DOI:
10.1039/D4SC04526B
(Edge Article)
Chem. Sci., 2024,
15, 15152-15159
Surefire generation of stannylpotassium: highly reactive stannyl anions and applications†
Received
8th July 2024
, Accepted 16th August 2024
First published on 22nd August 2024
Abstract
Organometallic reagents such as organolithium and Grignard reagents have long been esteemed in chemical synthesis for their exceptional reactivity. In contrast, the application of their sodium and potassium counterparts has been comparatively sluggish, notwithstanding their augmented reactivity stemming from their heightened ionic character. This inertia persists due to the constrained accessibility of these heavy alkali metal reagents. In this study, our focus was directed towards devising a convenient and pragmatic approach for fabricating heavy alkali metal-based reagents, particularly those grounded in potassium. Herein, we present a novel, direct method for generating stannylpotassium (Sn–K) reagents through the simple combination of readily available silylstannanes and t-BuOK. Subsequently, the generated Sn–K reagents were effectively harnessed for stannylative substitution of aryl halides, furnishing an array of arylstannanes straightforwardly under transition metal-free conditions. This application distinctly underscores the potential utility of highly reactive Sn–K species, hitherto sparingly tapped into within the realm of synthetic organic chemistry. Furthermore, our investigation confirms that Sn–K reagents manifest notably superior reactivity compared with their well-established stannyllithium (Sn–Li) counterparts. This heightened reactivity can be ascribed to the increasing ionic character of Sn–K, which was supported by computational experiments.
Introduction
The indispensable role of organometallic reagents as nucleophilic species in chemical synthesis is firmly established. Among the plethora of metallic elements, highly electropositive alkali and alkaline earth metals, notably lithium1,2 and magnesium,3 have been extensively harnessed across various synthetic endeavors, yielding a rich spectrum of organic compounds. This preference is attributed to their widespread availability and myriad advantages, including exceptionally high reactivity and versatility in reaction modalities. In accordance with Fajans' rule,4 organometallic reagents featuring heavy alkali metals as counter-cations tend to have a greater ionic character, thus implying heightened nucleophilicity and promising synthetic potential. Despite these prospects, their utilization in chemical synthesis has not always been pervasive, likely constrained by limited accessibility to such organometallic reagents. Notably, the contrast in reactivity between lithium and potassium-based organometallic species is stark: while the most commonly employed organolithium, n-BuLi, exhibits a half-life of 24 hours at 0 °C in THF,5 the infrequently utilized n-BuK swiftly undergoes reaction with THF, even at −30 °C (Fig. 1a).6
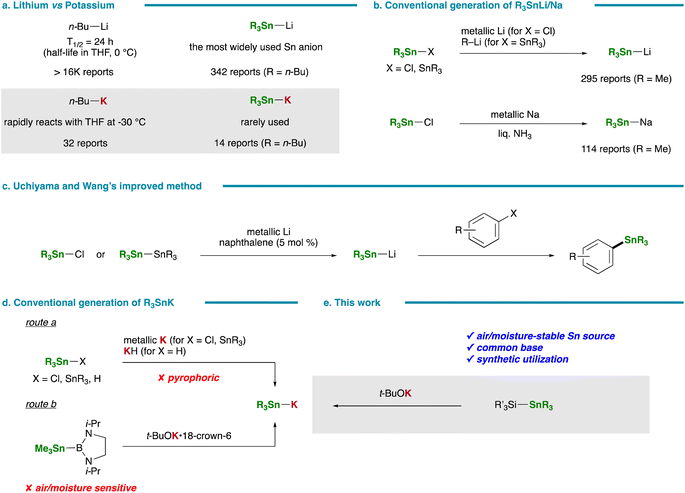 |
| | Fig. 1 Backgrounds and this work. (a) Lithium- and potassium-based nucleophilic species. (b) Conventional generation of R3Sn–Li and –Na reagents. (c) Uchiyama and Wang's improved method for generating R3Sn–Li reagents. (d) Conventional generation of R3Sn–K species. (e) This work: generation of R3Sn–K species from silylstannanes and t-BuOK. | |
A parallel scenario emerges in stannyl anion chemistry, where the application of stannyl anion species featuring various counter-cations (Sn–M: M = Li, Na, MgX, ZnX, Cu, etc.)7 facilitates direct and efficient routes to a broad spectrum of organostannanes of high synthetic importance. Notably, stannyl lithiums8–10 and sodiums11–13 have emerged as the most prevalent stannyl nucleophiles (Fig. 1a and b), albeit traditional methods for their generation often entail labor-intensive processes lacking in practicality. For instance, the synthesis of R3Sn–Na typically entails the direct reduction of R3Sn–Cl with metallic sodium in liquid ammonia, a procedure fraught with operational challenges such as the handling of ammonia gas under cryogenic conditions and the use of highly reactive metallic sodium (Fig. 1b).14,15 Similarly, established protocols for R3Sn–Li, involving either direct reduction of R3Sn–Cl with metallic lithium or the reaction of R3Sn–SnR3 with organolithium reagents, are beset by inefficiencies in generation and prone to side reactions.16,17 In 2015, Uchiyama and Wang addressed these limitations by employing naphthalene as a catalyst in the preparation of R3Sn–Li from metallic lithium and R3Sn–Cl, leading to notable improvements in efficiency and stability (Fig. 1c).18 Furthermore, the synthetic utility of this methodology was underscored by its successful application in stannylative substitution reactions of aryl halides, illustrating the potential of developing practical and efficient synthesis approaches to stannyl anion species for fostering novel tin-based synthetic transformations.
In stark contrast to the well-established preparations and subsequent transformations of R3Sn–Li/Na, the utilization of R3Sn–K in synthetic organic chemistry has been markedly scarce (Fig. 1a), despite its anticipated superior nucleophilicity arising from heightened ionicity. This disparity primarily stems from the lack of usability and practicality inherent in existing preparation methods: route a necessitates the use of hazardous, spontaneously combustible metallic potassium or potassium hydride as a potassium source,19–21 while route b requires stannylboranes, which are exceedingly sensitive to water and air,22 as a tin source (Fig. 1d). On the other hand, silicon- or aryl-stabilized stannyl potassiums [(R3Si)3Sn–K or ArSnK3] have been reported to be readily generated via the reaction of (R3Si)4Sn or ArSn(SiHMe2)3 with a common base, t-BuOK.23 These species have found utility in the synthesis of various compounds, including (R3Si)3Sn-substituted spirooligosilanes24 and tetraacylstannanes.25,26 Given this context, our focus was directed towards harnessing (triorganosilyl)stannanes, characterized by their stable and facile handling properties, as a tin source for generating R3Sn–K. While (triorganosilyl)stannanes have conventionally served as silylating/silylstannylating agents in transition metal-catalyzed reactions,27–32 we demonstrated for the first time their potential as a tin source for generating stannyl copper species (R3Sn–Cu) upon treatment with a copper alkoxide (R′OCu), which served as a key intermediate in the copper-catalyzed hydrostannylation of terminal alkynes.33 Herein, we unveil a straightforward and practical method for the preparation of R3Sn–K, achievable without the need for specialized equipment or stringent handling precautions, simply by mixing readily available (triorganosilyl)silylstannanes and t-BuOK, focusing mainly on elucidating the synthetic potential of this facile method (Fig. 1e). The in situ-generated R3Sn–K was demonstrated to react smoothly with a diverse array of aryl halides to furnish arylstannanes. Furthermore, the heightened reactivity of R3Sn–K, surpassing that of R3Sn–Li, aligns with expectations, given its markedly ionic nature inherent in the Sn–K bond.
Results and discussion
Generation of stannyl potassiums from (triorganosilyl)stannanes
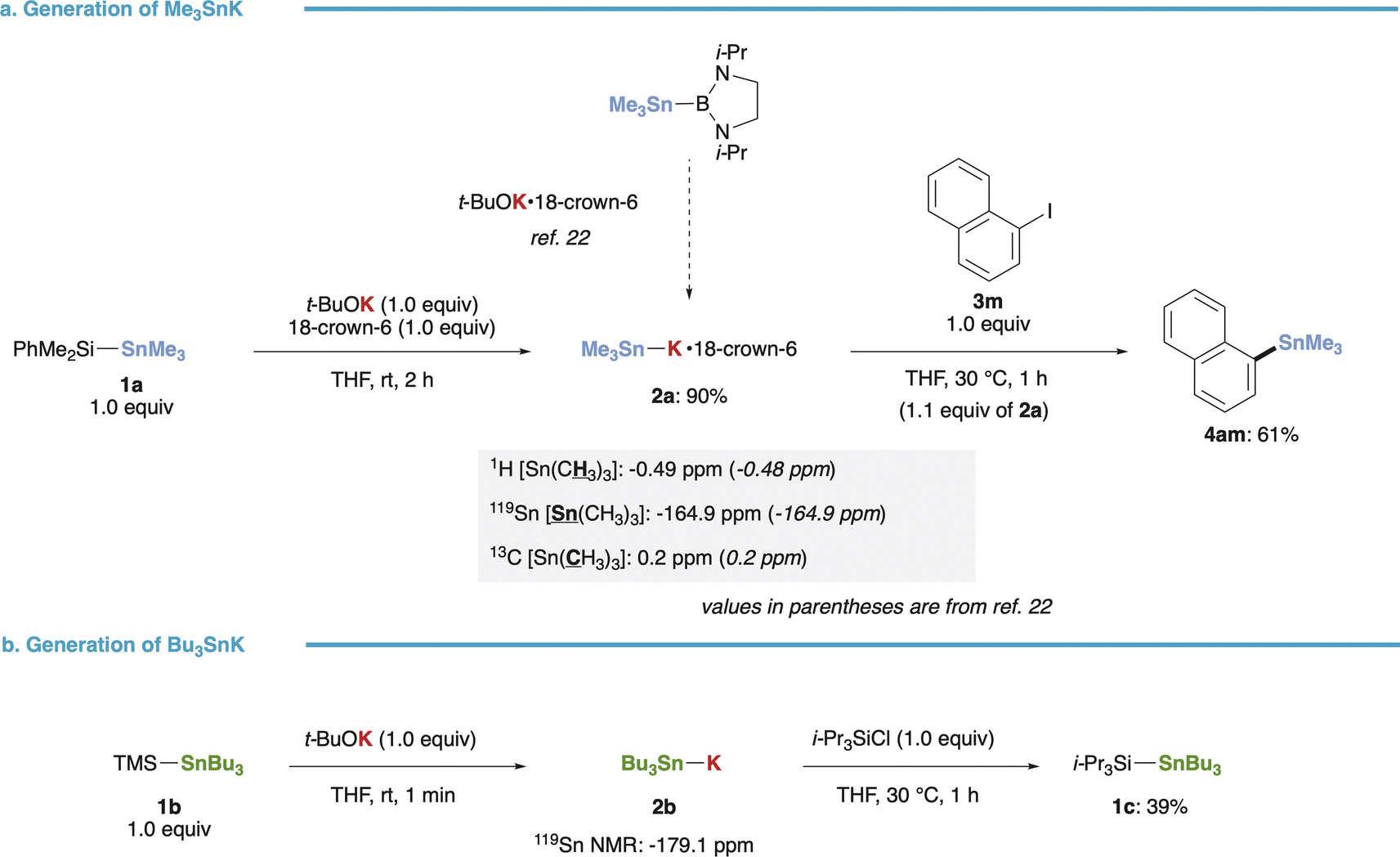
Our initial investigation focused on the synthesis of stannyl potassiums (R3Sn–K) from (triorganosilyl)stannanes. Treatment of (dimethylphenylsilyl)trimethylstannane (1a) with t-BuOK in the presence of 18-crown-6 yielded Me3Sn–K·18-crown-6 (2a) smoothly in 90% yield, as evidenced by NMR spectra (1H, 13C, 119Sn) consistent with those reported by Kleeberg, Grunenberg, and Xie (Fig. 2a).22 The resulting 2a exhibited sufficient nucleophilicity, as demonstrated by its reaction with 1-iodonaphthalene (3m) completing within 1 hour at 30 °C, affording the stannylated product (4am) in 61% yield. Furthermore, our method for preparing R3Sn–K was successfully extended to (trimethylsilyl)tributylstannane (1b), yielding Bu3Sn–K (2b) even in the absence of 18-crown-6. This was confirmed by the upfield-shifted signal (−179.1 ppm) observed in the 119Sn NMR spectrum.34 Subsequent addition of i-Pr3SiCl resulted in the formation of the trapped product (1c) in 39% yield (Fig. 2b).
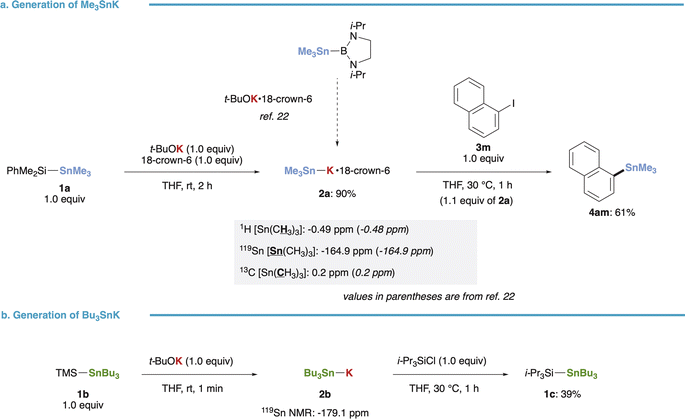 |
| | Fig. 2 Generation of R3Sn–K species from silylstannanes. (a) Generation, NMR analysis and reaction of Me3Sn–K. (b) Generation and reaction of Bu3Sn–K. | |
Optimization of reaction conditions for stannylation of aryl halides with R3Sn–K
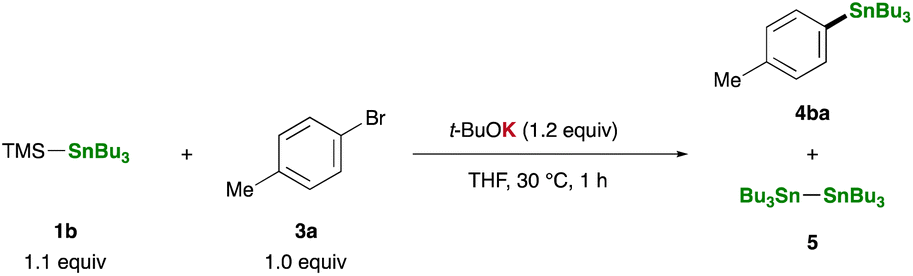
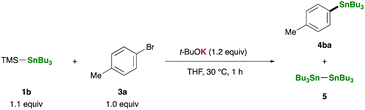
Equipped with our straightforward and practical method for generating R3Sn–K, we proceeded to investigate the reaction of TMS–SnBu3 (1b) with p-bromotoluene (3a) in THF using t-BuOK as an additive, with a focus on the synthetic utility of the in situ-generated R3Sn–K. Notably, the stannylative substitution reaction proceeded smoothly at 30 °C within 1 hour, affording tributyl(p-tolyl)stannane (4ba) in 54% yield (Table 1, entry 1). However, the reaction encountered sluggishness when conducted in other solvents such as toluene, DMF, acetonitrile, 1,4-dioxane, and DME, resulting in the formation of Bu3SnSnBu3(5) as a major side-product (entries 2–6). A counter-cation of bases significantly influenced the reaction outcome: no 4ba was observed when employing t-BuOLi or t-BuONa as additives (entries 7 and 8). Moreover, less basic potassium alkoxides (MeOK and EtOK) and fluoride ion (KF/18-crown-6), previously reported for generating stannyl anion species upon treatment with 1b,35 proved ineffective as additives (entries 9–11). Remarkably, the yield was substantially improved when p-iodotoluene (3b) was used instead of 3a, resulting in a 78% yield of 4ba while suppressing side-product formation (entries 12 and 13).
Table 1 Optimization of reaction conditionsa
|
|
| Entry |
Deviation from standard conditions |
Yieldb (%) |
|
4ba
|
5
|
|
Reaction conditions: 1b (0.2 mmol), 3a (0.2 mmol), t-BuOK (0.24 mmol, 1.2 equiv.), THF (0.67 mL), 30 °C, 1 h.
119Sn NMR yield determined by using tributyl(vinyl)stannane as an internal standard.
|
| 1 |
None |
54 |
0 |
| 2 |
Toluene instead of THF |
16 |
19 |
| 3 |
DMF instead of THF |
0 |
0 |
| 4 |
CH3CN instead of THF |
0 |
18 |
| 5 |
1,4-Dioxane instead of THF |
28 |
12 |
| 6 |
DME instead of THF |
5 |
26 |
| 7 |
t-BuOLi instead of t-BuOK |
0 |
0 |
| 8 |
t-BuONa instead of t-BuOK |
0 |
30 |
| 9 |
MeOK instead of t-BuOK |
10 |
18 |
| 10 |
EtOK instead of t-BuOK |
7 |
28 |
| 11 |
KF/18-crown-6 instead of t-BuOK |
0 |
32 |
| 12 |
p-iodotoluene (3b) instead of 3a |
71 |
0 |
| 13 |
3b instead of 3a, 1.1 equiv. of 1b/1.1 equiv. of t-BuOK |
78 |
0 |
Stannylation of various aryl halides
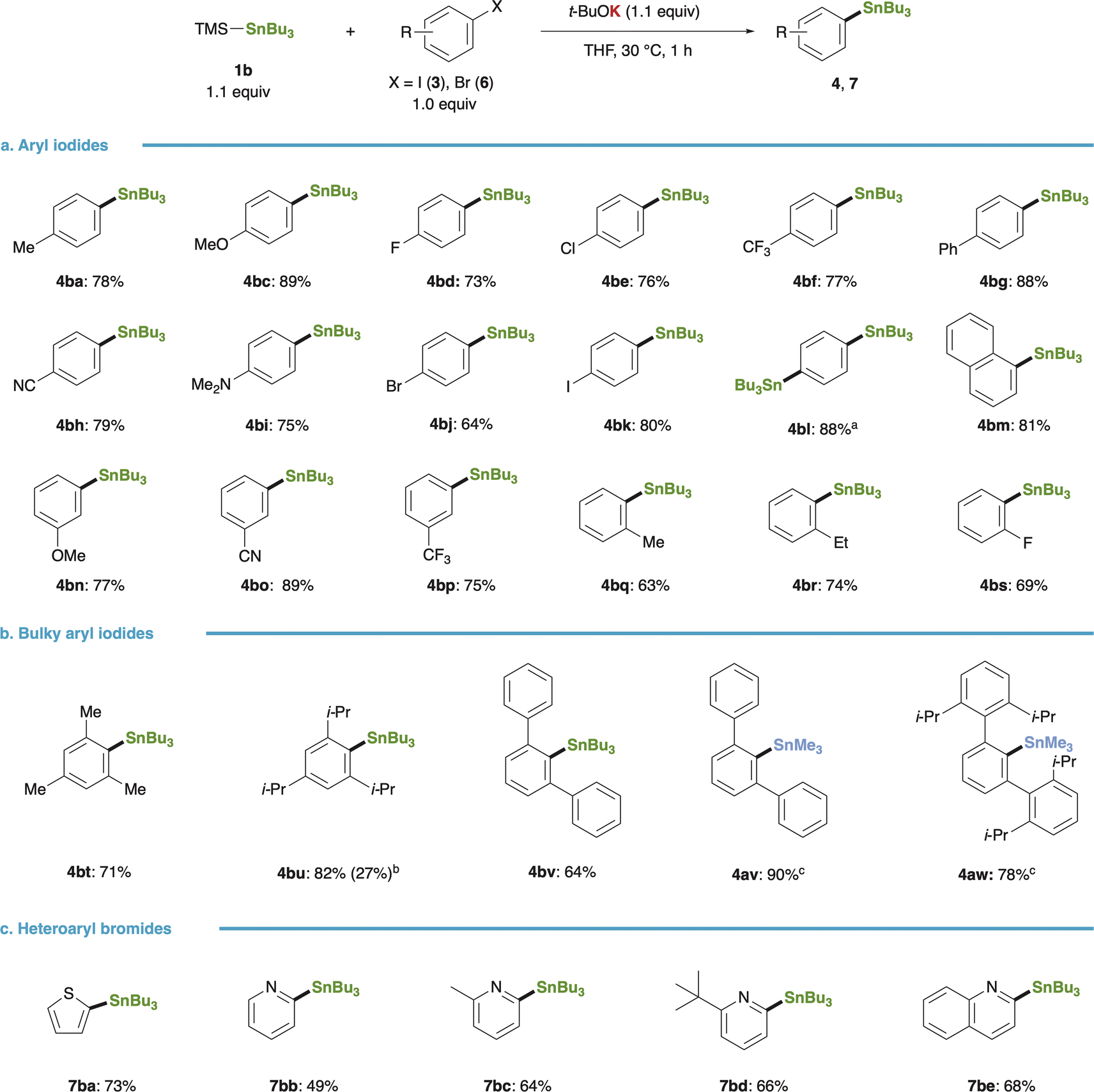
The substrate scope of the stannylation reaction with the in situ-generated Bu3Sn–K proved to be sufficiently broad (Fig. 3): a diverse array of aryl iodides bearing either electron-donating (3b, 3c, 3i) or -withdrawing groups (3d–3f, 3h) underwent efficient conversion to the respective arylstannanes upon mixing with 1b and t-BuOK, regardless of the electronic nature of the aryl rings (Fig. 3a). Chemoselective substitution occurred at the iodine site of 4-bromoiodobenzene, yielding 4-bromophenylstannane (4bj), while 4-iodophenylstannane (4bk) could be synthesized from 1,4-diiodobenzene; employing a twofold amount of 1b and t-BuOK led to the formation of a distannylated product (4bl). The superior reactivity of Sn–K over Sn–Li was evident in reactions involving bulky substrates such as mesityl (3t), 2,4,6-triisopropylphenyl (3u), and m-terphenyl (3v) iodides, providing good yields of arylstannanes (4bt–4bv, Fig. 3b), whereas the yield of the target product (4bu) dropped to 27% with its lithium counterpart. Additionally, the present method facilitated Me3Sn-installation using PhMe2Si–SnMe3 (1a), with the yield of 4av reaching 90%, and even extremely bulky aryl iodide (3w) was amenable to transformation into 4aw (78% yield), highlighting the exceptional nucleophilicity of Sn–K.
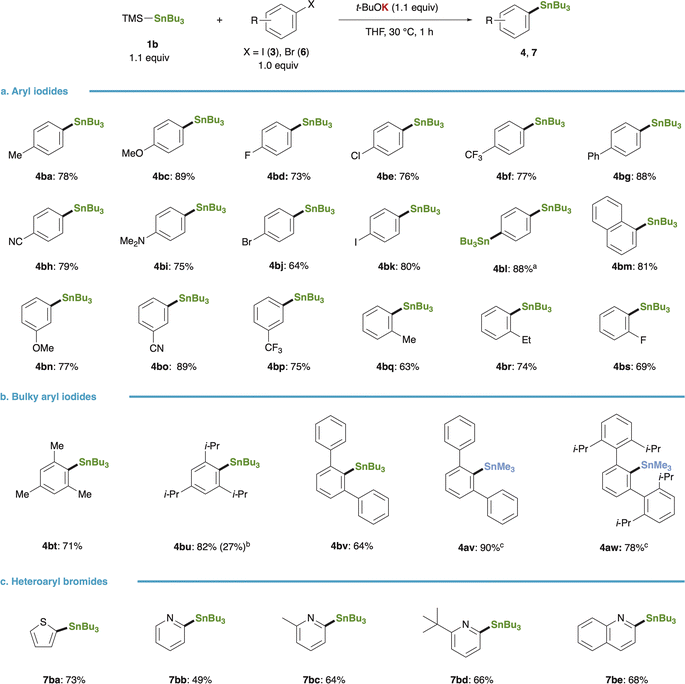 |
| | Fig. 3 Substrate scope of stannylation of aryl halides. (a) Stannylation of aryl iodides. (b) Stannylation of bulky aryl iodides. (c) Stannylation of heteroaryl bromides. Reaction conditions: 1b (0.22 mmol), 3 or 6 (0.20 mmol), t-BuOK (0.22 mmol), THF (0.67 mL), 30 °C, 1 h. Isolated yields were given. aThe reaction was carried out with 1,4-diiodobenzene (3k, 0.20 mmol) using 1b (0.44 mmol) and t-BuOK (0.44 mmol). bIsolated yield with Bu3SnLi under Uchiyama and Wang's conditions was given in parentheses. cPhMe2Si–SnMe3 (1a) was used instead of 1b. | |
Heteroaryl bromides (6a–6e) also engaged in the reaction efficiently, affording 2-thienyl- (7ba), 2-pyridyl- (7bb–7bd), and 2-quinolyl-stannane (7be) in yields ranging from 49% to 73% (Fig. 3c).
Mechanistic studies
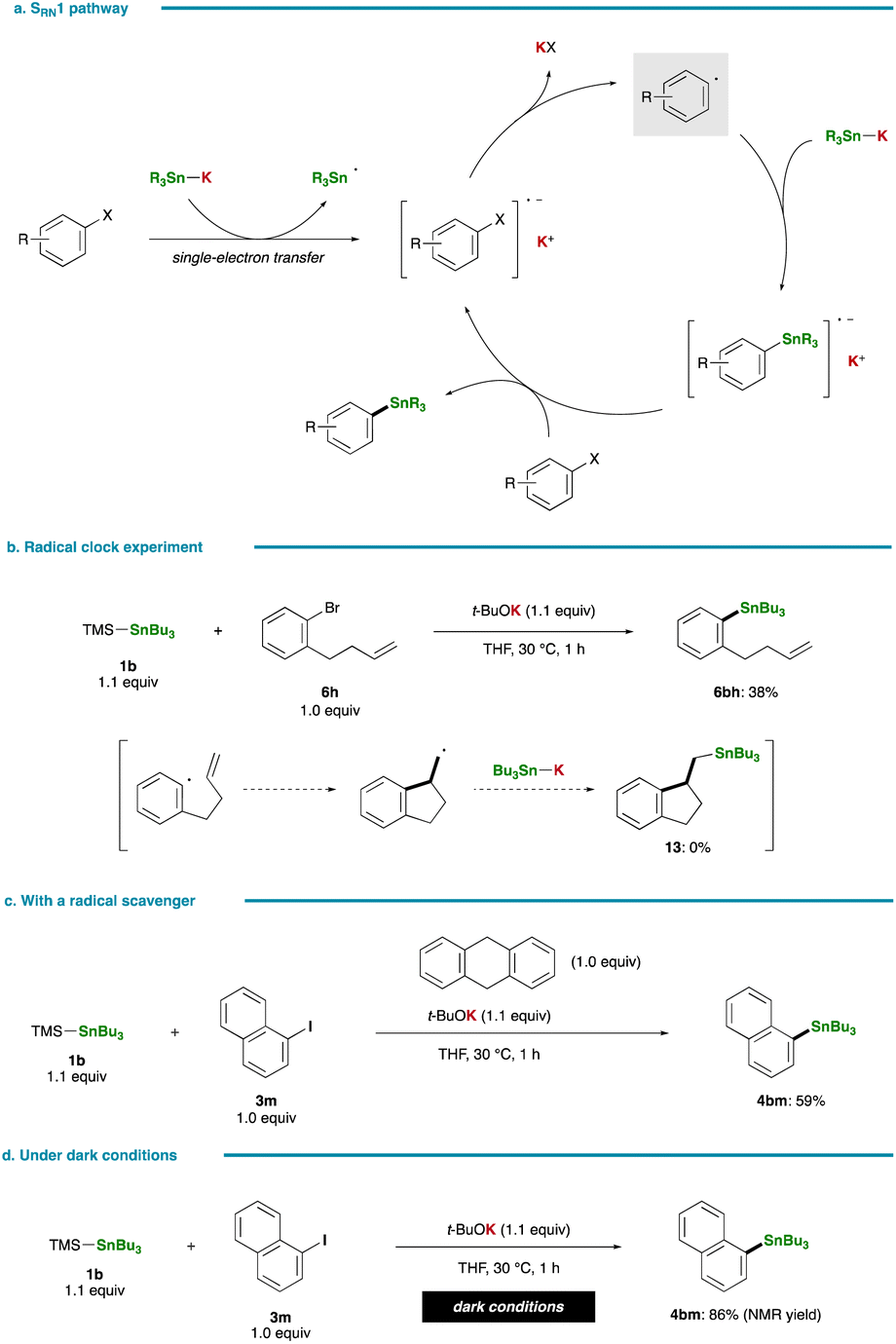
A plausible pathway for the stannylation, initiated by the generation of R3Sn–K via nucleophilic attack of t-BuOK on a silicon center of a silylstannane, is delineated in Fig. 4a. The co-production of a tert-butyl silyl ether (8) was confirmed in the stannylation of 3w (Fig. 4b). Subsequent halogen–metal exchange between the resulting R3Sn–K and an aryl halide, leading to the formation of an aryl anion, followed by its nucleophilic coupling with R3Sn–X, yields an arylstannane.13,36,37 The intermediary formation of an aryl anion is supported by treating 6f with isolated Me3Sn–K (2a′):38 an aryl anion intermediate (6f′) undergoes intramolecular cyclization at an ester carbonyl moiety prior to stannylation, resulting in the formation of 9 (Fig. 4c). Although the reaction of 2,4,6-tri-tert-butylphenyl bromide (6g) fails to yield the corresponding arylstannane, likely due to excessive steric hindrance, the preferential formation of a reduced product, 1,3,5-tri-tert-butylbenzene, also strongly implies the generation of an aryl anion intermediate (10′) that abstracts a C2 proton of THF.6 Notably, conducting the same reaction in THF-d8 indeed results in the formation of deuterated tri-tert-butylbenzene (10) with a deuterium incorporation ratio of 52% (Fig. 4c), providing further support for the proposed ionic pathway. While an initially formed tin-based by-product would be Bu3SnBr in the reaction of 6g, it was rapidly converted into Bu3SnOt-Bu and Bu3SnSnBu3 by reacting with remaining t-BuOK or Bu3Sn–K.39
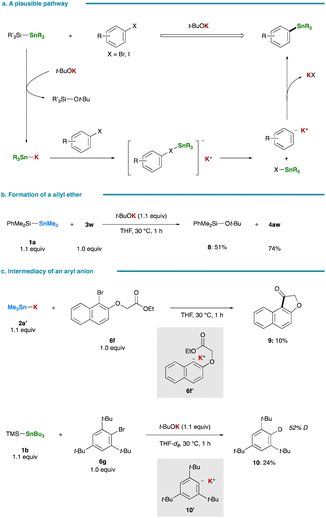 |
| | Fig. 4 Mechanistic studies. (a) A plausible pathway. (b) Formation of a silyl ether by-product. (c) Intermediacy of an aryl anion. | |
Fig. 5a presents an alternative pathway for the stannylation involving the formation of an aryl radical intermediate through single-electron transfer, a mechanism reported to be operative in certain stannylation reactions of aryl halides with Sn–Li/Na.36,37,40,41 Despite the propensity of the respective aryl radical to undergo rapid cyclization, as evidenced by a high rate constant for ring closure (k = 7.9 × 109 s−1 at 323 K),42 a radical clock experiment with 1-bromo-2-(but-3-en-1-yl)benzene (6h) yielded the simple substitution product (6bh, 38%; 11, 0%). This observation essentially rules out the possibility of the radical nucleophilic aromatic substitution pathway (SRN1) in the present stannylation (Fig. 5b). Moreover, this conclusion is reinforced by the smooth stannylation reaction conducted in the presence of a radical scavenger, 9,10-dihydroanthracene (Fig. 5c). In 2021, Nagashima and Uchiyama reported on the generation of a photoexcited triplet stannyl diradical that could serve as a key species in photoinduced stannylation of aryl halides.43 However, our reaction proceeded efficiently even under dark conditions, thereby excluding the involvement of a stannyl diradical species in the present case (Fig. 5d).
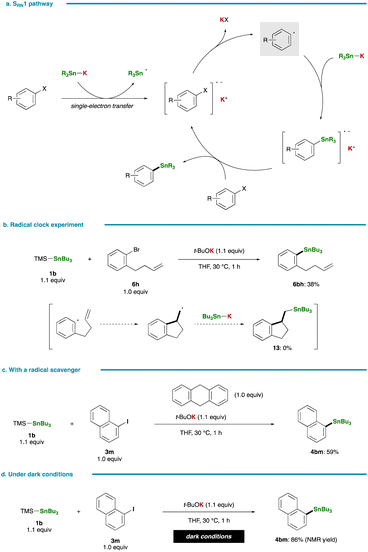 |
| | Fig. 5 Control experiments. (a) Stannylation via the SRN1 pathway. (b) Radical clock experiment. (c) Stannylation with a radical scavenger. (d) Stannylation under dark conditions. | |
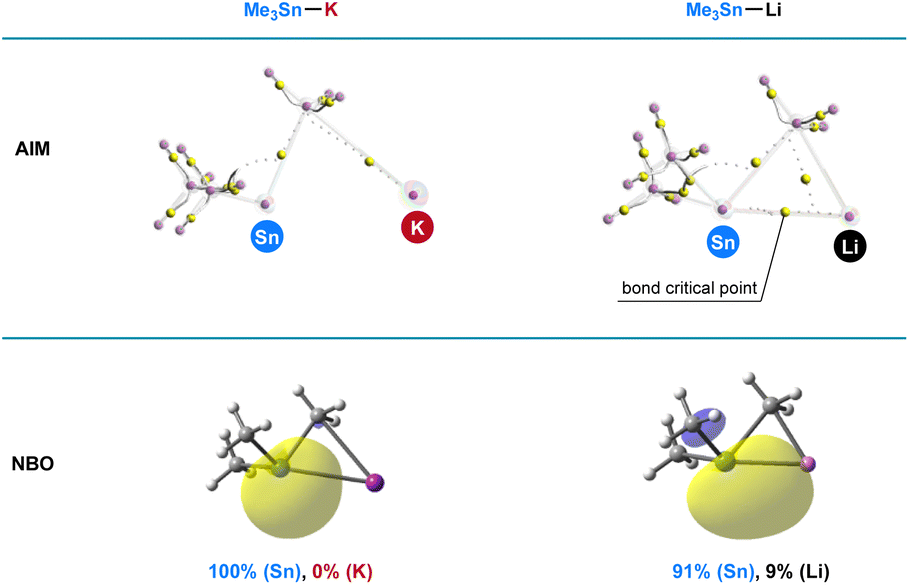
Computational studies
To illustrate the superior reactivity of R3Sn–K as stannyl nucleophiles compared with R3Sn–Li, population analysis employing the Atoms in Molecules (AIM) and Natural Bond Orbital (NBO) methods was conducted. Optimized structures of Me3Sn–K and Me3Sn–Li were obtained at the ωb97xd/6-31+G(d)/LANL2DZ level, and the bonding interactions between tin and alkali metal were thoroughly evaluated. As depicted in Fig. 6, AIM calculations revealed the absence of a bond critical point between tin and potassium in Me3Sn–K, while a bond critical point was observed in the tin–lithium bond, indicating a significantly greater ionic character in the tin–potassium bonding compared with the tin–lithium bonding. NBO analysis further corroborated these findings; the electron distribution between tin and lithium was approximately 91% (Sn) and 9% (Li), indicative of its partially covalent character in the tin–lithium bonding. In contrast, the electron density was fully polarized towards the Sn atom in Me3Sn–K. These results unequivocally demonstrate that the heightened reactivity of R3Sn–K observed in the stannylation of aryl halides can be attributed to the increased nucleophilicity arising from the pronounced ionic character in the tin–potassium bonding.
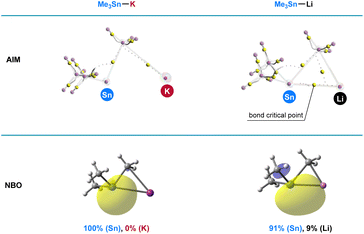 |
| | Fig. 6 Computational studies. Population analysis of Me3Sn–K and Me3Sn–Li. | |
Conclusion
We have developed a straightforward and practical method for preparing R3Sn–K from readily available silylstannanes and t-BuOK, presenting a promising avenue for its extensive utilization in tin-based synthetic transformations. Our methodology facilitated the facile transformation of diverse aryl halides into arylstannanes under mild conditions within 1 hour, simply by combining silylstannanes and t-BuOK. Moreover, R3Sn–K exhibited superior reactivity over commonly used R3Sn–Li, enabling the smooth stannylation of extremely bulky aryl halides. Detailed mechanistic studies unveiled an ionic pathway underlying the stannylation process, wherein an aryl anion intermediate is generated via halogen–metal exchange. Furthermore, the exceptional nucleophilicity of R3Sn–K can be ascribed to the highly ionic character induced by the potassium cation, which was substantiated by population analysis (NBO and AIM) of the Sn–K bond. This study unequivocally underscores the synthetic significance of highly nucleophilic R3Sn–K reagents, which have hitherto been underutilized in synthetic organic chemistry. It presents promising avenues for addressing challenging stannylation reactions that are otherwise difficult to achieve with conventional stannyl anions featuring a light alkali metal cation.
Data availability
The data that support the findings of this study are available in the ESI† of this article.
Author contributions
Y. H.: conceptualization, investigation, writing – original draft, review & editing. T. I.: investigation. K. N.: investigation. H. K.: investigation. M. N.: validation. T. T.: validation. H. Y.: conceptualization, funding acquisition, supervision, writing – original draft, review & editing.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was financially supported by JSPS KAKENHI Grant Numbers JP22H02079, JP23K18544. Financial support was also received from JST SPRING, Grant Number JPMJSP2132. The authors thank the Natural Science Center for Basic Research and Development, Hiroshima University (NBARD-00001) for technical assistance with the compound characterization. T. I. acknowledges the JSPS fellowship for young scientists. The computation was performed at the Research Center for Computational Science, Okazaki, Japan (Project: 22-IMS-C244 and 23-IMS-C184).
Notes and references
- J. M. Mallan and R. L. Bebb, Metalations by organolithium compounds, Chem. Rev., 1969, 69, 693–755 CrossRef.
- W. F. Bailey and J. J. Patricia, The mechanism of the lithium-halogen interchange reaction: a review of the literature, J. Organomet. Chem., 1988, 352, 1–46 CrossRef CAS.
- T. Banno, Y. Hayakawa and M. Umeno, Some applications of the Grignard cross-coupling reaction in the industrial field, J. Organomet. Chem., 2002, 653, 288–291 CrossRef CAS.
- K. Fajans, Electric forces and electronic configurations in carbon compounds, Chem. Eng. News Arch., 1949, 27, 900–904 CrossRef CAS.
- P. Stanetty and M. D. Mihovilovic, Half-lives of organolithium reagents in common ethereal solvents, J. Org. Chem., 1997, 62, 1514–1515 CrossRef CAS.
- R. Pi, W. Bauer, B. Brix, C. Schade and P. v. R. Schleyer,
n-Butylpotassium: preparation, synthetic use and NMR properties in hexane and tetrahydrofuran solution, J. Organomet. Chem., 1986, 306, C1–C4 CrossRef CAS.
-
A. G. Davies, Compounds with Tin-metal Bonds, in Organotin Chemistry, Wiley-VCH, 2004, pp 311–327 Search PubMed.
- J.-P. Quintard, G. Dumartin, C. Guerin, J. Dubac and A. Laporterie, Metallation des chlorosilanes par les trialkylstannylanions, J. Organomet. Chem., 1984, 266, 123–138 CrossRef CAS.
- T. Sato, M. Watanabe, T. Watanabe, Y. Onoda and E. Murayama, Trisubstituted stannyllithium as a double electron equivalent. Reaction with α,β-enones, J. Org. Chem., 1988, 53, 1894–1899 CrossRef CAS.
- S. Li, C. Lian, G. Yue, J. Zhang, D. Qiu and F. Mo, Transition metal free stannylation of alkyl halides: the rapid synthesis of alkyltrimethylstannanes, J. Org. Chem., 2022, 87, 4291–4297 CrossRef CAS PubMed.
- G. F. Smith, H. G. Kuivila, R. Simon and L. Sultan, Electron-transfer, halogen-metal exchange, and direct processes in formal nucleophilic substitutions on alkyl halides by trimethyltinsodium, J. Am. Chem. Soc., 1981, 103, 833–839 CrossRef CAS.
- E. C. Ashby, R. N. DePriest and W. Y. Su, Electron transfer in the reactions of alkyl halides with sodium trimethylstannate, Organometallics, 1984, 3, 1718–1727 CrossRef CAS.
- M. T. Lockhart, A. B. Chopa and R. A. Rossi, Reactions of haloarenes, haloheteroarenes and dihalobenzenes with triphenylstannyl anions in DMSO and acetonitrile, J. Organomet. Chem., 1999, 582, 229–234 CrossRef CAS.
- C. C. Yammal, J. C. Podestá and R. A. Rossi, Synthesis of asymmetrical aryl-tin compounds by cleavage of alkyl-tin bonds with sodium metal in liquid ammonia followed by SRN1 reactions with chloroarenes, J. Organomet. Chem., 1996, 509, 1–8 CrossRef CAS.
- A. B. Chopa, M. T. Lockhart and G. Silbestri, Synthesis of arylstannanes from arylamines, Organometallics, 2001, 20, 3358–3360 CrossRef CAS.
- H. Gilman and S. D. Rosenberg, The preparation of triphenyltin-lithium and some of its reactions, J. Am. Chem. Soc., 1952, 74, 531–532 CrossRef CAS.
- W. C. Still, Stannylation/destannylation. Preparation of α-alkoxy organolithium reagents and synthesis of dendrolasin via a carbinyl carbanion equivalent, J. Am. Chem. Soc., 1978, 100, 1481–1487 CrossRef CAS.
- D.-Y. Wang, C. Wang and M. Uchiyama, Stannyl-lithium: a facile and efficient synthesis facilitating further applications, J. Am. Chem. Soc., 2015, 137, 10488–10491 CrossRef CAS.
- K. Mochida, Reactions of trialkylstannane anions R3Sn− with arylstannanes ArSnR3, Bull. Chem. Soc. Jpn., 1987, 60, 3299–3306 CrossRef CAS.
- M. Wakasa and T. Kugita, Direct observation of a stannyl radical in the reactions of the tributylstannyl anion with alkyl halides by a stopped-flow technique, Organometallics, 1998, 17, 1913–1915 CrossRef CAS.
- T. Schollmeier, U. Englich, R. Fischer, I. Prass, K. Ruhlandt, M. Schürmann and F. Uhlig, Synthesis and reactivity of novel alkali metal stannides, Z. Naturforsch., 2004, 59, 1462–1470 CrossRef CAS.
- C. Kleeberg, J. Grunenberg and X. Xie, K–H3C and K–Sn interactions in potassium trimethylstannyl complexes: a structural, mechanochemical, and NMR study, Inorg. Chem., 2014, 53, 4400–4410 CrossRef CAS PubMed.
-
(a) R. Fischer, J. Baumgartner, C. Marschner and F. Uhlig, Tris(trimethylsilyl)stannyl alkali derivatives: syntheses and NMR spectroscopic properties, Inorg. Chim. Acta, 2005, 358, 3174–3182 CrossRef CAS;
(b) T. Tajima, M. Ikeda, M. Saito, K. Ishimura and S. Nagase, Synthesis, structure and reactions of a trianion equivalent, trilithiostannane, Chem. Commun., 2008, 6495–6497 RSC.
- J. Hlina, C. Mechtler, H. Wagner, J. Baumgartner and C. Marschner, Multiple silyl exchange reactions: a way to spirooligosilanes, Organometallics, 2009, 28, 4065–4071 CrossRef CAS.
- M. Mitterbauer, P. Knaack, S. Naumov, M. Markovic, A. Ovsianikov, N. Moszner and R. Liska, Acylstannanes: cleavable and highly reactive photoinitiators for radical photopolymerization at wavelengths above 500 nm with excellent photobleaching behavior, Angew. Chem., Int. Ed., 2018, 57, 12146–12150 CrossRef CAS PubMed.
- J. Radebner, A. Eibel, M. Leypold, N. Jungwirth, T. Pickl, A. Torvisco, R. Fischer, U. K. Fischer, N. Moszner, G. Gescheidt, H. Stueger and M. Haas, Tetraacylstannanes as long-wavelength visible-light photoinitiators with intriguing low toxicity, Chem. - Eur. J., 2018, 24, 8281–8285 CrossRef CAS PubMed.
- M. Kosugi, T. Ohya and T. Migita, Palladium catalyzed reaction of trimethylsilyltributyltin with α-halo ketones. Preparation of enol silyl ethers, Bull. Chem. Soc. Jpn., 1983, 56, 3539–3540 CrossRef CAS.
- B. L. Chenard, E. D. Laganis, F. Davidson and T. V. RajanBabu, Silyl stannanes: useful reagents for bis-functionalization of α,β-unsaturated ketones and acetylenes, J. Org. Chem., 1985, 50, 3666–3667 CrossRef CAS.
- B. L. Chenard and C. M. Van Zyl, Silylstannanes in organic synthesis. Scope and limitation of palladium-catalyzed reaction with acetylenes, J. Org. Chem., 1986, 51, 3561–3566 CrossRef CAS.
- T. N. Mitchell, R. Wickenkamp, A. Amamria, R. Dicke and U. Schneider, Formation and reactions of olefins with vicinal silyl and stannyl substituents, J. Org. Chem., 1987, 52, 4868–4874 CrossRef CAS.
- T. N. Mitchell, H. Killing, R. Dicke and R. Wickenkamp, Palladium-catalysed addition of the silicon–tin bond to alk-1-ynes and 1,1-dimethylallene, J. Chem. Soc. Chem. Commun., 1985, 354–355 RSC.
-
H. Yoshida, Product Subclass 6: Silyltin Reagents, in Science of Synthesis: Knowledge Updates 2022/3, ed. T. J. Donohoe, Z. Huang, C. Marschner and M. Oestreich, Thieme, 2022, pp 1–22 Search PubMed.
- H. Yoshida, A. Shinke, Y. Kawano and K. Takaki, Copper-catalyzed α-selective hydrostannylation of alkynes for the synthesis of branched alkenylstannanes, Chem. Commun., 2015, 51, 10616–10619 RSC.
- In sharp contrast to Me3Sn–K (2a and 2a′), isolation of Bu3Sn–K (2b) was of great difficulty; its degradation to Bu3Sn–SnBu3 was observed even using the glovebox technique. See ESI† for details.
- M. Mori, N. Isono and H. Wakamatsu, Utilization of the stannyl anionic species generated from Me3SiSnBu3 and X− in organic synthesis, Synlett, 1999, 269–280 CrossRef CAS.
- C. C. Yammal, J. C. Podesta and R. A. Rossi, Reactions of triorganostannyl ions with haloarenes in liquid ammonia. Competition between halogen-metal exchange and electron-transfer reactions, J. Org. Chem., 1992, 57, 5720–5725 CrossRef CAS.
- A. Postigo, S. E. Vaillard and R. A. Rossi, Mechanistic studies on the reactions of trimethylsilanide and trimethylstannylide ions with haloarenes in hexamethylphosphoramide, J. Organomet. Chem., 2002, 656, 108–115 CrossRef CAS.
- Me3Sn–K (2a′) could be generated and isolated in a similar way to the preparation of 2a. See ESI† for details.
- For the 119Sn NMR analysis of the reaction mixture, see ESI†.
- A. B. Chopa, M. T. Lockhart and G. Silbestri, Two-step synthesis of arylstannanes from phenols, Organometallics, 2000, 19, 2249–2250 CrossRef CAS.
- A. B. Chopa, M. T. Lockhart and G. F. Silbestri, Synthesis of bis(trimethylstannyl)aryl compounds via an SRN1 mechanism with intermediacy of monosubstitution products, Organometallics, 2002, 21, 5874–5878 CrossRef CAS.
- A. N. Abeywickrema and A. L. J. Beckwith, Rate constants for the cyclisation of some aryl radicals bearing unsaturated ortho-substituents, J. Chem. Soc. Chem. Commun., 1986, 464–465 RSC.
- K. Sakamoto, Y. Nagashima, C. Wang, K. Miyamoto, K. Tanaka and M. Uchiyama, Illuminating stannylation, J. Am. Chem. Soc., 2021, 143, 5629–5635 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data, DFT calculation data for AIM and NBO, copies of NMR spectra. See DOI: https://doi.org/10.1039/d4sc04526b |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Hiroto
Yoshida
and
Hiroto
Yoshida