
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast, efficient, narrowband room-temperature phosphorescence from metal-free 1,2-diketones: rational design and the mechanism†
Yosuke
Tani
 *ab,
Kiyoshi
Miyata
*c,
Erika
Ou
a,
Yuya
Oshima
a,
Mao
Komura
a,
Morihisa
Terasaki
a,
Shuji
Kimura
c,
Takumi
Ehara
c,
Koki
Kubo
c,
Ken
Onda
c and
Takuji
Ogawa
a
*ab,
Kiyoshi
Miyata
*c,
Erika
Ou
a,
Yuya
Oshima
a,
Mao
Komura
a,
Morihisa
Terasaki
a,
Shuji
Kimura
c,
Takumi
Ehara
c,
Koki
Kubo
c,
Ken
Onda
c and
Takuji
Ogawa
a
aDepartment of Chemistry, Graduate School of Science, Osaka University, Machikaneyama 1-1, Toyonaka, Osaka 560-0043, Japan. E-mail: y-tani@chem.sci.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita, Osaka 560-8531, Japan
cDepartment of Chemistry, Faculty of Science, Kyushu University, 744 Motooka, Nishi, Fukuoka 819-0395, Japan. E-mail: kmiyata@chem.kyushu-univ.jp
First published on 3rd June 2024
Abstract
We report metal-free organic 1,2-diketones that exhibit fast and highly efficient room-temperature phosphorescence (RTP) with high colour purity under various conditions, including solutions. RTP quantum yields reached 38.2% in solution under Ar, 54% in a polymer matrix in air, and 50% in crystalline solids in air. Moreover, the narrowband RTP consistently dominated the steady-state emission, regardless of the molecular environment. Detailed mechanistic studies using ultrafast spectroscopy, single-crystal X-ray structure analysis, and theoretical calculations revealed picosecond intersystem crossing (ISC) followed by RTP from a planar conformation. Notably, the phosphorescence rate constant kp was unambiguously established as ∼5000 s−1, which is comparable to that of platinum porphyrins (representative heavy-metal phosphor). This inherently large kp enabled the high-efficiency RTP across diverse molecular environments, thus complementing the streamlined persistent RTP approach. The mechanism behind the photofunction has been elucidated as follows: (1) the large kp is due to efficient intensity borrowing of the T1 state from the bright S3 state, (2) the rapid ISC occurs from the S1 to the T3 state because these states are nearly isoenergetic and have a considerable spin–orbit coupling, and (3) the narrowband emission results from the minimal geometry change between the T1 and S0 states. Such mechanistic understanding based on molecular orbitals, as well as the structure-RTP property relationship study, highlighted design principles embodied by the diketone planar conformer. The fast RTP strategy enables development of organic phosphors with emissions independent of environmental conditions, thereby offering alternatives to precious-metal based phosphors.
Introduction
Room-temperature phosphorescence (RTP) from metal-free organic molecules has been an area of intense research.1 Although classical RTP materials have been used for diverse applications, including organic light-emitting diodes (OLEDs) and bioimaging, they are mainly precious-metal complexes of Ir or Pt.2 Therefore, cost-effective, less-toxic, and sustainable metal-free alternatives are needed. However, intrinsic molecular RTP of metal-free phosphors has not rivalled that of metal complexes.Organic RTP must overcome several challenges stemming from the absence of heavy-metal atoms. The most severe is the inherently small phosphorescence transition probability (i.e., rate constant kp), causing poor RTP quantum yields (Φp). kp can be 104–105 s−1 for heavy-metal complexes,3 and only ∼10 s−1 or less for conventional organic compounds.1a Introducing heavy atoms, such as bromine, iodine, selenium, and tellurium, in conjunction with carbonyl functionalities, is a classical approach for enhancing kp.4 Despite its long history, however, an organic kp over ∼100 s−1 and/or a Φp over 1% in solutions are rarely observed (Fig. 1b, blue filled circles; Fig. S1 and Table S1†).5 The use of thiocarbonyls could provide a large kp in some cases (Fig. 1b, blue open circles).6 However, they are associated with high (photo)chemical reactivities and instability,7 thereby limiting their high Φp in perfluoroalkane solvents.6b The only practical way to improve Φp has been to reduce the nonradiative decay rate constant (knr). This approach has provided solid-state phosphors with RTP lasting for subseconds after excitation cessation (persistent RTP or afterglow).8 However, it inevitably restricts the organic phosphors to rigid media, such as crystals,9 or tailored host–guest systems with strong intermolecular interactions.10
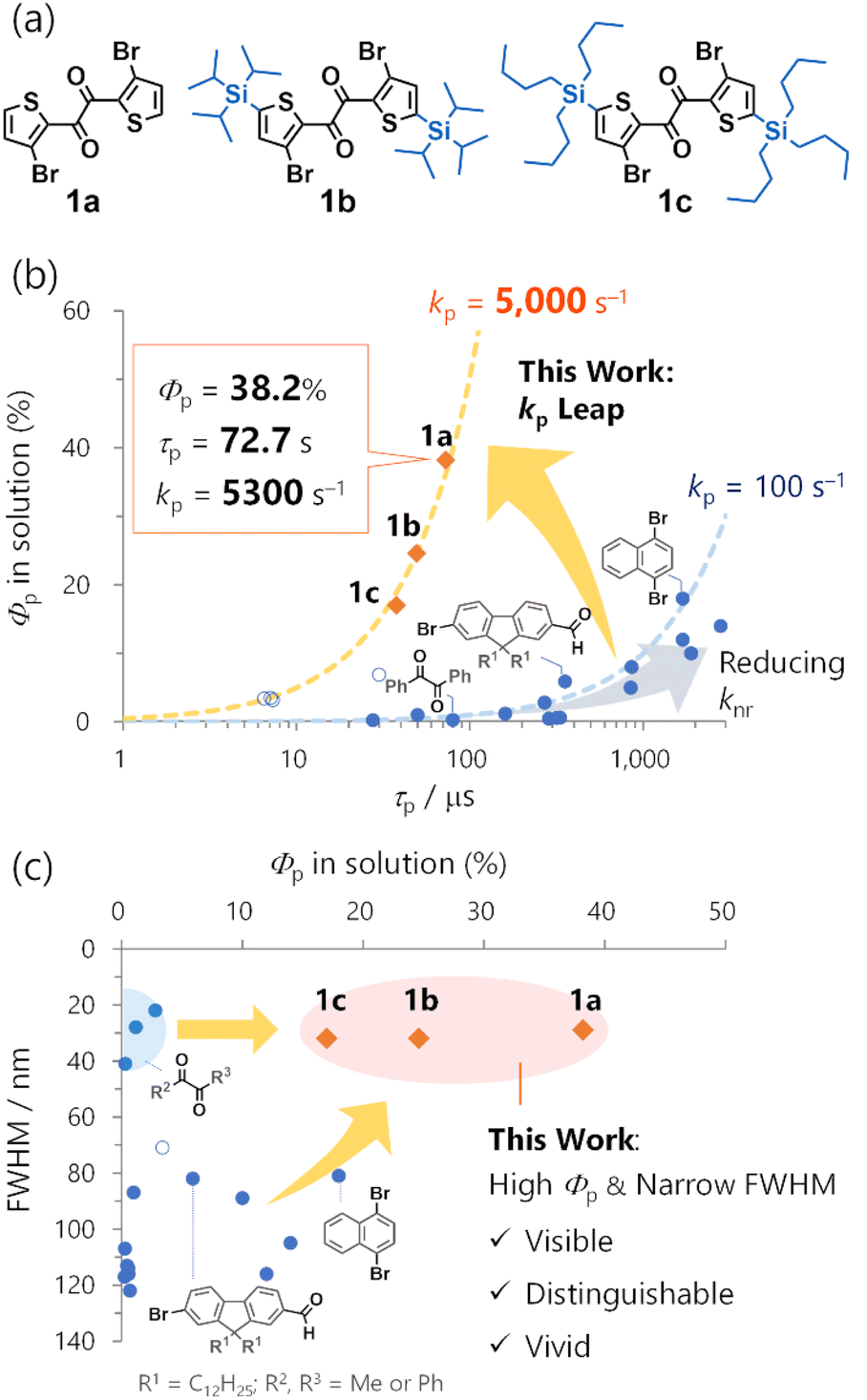 | ||
| Fig. 1 Comparison of representative metal-free organic phosphors (blue filled circles), thiocarbonyls (blue open circles), and 1 (orange diamonds) in solution. (a) Molecular structures of 1. (b) Room-temperature phosphorescence quantum yields (Φp) vs. lifetimes (τp); (c) full-width-at-half-maxima (FWHM) vs. Φp. Blue and orange broken lines in (b) represent kp = 100 and 5000 s−1, respectively, assuming unity intersystem crossing quantum yields. | ||
A promising direction for environmentally independent organic RTP could be the significant increase in kps. Complementary to the persistent RTP, fast RTP could have significant potential use in solutions or any non-rigid molecular environment, including biological conditions.11 Moreover, because fast RTP can avoid efficiency loss caused by the accumulation of the triplet excitons, it also could be used in optoelectronics such as OLEDs.12 However, the quest for fast RTP is facing difficulty so far. In 2022, a selenium-containing molecule (phenoxaselenine) was designed as a candidate for fast RTP; however, its RTP was weak even in a polymer matrix, and only the photoluminescence spectrum and intrinsic phosphorescence rate (ca. 4000 s−1) at 78 K were reported.13 Quite recently, organic ionic crystals were reported to exhibit fast RTP with impressive kp values up to ∼105 s−1.14 The authors ascribed the large kp to the external heavy atom effect of two appropriately arranged iodide counter anions. Nonetheless, such effects relied on the crystal packing, and no RTP was observed in solutions. Thus, molecular design for a large kp that enables RTP in solution is needed.
Low colour purity is another organic RTP challenge. Considering the luminescence of the same quantum efficiency, high-colour-purity emission has a sharp and intense spectral peak that is visible, distinguishable, and vivid. These are essential features for OLED displays and bioimaging.15 However, organic RTP is usually broad with 80–120 nm full-width at half-maxima (FWHMs) (Fig. 1b, Table S1†). Moreover, because of an inefficient intersystem crossing (ISC) from singlet to triplet states, organic phosphors often exhibit both RTP and fluorescence, further impairing colour purity. As a long-known exception, 1,2-diketones, such as biacetyl and benzil, exhibit narrowband RTP that consists of one main peak with a small FWHM value accompanied by weak vibronic bands.16 However, the RTP is feeble in solution. Thus, simultaneously achieving a high Φp and colour purity in organic RTP remains a tremendous challenge.
Using heteroaromatic 1,2-diketones, we previously developed a series of solid-state mechanoresponsive RTP materials,17 solvent-free liquid RTP materials,18 and photoresponsive RTP crystals (Fig. S3†).19 They exhibited RTP in non-rigid molecular environments, such as amorphous solids or solvent-free liquids, wherein conventional metal-free organic compounds rarely show RTP. Thus, our previous results not only demonstrated unique advantages of the 1,2-diketone-based materials in applications, but also implied large kps. However, the fundamental molecular RTP properties remain elusive due to the complexity of the condensed materials. While we have briefly reported some solution-phase RTP properties of thienyl diketones 1a and 1b,17a further investigations were necessary to evaluate kp quantitatively (Fig. 1a and S2†). Typically, kp is derived from experimental Φp, ISC quantum yield ϕISC, and RTP lifetime τp, expressed as kp = Φp/(ϕISC × τp). However, ϕISC was not evaluated in our prior research as we did not focus on kp. In addition, while we determined Φp and τp of 1b, the previous protocol did not assure consistent degassing for those measurements (i.e., the extent of oxygen quenching of RTP would be different; Fig. S2†). Consequently, these values cannot be employed to establish kp. Therefore, the mechanism governing the expected fast RTP remained unexplored, not to mention the absence of the molecular design principle for substantial kp.
Here, we disclose the inherent molecular RTP properties of the thienyl diketone derivatives 1, exhibiting high-efficiency narrowband RTP based on an exceptionally large kps of ∼5000 s−1 (Fig. 1). In particular, 1a exhibited 38.2% Φp in cyclohexane. To our knowledge, this is the highest efficiency for metal-free organic molecules in common solvents. Relative to benzil, the kp was increased by a factor of ∼100 from 39 s−1 to 5300 s−1, enabling a 100-time increase in Φp from 0.31% to 38% (Fig. 1a).16c Moreover, high Φp with a narrow ∼30 nm FWHM was observed in solution (Fig. 1b). The high efficiency and colour purity were also observed in various conventional polymer matrices and crystalline solids, indicating that the RTP was an inherent molecular feature. The RTP mechanism and origin were revealed through single-crystal X-ray structure analysis, time-resolved photoluminescence (TRPL) spectroscopy, transient absorption (TA) spectroscopy, time-resolved infrared (TRIR) spectroscopy, and quantum-chemical calculations. Furthermore, structure-RTP property relationship study considering molecular- and natural transition orbitals provided design principles for fast organic RTP. Our work demonstrates the potential of metal-free organic molecular materials to exhibit fast RTP comparable to precious-metal complexes. Complementary to persistent RTP, organic fast RTP is promising not only for application in solution but also in functional condensed materials as we have witnessed in our previous studies (Fig. S3†).
Results and discussion
RTP and related rate constants of 1a
We first examined the RTP properties in solution for the diketone 1a, which exhibits strong yellow emission (Fig. 2a and b). The steady-state photoluminescence spectrum of 1a in cyclohexane had a single, sharp emission peak at 560 nm accompanied by weak vibronic bands at 615 and ∼680 nm.20 The total photoluminescence quantum yield ΦPL was 38.2% under Ar and the lifetime was 72.7 μs, as determined with a protocol that assures consistent degassing for both measurements (Scheme S2†). TRPL spectra of 1a revealed that the emission emerged just after photoexcitation (within a <100 ps instrumental response function), and exhibited a long lifetime (Fig. S11†). Hence, the emission was phosphorescence, as reported previously for thienyl diketone analogs,17 and ISC occurred over a timescale less than the instrumental response.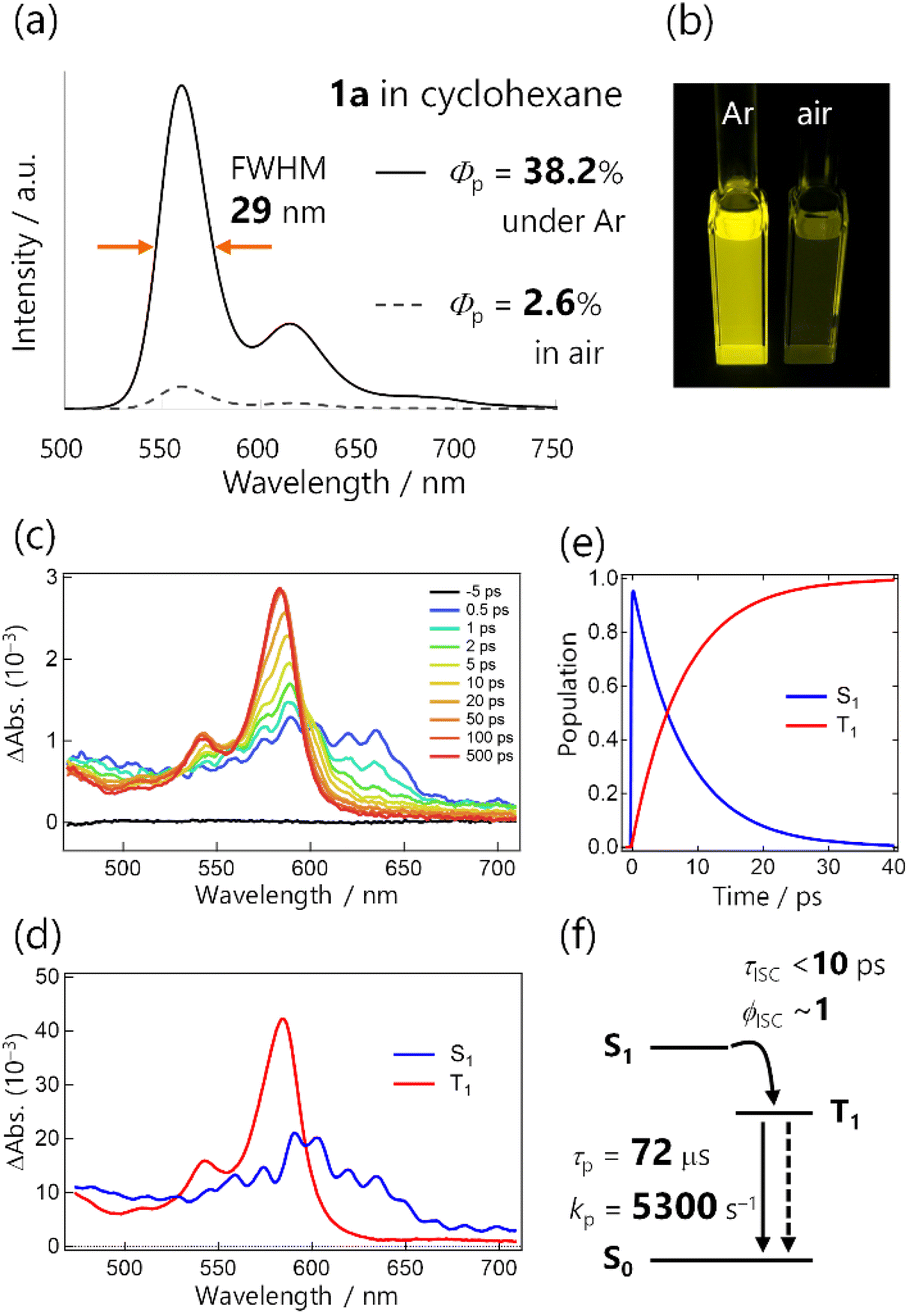 | ||
| Fig. 2 (a) Steady-state photoluminescence spectra of 1a in cyclohexane (4.4 × 10−6 M, excited at 368 nm). (b) Photograph of solutions under 365 nm excitation. (c) fs transient absorption spectra of 1a in cyclohexane excited at 355 nm. (d and e) Selected results from the global analysis based on a sequential model; (d) evolution-associated spectra and (e) corresponding concentration kinetics. Coherent artefact signals are omitted for clarity. (f) Schematic summary of the excited-state dynamics in 1a. | ||
To quantify the rapid ISC, we conducted femtosecond TA spectrum (fsTAS) measurement with ∼100 fs time resolution (Fig. 2c). The broad transient absorption over 450–650 nm changed to sharp spectra with a peak at 580 nm in a ∼10 ps timescale. We globally analysed the TAS by using a sequential model assuming two excited species, resulting in successful fitting with the time constant of the transition estimated to be 7.9 ps (Fig. 2d and e and S14†). We also conducted TAS measurements over nanosecond timescales (Fig. S13†), and observed that the TAS shape was unchanged up to the microsecond timescale. The long-lived spectral component was the lowest triplet excited state (T1 state), populated via ISC with a time constant of <10 ps for 1a in solution (Fig. 2f). Because the ISC outcompeted other relaxation processes, we can reasonably assume that the ISC quantum yield (ϕISC) was unity.
The total photoluminescence quantum yield can be expressed as ΦPL = Φf + Φp, where Φf is the fluorescence quantum yield. Given that ϕISC ∼ 1, phosphorescence dominates the total emission, and Φp ∼ ΦPL = 0.38. To the best of our knowledge,21–23 this is a record-breaking RTP efficiency for metal-free organic molecules in common solvents. In addition, with ϕISC ∼ 1 confirmed, Φp = ϕISCkpτp ∼ kpτp. For Φp = 0.38 and τp = 72.7 μs, kp was derived to be 5300 s−1, which greatly exceeded those of previously reported organic phosphors, except for thiocarbonyl compounds (Fig. 1a and Table S1†). Because of this exceptionally high kp, the RTP of 1a in solution was visible even in air (Φp = 2.6%, Fig. 2a and b, S10, Table S2, and ESI Movie 1†). We note that the estimation of kp in the literature often assumed ϕISC = 1 and Φp ∼ ΦPL without evaluating the time constant of ISC, and employed Φp and τp that were determined without assuring consistent degassing. This would be practical for qualitative purposes, but may cause significant errors in the kp value. Experimental evaluation of ϕISC and assuring consistent degassing are essential for the quantitative evaluation of kp.
The emission colour purity is also outstanding. Fluorescence was not discernible in the steady-state photoluminescence, and the FWHM of the RTP was only 29 nm for 1a (Fig. 2a and S10†). The RTP contained two other vibronic bands at around 615 and 680 nm; nonetheless, the first band at 560 nm covered 71% of the whole spectral area (Table S3†). This is distinct from other organic phosphors, which have broad and structure-less spectra with a 80–120 nm FWHM (Fig. 1c and Table S1†). Although there is a room for further improvement, the narrowband RTP with weak vibronic bands and the fluorescence-free nature represented a high colour purity emission; its coordinates (0.45, 0.54) are located almost at the edge of the chromaticity diagram of Commission internationale de l'éclairage (CIE) 1931 (Fig. 3b).
 | ||
| Fig. 3 (a) Steady-state photoluminescence (PL) spectra of 1a–1c in cyclohexane (10–4.4 × 10−6 M, excited at 368 nm) and (b) CIE1931 chromaticity diagram for their PL in cyclohexane. (c) Photophysical properties of 1a–1c in cyclohexane under Ar (10–4.4 × 10−6 M, excited at 368 nm). Φp, RTP quantum yields; FWHM, full-width-at-half-maxima; λem, emission maxima; τp, lifetimes; kp and knr, phosphorescence and nonradiative rate constants. (d and g) Steady-state PL spectra of (d) 1a–1c@PMMA (5 wt%, excited at 355–368 nm) and (g) a 1c crystal in cyclohexane (1.0 × 10−5 M, excited at 368 nm). (e and h) Photographs of (e) 1b@PMMA and (h) a 1c crystal under 365 nm excitation. (f) Φp in air and the FWHM of 1b-doped polymer films (5 wt%, excited at 350–375 nm). (i) ORTEP drawing of the crystal structure of 1c. Thermal ellipsoids are set at the 50% probability level. | ||
Substituent effect
Next, we investigated the effects of trialkylsilyl substituents on the RTP properties in solution. Trialkylsilyl substituents are known to perturb π-electronic systems through σ–π and/or σ*–π* conjugation (hyperconjugation). While these substituents sometimes improve the fluorescence efficiency, their effects on RTP properties have gained less attention.9d,24We introduced triisopropylsilyl (1b) and tributylsilyl groups (1c) onto the thienyl diketone core (Fig. 1a) and examined their properties in cyclohexane solution. The silylated derivatives 1b and 1c provided identical emission spectra with the maxima slightly red-shifted from 560 nm to 568 nm compared to those of 1a (Fig. 3a and S10†). The steady-state emissions were fluorescence-free and maintained a small FWHM (32 nm). This narrowband yellow emission had a CIE 1931 coordinate of (0.49, 051) (Fig. 3b).2c We also conducted fsTAS measurement for 1b (Fig. S15†) to analyse ISC; the main feature of the TAS was largely the same as that for 1a and the ISC time constants were 0.63 and 2.3 ps. The time constant is even faster than that of 1a and we can assume unity ϕISC for the silylated diketones. Φp for 1b and 1c was less than that for 1a, but still 24.6 and 17%, respectively (Fig. 3c). The decrease in Φp could be attributed to the doubled knr values, from 8500 s−1 for 1a to 15000 s−1 and 22000 s−1 for 1b and 1c, respectively. This was most likely because of the nonradiative decay accelerated by molecular motion involving the silyl groups. In contrast, kp values were only slightly changed, 5000 and 4500 s−1 for 1b and 1c. These results indicated that the large kp of 1 basically originated from the thienyl diketone core.
RTP of 1-doped polymer films in air
The huge kp of 1 enabled efficient RTP in air in the amorphous state using various conventional polymers as matrices (Fig. 3d–f and S20–S24 and Table S4†). The best Φp of 54% was achieved at room temperature in air for a poly(methyl methacrylate) (PMMA) film doped with 5-wt% 1a (Fig. 3d). The PMMA films doped with silylated diketones 1b and 1c also provided narrowband PL spectra similar to that of 1a and exhibited a high Φp of 48 and 40%, respectively (Fig. 3d and e). These were among the highest reported for polymer-based RTP systems doped with metal-free organic dyes.25 The reported systems with insufficient kp required a tailor-made approach that involves specific interactions between the polymers and dopants. Hydrogen bonding in poly(vinyl alcohol) was reported to be effective and widely used,10c–e but these interactions or polymers are difficult to use under highly humid conditions or in water. Because PMMA is resistant to water, the RTP properties of a 1c-doped PMMA film were not spoiled by water, exhibiting the same PL spectrum with a Φp of 35% even when submerged in water (Fig. S22†).26Moreover, phosphorescent films with Φp values of 38–18% were obtained by doping 1b into other five conventional polymers (Fig. 3f and S23, S24 and Table S4†). The emission maxima and FWHMs were almost unchanged, with colour purities independent of the polymers. Thus, RTP properties of the films were derived from inherent characteristics of 1.
We would like to emphasize that achieving efficient organic RTP in solution and in polymer matrices is challenging because nonradiative decay is not suppressed under these conditions. In such cases, the fast RTP (large kp) has a distinct advantage over persistent RTP with small kp. The same can be said for the RTP in non-crystalline aggregate states, such as the amorphous solid state and solvent-free liquid state. It becomes clear that our previous studies of mechanoresponsive RTP materials,17 solvent-free liquid RTP materials,18 and photoresponsive RTP crystals19 (Fig. S3†) demonstrated the usefulness and applications of fast RTP.
Conformation of RTP-emitting species
Considering that 1 has three successive single bonds in the diketone core, identifying the RTP-emitting conformation was important.16b–d,27 In our previous study, two distinct conformers of thienyl diketones were identified by single-crystal X-ray structure analysis.17a The 1a crystal exhibited a trans-planar (TP) conformation with respect to the dicarbonyl moiety, while the 1b crystal exhibited a skew conformation (Fig. S33†). Thus, the conformation in crystals varied based on the substituents due to subtle differences in the intermolecular interactions. Unfortunately, the 1a crystal was nonemissive, likely due to intermolecular electronic interactions. 1a exhibited one-dimensional columnar π-stacking with a 3.481 Å interplanar distance, and the absorption of the crystal had a long tail until over 700 nm (Fig. S34†). On the other hand, the 1b crystal emitted green RTP, which was different from the solution RTP. Therefore, further study was required to nail down the conformation of the yellow RTP-emitting species.In the present work, we found that crystals of 1c exhibited yellow RTP with a 50% Φp in air (Fig. 3g and h). The RTP decayed as a single exponential with a τp of 79 μs, and kp was 6000 s−1 (Fig. S25†). Most importantly, the PL spectrum was almost identical to that of 1c in solution, indicating emission from the same conformer (Fig. 3g). Single-crystal X-ray structure analysis revealed the TP conformation of 1c, with the thienyl diketone core spatially separated from neighbouring molecules by tributylsilyl substituents (Fig. 3i and S32†; no π-stacking, and the interplanar distance was 4.45 Å). Thus, the RTP from the 1c crystal, as well as 1 in solution, was unambiguously assigned to the monomer emission from its TP conformer. Notably, 1c represents a rare example of metal-free organic molecules exhibiting remarkable Φp in solution, in a polymer matrix, and in a crystal, with almost identical PL spectra.
To experimentally elucidate conformation dynamics in the excited state, TRIR spectroscopy was performed. Although we could not perform the TRIR measurement on 1a because of poor solubility, we managed to conduct the TRIR measurement on 1b in cyclohexane using a 267 nm wavelength pump light (Fig. S16†). The spectra converged to sharp spectra over 0.74 ps and 2.67 ps timescales, consistent with the time constants extracted from fsTAS of 1b (0.63 and 2.3 ps, Fig. S15†). Note that these time constants were comparable to those observed for 1a in the fsTAS measurement. This indicated that the excited-state dynamics were primarily affected by the thienyl diketone core, while the silyl groups had minor effects. The two timescales were attributed to the conformation change and ISC. More importantly, the converged TRIR spectra were consistent with simulated spectra, assuming a TP conformation in the T1 state (Fig. S17†). Thus, both the conformation changes and the ISC were ultrafast, and the RTP is confirmed to originate from the TP conformer.
Origin of the rapid ISC
Given that the ISC occurs at the TP conformation, we investigated the molecular origin of the rapid ISC by time-dependent density functional theory (TDDFT) calculations (Fig. 4 and S26†). In principle, ISC is fast when the energy gap is small and the spin–orbit coupling (SOC) matrix element is large.28 In the S1-optimized TP geometry, the S1 state was almost isoenergetic to the T3 state (energy gap <0.01 eV for 1a and 0.04 eV for 1b at the TDA/uCAM-B3LYP-D3/6-311G(d) level of theory).29 Moreover, the S1–T3 SOC matrix elements 〈S1|HSO|T3〉 for 1a and 1b were 125 and 112 cm−1, respectively. The substantial SOC matrix elements were consistent with El Sayed's rule; the electronic configurations of the S1 and T3 states were (n,π*) and (π,π*), respectively (Fig. S26†). These results strongly supported the ultrafast ISC, which occurred dominantly from the S1 to T3 states, followed by internal conversion to the T1 state.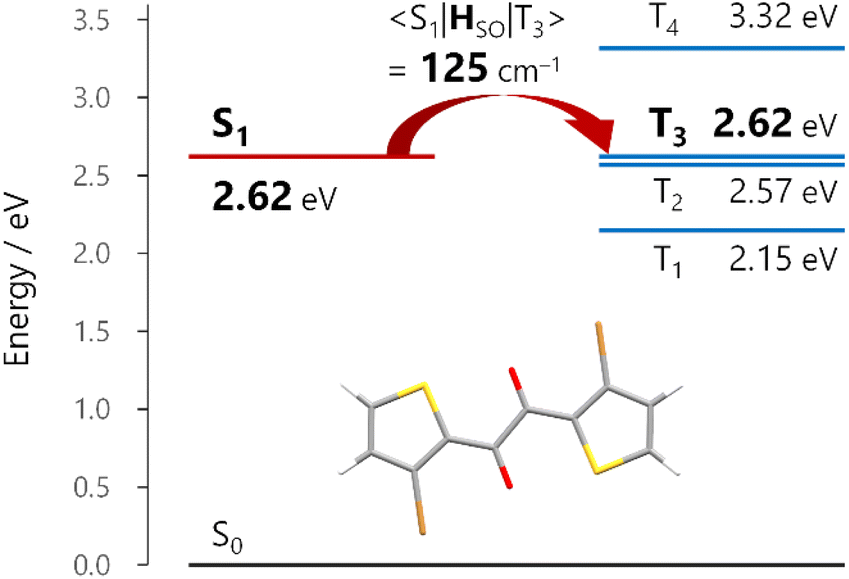 | ||
| Fig. 4 Energy diagram of 1a in the S1-optimized trans-planar (TP) geometry. Energies and the spin–orbit coupling matrix element between the S1 and T3 states 〈S1|HSO|T3〉 were calculated at the TDA/uCAM-B3LYP-D3/6-311G(d) level of theory. | ||
Origin of the substantial kp value
Based on concrete basis that the RTP comes from the TP conformer, we then investigated the origin of the outstanding kp. Theoretically, it was roughly based on intensity borrowing from the nth excited singlet states (Sn), as given by:28a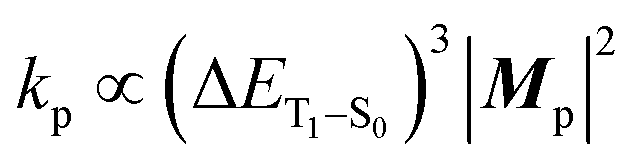 | (1) |
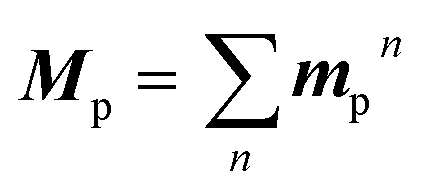 | (2) |
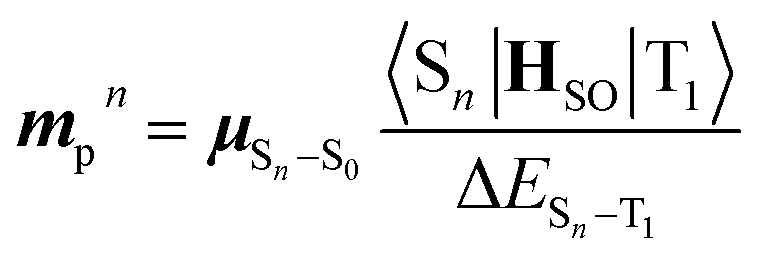 | (3) |
TDDFT calculations for the T1-optimized TP geometry of 1a yielded kp = 5400 s−1 (for n = 1–6), which excellently reproduced the experimental value of 5300 s−1. Remarkably, the T1 state strongly coupled with the S3 state (Fig. 5a); a large SOC matrix element 〈S3|HSO|T1〉 = 167 cm−1 and a reasonable ΔES3–T1 = 1.74 eV = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 provided a large mixing coefficient 〈S3|HSO|T1〉/ΔES3–T1 of 1.19 × 10−2. This coefficient indicates that the T1 state can borrow 1.19% of the transition dipole moment between the S3 and S0 states (i.e., μS3–S0), which is quite large for phosphorescence. Furthermore, the |μS3–S0| was as large as 4.68 D, thus realizing a large |mp3| = 56 × 10−3D. The contribution from mp3 corresponded to 93% of the total kp value and is obviously the source of the large kp (Fig. 6b, vide infra). Similar results were obtained for 1b, confirming that the origin of the substantial kp lies in the thienyl diketone core (Table S5†).
000 cm−1 provided a large mixing coefficient 〈S3|HSO|T1〉/ΔES3–T1 of 1.19 × 10−2. This coefficient indicates that the T1 state can borrow 1.19% of the transition dipole moment between the S3 and S0 states (i.e., μS3–S0), which is quite large for phosphorescence. Furthermore, the |μS3–S0| was as large as 4.68 D, thus realizing a large |mp3| = 56 × 10−3D. The contribution from mp3 corresponded to 93% of the total kp value and is obviously the source of the large kp (Fig. 6b, vide infra). Similar results were obtained for 1b, confirming that the origin of the substantial kp lies in the thienyl diketone core (Table S5†).
 | ||
| Fig. 5 (a) Energy diagram of 1a in the T1-optimized trans-planar (TP) geometry depicting the principal intensity-borrowing. Energies, the transition dipole moment μS3–S0, and the spin–orbit coupling matrix element 〈S3|HSO|T1〉 were calculated at the TDA/(u)CAM-B3LYP-D3/6-311G(d) level. (b) Principal natural transition orbitals (NTOs) for S3–S0 (left) and T1–S0 (right) transitions. | ||
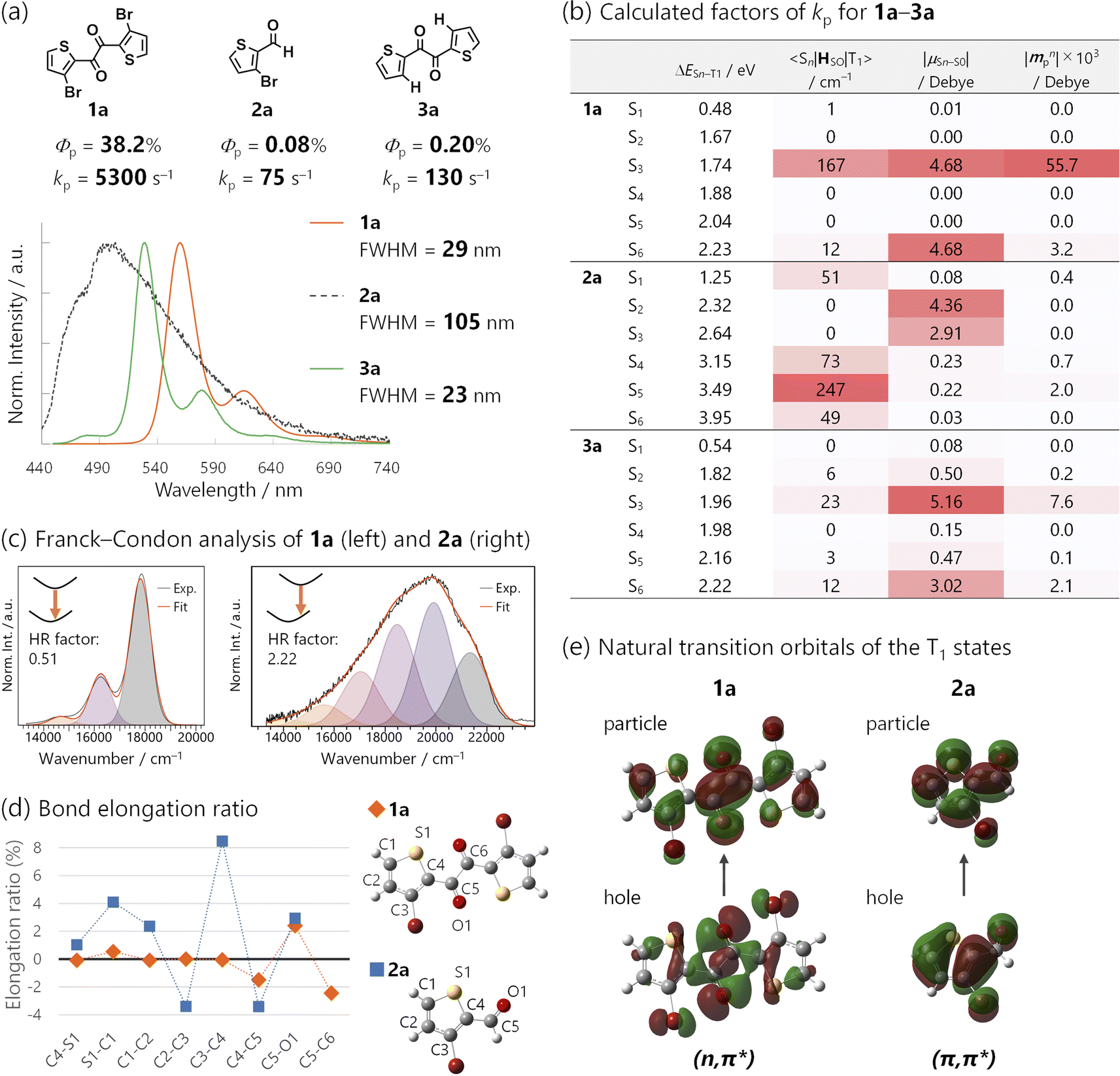 | ||
| Fig. 6 (a) Chemical structures of 1a, 2a, and 3a, and their Φp, estimated kp, steady-state photoluminescence spectra, and the corresponding FWHM in cyclohexane (1.0 × 10−5 M, 2.6 × 10−3 M, and 1.0 × 10−4 M, respectively) excited at 368 nm under Ar. kp was derived as kp = Φp/τp, assuming ϕISC = 1. (b) Calculated factors of kp for 1a–3a. (c) Franck–Condon analysis of the emission spectra of 1a (left) and 2a (right). Huang–Rhys (HR) factors were 0.51 and 2.22, respectively. (d) Bond-elongation ratio between the optimized geometries in the S0 and T1 states of 1a and 2a. (e) Natural transition orbitals of the T1 states for 1a and 2a. | ||
The natural transition orbitals (NTOs) of the S3 and T1 states provided an intuitive understanding of the large μS3–S0 and 〈S3|HSO|T1〉 (Fig. 5b). First, the S3 state (S3–S0 transition) had a pure (π,π*) configuration, where the NTOs delocalized over the entire molecule in a planar geometry, which was favourable for large μ. On the other hand, the T1 state had a pure (n,π*) configuration, i.e., it was not contaminated with (π,π*) character. The pure 3(n,π*) configuration enabled SOC with the pure 1(π,π*) state, consistent with El Sayed's rule.4a The pure (n,π*)/(π,π*) characters are noteworthy, because excited states with mixed (n,π*) and (π,π*) configurations diminish SOC.30 Most importantly, the hole NTOs of the S3 and T1 states around the heavy atoms, especially Br, were mutually perpendicular. This is ideal because the spin–orbit Hamiltonian HSO involves the orbital angular-momentum operator that rotates the orbitals by 90°.13,28a Thus, for the hole-constituting atomic orbital of Br, the in-plane σ-symmetric n orbital of T1 states (Fig. 5b bottom-right) became π-symmetric, producing a significant spatial integral with the out-of-plane p orbital of the S3 state (Fig. 5b bottom-left). The large spatial integral near the heavy nuclei boosts the heavy-atom effect, resulting in a large 〈S3|HSO|T1〉 = 167 cm−1. Thus, 1 embodies an ideal electronic structure for intensity-borrowing that enhances kp: (1) a planar π-system that produces large μ; (2) carbonyl groups in the π-plane that joined the pure 1(π,π*) and pure 3(n,π*) states; and (3) heavy atoms with electrons conjugated to both the π and σ-/n-systems to boost 〈Sn|HSO|T1〉.
Structure-RTP property relationship study
The significance of the diketone skeleton for the huge kp was further evident when comparing 1 with the corresponding bromoaldehyde 2 and diketone without Br atoms 3 (Fig. 6a and b for 1a, 2a, and 3a; Fig. S5† for 1b, 2b, and 3b bearing triisopropylsilyl groups; also see Table S2†). All the compounds exhibited RTP in degassed solutions; however, Φp of 1 was much higher than those of 2 and 3 regardless of the silyl substituents (38.2%, 0.08%, and 0.20% for 1a, 2a, and 3a; 24.6%, 0.22%, and 1.2% for 1b, 2b, and 3b, respectively).The superior Φp of 1 was attributed to its large kp, which was more than one order of magnitude larger than the estimated kps of 2 and 3 (Fig. 6a and Table S2†). TDDFT calculations reasonably reproduced this trend; the calculated kp for 1a, 2a, and 3a was 5400, 9, and 71 s−1, respectively (Fig. S27, S30 and S31†). The small kp of aldehyde 2 was because of the mismatched 〈Sn|HSO|T1〉 and μSn–S0 (Fig. 6b; see Table S4† for 1b, 2b, and 3b). Thus, the T1 state of 2 had a (π,π*) configuration (Fig. 6e), which coupled strongly to (n,π*) Sn states (e.g., n = 5; 〈S5|HSO|T1〉 was as large as 247 cm−1 for 2a). However, the (n,π*) configuration diminishes μSn–S0 (e.g., |μS5–S0| was only 0.22 D for 2a). Consequently, intensity-borrowing was insufficient, resulting in a small kp. Hence, a large 〈Sn|HSO|T1〉 (or a large mixing coefficient, 〈Sn|HSO|T1〉/ΔESn–T1) itself is insufficient and the combination with a large μSn–S0 in the same n-th singlet state is essential for achieving a huge kp.31 To meet this requirement, the T1 state should have a (n,π*) configuration. Indeed, the kp of Br-free diketone 3a was larger than that of Br-containing aldehyde 2a, because the T1 state of 3a had a (n,π*) configuration. Thus, the (n,π*) T1 state prefers coupling with (π,π*) Sn states, whose μSn–S0 can be sizable (e.g., n = 3, |μS3–S0| = 5.16 D for 3a; Fig. 6b). However, SOC between these states was not as effective as that for 1a because of the absence of the heavy atom, Br (e.g., 〈S3|HSO|T1〉 = 23 cm−1 for 3a). Therefore, intensity borrowing was less effective, yielding a lower kp than the brominated diketone 1a.
The side-on spatial arrangement of C–Br and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O was crucial for the heavy-atom effect because of better mutual alignment of the in-plane Br and O p orbitals. This resulted in large hole coefficients in the T1 state (Fig. 5b).32 However, the arrangement is energetically unfavourable due to electronic repulsion, because Br and O in the TP geometry of 1 were within the sum of their van der Waals radii according to the X-ray structure (Fig. S32†).17a,33,34 Indeed, DFT calculations for the aldehyde 2a indicated that the side-on conformation was less stable (by 3 kcal mol−1) for both S0 and T1 states than the conformation with oxygen pointing away from the Br (Fig. S28†).32 Interestingly, the TP geometry of 1 involved two-fold intramolecular S⋯O noncovalent chalcogen bonding interactions,35 with interaction energies as large as 7.74 kcal mol−1, according to the natural bonding orbital analysis of 1a.17a The chalcogen bonds stabilized the TP conformer and increased kp by forcibly fixing Br in the vicinity of carbonyl oxygens.
O was crucial for the heavy-atom effect because of better mutual alignment of the in-plane Br and O p orbitals. This resulted in large hole coefficients in the T1 state (Fig. 5b).32 However, the arrangement is energetically unfavourable due to electronic repulsion, because Br and O in the TP geometry of 1 were within the sum of their van der Waals radii according to the X-ray structure (Fig. S32†).17a,33,34 Indeed, DFT calculations for the aldehyde 2a indicated that the side-on conformation was less stable (by 3 kcal mol−1) for both S0 and T1 states than the conformation with oxygen pointing away from the Br (Fig. S28†).32 Interestingly, the TP geometry of 1 involved two-fold intramolecular S⋯O noncovalent chalcogen bonding interactions,35 with interaction energies as large as 7.74 kcal mol−1, according to the natural bonding orbital analysis of 1a.17a The chalcogen bonds stabilized the TP conformer and increased kp by forcibly fixing Br in the vicinity of carbonyl oxygens.
Origin of the narrowband emission
Finally, we investigated the origin of the narrowband emission of 1via a comparison of the 1a emission spectra with those of its aldehyde counterpart, 2a (Fig. 6a, c–e; see Fig. S18† for the comparison of 1b with 2b). Diketones 1a and 1b exhibited narrowband emissions (FWHM = 29 and 32 nm) while aldehydes 2a and 2b had broadband emissions (FWHM = 105 and 106 nm).The bandwidths could be strongly correlated with molecular geometry changes in the ground and excited states, which are quantified using the Huang–Rhys (HR) factor obtained from a Franck–Condon analysis of the spectra (Fig. 6c, S18, and Table S3†).15,36 The HR factor of diketone 1a (1b) was 0.51 (0.50), while that of aldehyde 2a (2b) was 2.22 (2.39). The smaller HR factors in 1 corresponded to smaller geometry changes in the T1-to-S0 transition, which were corroborated by DFT calculations. The results indicated that although diketone 1a and aldehyde 2a retained their planarity in the transition, bond length changes were small for 1a and large for 2a.
We visualized these bond length changes using elongation ratios, defined as (RT1 − RS1)/RS1, where RT1 and RS1 represent bond lengths in the T1-and S0-optimized geometries; a positive value indicates that the bond is longer in the T1 state (Fig. 6d). The bond lengths within the thiophene ring of diketone 1a exhibited minimal changes. Conversely, the absolute values of the elongation ratios were large in the case of aldehyde 2a (e.g., the bond length between 3C and 4C changed by 8.4%).
These considerable bond length changes in 2a were attributed to the (π,π*) character of the T1 state, as evident in the NTOs (Fig. 6e). The electronic transition from bonding to antibonding orbitals significantly affects the bond strength. In contrast, the NTOs of 1a were mainly localized on the 1,2-dicarbonyl moiety, and the electronic transition was nonbonding to antibonding, which led to minor changes in the bond lengths (Fig. 6d). Moreover, the transition in the diketones 1 had no net charge-transfer character because of the centrosymmetric geometry. This was in contrast to most aromatic (mono)carbonyl compounds, where 3(n,π*) states have a charge-transfer character from the carbonyl to the aromatic ring. These features of diketones minimized molecular geometry changes during phosphorescence emission. Therefore, the 1,2-diketone-based phosphor would be a promising platform for efficient narrowband RTP.
Conclusions
Efficient narrowband RTP from metal-free organic 3-bromo-2-thienyl diketones was observed in solutions, amorphous polymer matrices, and crystalline solids. A substantial phosphorescence rate constant of ∼5000 s−1 was experimentally confirmed, which was the key to outstanding RTP quantum yields in solution (38% under Ar) and in polymer films (up to 54% in air). Ultrafast spectroscopy and single-crystal X-ray structure analysis revealed that the fast RTP originated from the diketone planar conformation. Both experimental and theoretical analyses indicated that the planar conformer embodies ideal electronic structures for fast RTP:1. The planar geometry with a delocalized π-system
2. σ-Symmetric n-orbitals perpendicular to the π-system
3. Heavy atoms conjugated with both π- and n-electron systems
4. (n,π*) configuration of the T1 state
Fulfilling these points would be a promising design principle for molecules with large μSn–S0 and 〈Sn|HSO|T1〉 in the same Sn states, and results in a kp leap. In addition, the centrosymmetric nonbonding-to-antibonding electronic transition of the 1,2-diketone skeleton was the key to the narrowband emission. The fast RTP (large kp) is potentially advantageous for various applications, such as phosphorescence OLEDs and lasers, triplet-to-singlet conversion via Förster resonance energy transfer, bioimaging, and theranostics. However, such applications have been monopolized by precious-metal phosphors. We believe that the mechanistic elucidation paves the way for developing fast organic RTP, which could expand applications of metal-free organic materials.
Data availability
All experimental/computational procedures and data related to this article are provided in the ESI.†Author contributions
Y. T. project administration: lead; supervision: lead; conceptualisation: equal; funding acquisition: lead; investigation: lead; visualisation: lead; writing—original draft: lead; writing—review & editing: lead. K. M. project administration: lead; supervision: lead; conceptualisation: equal; funding acquisition: lead; investigation: supporting; visualisation: supporting; writing—original draft: supporting; writing—review & editing: lead. E. O., Y. O., M. T., and S. K. investigation: equal. M. K. investigation: equal; funding acquisition: supporting; writing—review & editing: supporting. T. E. and K. K. investigation: supporting. K. O. and T. O. resources: lead; supervision: supporting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (grant numbers JP23H03955, JP22H02159, JP20H05676, and JP19K15542). M. K. acknowledges a grant-in-aid for a JSPS Research Fellow (22J12961). Y. T. is grateful to the ENEOS Tonengeneral Research/Development Encouragement & Scholarship Foundation, the Izumi Science and Technology Foundation, the Toyota Physical and Chemical Research Institute, and the Yazaki Memorial Foundation for Science and Technology for the financial support. K. M. acknowledges Sumitomo Basic Science Research Projects, the Kyushu University Q-PIT Support Program for Young Researchers and Doctoral Students, and the Young Researchers Support Project, Faculty of Science, Kyushu University grant numbers 22-A5 (R4). The authors thank Dr Ken-ich Yamashita (Osaka University) for the lifetime measurements and Dr Yuki Kurashige (Kyoto University) and Dr Ryohei Kishi (Osaka University) for fruitful discussions on the quantum chemical calculations. Computations were performed at the Research Center for Computational Science, Okazaki, Japan (Project: 22-IMS-C153 and 23-IMS-C187). The experiments were partially performed at the Analytical Instrument Facility, Graduate School of Science, Osaka University, using research equipment shared in the MEXT project (JPMXS0441200023).Notes and references
- (a) S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef; (b) Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS.
- (a) P.-T. Chou and Y. Chi, Chem.–Eur. J., 2007, 13, 380–395 CAS; (b) Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 CAS; (c) C. Fan and C. Yang, Chem. Soc. Rev., 2014, 43, 6439–6469 RSC; (d) C.-P. Tan, Y.-M. Zhong, L.-N. Ji and Z.-W. Mao, Chem. Sci., 2021, 12, 2357–2367 CAS.
- (a) A. Bouskila, B. Drahi, E. Amouyal, I. Sasaki and A. Gaudemer, J. Photochem. Photobiol., A, 2004, 163, 381–388 CrossRef CAS; (b) A. K. Bansal, W. Holzer, A. Penzkofer and T. Tsuboi, Chem. Phys., 2006, 330, 118–129 CrossRef CAS; (c) A. Endo, K. Suzuki, T. Yoshihara, S. Tobita, M. Yahiro and C. Adachi, Chem. Phys. Lett., 2008, 460, 155–157 CrossRef CAS; (d) J. R. Sommer, A. H. Shelton, A. Parthasarathy, I. Ghiviriga, J. R. Reynolds and K. S. Schanze, Chem. Mater., 2011, 23, 5296–5304 CrossRef CAS.
- (a) N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, Sausalito, CA, 2009 Search PubMed; (b) N. S. e. Konstantin and A. B. Elena, Phys.-Usp., 2005, 48, 231 Search PubMed; (c) M. Kasha, J. Chem. Phys., 1952, 20, 71–74 CrossRef CAS; (d) D. S. McClure, J. Chem. Phys., 1949, 17, 905–913 CrossRef CAS; (e) D. R. Kearns and W. A. Case, J. Am. Chem. Soc., 1966, 88, 5087–5097 CrossRef CAS.
- (a) J. J. Donkerbroek, J. J. Elzas, C. Gooijer, R. W. Frei and N. H. Velthorst, Talanta, 1981, 28, 717–723 CrossRef CAS PubMed; (b) J. Xu, A. Takai, Y. Kobayashi and M. Takeuchi, Chem. Commun., 2013, 49, 8447–8449 RSC; (c) B. Ventura, A. Bertocco, D. Braga, L. Catalano, S. d'Agostino, F. Grepioni and P. Taddei, J. Phys. Chem. C, 2014, 118, 18646–18658 CrossRef CAS; (d) G. D. Gutierrez, G. T. Sazama, T. Wu, M. A. Baldo and T. M. Swager, J. Org. Chem., 2016, 81, 4789–4796 CrossRef CAS; (e) Z. Yu, Y. Wu, L. Xiao, J. Chen, Q. Liao, J. Yao and H. Fu, J. Am. Chem. Soc., 2017, 139, 6376–6381 CrossRef CAS; (f) H. Shu, H. Li, J. Rao, L. Chen, X. Wang, X. Wu, H. Tian, H. Tong and L. Wang, J. Mater. Chem. C, 2020, 8, 14360–14364 RSC.
- (a) A. Maciejewski and R. P. Steer, Chem. Rev., 1993, 93, 67–98 CrossRef CAS; (b) M. Szymanski, R. P. Steer and A. Maciejewski, Chem. Phys. Lett., 1987, 135, 243–248 CrossRef CAS; (c) M. Szymanski, A. Maciejewski and R. P. Steer, J. Phys. Chem., 1988, 92, 2485–2489 CrossRef CAS.
- J. D. Coyle, Tetrahedron, 1985, 41, 5393–5425 CrossRef CAS.
- (a) A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC; (b) W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS; (c) A. D. Nidhankar, Goudappagouda, V. C. Wakchaure and S. S. Babu, Chem. Sci., 2021, 12, 4216–4236 RSC.
- (a) D. B. Clapp, J. Am. Chem. Soc., 1939, 61, 523–524 CrossRef CAS; (b) W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS; (c) O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS; (d) M. Shimizu, R. Shigitani, M. Nakatani, K. Kuwabara, Y. Miyake, K. Tajima, H. Sakai and T. Hasobe, J. Phys. Chem. C, 2016, 120, 11631–11639 CrossRef CAS; (e) E. Hamzehpoor and D. F. Perepichka, Angew. Chem., Int. Ed., 2020, 59, 9977–9981 CAS; (f) A. Fermi, G. Bergamini, R. Peresutti, E. Marchi, M. Roy, P. Ceroni and M. Gingras, Dyes Pigm., 2014, 110, 113–122 CrossRef CAS; (g) Y. Wen, H. Liu, S. Zhang, Y. Gao, Y. Yan and B. Yang, J. Mater. Chem. C, 2019, 7, 12502–12508 RSC.
- (a) S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS; (b) M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Nat. Commun., 2015, 6, 8947 CrossRef PubMed; (c) M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Angew. Chem., Int. Ed., 2014, 53, 11177–11181 CrossRef CAS; (d) Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed; (e) H. Wu, W. Chi, Z. Chen, G. Liu, L. Gu, A. K. Bindra, G. Yang, X. Liu and Y. Zhao, Adv. Funct. Mater., 2019, 29, 1807243 CrossRef; (f) Z.-Y. Zhang, W.-W. Xu, W.-S. Xu, J. Niu, X.-H. Sun and Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 18748–18754 CrossRef CAS.
- W. Shao and J. Kim, Acc. Chem. Res., 2022, 55, 1573–1585 CrossRef CAS PubMed.
- (a) Z. Zhou, X. Xie, Z. Sun, X. Wang, Z. An and W. Huang, J. Mater. Chem. C, 2023, 11, 3143–3161 RSC; (b) H. Imahori, Y. Kobori and H. Kaji, Acc. Mater. Res., 2021, 2, 501–514 CrossRef CAS.
- W. Shao, H. Jiang, R. Ansari, P. M. Zimmerman and J. Kim, Chem. Sci., 2022, 13, 789–797 RSC.
- I. Partanen, O. Al-Saedy, T. Eskelinen, A. J. Karttunen, J. J. Saarinen, O. Mrózek, A. Steffen, A. Belyaev, P.-T. Chou and I. O. Koshevoy, Angew. Chem., Int. Ed., 2023, 62, e202305108 CrossRef CAS PubMed.
- J. M. Ha, S. H. Hur, A. Pathak, J.-E. Jeong and H. Y. Woo, NPG Asia Mater., 2021, 13, 53 CrossRef CAS.
- (a) H. L. J. Bäckström and K. Sandros, Acta Chem. Scand., 1960, 14, 48–62 CrossRef; (b) D. J. Morantz and A. J. C. Wright, J. Chem. Phys., 1971, 54, 692–697 CrossRef CAS; (c) L. Flamigni, F. Barigelletti, S. Dellonte and G. Orlandi, J. Photochem., 1983, 21, 237–244 CrossRef CAS; (d) A. K. Singh, D. K. Palit and J. P. Mittal, Chem. Phys. Lett., 2002, 360, 443–452 CrossRef CAS; (e) Y. Tsuboi, K. Okada, S. Ishizaka and N. Kitamura, Anal. Sci., 2005, 21, 303–308 CrossRef CAS PubMed; (f) Y. Gong, Y. Tan, H. Li, Y. Zhang, W. Yuan, Y. Zhang, J. Sun and B. Z. Tang, Sci. China: Chem., 2013, 56, 1183–1186 CrossRef CAS.
- (a) Y. Tani, M. Terasaki, M. Komura and T. Ogawa, J. Mater. Chem. C, 2019, 7, 11926–11931 RSC; (b) Y. Tani, M. Komura and T. Ogawa, Chem. Commun., 2020, 56, 6810–6813 RSC; (c) Y. Takewaki, T. Ogawa and Y. Tani, Front. Chem., 2022, 9, 812593 CrossRef.
- M. Komura, T. Ogawa and Y. Tani, Chem. Sci., 2021, 12, 14363–14368 RSC.
- M. Komura, H. Sotome, H. Miyasaka, T. Ogawa and Y. Tani, Chem. Sci., 2023, 14, 5302–5308 RSC.
- Vibronic band is a term for the band due to the transition from the vibrational ground state of an electronic excited state to different vibrational levels of the S0 (electronic ground) state. We have confirmed this assignment by TRPL measurements, where the whole spectrum decayed at the same rate (Fig. S12†)..
- In 2014, a family of organic boron complexes was reported to show RTP in solution with a quantum yield over 100% due to singlet fission. However, in 2022, another group evidently pointed out that the RTP was an artifact. They mentioned that “it is obvious that the error is caused by the limit of the dynamic range of the instrument,” and that the compound “exhibits solely a highly intense fluorescence in CH2Cl2 with no indication of phosphoresce and dual emission in the room temperature solution.” See ref. 22 for details..
- (a) M. Koch, K. Perumal, O. Blacque, J. A. Garg, R. Saiganesh, S. Kabilan, K. K. Balasubramanian and K. Venkatesan, Angew. Chem., Int. Ed., 2014, 53, 6378–6382 CrossRef CAS; (b) Y. Chen, C.-H. Wang, T.-C. Chou and P.-T. Chou, Angew. Chem., Int. Ed., 2022, 61, e202109224 CrossRef CAS.
- During the review process of this manuscript, X. Peng et al. published a report on an efficient organic RTP in solution. The total photoluminescence quantum yield (PLQY) in toluene was reported to be as high as 60% under N2 and 8% under O2; however, neither the RTP QY nor the intersystem crossing efficiency was investigated, and the RTP rate constant remains unclear. According to the reported fluorescence lifetimes and PL decay curve, total PLQY under N2 should include a considerable fluorescence contribution. These results highlight the significance of evaluating the intersystem crossing efficiency. See: X. Peng, P. Zou, J. Zeng, X. Wu, D. Xie, Y. Fu, D. Yang, D. Ma, B. Z. Tang and Z. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202405418 CrossRef CAS PubMed.
- (a) T. Nakashima, M. Shimada, Y. Kurihara, M. Tsuchiya, Y. Yamanoi, E. Nishibori, K. Sugimoto and H. Nishihara, J. Organomet. Chem., 2016, 805, 27–33 CrossRef CAS; (b) M. Shimizu, T. Kinoshita, R. Shigitani, Y. Miyake and K. Tajima, Mater. Chem. Front., 2018, 2, 347–354 RSC; (c) S. Hirata, Adv. Sci., 2019, 6, 1900410 CrossRef; (d) M. Shimizu, S. Nagano and T. Kinoshita, Chem.–Eur. J., 2020, 26, 5162–5167 CrossRef CAS.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS.
- (a) Z. He, W. Li, G. Chen, Y. Zhang and W.-Z. Yuan, Chin. Chem. Lett., 2019, 30, 933–936 CrossRef CAS; (b) J. Song, L. Ma, S. Sun, H. Tian and X. Ma, Angew. Chem., Int. Ed., 2022, 61, e202206157 CrossRef CAS; (c) M. Yasui, T. Fujihara, H. Ohtsu, Y. Wada, T. Shimada, Y. Zhu, M. Kawano, K. Hanaya, T. Sugai and S. Higashibayashi, Commun. Chem., 2023, 6, 245 CrossRef CAS PubMed; (d) Z. Chen, X. Chen, D. Ma, Z. Mao, J. Zhao and Z. Chi, J. Am. Chem. Soc., 2023, 145, 16748–16759 CrossRef CAS PubMed; (e) S. H. Lee, M. S. Valverde Paredes, P. M. Forster and D.-C. Lee, RSC Adv., 2024, 14, 6285–6291 RSC.
- (a) G. Baryshnikov, B. Minaev and H. Ågren, Chem. Rev., 2017, 117, 6500–6537 CrossRef CAS PubMed; (b) T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- M. J. G. Peach and D. J. Tozer, J. Phys. Chem. A, 2012, 116, 9783–9789 CrossRef CAS PubMed.
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS.
- (a) S. Hirata, J. Phys. Chem. Lett., 2018, 9, 4251–4259 CrossRef CAS; (b) I. Bhattacharjee and S. Hirata, Adv. Mater., 2020, 32, 2001348 CrossRef CAS.
- S. Sarkar, H. P. Hendrickson, D. Lee, F. DeVine, J. Jung, E. Geva, J. Kim and B. D. Dunietz, J. Phys. Chem. C, 2017, 121, 3771–3777 CrossRef CAS.
- One might expect the formation of halogen bonding (attractive interactions) in side-on arrangements. However, it is unlikely because the halogen bonding is a highly directional interaction between the lone pair of (in this case) carbonyl O and “sigma-hole” of Br that forms along the C–Br σ* bond. Indeed, there were no donor–acceptor interactions found between Br⋯O according to natural bonding orbital analysis of 1a..
- S. J. Ang, T. S. Chwee and M. W. Wong, J. Phys. Chem. C, 2018, 122, 12441–12447 CrossRef CAS.
- (a) D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS; (b) H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS PubMed; (c) R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010–2041 CrossRef CAS.
- M. de Jong, L. Seijo, A. Meijerink and F. T. Rabouw, Phys. Chem. Chem. Phys., 2015, 17, 16959–16969 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, physicochemical properties, computational details, and CIF files for the single-crystal X-ray structure analysis. CCDC 2269866. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02841d |
| This journal is © The Royal Society of Chemistry 2024 |