
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe clinical potential of L-oligonucleotides: challenges and opportunities
Victoria
Shearer†
,
Chen-Hsu
Yu†
,
Xuan
Han†
and
Jonathan T.
Sczepanski
 *
*
Department of Chemistry, Texas A&M University, College Station, Texas 77843, USA. E-mail: jon.sczepanski@chem.tamu.edu
First published on 21st October 2024
Abstract
Chemically modified nucleotides are central to the development of biostable research tools and oligonucleotide therapeutics. In this context, L-oligonucleotides, the synthetic enantiomer of native D-nucleic acids, hold great promise. As enantiomers, L-oligonucleotides share the same physical and chemical properties as their native counterparts, yet their inverted L-(deoxy)ribose sugars afford them orthogonality towards the stereospecific environment of biology. Notably, L-oligonucleotides are highly resistant to degradation by cellular nucleases, providing them with superior biostability. As a result, L-oligonucleotides are being increasingly utilized for the development of diverse biomedical technologies, including molecular imaging tools, diagnostic biosensors, and aptamer-based therapeutics. Herein, we present recent such examples that highlight the clinical potential of L-oligonucleotides. Additionally, we provide our perspective on the remaining challenges and practical considerations currently associated with the use of L-oligonucleotides and explore potential solutions that will lead to the broader adoption of L-oligonucleotides in clinical applications.
1 Introduction
Oligonucleotides (ONs) have become vital tools in biomedical research and in the clinic. Their capacity to be rationally engineered through Watson–Crick (WC) base pairing makes ON highly adaptable, whereas robust synthetic methods and commercial availability make them accessible to most laboratories. In research, oligonucleotides are ubiquitous, with uses ranging from PCR amplification to gene editing. In the clinic, ON-based therapeutics have emerged as a powerful approach to treat a range of diseases by targeting undruggable proteins,1,2 as well as the ever-growing list of disease associated non-coding RNAs.3–6 Indeed, the number of ON-based therapeutics being approved by the U.S. Food and Drug Administration (FDA) is on the rise, including antisense oligonucleotides (ASOs) and aptamers.7–9 ASOs and aptamers are both short, synthetic ONs that alter protein function either through sequence-specific hybridization with their mRNA or by directly binding to the protein target, respectively. Recent examples include the antisense oligonucleotide (ASO) Tofersen targeting SOD1 mRNA to treat amyotrophic lateral sclerosis (ALS)10 and the aptamer-based drug Avacincaptad pegol targeting complement protein C5 for the treatment of geographic atrophy,11 both of which were approved in 2023. Clinical applications of ONs are not without challenges, however. In particular, ONs are susceptible to degradation by cellular nucleases, which hampers their stability and overall effectiveness in biological environments. This vulnerability necessitates the use of chemical modifications that can resist enzymatic digestion, while also improving pharmacokinetic properties.9 Another concern is potential off-target interactions. While oligonucleotides reagents such as ASOs and aptamers are designed in vitro to be highly specific, they often have unintended off-target interactions within living organisms, leading to undesired consequences, including immune-stimulatory effects and cytotoxicity.12–15 For example, Mipomersen, an FDA approved ASO drug for familial hypercholesterolemia was withdrawn from the market in 2019 due to hepatotoxicity risks.16,17 These issues are compounded by the fact that ONs can have limited cellular uptake and endosomal escape, further restricting their therapeutic potential.18,19Significant progress has been made in addressing the limitations of ON-based therapeutics using chemical modifications and, thus, has been reviewed extensively.9,20–24 Indeed, all FDA approved ON therapeutics are at least partially modified, often containing phosphorothioate (PS) backbones and 2′-OH modifications, such as 2′-O-methyl (2′-OMe) and 2′-O-methoxyethyl (2′-MOE).9 Together, these modifications have been shown to increase nuclease resistance and cellular uptake, while reducing off-target interactions and toxicity.25–28 More recently, xeno nucleic acids (XNAs), which contain non-(deoxy)ribose sugar backbones, have emerged as attractive alternatives to traditional chemical modifications.29,30 Common XNAs include fluoroarabino nucleic acid (FANA), locked nucleic acid (LNA), threose nucleic acid (TNA), hexitol nucleic acid (HNA), and peptide nucleic acid (PNA). Like other modified ONs, many XNAs are capable of WC base pairing to native nucleic acids and, thus, retain the same programmability as their native counterparts.31–35 However, XNAs offer several additional benefits, including superior biostability, improved bioorthogonality, and increased functionality due to their expanded chemical and structural diversity.29,36,37 In particular, the utilization of XNA in DNAzymes has shown to improve their activity and stability.38–40 Given the advantages offered by XNAs, coupled with an expanding biochemical toolbox to support XNA research (e.g., XNA polymerases),41,42 XNAs are expected to play a major role in the development of future ON-based therapeutics.
One promising class of XNAs are L-DNA and L-RNA (i.e., L-ONs), which contain L-(deoxy)ribose sugar units (Fig. 1). L-ONs are mirror images (or enantiomers) of native D-DNA and D-RNA and, as such, are intrinsically orthogonal to the stereospecific environment of native biology. In particular, L-ONs are highly resistant to degradation by cellular nucleases, providing them with superior biostability.43,44 Unlike other chemically modified ONs and XNAs, L-ONs have the same physical and chemical properties as naturally occurring D-nucleic acids, providing an important benefit from a rational design perspective.45–47 Moreover, studies have shown that L-ONs are less susceptible to off-target interactions with endogenous biomacromolecules due, in part, to their inability to form contiguous WC base pairs with native D-nucleic acids.43,45,47 Clinical studies have also shown that L-ONs have very low, possibly negligible immunogenic potential.48 Given these properties, L-ONs have emerged as a promising platform for the development of biomedical technologies, including molecular imaging tools, diagnostic biosensors, and aptamer-based therapeutics (Fig. 1).48,49 Herein, we briefly highlight these and other recent examples that demonstrate the clinical potential of L-ONs. We then provide our perspective on remaining challenges and practical considerations currently associated with the use of L-ONs and explore potential solutions that may lead to the broader adoption of L-ONs in clinical applications.
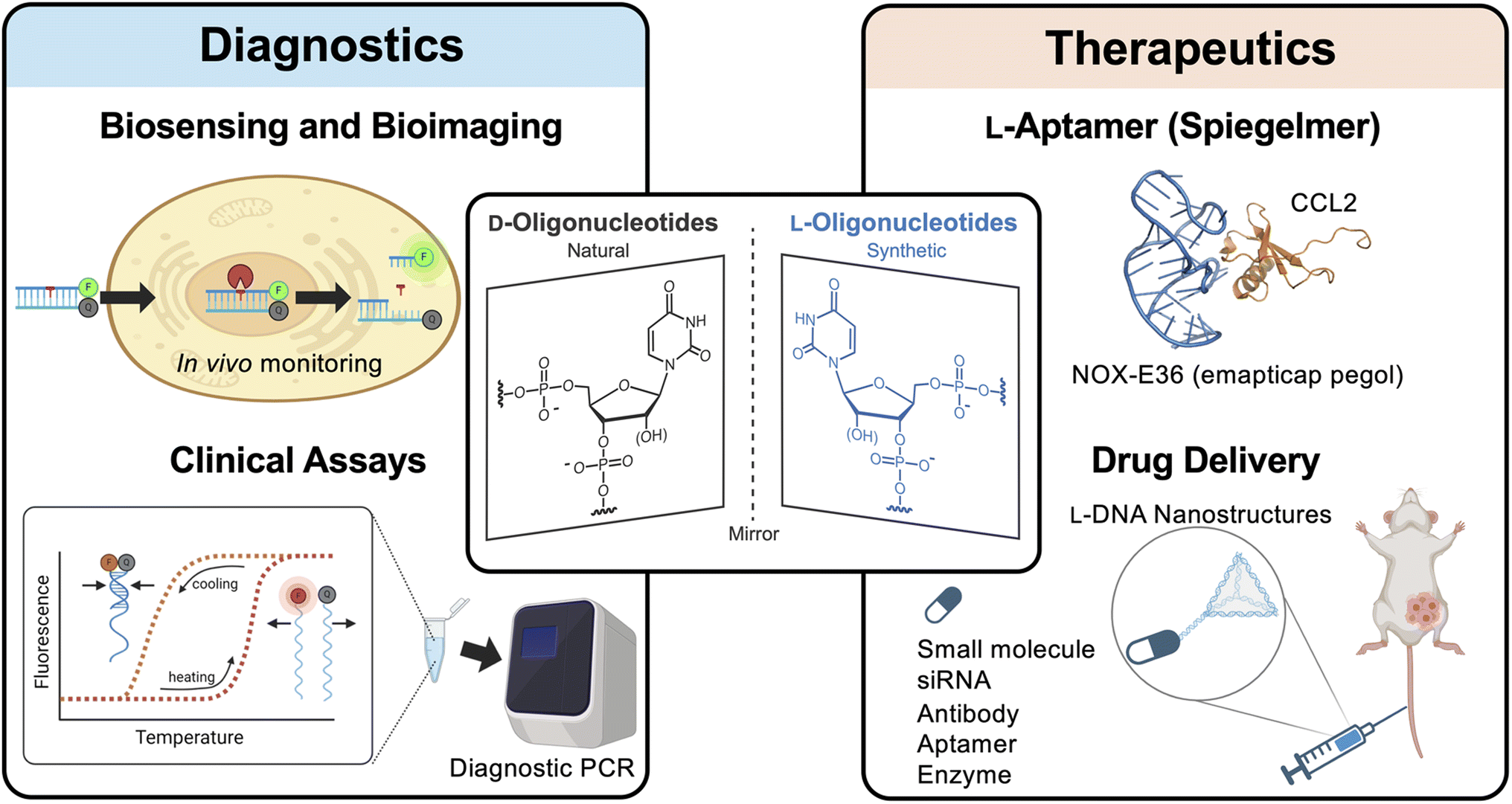 | ||
| Fig. 1 L-Oligonucleotides are the enantiomer of native D-oligonucleotides. Due to their beneficial properties and general bioorthogonality, L-oligonucleotides have been employed in a variety of biomedical technologies. Some examples are shown here. | ||
2 Recent advances of L-oligonucleotides in clinical applications
2.1 Spiegelmers: L-aptamer therapeutics
L-Aptamers (referred to as Spiegelmers) remain the most successful clinical application of L-ONs to date. As this topic has been reviewed previously,48–53 we provide a brief summary here for context. Aptamers are ON-based affinity reagents that are isolated through the process of SELEX (Systematic Evolution of Ligands by Exponential Enrichment)54–56 to bind molecular targets with high affinity and selectivity. Compared to protein antibodies, aptamers have many unique advantages, including ready availability through SELEX, high chemical stability and shelf life, easy chemical modification, small size, and inexpensive cost of production.57 These desirable properties make aptamers well-suited for a variety of biomedical applications, including diagnostic assays, drug delivery vehicles, and therapeutics.58–61 Indeed, the FDA has recognized the therapeutic potential of aptamers through the approval of pegaptanib sodium (Macugen) for macular degeneration62 and Avacincaptad pegol (Izervay) for the treatment of geographic atrophy secondary to age-related macular degeneration (AMD).11 These approvals underscore the growing significance of aptamers as therapeutics.Given the potential of aptamers in clinical applications, significant efforts have been made towards increasing their biostability and improving other pharmacological properties. Towards this end, L-ONs have proven to be a highly effective approach. L-ON aptamers can be generated using “mirror-image” SELEX (or “selection-reflection”).63–65 Here a D-ON library is evolved against the enantiomeric variant of the desired target enabling enzymatic amplification and sequencing of the ON library (Fig. 2). Once a suitable D-aptamer is identified, the corresponding L-aptamer is chemically synthesized and used to bind the desired, natural target. To date, Spiegelmers have been generated for a variety of targets, including small molecules, peptides, and proteins.64–67 Like D-aptamers, Spiegelmers fold into distinct structures that bind their targets with high affinity and selectivity.68,69 However, their inverted stereochemistry affords Spiegelmers high plasma stability and immunological passivity.70–74 Not surprisingly, Spiegelmers have shown promise as drugs, especially for targeting small secreted proteins, such as hormones and cytokines.48 A notable example is Olaptesed pegol (NOX-A12),73 which targets the C-X-C motif chemokine ligand 12 (CXCL12/SDF-1) that is involved in several aspects of tumor progression, including metastasis, angiogenesis, and survival.75 NOX-A12 is currently in phase 1/2 clinical trials as a combination therapy for hard-to-treat glioblastomas.76 When combined with radiotherapy and the anit-VEGF antibody Bevacizumab, NOX-A12 significantly improved survival rates and reduced tumor sizes in glioblastoma patients without causing dose-limiting toxicities, indicating a strong safety and efficacy profile.76–78 The success of NOX-A12 has shown that Spiegelmers have a promising future as therapeutics in clinical applications.
 | ||
| Fig. 2 Schematic depicting the “selection-reflection” process. The enantiomer of the intended target is used to select for D-ON aptamers. D-Aptamers with suitable binding properties are then synthesized in the L-form and used to bind the native target. Key steps of the SELEX process are highlighted, indicating their compatibility with L-ONs. | ||
In addition to proteins and peptides, Spiegelmers have recently shown promise in targeting pharmacologically relevant nucleic acids, such as RNA. Because WC base pairing is stereospecific, Spiegelmers must instead interact with native D-ONs through tertiary interactions (or shape), much like how proteins and antibodies recognize their targets. Despite the lack of complementarity, these so-called “cross-chiral” interactions between Spiegelmers and D-ONs occur with high affinity and excellent selectivity.79–82 This was recently exemplified by a Spiegelmer that was able to distinguish between the stem-loop II-like motif (s2m) RNA from SARS-CoV-2 and SARS-CoV-1, which differ by a single nucleotide in the target region.79 Importantly, Spiegelmers targeting structured RNAs have been shown to modulate RNA function through several modes of action, including by blocking functional RNA–protein interactions.81–85 For example, the Kwok lab has developed an L-RNA Spiegelmer targeting the human telomerase RNA G-quadruplex (hTERC) and demonstrated inhibition of telomerase activity in cell lysates.84 Given current challenges associated with the discovery of molecules capable of binding disease-relevant RNA structures with high affinity and selectivity, Spiegelmers, which can be readily obtained for most RNA targets, represent a promising approach to address this technological gap.
2.2 L-Oligonucleotides as drug delivery vehicles
ONs have emerged as a promising class of drug delivery vehicles. In particular, the programmability of WC base pairs allows for the self-assembly of DNA nanostructures having precise sizes, shapes, surface chemistries, and functions. As drug delivery platforms, these properties can be easily modulated to improve the therapeutic effectiveness of drugs by enhancing their solubility, improving distribution, and promoting cellular internalization.86,87 Towards the development of DNA nanostructure delivery platforms with improved biostability and drug efficacy, several groups have turned to L-DNA. Because D-DNA and L-DNA have identical physical properties, such as duplex thermostability, well-established principles for engineering self-assembling D-DNA nanostructures can be directly applied to L-DNA without further optimization, making L-DNA an ideal nucleic acid analogue from a design perspective.45–47 The use of L-DNA to engineer self-assembled nanostructures was first demonstrated by the Yan group, who also showed that L-DNA-based nanotubes are highly resistant to nuclease degradation.88 More recent studies have confirmed that L-DNA nanostructures resist exonuclease-mediated degradation for up to 24 hours in serum89 and are considerably more stable in live cells compared to those constructed using D-DNA.90,91 Subsequently, L-DNA nanostructures have been harnessed as versatile delivery platforms for small molecule drugs,92–95 anti-proliferating aptamers,90 enzymes,95,96 and siRNAs.91 Importantly, L-DNA nanostructures have been shown to be more effective in tumor-specific localization compared to nanostructures constructed from native D-DNA, 2′-OMe modified RNA, and 2′-fluoro modified RNA, which can be attributed to their superior serum stability, high rate of cancer cell uptake, and minimal macrophage uptake.95 For example, when intravenously injected into HeLa tumor-bearing mice, pyramid shaped L-DNA nanostructures showed a threefold higher accumulation in tumors compared to the liver, showcasing superior relative tumor specificity compared to other reported nanoparticle delivery vehicles.95 Taken together, L-DNA nanostructures represent a promising approach for drug delivery, especially for targeting the tumor environment, and we encourage future growth in this area.2.3 L-Oligonucleotide-based biosensors
L-ONs also have clinical applications in the fields of sensors and diagnostics. Many L-ON-based biosensors have been created by simply inverting the stereochemistry of existing D-DNA and D-RNA designs. One prominent example of this is the molecular beacon (MB).97 MBs are hairpin-shaped ONs with an internally quenched fluorophore that can be activated upon denaturation of the MB structure, for example, upon heating or binding to a complementary sequence. Due to their predictable design, ease of synthesis, high sensitivity, and rapid response, MBs have found broad utility in a variety of bioanalytical and biomedical applications.98–102 Construction of MB using L-ONs offers a straightforward strategy to generate biostable and bioorthogonal molecular sensors. For example, L-ON-based MB probes have been used to image stimuli-dependent temperature changes in live cells103,104 and to accurately monitor melting and annealing during PCR in real-time, facilitating the development of highly sensitive and selective PCR-based diagnostic assays.105,106 Ligand-dependent DNAzymes are also amenable to this “inversion” approach, assuming the analyte is achiral. Indeed, mirrored versions of several metal ion-dependent DNAzyme sensors have been reported,107–109 which show dramatically improved stability and functionality compared to their D-ON counterparts when employed in live-cell imaging assays.108While simply inverting the stereochemistry of known ON-based sensors allows for straightforward construction of more robust probes, this approach is limited to achiral analytes. This is due to the principle of reciprocal chiral substrate specificity,110 which dictates that stereochemical inversion of a chiral receptor (i.e., the sensor) also requires inversion of the chiral ligand. Thus, D-ON-based sensors that rely on interactions with chiral analytes, such as proteins and nucleic acids, are unable to engage these ligands upon stereochemical inversion of their backbones. To overcome this limitation, several groups have turned to “chimeric” ONs constructed from both D- and L-DNA. Here, a region of natural D-DNA is embedded within (or linked to) the L-DNA-based probe to enable its engagement with native ligands. For example, chimeric MBs have been constructed with L-DNA stems and D-DNA loops (Fig. 3a).111 Use of L-DNA in the stem provides improved stability and reduced off-target interactions, whereas use of D-DNA in the loop enables hybridization of the probe with a target nucleic acid sequence. A similar chimeric approach has been used to interface L-DNA-based fluorescence in situ hybridization (FISH) probes with target RNA sequences in fixed cells, enabling the sensitive imaging of endogenous mRNA and microRNA biomarkers within single cells.112 Recently, we developed chimeric D/L-DNA probes for imaging specific DNA repair activities in living cells, demonstrating that the chimeric approach can also be used to interface L-ON-based probes with native proteins (Fig. 3b).113 These probes consist of an L-DNA duplex containing a centrally positioned region of D-DNA with the target lesion, therefore enabling recognition by native repair enzymes. Upon recognition and repair of the lesion by the corresponding repair enzyme(s), the probe disassembles giving rise to a fluorescent signal. We showed that these chimeric probes can be used to monitor relative DNA repair activity, evaluate the efficiency of inhibitors, and study enzyme mutants in live cells. Given the involvement of DNA damage and repair pathways in human diseases and aging,114–116 these probes should enable a broad spectrum of clinical applications.
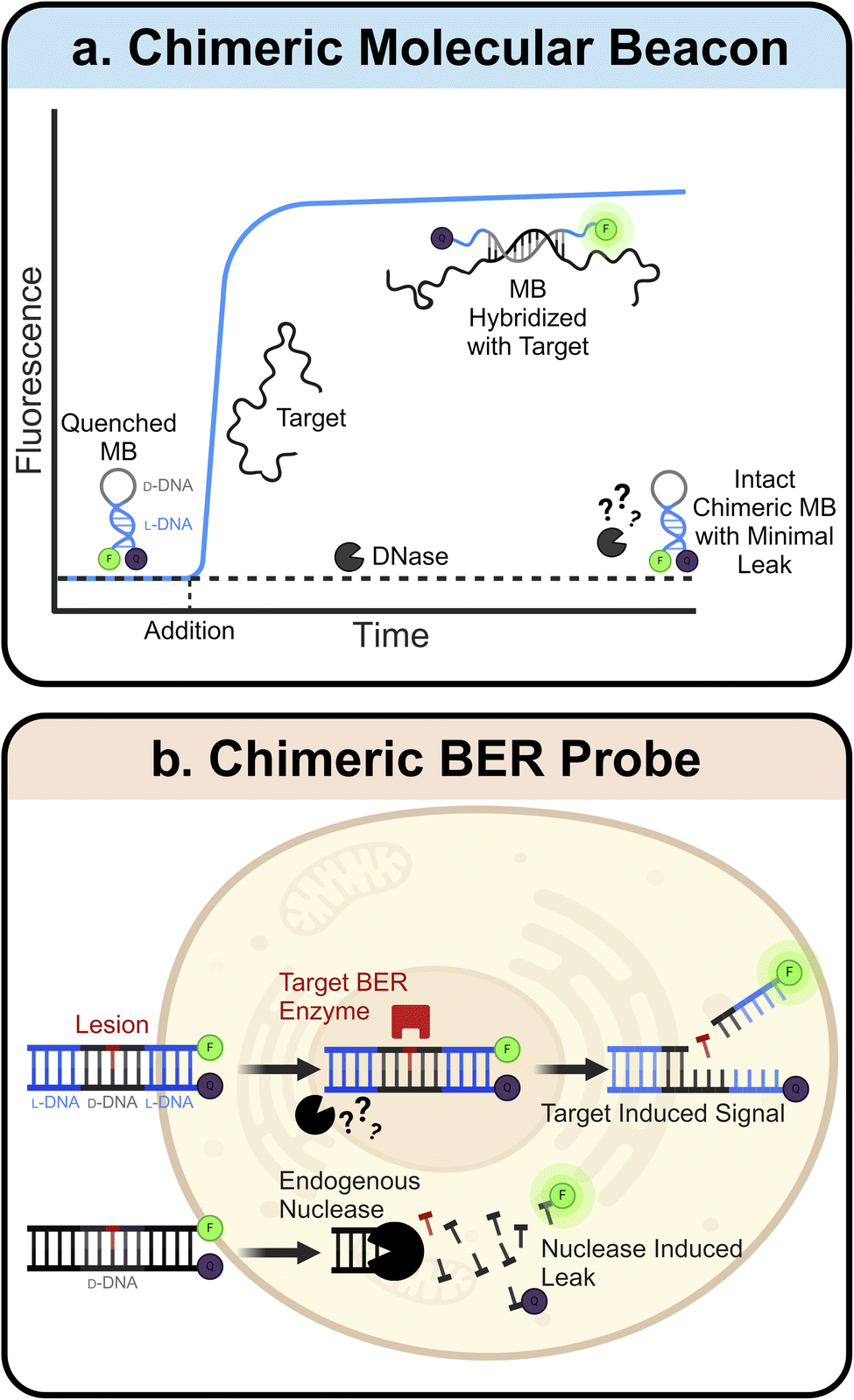 | ||
| Fig. 3 Chimeric D/L-DNA probes. (a) Schematic depiction of the chimeric molecular beacon. D-DNA binding domain (grey) is protected by an L-DNA stem (blue), adding superior biostability to the existing design. Blue curve represents target induced signal increase, while the black dotted line indicates minimal leak due to resistance to nuclease degradation. (b) Schematic depiction of the chimeric BER probe. The D-DNA domain (black) containing a lesion is protected by flanking domains of L-DNA (blue), allowing for detection of target BER enzyme activity in live cells with minimal background. | ||
The field of electrochemical aptamer-based (E-AB) sensors is another area where L-ONs are starting to have an impact. E-AB sensors rely on ON-based aptamers to translate the presence of a specific ligand into an electrochemical signal.117 Although E-AB sensors have been used successfully for continuous, real-time measurement of specific molecular targets within living animals, their in vivo operation is limited to a few hours due, in part, to degradation of the aptamer component.118–120 Use of L-ON-based aptamers presents a straightforward solution to this problem. Indeed, the Arroyo-Currás lab recently showed that conversion of a D-DNA aptamer into its enantiomeric form does not affect performance of E-AB sensor signalling, at least where achiral ligands are concerned.121 Furthermore, they showed that L-DNA-based E-AB sensors are significantly more resistant to nuclease-dependent signal loss than those employing D-DNA, indicating that the use of L-ONs is a promising approach for prolonging the lifespan of invasive E-AB sensors of future clinical applications.
3 Perspectives and outlook
3.1 Overcoming the bioorthogonality of L-oligonucleotides
While the bioorthogonality of L-oligonucleotides (e.g., nuclease resistance) is often touted as a key advantage of this polymer, it also represents the key weakness, which limits the types of applications that can be currently accessed and hinders the broader adoption of L-oligonucleotides into the clinic. This section will discuss recent efforts to overcome the practical challenges associated with using this bio-orthogonal polymer and our viewpoint on where future initiatives should be focused, providing a blueprint for bringing L-oligonucleotides to the forefront of biomedical technologies.3.1.1.1 Polymerases. Perhaps the most glaring challenge of working with L-ONs is their incompatibility with native DNA/RNA polymerases, precluding straightforward enzymatic synthesis and amplification of L-ONs in the laboratory. In particular, the polymerase chain reaction (PCR), the cornerstone of biomedical research and medical diagnostics, is mostly unavailable to L-ON researchers, severely limiting the types of applications that can be accessed using this powerful nucleic acid analogue. Furthermore, in the absence of enzymatic methods, L-ONs are almost exclusively prepared using solid-phase phosphoramidite chemistry, which imposes a limit on the length and quality of L-ONs that can be obtained. While there is not yet an ideal solution to this problem, significant progress has been made towards the enzymatic synthesis of L-ONs.
3.1.1.1.1 Mirror-image polymerase constructed of D-amino acids. The most obvious method to resolve compatibility issues between L-ONs and natural polymerases is to also invert the chirality of enzymes. Indeed, early studies on mirror-image proteins consisting of D-amino acids demonstrated their capability to bind and process mirror-image substrates, such as the D-HIV-1 protease.110,122,123 Thus, it is only natural that researchers considered inverting the stereochemistry of natural polymerase enzymes to facilitate L-ON synthesis. These efforts were pioneered by Zhu and coworkers, who performed the total chemical synthesis of the enantiomer of the smallest known DNA polymerase, African swine fever virus polymerase X (ASFV pol X, 20 kDa).124 This 174-residue protein was assembled via native chemical ligation (NCL) of three D-peptide fragments obtained via solid-phase synthesis. Using this D-amino acid polymerase, the authors successfully demonstrated the template-directed polymerization of L-DNA and transcription of L-RNA for the first time.124 Building on these results and employing more refined synthetic methods, researchers have pursued the synthesis of larger mirror-image polymerases having greater processivity, enhanced fidelity, and thermal stability. Indeed, mirror-image versions of Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4) analogue (40.8 kDa),125–127Pyrococcus furiosus (Pfu) DNA polymerase (90 kDa),128 and T7 RNA polymerase (100 kDa)129 have now been reported. This progress has enabled the assembly and amplification of gene-sized L-DNA fragments and transcription of full-length ribosomal L-RNAs,129 bringing the field one step closer to a mirror-image ribosome-based translation system. While this progress is both promising and inspiring, the chemical approaches used to synthesize large mirror-image polymerases remain highly specialized, labor intensive, costly, and have proven difficult to scale. Thus, they are not practical solutions for the average researcher, at least not yet.
3.1.1.1.2 Cross-chiral polymerases: a more practical approach?. An alternative path for polymerization of L-ONs that warrants more attention is the use of cross-chiral enzymes, i.e., enzymes composed of native polymers (D-ONs and L-amino acids) that can act directly on L-ONs (Fig. 4). Such enzymes could be readily prepared using standard biochemical and molecular biology techniques, making them much more accessible to a broader range of researchers. Indeed, some progress has been made towards this goal. In 2014, in vitro evolution was used to discover a cross-chiral ligase ribozyme.130 The D-RNA enzyme was capable of joining two L-RNA stands on a complementary L-RNA template, demonstrating for the first time that cross-chiral synthesis of L-ONs is possible. Further evolution of this D-RNA enzyme aimed at generating a general and processive cross-chiral polymerase yielded variants capable of assembling long L-RNAs from a mixture of L-trinucleotide building blocks.131,132 Furthermore, the cross-chiral polymerase ribozyme was used to perform exponential amplification of L-RNA, providing a proof-of-principle for cross-chiral ribo-PCR amplification of L-RNA. Despite its promise, however, this enzyme is still quite early in its evolutionary trajectory and therefore stills suffers from slow catalytic activity and processivity of polymerization, limiting the size of L-RNAs that it can produce. Nevertheless, given the progress that has already been made, we expect that further in vitro evolution could ultimately yield cross-chiral polymerase ribozymes with activity comparable to native protein enzymes. The success of the cross-chiral polymerase ribozyme also begs the question: Is cross-chiral polymerization possible for proteins of native chirality (Fig. 4)? There have been examples of some proteins that have ambidextrous operating capabilities. For example, the bacterial GroEL/ES chaperone protein can surprisingly fold a D-protein.133 Furthermore, many polymerases have been engineered to tolerate non-native sugar backbones, including 2′-fluoroarabino nucleic acid (FANA),134 arabino nucleic acid (ANA),41 hexitol nucleic acid (HNA),135 threose nucleic acid (TNA),136–138 and phosphonomethylthreosyl nucleic acid (PMT).139 Therefore, it is not unreasonable to expect that native polymerase could be evolved to accommodate L-ONs. The primary obstacle, however, may be the opposite (left-handed) helicity of L-ONs. This is evident in the crystal structure of Dpo4 polymerase bound to L-DNA, which shows that accommodation of L-DNA would require a major reconfiguration of the active site and DNA-binding surfaces.140 However, as rational and high-throughput protein engineering and screening methods continue to advance,141 it may become possible to completely invert the substrate specificity of native polymerase enzymes.
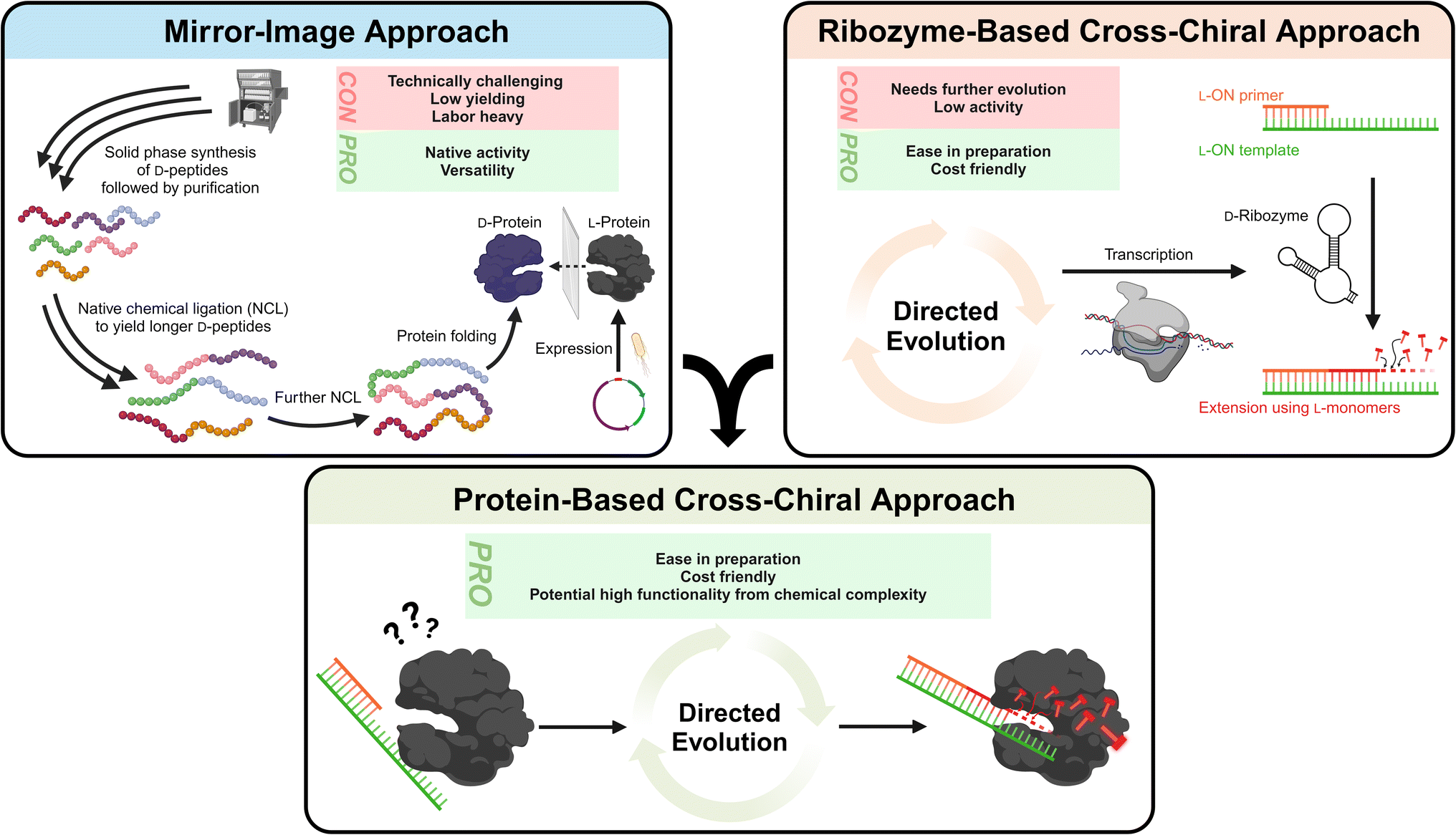 | ||
| Fig. 4 Current strategies to expand the L-ON toolkit include inverting the chirality of native protein enzymes (i.e., the mirror-image approach) and the development of native D-RNA ribozymes that can act directly on L-ONs (i.e., the ribozyme-based cross-chiral approach). Each approach has its pros and cons. We encourage integration of these two approaches, whereby directed evolution is used to generate native protein enzymes that can act directly on L-ONs, as an alternative strategy with several practical advantages. The example shown here is for polymerases, but this idea can be applied to other enzymatic functions as well (ligases, nucleases, etc.). | ||
It is clear that the future of L-ONs rests, in part, on the ability to routinely synthesize and amplify L-DNA and transcribe L-RNA through enzymatic means, which will provide greater accessibility of these polymers to researchers and open the door to exciting new technologies, including those unforeseen. Routine synthesis of long L-ONs would, for example, enable construction of bioorthogonal versions of many promising riboswitch-based biosensors in support of bioimaging and diagnostic applications142–144 and allow for the construction of larger, more complex L-DNA nanotechnologies with potential applications in drug delivery.94 Interestingly, the hypothetical cross-chiral acting polymerases discussed above could be expressed in living organisms and used to genetically encode robust L-ON-based technologies (e.g., biosensors) or even establish an orthogonal L-DNA/RNA replication system in the host, allowing for accelerated and continuous evolution of functional L-ONs.145 Perhaps the area that would benefit the most (and most immediately) from routine polymerization and amplification of L-ONs is in vitro selection of functional L-ONs, such as Spiegelmers and L-ON-based enzymes. This is discussed in detail below (Section 3.1.2).
3.1.1.2 Ligases. Efficient ligation is also an important roadblock towards the clinical advancement of L-ONs. As with polymerases, ligases are ubiquitous in biomedical research and have important applications in medical diagnosis.146 For example, DNA amplification by the ligase chain reaction (LCR) has emerged as powerful platform for genotyping applications, such as the detection of gene mutations and viral/bacterial pathogens.147 In the absence of tools capable of ligating L-ONs, development of more robust versions of these technologies using L-DNA is out of reach. Furthermore, ligation represents another potential solution to the L-ON synthesis problem by allowing long L-ONs to be assembled from shorter fragments and amplified via mirror-image LCR.130 Ligation also opens the door to diverse bioconjugation techniques and other ON manipulations not currently feasible using L-ONs. 3′ terminal labelling of L-RNA is one example.148,149 In the long run, L-ON ligation could facilitate the isothermal assembly of gene-size L-DNA fragments in support of a D-amino acid protein production using mirror-image ribosomes,150 and may prove to be essential for next-generation sequencing (NGS) of L-ONs, either for sample preparation or for the sequencing reaction itself (e.g., sequencing by ligation).151
Realizing the need for and implications of tools that are capable of ligating L-ONs, researchers have pursued several strategies to overcome the current deficit. While purely chemical approaches, such as phosphorothioate (PS) and “click” chemistry-based ligation,152–154 may prove useful for ligating L-ONs, we focus our attention on enzymatic approaches as the most promising pathway forward. Similar to polymerases, the Hoheisel group synthesized the D-amino acid version of the DNA-ligase of H. influenzae (D-LigA; 252 amino acids) and demonstrated efficient ligation activity on L-DNA substrates using an L-ATP cofactor.155 They also demonstrated ligation-mediated assembly of a 300-mer double-stranded L-DNA from 18 shorter L-DNA oligonucleotides, providing a proof-of-principle for ligase-mediated assembly of gene-size L-DNAs. Although promising, this approach still relies on the synthesis of D-amino acid proteins and, as discussed above with polymerases, is not yet practical for the average researcher. With this in mind, our group developed a strategy to ligate L-ON using native protein ligases. Here, two strands of L-RNA were joined by native T4 RNA ligase by incorporating D-ribonucleotides at the ligation junction.156 By carefully positioning these D-RNA ligation junctions at non-critical positions within the otherwise all L-RNA structure, we demonstrated that this approach could be used to assemble functional L-RNAs of considerable length, including an L-RNA version of a 124 nt theophylline biosensor that retained activity in serum. The ability to prepare nuclease resistant versions of RNA-based sensors using this straightforward approach greatly expands the utility of such technologies for applications in molecular sensing and imaging. It is important to note that L-ON ligation needs not be limited to the use of protein enzymes. As we discussed above, in vitro evolution was used to generate a cross-chiral D-RNA ribozyme that is capable of joining two or more strands of L-RNA. With further in vitro evolution to improve activity, such ribozymes should provide an attractive approach for routine L-ON ligation, as large quantities of the D-RNA enzyme can be easily generated using in vitro transcription. In vitro directed evolution could also be used to obtain cross-chiral protein ligases, either by engineering known ligases to accept L-ON substrates or by being created de novo using mRNA display or similar strategies.157 Interestingly, these cross-chiral L-ON protein ligases could serve as a starting point for the generation of cross-chiral L-ON polymerase activity through further evolution, as has been demonstrated previously with ribozyme ligases.132,158
3.1.1.3 Nucleases. Many important biomedical applications rely on the ability to readily break phosphodiester linkages between nucleotides. Nucleases have found widespread use in biomedical applications that require precise manipulation of ONs, including restriction digestion, molecular interaction probing, and sequencing.159–161 Programmable nucleases, such as CRISPR/Cas9, are powerful gene editing tools with broad applications for the prevention or treatment of numerous diseases.162 As the field of L-ONs matures and the ability to amplify and replicate L-ONs becomes routine, inevitably too will the need for nucleases to support a growing list of applications. There are several purely chemical approaches that can be used to cleave L-ONs directly. For example, the group of reactions used during Maxam Gilbert sequencing allow for nucleotide-specific cleavage of DNA irrespective of stereochemistry.163 This was recently demonstrated through the sequencing of a 55 nt long L-DNA.164 However, the future surely lies in the development of programmable nucleases capable of cleaving any L-ON sequence with precision. One potential solution is the use of RNA- and DNA-cleaving DNAzymes.165,166 These DNAzymes typically consist of a defined catalytic core flanked by two binding arms that can be exchanged to recognize most substrates through complementary interactions. Thus, they can be readily synthesized in their mirror-image form and programmed to cleave most L-RNA and L-DNA in a sequence-specific manner. Similarly, one can synthesize protein nucleases in their mirror-image form. In 2015, several groups reported the synthesis of mirror-image B. amyloliquefaciens ribonuclease (barnase) and demonstrated its nuclease activity toward L-RNA.67,167 Moving forward, it will be important to demonstrate this approach using programable nucleases, such as Cas9 and TALENs (transcription activator-like effector nucleases). This will not be without its challenges, however, as chemical synthesis of the enantiomeric forms of these systems, which are at least 600 amino acids, will require a Herculean amount of effort. Instead, we again encourage the community to explore the development of cross-chiral enzymes capable of cleaving L-ONs. In particular, the successful in vitro evolution of a cross-chiral D-RNA ribozyme that can join L-RNA bodes well for evolving the reverse activity using similar methods.130 Beyond expanding the L-ON toolbox, we expect that cross-chiral nucleases will have immediate clinical utility. For example, L-DNA versions of cross-chiral RNA-cleaving DNAzymes could be used in gene silencing applications by targeting endogenous mRNAs (e.g., oncogenes) for degradation. This approach represents a promising strategy to improve the biostability and other pharmacological properties of DNAzyme therapeutics and warrants further attention in the future.
Very recently, the direct in vitro selection of L-aptamers using mirror-image polymerases has been reported.170 This selection process utilized an engineered version of D-Dpo4 DNA polymerase (D-Dpo4-5m) for PCR amplification of an L-DNA library following affinity enrichment steps. The target for this selection was native human thrombin, which would be challenging to synthesize in the opposite handedness due to its large size (294 amino acids) and intensive glycosylation.171 After nine rounds of selection, two L-DNA aptamers were identified and shown to bind native human thrombin with Kd values in the low nanomolar range. This groundbreaking study proved that, with the appropriate toolbox, the direct in vitro selection of L-ONs is possible. Nevertheless, this report also highlighted some of the remaining challenges. Producing useable quantities of high-fidelity mirror-image DNA polymerases, which tend to be quite large, still presents a significant synthetic challenge. Consequently, this work utilized the shorter, more synthetically tractable D-Dpo4-5m, which suffers from low fidelity and suboptimal amplification efficiency, especially for long DNA sequences, that together could hinder the amplification of rare sequences in the pool. Sequencing of enriched L-DNA pools is another technical hurdle highlighted by this work. In the absence of bacterial cloning or high-throughput L-DNA sequencing, individual L-DNA sequences from the enriched pool were isolated by denaturing gradient gel electrophoresis (DGGE), which separates different DNA sequences of similar lengths based on their different melting temperatures.172 Individual bands were then isolated, amplified by mirror-image PCR using D-Dpo4-5m, and sequenced by the phosphorothioate approach with L-deoxynucleoside α-thiotriphosphates (L-dNTPαSs) and cleavage by 2-iodoethanol.128 This approach is extremely labor intensive and does not guarantee single-sequence resolution, especially for sequences with similar melting temperatures. Moreover, it dictates that many rounds of selection be carried out, as sequence isolation by DGGE requires a high level of convergence within the L-DNA pools. Nevertheless, the progress already made provides confidence that these technical hurdles can be overcome. For example, the accuracy and throughput of L-DNA sequencing could be improved by adopting mirror-image versions of nanopore sequencing or massively parallel sequencing-by-synthesis techniques using the appropriate mirror-image toolbox,173,174 thereby increasing the overall efficiency and practicality of the L-ON selection process.
Direct in vitro selection/evolution of L-ONs using straightforward and practical methods would open the door to a number of exciting opportunities for researchers and clinicians. For example, without the need to generate the enantiomer of the desired target, as is currently required for mirror-image SELEX, the pool of clinically relevant molecules that could be targeted by Spiegelmers is virtually limitless. Even whole cells could be targeted with relative ease to discover binders, inhibitors, and biomarkers without pre-existing knowledge of the target itself. Cell-based SELEX approaches would also benefit from the biostability of L-ON pools. Given the ability of Spiegelmers to evade biodegradation during storage and use, coupled with their convenient production, we expect that as the repertoire of Spiegelmers grows, so too will their utility in applications currently dominated by traditional aptamers and antibodies. Potential applications of Spiegelmers in nuclease-rich environments could include enzyme-linked immunosorbent assays (ELISAs), immunohistochemistry, and drug delivery (using cell-targeting Spiegelmers). Furthermore, the ability to directly isolate L-aptamer-based molecular switches that undergo a binding-induced conformational change (e.g., via “Capture SELEX”)175 should fuel advances in the areas of biosensing and diagnostics, such as the development of more robust E-AB sensors for invasive molecular monitoring.176 Beyond Spiegelmers, direct L-ON selection schemes are expected to find applications in the discovery of L-ribozymes and L-DNAzymes that are capable of catalyzing the manipulation (e.g., cleaving, tagging, etc.) of clinically relevant proteins and nucleic acids.
While indirect approaches for interfacing D- and L-ON are promising, future efforts should be directed towards the establishment of a “heterochiral code” that would enable the rational design of stable heterochiral complexes in a sequence-specific manner. In vitro selection methods already allow for direct D- and L-ON interactions to be discovered from random sequence space.80,81,85 With more examples, as well as structural information, patterns may emerge that point to an alternative set of “heterochiral” base pairing rules. Indeed, molecular dynamics simulations reveal numerous recurring geometric patterns that suggest potential structural motifs between heterochiral RNA strands,177 indicating that computational modeling and machine learning could be employed for discovering novel sequence-specific interactions between the enantiomers. It is important to note that the sequence-specific interface between D- and L-ONs does not necessarily have to adhere to the traditional anti-parallel duplex model. Alternative structural arrangements, such as parallel-stranded duplexes, triplexes, and patterned mismatches, as well as the use of chemically modified nucleotides with alternative base pairing properties, should all be considered when exploring the potential for sequence-specific cross-chiral interfacing. Overall, the ability to rationally design stable heterochiral complexes in a sequence-specific manner would represent a major breakthrough in the field of L-ONs, enabling the integration of this polymer into a number of clinically relevant applications.
3.2 Biological interactions of L-oligonucleotides
If L-ONs are to be routinely employed in a clinical setting, then it is imperative that we understand how they interact with biology and their potential consequences. Indeed, extensive in vivo investigations into the mechanisms of action and off-target interactions of D-ONs, such as ASOs26,184–187 and aptamers,15,61 have greatly benefited the clinical translation of ON-based therapeutics. In contrast, despite the enormous promise of Spiegelmer therapeutics, we are still just beginning to understand how L-ONs behave in living systems, how they interact with these environments, and the potential consequences. Although careful in vivo pharmacological studies have been carried out on Spiegelmer therapeutics (and have been reviewed elsewhere),48 the vast majority of these Spiegelmers have been developed against extracellular targets and, consequently, little attention has been paid to the behavior of L-ON at the intracellular level. As the field of L-ON-based therapeutics matures and shifts towards intracellular targets, it will become increasingly important to understand how L-ONs behave within the complex environment of the cell. Therefore, we focus our discussion below on the current understanding of how L-oligonucleotides behave inside cells, the known and potential off-target effects, and where we believe future efforts should be focused with an eye toward broader clinical adoption.| Nucleic acid | Features | Delivery method | Cell line | Localization | Reference |
|---|---|---|---|---|---|
| L-RNA | Single stranded G-rich | Transfection | HeLa | Nucleoplasm, nucleolus, nuclear foci | 188 |
| Double stranded G-rich | Transfection | HeLa | Nucleoplasm, nucleolus | 188 | |
| G-quadruplex | Transfection | HeLa | Nucleoplasm, nucleolus, nuclear foci | 188 | |
| G-quadruplex | Cell penetrating peptide | HeLa | Cytosol | 189 | |
| A-rich | Transfection | HeLa | Nucleoplasm | 188 | |
| Double stranded; mixed-sequence aptamers | Cholesterol | HeLa | Cytosol | 181 | |
| L-DNA | Duplex | Transfection | HeLa | Nucleus | 180 |
| Duplex | Direct uptake | SCC7 | Cytosol | 93 | |
| Duplex; D/L chimeric | Transfection | HeLa | Nucleoplasm | 113 | |
| 5′-L-DNA capped D-DNA duplex | Transfection | HeLa | Cytosol, nucleus | 190 | |
| G-quadruplex | Transfection | HeLa | Nucleoplasm | 188 | |
| ATP aptamer | Graphene oxide complex | HeLa | Cytosol | 191 | |
| L-DNA nano-structure | Tetrahedron | Direct uptake | HeLa | Cytosol | 92 |
| Pyramid, triangular prism, cube and rugby ball-like construct | Direct uptake | HeLa, RAW264.7 | Cytosol | 95 |
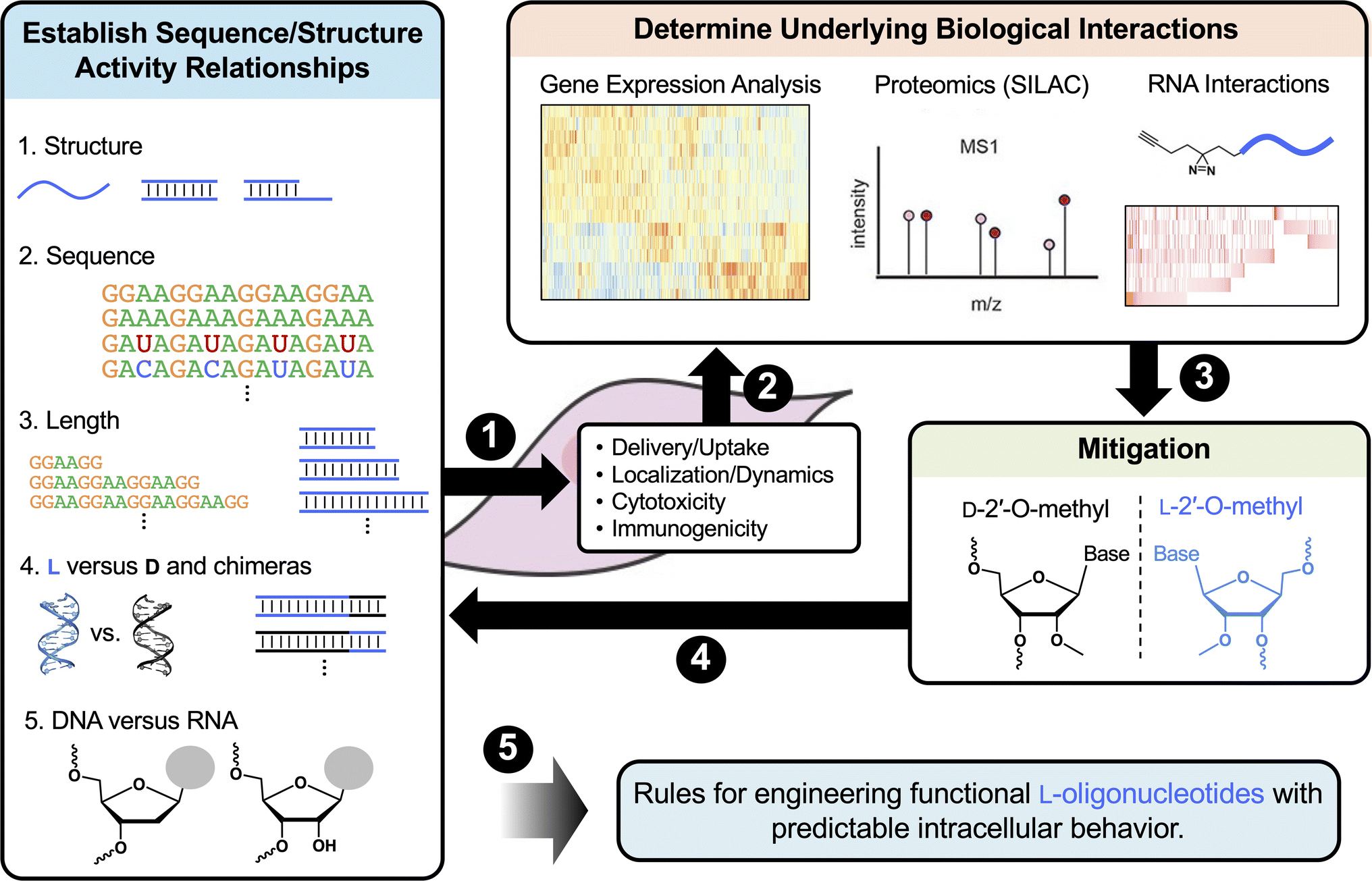 | ||
| Fig. 5 Proposed workflow for characterizing the intracellular behaviors of L-ONs. (1) Determine the impact of length, sequence, structure, and sugar chemistry on cellular uptake, distribution, cytotoxicity, and immunogenicity. (2) Determine the biological interactions underlying key behaviors. (3) Use information gained from steps 1 and 2 to devise strategies to mitigate unwanted effects (e.g., 2′-OMe to mitigate protein interactions) and verify through repeated analysis (4). (5) Collectively, these data will contribute to the establishment of rules for rationally designing L-ONs with predictable intracellular behaviors. | ||
First, it will be important to further characterize how common features such as the length, sequence, structure, and sugar chemistry influence the cellular uptake and distribution of L-ONs. It will also be important to establish detailed structure toxicity/immunogenicity relationships. Such information can be used to predict toxicity from sequence and will potentially allow for toxic motifs, such as single-stranded G-rich sequences, to be eliminated at the design stage. These studies may also reveal previously unrecognized therapeutic opportunities for L-ONs. For example, a better understanding of L-ON immunogenicity could lead to the development of L-ON-based vaccine adjuvants having superior biostability and reduced off-target hybridization compared to current immunomodulatory ONs.196
Second, it is critical that we map the interactions of L-ONs with cellular proteins, especially for cytotoxic sequences. Mass spectrometry (MS)-based proteomics techniques, such as stable isotope labeling by amino acids in cell culture (SILAC),197 have been shown to be an efficient assay for detecting interactions of D-ONs with proteins and could be readily adopted for the detection and quantitation of the L-ON–interacting proteome.198,199 Such information will not only shed light on the mechanistic basis for observed intracellular behaviors of L-ONs, but may also inform strategies to mitigate off-target cytotoxicity. For example, studies have shown that the cytotoxicity resulting from interaction of D-ONs with nuclear paraspeckle proteins can be mitigated via the incorporation of 2′-OMe modifications.184 If similar interactions are observed for cytotoxic L-RNAs, as proposed, it would be very interesting to test whether this mitigation strategy translates across the chiral mirror.
Third, although canonical WC base pairing is not possible between D- and L-ONs, potential interactions between them should not be overlooked. Indeed, previous studies suggest that homopolymers between D- and L-ONs can form stable complexes, particularly in the context of RNA, which has a greater propensity for binding the enantiomer of its complement than DNA.200–202 The greater cytotoxicity found for L-RNA compared to L-DNA of the same sequence may be explained, in part, by these interactions.188 However, further investigations are needed to determine the extent to which L-ONs interact with endogenous ONs and the consequences of these interactions, for which almost nothing is known. Cross-linking and immunoprecipitation (CLIP)-based approaches provide one potential strategy to map the interactions between L-ON and endogenous nucleic acids.203 For example, in situ crosslinking of biotinylated L-ONs with endogenous nucleic acids would permit selective pulldown and identification of these interactions via high-throughput sequencing.
Finally, even with a comprehensive understanding of L-ON behavior in hand, intracellular applications of L-ONs, especially L-ON-based therapeutics, will require effective delivery strategies. For most research applications, ONs can be delivered into cells using common liposome-based transfection reagents, regardless of chirality. Indeed, work from our lab and others have shown that L-ONs are efficiently delivered into diverse cells lines (e.g., HeLa, MCF7, A375, HEK293T) using Lipofectamine and related reagents.113,180,188,193 While this approach is sufficient for basic research and preclinical studies, it is not ideal for most therapeutic applications. Instead, we should look to the many promising delivery systems currently being employed for D-ON-based therapeutics, such as direct conjugation to carriers (e.g., peptides, lipids, receptor ligands, etc.) or incorporation into lipid-derived nanoparticles (LNPs), all of which function independent of the chirality of the ON.21 Thus, we expect that they can also be applied directly to L-ONs with little optimization. For example, cholesterol conjugation, which has been used extensively to enhance siRNA delivery,204 was shown to facilitate efficient cellular uptake of L-RNA-based molecular probes.181 Similarly, conjugating a cell-penetrating peptide to the G-quadruplex-targeting Spiegelmer L-Apt.4-1c resulted in cellular uptake efficiencies similar to traditional transfection reagents.189 Formulations of Spiegelmers with branched polyethyleneimine (PEI) have also been shown to permit efficient cellular delivery and targeting of intracellular proteins.205 The ability to borrow proven delivery strategies from the D-ON therapeutics field is encouraging for the future development of therapeutic L-ONs.
4 Conclusions and outlook
Recent advancements have put a spotlight on ON-based therapeutics. The COVID-19 pandemic and the first approved mRNA vaccines have highlighted the power of nucleic acid-based therapeutic strategies.206,207 This is further exemplified by the growing list of approved ASO drugs for diseases that have long lacked treatment options.1,2,7 Nevertheless, native nucleic acids and their chemically modified counterparts have well-recognized limitations that L-ONs are poised to overcome. Indeed, the superior biostability and low immunogenicity of L-ONs have already been exploited to develop Spiegelmer therapeutics, with several examples advancing to clinical trials.208 Even with the advantages offered by L-ONs, however, major challenges remain. Herein, we have provided our perspective on these challenges and explored potential solutions that we believe will pave the way toward the broader adoption of L-ONs in clinical applications:(1) An expanded L-ON toolkit will allow for more practical synthesis and manipulation of L-ONs, while enabling powerful applications such as direct in vitro selection of Spiegelmer therapeutics and other functional L-ONs. Future focus: we encourage researchers to explore the development of cross-chiral enzymes, i.e., enzymes composed of native polymers (D-ONs and L-amino acids) that can act directly on L-ONs (Fig. 4). Compared to mirror-image enzymes, which must be produced synthetically, cross-chiral enzymes could be readily prepared using standard biochemical and molecular biology techniques, making these tools much more accessible to a broader range of researchers.
(2) Establishment of a “heterochiral code” will enable the rational design of stable heterochiral complexes in a sequence-specific manner, facilitating the use of L-ONs in common hybridization-based technologies, such as ASOs. Future focus: in vitro selection methods already allow for direct D- and L-ON interactions to be discovered from random sequence space. Pairing these methods (and their datasets) with machine learning approaches may allow for prediction of heterochiral nucleic acid interactions. Such efforts will be aided by structural studies of heterochiral complexes.
(3) An improved understanding of the behavior of L-ONs inside cells will allow for the engineering of functional L-ONs, such as Spiegelmer therapeutics, with predictable intracellular behaviors, a critical step towards expanding the therapeutic potential of L-ONs. Future focus: efforts should be directed at characterizing how common features such as the length, sequence, structure, and chemical modifications impact the cellular uptake and distribution of L-ONs. Establishing protein and nucleic acid interactomes of L-ONs will provide the mechanistic basis for observed intracellular behaviors and inform strategies to mitigate off-target effects.
We hope that the challenges and opportunities discussed herein will inspire nucleic acid researchers and serve as a roadmap for future investigations. Indeed, given the unique advantages offered by L-ONs and the recent clinical success of Spiegelmers, we expect that interest in this area will continue to grow, bringing about unique and exciting solutions to each of the challenges discussed herein. Success in this regard will undoubtedly lead to a rapid expansion of L-ON-based technologies with tremendous potential to advance biomedical research and improve patient care.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Victoria Shearer: writing – original draft, writing – review & editing. Chen-Hsu Yu: writing – original draft, writing – review & editing. Xuan Han: writing – original draft, writing – review & editing. Jonathan Sczepanski: writing – original draft, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Institute of General Medical Sciences (R35GM124974) of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- D. Bartolucci, A. Pession, P. Hrelia and R. Tonelli, Precision Anti-Cancer Medicines by Oligonucleotide Therapeutics in Clinical Research Targeting Undruggable Proteins and Non-Coding RNAs, Pharmaceutics, 2022, 14, 1453 CrossRef PubMed.
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, The current landscape of nucleic acid therapeutics, Nat. Nanotechnol., 2021, 16, 630–643 CrossRef.
- F. M. Foss, C. Querfeld, P. Porcu, Y. H. Kim, T. Pacheco, A. S. Halwani, J. DeSimone, B. M. William, A. G. Seto, J. Ruckman, M. L. Landry, A. L. Jackson, L. A. Pestano, B. A. Dickinson, M. Sanseverino, D. M. Rodman, P. Rubin, G. S. Gordon and W. S. Marshall, Phase 1 trial evaluating MRG-106, a synthetic inhibitor of microRNA-155, in patients with cutaneous t-cell lymphoma (CTCL), J. Clin. Oncol., 2017, 35, 7564 Search PubMed.
- D. S. Hong, Y. K. Kang, M. Borad, J. Sachdev, S. Ejadi, H. Y. Lim, A. J. Brenner, K. Park, J. L. Lee, T. Y. Kim, S. Shin, C. R. Becerra, G. Falchook, J. Stoudemire, D. Martin, K. Kelnar, H. Peltier, V. Bonato, A. G. Bader, S. Smith, S. Kim, V. O'Neill and M. S. Beg, Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours, Br. J. Cancer, 2020, 122, 1630–1637 CrossRef CAS.
- B. D. Adams, C. Parsons, L. Walker, W. C. Zhang and F. J. Slack, Targeting noncoding RNAs in disease, J. Clin. Invest., 2017, 127, 761–771 CrossRef PubMed.
- M. C. Jiang, J. J. Ni, W. Y. Cui, B. Y. Wang and W. Zhuo, Emerging roles of lncRNA in cancer and therapeutic opportunities, Am. J. Cancer Res., 2019, 9, 1354–1366 Search PubMed.
- B. P. Vinjamuri, J. Pan and P. Peng, A Review on Commercial Oligonucleotide Drug Products, J. Pharm. Sci., 2024, 113, 1749–1768 CrossRef PubMed.
- D. Al Shaer, O. Al Musaimi, F. Albericio and B. G. de la Torre, 2023 FDA TIDES (Peptides and Oligonucleotides) Harvest, Pharmaceuticals, 2024, 17, 243 CrossRef PubMed.
- M. Egli and M. Manoharan, Chemistry, structure and function of approved oligonucleotide therapeutics, Nucleic Acids Res., 2023, 51, 2529–2573 CrossRef.
- FDA approves treatment of amyotrophic lateral sclerosis associated with a mutation in the SOD1 gene, https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-treatment-amyotrophic-lateral-sclerosis-associated-mutation-sod1-gene, (accessed July, 2024).
- A. Mullard, FDA approves second RNA aptamer, Nat. Rev. Drug Discovery, 2023, 22, 774 Search PubMed.
- A. Goyenvalle, C. Jimenez-Mallebrera, W. van Roon, S. Sewing, A. M. Krieg, V. Arechavala-Gomeza and P. Andersson, Considerations in the Preclinical Assessment of the Safety of Antisense Oligonucleotides, Nucleic Acid Ther., 2023, 33, 1–16 CrossRef PubMed.
- M. M. Janas, Y. Jiang, M. K. Schlegel, S. Waldron, S. Kuchimanchi and S. A. Barros, Impact of Oligonucleotide Structure, Chemistry, and Delivery Method on In Vitro Cytotoxicity, Nucleic Acid Ther., 2017, 27, 11–22 CrossRef CAS PubMed.
- K. S. Frazier, Antisense oligonucleotide therapies: the promise and the challenges from a toxicologic pathologist's perspective, Toxicol. Pathol., 2015, 43, 78–89 CrossRef PubMed.
- L. Kelly, K. E. Maier, A. Yan and M. Levy, A comparative analysis of cell surface targeting aptamers, Nat. Commun., 2021, 12, 6275 CrossRef PubMed.
- H. Wu, A. Wahane, F. Alhamadani, K. Zhang, R. Parikh, S. Lee, E. M. McCabe, T. P. Rasmussen, R. Bahal, X. B. Zhong and J. E. Manautou, Nephrotoxicity of marketed antisense oligonucleotide drugs, Curr. Opin. Toxicol., 2022, 32, 100373 CrossRef PubMed.
- J. S. Parham and A. C. Goldberg, Mipomersen and its use in familial hypercholesterolemia, Expert Opin. Pharmacother., 2019, 20, 127–131 CrossRef PubMed.
- J. P. Bost, H. Barriga, M. N. Holme, A. Gallud, M. Maugeri, D. Gupta, T. Lehto, H. Valadi, E. K. Esbjorner, M. M. Stevens and S. El-Andaloussi, Delivery of Oligonucleotide Therapeutics: Chemical Modifications, Lipid Nanoparticles, and Extracellular Vesicles, ACS Nano, 2021, 15, 13993–14021 CrossRef.
- S. M. Hammond, A. Aartsma-Rus, S. Alves, S. E. Borgos, R. A. M. Buijsen, R. W. J. Collin, G. Covello, M. A. Denti, L. R. Desviat, L. Echevarria, C. Foged, G. Gaina, A. Garanto, A. T. Goyenvalle, M. Guzowska, I. Holodnuka, D. R. Jones, S. Krause, T. Lehto, M. Montolio, W. Van Roon-Mom and V. Arechavala-Gomeza, Delivery of oligonucleotide-based therapeutics: challenges and opportunities, EMBO Mol. Med., 2021, 13, e13243 CrossRef PubMed.
- S. Ochoa and V. T. Milam, Modified Nucleic Acids: Expanding the Capabilities of Functional Oligonucleotides, Molecules, 2020, 25, 4659 CrossRef PubMed.
- T. C. Roberts, R. Langer and M. J. A. Wood, Advances in oligonucleotide drug delivery, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef PubMed.
- A. Curreri, D. Sankholkar, S. Mitragotri and Z. Zhao, RNA therapeutics in the clinic, Bioeng. Transl. Med., 2023, 8, e10374 CrossRef PubMed.
- A. Khvorova and J. K. Watts, The chemical evolution of oligonucleotide therapies of clinical utility, Nat. Biotechnol., 2017, 35, 238–248 CrossRef PubMed.
- V. K. Sharma and J. K. Watts, Oligonucleotide therapeutics: chemistry, delivery and clinical progress, Future Med. Chem., 2015, 7, 2221–2242 CrossRef PubMed.
- C. Kratschmer and M. Levy, Effect of Chemical Modifications on Aptamer Stability in Serum, Nucleic Acid Ther., 2017, 27, 335–344 CrossRef PubMed.
- S. T. Crooke, S. Wang, T. A. Vickers, W. Shen and X.-h. Liang, Cellular uptake and trafficking of antisense oligonucleotides, Nat. Biotechnol., 2017, 35, 230–237 CrossRef CAS PubMed.
- F. Debart, S. Abes, G. Deglane, H. M. Moulton, P. Clair, M. J. Gait, J. J. Vasseur and B. Lebleu, Chemical modifications to improve the cellular uptake of oligonucleotides, Curr. Trends Med. Chem., 2007, 7, 727–737 CrossRef PubMed.
- C. Wilson and A. D. Keefe, Building oligonucleotide therapeutics using non-natural chemistries, Curr. Opin. Chem. Biol., 2006, 10, 607–614 CrossRef.
- K. Morihiro, Y. Kasahara and S. Obika, Biological applications of xeno nucleic acids, Mol. BioSyst., 2017, 13, 235–245 RSC.
- J. C. Chaput, Redesigning the Genetic Polymers of Life, Acc. Chem. Res., 2021, 54, 1056–1065 CrossRef PubMed.
- Q. Wang, L. Chen, Y. Long, H. Tian and J. Wu, Molecular Beacons of Xeno-Nucleic Acid for Detecting Nucleic Acid, Theranostics, 2013, 3, 395–408 CrossRef PubMed.
- M. Ali, R. Neumann and W. Ensinger, Sequence-Specific Recognition of DNA Oligomer Using Peptide Nucleic Acid (PNA)-Modified Synthetic Ion Channels: PNA/DNA Hybridization in Nanoconfined Environment, ACS Nano, 2010, 4, 7267–7274 CrossRef.
- H. D. Winter, E. Lescrinier, A. V. Aerschot and P. Herdewijn, Molecular Dynamics Simulation To Investigate Differences in Minor Groove Hydration of HNA/RNA Hybrids As Compared to HNA/DNA Complexes, J. Am. Chem. Soc., 1998, 120, 5381–5394 Search PubMed.
- I. Anosova, E. A. Kowal, N. J. Sisco, S. Sau, J. Y. Liao, S. Bala, E. Rozners, M. Egli, J. C. Chaput and W. D. Van Horn, Structural Insights into Conformation Differences between DNA/TNA and RNA/TNA Chimeric Duplexes, ChemBioChem, 2016, 17, 1705–1708 CrossRef PubMed.
- R. El-Khoury and M. J. Damha, 2′-Fluoro-arabinonucleic Acid (FANA): A Versatile Tool for Probing Biomolecular Interactions, Acc. Chem. Res., 2021, 54, 2287–2297 CrossRef CAS PubMed.
- T. Tu, S. Huan, G. Ke and X. Zhang, Functional Xeno Nucleic Acids for Biomedical Application, Chem. Res. Chin. Univ., 2022, 38, 912–918 CrossRef CAS.
- H. Asanuma, Y. Kamiya, H. Kashida and K. Murayama, Xeno nucleic acids (XNAs) having non-ribose scaffolds with unique supramolecular properties, Chem. Commun., 2022, 58, 3993–4004 RSC.
- Y. Wang, K. Nguyen, R. C. Spitale and J. C. Chaput, A biologically stable DNAzyme that efficiently silences gene expression in cells, Nat. Chem., 2021, 13, 319–326 CrossRef PubMed.
- T. N. Malik and J. C. Chaput, XNA enzymes by evolution and design, Curr. Res. Chem. Biol., 2021, 1, 100012 CrossRef.
- A. I. Taylor, C. J. K. Wan, M. J. Donde, S. Y. Peak-Chew and P. Holliger, A modular XNAzyme cleaves long, structured RNAs under physiological conditions and enables allele-specific gene silencing, Nat. Chem., 2022, 14, 1295–1305 CrossRef PubMed.
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S.-Y. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Synthetic Genetic Polymers Capable of Heredity and Evolution, Science, 2012, 336, 341–344 CrossRef PubMed.
- E. Medina, E. J. Yik, P. Herdewijn and J. C. Chaput, Functional Comparison of Laboratory-Evolved XNA Polymerases for Synthetic Biology, ACS Synth. Biol., 2021, 10, 1429–1437 CrossRef PubMed.
- N. C. Hauser, R. Martinez, A. Jacob, S. Rupp, J. D. Hoheisel and S. Matysiak, Utilising the left-helical conformation of L-DNA for analysing different marker types on a single universal microarray platform, Nucleic Acids Res., 2006, 34, 5101–5111 CrossRef PubMed.
- H. Urata, E. Ogura, K. Shinohara, Y. Ueda and M. Akagi, Synthesis and properties of mirror-image DNA, Nucleic Acids Res., 1992, 20, 3325–3332 CrossRef CAS PubMed.
- M. Szabat, D. Gudanis, W. Kotkowiak, Z. Gdaniec, R. Kierzek and A. Pasternak, Thermodynamic Features of Structural Motifs Formed by beta-L-RNA, PLoS One, 2016, 11, e0149478 CrossRef PubMed.
- H. Urata, K. Shinohara, E. Ogura, Y. Ueda and M. Akagi, Mirror-Image DNA, J. Am. Chem. Soc., 1991, 113, 8174–8175 CrossRef.
- K. Hoehlig, L. Bethge and S. Klussmann, Stereospecificity of Oligonucleotide Interactions Revisited: No Evidence for Heterochiral Hybridization and Ribozyme/DNAzyme Activity, PLoS One, 2015, 10, e0115328 CrossRef PubMed.
- A. Vater and S. Klussmann, Turning mirror-image oligonucleotides into drugs: the evolution of Spiegelmer therapeutics, Drug Discovery Today, 2015, 20, 147–155 Search PubMed.
- B. E. Young, N. Kundu and J. T. Sczepanski, Mirror-Image Oligonucleotides: History and Emerging Applications, Chem.–Eur. J., 2019, 25, 7981–7990 CrossRef PubMed.
- S. Hoffmann, J. Hoos, S. Klussmann and S. Vonhoff, RNA aptamers and spiegelmers: synthesis, purification, and post-synthetic PEG conjugation, Curr. Protoc. Nucleic Acid Chem., 2011, 46, 4.46.41–44.46.30 Search PubMed.
- A. Vater and S. Klussmann, Toward third-generation aptamers: Spiegelmers and their therapeutic prospects, Curr. Opin. Drug Discovery Dev., 2003, 6, 253–261 Search PubMed.
- D. Eulberg, F. Jarosch, S. Vonhoff and S. Klussmann, in The Aptamer Handbook, 2006, pp. 417–442, DOI:10.1002/3527608192.ch18.
- D. Eulberg and S. Klussmann, Spiegelmers: biostable aptamers, ChemBioChem, 2003, 4, 979–983 Search PubMed.
- C. Tuerk and L. Gold, Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase, Science, 1990, 3, 505–510 CrossRef PubMed.
- G. F. Joyce, Amplification, mutation and selection of catalytic RNA, Gene, 1989, 82, 83–87 Search PubMed.
- A. D. Ellington and J. W. Szostak, In vitro Selection of Rna Molecules That Bind Specific Ligands, Nature, 1990, 346, 818–822 Search PubMed.
- V. Thiviyanathan and D. G. Gorenstein, Aptamers and the next generation of diagnostic reagents, Proteomics: Clin. Appl., 2012, 6, 563–573 Search PubMed.
- L. Li, S. Xu, H. Yan, X. Li, H. S. Yazd, X. Li, T. Huang, C. Cui, J. Jiang and W. Tan, Nucleic Acid Aptamers for Molecular Diagnostics and Therapeutics: Advances and Perspectives, Angew. Chem., Int. Ed. Engl., 2021, 60, 2221–2231 CrossRef PubMed.
- G. Zhu and X. Chen, Aptamer-based targeted therapy, Adv. Drug Delivery Rev., 2018, 134, 65–78 CrossRef PubMed.
- S. Ni, Z. Zhuo, Y. Pan, Y. Yu, F. Li, J. Liu, L. Wang, X. Wu, D. Li, Y. Wan, L. Zhang, Z. Yang, B. T. Zhang, A. Lu and G. Zhang, Recent Progress in Aptamer Discoveries and Modifications for Therapeutic Applications, ACS Appl. Mater. Interfaces, 2021, 13, 9500–9519 CrossRef PubMed.
- S. M. Nimjee, R. R. White, R. C. Becker and B. A. Sullenger, Aptamers as Therapeutics, Annu. Rev. Pharmacol. Toxicol., 2017, 57, 61–79 CrossRef PubMed.
- E. W. M. Ng, D. T. Shima, P. Calias, E. T. Cunningham, D. R. Guyer and A. P. Adamis, Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease, Nat. Rev. Drug Discovery, 2006, 5, 123–132 CrossRef PubMed.
- A. Nolte, S. Klussmann, R. Bald, V. A. Erdmann and J. P. Furste, Mirror-design of L-oligonucleotide ligands binding to L-arginine, Nat. Biotechnol., 1996, 14, 1116–1119 CrossRef PubMed.
- K. P. Williams, X. H. Liu, T. N. Schumacher, H. Y. Lin, D. A. Ausiello, P. S. Kim and D. P. Bartel, Bioactive and nuclease-resistant L-DNA ligand of vasopressin, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 11285–11290 CrossRef PubMed.
- S. Klussmann, A. Nolte, R. Bald, V. A. Erdmann and J. P. Fürste, Mirror-image RNA that binds D-adenosine, Nat. Biotechnol., 1996, 14, 1112–1115 CrossRef PubMed.
- W. G. Purschke, F. Radtke, F. Kleinjung and S. Klussmann, A DNA Spiegelmer to staphylococcal enterotoxin B, Nucleic Acids Res., 2003, 31, 3027–3032 CrossRef PubMed.
- C. Olea, J. Weidmann, P. E. Dawson and G. F. Joyce, An L-RNA Aptamer that Binds and Inhibits RNase, Chem. Biol., 2015, 22, 1437–1441 CrossRef CAS PubMed.
- L. Yatime, C. Maasch, K. Hoehlig, S. Klussmann, G. R. Andersen and A. Vater, Structural basis for the targeting of complement anaphylatoxin C5a using a mixed L-RNA/L-DNA aptamer, Nat. Commun., 2015, 6, 6481 CrossRef CAS PubMed.
- D. Oberthur, J. Achenbach, A. Gabdulkhakov, K. Buchner, C. Maasch, S. Falke, D. Rehders, S. Klussmann and C. Betzel, Crystal structure of a mirror-image L-RNA aptamer (Spiegelmer) in complex with the natural L-protein target CCL2, Nat. Commun., 2015, 6, 6923 CrossRef PubMed.
- M. Boyce, S. Warrington, B. Cortezi, S. Zöllner, S. Vauléon, D. W. Swinkels, L. Summo, F. Schwoebel and K. Riecke, Safety, pharmacokinetics and pharmacodynamics of the anti-hepcidin Spiegelmer lexaptepid pegol in healthy subjects, Br. J. Pharmacol., 2016, 173, 1580–1588 CrossRef CAS.
- J. Hoellenriegel, D. Zboralski, C. Maasch, N. Y. Rosin, W. G. Wierda, M. J. Keating, A. Kruschinski and J. A. Burger, The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization, Blood, 2014, 123, 1032–1039 CrossRef CAS.
- H. Ludwig, K. Weisel, M. T. Petrucci, X. Leleu, A. M. Cafro, L. Garderet, C. Leitgeb, R. Foa, R. Greil, I. Yakoub-Agha, D. Zboralski, S. Vauleon, T. Dummler, D. Beyer, A. Kruschinski, K. Riecke, M. Baumann and M. Engelhardt, Olaptesed pegol, an anti-CXCL12/SDF-1 Spiegelmer, alone and with bortezomib-dexamethasone in relapsed/refractory multiple myeloma: a Phase IIa Study, Leukemia, 2017, 31, 997–1000 CrossRef CAS.
- A. Vater, J. Sahlmann, N. Kroger, S. Zollner, M. Lioznov, C. Maasch, K. Buchner, D. Vossmeyer, F. Schwoebel, W. G. Purschke, S. Vonhoff, A. Kruschinski, K. Hubel, M. Humphrey, S. Klussmann and F. Fliegert, Hematopoietic stem and progenitor cell mobilization in mice and humans by a first-in-class mirror-image oligonucleotide inhibitor of CXCL12, Clin. Pharmacol. Ther., 2013, 94, 150–157 CrossRef CAS PubMed.
- B. Wlotzka, S. Leva, B. Eschgfäller, J. Burmeister, F. Kleinjung, C. Kaduk, P. Muhn, H. Hess-Stumpp and S. Klussmann, In vivo properties of an anti-GnRH Spiegelmer: an example of an oligonucleotide-based therapeutic substance class, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8898–8902 CrossRef CAS PubMed.
- I. Kryczek, S. Wei, E. Keller, R. Liu and W. Zou, Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis, Am. J. Physiol.: Cell Physiol., 2007, 292, C987–C995 CrossRef CAS PubMed.
- F. A. Giordano, J. P. Layer, S. Leonardelli, L. L. Friker, R. Turiello, D. Corvino, T. Zeyen, C. Schaub, W. Müller, E. Sperk, L. C. Schmeel, K. Sahm, C. Oster, S. Kebir, P. Hambsch, T. Pietsch, S. Bisdas, M. Platten, M. Glas, C. Seidel, U. Herrlinger and M. Hölzel, L-RNA aptamer-based CXCL12 inhibition combined with radiotherapy in newly-diagnosed glioblastoma: dose escalation of the phase I/II GLORIA trial, Nat. Commun., 2024, 15, 4210 Search PubMed.
- TME pharma announces FDA clearance of investigational new drug (IND) application for NOX-A12 phase 2 trial in brain cancer, https://www.tmepharma.com/downloads/pressrel/2024-03-05_TME_NOX-A12_GBM_IND_Approval_EN.pdf, (accessed July 2024).
- TME pharma announces 33% of patients receiving NOX-A12 in combination with bevacizumab and radiotherapy achieve two-year survival in gloria phase 1/2 trial in brain cancer, https://www.tmepharma.com/downloads/pressrel/2024-04-23_TME_GBM_2-Year_Survival_EN.pdf, (accessed July 2024).
- J. Li and J. T. Sczepanski, Targeting a conserved structural element from the SARS-CoV-2 genome using L-DNA aptamers, RSC Chem. Biol., 2022, 3, 79–84 Search PubMed.
- S. Dey and J. T. Sczepanski, In vitro selection of L-DNA aptamers that bind a structured D-RNA molecule, Nucleic Acids Res., 2020, 48, 1669–1680 CrossRef.
- J. T. Sczepanski and G. F. Joyce, Specific Inhibition of MicroRNA Processing Using L-RNA Aptamers, J. Am. Chem. Soc., 2015, 137, 16032–16037 CrossRef PubMed.
- J. T. Sczepanski and G. F. Joyce, Binding of a Structured D-RNA Molecule by an L-RNA Aptamer, J. Am. Chem. Soc., 2013, 135, 13290–13293 Search PubMed.
- D. Ji, K. Lyu, H. Zhao and C. K. Kwok, Circular L-RNA aptamer promotes target recognition and controls gene activity, Nucleic Acids Res., 2021, 49, 7280–7291 Search PubMed.
- M. I. Umar and C. K. Kwok, Specific suppression of D-RNA G-quadruplex–protein interaction with an L-RNA aptamer, Nucleic Acids Res., 2020, 48, 10125–10141 Search PubMed.
- D. Ji, J. H. Yuan, S. B. Chen, J. H. Tan and C. K. Kwok, Selective targeting of parallel G-quadruplex structure using L-RNA aptamer, Nucleic Acids Res., 2023, 51, 11439–11452 CrossRef PubMed.
- Q. Hu, H. Li, L. Wang, H. Gu and C. Fan, DNA Nanotechnology-Enabled Drug Delivery Systems, Chem. Rev., 2019, 119, 6459–6506 Search PubMed.
- M. Kumar, A. Jha and B. Mishra, DNA-Based Nanostructured Platforms as Drug Delivery Systems, Chem Bio Eng., 2024, 1, 179–198 Search PubMed.
- C. X. Lin, Y. G. Ke, Z. Li, J. H. Wang, Y. Liu and H. Yan, Mirror Image DNA Nanostructures for Chiral Supramolecular Assemblies, Nano Lett., 2009, 9, 433–436 Search PubMed.
- T. L. Mallette and M. R. Lakin, Protecting Heterochiral DNA Nanostructures against Exonuclease-Mediated Degradation, ACS Synth. Biol., 2022, 11, 2222–2228 Search PubMed.
- K. R. Kim, T. Lee, B. S. Kim and D. R. Ahn, Utilizing the bioorthogonal base-pairing system of L-DNA to design ideal DNA nanocarriers for enhanced delivery of nucleic acid cargos, Chem. Sci., 2014, 5, 1533–1537 RSC.
- H. B. D. Thai, K. R. Kim, K. T. Hong, T. Voitsitskyi, J. S. Lee, C. Mao and D. R. Ahn, Kidney-Targeted Cytosolic Delivery of siRNA Using a Small-Sized Mirror DNA Tetrahedron for Enhanced Potency, ACS Cent. Sci., 2020, 6, 2250–2258 CrossRef PubMed.
- J. H. Kang, K. R. Kim, H. Lee, D. R. Ahn and Y. T. Ko, In vitro and in vivo behavior of DNA tetrahedrons as tumor-targeting nanocarriers for doxorubicin delivery, Colloids Surf., B, 2017, 157, 424–431 CrossRef PubMed.
- A. Y. Lee, K. R. Kim, J. H. Yu and D. R. Ahn, L-DNA linear duplex: An efficient drug delivery carrier with a simple structure, J. Ind. Eng. Chem., 2019, 74, 187–192 Search PubMed.
- K. R. Kim, H. Y. Kim, Y. D. Lee, J. S. Ha, J. H. Kang, H. Jeong, D. Bang, Y. T. Ko, S. Kim, H. Lee and D. R. Ahn, Self-assembled mirror DNA nanostructures for tumor-specific delivery of anticancer drugs, J. Controlled Release, 2016, 243, 121–131 CrossRef PubMed.
- K. R. Kim, S. J. Kang, A. Y. Lee, D. Hwang, M. Park, H. Park, S. Kim, K. Hur, H. S. Chung, C. Mao and D. R. Ahn, Highly tumor-specific DNA nanostructures discovered by in vivo screening of a nucleic acid cage library and their applications in tumor-targeted drug delivery, Biomaterials, 2019, 195, 1–12 CrossRef CAS.
- K. R. Kim, D. Hwang, J. Kim, C. Y. Lee, W. Lee, D. S. Yoon, D. Shin, S. J. Min, I. C. Kwon, H. S. Chung and D. R. Ahn, Streptavidin-mirror DNA tetrahedron hybrid as a platform for intracellular and tumor delivery of enzymes, J. Controlled Release, 2018, 280, 1–10 CrossRef CAS.
- S. Tyagi and F. R. Kramer, Molecular beacons: probes that fluoresce upon hybridization, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS PubMed.
- G. Yang, T. Song, M. Wang, M. Y. Li, Q. Q. Su, Z. X. Xie, X. X. Xie, H. X. Zhang, Y. Feng, C. H. Wu, Y. Y. Liu and H. Yang, Recent Advancements in Nanosystem-Based Molecular Beacons for RNA Detection and Imaging, ACS Appl. Nano Mater., 2022, 5, 3065–3086 CrossRef CAS.
- N. Bidar, M. Amini, F. Oroojalian, B. Baradaran, S. S. Hosseini, M. A. Shahbazi, M. Hashemzaei, A. Mokhtarzadeh, M. R. Hamblin and M. de la Guardia, Molecular beacon strategies for sensing purpose, TrAC, Trends Anal. Chem., 2021, 134, 116143 CrossRef CAS.
- J. Zheng, R. Yang, M. Shi, C. Wu, X. Fang, Y. Li, J. Li and W. Tan, Rationally designed molecular beacons for bioanalytical and biomedical applications, Chem. Soc. Rev., 2015, 44, 3036–3055 RSC.
- S. X. Han, X. Jia, J. L. Ma and Q. Zhu, Molecular beacons: a novel optical diagnostic tool, Arch. Immunol. Ther. Exp., 2013, 61, 139–148 CrossRef.
- S. Mao, Y. Ying, R. Wu and A. K. Chen, Recent Advances in the Molecular Beacon Technology for Live-Cell Single-Molecule Imaging, iScience, 2020, 23, 101801 CrossRef PubMed.
- F. Lang, F. Ronicke and H. A. Wagenknecht, Cell-resistant wavelength-shifting molecular beacons made of L-DNA and a clickable L-configured uridine, Org. Biomol. Chem., 2024, 22, 4568–4573 RSC.
- G. Ke, C. Wang, Y. Ge, N. Zheng, Z. Zhu and C. J. Yang, L-DNA molecular beacon: a safe, stable, and accurate intracellular nano-thermometer for temperature sensing in living cells, J. Am. Chem. Soc., 2012, 134, 18908–18911 CrossRef PubMed.
- E. M. Euliano, A. N. Hardcastle, C. M. Victoriano, W. E. Gabella, F. R. Haselton and N. M. Adams, Multiplexed Adaptive RT-PCR Based on L-DNA Hybridization Monitoring for the Detection of Zika, Dengue, and Chikungunya RNA, Sci. Rep., 2019, 9, 11372 CrossRef.
- N. Spurlock, W. E. Gabella, D. J. Nelson, D. T. Evans, M. E. Pask, J. E. Schmitz and F. R. Haselton, Implementing L-DNA analogs as mirrors of PCR reactant hybridization state: theoretical and practical guidelines for PCR cycle control, Anal. Methods, 2024, 16, 2840–2849 RSC.
- H. Liang, S. Xie, L. Cui, C. Wu and X. Zhang, Designing a Biostable L-DNAzyme for Lead(II) Ion Detection in Practical Samples, Anal. Methods, 2016, 8, 7260–7264 RSC.
- L. Cui, R. Peng, T. Fu, X. Zhang, C. Wu, H. Chen, H. Liang, C. J. Yang and W. Tan, Biostable L-DNAzyme for Sensing of Metal Ions in Biological Systems, Anal. Chem., 2016, 88, 1850–1855 CrossRef PubMed.
- L. Yuan, T. Tian, Y. Chen, Z. Zhang and X. Zhou, An L-DNA G-quadruplex: application for peroxidase DNAzyme, Nucleosides, Nucleotides Nucleic Acids, 2013, 32, 589–598 CrossRef PubMed.
- R. C. Milton, S. C. Milton and S. B. Kent, Total chemical synthesis of a D-enzyme: the enantiomers of HIV-1 protease show reciprocal chiral substrate specificity [corrected], Science, 1992, 256, 1445–1448 CrossRef PubMed.
- Y. Kim, C. J. Yang and W. Tan, Superior structure stability and selectivity of hairpin nucleic acid probes with an L-DNA stem, Nucleic Acids Res., 2007, 35, 7279–7287 Search PubMed.
- M. Ogata, G. Hayashi, A. Ichiu and A. Okamoto, L-DNA-tagged fluorescence in situ hybridization for highly sensitive imaging of RNAs in single cells, Org. Biomol. Chem., 2020, 18, 8084–8088 RSC.
- W. Zhong and J. T. Sczepanski, Chimeric D/L-DNA Probes of Base Excision Repair Enable Real-Time Monitoring of Thymine DNA Glycosylase Activity in Live Cells, J. Am. Chem. Soc., 2023, 145, 17066–17074 CrossRef PubMed.
- R. Huang and P. K. Zhou, DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy, Signal Transduction Targeted Ther., 2021, 6, 254 Search PubMed.
- T. Visnes, M. Grube, B. M. F. Hanna, C. Benitez-Buelga, A. Cazares-Korner and T. Helleday, Targeting BER enzymes in cancer therapy, DNA Repair, 2018, 71, 118–126 CrossRef PubMed.
- A. Carusillo and C. Mussolino, DNA Damage: From Threat to Treatment, Cells, 2020, 9, 1665 CrossRef PubMed.
- Y. Xiao, A. A. Lubin, A. J. Heeger and K. W. Plaxco, Label-free electronic detection of thrombin in blood serum by using an aptamer-based sensor, Angew. Chem., Int. Ed. Engl., 2005, 44, 5456–5459 CrossRef PubMed.
- A. M. Downs and K. W. Plaxco, Real-Time, In Vivo Molecular Monitoring Using Electrochemical Aptamer Based Sensors: Opportunities and Challenges, ACS Sens., 2022, 7, 2823–2832 CrossRef PubMed.
- L. R. Schoukroun-Barnes, F. C. Macazo, B. Gutierrez, J. Lottermoser, J. Liu and R. J. White, Reagentless, Structure-Switching, Electrochemical Aptamer-Based Sensors, Annu. Rev. Anal. Chem., 2016, 9, 163–181 CrossRef PubMed.
- N. Arroyo-Currás, P. Dauphin-Ducharme, K. Scida and J. L. Chávez, From the beaker to the body: translational challenges for electrochemical, aptamer-based sensors, Anal. Methods, 2020, 12, 1288–1310 RSC.
- A. Shaver, N. Kundu, B. E. Young, P. A. Vieira, J. T. Sczepanski and N. Arroyo-Currás, Nuclease Hydrolysis Does Not Drive the Rapid Signaling Decay of DNA Aptamer-Based Electrochemical Sensors in Biological Fluids, Langmuir, 2022, 37, 5213–5221 CrossRef PubMed.
- L. Zhao and W. Lu, Mirror image proteins, Curr. Opin. Chem. Biol., 2014, 22, 56–61 CrossRef PubMed.
- K. Harrison, A. S. Mackay, L. Kambanis, J. W. C. Maxwell and R. J. Payne, Synthesis and applications of mirror-image proteins, Nat. Rev. Chem, 2023, 7, 383–404 CrossRef PubMed.
- Z. M. Wang, W. L. Xu, L. Liu and T. F. Zhu, A synthetic molecular system capable of mirror-image genetic replication and transcription, Nat. Chem., 2016, 8, 698–704 CrossRef PubMed.
- W. J. Jiang, B. C. Zhang, C. Y. Fan, M. Wang, J. X. Wang, Q. Deng, X. Y. Liu, J. Chen, J. S. Zheng, L. Liu and T. F. Zhu, Mirror-image polymerase chain reaction, Cell Discovery, 2017, 3, 17037 CrossRef CAS PubMed.
- M. Wang, W. J. Jiang, X. Y. Liu, J. X. Wang, B. C. Zhang, C. Y. Fan, L. Liu, G. Pena-Alcantara, J. J. Ling, J. Chen and T. F. Zhu, Mirror-Image Gene Transcription and Reverse Transcription, Chem, 2019, 5, 848–857 CAS.
- A. Pech, J. Achenbach, M. Jahnz, S. Schulzchen, F. Jarosch, F. Bordusa and S. Klussmann, A thermostable D-polymerase for mirror-image PCR, Nucleic Acids Res., 2017, 45, 3997–4005 CrossRef CAS PubMed.
- C. Fan, Q. Deng and T. F. Zhu, Bioorthogonal information storage in L-DNA with a high-fidelity mirror-image Pfu DNA polymerase, Nat. Biotechnol., 2021, 39, 1548–1555 CrossRef CAS.
- Y. Xu and T. F. Zhu, Mirror-image T7 transcription of chirally inverted ribosomal and functional RNAs, Science, 2022, 378, 405–412 CrossRef CAS PubMed.
- J. T. Sczepanski and G. F. Joyce, A cross-chiral RNA polymerase ribozyme, Nature, 2014, 515, 440–442 CrossRef CAS.
- G. A. L. Bare and G. F. Joyce, Cross-Chiral, RNA-Catalyzed Exponential Amplification of RNA, J. Am. Chem. Soc., 2021, 143, 19160–19166 CrossRef CAS PubMed.
- K. F. Tjhung, J. T. Sczepanski, E. R. Murtfeldt and G. F. Joyce, RNA-Catalyzed Cross-Chiral Polymerization of RNA, J. Am. Chem. Soc., 2020, 142, 15331–15339 CrossRef CAS PubMed.
- M. T. Weinstock, M. T. Jacobsen and M. S. Kay, Synthesis and folding of a mirror-image enzyme reveals ambidextrous chaperone activity, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11679–11684 CrossRef CAS PubMed.
- C. G. Peng and M. J. Damha, Polymerase-directed synthesis of 2′-deoxy-2′-fluoro-β-D-arabinonucleic acids, J. Am. Chem. Soc., 2007, 129, 5310–5311 CrossRef CAS PubMed.
- C. Samson, P. Legrand, M. Tekpinar, J. Rozenski, M. Abramov, P. Holliger, V. B. Pinheiro, P. Herdewijn and M. Delarue, Structural Studies of HNA Substrate Specificity in Mutants of an Archaeal DNA Polymerase Obtained by Directed Evolution, Biomolecules, 2020, 10, 1647 CrossRef CAS.
- J. K. Ichida, A. Horhota, K. Y. Zou, L. W. McLaughlin and J. W. Szostak, High fidelity TNA synthesis by therminator polymerase, Nucleic Acids Res., 2005, 33, 5219–5225 CrossRef CAS.
- N. Chim, C. H. Shi, S. P. Sau, A. Nikoomanzar and J. C. Chaput, Structural basis for TNA synthesis by an engineered TNA polymerase, Nat. Commun., 2017, 8, 1810 CrossRef.
- M. R. Dunn and J. C. Chaput, Reverse Transcription of Threose Nucleic Acid by a Naturally Occurring DNA Polymerase, ChemBioChem, 2016, 17, 1804–1808 CrossRef CAS PubMed.
- M. Hajjar, N. Chim, C. Liu, P. Herdewijn and J. C. Chaput, Crystallographic analysis of engineered polymerases synthesizing phosphonomethylthreosyl nucleic acid, Nucleic Acids Res., 2022, 50, 9663–9674 CrossRef CAS PubMed.
- J. An, J. Choi, D. Hwang, J. Park, C. W. Pemble, T. H. M. Duong, K. R. Kim, H. Ahn, H. S. Chung and D. R. Ahn, The crystal structure of a natural DNA polymerase complexed with mirror DNA, Chem. Commun., 2020, 56, 2186–2189 RSC.
- R. J. McLure, S. E. Radford and D. J. Brockwell, High-throughput directed evolution: a golden era for protein science, Trends Chem., 2022, 4, 378–391 CrossRef CAS.
- S. Manna, J. Truong and M. C. Hammond, Guanidine Biosensors Enable Comparison of Cellular Turn-on Kinetics of Riboswitch-Based Biosensor and Reporter, ACS Synth. Biol., 2021, 10, 566–578 CrossRef CAS PubMed.
- M. You, J. L. Litke and S. R. Jaffrey, Imaging metabolite dynamics in living cells using a Spinach-based riboswitch, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E2756–E2765 CAS.
- S. K. Dey, G. S. Filonov, A. O. Olarerin-George, B. T. Jackson, L. W. S. Finley and S. R. Jaffrey, Repurposing an adenine riboswitch into a fluorogenic imaging and sensing tag, Nat. Chem. Biol., 2022, 18, 180–190 CrossRef CAS.
- M. Schmidt, Xenobiology: a new form of life as the ultimate biosafety tool, BioEssays, 2010, 32, 322–331 CrossRef CAS PubMed.
- S. Shuman, DNA ligases: progress and prospects, J. Biol. Chem., 2009, 284, 17365–17369 Search PubMed.
- A. A. Gibriel and O. Adel, Advances in ligase chain reaction and ligation-based amplifications for genotyping assays: Detection and applications, Mutat. Res., Rev. Mutat. Res., 2017, 773, 66–90 CrossRef CAS PubMed.
- T. E. England, A. G. Bruce and O. C. Uhlenbeck, in Methods in Enzymology, ed. L. Grossman and K. Moldave, Academic Press, 1980, vol. 65, pp. 65–74 Search PubMed.
- R. W. Richardson and R. I. Gumport, Biotin and fluorescent labeling of RNA using T4 RNA ligase, Nucleic Acids Res., 1983, 11, 6167–6184 CrossRef CAS PubMed.
- S. de Kok, L. H. Stanton, T. Slaby, M. Durot, V. F. Holmes, K. G. Patel, D. Platt, E. B. Shapland, Z. Serber, J. Dean, J. D. Newman and S. S. Chandran, Rapid and reliable DNA assembly via ligase cycling reaction, ACS Synth. Biol., 2014, 3, 97–106 CrossRef CAS.
- W. J. Ansorge, Next-generation DNA sequencing techniques, New Biotechnol., 2009, 25, 195–203 CrossRef CAS.
- F. Müggenburg and S. Müller, Azide-modified Nucleosides as Versatile Tools for Bioorthogonal Labeling and Functionalization, Chem. Rec., 2022, 22, e202100322 CrossRef PubMed.
- A. H. El-Sagheer and T. Brown, Click Nucleic Acid Ligation: Applications in Biology and Nanotechnology, Acc. Chem. Res., 2012, 45, 1258–1267 CrossRef CAS.
- H. Maruyama, R. Oikawa, M. Hayakawa, S. Takamori, Y. Kimura, N. Abe, G. Tsuji, A. Matsuda, S. Shuto, Y. Ito and H. Abe, Chemical ligation of oligonucleotides using an electrophilic phosphorothioester, Nucleic Acids Res., 2017, 45, 7042–7048 CrossRef CAS.
- J. Weidmann, M. Schnölzer, P. E. Dawson and J. D. Hoheisel, Copying Life: Synthesis of an Enzymatically Active Mirror-Image DNA-Ligase Made of D-Amino Acids, Cell Chem. Biol., 2019, 26, 645–651 CrossRef CAS PubMed.
- C. H. Yu, A. M. Kabza and J. T. Sczepanski, Assembly of long L-RNA by native RNA ligation, Chem. Commun., 2021, 57, 10508–10511 RSC.
- B. Seelig and J. W. Szostak, Selection and evolution of enzymes from a partially randomized non-catalytic scaffold, Nature, 2007, 448, 828–831 CrossRef CAS PubMed.
- W. K. Johnston, P. J. Unrau, M. S. Lawrence, M. E. Glasner and D. P. Bartel, RNA-Catalyzed RNA Polymerization: Accurate and General RNA-Templated Primer Extension, Science, 2001, 292, 1319–1325 CrossRef CAS PubMed.
- R. J. Roberts, How restriction enzymes became the workhorses of molecular biology, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5905–5908 CrossRef CAS.
- Y. Peng, T. J. Soper and S. A. Woodson, in Bacterial Regulatory RNA Methods and Protocols, ed. K. C. Keiler, Humana Press, Totowa, NJ, 2012, vol. 905, pp. 213–224 Search PubMed.
- A. P. Boyle, S. Davis, H. P. Shulha, P. Meltzer, E. H. Margulies, Z. Weng, T. S. Furey and G. E. Crawford, High-Resolution Mapping and Characterization of Open Chromatin across the Genome, Cell, 2008, 132, 311–322 CrossRef CAS.
- T. Li, Y. Yang, H. Qi, W. Cui, L. Zhang, X. Fu, X. He, M. Liu, P.-f. Li and T. Yu, CRISPR/Cas9 therapeutics: progress and prospects, Signal Transduction Targeted Ther., 2023, 8, 36 CrossRef PubMed.
- A. M. Maxam and W. Gilbert, A new method for sequencing DNA, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 560–564 CrossRef CAS PubMed.
- X. Liu and T. F. Zhu, Sequencing Mirror-Image DNA Chemically, Cell Chem. Biol., 2018, 25, 1151–1156 CrossRef CAS PubMed.
- M. Liu, D. Chang and Y. Li, Discovery and Biosensing Applications of Diverse RNA-Cleaving DNAzymes, Acc. Chem. Res., 2017, 50, 2273–2283 CrossRef CAS PubMed.
- M. Hollenstein, DNA Catalysis: The Chemical Repertoire of DNAzymes, Molecules, 2015, 20, 20777–20804 CrossRef CAS PubMed.
- A. A. Vinogradov, E. D. Evans and B. L. Pentelute, Total synthesis and biochemical characterization of mirror image barnase, Chem. Sci., 2015, 6, 2997–3002 RSC.
- K. Sefah, D. Shangguan, X. Xiong, M. B. O'Donoghue and W. Tan, Development of DNA aptamers using Cell-SELEX, Nat. Protoc., 2010, 5, 1169–1185 CrossRef CAS PubMed.
- M. Kohlberger and G. Gadermaier, SELEX: Critical factors and optimization strategies for successful aptamer selection, Biotechnol. Appl. Biochem., 2022, 69, 1771–1792 CrossRef CAS PubMed.
- J. Chen, M. Y. Chen and T. F. Zhu, Directed evolution and selection of biostable L-DNA aptamers with a mirror-image DNA polymerase, Nat. Biotechnol., 2022, 40, 1601–1609 CrossRef CAS.
- W. Bode, I. Mayr, U. Baumann, R. Huber, S. R. Stone and J. Hofsteenge, The refined 1.9 A crystal structure of human alpha-thrombin: interaction with D-Phe-Pro-Arg chloromethylketone and significance of the Tyr-Pro-Pro-Trp insertion segment, EMBO J., 1989, 8, 3467–3475 CrossRef CAS.
- G. Muyzer and K. Smalla, Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology, Antonie van Leeuwenhoek, 1998, 73, 127–141 CrossRef CAS PubMed.
- J. Pugh, in Nanopore Sequencing: Methods and Protocols, ed. K. Arakawa, Springer US, New York, NY, 2023, vol. 2632, pp. 3–14 Search PubMed.
- M. Uhlen and S. R. Quake, Sequential sequencing by synthesis and the next-generation sequencing revolution, Trends Biotechnol., 2023, 41, 1565–1572 CrossRef CAS PubMed.
- R. Stoltenburg, N. Nikolaus and B. Strehlitz, Capture-SELEX: Selection of DNA Aptamers for Aminoglycoside Antibiotics, J. Anal. Methods Chem., 2012, 2012, 415697 Search PubMed.
- M. Jarczewska, Ł. Górski and E. Malinowska, Electrochemical aptamer-based biosensors as potential tools for clinical diagnostics, Anal. Methods, 2016, 8, 3861–3877 RSC.
- M. Dudek and J. Trylska, Molecular Dynamics Simulations of L-RNA Involving Homo- and Heterochiral Complexes, J. Chem. Theory Comput., 2017, 13, 1244–1253 CrossRef CAS.
- G. H. Zerze, F. H. Stillinger and P. G. Debenedetti, Thermodynamics of DNA Hybridization from Atomistic Simulations, J. Phys. Chem. B, 2021, 125, 771–779 CrossRef CAS.
- A. M. Kabza, B. E. Young and J. T. Sczepanski, Heterochiral DNA Strand-Displacement Circuits, J. Am. Chem. Soc., 2017, 139, 17715–17718 CrossRef CAS PubMed.
- W. Zhong and J. T. Sczepanski, Direct Comparison of D-DNA and L-DNA Strand-Displacement Reactions in Living Mammalian Cells, ACS Synth. Biol., 2021, 10, 209–212 CrossRef CAS PubMed.
- W. Zhong and J. T. Sczepanski, A Mirror Image Fluorogenic Aptamer Sensor for Live-Cell Imaging of MicroRNAs, ACS Sens., 2019, 4, 566–570 CrossRef CAS PubMed.
- B. E. Young and J. T. Sczepanski, Heterochiral DNA Strand-Displacement Based on Chimeric D/L-Oligonucleotides, ACS Synth. Biol., 2019, 8, 2756–2759 CrossRef CAS.
- T. L. Mallette, M. N. Stojanovic, D. Stefanovic and M. R. Lakin, Robust Heterochiral Strand Displacement Using Leakless Translators, ACS Synth. Biol., 2020, 9, 1907–1910 CrossRef CAS PubMed.
- W. Shen, C. L. De Hoyos, M. T. Migawa, T. A. Vickers, H. Sun, A. Low, T. A. Bell III, M. Rahdar, S. Mukhopadhyay, C. E. Hart, M. Bell, S. Riney, S. F. Murray, S. Greenlee, R. M. Crooke, X. H. Liang, P. P. Seth and S. T. Crooke, Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index, Nat. Biotechnol., 2019, 37, 640–650 CrossRef CAS.
- S. T. Crooke, T. A. Vickers and X. H. Liang, Phosphorothioate modified oligonucleotide-protein interactions, Nucleic Acids Res., 2020, 48, 5235–5253 CrossRef.
- F. Wang, E. Calvo-Roitberg, J. M. Rembetsy-Brown, M. Fang, J. Sousa, Z. J. Kartje, P. M. Krishnamurthy, J. Lee, M. R. Green, A. A. Pai and J. K. Watts, G-rich motifs within phosphorothioate-based antisense oligonucleotides (ASOs) drive activation of FXN expression through indirect effects, Nucleic Acids Res., 2022, 50, 12657–12673 CrossRef CAS.
- A. J. Pollak, L. Zhao and S. T. Crooke, Systematic Analysis of Chemical Modifications of Phosphorothioate Antisense Oligonucleotides that Modulate Their Innate Immune Response, Nucleic Acid Ther., 2023, 33, 95–107 CrossRef CAS PubMed.
- C. H. Yu and J. T. Sczepanski, The influence of chirality on the behavior of oligonucleotides inside cells: revealing the potent cytotoxicity of G-rich L-RNA, Chem. Sci., 2023, 14, 1145–1154 RSC.
- K. Zhang, Q. Nie, T. Chi-Kong Lau and C. Kit Kwok, Rational Design of L-RNA Aptamer-Peptide Conjugate for Efficient Cell Uptake and G-quadruplex-Mediated Gene Control, Angew. Chem., Int. Ed. Engl., 2024, 63, e202310798 CrossRef CAS PubMed.
- T. L. Mallette, D. S. Lidke and M. R. Lakin, Heterochiral modifications enhance robustness and function of DNA in living human cells, ChemBioChem, 2024, 25, e202300755 CrossRef CAS PubMed.
- X. N. Feng, Y. X. Cui, J. Zhang, A. N. Tang, H. B. Mao and D. M. Kong, Chiral Interaction Is a Decisive Factor To Replace D-DNA with L-DNA Aptamers, Anal. Chem., 2020, 92, 6470–6477 CrossRef CAS PubMed.
- G. Chatterjee, Y. J. Chen and G. Seelig, Nucleic Acid Strand Displacement with Synthetic mRNA Inputs in Living Mammalian Cells, ACS Synth. Biol., 2018, 7, 2737–2741 CrossRef CAS.
- J. C. Lai, B. D. Brown, A. M. Voskresenskiy, S. Vonhoff, S. Klussman, W. H. Tan, M. Colombini, R. Weeratna, P. Miller, L. Benimetskaya and C. A. Stein, Comparison of D-G3139 and its enantiomer L-G3139 in melanoma cells demonstrates minimal but dramatic chiral dependency, Mol. Ther., 2007, 15, 270–278 CrossRef CAS PubMed.
- C. E. Deckard and J. T. Sczepanski, Polycomb repressive complex 2 binds RNA irrespective of stereochemistry, Chem. Commun., 2018, 54, 12061–12064 RSC.
- K. A. Fitzgerald and J. C. Kagan, Toll-like Receptors and the Control of Immunity, Cell, 2020, 180, 1044–1066 CrossRef CAS.
- N. Kayraklioglu, B. Horuluoglu and D. M. Klinman, in DNA Vaccines, Humana, New York, NY, 2021, vol. 2197, pp. 51–85 Search PubMed.
- H. C. Harsha, H. Molina and A. Pandey, Quantitative proteomics using stable isotope labeling with amino acids in cell culture, Nat. Protoc., 2008, 3, 505–516 CrossRef CAS.
- A. Hanswillemenke, D. T. Hofacker, M. Sorgenfrei, C. Fruhner, M. Franz-Wachtel, D. Schwarzer, B. Macek and T. Stafforst, Profiling the interactome of oligonucleotide drugs by proximity biotinylation, Nat. Chem. Biol., 2024, 20, 555–565 CrossRef CAS PubMed.
- G. Mittler, F. Butter and M. Mann, A SILAC-based DNA protein interaction screen that identifies candidate binding proteins to functional DNA elements, Genome Res., 2009, 19, 284–293 CrossRef CAS PubMed.
- G. W. Ashley, Modeling, Synthesis, and Hybridization Properties of (L)-Ribonucleic Acid, J. Am. Chem. Soc., 1992, 114, 9731–9736 CrossRef CAS.
- A. Garbesi, M. L. Capobianco, F. P. Colonna, M. Maflini, D. Niccoiai and L. Tondelli, Chirally-Modifiedoligonucleotides and the Control of Gene Expression. The Case of L-DNAS And-RNAS, Nucleosides Nucleotides, 1998, 17, 1275–1287 CrossRef CAS.
- D. D'Alonzo, A. Guaragna and G. Palumbo, Exploring the role of chirality in nucleic acid recognition, Chem. Biodiversity, 2011, 8, 373–413 CrossRef PubMed.
- M. Hafner, M. Katsantoni, T. Köster, J. Marks, J. Mukherjee, D. Staiger, J. Ule and M. Zavolan, CLIP and complementary methods, Nat. Rev. Methods Primers, 2021, 1, 20 CrossRef CAS.
- T. Shmushkovich, K. R. Monopoli, D. Homsy, D. Leyfer, M. Betancur-Boissel, A. Khvorova and A. D. Wolfson, Functional features defining the efficacy of cholesterol-conjugated, self-deliverable, chemically modified siRNAs, Nucleic Acids Res., 2018, 46, 10905–10916 CrossRef CAS.
- C. Maasch, A. Vater, K. Buchner, W. G. Purschke, D. Eulberg, S. Vonhoff and S. Klussmann, Polyetheylenimine-polyplexes of Spiegelmer NOX-A50 directed against intracellular high mobility group protein A1 (HMGA1) reduce tumor growth in vivo, J. Biol. Chem., 2010, 285, 40012–40018 CrossRef CAS PubMed.
- L. R. Baden, H. M. El Sahly, B. Essink, K. Kotloff, S. Frey, R. Novak, D. Diemert, S. A. Spector, N. Rouphael, C. B. Creech, J. McGettigan, S. Khetan, N. Segall, J. Solis, A. Brosz, C. Fierro, H. Schwartz, K. Neuzil, L. Corey, P. Gilbert, H. Janes, D. Follmann, M. Marovich, J. Mascola, L. Polakowski, J. Ledgerwood, B. S. Graham, H. Bennett, R. Pajon, C. Knightly, B. Leav, W. Deng, H. Zhou, S. Han, M. Ivarsson, J. Miller, T. Zaks and C. S. Group, Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine, N. Engl. J. Med., 2021, 384, 403–416 CrossRef CAS.
- F. P. Polack, S. J. Thomas, N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, J. L. Perez, G. Perez Marc, E. D. Moreira, C. Zerbini, R. Bailey, K. A. Swanson, S. Roychoudhury, K. Koury, P. Li, W. V. Kalina, D. Cooper, R. W. Frenck Jr, L. L. Hammitt, O. Tureci, H. Nell, A. Schaefer, S. Unal, D. B. Tresnan, S. Mather, P. R. Dormitzer, U. Sahin, K. U. Jansen, W. C. Gruber and C. C. T. Group, Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine, N. Engl. J. Med., 2020, 383, 2603–2615 CrossRef CAS PubMed.
- TME Pharma Pipeline, https://www.tmepharma.com/index.php?option=com_content%26view=article%26id=15%26Itemid=503, (accessed July, 2024).
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |