Situ UV–Vis Spectroscopy, Energy Fuels, 2023, 37(13), 9411–9418, DOI:10.1021/acs.energyfuels.3c00333.
- S. C. M. Mizuno, S. Dulnee, T. C. P. Pereira, R. J. Passini, E. A. Urquieta-Gonzalez, J. M. R. Gallo, J. B. O. Santos and J. M. C. Bueno, Stepwise Methane to Methanol Conversion: Effect of Copper Loading on the Formation of Active Species in Copper-Exchanged Mordenite, Expand. Catal. Front., 2021, 381, 13–25, DOI:10.1016/j.cattod.2020.11.027.
- J. Pokhrel and D. F. Shantz, Continuous Partial Oxidation of Methane to Methanol over Cu-SSZ-39 Catalysts, J. Catal., 2023, 421, 300–308, DOI:10.1016/j.jcat.2023.03.021.
- J. Ohyama, Y. Tsuchimura, A. Hirayama, H. Iwai, H. Yoshida, M. Machida, S. Nishimura, K. Kato and K. Takahashi, Relationships among the Catalytic Performance, Redox Activity, and Structure of Cu-CHA Catalysts for the Direct Oxidation of Methane to Methanol Investigated Using In Situ XAFS and UV–Vis Spectroscopies, ACS Catal., 2022, 12(4), 2454–2462, DOI:10.1021/acscatal.1c05559.
- M. A. Artsiusheuski, J. A. van Bokhoven and V. L. Sushkevich, Structure of Selective and Nonselective Dicopper (II) Sites in CuMFI for Methane Oxidation to Methanol, ACS Catal., 2022, 12(24), 15626–15637, DOI:10.1021/acscatal.2c05299.
- J. Ohyama, Y. Tsuchimura, H. Yoshida, M. Machida, S. Nishimura and K. Takahashi, Bayesian-Optimization-Based Improvement of Cu-CHA Catalysts for Direct Partial Oxidation of CH4, J. Phys. Chem. C, 2022, 126(46), 19660–19666, DOI:10.1021/acs.jpcc.2c04229.
- F. Göltl, S. Bhandari and M. Mavrikakis, Thermodynamics Perspective on the Stepwise Conversion of Methane to Methanol over Cu-Exchanged SSZ-13, ACS Catal., 2021, 11(13), 7719–7734, DOI:10.1021/acscatal.1c00691.
- J. Ohyama, A. Hirayama, N. Kondou, H. Yoshida, M. Machida, S. Nishimura, K. Hirai, I. Miyazato and K. Takahashi, Data Science Assisted Investigation of Catalytically Active Copper Hydrate in Zeolites for Direct Oxidation of Methane to Methanol Using H2O2, Sci. Rep., 2021, 11(1), 2067, DOI:10.1038/s41598-021-81403-4.
- K. Kvande, M. Mawanga, S. Prodinger, B. G. Solemsli, J. Yang, U. Olsbye, P. Beato, E. A. Blekkan and S. Svelle, Microcalorimetry on Cu-MCM-22 Reveals Structure–Activity Relationships for the Methane-to-Methanol Reaction, Ind. Eng. Chem. Res., 2023, 62(28), 10939–10950, DOI:10.1021/acs.iecr.3c00988.
- E. Borfecchia, K. A. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. V. Soldatov, S. Bordiga and C. Lamberti, Revisiting the Nature of Cu Sites in the Activated Cu-SSZ-13 Catalyst for SCR Reaction, Chem. Sci., 2015, 6(1), 548–563, 10.1039/C4SC02907K.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, Catalysis in a Cage: Condition-Dependent Speciation and Dynamics of Exchanged Cu Cations in SSZ-13 Zeolites, J. Am. Chem. Soc., 2016, 138(18), 6028–6048, DOI:10.1021/jacs.6b02651.
- A. J. Heyer, D. Plessers, J. Ma, B. E. R. Snyder, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Magnetic Exchange Coupling in Zeolite Copper Dimers and Its Contribution to Methane Activation, J. Am. Chem. Soc., 2024, 146, 6061–6071, DOI:10.1021/jacs.3c13295.
- A. A. Kolganov, A. A. Gabrienko and A. G. Stepanov, Reaction of Methane with Benzene and CO on Cu-Modified ZSM-5 Zeolite Investigated by 13C MAS NMR Spectroscopy, Chem. Phys. Lett., 2023, 810, 140188, DOI:10.1016/j.cplett.2022.140188.
- C. Zhou, S. Li, S. He, Z. Zhao, Y. Jiao and H. Zhang, Temperature-Dependant Active Sites for Methane Continuous Conversion to Methanol over Cu-Zeolite Catalysts Using Water as the Oxidant, Fuel, 2022, 329, 125483, DOI:10.1016/j.fuel.2022.125483.
- H. Zhang, P. Han, D. Wu, C. Du, J. Zhao, K. H. L. Zhang, J. Lin, S. Wan, J. Huang, S. Wang, H. Xiong and Y. Wang, Confined Cu-OH Single Sites in SSZ-13 Zeolite for the Direct Oxidation of Methane to Methanol, Nat. Commun., 2023, 14(1), 7705, DOI:10.1038/s41467-023-43508-4.
- Y. Deng, H. Lei, U. Simon, D. Ye and P. Chen, Transition-Metal-Promoted Cu-SSZ-13 Catalysts for Continuous Selective Oxidation of Methane to Methanol Using Molecular Oxygen, ACS Catal., 2024, 14(1), 292–298, DOI:10.1021/acscatal.3c04603.
- N. V. Kolesnichenko, Y. M. Snatenkova, T. I. Batova, O. V. Yashina and K. B. Golubev, Oxidative Carbonylation of Methane to Acetic Acid over Micro-Mesoporous Rhodium-Modified Zeolites, Microporous Mesoporous Mater., 2022, 330, 111581, DOI:10.1016/j.micromeso.2021.111581.
- R. Xu, N. Liu, C. Dai, Y. Li, J. Zhang, B. Wu, G. Yu and B. Chen, H2O-Built Proton Transfer Bridge Enhances Continuous Methane Oxidation to Methanol over Cu-BEA Zeolite, Angew. Chem., Int. Ed., 2021, 60(30), 16634–16640, DOI:10.1002/anie.202105167.
- X. Guan, Y. Wang, X. Liu, H. Du, X. Guo and Z. Zhang, Enhancing the Activity of Cu-MOR by Water for Oxidation of Methane to Methanol, Catalysts, 2023, 13, 1066, DOI:10.3390/catal13071066.
- L. Sun, Y. Wang, C. Wang, Z. Xie, N. Guan and L. Li, Water-Involved Methane-Selective Catalytic Oxidation by Dioxygen over Copper Zeolites, Chem, 2021, 7(6), 1557–1568, DOI:10.1016/j.chempr.2021.02.026.
- J. Ohyama, A. Hirayama, Y. Tsuchimura, N. Kondou, H. Yoshida, M. Machida, S. Nishimura, K. Kato, I. Miyazato and K. Takahashi, Catalytic Direct Oxidation of Methane to Methanol by Redox of Copper Mordenite, Catal. Sci. Technol., 2021, 11(10), 3437–3446, 10.1039/D1CY00125F.
- L. P. M. Freitas, A. A. Espírito Santo, T. C. Lourenço, J. L. F. Da Silva and G. T. Feliciano, Steric and Electrostatic Effects on the Diffusion of CH4/CH3OH in Copper-Exchanged Zeolites: Insights from Enhanced Sampling Molecular Dynamics and Free Energy Calculations, Langmuir, 2021, 37(26), 8014–8023, DOI:10.1021/acs.langmuir.1c01078.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y.-S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, Bioinspired Metal–Organic Framework Catalysts for Selective Methane Oxidation to Methanol, J. Am. Chem. Soc., 2018, 140(51), 18208–18216, DOI:10.1021/jacs.8b11525.
- T. Jia, W. Wang, L. Zhang, D. Zeng, J. Wang and W. Wang, An Efficient Strategy for the Partial Oxidation of Methane into Methanol over POM-Immobilized MOF Catalysts under Ambient Conditions, Appl. Catal., B, 2024, 340, 123168, DOI:10.1016/j.apcatb.2023.123168.
- N. Antil, M. Chauhan, N. Akhtar, R. Kalita and K. Manna, Selective Methane Oxidation to Acetic Acid Using Molecular Oxygen over a Mono-Copper Hydroxyl Catalyst, J. Am. Chem. Soc., 2023, 145(11), 6156–6165, DOI:10.1021/jacs.2c12042.
- X. Feng, Y. Song, J. S. Chen, Z. Xu, S. J. Dunn and W. Lin, Rational Construction of an Artificial Binuclear Copper Monooxygenase in a Metal–Organic Framework, J. Am. Chem. Soc., 2021, 143(2), 1107–1118, DOI:10.1021/jacs.0c11920.
- J. Meyet, A. Ashuiev, G. Noh, M. A. Newton, D. Klose, K. Searles, A. P. van Bavel, A. D. Horton, G. Jeschke, J. A. van Bokhoven and C. Copéret, Methane-to-Methanol on Mononuclear Copper(II) Sites Supported on Al2O3: Structure of Active Sites from Electron Paramagnetic Resonance, Angew. Chem., Int. Ed., 2021, 60(29), 16200–16207, DOI:10.1002/anie.202105307.
- Y.-Q. Chen, R.-X. Zhang, M. Ren, Y.-X. Jin, W.-J. Wang, J.-Y. Feng, Z.-H. Gao, Z.-F. Yan, Y.-M. Liu, W. Huang, L. Liu and Z.-J. Zuo, Photo-Assisted Thermal Catalysis for Methanol Synthesis from Methane Oxidation on Cu-MOR/g-C3N4, Fuel, 2023, 340, 127525, DOI:10.1016/j.fuel.2023.127525.
- T. Xu, Z. Jiang, P. Liu, H. Chen, X. Lan, D. Chen, L. Li and Y. He, Immobilization of Oxygen Atoms in the Pores of Microporous Metal–Organic Frameworks for C2H2 Separation and Purification, ACS Appl. Nano Mater., 2020, 3(3), 2911–2919, DOI:10.1021/acsanm.0c00162.
- G. Li, H. Wang, Q. Li, X. Zhang, Y. Qin, Y. Bi and L. Song, Regulation of the Nature and Sites of Copper Species in CuNaY Zeolites for Ethylene and Ethane Separation, New J. Chem., 2023, 47(12), 5650–5658, 10.1039/D3NJ00168G.
- L. Luo, X. Han, K. Wang, Y. Xu, L. Xiong, J. Ma, Z. Guo and J. Tang, Nearly 100% Selective and Visible-Light-Driven Methane Conversion to Formaldehyde via. Single-Atom Cu and Wδ+, Nat. Commun., 2023, 14(1), 2690, DOI:10.1038/s41467-023-38334-7.
- M. L. Bols, J. Ma, F. Rammal, D. Plessers, X. Wu, S. Navarro-Jaén, A. J. Heyer, B. F. Sels, E. I. Solomon and R. A. Schoonheydt, In Situ UV–Vis–NIR Absorption Spectroscopy and Catalysis, Chem. Rev., 2024, 124(5), 2352–2418, DOI:10.1021/acs.chemrev.3c00602.
- H. Huang, X. Jing, J. Deng, C. Meng and C. Duan, Enzyme-Inspired Coordination Polymers for Selective Oxidization of C(Sp3)–H Bonds via Multiphoton Excitation, J. Am. Chem. Soc., 2023, 145(4), 2170–2182, DOI:10.1021/jacs.2c09348.
| This journal is © The Royal Society of Chemistry 2024 |