
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioisomers containing triarylboron-based motifs as multi-functional photoluminescent materials: from dual-mode delayed emission to pH-switchable room-temperature phosphorescence†
Ramar
Arumugam‡
 a,
Akkarakkaran Thayyil
Muhammed Munthasir‡
b,
Ramkumar
Kannan
a,
Dipanjan
Banerjee
c,
Pagidi
Sudhakar
a,
Venugopal Rao
Soma
c,
Pakkirisamy
Thilagar
*b and
Vadapalli
Chandrasekhar
*a
a,
Akkarakkaran Thayyil
Muhammed Munthasir‡
b,
Ramkumar
Kannan
a,
Dipanjan
Banerjee
c,
Pagidi
Sudhakar
a,
Venugopal Rao
Soma
c,
Pakkirisamy
Thilagar
*b and
Vadapalli
Chandrasekhar
*a
aTata Institute of Fundamental Research, Hyderabad 500046, India. E-mail: vc@tifrh.res.in
bDepartment of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore 560012, India. E-mail: thilagar@iisc.ac.in
cSchool of Physics and DIA-CoE (ACRHEM), University of Hyderabad, Hyderabad 500046, India
First published on 7th October 2024
Abstract
Triarylboron compounds have been established as promising candidates in optoelectronic applications. However, realizing multi-functional properties in triaryl boron-based materials remains challenging. Herein, we present two regioisomers, 1 and 2, designed judiciously by connecting a dimethylamino donor and a dimesitylboryl acceptor at 1,4 and 2,6-positions of the naphthalene spacer, respectively. Both compounds 1 and 2 display simultaneous, delayed fluorescence and persistent room-temperature phosphorescence (580 nm, τav = 168 ms, Φ = 76% for 1; 550 nm, τav = 129 ms, Φ = 88% for 2 in 1 wt% PMMA), with the delayed fluorescence bands being sensitive to doping concentration (in PMMA). Notably, compound 1 in 1 wt% PMMA films demonstrates a reversibly switchable single-molecule phosphorescence from orange (580 nm) to green (λPh = 550 nm, τav = 42 ms) in response to pH, which can be utilized for anti-counterfeiting applications. These results were further corroborated by studying the respective cationic salts 1-OTF and 2-OTF. Moreover, 1 and 2 exhibited blue-shifted fluorescence in response to mechanical pressure. Compound 2 also showed three-photon (σ3P) absorption properties which were better compared to those of compound 1.
Introduction
Main group element-containing organic materials have received a lot of attention due to their promising photophysical properties1–10 and potential applications in organic light-emitting diodes (OLEDs),10–13 organic field effect transistors (OFETs),14–18 optical switches,19–22 organic solar cells,23–26 bio-imaging27–29 and sensors.30–34 In recent years, research on organic materials that show thermally activated delayed fluorescence (TADF) has been rapidly progressing as it offers 100% internal quantum efficiency which has led to the development of high-performance metal-free OLEDs.35–38 TADF occurs from the excited singlet state (S1) prior to the intersystem crossing (ISC) and reverse intersystem crossing (rISC) processes between excited singlet S1 and triplet states (Tn, n ≥ 1). Another delayed emission that involves triplet states is phosphorescence/afterglow in which radiative decay occurs from excited triplet (T1) to the singlet ground state (S0).39–41 Numerous ultralong room temperature phosphorescence (RTP) organic materials were reported by employing different strategies such as hydrogen bonding, incorporation of aromatic carbonyls, H-aggregation, doping in polymer matrices, external heavy atom effects, crystal packing, and incorporating heteroatoms with lone pairs of electrons.42–47 Such strategies were found to significantly suppress the molecular vibrations and enhance the phosphorescence efficiency from the triple state. Besides, heavy atoms increase the spin–orbit coupling and increase the ISC rate from S1 to Tn. Among different strategies, doping chromophores in rigid polymer matrices has been extensively used because the polymer matrix can not only suppress/arrest the molecular vibrations significantly and disperse the chromophores but also reduce the triplet oxygen diffusion which is detrimental to the triplet state. RTP materials have found applications in sensors, anti-counterfeiting, information storage, and imaging.31,48–51Among a wide variety of organic RTP materials, triarylboron (TAB) compounds offer intriguing bright fluorescence because of the presence of an empty p-orbital on boron which allows enhanced π-conjugation. In spite of this promise, studies on phosphorescence in triarylboron compounds have been relatively sparse.45,52–57 Recently, Marder and co-workers reported TAB compounds that showed efficient RTP resulting from an ISC from (σ, Bp) → (π, Bp) and C–H⋯C interactions in the crystalline state.56 Xu et al. reported a series of molecules with different Lewis acidity and basicity that showed tunable ultralong organic phosphorescence.58 Zhao and co-workers reported efficient room-temperature phosphorescence (RTP) in the amorphous state from boron-containing biphenyl derivatives owing to intramolecular π–π and B⋯N electronic interactions and the presence of bromine atoms.45 Thilagar et al. reported the impact of branching on the RTP and lifetime of TAB-based compounds.59 Very recently, Ding et al. reported TAB containing donor (D)–π–acceptor (A) materials that showed time-dependent photo-activated room-temperature phosphorescence.60
A subset of such interesting photoluminescent molecules exhibited dual delayed emissions such as TADF and RTP. For instance, Wang et al. reported a carbazole-type crystalline compound with persistent RTP and TADF emissions or time-dependent afterglow color.61 Yang et al. reported an indolocarbazole-doped polyvinyl alcohol (PVA) film which showed TADF and RTP emissions and then used it as temperature-sensitive security ink for multilevel anti-counterfeiting and information encryption.62 Recently, Thilagar and co-workers reported TADF and single molecular RTP in a BN-based molecular system.54 Very recently, Wagner and co-workers reported laterally substituted 9,10-dimesityl-9,10-diboraanthracenes that showed both TADF and RTP.63 Although dual-mode delayed emission finds fundamental importance and useful applications like anti-counterfeiting and information encryption, however, achieving both TADF and single molecular RTP in a single emitter is a challenging task.
As part of our ongoing exploration of TAB-based RTP materials, we were interested in tuning the excited state properties by molecular engineering, i.e., by decorating the boron acceptor and amino donor on the different positions of the naphthalene spacer. Accordingly, we have designed and synthesized two regioisomers, 1 and 2, containing dimethylamino donor and dimesitylboryl acceptor at 1,4 and 2,6-positions of the naphthalene spacer, respectively. The boron–nitrogen system is well known for its delayed fluorescence behavior, while naphthalene derivatives are recognized for their inherent phosphorescence. By incorporating both systems into a single unit, dual emission can be achieved, utilizing the unique features of each. Additionally, the two distinct transition dipole moments in naphthalene provide directionality to the substituents, resulting in distinct electronic properties based on the substitution position. As anticipated, these regioisomers exhibited dual-mode delayed fluorescence and room-temperature phosphorescence (RTP) emission, along with multi-stimuli responsive optical properties (mechanical pressure, acid, and base). These properties were utilized to develop a rewritable ink (1 wt% PMMA) for anti-counterfeiting applications. Furthermore, compound 2 exhibited superior three-photon non-linear optical properties compared to 1. All these intriguing results are discussed in this manuscript.
Results and discussion
Synthesis and characterization
The precursors 4-bromo-N,N-dimethylnaphthalen-1-amine (P1)64 and 6-bromo-N,N-dimethylnaphthalen-2-amine (P2)65 were synthesized following a modified literature protocol (ESI Scheme S1†). Lithiation of P1 and P2 using n-butyllithium in diethyl ether at −78 °C, followed by trapping the C-centered carbanion with dimesitylfluoroborane, afforded target compounds 1 and 2 in 65% and 60% yields, respectively (Scheme 1). These compounds were characterized by 1H, 13C and 11B NMR and high-resolution mass spectrometry (ESI Fig. S1–S8†). The 1H NMR spectra of both compounds show two singlets corresponding to ortho (12H) and para (6H) methyl protons. This indicates that the two mesityl groups are in the same chemical environment and are not influenced by the position of the naphthylamine unit. In contrast, the substitution position significantly affects the 1H resonances of the naphthalene spacer. The 11B resonance in 1 is deshielded (70.2 ppm) compared to 2 (68.5 ppm). The variation of 11B chemical shifts in 1 and 2 suggests that different substitution on the naphthyl amine motif has significant electronic influence and thus, these molecules were expected to show distinct optical features. | ||
| Scheme 1 Synthesis of 1 and 2. | ||
Single crystal X-ray diffraction studies
Single crystals of 1 and 2 suitable for X-ray diffraction studies were obtained from a slow evaporation of solutions of these compounds in dichloromethane and n-hexane solvent mixture. Compound 1 crystallizes in an orthorhombic lattice with a P212121 space group, while 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (ESI Table S1†). The molecular structures of 1 and 2 are given in Fig. 1a and b. The boron center in 1 and 2 adopted a trigonal planar arrangement with sums of total bond angles being 360°, which is consistent with the results observed in 4-(dimesitylboraneyl)-N,N-dimethylaniline (R1)66 (ESI Table S2 and Fig. S8a†). The nitrogen center in 2 adopted a trigonal planar arrangement. In contrast, the nitrogen center in 1 adopted a pyramidal geometry with sums of total bond angles being ∼343° which can be attributed to the steric repulsions between the methyl groups on N and adjacent hydrogen atoms of the naphthalene ring. The B–C(Nap) bond length is nearly the same in 1 and 2; however, the N–C(Nap) bond length is longer in the case of 1 (1.409(5) Å) compared to 2 (1.371(2) Å). These bond length parameters suggest that the D–A interaction in 1 is expected to be weaker than in 2. The dihedral angle (θBN) between the B3C and N3C planes in 2 (32.2°) is smaller than that in 1 (57.4°) (ESI Table S2 and Fig. S9–S11†). Similarly, the dihedral angle between NMe2 and naphthyl planes in 2 (3.2°) is lower than that in 1 (61.1°). Compound 1 showed weak C–H⋯π interactions between the mesityl proton of one molecule and the mesityl-π cloud of the adjacent molecule (3.140 Å) (ESI Fig. S12†). In contrast, 2 showed dense packing due to its nearly planar arrangement of NMe2 and the naphthalene ring, enabling two molecules to stack in an antiparallel fashion, resulting in N⋯π interaction (3.440 Å) in addition to C–H⋯π interactions (3.069 Å and 3.210 Å) (ESI Fig. S13†). These interactions lead to a dense 3D packing in 2 compared to 1, which can directly affect their photophysical properties in the solid state, as discussed in the following sections.
(ESI Table S1†). The molecular structures of 1 and 2 are given in Fig. 1a and b. The boron center in 1 and 2 adopted a trigonal planar arrangement with sums of total bond angles being 360°, which is consistent with the results observed in 4-(dimesitylboraneyl)-N,N-dimethylaniline (R1)66 (ESI Table S2 and Fig. S8a†). The nitrogen center in 2 adopted a trigonal planar arrangement. In contrast, the nitrogen center in 1 adopted a pyramidal geometry with sums of total bond angles being ∼343° which can be attributed to the steric repulsions between the methyl groups on N and adjacent hydrogen atoms of the naphthalene ring. The B–C(Nap) bond length is nearly the same in 1 and 2; however, the N–C(Nap) bond length is longer in the case of 1 (1.409(5) Å) compared to 2 (1.371(2) Å). These bond length parameters suggest that the D–A interaction in 1 is expected to be weaker than in 2. The dihedral angle (θBN) between the B3C and N3C planes in 2 (32.2°) is smaller than that in 1 (57.4°) (ESI Table S2 and Fig. S9–S11†). Similarly, the dihedral angle between NMe2 and naphthyl planes in 2 (3.2°) is lower than that in 1 (61.1°). Compound 1 showed weak C–H⋯π interactions between the mesityl proton of one molecule and the mesityl-π cloud of the adjacent molecule (3.140 Å) (ESI Fig. S12†). In contrast, 2 showed dense packing due to its nearly planar arrangement of NMe2 and the naphthalene ring, enabling two molecules to stack in an antiparallel fashion, resulting in N⋯π interaction (3.440 Å) in addition to C–H⋯π interactions (3.069 Å and 3.210 Å) (ESI Fig. S13†). These interactions lead to a dense 3D packing in 2 compared to 1, which can directly affect their photophysical properties in the solid state, as discussed in the following sections.
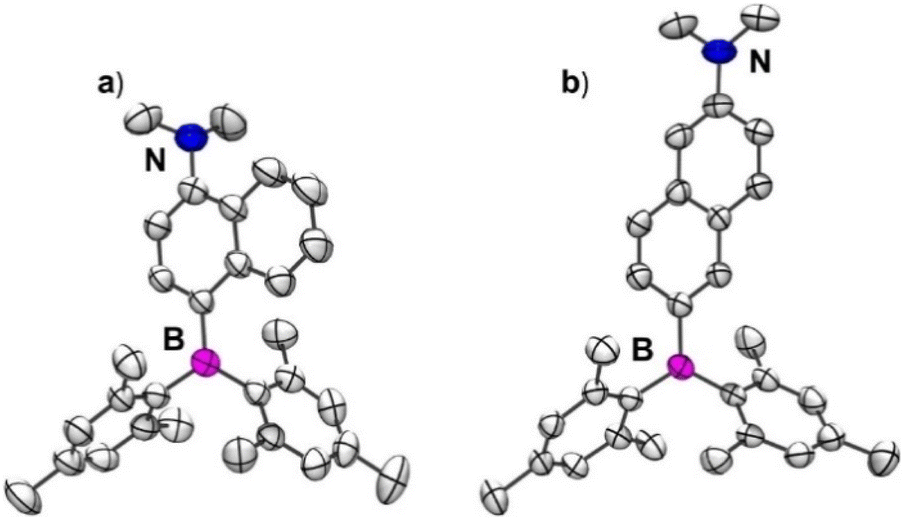 | ||
| Fig. 1 ORTEP representation of the molecular structure of (a) 1 and (b) 2 with a 50% probability of thermal ellipsoids. Atom color codes: carbon (gray), nitrogen (blue), boron (magenta). All the hydrogen atoms are removed for clarity. | ||
Photophysical and theoretical studies
Solution state absorption and emission studies
The absorption spectra of 1 and 2 in n-hexane revealed two absorption bands (Fig. 2a). Compound 1 showed absorption bands at 370 and 320 nm, while 2 exhibited bands at 380 and 297 nm, along with a shoulder at 323 nm. To understand the electronic nature of these bands, absorption spectra were recorded in solvents of different polarities (ESI Fig. S14†). The results obtained revealed that the high-energy bands were nearly insensitive to solvent polarity, while the broad low-energy bands showed bathochromic shifts in polar solvents. This indicated that the high-energy bands arise from local electronic transitions within the D/A or spacer, while the low-energy bands result from an intramolecular charge transfer (ICT) transition from the NMe2 donor to the boryl acceptor, which is sensitive to the solvent polarity. The redshift (10 nm) of the low-energy band in 2 compared to 1 can be attributed to the extended conjugation and strong donor–acceptor interaction in the former compared to the latter.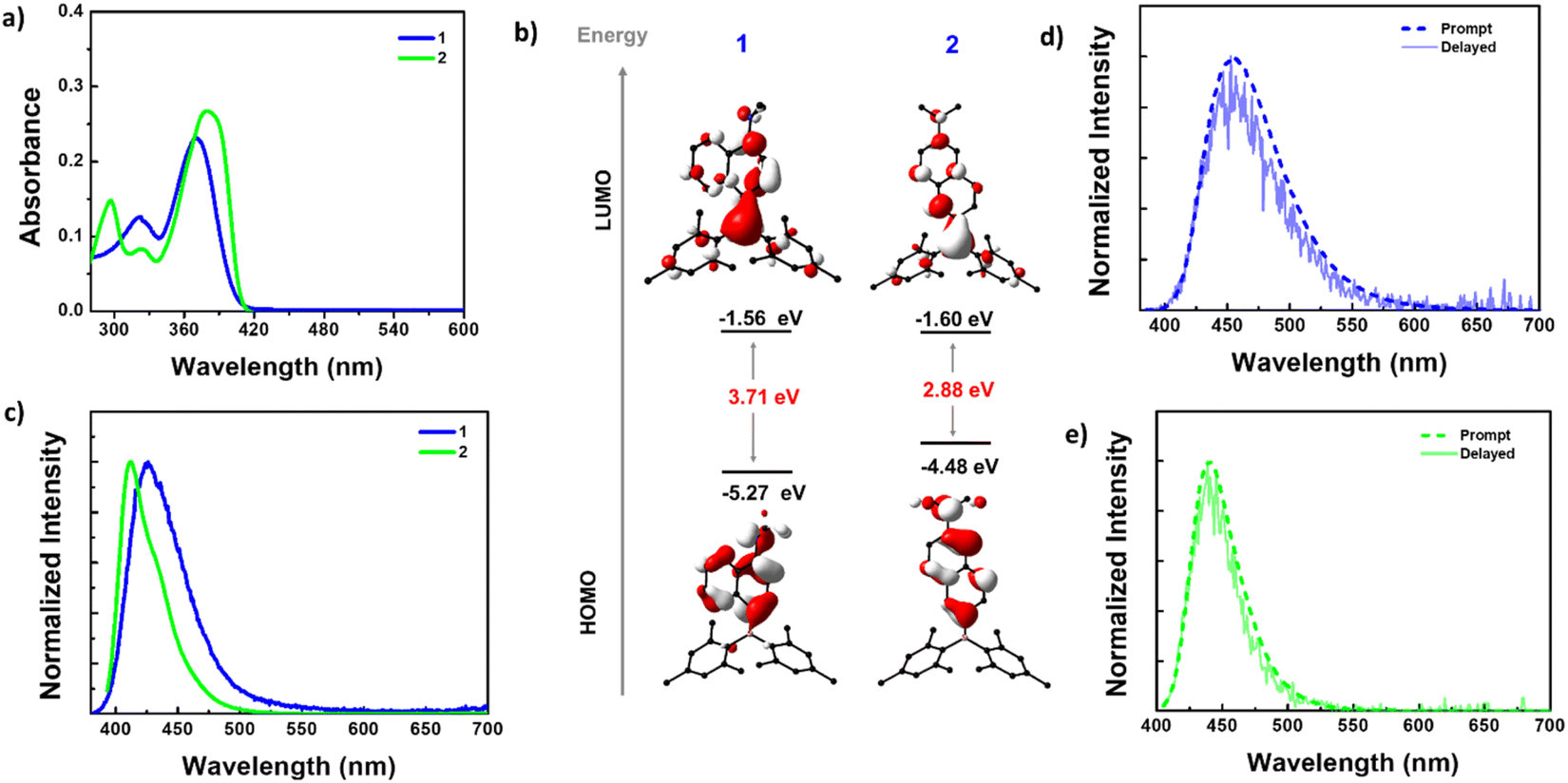 | ||
| Fig. 2 (a) UV-visible absorption spectra for 1 and 2 in hexane (conc. 10−5 M). (b) Frontier molecular orbitals (iso value = 0.04) generated from the ground state [S0] geometries using the B3LYP/6-31G(d,p) level of theory. (c) Steady-state photoluminescence (PL) spectra for 1 and 2 in hexane (conc. 10−5 M) at λex = 380 nm. Prompt and delayed (20 μs delay) PL spectra for (d) 1 and (e) 2 in degassed toluene (conc. 10−5 M) at λex = 380 nm. | ||
Furthermore, the ground state geometry of 1 and 2 was optimized using density functional theory (DFT), and vertical transitions were simulated using time-dependent DFT (TD-DFT) calculations to shed more light on the nature of absorption bands (Fig. 2b, ESI Fig. S15–S17, Tables S4 and S5†). The ground state-optimized geometries closely resembled the molecular structures obtained by X-ray analysis (ESI Table S3†). The highest occupied molecular orbitals (HOMOs) of both 1 and 2 are localized on the NMe2 donor and naphthalene spacer, while the lowest unoccupied molecular orbitals (LUMOs) are distributed on the entire molecular skeleton with a significant contribution from the boron center. The HOMO energy level of 1 (−5.27 eV) is more stabilized than that of 2 (−4.48 eV). The LUMO energy level of 2 (−1.60 eV) is slightly stabilized compared with that of 1 (−1.56 eV). Consequently, a lower band gap was observed for 2 (2.88 eV) than for 1 (3.71 eV). These bandgap trends were reflected in their experimental absorption spectra.
In n-hexane solution, 1 exhibited a single broad emission with a maximum at 423 nm (FWHM = 52 nm), while 2 displayed a sharper emission at 412 nm (FWHM = 37 nm) with a shoulder at 430 nm (Fig. 2c). Unlike the absorption spectra, the PL maxima of 2 are blue-shifted compared to 1, which can be attributed to the larger structural reorganization of 1 in the excited state. The PL spectra of both the compounds showed a bathochromic shift in polar solvents indicating that the emission originates from a polar charge transfer state (ESI Fig. S18–S20†). From time-resolved PL decay, compound 2 showed a mono-exponential decay with a lifetime (τ) of 2.94 ns @ 412 nm while 1 displayed bi-exponential decay with an average lifetime (τav) of 0.73 ns @ 423 nm (ESI Table S6†). The PLQY (Φ) of these compounds in different solvents is in line with the intensity variation [ΦPL = 6.90% for 1 and 71.60% for 2 in hexane, 33.25% for 1 and 86.74% for 2 in DMSO] (ESI Table S6†). The excitation spectra recorded for both compounds reproduced the absorption spectra in respective solvents, indicating that the same species are present in the ground and excited states (ESI Fig. S21†).
We have compared the absorption and emission features of the precursors (P1 and P2) with those of compounds 1 and 2 to understand the changes in their absorption and emission features upon the covalent linking of boryl units to the precursors (ESI Fig. S22†). Interestingly, the emission and absorption characteristics of P1 differ significantly from those of 1, while P2 has almost identical features to 2, in dilute hexane solutions. For P1, the absorption spectrum shows a single broadband around 320 nm, similar to the higher energy band in compound 1 (ESI Fig. S22a†). These findings indicate that the higher energy absorption band corresponds to a local transition within the naphthylamine unit, while the lower energy band arises due to CT from the donor naphthylamine to the boryl acceptor. These differences are also evident in the emission spectra, where compound 1 shows a redshift (λem = 423 nm) compared to P1 (λem = 400 nm). In contrast, the changes in 2 compared to P2 are minimal. Both P2 and compound 2 exhibit two sets of absorption bands, with the higher energy bands being identical. However, the lower energy band and emission band in P2 are blue-shifted by 10 nm compared to 2 (ESI Fig. S22b†). These results are further supported by the computational results; the band gap found for P1 (4.29 eV) and 1 (3.71 eV) differs largely, whereas P2 (2.72 eV) and 2 (2.88 eV) have very minimal changes (ESI Fig. S22c†).
To validate the geometrical reorganization in the excited state, the PL spectra of these compounds were recorded in various viscous solvents, as viscosity is a key factor influencing molecular motion. PL studies were performed in n-hexane (non-polar and non-viscous) and paraffin (non-polar and viscous), methanol (polar and non-viscous), and ethylene glycol (polar and viscous) (ESI Fig. S23†). It is evident from the spectra that there is a significant increase in PL intensity in paraffin over n-hexane in the case of 1. However, this increment is very minimal in the case of compound 2. Conversely, upon changing solvents from methanol to ethylene glycol, the emission intensity of compound 1 is reduced, while compound 2 shows a slight rise in intensity. These results indicate that considerable structural reorganization happens for 1 in the excited state in comparison to 2.
Furthermore, the excited singlet state (S1) geometry of 1 and 2 was optimized and the structural parameters were compared with the ground state structures (ESI Table S3†). The results suggest that the molecules undergo significant geometrical changes, which can be understood by monitoring the change in the dihedral angle between D–A/A–S/D–S (S = spacer) and bond parameters as tabulated in ESI Table S3.† Compound 1 exhibits a highly twisted geometry in the ground state compared to 2. Upon excitation, both molecules undergo twisting around the B–C bond, creating a greater twist angle between the acceptor and the naphthalene–NMe2 (S–D) group. These significant changes in compound 2 result in a decrease in the co-planarity that the molecule has in the ground state, leading to destabilization of the state and resulting in a slightly blue-shifted emission. These molecular motions are arrested in the viscous solvent, resulting in an enhancement of intensity in the case of paraffin compared to hexane. In hexane, the molecule can dissipate its excited energy non-radiatively due to intramolecular motion.
The D–π–A systems are well known for their delayed luminescence (DF) behavior, so we conducted delayed luminescence studies of 1 and 2 in the solution state. Time-gated spectra (delayed time = 20 μs) and lifetimes were collected for these compounds in degassed toluene (Fig. 2d and e). As anticipated, the prompt fluorescence (PF) spectra of both compounds perfectly matched the delayed spectra, indicating the delayed fluorescence nature of these emissions. Furthermore, the lifetime of the DF is in the microsecond range. These longer lifetimes of the DF can be attributed to the involvement of the triplet state, as observed in many DF systems (ESI Fig. S24b, d, S25b and d†).
To confirm the involvement of the triplet excited state, the PL spectra were collected under different atmospheres (ESI Fig. S24 and S25†). The results indicate that both the prompt and delayed spectra exhibit a drastic decrease in intensity in the presence of oxygen, which is attributed to the quenching of the triplet state by oxygen. Furthermore, prompt lifetime and total photoluminescence quantum yield (PLQY) also reduced drastically under oxygen-rich conditions [N2ΦPL = 47.99% for 1 and 93.48% for 2, O2ΦPL 24.04% for 1, and 53.34% for 2] (ESI Table S7†). These findings support the delayed fluorescence characteristics of these compounds.
To comprehensively understand the impact of the positions of D and A on the naphthalene spacer, we thoroughly investigated emission parameters such as lifetime, quantum yield, and various radiative and non-radiative decay rates for 1 and 2 (Table 1). The average fluorescence lifetime for compound 2 [τav = 3.52 ns] is slightly lower compared to that for compound 1 [τav = 4.57 ns], while the delayed fluorescence lifetime is almost the same (Table 1). Interestingly, the photoluminescence quantum yield for compound 2 (93.48%) is significantly higher compared to that for compound 1 (47.99%). The higher Φ for 2 is due to the smaller non-radiative decay rate constant of 10.89 × 106 s−1 than that of 1 (56.82 × 106 s−1) (Table 1).
| Compound | λ ex [nm] | λ em [nm] | τ PF [ns] | τ DF [μs] | ϕ total [%] | ϕ PF [%] | ϕ DF [%] | k PF [107 s−1] | k DF [104 s−1] | k ICS [107 s−1] | k rICS [104 s−1] | k nr(S1) [106 s−1] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a τ PF and τDF are the average fluorescence and delayed fluorescence lifetime. ϕtotal is total Φ, ϕPF, and ϕDF for prompt and delayed fluorescence quantum yields, kPF, kDF, kICS, krISCknr(S1) are the fluorescence decay, delayed fluorescence decay, intersystem crossing, reverse intersystem crossing, non-radiative decay rate constants respectively. λex, and λem, are the excitation and emission wavelengths respectively. | ||||||||||||
| 1 | 375 | 455 | 4.57 | 5.23 | 47.99 | 24.04 | 23.95 | 5.26 | 4.58 | 10.94 | 38.09 | 56.82 |
| 2 | 395 | 440 | 3.52 | 5.49 | 93.48 | 53.34 | 40.14 | 15.05 | 7.28 | 12.27 | 18.21 | 10.89 |
Solid-state emission studies
In contrast to the solution state, the solid-state photoluminescence spectra of 1 exhibit dual emission bands: a narrow emission band at 450 nm and a broad emission band at 520 nm, while compound 2 shows a broad emission band centered at 470 nm with a minor shoulder at 520 nm in the solid state (Fig. 3a, ESI Fig. S26 and S27†). The FWHM of the emission bands is found to be 38 nm for 1 and 50 nm for 2. Compound 1 has the average lifetime τav = 3.36 ns at 450 nm and 16.23 ns at 520 nm while 2 has τav = 5.13 ns at 470 nm and 11.60 ns at 520 nm indicating that the emission originates from the singlet state (ESI Fig. S31, Tables S8 and S9†).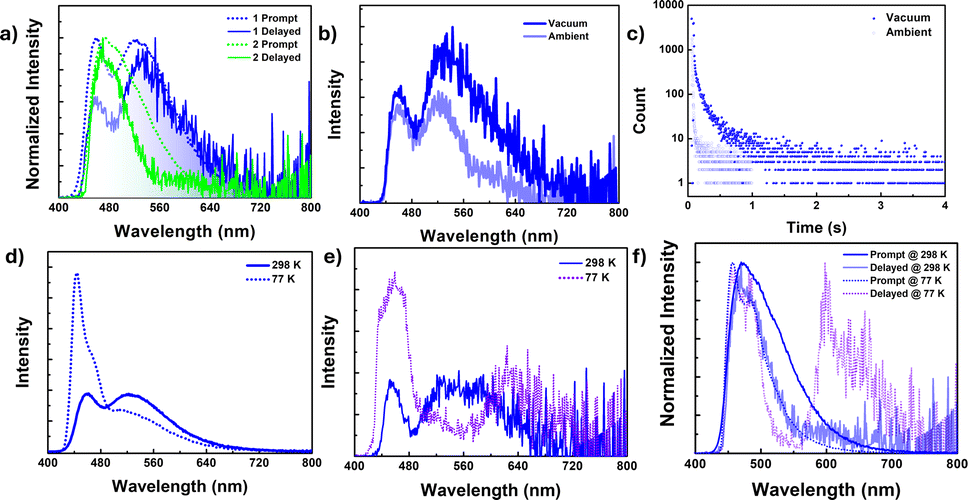 | ||
| Fig. 3 (a) Prompt and delayed (50 μs delay) PL spectra for 1 and 2 in solid state (as prepared) [λex = 380 nm], (b) time-gated PL spectra (50 μs delay) and (c) delayed fluorescence lifetime decay [λem = 520 nm] for 1 (as prepared) [λex = 380 nm] at 298 K under vacuum (absence of oxygen) and ambient conditions (presence of oxygen). (d) Fluorescence spectra and (e) time-gated spectra [50 μs delay] for 1 under a vacuum atmosphere at 298 K and 77 K upon λex = 380 nm. (f) Normalized prompt and delayed spectra [50 μs delay] for 2 under a vacuum atmosphere at 298 K and 77 K upon λex = 380 nm. | ||
To resolve any ambiguity regarding the origin of the emission, we conducted delayed emission studies in the solid state. The delayed emission spectra of both compounds overlapped smoothly with the prompt (fluorescence) spectra and showed a lifetime in the microsecond range (Fig. 3a and ESI Fig. S30 and S32†). Additionally, to confirm the involvement of the triplet state, we performed both steady-state and time-gated measurements under oxygen-free (vacuum) conditions (Fig. 3b, c and S30–S33†). For compound 1, the band at ∼520 nm experiences significant emission quenching in the presence of oxygen, whereas the emission band at 450 nm is minimally affected. A similar trend is observed in the delayed emission lifetime as well. However, we were unable to record the emission lifetime of the band centered at 450 nm under ambient conditions. The delayed lifetime found at the 450 nm band [τav = 9.43 μs] is considerably shorter compared to the lifetime found for the band at 520 nm [τav = 206.11 μs] under vacuum. These results indicate that the two emission bands observed for 1 in the solid have different origins, with the lower energy band having a significant contribution from the triplet state. For compound 2, the emission band centered at 470 nm shows considerable enhancement in intensity, and an additional red-shifted band arises at 610 nm under vacuum (in the absence of oxygen). Lifetime measurements conclude that the newly arisen emission bands have very high lifetimes in milliseconds [τav = 363.17 ms] and are attributed to phosphorescence originating from the triplet state. On the other hand, the 470 nm band showed a lifetime in microseconds [5.93 μs] under ambient conditions and increased more than thrice under vacuum (absence of oxygen) [τav = 17.60 μs]. Thus, the band at 470 nm is ascribed to DF.
To gain further insight into the emission process, we investigated the emission spectra and lifetimes of these compounds in the solid state at different temperatures. Upon lowering the temperature to 77 K, the PL intensity of the high-energy band (∼450 nm) in 1 increased with a strong vibrational progression, while the lower-energy broadband at 520 nm drastically reduced its emission intensity in the steady-state measurement, which was reflected in the lifetime as well [τav = 14.70 ns at 77 K and 16.58 ns at 298 K] (Fig. 3d and ESI Fig. S34†). However, in the time-gated spectra, the lower-energy band at 520 nm completely vanished, indicating that the lower-energy band observed for compound 1 originated from a thermally assisted process (TADF) (Fig. 3e). Additionally, there is a weak redshifted band at 620 nm, and the lifetime corresponding to this band is in milliseconds [τav = 242.82 ms], proving that the redshifted bands are phosphorescent in nature (ESI Fig. S35, S36, Tables S8 and S9†). On the other hand, for compound 2, both the prompt and delayed bands (delayed fluorescence band at 470 nm and phosphorescence band centered at 645 nm) increased their emission intensity with structured patterns, along with a sharp rise in their lifetime values (Fig. 3f, ESI Fig. S37–S39, Tables S8 and S9†).
In short, compound 1 exhibits dual emission behavior in the solid state, with emission bands observed at 450 nm and 520 nm. Among these two bands, the emission at 450 nm is fluorescence in nature, originating from the locally excited singlet state (S), which is less affected by the presence of oxygen and increases in intensity with a decrease in temperature. The second band at 520 nm is a thermally activated delayed fluorescence (TADF) band, originating from a stable singlet state (S*) following efficient thermally assisted reverse intersystem crossing (rISC) from the triplet state. This band is highly sensitive to oxygen and is quenched at low temperatures, allowing the triplet excitons to decay radiatively, resulting in a phosphorescence band at 620 nm at 77 K. On the other hand, compound 2 exhibits a weak room temperature phosphorescence band at 610 nm from a low-energy triplet state with a lifetime in milliseconds under vacuum conditions, along with a delayed fluorescence band at 470 nm. Both these bands are found to increase their intensity upon decreasing in temperature. Hence, the delayed fluorescence emission is likely to occur by utilizing isoenergetic high-lying triplet states (Tn) for rISC, which do not require temperature assistance. These interpretations are supported by comprehensive theoretical analyses, which will be discussed later in the manuscript.
Single-molecular persistent room temperature phosphorescence (pRTP)
The pristine samples of 1 and 2 exhibited no or very weak phosphorescence, which was attributed to crystallization-induced triplet quenching or overall concentration quenching. However, recent studies indicated that naphthalene derivatives could display efficient single molecular persistent room-temperature phosphorescence (RTP).54 In this context, we conducted PL studies of these compounds doped in the PMMA matrix with varying concentrations (1, 10, and 50 wt%) of 1 and 2 (Fig. 4 and ESI S40–S43†). It was observed that both compounds displayed efficient persistent phosphorescence (pRTP) at room temperature in the absence of oxygen (vacuum), with lifetimes in the millisecond range. This phosphorescence was quenched as the doping concentration increased from 1 to 50 wt% and was absent in the neat film, indicating the single molecular origin of the phosphorescence emission. The prompt spectra for compound 1 (450 nm) in the 1 wt% thin film showed a single emission band, similar to the high-energy band observed in the solid state (450 nm) (see Fig. 4a). With increasing concentration, a slight redshift occurred, accompanied by broadening of the spectra, particularly at the tail end (Fig. 4a). These findings provided insight into the dual-band origin for compound 1 in the solid state, indicating that the low-energy band stemmed from a single molecule, exhibiting fluorescence, while the redshifted broadband is of intermolecular origin, demonstrating TADF nature as discussed vide supra.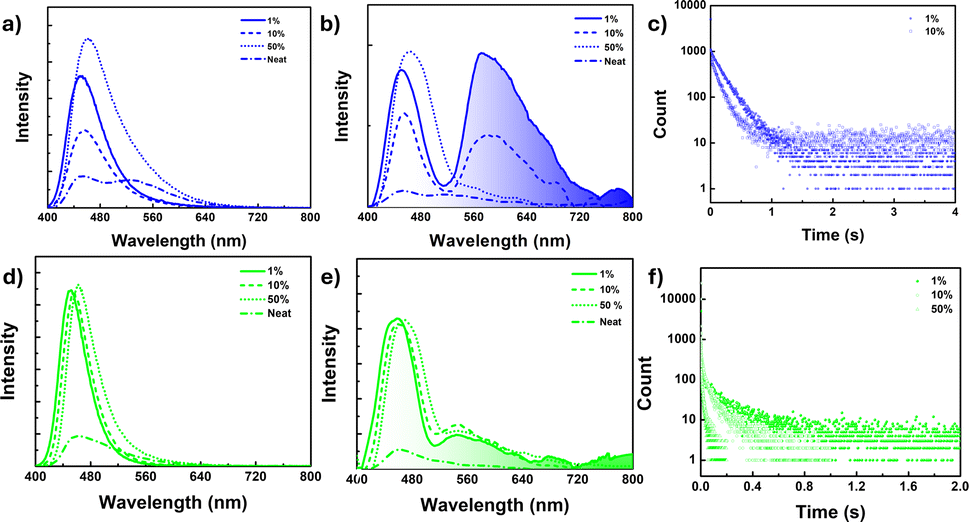 | ||
| Fig. 4 (a and d) Fluorescence spectra, (b and e) delayed luminescence spectra [50 μs delay] of the neat film and doped PMMA film (λex = 380 nm) under vacuum at 298 K for 1 (a and b) and 2 (d and e) [spectra smoothened up to 50 pt for clarity, unsmoothed spectra in ESI Fig. S40c and 42c†], (c) phosphorescence lifetime decay for the 1 doped PMMA film (1 wt%, 10 wt%) at λex = 380 nm, λem = 580 nm. (f) 2 doped PMMA film (1 wt%, 10 wt% and 50 wt%) at λex = 380 nm, λem = 540 nm under vacuum at 298 K. | ||
The delayed spectra for doped matrices (1 and 10 wt% in PMMA) exhibited a redshifted structured band at 580 nm, along with a high energy band (450 nm) overlapping with the prompt fluorescence (Fig. 4b). Moreover, the 580 nm band is quenched at the 50 wt% doped matrix level and disappears in the neat film. A similar trend was observed in their decay profiles, with slightly higher lifetimes observed for 1 at the 1 wt% doping level [τav = 168.1 ms for 1 wt% and τav = 133.1 ms for 10 wt%] (Fig. 4c). Additionally, the excitation spectra corresponding to the emission band at 1 and 10 wt% doping levels reproduced their absorption spectra, further confirming their single molecular origin (ESI Fig. S40b†).
A similar phosphorescence pattern was observed for compound 2 as well; however, the observed PL band was weak and slightly blue-shifted (λem = 550 nm) compared to compound 1 (580 nm) (Fig. 4d–f). Furthermore, for compound 2, phosphorescence bands were present even at 50 wt%, exhibiting marginal changes in the intensity with variations in doping concentration and disappearing in the neat film (Fig. 4e). A slight redshift in delayed fluorescence spectra was noted for 2 with increasing doping concentration. However, compound 2 did not fully restore its pristine spectra even at 50 wt%; consequently, the delayed fluorescence spectra in the neat film was slightly blue-shifted compared to 50 wt%. While there was only a marginal intensity difference observed for the phosphorescence band, a notable decrease in the lifetime decay was observed upon increasing the doping concentration (τav = 129.26 ms at 1 wt%, 67.40 ms at 10 wt%, 8.72 ms at 50 wt%) (Fig. 4f and ESI Fig. S43†).
To gain an unambiguous understanding of the involvement of the triplet state, we conducted PL measurements for these films (both doped and neat) under air and vacuum at 298 K and low temperature (77 K) under vacuum for the 1 wt% doped film for both compounds (ESI Fig. S44–S56†). The phosphorescence band of 1 and 2 (1 wt%, 10 wt%, and 50 wt%) completely disappeared when exposed to air, with only the delayed fluorescence band persisting, albeit with a slightly reduced lifetime.
The oxygen sensitivity of these bands (delayed fluorescence and phosphorescence bands) indicated the involvement of the triplet state in the emission process (ESI Tables S10–S13†). Furthermore, at low temperatures (77 K), the 1 wt% doped films of 1 and 2 showed stronger luminescence and slower decay than under ambient conditions. The increment in the phosphorescence intensity was almost double in the case of 1, whereas for 2, these changes were marginal (ESI Fig. S51c, S55c and Tables S10–S13†). The lowering of the temperature arrested molecular motion, which reduced the nonradiative decay channels from the excited state (both from singlet and triplet), resulting in the enhancement of both fluorescence and phosphorescence emission intensity as well as lifetime.
In summary, compound 1 exhibited a dual emission band in the solid state, with the higher energy band (∼450 nm) being fluorescence in nature (τav = 3.36 ns) and originating from a single molecular species, as evident in the 1 wt% doped PMMA. These bands remained consistent with the emission bands observed in toluene, allowing us to assign their origin to the singlet locally excited (1LE) state (see Fig. 5a–c). The lower energy band (∼520 nm) was only present in the solid or neat film, with its appearance observed as a broad tailing of spectra upon increasing the compound's doping concentration in the PMMA matrix. This indicated an intermolecular origin due to close packing in the solid/neat film. Additionally, this band experienced significant intensity loss with decreased temperature or in the presence of oxygen, possessing a lifetime in microseconds (τav = 206.11 μs, and 15.99 μs in the absence and presence of oxygen, respectively) at room temperature (ESI Table S9†). These findings suggested the emission band at ∼520 nm is TADF, involving efficient triplet harvesting via rISC. Furthermore, these bands closely resembled the emission bands in polar solvents such as methanol and exhibited a significantly lower fluorescence lifetime than the higher energy band, collectively pointing to the CT nature of the state [1CT] (Fig. 5a–c). Moreover, compound 1, upon lowering the temperature, resulted in the appearance of redshifted phosphorescence, which disappeared with the concomitant rise of the TADF band at 520 nm, suggesting probable triplet-up conversion. This up-conversion was arrested at low temperatures, leading to phosphorescence emission from the triplet state (ΔEST = 0.39 eV). Additionally, the weak and redshifted nature of this phosphorescence band (630 nm) indicated its origin from a CT triplet state of intermolecular origin (3CT). On the other hand, the stronger and more structured phosphorescence band observed at 540 nm in the doped film (1 and 10 wt%) originated mainly from a triplet state of single molecular origin (3LE) (Fig. 5c).
 | ||
| Fig. 5 (a and d) Fluorescence spectra, (b and e) time gated spectra [50 μs delay] [smoothened up to 50 pt for clarity, unsmoothed spectra in ESI Fig. S34, S37, S40, and S42†] for (a and c) 1 and (b and d) 2 in the solid state (as prepared solid) at 298 K and 77 K, as a neat film and a doped film in the PMMA matrix (1 wt%) under vacuum [λex = 380 nm]. Plausible emission mechanism involved in (c) 1 and (f) 2 respectively. | ||
Compound 2 exhibited distinct emission characteristics, manifesting a broad single emission band centered at 470 nm in the solid state. Additionally, it displayed a weak RTP band at 580 nm, which became more prominent with vibronic progression at low temperatures (Fig. 5d–f). Furthermore, the time-gated PL spectra confirmed the presence of a DF band at 470 nm, showing greater intensity and lifetime at low temperatures, thereby ruling out TADF.63,67 This DF process likely occurred via vibronic coupling between the emissive singlet state and the higher energy triplet manifold with a small energy gap.63,67 Similar to doped thin films of compound 1, compound 2 also exhibited a phosphorescence band in the region of 560–700 nm, corresponding to the single molecular RTP peaks observed for reported naphthalene-based phosphors. However, the neat film of these compounds could not replicate their solid-state PL features or delayed fluorescence nature, attributed to the distinct tight molecular packing in the solid state compared to the thin film (Fig. 5f).
Excited state theoretical studies
To understand the nature of the excited states involved in DF/RTP processes, we conducted a detailed theoretical investigation including geometry optimization in an excited state (S1 and T1), spin-orbital coupling constants (ξSOC) between singlet and triplet manifolds, vertical transitions, and natural transition orbitals (NTOs) [S1, Tn (n = 1–5)] (ESI Fig. S57–S63 and Tables S14–S17†). For the S1 state of compound 1, the hole is located on the naphthalene and NMe2 moieties while the particle is located largely on boron with a minor contribution from the naphthalene spacer, indicating that the CT character with the very minor LE character dominates the S1 state. In the case of T1, the hole and particle are concentrated on the naphthalene and NMe2 moieties with additional contribution from B–C(Nap) indicative of the LE character (ESI Fig. S62 and S63†). Higher triplet states possess an LE character. A similar kind of NTO distribution is observed for 2 also, where the S1 state illustrates its CT character, the T1 state as LE, and the higher triplet state as LE.There was considerable overlap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), resulting in a relatively high ΔES1–T1 for 1 (0.87 eV) and 2 (0.83 eV), making efficient rISC realization impossible. Nonetheless, there were high-lying triplet states, such as T2 or T3, which were energetically closer to S1, enabling efficient intersystem crossing. For compound 1, the results revealed that S1 to both T2 and T3 transitions were within the energy constraints to realize efficient spin crossover, and both were of LE nature. However, the spin-orbital coupling (SOC) constant values for the S1 to T2 transition were high (1.228 cm−1), suggesting a higher priority for T2 over T3. There is a large energy gap (0.74 eV) between the T1 and T2 states. Therefore, there is a competing pathway between DF and phosphorescence. Presumably, these energy level alignments could be the reason for the simultaneous realization of DF and RTP from a mono-molecular species (ESI Fig. S60, S62 and Table S15†). Compound 2 exhibited a similar scenario, with an inaccessible T1 but high-lying T2 and T3 in energetically favorable positions, as seen in the ESI Fig. S59, S61 and Table S15.†
Mechanochromism
Mechanochromic materials have potential applications in optical storage, security papers, memory devices, anti-counterfeiting and sensors.31,48–51 Generally, color change or emission shift is due to the disruption of ordered molecular arrangement (molecular packing) upon mechanical force. The distinct crystal packing of compounds 1 and 2 prompted us to investigate their optical response toward mechanical force as it disrupts molecular arrangements. Interestingly, crystals of 1 and 2 showed blue-shifted emission, which is quite uncommon in mechanochromic materials. The greenish yellow color crystals of 1 showed dual emission with λPL of 460 (τav = 3.11 ns) and 530 (τav = 19.60 ns) nm, upon grinding, the low energy band disappeared while the high energy band was slightly blue-shifted (453 nm, τav = 3.09 ns) which led to blue color of the ground form under UV light irradiation as seen in Fig. 6b. The lower energy band (530 nm) stems from the intermolecular origin (vide infra) in the solid state, and mechanical force disrupts such sort of molecular arrangement, leading to a single molecular emission (460 nm) as it is close to the 1 wt% doped PMMA film (450 nm). Attempts to check the reversibility from ground form to crystals either by heating or solvent fuming were unsuccessful. The ground form was converted back to crystals through recrystallization from slow evaporation of the DCM/hexane solvent mixture.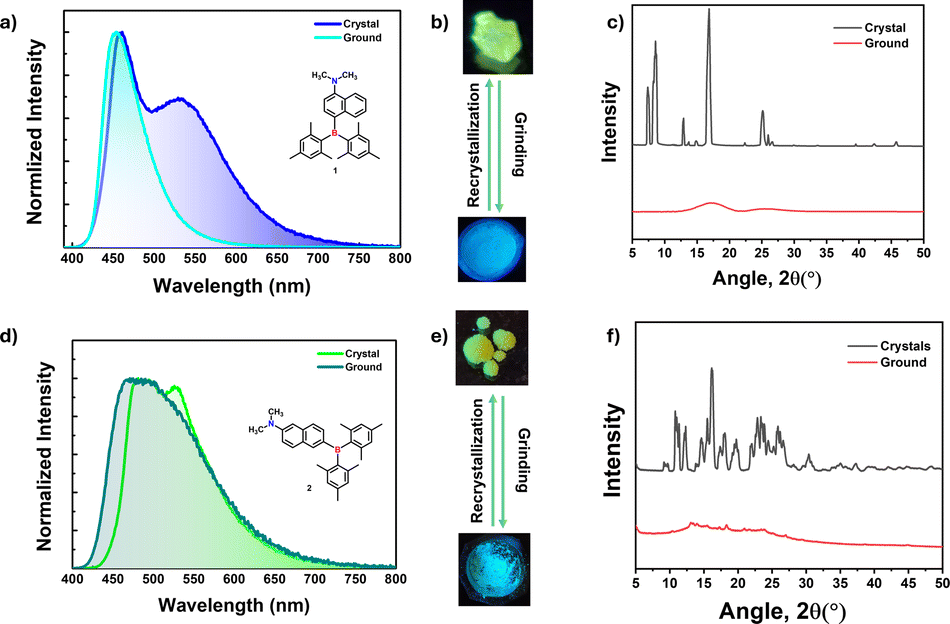 | ||
| Fig. 6 Steady-state PL spectra for (a) 1 and (d) 2 in crystal and grounded form along with their (b and e) photographs before and after grinding. PXRD pattern for (c) 1 and (f) 2 before and after grinding. | ||
Powder X-ray measurements were recorded to understand the mechanochromic behavior. From PXRD spectra, the crystals showed fine reflection peaks, indicating the well-ordered molecular arrangement, while the ground form showed diffuse spectra with no reflections, indicating the amorphous nature (Fig. 6c). The result further confirms that the emission color change is due to a phase transition from crystalline to amorphous. Akin to 1, compound 2 also showed similar behavior. The yellowish green color crystals showed emission maxima at 490 nm (τav = 4.51 ns) with λPL of 526 nm (τav = 8.83 ns) shoulder band switches to a slightly blue-shifted broad band with an emission maximum at of 480 nm (τav = 5.16 ns) (ESI Fig. S64, S65 and Table S18†). Similar to compound 1, the mechanochromic behavior of compound 2 is also irreversible. The PXRD pattern for crystals and ground forms of 2 confirms the phase transition from crystalline to amorphous like 1 (Fig. 6f).
Demonstration of afterglow and its response towards acid/base and anti-counterfeiting applications
After successfully demonstrating pRTP from the doped film, we have exploited the pRTP nature of 1 as a phosphorescence ink for data storage and other applications. The 1 wt% doped PMMA ink of 1 was used to write the letters “TIFR IPC” on a glass slide. These letters exhibited blue color fluorescence under UV light. Once the UV light was turned off, the letters showed persistent orange color room temperature phosphorescence (λex = 365 nm) under vacuum (Fig. 7a). Furthermore, we have used 1 wt% and 50 wt% doped PMMA ink for anti-counterfeiting applications because the former exhibited bright afterglow which can be seen by the naked eye while the latter cannot be seen. For the encryption process, in digital number code format “8888” the alphanumeric secret code “293P” is encrypted using 1 wt% doped PMMA ink while the rest of the area in “8888” is written using 50 wt% doped PMMA ink. Under UV light, all digits “8888” showed bright blue color fluorescence (Fig. 7b). For the decryption, just turning off the UV light resulted in the appearance of an orange color alphanumeric secret code “293P”, showcasing its potential in anti-counterfeiting applications. | ||
| Fig. 7 Digital photographs of (a) writing done with ink made from a mixture of PMMA and 1 (1 wt%) under UV on and off conditions. (b) Anti-counterfeiting demonstration using ink made from a mixture of PMMA and 1 (1 wt% and 50 wt%) compound 1 utilizing single molecular pRTP emission features of 1. | ||
Numerous chromophores exhibited fluorescence on and off behavior or fluorescence emission change upon exposure to acid and base. However, the phosphorescence response towards acid/base was underexplored. The acid-sensitive amino donors in our compounds inspired us to explore their fluorescence and phosphorescence response towards acid and base. The PMMA film (1 wt%) of 1 showed blue fluorescence (λPL = 450 nm, τav = 5.3 ns) and orange color phosphorescence under vacuum (λPh = 580 nm, τ = 168 ms) (Fig. 8 and ESI Fig. S66 and Table S19†). When the films were exposed to trifluoroacetic acid vapors (TFA), it was found that the fluorescence emission (λPL = 400 nm, τav = 3.1 ns) is blue-shifted with the reduced intensity, as seen in Fig. 8. This is due to the protonation of amino donors, which results in a decrease in intramolecular charge transfer. Interestingly, the protonated form showed persistent greenish-yellow RTP emission (λPh = 550 nm, τ = 42 ms) under the vacuum. Further exposure to ammonia vapor immediately brings it back to its original form. This pH-sensitive behavior is illustrated by writing the letters “11B” on the glass slide, as shown in Fig. 8e.
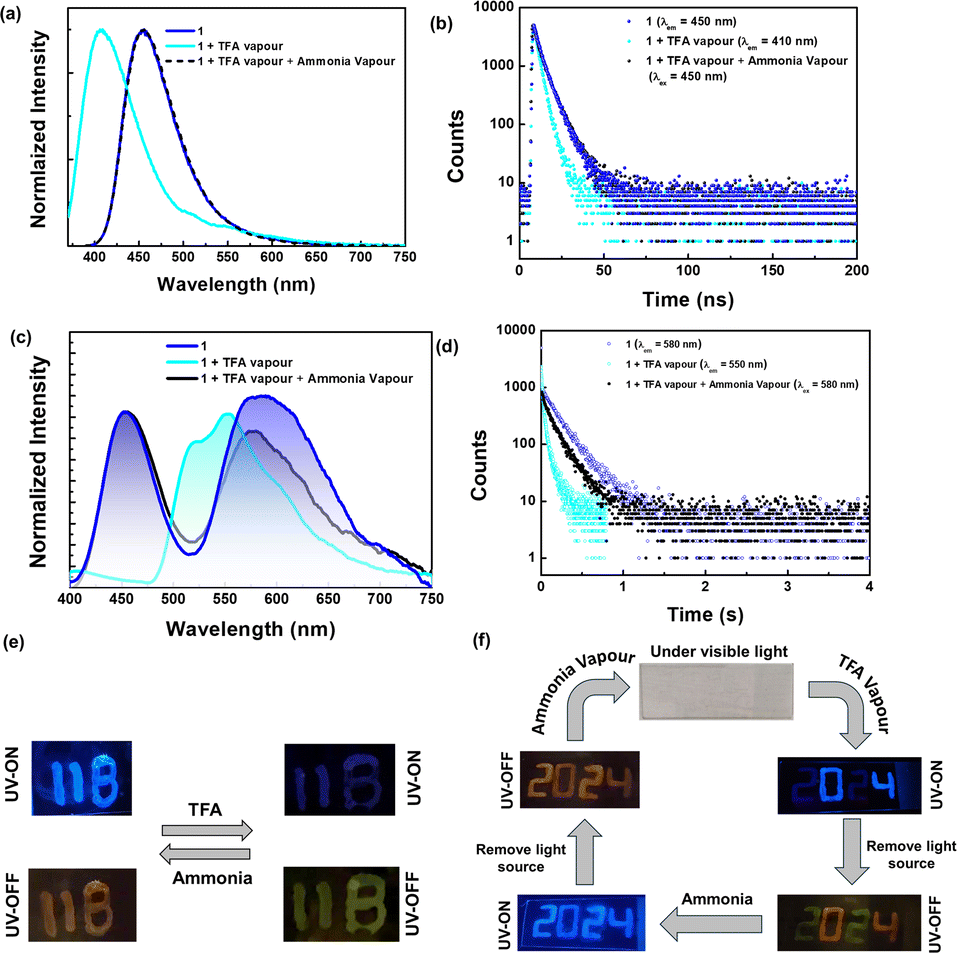 | ||
| Fig. 8 (a) Fluorescence spectra and (c) phosphorescence spectra [smoothened up to 50 pt for clarity, unsmoothed spectra in ESI Fig. S66d†] (b and d) time-resolved decay for 1 doped in the PMMA film (1 wt%). Photographs illustrating (e) pH-sensitive emission features of the doped film (1 wt% in PMMA) and (f) the anti-counterfeiting application of the 1 film (1 wt% in PMMA) by exploring pH-sensitive emission features. | ||
Compound 2 also exhibits a response to changes in pH similar to 1. While the fluorescence response of compound 2 resembles that of compound 1, the phosphorescence bands remain unchanged upon exposure to TFA. However, the phosphorescence lifetime values of compound 2 decreased significantly upon exposure to acid. In addition, neither the fluorescence emission nor the phosphorescence lifetime returns completely to their original states after treatment with a base (ESI Fig. S67 and Table S20†). Due to the weak phosphorescence emission of compound 2, images under UV-off conditions could not be captured, and therefore, only images depicting fluorescence switching are included in Fig. S67.† Additionally, we attempted emission switching using inorganic acid vapor, such as HCl.
We observed similar changes in emission, but the process took significantly longer (>2 h) compared to TFA vapor (<5 min) (ESI Fig. S68†). The pH-sensitive phosphorescence of 1 is used for anti-counterfeiting applications. The 1 wt% doped PMMA ink is used to write the number code “2024”. Under daylight, this code is not visible; however, it showed blue fluorescence under UV light (ex = 365 nm). Then, the numbers 0 and 4 are masked with aluminum foil and the numbers 2 and 2 are exposed to acid vapour. Under UV light, only the numbers 0 and 4 showed intense fluorescence, while 2 and 2 appeared with faded color. Once the UV light was turned off, 0 and 4 showed orange color phosphorescence, and 2 and 2 showed green color room temperature phosphorescence under vacuum. Upon base treatment, all numbers showed orange color room temperature phosphorescence and blue fluorescence.
The different fluorescence and phosphorescence responses of this compound can be used for real-time anti-counterfeiting applications.
To strongly advocate for the switchable room temperature phosphorescence (RTP) triggered by acid vapor, we endeavored to produce protonated derivatives of compounds 1 and 2. Unfortunately, our attempts were unsuccessful. Instead, we achieved the successful synthesis of methylated derivatives, referred to as compounds 1-OTF and 2-OTF henceforth. Compounds 1-OTF and 2-OTF were synthesized by methylating the NMe2 donor for 1 and 2 using MeOTF in diethyl ether. Detailed synthesis schemes and other characterization data are included in the ESI (Scheme S2, Fig. S69–S75 and Table S21).† Prior to the comparison of photophysical studies, we compare the structural/geometrical parameters of 1-OTF and 2-OTF with those of 1 and 2, respectively, using the data obtained either from crystal (in the case of 1-OTF and 1) or from ground state optimized geometry (in the case of 2-OTF and 2). It is clear from the crystal/ground optimized structure that the boron center is affected very minimally upon methylation on the N center, as we can see from the marginal geometrical changes around the B center [θS–A = 43.65°, 27.13° and B–C bond length 1.590 Å, 1.578 Å for 1-OTF and 2-OTF respectively]. However, considerable geometrical changes occur around the nitrogen center as reflected in the higher N–C bond length (1.526 Å, 1.514 Å for 1-OTF and 2-OTF) and dihedral angle related to donor unit [θS–D = 85.59°, 87.97° and θD–A = 77.34°, 89.11° for 1-OTF and 2-OTF respectively] (ESI Table S22†). These geometrical changes make the molecules (1-OTF, 2-OTF) more twisted than those of their neutral derivatives (1 and 2) in vide supra.
Furthermore, these geometrical changes perturbed their electronic structure. As seen from the FMO diagram, the salts (1-OTF and 2-OTF) have distinct FMOs compared to neutral derivatives (1 and 2), where the HOMO is solely concentrated on the OTF moiety and LUMO on the boryl aniline unit. The new orbital distribution changes D–A architecture, where the OTF unit acts as a donor and whole boryl aniline as an acceptor, resulting in a stronger charge transfer character in the salt compared to the neutral analogs. The same can be confirmed by the drastic increase in the ground state dipole moment in 1-OTF (14.9 D) and 2-OTF (15.2 D) compared to 1 (1.7160 D) and 2 (4.1916 D) respectively. Upon comparing FMO's energy, both the HOMO and LUMO are stabilized; however, the extent of stabilization in the LUMO is more compared to the HOMO, though the overall stabilization occurs in 1-OTF and 2-OTF compared to 1 and 2. In addition, the band gap is slightly reduced in cases 1-OTF (3.58 eV) compared to 1 (3.71 eV), and the reverse trend is observed for 2 (2.88 eV) and its 2-OTF (3.66 eV).
Following successful synthesis and geometrical analysis, we compared the emission properties of doped films of 1-OTF and 2-OTF (1 wt% and neat) with those of 1 and 2 (ESI Fig. S76–S85, Tables S23 and S24†). As expected, the 1 wt% doped film of compound 1-OTF exhibits almost similar phosphorescence features (λmax = 560 nm) to that of the TFA-exposed 1 wt% doped film of compound 1. However, a significant increase in phosphorescence lifetime is observed for 1-OTF (τav = 154.58 ms) compared to 1 (τav = 41.98 ms) (Fig. 9 and ESI Fig. S76b, S78 and S80†). Conversely, a slight redshift is observed for the fluorescence band of 2-OTF (λmax = 440 nm), attributed to the change in donor strength from amine (NMe2) to ammonium salt (NMe3+) that results from the reduction in band gap as mentioned above (Fig. 9 and ESI Fig. S81b, S83, S85, Tables S23 and S24†). In contrast, the photoluminescence (PL) features of neat films of 1-OTF differ distinctly from those of 1. The fluorescence spectra of 1-OTF (λmax = 430 nm) in the neat film are blue-shifted compared to those of the neat film of 1 (λmax = 450 nm) (ESI Fig. S76a, d, S77 and S79†). Notably, phosphorescence bands in 1-OTF are observed even in the neat film (λmax = 610 nm) compared to 1, with a slight redshift and lower lifetime (τav = 4.04 ms) compared to the 1 wt% doped film of 1-OTF. The appearance of phosphorescence bands in the neat film and the longer phosphorescence lifetime in compound 1-OTF can be attributed to the efficient spin-flip process occurring within the cationic salt. This is due to its elevated spin–orbit coupling (SOC), a trait commonly found in emissive organic ionic species (ESI Table S25†). Similar changes were observed for 2-OTF compared to 2 (ESI Fig. S81–S85 and Table S25†). Furthermore, to support the claim, we have compared the energy levels of the singlet and triplet manifolds involved in the emission process (ESI Fig. S86†). Both the singlet and triplet manifolds are greatly stabilized in the case of 1-OTF; however, changes are minimal for 2-OTF compared to 2.
 | ||
| Fig. 9 (a) Molecular structure for compound 1-OTF, (b) ground state optimized geometry for compound 2-OTF and (c) FMO's for compounds 1-OTF and 2-OTF generated from geometry optimized structure (corresponding coordinates are taken from the crystal structure in the case of 1-OTF) using DFT at B3LYP and 631g(d,p) level of theory. (d) Prompt and delayed spectra under vacuum [absence of oxygen] for 1-OTF, 1, and 1(TFA) [1 after being exposed to TFA] in the PMMA matrix (1 wt%) [λex = 380 nm], (e) 2-OTF, 2, and 2(TFA) [1 after being exposed to TFA] in the PMMA matrix (1 wt%) [λex = 380 nm]. | ||
Non-linear optical properties
TAB-containing compounds with donor substituents showed prominent non-linear optical properties because of their intramolecular CT characteristics.68–72 For example, Marder and co-workers have made a seminal contribution to the two-photon absorption properties of several TAB-based donor–acceptor compounds.66,73–77 The present two compounds (1 and 2) have different degrees of CT characteristics which inspired us to investigate their non-linear optical properties, in particular, three-photon absorption properties. Fig. 10a–d illustrate the open-aperture and closed-aperture Z-scan data of compounds 1 and 2. Fig. 10a shows both two-photon absorption (2PA; green-colored line) and three-photon absorption (3PA; red-colored line) fits. The 3PA fits are a perfect match to the experimental data. The magnitudes of the extracted 3PA coefficients were 3.0 × 10−23 cm3 W−2 and 3.9 × 10−23 cm3 W−2, respectively (ESI Table S26†). Complete details of the calculations of various NLO coefficients have been presented in our earlier works.78,79 The corresponding 3PA cross-sections were calculated to be ∼10−78 cm6 s2. Compound 2 has depicted a decisive enhancement of ∼33% when compared to the magnitude of compound 1. This can be attributed to both molecules' ground state dipole moments of 1.72 D for compound 1 and 4.19 D for compound 2 obtained from theoretical calculations (ESI Table S14†).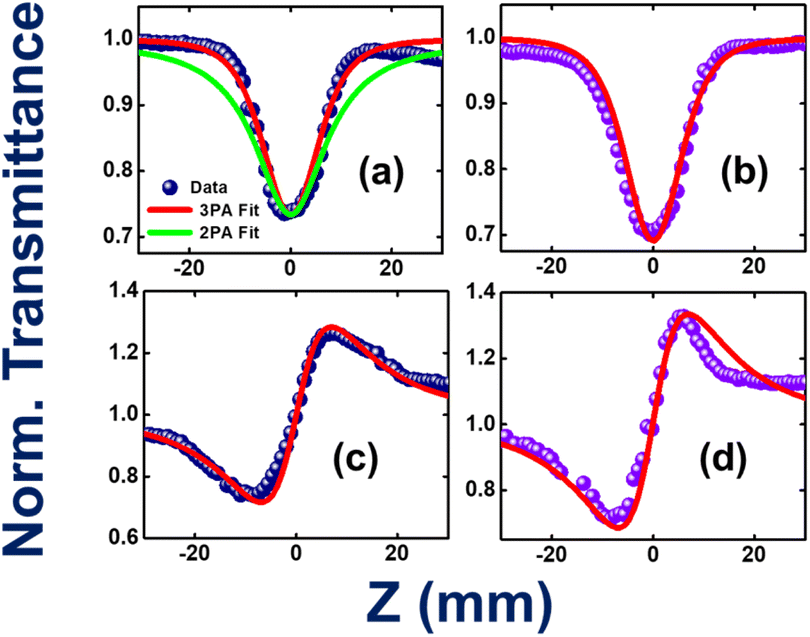 | ||
| Fig. 10 (a) Open aperture Z-scan data of compound 1. Both two-photon absorption (green-colored line) and three-photon absorption (red-colored line) fits are included in the graph. (b) Open aperture Z-scan data and fits of compound 2. (c) Closed aperture Z-scan data of compound 1 (d) closed aperture Z-scan data of compound 1. Solid symbols represent the experimental data, while the solid lines represent the theoretical fits to the data. 800 nm, 1 kHz, ∼70 femtosecond pulses were used to obtain this data. | ||
Zhu et al. have investigated the structure–property relationships of stilbene-based dipolar/quadrupolar chromophores using essential-state models for 3PA.80 They claim that for dipolar systems, they observed an important role played by the change in dipole moment (Dm). This dipole moment change occurred between the ground and 3PA active states. They predicted 3PA cross-sections of 10−78 to 10−80 cm6 s2 in their case. Fig. 10c and d depict the closed aperture data of compounds 1 and 2, respectively.
The magnitude of NLO coefficients depends inherently on the input pulse duration, excitation wavelength, and the availability of the high-lying excited states. In the present case, both the compounds have high-lying states conducive for 3PA [excitation at 800 nm (2.55 eV) to 3PA near 266 nm (4.66 eV)]. The magnitudes of the nonlinear refraction (n2) coefficients were extracted to be 4.9 × 10−16 cm2 W−1 and 5.6 × 10−16 cm2 W−1, respectively. The 3PA coefficients of the present compounds are superior to those of some recently reported novel organic compounds, such as porphyrin–phthalimide-based donor–acceptor systems,81 and comparable to structurally unconstrained green fluorescence protein (GFP) chromophores.82 The solvent contribution (dichloromethane, DCM) was also investigated in similar experimental conditions, and the data is presented in ESI Fig. S87.† The magnitude of solvent NLO coefficients was much smaller (more than one order of magnitude lower than the solute) and was, therefore, ignored. The errors in the above measurements were estimated to be <10% due to various factors, such as errors in the estimation of the peak intensity, input laser fluctuations, data analysis, etc.
Conclusions
We have reported two regioisomers 1 and 2 by strategically placing a TAB acceptor and an amine donor around a naphthalene π-spacer, resulting in a D–π–A system with intriguing multi-functional photophysical properties. Changing the substitution position of the donor NMe2 and acceptor BMes2 around the naphthalene spacer from 1,4 to 2,6 shifted the emission process from TADF to RTP in the solid state by finely adjusting the triplet and singlet manifolds. Compounds 1 and 2 exhibited single molecular persistent RTP in doped films in which compound 1 showed persistent RTP compared to compound 2. Interestingly, the phosphorescence bands of these compounds are sensitive to acid and base vapor, with compound 1 showing reversible switching of its pRTP band from orange to greenish yellow upon exposure to acid/base vapor. To the best of our knowledge, this is the first report on the TAB system that shows pH-sensitive phosphorescence switching. Furthermore, the switchable long pRTP behavior of compound 1 was utilized to develop an ink for advanced anti-counterfeiting applications. Both compounds displayed hypsochromically shifted mechanochromism. Additionally, the three-photon absorption properties of these regio-isomers were explored, revealing higher values observed for 2 than 1. This molecular engineering strategy opens avenues for developing multi-functional TAB-based compounds for various applications.Data availability
The data that support the findings of this study are available in the ESI† of this article. Details on the synthesis and structural characterization, photo-physical, results from DFT and TD-DFT and RTP crystallographic data (2288523 for 1, 2288524 for 2, and 2291487 for 1-OTF) are provided.Author contributions
The project was designed by VC and PT. RA and MMAT contributed equally. Synthesis and characterization by RA and RK. Photophysical characterization and mechanochromism studies by RA, MMAT, and PS, Nonlinear Optical properties by DB and VR Soma. All authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Prof. V. C. is thankful to the Department of Science and Technology, New Delhi, India, for a National J. C. Bose Fellowship. R. A., R. K., and P. S. are thankful to the TIFR Hyderabad from the Department of Atomic Energy (DAE), India. MMAT thanks CSIR India for the research fellowship (09/079(2806)/2019-EMR-1). Prof. PT is thankful to SERB (EEQ/2023/000903) and IISc for the financial support.References
- A. M. Priegert, B. W. Rawe, S. C. Serin and D. P. Gates, Polymers and the p-block elements, Chem. Soc. Rev., 2016, 45, 922–953 RSC.
- K. Liu, Z. Jiang, R. A. Lalancette, X. Tang and F. Jäkle, Near-Infrared-Absorbing B–N Lewis Pair-Functionalized Anthracenes: Electronic Structure Tuning, Conformational Isomerism, and Applications in Photothermal Cancer Therapy, J. Am. Chem. Soc., 2022, 144, 18908–18917 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Synthesis of conjugated polymers for organic solar cell applications, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- S. Kuila and S. J. George, Phosphorescence Energy Transfer: Ambient Afterglow Fluorescence from Water-Processable and Purely Organic Dyes via Delayed Sensitization, Angew. Chem., Int. Ed., 2020, 59, 9393–9397 CrossRef CAS PubMed.
- Y. J. Cheng, S. H. Yang and C. S. Hsu, Synthesis of Conjugated Polymers for Organic Solar Cell Applications, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- T. W. Hudnall and F. P. Gabbaï, Ammonium boranes for the selective complexation of cyanide or fluoride ions in water, J. Am. Chem. Soc., 2007, 129, 11978–11986 CrossRef CAS PubMed.
- K. Parab, K. Venkatasubbaiah and F. Jäkle, Luminescent triarylborane-functionalized polystyrene: synthesis, photophysical characterization, and anion-binding studies, J. Am. Chem. Soc., 2006, 128, 12879–12885 CrossRef CAS.
- J.-F. Chen, X. Yin, K. Zhang, Z. Zhao, S. Zhang, N. Zhang, N. Wang and P. Chen, Pillar [5] arene-based dual chiral organoboranes with allowed host-guest chemistry and circularly polarized luminescence, J. Org. Chem., 2021, 86, 12654–12663 CrossRef CAS.
- A. Fukazawa and S. Yamaguchi, Ladder π-Conjugated Materials Containing Main-Group Elements, Chem.–Asian J., 2009, 4, 1386–1400 CrossRef CAS PubMed.
- D. Chen, S.-J. Su and Y. Cao, Nitrogen heterocycle-containing materials for highly efficient phosphorescent OLEDs with low operating voltage, J. Mater. Chem. C, 2014, 2, 9565–9578 RSC.
- M. Mamada, M. Hayakawa, J. Ochi and T. Hatakeyama, Organoboron-based multiple-resonance emitters: synthesis, structure–property correlations, and prospects, Chem. Soc. Rev., 2024, 53, 1624–1692 RSC.
- P. Sudhakar, A. K. Gupta, D. B. Cordes and E. Zysman-Colman, Thermally activated delayed fluorescence emitters showing wide-range near-infrared piezochromism and their use in deep-red OLEDs, Chem. Sci., 2024, 15, 545–554 RSC.
- R. W. Weerasinghe, S. M. Suresh, D. Hall, T. Matulaitis, A. M. Slawin, S. Warriner, Y. T. Lee, C. Y. Chan, Y. Tsuchiya and E. Zysman-Colman, A Boron, Nitrogen, and Oxygen Doped π-Extended Helical Pure Blue Multiresonant Thermally Activated Delayed Fluorescent Emitter for Organic Light Emitting Diodes That Shows Fast kRISC Without the Use of Heavy Atoms, Adv. Mater., 2024, 36, 2402289 CrossRef CAS PubMed.
- T. Huang, Z. Ding, H. Liu, P.-A. Chen, L. Zhao, Y. Hu, Y. Yao, K. Yang and Z. Zeng, Electron-transporting boron-doped polycyclic aromatic hydrocarbons: facile synthesis and heteroatom doping positions-modulated optoelectronic properties, Chin. Chem. Lett., 2024, 35, 109117 CrossRef CAS.
- H.-S. Kim, Y.-H. Kim, T.-H. Kim, Y.-Y. Noh, S. Pyo, M. H. Yi, D.-Y. Kim and S.-K. Kwon, Synthesis, and studies on 2-hexylthieno [3, 2-b] thiophene end-capped oligomers for OTFTs, Chem. Mater., 2007, 19, 3561–3567 CrossRef CAS.
- E. Lim, B.-J. Jung, J. Lee, H.-K. Shim, J.-I. Lee, Y. S. Yang and L.-M. Do, Thin-Film Morphologies and Solution-Processable Field-Effect Transistor Behavior of a Fluorene–Thieno[3,2-b]thiophene-Based Conjugated Copolymer, Macromolecules, 2005, 38, 4531–4535 CrossRef CAS.
- P. Liu, Y. Wu, H. Pan, Y. Li, S. Gardner, B. S. Ong and S. Zhu, Novel high-performance liquid-crystalline organic semiconductors for thin-film transistors, Chem. Mater., 2009, 21, 2727–2732 CrossRef CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner and W. Zhang, Liquid-crystalline semiconducting polymers with high charge-carrier mobility, Nat. Mater., 2006, 5, 328–333 CrossRef CAS.
- Z. M. Hudson and S. Wang, Metal-containing triarylboron compounds for optoelectronic applications, Dalton Trans., 2011, 40, 7805–7816 RSC.
- Z. M. Hudson, S. B. Zhao, R. Y. Wang and S. Wang, Switchable Ambient-Temperature Singlet–Triplet Dual Emission in Nonconjugated Donor–Acceptor Triarylboron–PtII Complexes, Chem.–Eur. J., 2009, 15, 6131–6137 CrossRef CAS PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Rapid and reversible shape changes of molecular crystals on photoirradiation, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- Y. Yu, C. Dong, A. F. Alahmadi, B. Meng, J. Liu, F. Jäkle and L. Wang, A p-π* conjugated triarylborane as an alcohol-processable n-type semiconductor for organic optoelectronic devices, J. Mater. Chem. C, 2019, 7, 7427–7432 RSC.
- F. Liu, J. Liu and L. Wang, Effect of fluorine substitution in organoboron electron acceptors for photovoltaic application, Org. Chem. Front., 2019, 6, 1996–2003 RSC.
- S. K. Sarkar, L. J. Kang, U. K. Pandey, C. K. Luscombe and P. Thilagar, Triarylborane-BODIPY conjugate: an efficient non-fullerene electron acceptor for bulk heterojunction organic solar cell, Sol. Energy, 2021, 230, 242–249 CrossRef CAS.
- H. Xu, J. Han, S. Chen, Y. Liu, L. H. Hernandez, J. Bertrandie, M. Babics, S. Alam, D. R. Villalva and S. H. K. Paleti, Dissecting the structure-stability relationship of Y-series electron acceptors for real-world solar cell applications, Joule, 2023, 7, 2135–2151 CrossRef CAS.
- Y. Yu, B. Meng, F. Jäkle, J. Liu and L. Wang, Molecular acceptors based on a triarylborane core unit for organic solar cells, Chem.–Eur. J., 2020, 26, 873–880 CrossRef CAS PubMed.
- Í. A. Bozzi, L. A. Machado, E. B. Diogo, F. G. Delolo, L. O. Barros, G. A. Graça, M. H. Araujo, F. T. Martins, L. F. Pedrosa and L. C. da Luz, Electrochemical Diselenation of BODIPY Fluorophores for Bioimaging Applications and Sensitization of 1O2, Chem.–Eur. J., 2024, 30, e202303883 CrossRef PubMed.
- S. K. Sarkar, M. Pegu, S. K. Behera, S. K. Narra and P. Thilagar, Aggregation-Induced and Polymorphism-Dependent Thermally Activated Delayed Fluorescence (TADF) Characteristics of an Oligothiophene: Applications in Time-Dependent Live Cell Multicolour Imaging, Chem.–Asian J., 2019, 14, 4588–4593 CrossRef CAS.
- Q. Zhao, C. Huang and F. Li, Phosphorescent heavy-metal complexes for bioimaging, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- K. Jamuna, S. Gayathri, S. Sivakumar and B. Ashokkumar, Design and development of a fluorometric and colorimetric sensor for toxic cyanide detection by pyridinium scaffolds: live cell imaging and real sample analysis, Sens. Diagn., 2023, 2, 337–346 RSC.
- O. S. Ipek, S. Topal and T. Ozturk, Synthesis, characterization and sensing properties of donor-acceptor systems based dithieno [3, 2-b; 2′, 3′-d] thiophene and boron, Dyes Pigm., 2021, 192, 109458 CrossRef CAS.
- S. Pagidi, N. K. Kalluvettukuzhy and P. Thilagar, Triarylboron Anchored Luminescent Probes: Selective Detection and Imaging of Thiophenols in the Intracellular Environment, Langmuir, 2018, 34, 8170–8177 CrossRef CAS.
- H. K. Sadhanala, S. Pagidi and A. Gedanken, High quantum yield boron-doped carbon dots: a ratiometric fluorescent probe for highly selective and sensitive detection of Mg2+ ions, J. Mater. Chem. C, 2021, 9, 1632–1640 RSC.
- H. K. Sadhanala, S. Pagidi, S. Yadav, M. Beiderman, I. Grinberg, D. Fixler and A. Gedanken, Boron-doped Carbon Dots with Surface Oxygen Functional Groups as a Highly Sensitive and Label-free Photoluminescence Probe for the Enhanced Detection of Mg2+ Ions, ChemistrySelect, 2022, 7, e202201261 CrossRef CAS.
- C. Ippen, W. Guo, D. Zehnder, D. Kim, J. Manders, D. Barrera, B. Newmeyer, D. Hamilton, C. Wang and C. Hotz, High efficiency heavy metal free QD-LEDs for next generation displays, J. Soc. Inf. Disp., 2019, 27, 338–346 CrossRef CAS.
- H. Sasabe and J. Kido, Development of high performance OLEDs for general lighting, J. Mater. Chem. C, 2013, 1, 1699–1707 RSC.
- X. Wang, T. Ishwara, W. Gong, M. Campoy-Quiles, J. Nelson and D. D. Bradley, High-performance metal-free solar cells using stamp transfer printed vapor phase polymerized poly (3, 4-ethylenedioxythiophene) top anodes, Adv. Funct. Mater., 2012, 22, 1454–1460 CrossRef CAS.
- Z. Zhou, X. Xie, Z. Sun, X. Wang, Z. An and W. Huang, Recent advances in metal-free phosphorescent materials for organic light-emitting diodes, J. Mater. Chem. C, 2023, 11, 3143–3161 RSC.
- D. G. Congrave, B. H. Drummond, P. J. Conaghan, H. Francis, S. T. Jones, C. P. Grey, N. C. Greenham, D. Credgington and H. Bronstein, A simple molecular design strategy for delayed fluorescence toward 1000 nm, J. Am. Chem. Soc., 2019, 141, 18390–18394 CrossRef CAS.
- Q. Xue and G. Xie, Thermally activated delayed fluorescence beyond through-bond charge transfer for high-performance OLEDs, Adv. Opt. Mater., 2021, 9, 2002204 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Recent advances in organic thermally activated delayed fluorescence materials, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- B. Ding, X. Ma and H. Tian, Recent Advances of Pure Organic Room Temperature Phosphorescence Based on Functional Polymers, Acc. Mater. Res., 2023, 4, 827–838 CrossRef CAS.
- S. Jena, A. T. M. Munthasir and P. Thilagar, Unraveling Ultralong Phosphorescence in Ar2PO(H): n(O) → σ*(P-C) Transitions in Ar2PO(H) Stabilize Triplet States Better than n(P) → σ*(P-C) in Ar3P, J. Mater. Chem. C, 2023, 127, 9918–9930 CAS.
- S. Jena, A. T. M. Munthasir and P. Thilagar, Ultralong room temperature phosphorescence and ultraviolet fluorescence from simple triarylphosphine oxides, J. Mater. Chem. C, 2022, 10, 9124–9131 RSC.
- J.-L. Ma, H. Liu, S.-Y. Li, Z.-Y. Li, H.-Y. Zhang, Y. Wang and C.-H. Zhao, Metal-free room-temperature phosphorescence from amorphous triarylborane-based biphenyl, Organometallics, 2020, 39, 4153–4158 CrossRef CAS.
- C. Si, T. Wang, A. K. Gupta, D. B. Cordes, A. M. Slawin, J. S. Siegel and E. Zysman-Colman, Room-Temperature Multiple Phosphorescence from Functionalized Corannulenes: Temperature Sensing and Afterglow Organic Light-Emitting Diode, Angew. Chem., Int. Ed., 2023, 62, e202309718 CrossRef CAS PubMed.
- J. Yang, M. Fang and Z. Li, Stimulus-responsive room temperature phosphorescence materials: internal mechanism, design strategy, and potential application, Acc. Mater. Res., 2021, 2, 644–654 CrossRef CAS.
- K. Isoda, M. Matsubara, A. Ikenaga, Y. Akiyama and Y. Mutoh, Reversibly/irreversibly stimuli-responsive inks based on N-heteroacene liquids, J. Mater. Chem. C, 2019, 7, 14075–14079 RSC.
- K. Lou, Z. Hu, H. Zhang, Q. Li and X. Ji, Information storage based on stimuli-responsive fluorescent 3D code materials, Adv. Funct. Mater., 2022, 32, 2113274 CrossRef CAS.
- X.-L. Lu and M. Xia, Multi-stimuli response of a novel half-cut cruciform and its application as a security ink, J. Mater. Chem. C, 2016, 4, 9350–9358 RSC.
- Y. Ma, Y. Yu, J. Li, S. Liu, W. Huang and Q. Zhao, Stimuli-responsive photofunctional materials for green and security printing, InfoMat, 2021, 3, 82–100 CrossRef CAS.
- G. Li, X. Yang, J. Miao, Y. Cui, G. Sun and Y. Li, Substitution position regulating the excitation-dependent fluorescence–phosphorescence emission of formylphenylboronic acids for single-component white light and anticounterfeiting, CrystEngComm, 2023, 25, 5479–5485 RSC.
- Q. Liao, Q. Li and Z. Li, Substituent Effects in Organic Luminogens with Room Temperature Phosphorescence, ChemPhotoChem, 2021, 5, 694–701 CrossRef CAS.
- R. P. Nandi, N. K. Kalluvettukuzhy, S. Pagidi and P. Thilagar, Molecular Persistent Room-Temperature Phosphorescence from Tetraarylaminoboranes, Inorg. Chem., 2023, 62, 1122–1134 CrossRef CAS.
- Z. Wang, X. Cheng, Y. Xie, S. Liu, M. Dong, J. Zhao, F. Liang, Z. An and W. Huang, Recent Advances in Organic Room-Temperature Phosphorescence of Heteroatom (B/S/P)-Containing Chromophores, CCS Chem., 2023, 5, 292–309 CrossRef CAS.
- Z. Wu, J. Nitsch, J. Schuster, A. Friedrich, K. Edkins, M. Loebnitz, F. Dinkelbach, V. Stepanenko, F. Würthner, C. M. Marian, L. Ji and T. B. Marder, Persistent Room Temperature Phosphorescence from Triarylboranes: A Combined Experimental and Theoretical Study, Angew. Chem., Int. Ed., 2020, 59, 17137–17144 CrossRef CAS PubMed.
- Y. Xie, S. Dai, Y. Wang, X. Wang, Y. Sun, Z. Ju, R. Fang, B. Zhang, J. Wu and X. Zhang, Structural and mechanistic studies of excitation- and temperature-tunable multicolor luminescence of triarylborane, CrystEngComm, 2023, 25, 2204–2212 RSC.
- L. Xu, K. Zhou, X. Qiu, B. Rao, D. Pei, A. Li, Z. An and G. He, Tunable ultralong organic phosphorescence modulated by main-group elements with different Lewis acidity and basicity, J. Mater. Chem. C, 2020, 8, 14740–14747 RSC.
- S. Pagidi, N. K. Kalluvettukuzhy and P. Thilagar, Effect of Branching on the Delayed Fluorescence and Phosphorescence of Simple Borylated Arylamines, Inorg. Chem., 2020, 59, 3142–3151 CrossRef CAS PubMed.
- H. Ding, Y. Sun, M. Tang, J. Wen, S. Yue, Y. Peng, F. Li, L. Zheng, S. Wang and Y. Shi, Time-dependent photo-activated aminoborane room-temperature phosphorescence materials with unprecedented properties: simple, versatile, multicolor-tuneable, water resistance, optical information writing/erasing, and multilevel data encryption, Chem. Sci., 2023, 14, 4633–4640 RSC.
- J. X. Wang, Y. G. Fang, C. X. Li, L. Y. Niu, W. H. Fang, G. Cui and Q. Z. Yang, Time-Dependent Afterglow Color in a Single-Component Organic Molecular Crystal, Angew. Chem., 2020, 132, 10118–10122 CrossRef.
- Y. Yang, Y. Liang, Y. Zheng, J. A. Li, S. Wu, H. Zhang, T. Huang, S. Luo, C. Liu and G. Shi, Efficient and Color-Tunable Dual-Mode Afterglow from Large-Area and Flexible Polymer-Based Transparent Films for Anti-Counterfeiting and Information Encryption, Angew. Chem., 2022, 134, e202201820 CrossRef.
- J. Jovaišaitė, S. Kirschner, S. Raišys, G. Kreiza, P. Baronas, S. Juršėnas and M. Wagner, Diboraanthracene-Doped Polymer Systems for Colour-Tuneable Room-Temperature Organic Afterglow, Angew. Chem., Int. Ed., 2023, 62, e202215071 CrossRef.
- K. Kumar, R. J. Tepper, Y. Zeng and M. B. Zimmt, Syntheses of Rigid and Semirigid Molecules for Investigations of Photoinduced Electron Transfer Reactions, J. Org. Chem., 1995, 60, 4051–4066 CrossRef CAS.
- X. Liu, M. Xiang, Z. Tong, F. Luo, W. Chen, F. Liu, F. Wang, R.-Q. Yu and J.-H. Jiang, Activatable Fluorescence Probe via Self-Immolative Intramolecular Cyclization for Histone Deacetylase Imaging in Live Cells and Tissues, Anal. Chem., 2018, 90, 5534–5539 CrossRef CAS PubMed.
- Z. Yuan, C. D. Entwistle, J. C. Collings, D. Albesa-Jové, A. S. Batsanov, J. A. Howard, N. J. Taylor, H. M. Kaiser, D. E. Kaufmann, S. Y. Poon, W. Y. Wong, C. Jardin, S. Fathallah, A. Boucekkine, J. F. Halet and T. B. Marder, Synthesis, Crystal Structures, Linear and Nonlinear Optical Properties, and Theoretical Studies of (p-R-Phenyl)-, (p-R-Phenylethynyl)-, and (E)-[2-(p-R-Phenyl)ethenyl]dimesitylboranes and Related Compounds, Chem.–Eur. J., 2006, 12, 2758–2771 CrossRef CAS PubMed.
- F. Sadiq, Z. Wang, Y. Hou, J. Zhao, A. Elmali, D. Escudero and A. Karatay, Thienyl/phenyl bay-substituted perylenebisimides: intersystem crossing and application as heavy atom-free triplet photosensitizers, Dyes Pigm., 2021, 184, 108708 CrossRef CAS.
- A. Amar, A. Elkechai, J.-F. Halet, F. Paul and A. Boucekkine, Two-photon absorption of dipolar and quadrupolar oligothiophene-cored chromophore derivatives containing terminal dimesitylboryl moieties: a theoretical (DFT) structure–property investigation, New J. Chem., 2021, 45, 15074–15081 RSC.
- P. Chen, A. S. Marshall, S. H. Chi, X. Yin, J. W. Perry and F. Jäkle, Luminescent Quadrupolar Borazine Oligomers: Synthesis, Photophysics, and Two-Photon Absorption Properties, Chem.–Eur. J., 2015, 21, 18237–18247 CrossRef CAS.
- Z. q. Liu, Q. Fang, D. Wang, D. x. Cao, G. Xue, W. t. Yu and H. Lei, Trivalent Boron as an Acceptor in Donor–π–Acceptor-Type Compounds for Single- and Two-Photon Excited Fluorescence, Chem.–Eur. J., 2003, 9, 5074–5084 CrossRef CAS PubMed.
- S. Sa, V. Mukundam, A. Kumari, R. Das and K. Venkatasubbaiah, Synthesis of pyrazole anchored three-coordinated organoboranes and their application in the detection of picric acid, Dalton Trans., 2021, 50, 6204–6212 RSC.
- R. Stahl, C. Lambert, C. Kaiser, R. Wortmann and R. Jakober, Electrochemistry and Photophysics of Donor-Substituted Triarylboranes: Symmetry Breaking in Ground and Excited State, Chem.–Eur. J., 2006, 12, 2358–2370 CrossRef CAS.
- J. C. Collings, S. Y. Poon, C. Le Droumaguet, M. Charlot, C. Katan, L. O. Pålsson, A. Beeby, J. A. Mosely, H. M. Kaiser, D. Kaufmann, W. Yeung Wong, M. B. Desce and T. B. Marder, The Synthesis and One- and Two-Photon Optical Properties of Dipolar, Quadrupolar and Octupolar Donor–Acceptor Molecules Containing Dimesitylboryl Groups, Chem.–Eur. J., 2009, 15, 198–208 CrossRef CAS.
- S. Griesbeck, E. Michail, F. Rauch, H. Ogasawara, C. Wang, Y. Sato, R. M. Edkins, Z. Zhang, M. Taki, C. Lambert, S. Yamaguchi and T. B. Marder, The Effect of Branching on the One- and Two-Photon Absorption, Cell Viability, and Localization of Cationic Triarylborane Chromophores with Dipolar versus Octupolar Charge Distributions for Cellular Imaging, Chem.–Eur. J., 2019, 25, 13164–13175 CrossRef CAS.
- S. Griesbeck, E. Michail, C. Wang, H. Ogasawara, S. Lorenzen, L. Gerstner, T. Zang, J. Nitsch, Y. Sato, R. Bertermann, M. Taki, C. Lambert, S. Yamaguchi and T. B. Marder, Tuning the π-bridge of quadrupolar triarylborane chromophores for one- and two-photon excited fluorescence imaging of lysosomes in live cells, Chem. Sci., 2019, 10, 5405–5422 RSC.
- S. Griesbeck, Z. Zhang, M. Gutmann, T. Lühmann, R. M. Edkins, G. Clermont, A. N. Lazar, M. Haehnel, K. Edkins, A. Eichhorn, M. B. Desce, L. Meinel and T. B. Marder, Water-Soluble Triarylborane Chromophores for One- and Two-Photon Excited Fluorescence Imaging of Mitochondria in Cells, Chem.–Eur. J., 2016, 22, 14701–14706 CrossRef CAS PubMed.
- L. Ji, R. M. Edkins, L. J. Sewell, A. Beeby, A. S. Batsanov, K. Fucke, M. Drafz, J. A. Howard, O. Moutounet, F. Ibersiene, A. Boucekkine, E. Furet, Z. Liu, J. F. Halet, C. Katan and T. B. Marder, Experimental and Theoretical Studies of Quadrupolar Oligothiophene-Cored Chromophores Containing Dimesitylboryl Moieties as π-Accepting End-Groups: Syntheses, Structures, Fluorescence, and One- and Two-Photon Absorption, Chem.–Eur. J., 2014, 20, 13618–13635 CrossRef CAS.
- D. Banerjee, S. S. B. Moram, C. Byram, J. Rathod, T. Jena, G. K. Podagatlapalli and V. R. Soma, Plasmon-enhanced ultrafast and tunable thermo-optic nonlinear optical properties of femtosecond laser ablated TiO2 and Silver-doped TiO2 nanoparticles, Appl. Surf. Sci., 2021, 569, 151070 CrossRef CAS.
- T. Sarma, P. K. Panda, P. Anusha and S. V. Rao, Dinaphthoporphycenes: Synthesis and Nonlinear Optical Studies, Org. Lett., 2011, 13, 188–191 CrossRef CAS PubMed.
- L. Zhu, Y. Yi, Z. Shuai, J.-L. Brédas, D. Beljonne and E. Zojer, Structure-property relationships for three-photon absorption in stilbene-based dipolar and quadrupolar chromophores, J. Chem. Phys., 2006, 125, 044101 CrossRef.
- M. S. Ahmed, C. Biswas, B. Bhavani, S. Prasanthkumar, D. Banerjee, V. Kumar, P. Chetti, L. Giribabu, V. R. Soma and S. S. K. Raavi, Metalated porphyrin-napthalimide based donor-acceptor systems with long-lived triplet states and effective three-photon absorption, J. Photochem. Photobiol., A, 2023, 435, 114324 CrossRef CAS.
- M. S. Ahmed, C. Biswas, D. Banerjee, P. Chetti, J.-S. Yang, V. R. Soma and S. S. K. Raavi, Femtosecond Third-Order Non-Linear Optical Properties of Unconstrained Green Fluorescence Protein Chromophores, Front. Phys., 2022, 10, 914135 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental procedure details and photophysical and theoretical data. CCDC 2288523 for 1, 2288524 for 2 and 2291487 for 1-OTF. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05656f |
| ‡ RA and MMAT contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |