
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective hydrogenolysis of the Csp2–O bond in the furan ring using hydride–proton pairs derived from hydrogen spillover†
Fangfang
Peng
a,
Bin
Zhang
a,
Runyao
Zhao
ab,
Shiqiang
Liu
a,
Yuxuan
Wu
ab,
Shaojun
Xu
 c,
Luke L.
Keenan
d,
Huizhen
Liu
ab,
Qingli
Qian
ab,
Tianbin
Wu
a,
Haijun
Yang
a,
Zhimin
Liu
ab,
Jikun
Li
*abe,
Bingfeng
Chen
*a,
Xinchen
Kang
*ab and
Buxing
Han
*abf
c,
Luke L.
Keenan
d,
Huizhen
Liu
ab,
Qingli
Qian
ab,
Tianbin
Wu
a,
Haijun
Yang
a,
Zhimin
Liu
ab,
Jikun
Li
*abe,
Bingfeng
Chen
*a,
Xinchen
Kang
*ab and
Buxing
Han
*abf
aBeijing National Laboratory for Molecular Sciences, CAS Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Center for Carbon Neutral Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: hanbx@iccas.ac.cn; kangxinchen@iccas.ac.cn; chenbf@iccas.ac.cn; jikunli@iccas.ac.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 101408, P. R. China
cDepartment of Chemical Engineering, School of Engineering, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
dDiamond Light Source, Harwell Science Campus, Oxfordshire, OX11 0DE, UK
eBeijing National Laboratory for Molecular Sciences, Key Laboratory of Photochemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China
fShanghai Key Laboratory of Green Chemistry and Chemical Processes, State Key Laboratory of Petroleum Molecular & Process Engineering, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai, 200062, China
First published on 28th October 2024
Abstract
Selective hydrogenolysis of biomass-derived furanic compounds is a promising approach for synthesizing aliphatic polyols by opening the furan ring. However, there remains a significant need for highly efficient catalysts that selectively target the Csp2–O bond in the furan ring, as well as for a deeper understanding of the fundamental atomistic mechanisms behind these reactions. In this study, we present the use of Pt–Fe bimetallic catalysts supported on layered double hydroxides [PtFex/LDH] for the hydrogenolysis of furanic compounds into aliphatic alcohols, achieving over 90% selectivity toward diols and triols. Our findings reveal that the synergy between Pt nanoparticles, atomically dispersed Pt sites and the support facilitates the formation of hydride-proton pair at the Ptδ+⋯O2− Lewis acid–base unit of PtFex/LDH through hydrogen spillover. The hydride specifically targets the Csp2–O bond in the furan ring, initiating an SN2 reaction and ring cleavage. Moreover, the presence of Fe improves the yield of desired alcohols by inhibiting the adsorption of vinyl groups, thereby suppressing the hydrogenation of the furan ring.
Introduction
Due to the depletion of fossil resources and the importance of carbon cycling, the conversion of biomass into valuable chemicals has received much attention.1–3 Furanic compounds, such as furfural (FFR), furfural alcohol (FA) and 5-hydroxymethylfurfural (HMF), derived from the cellulose hydrolysis, represent crucial platform molecules in biomass conversion. Hydrogenolysis of furanic compounds, achieved through the cleavage of Csp2–O bonds in the furan ring, is a highly effective method for producing diols or triols, which are crucial precursors for a range of applications, including polyurethanes, polyesters, polymeric plasticizers and low-toxic microbicides.4–6 Supported Pt catalysts have proven effective for the selective hydrogenolysis of furanic compounds, including FFR, FA, HMF, furancarboxylic acids with factors such as composition, metal dispersion, and support all playing significant roles in overall catalytic performance.7–14 Despite decades of extensive research aimed at improving the selectivity of diols or triols, achieving a comprehensive understanding of the fundamental atomistic mechanisms that govern the cleavage of Csp2–O bonds in the furan ring remains a critical objective.In addition to the active sites of catalysts, the selectivity for ring-opening products is also associated with the active hydrogen species. In catalytic hydrogenation reactions, the dissociation of H2 determines both activity and selectivity. It is widely accepted that on extended metal surfaces (metal–metal sites), H2 dissociation tends to favor the homolytic pathway. In contrast, when the local coordination structures of the metal centers involve Lewis acid–base units, H2 molecules are more inclined to undergo heterolytic dissociation at these sites, forming hydride–proton pairs.15–17 The resulting hydride–proton pairs facilitate the hydrogenation of polar bonds since polar bonds are excellent acceptors for both hydrides and protons.15,16 However, the relatively high energy barrier associated with the direct heterolytic pathway may lead to a sluggish hydrogenation kinetics compared with the barrierless homolytic dissociation of H2 on metal ensembles.15 Hydrogen spillover involves the homolytic dissociation of H2 on metals, where the resulting active H* species migrate to either the support or the metal sites. This migration can lead to charge separation into protons and/or hydrides.18–21 Specifically, we can infer from the local coordination structures of H2 heterolytic dissociation that proton and hydride pairs could form through charge separation of the active H* species when Lewis acid–base units are present on the catalyst.
Over the past decades, layered double oxides (LDOs) and hydroxides (LDHs) have made significant contributions to high-value upgrading reactions of biomass resources.22 Due to the availability of acid–base sites, they are excellent candidates for generating protons and hydrides via spillover. Herein, we developed Mg,Al-LDH-supported Pt/Fe catalysts (PtFex/LDH) for the hydrogenolysis of biomass-derived furanic compounds into their corresponding ring-opening alcohol products, achieving diol/triol yields exceeding 90% with complete conversion of the furanic compounds. The hydrogenolysis process of the Csp2–O bond was elucidated through spectroscopic measurements, Kinetic isotope experiment analysis, and density functional theory (DFT) calculations, highlighting the role of hydrogen species—H+ and H− pairs—in opening the unsaturated furan ring. The findings suggest that this selective hydrogenolysis follows an SN2 mechanism, with H− acting as the nucleophile. Furthermore, systematic characterization and control experiments revealed the synergy between Pt nanoparticles (NPs) and Ptδ+⋯O2− Lewis acid–base units, as well as the role of Fe in facilitating the ring-opening of furan compounds.
Results and discussion
Catalyst characterizations
The catalysts were synthesized using Mg,Al-LDH as the support precursor. The crystal phase of LDH is identified as Mg6Al2(OH)18·4.5H2O (PDF#35-0965) (Fig. 1A), and it exhibits irregular nanosheet morphology, as shown from the scanning electron microscopy (SEM) images (Fig. S1†). After calcination and H2 reduction treatment, the LDH converted to mixed Mg/Al oxide [(Mg,Al)O]. Catalysts with different Fe/Pt atomic ratio were prepared and the Fe/Pt atomic ratios were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES), as shown in Table S1.† We denote the as-prepared catalyst PtFex/(Mg,Al)O (x = Fe/Pt atomic ratio), which can revert to PtFex/LDH under hydration conditions owing to the “memory effect”,23 as confirmed by the X-ray diffraction (XRD) pattern (Fig. 1A). High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images and elemental distribution mappings of the PtFe0.7/(Mg,Al)O demonstrate the uniform dispersion of Pt and Fe on the support (Fig. 1B). Pt in Pt/(Mg,Al)O is found to exist in both NPs and atomically dispersed states, as observed through aberration-corrected HAADF-STEM (Fig. S2†). The PtFe0.7/(Mg,Al)O exhibits a similar morphology to Pt/(Mg,Al)O (Fig. 1C), indicating that the addition of Fe does not alter or influence the state of Pt. Scanning transmission electron microscopy with energy dispersive X-ray spectroscopy (STEM-EDX) line-scanning analysis of PtFe0.7/(Mg,Al)O demonstrates the presence of Fe deposition on the Pt surface (Fig. S3†).12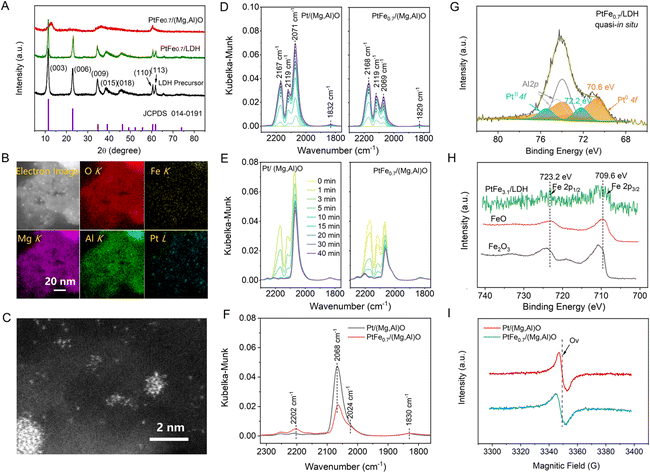 | ||
| Fig. 1 (A) XRD patterns of LDH precursor as well as PtFe0.7/(Mg,Al)O catalyst before and after water treatment. (B) HAADF-STEM image and elemental distribution mappings of PtFe0.7/(Mg,Al)O. (C) Aberration-corrected HAADF-STEM image of PtFe0.7/(Mg,Al)O. (D and E) The dependence of DRIFTS spectra for CO adsorption (D) and desorption (E) on time over Pt/(Mg,Al)O and PtFe0.7/(Mg,Al)O. (F) Comparison of DRIFTS spectra over Pt/(Mg,Al)O and PtFe0.7/(Mg,Al)O after CO adsorption and followed by a Ar flow of 40 min. (G) Quasi-in situ Pt 4f XPS spectra of PtFe0.7/LDH. (H) Quasi-in situ Fe 2p XPS spectra of PtFe3.1/LDH and FeOx references. (I) EPR spectra of Pt/(Mg,Al)O and PtFe0.7/(Mg,Al)O. | ||
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was conducted over Pt/(Mg,Al)O and PtFe0.7/(Mg,Al)O using CO as the probe to further explore the structure of the catalysts (Fig. 1D–F). As seen in Fig. 1D, the doublet peaks at approximately 2167 cm−1 and 2119 cm−1 are attributed to CO gas.24,25 Similar features of CO adsorption bands are also observed in samples without Pt, such as Fe/(Mg,Al)O and (Mg,Al)O (Fig. S4†). The band at around 2070 cm−1 is assigned to CO linearly adsorbed on Pt metal sites, while the shoulder at approximately 2024 cm−1 corresponds to CO adsorbed on lower-coordination Pt metal sites.26–28 The decrease in intensity of the CO adsorption band at 2070 cm−1 indicates a significant reduction in the exposed Pt metal sites in PtFe0.7/(Mg,Al)O compared with Pt/(Mg,Al)O. This may be attributed to the Fe oxide/hydroxide species deposited on the surface of Pt nanoparticles, which obscure partial active sites of Pt,29 which aligns with the STEM-EDX line-scanning results (Fig. S3†). The weak peak at around 1830 cm−1 is assigned to bridge-bonded CO adsorption on Pt. In DRIFTS spectra of CO desorption (Fig. 1E and F), the peak at 2202 cm−1 is associated with CO bound to cationic Pt sites,24,30 which is more prominent in PtFe0.7/(Mg,Al)O than in Pt/(Mg,Al)O.
Quasi-in situ X-ray photoelectron spectroscopy (XPS) analysis of PtFex/LDH (the hydrothermally treated PtFex/(Mg,Al)O samples) was further conducted in an anaerobic environment. Pt 4f spectrum clearly demonstrates that both Pt0 and Pt2+ are present on the surface of these catalysts (Fig. 1G).31 Compared with Pt/LDH, the binding energies (BE) of Pt0 in PtFex/LDH exhibit a slight positive shift (Fig. S5†), indicating a lower electron density of surface Pt. This phenomenon could be attributed to electron transfer from Pt clusters to FeOxvia Pt–O–Fe bonding.29 Notably, the oxidation state of Pt in PtFe0.7/LDH aligns with that observed in PtFe0.7/(Mg,Al)O sample (Fig. S6†). Additionally, the valence state of Fe is +2 under the anaerobic environment (Fig. 1H) but +3 in air (Fig. S7†). X-ray absorption spectroscopy (XAS) were employed to analyze the local electronic and geometric structures of PtFex/(Mg, Al)O. The Pt L3-edge extended X-ray absorption fine structure (EXAFS) spectra of PtFex/(Mg,Al)O exhibits two peaks at 2.0 and 2.7 Å, corresponding to the Pt–O and Pt–Pt bonds, respectively (Fig. S8, S9, and Table S2†), further affirming the co-existence of Pt single sites and clusters in PtFex/(Mg,Al)O. The Fe K-edge EXAFS spectra of PtFex/(Mg,Al)O (Fig. S10, S11 and Table S2†) also provide evidence for the absence of a Pt–Fe bond, confirming that no Pt–Fe alloy is formed.32,33 Electron paramagnetic resonance (EPR) spectroscopy were employed to characterize the paramagnetic centers of the catalysts. EPR signals of oxygen vacancies (Ov) were observed at 3350 G in our catalysts (Fig. 1I).
Hydrogenolysis of furanic compounds
The hydrogenolysis reaction was performed using FA as the substrate to investigate the selective cleavage of Csp2–O bond in furan ring over PtFex/(Mg,Al)O catalysts. As aforementioned, under hydration conditions, (Mg,Al)O can be converted into LDH (Fig. 1A). Consequently, we refer to the catalysts as PtFex/LDH during the reaction. The reaction was conducted under a H2 pressure of 1.0 MPa in water at 150 °C. As seen in Table 1, FA achieves a complete conversion within 5.5 h, with selectivity toward 1,2-pentanediol (1,2-PeD) and diols of 82.0% and 90.9%, respectively, marking one of the most promising results to date (Table S3†). Compared with the PtFe0.7/LDH catalyst, Pt/LDH exhibits similar activity but lower selectivity towards diols, while Fe/LDH exhibits only 1.4% FA conversion. This suggests that Pt is the primary active component for FA conversion, and Fe enhances the selectivity towards ring-opening products. This is because Fe reduces the electron density on the Pt surface (Fig. S5†),31 thereby improving the selectivity for C–O bond hydrogenolysis.|
|
||||||
|---|---|---|---|---|---|---|
| Catalyst | Conv. (%) | Selectivity (%) | ||||
| m | n | f | e | m + n | ||
| a Reaction conditions: 80 μL FA, 30 mg catalyst, 2.0 mL water, 1.0 MPa H2, 150 °C, 5.5 h. Pentanol including 1-pentanol and 2-pentanol. b The solvent was 2.0 mL mixture of ethanol and water (v/v = 1/1). c The solvent was 2.0 mL ethanol. | ||||||
| PtFe0.7/LDH | 100 | 82.0 | 8.9 | 4.8 | 2.3 | 90.9 |
| Pt/LDH | 100 | 70.1 | 9.1 | 1.5 | 16.3 | 79.2 |
| Fe/LDH | 1.4 | — | — | — | — | — |
| PtCo0.8/LDH | 100 | 60.8 | 7.5 | 14.0 | 13.2 | 68.3 |
| PtNi0.7/LDH | 100 | 17.0 | 10.4 | 1.0 | 69.3 | 27.4 |
| PtMn0.7/LDH | 12.1 | 28.2 | 15.0 | 2.9 | 30.5 | 43.2 |
| PtFe0.7/LDHb | 59.1 | 73.8 | 18.2 | 6.3 | 1.7 | 92 |
| PtFe0.7/LDHc | 12.6 | 37.7 | 18.5 | 3.2 | 24.0 | 56.2 |
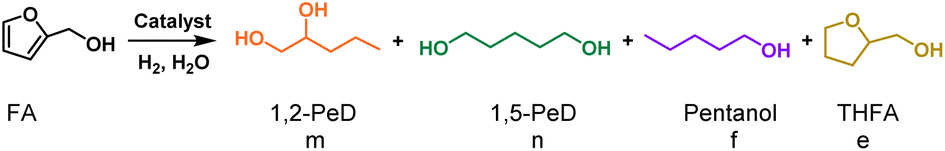
Furthermore, when Fe in PtFe0.7/LDH is substituted with other transition metals including Co, Ni and Mn, the conversion of FA or the selectivity of diols decreases. Specifically, the conversion of FA over PtMn0.7/LDH is only 12.1%, while the selectivity of diols is only 27.4% over PtNi0.7/LDH. The influence of the Fe/Pt ratio on the catalytic performance was further investigated (Fig. 2A). With increasing Fe content in PtFex/(Mg,Al)O, the formation of the ring hydrogenation product tetrahydrofurfuryl alcohol (THFA) is suppressed, leading to increased selectivity toward 1,2-PeD. However, with further increasing Fe content, the conversion of FA noticeably decreases, indicating that the excessive Fe may reduce the activity of the catalyst. Recycling experiments were conducted to assess the stability of the PtFe0.7/LDH catalyst.34 The selectivity and conversion results indicate that the catalyst remains relatively stable over three cycles (Fig. S12).†
 | ||
| Fig. 2 (A) FA hydrogenolysis over various PtFex/LDH catalysts. (B) Plot of content of different species vs. time over PtFe0.7/LDH catalyst. (C) Plot of content of different species vs. time over Pt/LDH catalyst. (D–F) Primary isotope effect for the hydrogenolysis of FA to 1,2-PeD (D), 1,5-PeD (E) and THFA (F). The reaction conditions in A–C: 80 μL FA, 30 mg catalyst, 2.0 mL water, 1.0 MPa H2, 150 °C. The reaction conditions in D–F: 80 μL FA, 30 mg catalyst, 2.0 mL H2O (or D2O), 1.0 MPa H2 (or D2), 150 °C. | ||
Fig. 2B and C illustrates the time-dependent FA conversion over Pt/LDH and PtFe0.7/LDH. 1,2-PeD remains the dominant product consistently over Pt/LDH and PtFe0.7/LDH. Notably, the cyclic hydrogenation product THFA is significantly inhibited by Fe introduction. This effect could be attributed to the reduced electron density of Pt nanoparticles (Fig. S5)† in the presence of Fe, which hinders the adsorption of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and thereby suppresses ring hydrogenation.29,31 Kinetic isotope effect (KIE) experiments, which compare the reaction rates using H2 and D2, or H2O and D2O, were conducted by replacing hydrogen (H) with deuterium (D) either in water (KIEH2O/D2O = kH2O/kD2O) or in H2 (KIEH2/D2 = kH2/kD2) (Fig. 2D–F). The apparent KIEH2/D2 value is approximately unity in both ring-opening and ring-hydrogenation reactions. Gas chromatography-mass spectrometer (GC-MS) (Fig. S13 and S14†) and nuclear magnetic resonance (NMR) (Fig. S15†) spectra indicate that D is distributed across almost all carbon atoms of the products. Considering the ∼16
C bonds and thereby suppresses ring hydrogenation.29,31 Kinetic isotope effect (KIE) experiments, which compare the reaction rates using H2 and D2, or H2O and D2O, were conducted by replacing hydrogen (H) with deuterium (D) either in water (KIEH2O/D2O = kH2O/kD2O) or in H2 (KIEH2/D2 = kH2/kD2) (Fig. 2D–F). The apparent KIEH2/D2 value is approximately unity in both ring-opening and ring-hydrogenation reactions. Gas chromatography-mass spectrometer (GC-MS) (Fig. S13 and S14†) and nuclear magnetic resonance (NMR) (Fig. S15†) spectra indicate that D is distributed across almost all carbon atoms of the products. Considering the ∼16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 total H/D ratio in the system, exchange between H and D likely occurs during hydrogen spillover (see below) before hydrogenation. The apparent KIEH2O/D2O values for THFA production and the ring-opening reaction are noticeably different, with values of ∼2 and >5, respectively. The higher KIEH2O/D2O value for the ring-opening reaction suggests that it is more likely to involve a high-barrier hydrogen transfer process than THFA production.
:1 total H/D ratio in the system, exchange between H and D likely occurs during hydrogen spillover (see below) before hydrogenation. The apparent KIEH2O/D2O values for THFA production and the ring-opening reaction are noticeably different, with values of ∼2 and >5, respectively. The higher KIEH2O/D2O value for the ring-opening reaction suggests that it is more likely to involve a high-barrier hydrogen transfer process than THFA production.
Hydrogenolysis reactions of other furanic compounds, including furan, furfural (FFR) and 5-hydroxymethylfurfural (HMF), were also conducted over the PtFe0.7/LDH catalyst. As anticipated, high selectivity towards ring-opened alcohols was achieved (Fig. S16–S19 and Table S4†). Furanic compounds with oxygenated side chains, such as FA, FFR and HMF, exhibit higher selectivity toward ring-opened products, benefiting from the oxygenated side chain preferentially absorbing on the catalyst surface. These catalytic results confirm that PtFe0.7/LDH is indeed a highly active and selective catalyst for the hydrogenolysis of various furanic compounds to polyols. Particularly, it is observed that FFR is fully converted into FA over PtFe0.7/LDH within 30 min (Fig. S20†), demonstrating the high activity of hydrogen species for the hydrogenation of polar CO bond.
Hydrogen spillover and Pt–hydrides
It has been established that H2 activation plays a crucial role in determining the selectivity of the catalysts for hydrogenation/hydrogenolysis.15 To distinguish H2 dissociation on Pt NPs or on Pt single sites, CO poisoning experiments were conducted over Pt/LDH and PtFe0.7/LDH catalysts. Upon the addition of 0.2 MPa CO, the conversion of FA decreases to 1.2% and 8% over Pt/LDH and PtFe0.7/LDH, respectively (Fig. S21†). Given that single metal sites exhibit higher CO tolerance compared with metal NPs,35 the reduced activity of catalysts can be attributed to the poisoned Pt NPs hindering H2 dissociation. Therefore, it can be speculated that H2 molecules are dissociated into H atoms on Pt NPs, which may subsequently spill over to the LDH support and Pt single atom sites.36 H2 spillover experiments were conducted by the reduction of WO3. The color of WO3 sample changed from yellow to black after being mixed with Pt/(Mg,Al)O or PtFex/(Mg,Al)O treated with hydrogen, demonstrating the occurrence of H2 spillover (Fig. 3A). This is because H atom can readily react with yellow WO3 to form black HxWO3 when spillover occurs.37 The presence of oxygen vacancies on the (Mg,Al)O support may facilitate the hydrogen spillover process.38,39 | ||
| Fig. 3 (A) Photographs of samples before and after treatment with H2 at 30 °C for 10 min. (B) DRIFTS spectra in H2 over Pt/(Mg,Al)O and PtFe0.7/(Mg, Al) O (C) catalysts. (D) Hydrogenolysis of various furanic substrates with different double bonds over PtFe0.7/(Mg,Al)O catalyst. Reaction conditions: 80 μL furanic compounds, 30 mg catalyst, 2.0 mL water, 1.0 MPa H2, 150 °C, 5.5 h. (E) The LUMO Löwdin populations of C2 and C5 in protonated furanic compounds and LUMO energy in protonated and unprotonated furanic compounds. (F) LUMOs of different furanic compounds, contoured at 0.029 a.u. | ||
An in situ diffuse reflection infrared Fourier transform spectroscopy (DRIFTS) study in H2 over Pt/(Mg,Al)O and PtFe0.7/(Mg,Al)O catalysts was conducted to analyze the surface species (Fig. 3B and C). The bands observed in the range of 2800∼3700 cm−1 and around 1600 cm−1 are assigned to hydroxyl stretching vibrations and water bending vibrations, respectively, which derives from the reduction of the supported PtOx phase.40,41 It seems that the addition of Fe reduces the intensity of the hydroxyl and water peaks. According to the previous reports,41,42 the peaks at ∼2050 cm−1 and ∼1950 cm−1 are related to strongly adsorbed linear Pt–H species. The difference between these two strongly adsorbed species might be related to the type of environment around the hydride. It has been proven that under negative fields, the stretching frequency of Pt–H can shift to a lower frequency.43 As shown in Fig. 3B and C, compared with Pt/(Mg,Al)O, PtFe0.7/(Mg,Al)O exhibits a higher ratio of low-frequency Pt–H to high-frequency Pt–H, indicating that the addition of Fe promotes the generation of more negatively charged Pt–H species.
From the experimental observations, we propose that H2 molecules first undergo barrierless homolysis activation on metallic Pt NPs of PtFex/(Mg,Al)O catalysts, forming neutral active H* species. In the presence of Ptδ+⋯O2− Lewis acid–base sites, these neutral active H* species can then migrate either to Pt single sites, producing Pt–Hδ− species, or interact with oxygen species, producing O–H+ species.44 Therefore, Pt–Hδ− and O–H+ likely function as active species for the ring-opening reaction. Low conversion and diols selectivity are observed when the reaction is conducted in ethanol or a water/ethanol mixture (Table 1), suggesting that water is crucial for the hydrogenation/hydrogenolysis of FA, likely due to its role in hydrogen spillover and proton transfer.18,45–49
Mechanistic analysis
To further clarify the reaction mechanism, the hydrogenolysis reaction of furan rings with different double bonds were conducted (Fig. 3D). The resulting order of ring-opening selectivity is furan > 2,3-dihydrofuran > 2,5-dihydrofuran > tetrahydrofuran = 0. DFT calculations show that the lowest unoccupied molecular orbital (LUMO) contains a higher contribution from C2 p-orbitals as well as significant C–O π antibonding character when a double bond is adjacent to the oxygen of the furan ring (Fig. 3E and F). This makes the carbon atom (C2) next to the oxygen more susceptible to nucleophilic attack by the hydride, resulting in the cleavage of the Csp2–O bond through an SN2 reaction.As mentioned above, a significant normal KIE is observed (KIEH2O/D2O > 5) in the ring-opening reaction (Fig. 2D and E). Such a large H–D KIE in hydrogenation has also been observed on a Pd single atom catalyst (kH/kD = 5.75),17 suggesting that the rate-determining step is proton transfer rather than the hydrogen atom or hydride transfer from the Pt–Hδ− motif. Otherwise, an inverse KIE would be observed as the force constant of Pt–H bond is smaller than that of C–H bond.50–52 By comparison, for the ring hydrogenation to THFA, a smaller KIE (KIEH2O/D2O ∼2) is observed, which may be attributed to a weighted average of two pathways: one involving neutral hydrogen (H*) on the Pt nanoparticle surface, and the other involving polar hydrogen (Hδ− and H+). The former pathway is dominant due to the nonpolar nature of the CC bond.47
Based on the experimental and theoretical evidence, we propose the reaction mechanism for the ring-opening of FA over PtFex/LDH as illustrated in Fig. 4. The FA hydrogenolysis starts from the barrierless homolysis activation of H2 on the Pt NPs, and then the active H* species migrate to the Pt single sites and the LDH support, producing Pt–Hδ− and O–H+ (proton) species at the Ptδ+⋯O2− Lewis acid–base unit of PtFex/LDH.44 It should be noted that in an aqueous solution, H+ may migrate to other oxygen atoms on the support or form hydrated protons during the transfer process. DFT calculations indicate a reduction in the LUMO energy after the furanic compounds are protonated (Fig. 3E and S22†). This suggests that the reaction is initiated by the proton combining with O on the furan ring, and subsequently Hδ− attacks the carbon atom of the Csp2–O bond in the furan ring, initiating an SN2 reaction that cleaves the furan ring. Upon completion of the ring-opening, the reaction intermediates form unsaturated alcohols. Subsequent hydrogenation of these unsaturated alcohols yields the final desired alcohol products.
 | ||
| Fig. 4 Proposed mechanism for the ring-opening of FA over PtFex/LDH catalyst. | ||
Further examination of the DFT results reveals the Löwdin population of the LUMO on the carbon atom with oxygenated side chain is lower compared with that without such a chain (Fig. S22†), leading to Hδ− predominantly targeting the carbon atom of the Csp2–O bond furthest from the hydroxymethyl group. The steric hindrance caused by the hydroxymethyl group may be another reason for the higher selectivity of 1,2-PeD over 1,5-PeD.53 The addition of Fe enhances catalytic selectivity by facilitating the formation of Fe oxide/hydroxide species on the Pt surface, which inhibit the adsorption of vinyl groups and consequently suppress the hydrogenation of the furan ring.29
Conclusions
In this work, we developed PtFex/(Mg,Al)O [PtFex/LDH under hydrothermal conditions] catalysts for the selective hydrogenolysis of the Csp2–O bond in the furan ring to polyols. Over 90% selectivity for diols or triols can be achieved at complete conversion of the furanic compounds using the PtFe0.7/LDH catalyst. During the reaction, H2 dissociates on Pt NPs, and active hydrogen atoms migrate to the Ptδ+⋯O2− unit of PtFex/LDH to produce Pt–Hδ− and O–H+ pairs via spillover. The resulting Pt–Hδ− and O–H+ serve as active species for cleaving the Csp2–O bond through SN2 reaction, facilitating the ring-opening of furanic compounds. The introduction of Fe inhibits the adsorption of vinyl groups on the Pt surface, thereby suppressing the hydrogenation of the furan ring. This study underscores the synergistic interplay among NPs, single metal sites and Lewis basic sites on the support in achieving the selective hydrogenation of furanic compounds. Additionally, the regioselectivity of ring-opening reaction are elucidated at the molecular level. We anticipate that these findings will provide valuable guidance for designing catalysts for other selective hydrogenation reactions.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
FFP: performed the experiments and analysis, as well as wrote the manuscript. BXH, XCK and BFC: funding acquisition, supervision and edited the manuscript. JKL: performed the computations, wrote the computational methodology, and participate in spectroscopic and mechanistic analysis. BZ: participate in DRIFT data analysis. RYZ, SJX and LLK: performed the EXAFS data fitting and analysis. Other people: participated in discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Key Research and Development Program of China (2023YFA1506804, 2023YFA1508103), National Natural Science Foundation of China (22273108, 22121002, 22273111), Beijing Natural Science Foundation (2222043), CAS Project for Young Scientists in Basic Research (YSBR-050), ICCAS program for Carbon Neutral Chemistry (CCNC-202403) and Youth Fund of National Natural Science Foundation (223002209). The authors gratefully acknowledge the cooperation of the beamline scientists at 1W1B and 4B9A Beijing Synchrotron Radiation Facility, China.References
- K. Lee, Y. X. Jing, Y. Q. Wang and N. Yan, Nat. Rev. Chem, 2022, 6, 635–652 CrossRef PubMed.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. L. Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- Y. Nakagawa, M. Tamura and K. Tomishige, ACS Catal., 2013, 3, 2655–2668 CrossRef CAS.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS PubMed.
- N. Enjamuri and S. Darbha, Catal. Rev., 2020, 62, 566–606 CrossRef CAS.
- T. Mizugaki, T. Yamakawa, Y. Nagatsu, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, ACS Sustain. Chem. Eng., 2014, 2, 2243–2247 CrossRef CAS.
- Y. R. Zhu, W. F. Zhao, J. Zhang, Z. An, X. D. Ma, Z. J. Zhang, Y. T. Jiang, L. R. Zheng, X. Shu, H. Y. Song, X. Xiang and J. He, ACS Catal., 2020, 10, 8032–8041 CrossRef CAS.
- T. Tong, X. H. Liu, Y. Guo, M. N. Banis, Y. F. Hu and Y. Q. Wang, J. Catal., 2018, 365, 420–428 CrossRef CAS.
- R. F. Ma, X. P. Wu, T. Tong, Z. J. Shao, Y. Q. Wang, X. H. Liu, Q. N. Xia and X. Q. Gong, ACS Catal., 2017, 7, 333–337 CrossRef CAS.
- T. Tong, Q. N. Xia, X. H. Liu and Y. Q. Wang, Catal. Commun., 2017, 101, 129–133 CrossRef CAS.
- C. Cao, W. X. Guan, Q. Y. Liu, L. Li, Y. Su, F. Liu, A. Q. Wang and T. Zhang, Green Chem., 2024, 26, 6511–6519 RSC.
- T. Asano, H. Takagi, Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem., 2019, 21, 6133–6145 RSC.
- Q. H. Sun, S. Wang and H. C. Liu, ACS Catal., 2019, 9, 11413–11425 CrossRef CAS.
- D. R. Aireddy and K. L. Ding, ACS Catal., 2022, 12, 4707–4723 CrossRef CAS.
- H. T. Cai, R. Schimmenti, H. Y. Nie, M. Mavrikakis and Y. H. C. Chin, ACS Catal., 2019, 9, 9418–9437 CrossRef CAS.
- P. X. Liu, Y. Zhao, R. X. Qin, S. G. Mo, G. X. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. D. Zang, B. H. Wu, G. Fu and N. F. Zheng, Science, 2016, 352, 797–801 CrossRef CAS PubMed.
- J. Shangguan and Y. H. C. Chin, ACS Catal., 2019, 9, 1763–1778 CrossRef CAS.
- J. Im, H. Shin, H. Jang, H. Kim and M. Choi, Nat. Commun., 2014, 5, 3370 CrossRef PubMed.
- M. J. Hülsey, V. Fung, X. D. Hou, J. S. Wu and N. Yan, Angew. Chem., Int. Ed., 2022, 61, e202208237 CrossRef PubMed.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- Y. S. Yang, Z. Ren, S. J. Zhou and M. Wei, ACS Catal., 2021, 11, 6440–6454 CrossRef CAS.
- X. C. Zhu, C. P. Chen, H. R. Suo, Q. Wang, Y. X. Shi, D. O'Hare and N. S. Cai, Energy, 2019, 167, 960–969 CrossRef CAS.
- L. DeRita, S. Dai, K. Lopez-Zepeda, N. Pham, G. W. Graham, X. Q. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 14150–14165 CrossRef CAS PubMed.
- X. I. Pcrcira-Hcrnándcz, A. DeLaRiva, V. Muravev, D. Kunwar, H. Xiong, B. Sudduth, M. Engelhard, L. Kovarlk, E. J. M. Hcnscn, Y. Wang and A. K. Datye, Nat. Commun., 2019, 10, 1358 CrossRef PubMed.
- M. J. Kale and P. Christopher, ACS Catal., 2016, 6, 5599–5609 CrossRef CAS.
- R. K. Brandt, M. R. Hughes, L. P. Bourget, K. Truszkowska and R. G. Greenler, Surf. Sci., 1993, 286, 15–25 CrossRef CAS.
- M. J. Kappers and J. H. Vandermaas, Catal. Lett., 1991, 10, 365–373 CrossRef CAS.
- Y. Wang, R. X. Qin, Y. K. Wang, J. Ren, W. T. Zhou, L. Y. Li, J. Ming, W. Y. Zhang, G. Fu and N. F. Zheng, Angew. Chem., Int. Ed., 2020, 59, 12736–12740 CrossRef CAS PubMed.
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Science, 2015, 350, 185–189 CrossRef CAS PubMed.
- S. X. Bai, L. Z. Bu, Q. Shao, X. Zhu and X. Q. Huang, J. Am. Chem. Soc., 2018, 140, 8384–8387 CrossRef CAS PubMed.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- S. Mukerjee, S. Srinivasan, M. P. Soriaga and J. Mcbreen, J. Electrochem. Soc., 1995, 142, 1409–1422 CrossRef CAS.
- S. L. Scott, ACS Catal., 2018, 8, 8597–8599 CrossRef CAS.
- H. F. Qi, J. Yang, F. Liu, L. L. Zhang, J. Y. Yang, X. Y. Liu, L. Li, Y. Su, Y. F. Liu, R. Hao, A. Q. Wang and T. Zhang, Nat. Commun., 2021, 12, 3295 CrossRef CAS PubMed.
- M. Xiong, Z. Gao and Y. Qin, ACS Catal., 2021, 11, 3159–3172 CrossRef CAS.
- C. T. Wang, E. Guan, L. Wang, X. F. Chu, Z. Y. Wu, J. Zhang, Z. Y. Yang, Y. W. Jiang, L. Zhang, X. J. Meng, B. C. Gates and F. S. Xiao, J. Am. Chem. Soc., 2019, 141, 8482–8488 CrossRef CAS PubMed.
- R. Prins, V. K. Palfi and M. Reiher, J. Phys. Chem. C, 2012, 116, 14274–14283 CrossRef CAS.
- R. Prins, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS PubMed.
- D. H. Lenz and W. C. Conner, J. Catal., 1988, 112, 116–125 CrossRef CAS.
- M. Carosso, E. Vottero, A. Lazzarini, S. Morandi, M. Manzoli, K. A. Lomachenko, M. J. Ruiz, R. Pellegrini, C. Lamberti, A. Piovano and E. Groppo, ACS Catal., 2019, 9, 7124–7136 CrossRef CAS.
- D. Palecek, G. Tek, J. G. Lan, M. Iannuzzi and P. Hamm, J. Phys. Chem. Lett., 2018, 9, 1254–1259 CrossRef CAS PubMed.
- M. Tomonari and O. Sugino, Chem. Phys. Lett., 2007, 437, 170–175 CrossRef CAS.
- X. Deng, B. Qin, R. Z. Liu, X. T. Qin, W. L. Dai, G. J. Wu, N. J. Guan, D. Ma and L. D. Li, J. Am. Chem. Soc., 2021, 143, 20898–20906 CrossRef CAS PubMed.
- X. J. Zhao, J. Wang, L. Z. Lian, G. J. Zhang, P. An, K. Zeng, H. C. He, T. C. Yuan, J. H. Huang, L. Q. Wang and Y. N. Liu, ACS Catal., 2023, 13, 2326–2334 CrossRef CAS.
- L. R. Merte, G. W. Peng, R. Bechstein, F. Rieboldt, C. A. Farberow, L. C. Grabow, W. Kudernatsch, S. Wendt, E. Lægsgaard, M. Mavrikakis and F. Besenbacher, Science, 2012, 336, 889–893 CrossRef CAS PubMed.
- Z. Zhao, R. Bababrik, W. H. Xue, Y. P. Li, N. M. Briggs, D. T. Nguyen, U. Nguyen, S. P. Crossley, S. W. Wang, B. Wang and D. E. Resasco, Nat. Catal., 2019, 2, 431–436 CrossRef CAS.
- Q. Deng, R. Zhou, Y. C. Zhang, X. Li, J. H. Li, S. B. Tu, G. Sheng, J. Wang, Z. L. Zeng, T. Yoskamtorn and S. C. E. Tsang, Angew. Chem., Int. Ed., 2023, 62, e202211461 CrossRef CAS PubMed.
- X. Li, Z. K. Tong, S. Zhu, Q. Deng, S. X. Chen, J. Wang, Z. L. Zeng, Y. L. Zhang, J. J. Zou and S. G. Den g, J. Catal., 2022, 405, 363–372 CrossRef CAS.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, 2nd edn, 2006 Search PubMed.
- F. F. Peng, J. F. Xiang, H. S. Qin, B. F. Chen, R. Duan, W. L. Zhao, S. Q. Liu, T. B. Wu, W. L. Yuan, Q. Li, J. K. Li, X. C. Kang and B. X. Han, J. Am. Chem. Soc., 2023, 145, 23905–23909 CrossRef CAS PubMed.
- T. F. Liu, M. Y. Guo, A. Orthaber, R. Lomoth, M. Lundberg, S. Ott and L. Hammarström, Nat. Chem., 2018, 10, 881–887 CrossRef CAS PubMed.
- K. Hayashi, P. V. Sushko, Y. Hashimoto, A. L. Shluger and H. Hosono, Nat. Commun., 2014, 5, 3515 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05751a |
| This journal is © The Royal Society of Chemistry 2024 |