
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pnictogen-based vanadacyclobutadiene complexes†
Mehrafshan G.
Jafari
 a,
John B.
Russell
a,
Hwan
Myung
bc,
Seongyeon
Kwon
cb,
Patrick J.
Carroll
a,
Michael R.
Gau
a,
Mu-Hyun
Baik
*cb and
Daniel J.
Mindiola
*a
a,
John B.
Russell
a,
Hwan
Myung
bc,
Seongyeon
Kwon
cb,
Patrick J.
Carroll
a,
Michael R.
Gau
a,
Mu-Hyun
Baik
*cb and
Daniel J.
Mindiola
*a
aDepartment of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. E-mail: mindiola@sas.upenn.edu
bDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea
cCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea
First published on 1st November 2024
Abstract
The reactivity of the V![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CtBu multiple bonds in the complex (dBDI)VCtBu(OEt2) (C) (dBDI2− = ArNC(CH3)CHC(CH2)NAr, Ar = 2,6-iPr2C6H3) with unsaturated substrates such as NCR (R = Ad or Ph) and PCAd leads to the formation of rare 3d transition metal compounds featuring α-aza-vanadacyclobutadiene, (dBDI)V(κ2-C,N-tBuCC(R)N) (R = Ad, 1; R = Ph, 2) and β-phospha-vanadacyclobutadiene moieties, (dBDI)V(κ2-C,C-tBuCPCAd) (3). Complexes 1–3 are characterized using multinuclear and multidimensional NMR spectroscopy, including the preparation of the 50% 15N-enriched isotopologue (dBDI)V(κ2-C,N-tBuCC(Ad)15N) (1-15N). Solid-state structural analysis is used to determine the dominant resonance structures of these unique pnictogen-based vanadacyclobutadienes. A systematic comparison with the known vanadacyclobutadiene (dBDI)V(κ2-C,C-tBuCC(H)CtBu) (4) is also presented. Theoretical investigations into the electronic structure of 2–4 highlight the crucial role of unique V–heteroatom interactions in stabilizing the vanadacyclobutadienes and identify the most dominant resonance structures.
CtBu multiple bonds in the complex (dBDI)VCtBu(OEt2) (C) (dBDI2− = ArNC(CH3)CHC(CH2)NAr, Ar = 2,6-iPr2C6H3) with unsaturated substrates such as NCR (R = Ad or Ph) and PCAd leads to the formation of rare 3d transition metal compounds featuring α-aza-vanadacyclobutadiene, (dBDI)V(κ2-C,N-tBuCC(R)N) (R = Ad, 1; R = Ph, 2) and β-phospha-vanadacyclobutadiene moieties, (dBDI)V(κ2-C,C-tBuCPCAd) (3). Complexes 1–3 are characterized using multinuclear and multidimensional NMR spectroscopy, including the preparation of the 50% 15N-enriched isotopologue (dBDI)V(κ2-C,N-tBuCC(Ad)15N) (1-15N). Solid-state structural analysis is used to determine the dominant resonance structures of these unique pnictogen-based vanadacyclobutadienes. A systematic comparison with the known vanadacyclobutadiene (dBDI)V(κ2-C,C-tBuCC(H)CtBu) (4) is also presented. Theoretical investigations into the electronic structure of 2–4 highlight the crucial role of unique V–heteroatom interactions in stabilizing the vanadacyclobutadienes and identify the most dominant resonance structures.
Introduction
Metallacyclobutadienes (MCBDs) are proposed intermediates, and in some cases, isolable species in alkylidyne-alkyne cross-metathesis reactions.1–22 However, when a metal alkylidyne (MCR), reacts with a nitrile (NCR), or a phosphaalkyne (PCR), cross-metathesis proceeds via the formation of pnictogen-based heterometallacyclobutadiene. In the case of NCR cycloaddition across the MC bond, the reaction yields an α-aza-metallacyclobutadiene (α-N-MCBD, top left of Fig. 1).23,24 Such species are proposed as intermediates not only in cross-metathesis of a MCR with a NCR, but also in the exchange of a nitride MN with the corresponding alkyne (RCCR).25,26 To date, the isolation of an α-N-MCBD scaffold has been reported only by us with titanium, in the form of the complex (PNP)Ti(κ2-C,N-tBuCC(R)N)23 (R = tBu, Ad; Ad = 1-adamantyl; PNP = N[2-PiPr2-4-methylphenyl]2−). Although the latter species could not be structurally confirmed via single crystal X-ray diffraction analysis (scXRD), a combination of 15N isotopic labelling, NMR spectroscopy, and computational studies, strongly supported the existence of a planar, four-membered ring.
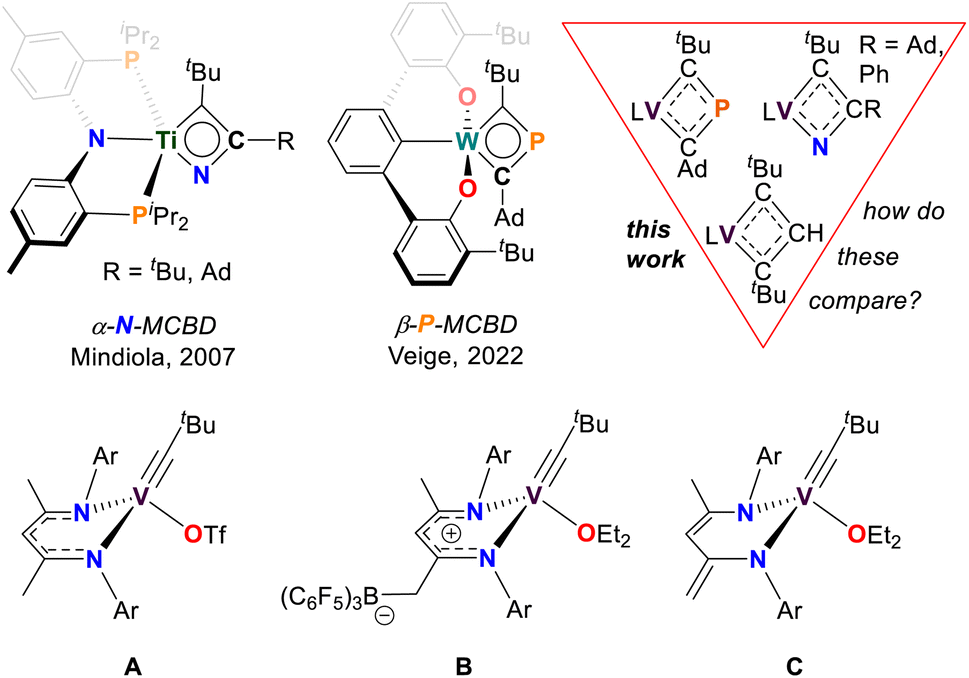 | ||
| Fig. 1 Top: Isolable pnictogen-based metallacyclobutadienes (MCBD) complexes and the vanadacyclobutadiene (VCBD) species reported in this work (L = dBDI2−). Bottom: Selected [VV] alkylidyne complexes, with complex C being the focus of the present study. | ||
Given the isolobal relationship between C− and P (or RC and P), one would anticipate PCR, to undergo metathesis reactions with MCR in a manner like alkynes. In this context, Hill and co-workers have explored the reactivity of MCR with PCR,27,28 but it was only recently that Veige and co-workers successfully isolated a monometallic β-phospha-metallacyclobutadiene (β-P-MCBD) scaffold.29 In their study, a β-P-MCBD scaffold (middle top of Fig. 1) in complex (tBuOCO)W(κ2-C,C-tBuCPCAd) (tBuOCO3− = ipso-C6H3[2,6-(C6H3-o-tBu)2]) was formed via insertion of a CtBu fragment into a side-on bound, 1-adamantyl phosphaethyne, PCAd ligand. Due to the electronegativity difference between N (3.04) and P (2.19) on the Pauling scale, the regioselectivity in the cycloaddition step of the pnictogen based alkyne, PnCR (Pn = pnictogen) across the MC bond was reversed. Additionally, Hard–Soft Acid–Base (HSAB) considerations could also be applied to these differences suggesting that the hard nitrogen atom would preferentially bind to the hard [TiIV] nucleus while the soft phosphorus atom would not want to interact with the hard [WVI] nucleus.
Heteroatom containing MCBDs are seldom reported, and their chemistry remains largely unexplored, with a few notable exceptions shown in Fig. 1.29–39 In the context of 3d transition metals, alkylidyne cross-metathesis reactions involving vanadium are virtually unknown, since MCR motifs have been rarely documented with this class of metal ions.23,40–56 There are only a few examples of stable vanadium Schrock-like carbyne complexes known.41,47,57 Notably, vanadium alkylidynes (VCR) (A–C in Fig. 1), are catalysts for the formation of cyclic polyphenylacetylene.58 It was suggested that the initiation step of the polymerization involved a [2+2] cycloaddition of the VCR fragment and a phenylacetylene (PhCCH) to form a vanadacyclobutadiene (VCBD). This was further corroborated through our isolation of the VCBD (dBDI)V(κ2-C,C-tBuCC(H)CtBu)58 (4), (dBDI2− = ArNC(CH3)CHC(CH2)NAr, Ar = 2,6-iPr2C6H3) (top right of Fig. 1). Noting that the barrier for metathesis was quite low for complex C (21 kcal mol−1) and given that no structurally characterized examples of an α-N-MCBD exist, and only a single structural example of a β-P-MCBD is known, we sought to explore this rare ligand class with the VCR motif and compare their structural topology.
In this study, we report VCBD complexes that incorporate pnictogens (Pn) and compare them to the known carbon-only vanadacyclobutadiene (4), considering the isolobal relationship between CH and P/N. Although these scaffolds are generally represented as fully delocalized, substantial differences in the electronic structure are expected due to the disparity in electronegativities between N (3.04), C (2.55), and P (2.19). Indeed, cycloaddition intermediates between MC motifs and PnC bonds of heteroalkynes have rarely been isolated, and with the increasing interest in the chemistry of four-membered hetero-metallacycles,29,33,59–68 we decided to investigate the reactivity of VCR with PnCR. Herein, we present the successful isolation of the first α-aza-vanadacyclobutadiene (α-N-VCBD) and a rarely reported β-phospha-vanadacyclobutadiene (β-P-VCBD) complexes, complete with single crystal X-ray diffraction (scXRD) studies. We performed a systematic comparison between these complexes, focusing on their spectroscopic features and structural metrics to evaluate the influence of a heteroatom substitution. Additionally, density functional theory (DFT) calculations were employed to assess the impact of the heteroatoms on the electronic and resonance structures of these complexes.
Results and discussion
Among the vanadium alkylidynes A–C, complex C (Fig. 1) stands out as the most suitable reagent for various reactions, due to its stability against intramolecular degradation and its enhanced reactivity, which is attributed to the lability of the diethyl ether (Et2O) ligand. Consequently, we directed our efforts toward investigating the reactivity of this complex in cycloaddition reactions with unsaturated substrates, such as PnCR (Pn = N, R = Ad, Ph; Pn = P, R = Ad), to determine whether they form stable Pn-based VCBDs. These substrates were selected to probe the nucleophilic nature of the alkylidyne carbon and to examine the differences in bond formation between the more electronegative N and the more electropositive P. We envisioned that gaining structural insights into these rare heteroatom-substituted MCBD fragments, by exchanging isolobal moieties CH, N, and P, would allow us to compare their electronic properties.
Synthesis and characterization of the α-N-VCBD scaffold
We anticipated that the higher electronegativity of nitrogen (3.04) would favor the formation of a C–C bond between the alkylidyne carbon and the nitrile carbon atoms.69,70 As expected, treating C with NCR (R = Ad, Ph) in pentane, deuterated benzene (C6D6), or toluene (for 2) at room temperature for one hour resulted in the formation of α-N-VCBD complexes (dBDI)V(κ2-C,N-tBuCC(R)N) (R = Ad, 1, 49% yield; Ph, 2, 89% yield), as illustrated in the top portion of Scheme 1. For NCAd, the reaction yielded a brown-colored complex, 1. However, the formation of 1 required an excess amount of nitrile and dilute reaction conditions to attain full conversion. Repetitive evacuation of the side product, Et2O, over the course of the reaction also promoted the formation of 1. In contrast, the reaction of C with one equivalent of NCPh proceeded almost immediately, forming purple-red complex 2 in nearly quantitative yields (89% isolated).
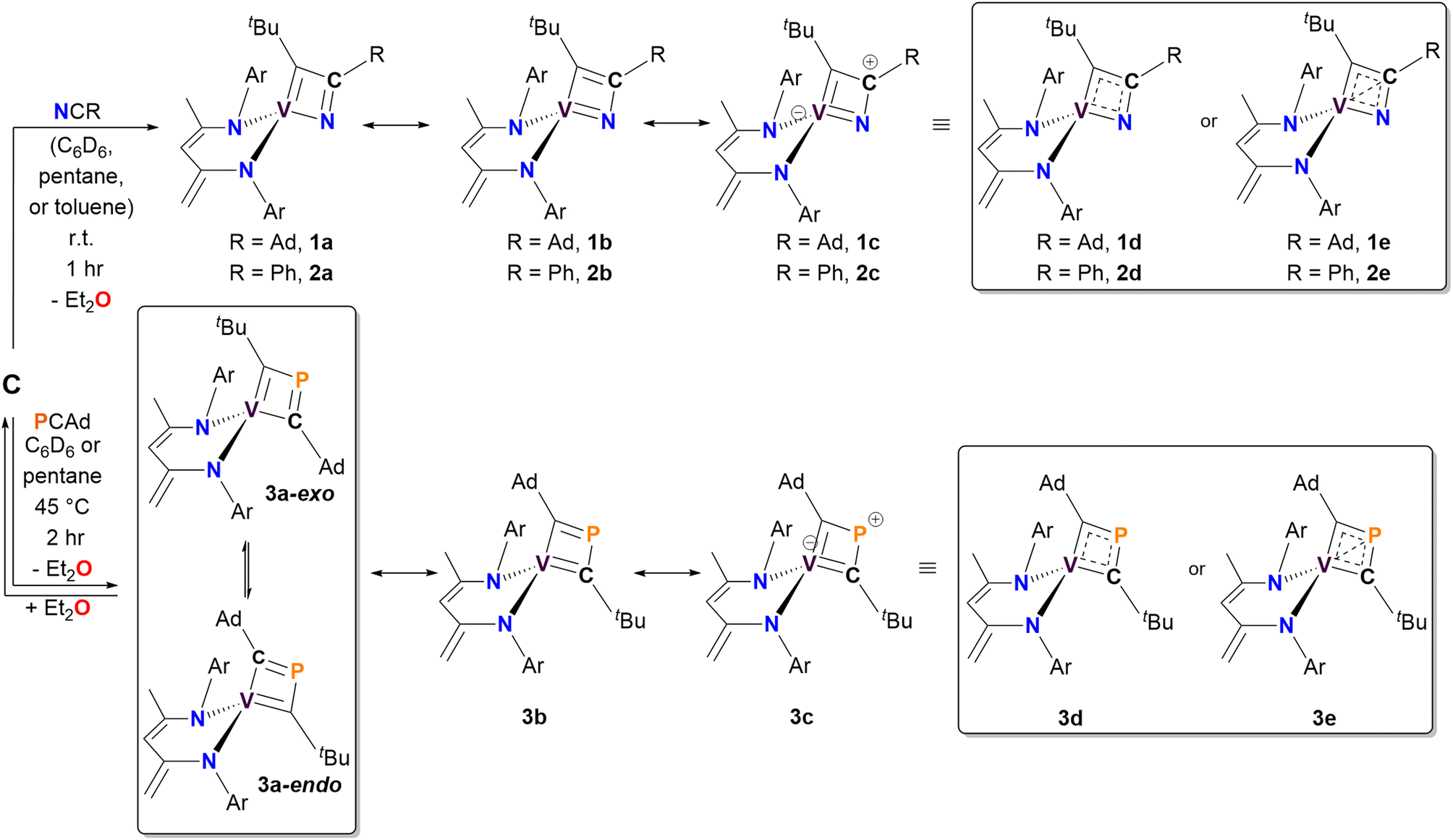 | ||
| Scheme 1 Synthesis of complexes 1–3via the [2+2]-cycloaddition of the vanadium alkylidyne C with pnictogen-containing alkynes PnCR (Pn = N, R = Ad or Ph; Pn = P, R = Ad). Potential resonance structures and charge delocalized structures for each complex are also illustrated. | ||
Both complexes 1 and 2 exhibit characteristic resonances in the 1H NMR spectrum for the methylene moiety of the bis-anilido ligand (dBDI2−), showing a virtual pair of doublets at 3.76 and 3.29 ppm for 1 (Fig. S2†) and at 3.75 and 3.25 ppm for 2 (Fig. S9†). The resulting 1H–13C HSQC experiment on complex 2 (Fig. S13†) revealed that two inequivalent proton resonances correspond to a single carbon resonance at 88.65 ppm. The corresponding 13C{1H} DEPT-135 spectrum of 2 (Fig. S11†) revealed the carbon resonance at 88.65 ppm has sp2 like character, leading us to assign the methylene (CH2) fragment as this resonance. Like 1, the 1H and 13C{1H} NMR spectra of 2 reveal extensive overlap of resonances in the 1H NMR spectrum, indicating the presence of structurally similar but magnetically distinct compounds. The NMR spectral data for the less sterically hindered complex 2 are less complicated as only a single isomer is present, allowing for a complete assignment of resonances. For instance, the β-carbon in the four-membered ring of 2 was observed at 157.9 ppm in the 13C{1H} NMR spectrum (Fig. S10†), a shift further upfield than the previously reported titanium α-N-MCBD derivative (PNP)Ti(κ2-C,N-tBuCC(Ad)N), which resonates at 240.5 ppm.23 Overall, the 1H and 13C{1H} NMR spectral data for complexes 1 and 2 are in accord with these compounds possessing C1 symmetry, as indicated by the presence of four inequivalent isopropyl methine resonances for the two aryl groups and two α-carbons for the chelating bis-anilido ligand. A 1H–1H EXSY NMR spectrum of 2 (Fig. 2A) also reveals the fluxionality of the molecule at room temperature, indicated by extensive off-diagonal couplings of protons, thus corroborating why two diastereomers are unobservable on the NMR time scale at room temperature. Moreover, the 51V NMR of 1 (Inset Fig. 2B) and 2 (Fig. S15†) show a single resonance feature at 381.32 ppm (Δν1/2 = 339.2 Hz) and 344.76 ppm (Δν1/2 = 276.0 Hz), respectively.
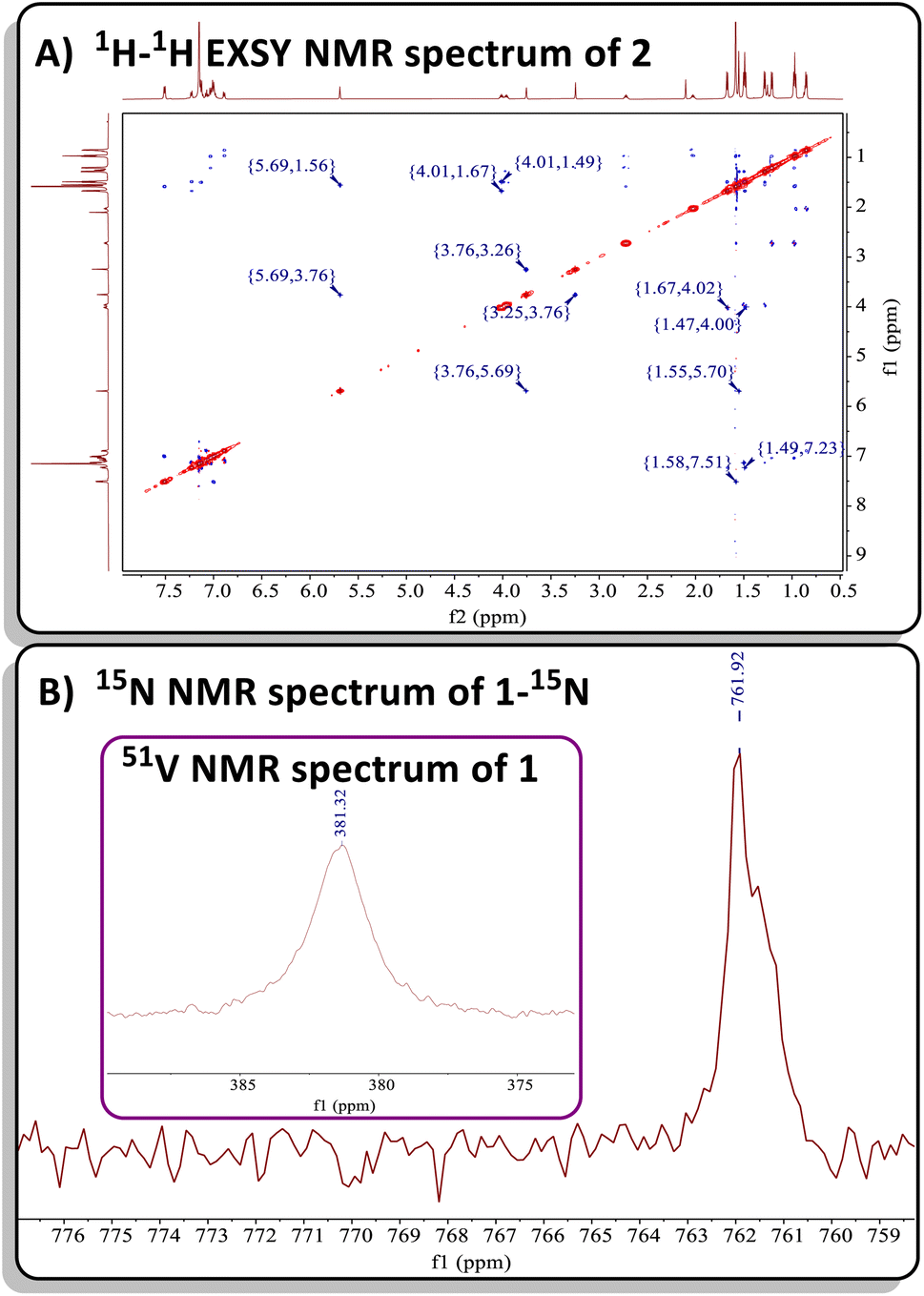 | ||
| Fig. 2 (A) 1H–1H EXSY NMR spectrum of 2 showcasing its fluxionality at room temperature. (B) 15N NMR spectrum of 50% 15N enriched complex 1-15N. Inset: 51V NMR spectrum of 1. | ||
Due to the lipophilic nature of complex 1, numerous attempts to obtain single crystals were unsuccessful. Consequently, we turned to the ∼50% 15N enriched isotopologue, 15NCAd, prepared using the method reported by Johnson and co-workers.71 Previous reports have demonstrated that M–N multiple bonding with a planar, sp2-like nitrogen leads to a downfield shift in the 15N NMR resonance, compared to a more pyramidalized, sp3-like nitrogen involved in a plausible tetrahedrane MCBD structure.23,72 Treatment of 50% enriched 15NCAd with complex C, followed by an analogous workup, resulted in the isolation of the isotopologue (dBDI)V(κ2-C,N-tBuCC(Ad)15N) (1-15N). Subsequent 15N NMR spectral analysis revealed a highly downfield resonance at ∼761 ppm, referenced to 15NCAd at 242 ppm versus NH3 (l) at 0 ppm at 27 °C (Fig. 2B).4,69 This value is consistent with the 15N chemical shift observed for the titanium α-N-MCBD complex, (PNP)Ti(κ2-C,N-tBuCC(Ad)15N) (15N NMR: 672.6 ppm at 55 °C), suggesting that the nitrogen atom in 1-15N is likely sp2-hybridized, forming a planar, four-membered MCCN ring scaffold.23 In contrast to the niobium methylidyne complex (PNP)NbCH(OAr), which undergoes cross-metathesis with [NCR] to produce (PNP)NbN(OAr) and HCCR (R = tBu, Ad),24 the four-coordinate complexes 1 and 2 did not undergo [2+2]-cycloreversion and subsequent expulsion of the alkyne tBuCCR (R = Ad, Ph). Computational studies have revealed that the dissociation of Et2O from C involves a transition state with an energy barrier of 26 kcal mol−1, while the transient alkylidyne species {(dBDI)VCtBu} is approximately 21 kcal mol−1 higher in energy than its precursor.58 Therefore, the elimination of alkyne from 1 or 2 is improbable, as it would result in the formation of a highly reactive three-coordinate vanadium nitride {(dBDI)VN} fragment. Additionally, theoretical studies on all carbon based MCBD complexes have suggested that the retro [2+2] cycloaddition reaction is unfavourable for group 4 and 5 MCBDs.73 Notably, when an excess amount of 15NCAd was added to a C6D6 solution of complex 1, no formation of 1-15N was observed (monitored by 15N NMR spectroscopy over 24 hours), further indicating that the cycloreversion process is unfavourable under these conditions.
Unlike complex 1, complex 2 can be crystallized as single crystals from a pentane/toluene vapor-diffused mixture cooled to −35 °C. scXRD analysis revealed that complex 2 crystallizes in the monoclinic and centrosymmetric space group P2(1)/n. Fig. 3A depicts the structure of 2, which manifests C1 symmetry, consistent with its solution-phase NMR spectra. The structure shows a short V1–N3 distance of 1.697(1) Å, indicating a strong V–N interaction, while the relatively long N3–C31 distance of 1.482(2) Å suggests a smaller contribution of the 2a resonance structure compared to 2b (Scheme 1). The V1–C30 distance of 1.930(2) Å is notably longer compared to typical V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds40,74–91 (1.7 Å to 1.9 Å), reported in the Cambridge structural database (CCDC), further supporting the dominance of resonance structure 2b over 2a. Additionally, the observed C30–C31 bond length of 1.369(2) Å is indicative of the CC double bond character, which aligns poorly with an extreme canonical form 2c. Thus, the crystallographic data supports the view that 2b is the most representative resonance structure of 2 compared to 2a or the extreme form 2c.
C double bonds40,74–91 (1.7 Å to 1.9 Å), reported in the Cambridge structural database (CCDC), further supporting the dominance of resonance structure 2b over 2a. Additionally, the observed C30–C31 bond length of 1.369(2) Å is indicative of the CC double bond character, which aligns poorly with an extreme canonical form 2c. Thus, the crystallographic data supports the view that 2b is the most representative resonance structure of 2 compared to 2a or the extreme form 2c.
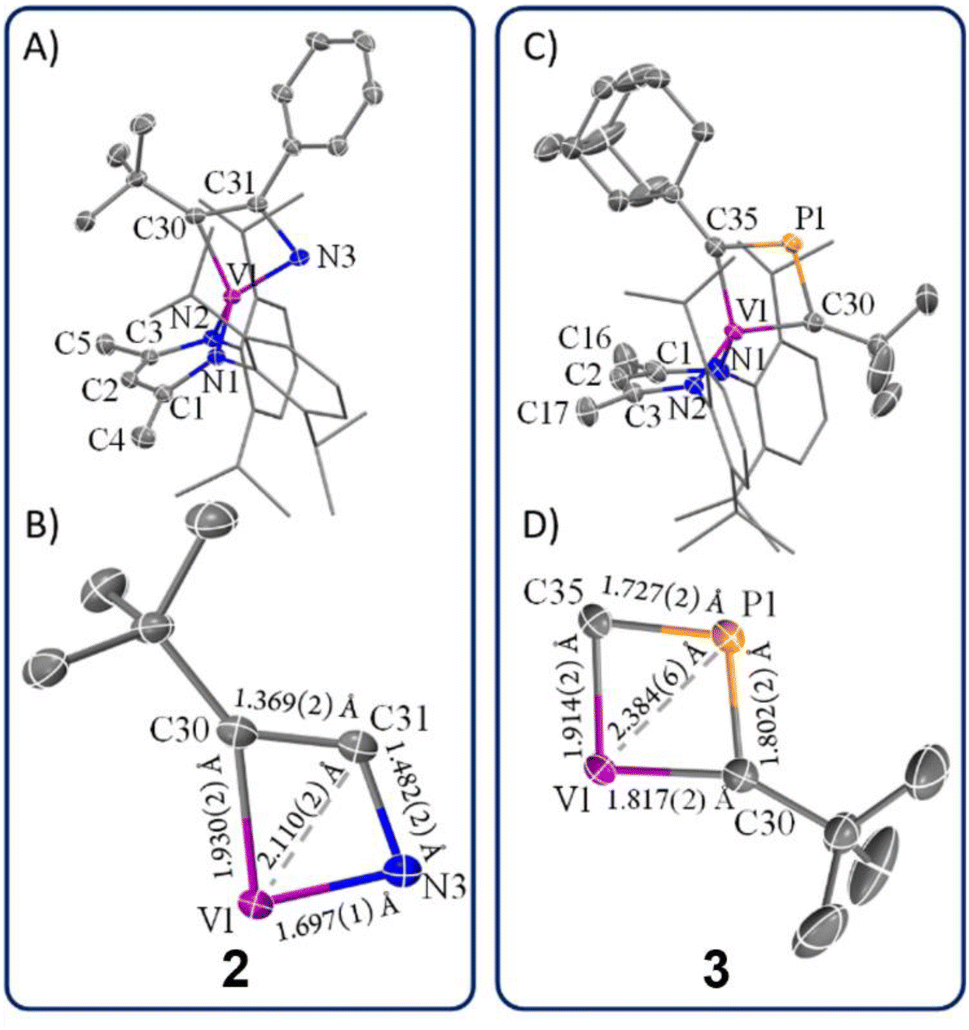 | ||
| Fig. 3 (A) Structural representation of 2 with thermal ellipsoids at 50% probability level and hydrogen atoms omitted for clarity. (B) A closer examination of bond distances in the α-N-VCBD motif in 2. (C) Structural representation of 3 with thermal ellipsoids at 50% probability level and hydrogen atoms omitted for clarity. (D) A closer examination of bond distances in the β-P-VCBD motif in 3. | ||
Fig. 3B provides a close-up view of the metallacyclic framework. The short V1–C31 distance of 2.110(2) Å in complex 2 hints at a possible interaction between vanadium and the β-carbon, suggesting a more delocalized structure such as 2d or even one with delocalized bonding involving a V–Cβ interaction (2e). The [VNCC] ring is further revealed as a nearly planar with the V1–C30–C31–N3 torsion angle of 1.0(4)°. However, the metallacycle deviates from perfect square geometry, as evidenced by internal angles: V1–C30–C31 (77.4(4)°), C30–C31–N3 (116.2(0)°), C31–N3–V1 (82.8(4)°), and N3–V1–C30 (83.5(0)°). To the best of our knowledge, complex 2 represents the only structural study of an α-N-MCBD scaffold, making these observations particularly significant in understanding the bonding and geometric properties of such complexes.
Synthesis and characterization of the β-P-VCBD scaffold
The reaction of C with PCAd in pentane or C6D6 at 45 °C for two hours produced a new vanadium product in 61% isolated yield, identified as the β-P-VCBD complex (dBDI)V(κ2-C,C-tBuCPCAd) (3), based on a combination of spectroscopic and structural data (Scheme 1, bottom). While one might expect Et2O to be a labile ligand in complex C, achieving full conversion to 3 requires not only an excess of PCAd but also dilute reaction conditions. This observation suggests that the cycloaddition and cycloreversion of the phosphaalkyne are in equilibrium, even in the presence of a weak donor ligand such as Et2O. According to spectroscopic and structural studies (vide infra), complex 3 exhibits overall C1 symmetry due to the absence of a σ plane or C2 elements in the dBDI2− scaffold, indicating that the vanadium center in complex 3 is chiral. The 1H NMR spectrum of 3 (Fig. S18†) displays two pairs of inequivalent methylene hydrogens at 3.80 and 2.91 ppm, which correlates to a carbon resonance at 82.08, based on a 1H–13C HSQC spectroscopic experiment (Fig. S22 and S23†). Two distinct signals for the methine CH fragment of the bis-anilido backbone were also observed through the 1H–13C HSQC experiment at 5.75 and 5.69 ppm. In addition, the 51V NMR spectrum reveals two distinct resonances (220.0, Δν1/2 = 371 Hz; 223.9 ppm, Δν1/2 = 250 Hz), indicating the presence of two magnetically inequivalent but structurally similar species in solution92 (inset in Fig. 4), which is corroborated by the appearance of many more resonances in the 13C NMR spectrum (Fig. S19†). The 31P{1H} NMR spectrum revealed only one diagnostic feature at 151 ppm, and this resonance is quite broad (Δν1/2 = 437 Hz, Fig. 4). The broadening of this 31P{1H} NMR resonance, which is attributed to the β-P atom, most likely stems from coupling with the quadrupolar 51V nucleus (I = 7/2, 99.75%) as well as dynamic phenomena involving the two diastereomers of a species with C1 symmetry. Variable–Temperature (VT) NMR studies at elevated temperatures did not reveal the interconversion and coalescence of the two species, as this complex slowly decomposes above 55 °C (Fig. S20†). Additionally, it should be noted that Piers and co-workers have reported similar dynamic behavior involving the formation of endo/exo diastereomers in zwitterionic systems such as ([ArNCtBu]2CH)Sc(CH3)[CH3B(C6F5)3] (Ar = 2,6-iPr2C6H3).72,93 The reaction to form 3 is regioselective, but it is most likely non-stereospecific, depending on whether the PCAd approaches the non-planar {(dBDI)VCtBu} framework from the exo or endo side to ultimately cycloadd across the VCtBu ligand in C (Scheme 1). Adding to this complexity, this cycloaddition process is reversible, likely leading to the decomposition of {(dBDI)VCtBu}. Based on VT NMR studies, these two conformers do not interconvert below the decomposition temperature, likely due to steric hindrance imposed by bulky substituents (e.g., Ar, tBu, and Ad groups).
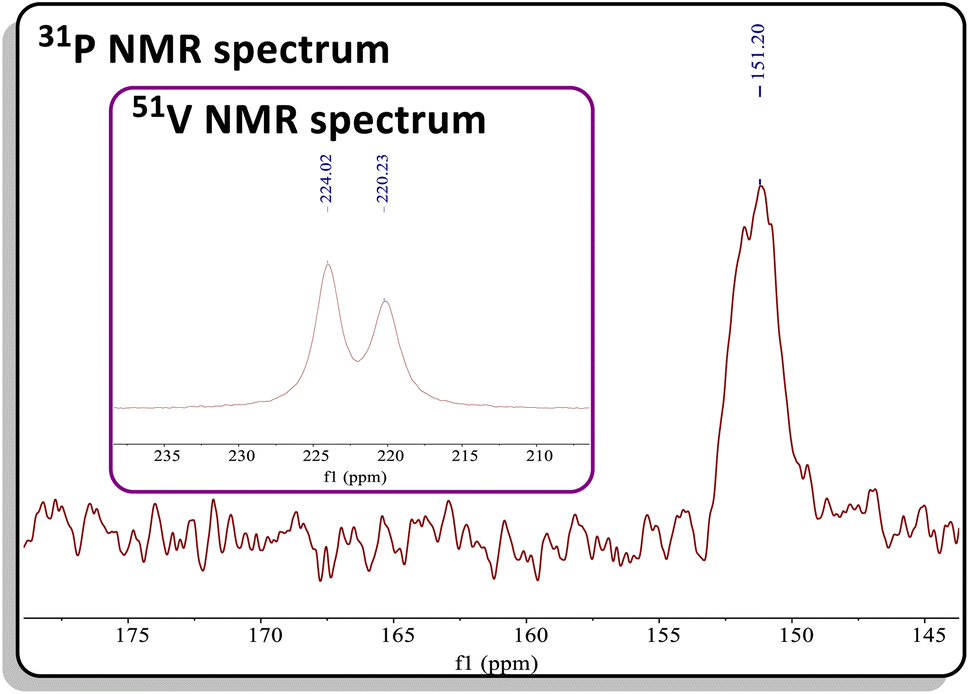 | ||
| Fig. 4 31P NMR spectrum of 3. Inset: 51V NMR spectrum of 3 showing two resonances with similar chemical shifts. | ||
The solid-state structure of 3 is shown in Fig. 3C. Upon inspection, one can observe the short but varying V–C(tBu) (1.817(2) Å) and V–C(Ad) (1.914(2) Å) distances, both of which fall within the range of VC double bond lengths when compared to previously reported and structurally characterized compounds with VC double bonds compiled in the CCDC.40,74–91 Additionally, the short Cα–P distances (P1–C30, 1.802(2) Å; P1–C35, 1.727(2) Å) are comparable to those observed in structurally characterized phosphaalkenes reported in the CCDC (1.65–1.75 Å).94 Notably, the V1–P1 distance of 2.384(6) Å is much shorter than the sum of the van der Waals radii,95 which tantalizingly suggests the potential interaction between the metal center and P atom; however, such an interaction is likely to be extremely fragile, akin to those described for isolobal κ2-C,C-deprotiometallacyclobutadiene (dMCBD) scaffolds.2,10,73,96–100 A close examination of the [VCPC] ring in Fig. 3D reveals a relatively planar but more kite-like framework, with angles V1–C30–P1 (82.4(3)°), C30–P1–C35 (101.6(2)°), P1–C35–V1 (81.6(6)°), and C35–V1–C30 (94.2(9)°). The V1–C30–P1–C35 torsion angle of 0.3(0)° further emphasizes the planarity of the four-membered ring in 3 compared to 2. Given the ability of the double bonds to delocalize within the four-membered ring, complex 3 can be described as an average of two possible canonical forms, 3a and 3b, as indicated in Scheme 1, but with some contribution from the more puckered resonance structure 3c, which leads to a shortening of the vanadium distance to all other atoms in the metallacycle. Akin to complex 2, resonance structure 3c represents a more extreme scenario where the β-P could possess some formal cationic character while interacting with the electron-rich metal center. Scheme 1 also depicts a more delocalized resonance structure, 3d and 3e, similar to how the MCBD and dMCBD complex has been described in the literature for Mo,2,16,98 W,96,100 and more recently group four36,101–103 and five58,104 transition metals. It is also noteworthy that in the solid-state structure of this molecule, the Ad group points towards the dBDI2− ligand, in accord with the endo isomer shown in Scheme 1.
Impact of the heteroatom substitution on the structures of 2–4
In our recent studies, we synthesized the VCBD complex (dBDI)V(κ2-C,C-tBuCC(H)CtBu) (4) by treating complex C with the terminal alkyne HCCtBu.58 The solid-state structure confirmed the diamond-like shape of the VCBD moiety, with the V–Cα bond lengths of 1.891(7) and 1.788(9) Å. The Cα–Cβ distances of 1.464(1) and 1.410(1) Å are between typical C–C single bonds and CC double bonds. The relatively short V–Cβ distance of 2.004(8) Å, compared to 2.110(2) Å in complex 2 and 2.384(6) Å in complex 3, suggests a potential interaction between the vanadium center and the β-carbon.91
With all VCBD complexes supported with the same dBDI ligand structurally confirmed, we can perform a meaningful comparison of their geometries. Table 1 provides a comparative analysis of these scaffolds, emphasizing key metrical parameters for the VCBD. One striking observation is that the V–Xα bond in complex 2 is significantly shorter than in complexes 3 and 4, underscoring the strong interaction between the vanadium center and the α-N atom. The stronger interaction is attributed to the higher electronegativity of the nitrogen atom, which likely facilitates a stronger bond with vanadium. Conversely, there is a gradual decrease in the V–Cα bond length from 2 to 4, suggesting an increasing contribution of the resonance structure featuring the VCα double bond. Moreover, the V–Xβ bond in complex 3 is significantly longer than in complexes 2 and 4, which can be attributed to the larger covalent radius for phosphorus when compared to carbon. When comparing the Cα–Xβ–Xα angle, complex 3 stands out as the closest to a square geometry, with this angle being close to 90°. On the other hand, complex 4, despite its symmetrical bonding nature, deviates the most from the square geometry with an angle of 124.3°. The notable differences in these structural parameters suggest that the inclusion of heteroatoms significantly alters the electronic properties of the VCBDs. These changes likely influence the reactivity and stability of these complexes, providing insights into the role of heteroatoms in modulating the electronic structures of the VCBDs.
|
|
|||
|---|---|---|---|
| 2 (Xα = N; Xβ = C) | 3 (Xα = C; Xβ = P) | 4 (Xα = Xβ = C) | |
| V–Cα (Å) | 1.930(2) | 1.817(2) | 1.788(7) |
| V–Xα (Å) | 1.671(1) | 1.914(2) | 1.891(7) |
| V–Xβ (Å) | 2.110(2) | 2.384(6) | 2.004(8) |
| Cα–Xβ (Å) | 1.369(2) | 1.802(2) | 1.464(1) |
| Xα–Xβ (Å) | 1.482(2) | 1.727(2) | 1.410(1) |
| V–Cα–Xβ (°) | 77.4(4) | 82.4(9) | 75.3(4) |
| V–Xα–Xβ (°) | 82.9(1) | 81.7(8) | 73.1(4) |
| Cα–Xβ–Xα (°) | 116.3(1) | 101.6(1) | 124.3(7) |
| Xα–V–Cα (°) | 83.5(1) | 94.3(9) | 87.3(3) |
Theoretical investigation on the electronic and resonance structures of 2–4
To further investigate the impact of heteroatom substitution on the electronic structures of the VCBDs, we performed DFT calculations at the PBE-D3(BJ)/TZ2P//DZP level of theory.105–109Fig. 5 illustrates the frontier orbitals of three complexes, highlighting primary interactions involving the VCBD ligands, identified as π, σ, and σ-bonding orbitals. The energy level of the lowest σ-bonding orbital follows the anticipated electronegativity trend of P, C, and N atoms, with significant stabilization observed in complex 2, indicating a strong V–N bond. Interestingly, an interaction between the β-P atom and the vanadium center is observed in the HOMO–2 of 3, stabilizing its energy level below that of 4. This interaction is characterized as a dative bond, where the lone pair of the β-P atom donates to the vacant d-orbital of the metal center, which is not seen in 2 and 4 due to the presence of β-(C–R) bonds (R = Ph (2) and H (4)). These findings suggest that the substitution of heteroatoms results in unique bonding interactions, where in the case of P, the V–P interaction contributes some to the stabilization of the four membered-ring. The remaining d-orbitals on vanadium form σ-bonds with the dBDI2− ligand (Fig. S37†).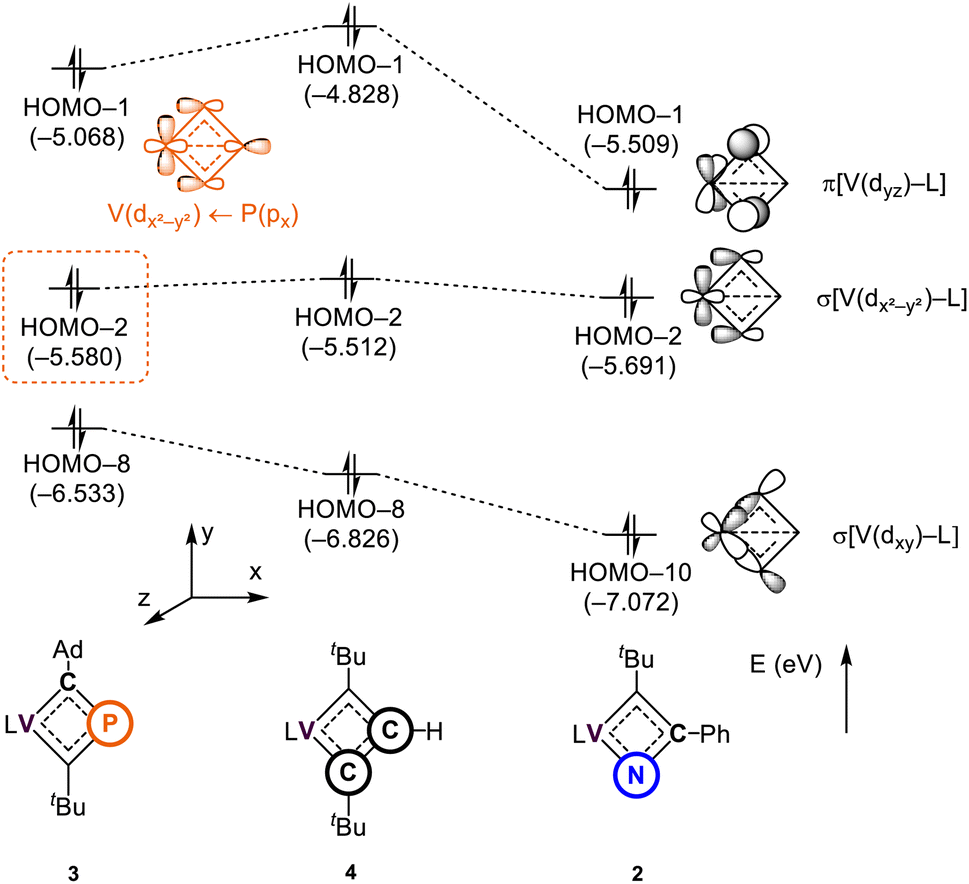 | ||
| Fig. 5 Molecular orbitals of 2–4 interacting between the metal center and the vanadacyclobutadiene scaffolds. L = dBDI2− = ArNC(CH3)CHC(CH2)NAr, Ar = 2,6-iPr2C6H3. | ||
As shown in Scheme 1, the ambiguous bonding nature within the VCBD scaffolds leads to numerous possible resonance- and charge delocalized-structures for these complexes. To identify the most contributing resonance structure, we analyzed the Nalewajski-Mrozek bond order,110 which offers a valence bond interpretation of DFT results, similar to the Mayer bond order analysis111 (Fig. S39†). The corresponding bond orders and resonance structures are summarized in Fig. 6.
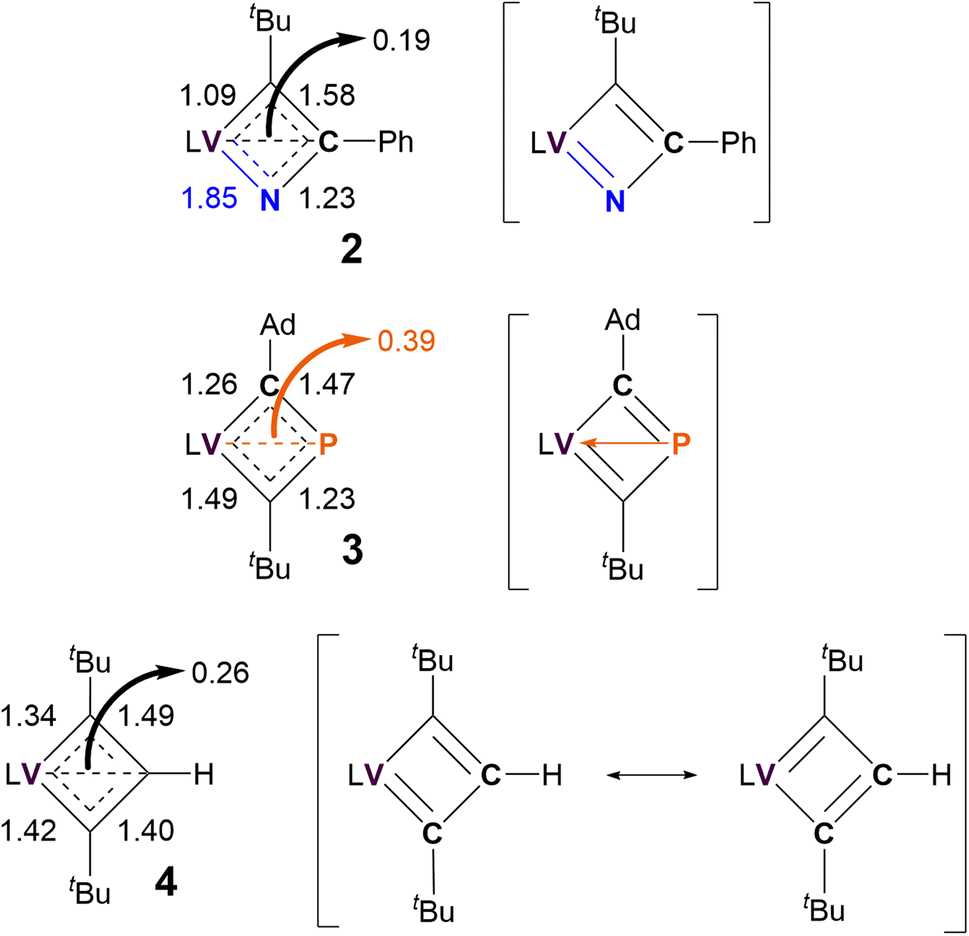 | ||
| Fig. 6 Nalewajski-Mrozek bond orders of VCBD scaffolds and most probable resonance structures of 2–4. L = dBDI2− = ArNC(CH3)CHC(CH2)NAr, Ar = 2,6-iPr2C6H3. | ||
Unsurprisingly, the resonance between the two Lewis valence structures of the VCBD is strongest in complex 4, with the computed bond order of 1.45 being very close to the ideal resonance value of 1.5. Note, that the calculated numbers in Fig. 6 show a slight variation (1.49/1.40) due to deviations of the molecular structure from the ideal C2v symmetry. Incorporating a strongly donating imido-like functionality in complex 2 distorts the resonance form much more toward a localized Lewis valence form where a VN double bond character is emphasized, with a calculated bond order of 1.85. Consequently, the N–C bond has lost a notable amount of its double bond character and shows a bond order of only 1.23. Thus, the localized valence structure is a much more appropriate depiction of bonding in 2 than 4, suggesting that resonance form 2b in Scheme 1, is the most reasonable, which agrees with our scXRD analysis and optimized structure obtained via computational studies. In 3, the P–C bonds show an average bond order of 1.35, with the individual bond orders being 1.47 and 1.23 due to structural distortions caused by the Ad and tBu ligands. Thus, a localized Lewis structure is also a good representation for 3, but only for steric reasons. Interestingly, all three complexes show notable α,β-[CCC/CPC/NCC] agostic interactions112 that can be envisioned as direct donation from the β-atom of the metallacycle into the metal center. The formal bond orders of this interaction are 0.26, 0.39 and 0.19 for complexes 4, 3, and 2, respectively. This trend for the 3-center-4-electron interaction is easy to understand, given that the C–P bonds are more polarizable than C–C, while C–N is less polarizable than C–C. This trend suggests that complex 3 should be illustrated with charge delocalization around the four membered ring with some electronic interaction between the vanadium and the β-phosphorous atom, i.e. complex 3e in Scheme 1. Different electron partitioning schemes, such as atoms in molecules113,114 and the electron localization function115 analysis paint a similar picture of the electronic structure and are shown in the ESI (Fig. S40–S42).† Thus, from a purely electronic viewpoint, β-P-VCBD is best prepared to engage in C–P bond activation reactions, followed by α-N-VCBD, while the purely carbon based VCBD should in principle be the least reactive species in the series. These Lewis valence structures that best reflect the underlying electronic structure of the three complexes are shown in Fig. 6.
Conclusions
In this study, the vanadium alkylidyne complex C has been demonstrated to undergo regiospecific [2 + 2]-cycloaddition with NCR (R = Ad or Ph) and PCAd. Based on the 15N NMR chemical shift observed in 50% 15N-enriched sample of 1, we anticipate that its geometry is like that of the Ph derivative 2, consistent with a planar α-N-VCBD where nitrogen is directly coordinated to the metal center. In the case of 3, the more electropositive phosphorus atom causes a reversal in regioselectivity, resulting in the formation of a β-P-VCBD scaffold. No evidence of cross-metathesis was observed for these complexes. The successful identification of the crystal structures of 2 and 3 allowed for a systematic comparison of their geometries with the all-carbon analogue 4, which was structurally confirmed in a previous report. This series of complexes reveals significant differences in the bonding nature between the metal center and adjacent atoms, as well as variations in the shape of the VCBD scaffolds, indicating different distributions of electron density within these metallacyclic framework. Theoretical investigations into the electronic and resonance structures of 2 and 3 suggest that the unique V–N and V–P interactions enhance the stability of the VCBD scaffolds, resulting in a single dominant resonance structure, in contrast to their analogue 4. We are currently exploring the reactivity of these metallacycles, with a particular interest in the weak V–Xβ interactions and the Lewis basicity of the phosphorus atom in 3.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
MGJ: writing – review & editing, writing – original draft, methodology, investigation, formal analysis. JBR: writing – review & editing, writing – original draft, methodology, investigation, formal analysis. HM: formal analysis, investigation, methodology, visualization, writing – original draft, writing – review & editing; SK: formal analysis, investigation, methodology, visualization, writing – original draft, writing – review & editing. PJC: investigation, formal analysis. MRG: investigation, formal analysis. MHB: funding acquisition, resources, supervision, writing – review & editing. DJM: writing – review & editing, supervision, funding acquisition.Conflicts of interest
The authors declare no competing conflict of interest.Acknowledgements
We thank Professor Takashi Kurogi for insightful discussions, the University of Pennsylvania, and the US National Science Foundation (CHE1764329 and CHE2154620 to D. J. M.) for financial support of this research. The authors also acknowledge the NIH supplements award 3R01GM118510-03S1 and 3R01GM087605-06S1 and financial support of the Vagelos Institute for Energy Sciences and Technology (VIEST) for the purchase of NMR instrument NEO600. J.B.R thanks VIEST for a predoctoral fellowship.We thank the Institute for Basic Science in Korea for financial support (IBS-R010-A1).Notes and references
- R. R. Schrock, J. H. Freudenberger, M. L. Listemann and L. G. McCullough, Recent advances in the chemistry of well-defined olefin and acetylene metathesis catalysts, J. Mol. Catal., 1985, 28, 1–8 CrossRef CAS.
- L. G. McCullough, R. R. Schrock, J. C. Dewan and J. C. Murdzek, Multiple metal-carbon bonds. 38. Preparation of trialkoxymolybdenum(VI) alkylidyne complexes, their reactions with acetylenes, and the x-ray structure of Mo[C3(CMe3)2][OCH(CF3)2](C5H5N)2, J. Am. Chem. Soc., 1985, 107, 5987–5998 CrossRef CAS.
- M. R. Churchill and J. W. Ziller, Crystal structure of (η5-C5H5)W[C(Ph)C(CMe3)C(Ph)]Cl2, a molecule possessing a localized, non-planar tungstenacyclobutadiene ring, J. Organomet. Chem., 1985, 279, 403–412 CrossRef CAS.
- R. R. Schrock, I. A. Weinstock, A. D. Horton, A. H. Liu and M. H. Schofield, Preparation of rhenium(VII) monoimido alkylidyne complexes and metathesis of acetylenes via rhenacyclobutadiene intermediates, J. Am. Chem. Soc., 1988, 110, 2686–2687 CrossRef CAS.
- L. L. Padolik, J. C. Gallucci and A. Wojcicki, Fischer-type rhenacyclobutadiene complexes: synthesis, structure, and nucleophilic addition/substitution and oxidation reactions, J. Am. Chem. Soc., 1993, 115, 9986–9996 CrossRef CAS.
- A. Fürstner and P. W. Davies, Alkyne metathesis, Chem. Commun., 2005, 2307–2320 RSC.
- W. Zhang and J. S. Moore, Alkyne Metathesis: Catalysts and Synthetic Applications, Adv. Synth. Catal., 2007, 349, 93–120 CrossRef CAS.
- S. Beer, C. G. Hrib, P. G. Jones, K. Brandhorst, J. Grunenberg and M. Tamm, Efficient Room-Temperature Alkyne Metathesis with Well-Defined Imidazolin-2-iminato Tungsten Alkylidyne Complexes, Angew. Chem., Int. Ed., 2007, 46, 8890–8894 CrossRef CAS PubMed.
- M. E. O'Reilly, I. Ghiviriga, K. A. Abboud and A. S. Veige, Unusually stable tungstenacyclobutadienes featuring an ONO trianionic pincer-type ligand, Dalton Trans., 2013, 42, 3326–3336 RSC.
- A. Furstner, Alkyne metathesis on the rise, Angew. Chem., Int. Ed., 2013, 52, 2794–2819 CrossRef PubMed.
- R. Ramírez-Contreras, N. Bhuvanesh and O. V. Ozerov, Cycloaddition and C–H Activation Reactions of a Tantalum Alkylidyne, Organometallics, 2015, 34, 1143–1146 CrossRef.
- P. R. Remya and C. H. Suresh, Planar tetracoordinate carbon in tungstenacyclobutadiene from alkyne metathesis and expanded structures, Dalton Trans., 2016, 45, 1769–1778 RSC.
- C. Bittner, H. Ehrhorn, D. Bockfeld, K. Brandhorst and M. Tamm, Tuning the Catalytic Alkyne Metathesis Activity of Molybdenum and Tungsten 2,4,6-Trimethylbenzylidyne Complexes with Fluoroalkoxide Ligands OC(CF3)nMe3–n (n = 0–3), Organometallics, 2017, 36, 3398–3406 CrossRef CAS.
- D. P. Estes, C. P. Gordon, A. Fedorov, W.-C. Liao, H. Ehrhorn, C. Bittner, M. L. Zier, D. Bockfeld, K. W. Chan, O. Eisenstein, C. Raynaud, M. Tamm and C. Copéret, Molecular and Silica-Supported Molybdenum Alkyne Metathesis Catalysts: Influence of Electronics and Dynamics on Activity Revealed by Kinetics, Solid-State NMR, and Chemical Shift Analysis, J. Am. Chem. Soc., 2017, 139, 17597–17607 CrossRef CAS PubMed.
- M. Cui, R. Lin and G. Jia, Chemistry of Metallacyclobutadienes, Chem.–Asian J., 2018, 13, 895–912 CrossRef CAS PubMed.
- H. Ehrhorn, D. Bockfeld, M. Freytag, T. Bannenberg, C. E. Kefalidis, L. Maron and M. Tamm, Studies on Molybdena- and Tungstenacyclobutadiene Complexes Supported by Fluoroalkoxy Ligands as Intermediates of Alkyne Metathesis, Organometallics, 2019, 38, 1627–1639 CrossRef CAS.
- H. Ehrhorn and M. Tamm, Well-Defined Alkyne Metathesis Catalysts: Developments and Recent Applications, Chem.–Eur. J., 2019, 25, 3190–3208 CrossRef CAS PubMed.
- J. Hillenbrand, M. Leutzsch, C. P. Gordon, C. Copéret and A. Fürstner, 183W NMR Spectroscopy Guides the Search for Tungsten Alkylidyne Catalysts for Alkyne Metathesis, Angew. Chem., Int. Ed., 2020, 59, 21758–21768 CrossRef CAS PubMed.
- P. M. Hauser, M. van der Ende, J. Groos, W. Frey, D. Wang and M. R. Buchmeiser, Cationic Tungsten Alkylidyne N-Heterocyclic Carbene Complexes: Synthesis and Reactivity in Alkyne Metathesis, Eur. J. Inorg. Chem., 2020, 2020, 3070–3082 CrossRef CAS.
- A. Haack, J. Hillenbrand, M. Leutzsch, M. van Gastel, F. Neese and A. Fürstner, Productive Alkyne Metathesis with “Canopy Catalysts” Mandates Pseudorotation, J. Am. Chem. Soc., 2021, 143, 5643–5648 CrossRef CAS PubMed.
- R. R. Thompson, M. E. Rotella, X. Zhou, F. R. Fronczek, O. Gutierrez and S. Lee, Impact of Ligands and Metals on the Formation of Metallacyclic Intermediates and a Nontraditional Mechanism for Group VI Alkyne Metathesis Catalysts, J. Am. Chem. Soc., 2021, 143, 9026–9039 CrossRef CAS PubMed.
- Y. Cai, Y. Hua, Z. Lu, J. Chen, D. Chen and H. Xia, Metallacyclobutadienes: Intramolecular Rearrangement from Kinetic to Thermodynamic Isomers, Adv. Sci., 2024, 11, 2403940 CrossRef CAS PubMed.
- B. C. Bailey, A. R. Fout, H. Fan, J. Tomaszewski, J. C. Huffman, J. B. Gary, M. J. Johnson and D. J. Mindiola, Snapshots of an alkylidyne for nitride triple-bond metathesis, J. Am. Chem. Soc., 2007, 129, 2234–2235 CrossRef CAS PubMed.
- T. Kurogi, P. J. Carroll and D. J. Mindiola, A Terminally Bound Niobium Methylidyne, J. Am. Chem. Soc., 2016, 138, 4306–4309 CrossRef CAS PubMed.
- A. M. Geyer, R. L. Gdula, E. S. Wiedner and M. J. Johnson, Catalytic nitrile-alkyne cross-metathesis, J. Am. Chem. Soc., 2007, 129, 3800–3801 CrossRef CAS PubMed.
- A. M. Geyer, E. S. Wiedner, J. B. Gary, R. L. Gdula, N. C. Kuhlmann, M. J. Johnson, B. D. Dunietz and J. W. Kampf, Synthetic, mechanistic, and computational investigations of nitrile-alkyne cross-metathesis, J. Am. Chem. Soc., 2008, 130, 8984–8999 CrossRef CAS PubMed.
- A. F. Hill, J. A. K. Howard, T. P. Spaniol, F. G. A. Stone and J. Szameitat, Reactions of Mono- and Dinuclear Metal-Alkylidyne Complexes with Phosphaalkynes: C≡P and C≡Mo Metathesis, Angew. Chem., Int. Ed. Engl., 1989, 28, 210–211 CrossRef.
- Hill and coworkers, were however able to isolate a β-phosphametallacycle supported by a bridging iron tricarbonyl fragment as described in ref. 27.
- V. K. Jakhar, A. M. Esper, I. Ghiviriga, K. A. Abboud, C. Ehm and A. S. Veige, Isolation of an Elusive Phosphametallacyclobutadiene and Its Role in Reversible Carbon−Carbon Bond Cleavage, Angew. Chem., Int. Ed., 2022, 61, e202203073 CrossRef CAS PubMed.
- S. S. A. Juaid, D. Carmichael, P. B. Hitchcock, A. Marinetti, F. Mathey and J. F. Nixon, Reactions of zero- and di-valent platinum metal complexes with phosphirene ring systems. Crystal structures of cis-[PtCl2(PEt3)(PPhCPh=CPh)], cis-[Ptl(PPh3)(CPh=CPhPPhMe)] and [(Et3P)2Pt(CPh=CPhPPh)W(CO)5], J. Chem. Soc., Dalton Trans., 1991, 905–915 RSC.
- F. G. N. Cloke, P. B. Hitchcock, J. F. Nixon, D. James Wilson and P. Mountford, One- and two-step [2+2] cycloaddition reactions of group 4 imides with the phosphaalkyne ButCP. Crystal and molecular structures of [Zr(η5-C5H5)2(PCButNC6H3Me2-2,6)] and [TiCl2(P2C2But2NBut)(py)] (py) = pyridine), Chem. Commun., 1999, 661–662 RSC.
- S. Roy, E. D. Jemmis, A. Schulz, T. Beweries and U. Rosenthal, Theoretical Evidence of the Stabilization of an Unusual Four-Membered Metallacycloallene by a Transition-Metal Fragment, Angew. Chem., Int. Ed., 2012, 51, 5347–5350 CrossRef CAS PubMed.
- S. Roy, U. Rosenthal and E. D. Jemmis, Metallacyclocumulenes: A Theoretical Perspective on the Structure, Bonding, and Reactivity, Acc. Chem. Res., 2014, 47, 2917–2930 CrossRef CAS PubMed.
- F. Reiß, M. Reiß, A. Spannenberg, H. Jiao, D. Hollmann, P. Arndt, U. Rosenthal and T. Beweries, Titanocene Silylpropyne Complexes: Promising Intermediates en route to a Four-Membered 1-Metallacyclobuta-2,3-diene?, Chem.–Eur. J., 2017, 23, 14158–14162 CrossRef PubMed.
- U. Rosenthal, A Ghost Trapped: Realization of the 1-Titanacyclobuta-2,3-diene as the First Four-Membered Group 4 Metallacycloallene, Eur. J. Inorg. Chem., 2019, 2019, 3456–3461 CrossRef CAS.
- F. Reiß, M. Reiß, J. Bresien, A. Spannenberg, H. Jiao, W. Baumann, P. Arndt and T. Beweries, 1-Titanacyclobuta-2,3-diene – an elusive four-membered cyclic allene, Chem. Sci., 2019, 10, 5319–5325 RSC.
- T. Yoshimoto, H. Hashimoto, M. Ray, N. Hayakawa, T. Matsuo, J. Chakrabarti and H. Tobita, Products of [2+2] Cycloaddition between a W≡Si Triple-bonded Complex and Alkynes: Isolation, Structure, and Non-classical Bonding Interaction, Chem. Lett., 2020, 49, 311–314 CrossRef CAS.
- H. Lang, L. Zsolnai and G. Huttner, Metallorganische π-Liganden: η4-1-Phospha-2-ferracyclobutadien-Komplexe, Chem. Ber., 1985, 118, 4426–4432 CrossRef CAS.
- J. B. Russell, M. G. Jafari, J. H. Kim, B. Pudasaini, A. Ozarowski, J. Telser, M.-H. Baik and D. J. Mindiola, Ynamide and Azaalleneyl. Acid-Base Promoted Chelotropic and Spin-State Rearrangements in a Versatile Heterocumulene [(Ad)NCC(tBu)]-, Angew. Chem., Int. Ed., 2024, 63, e202401433 CrossRef CAS PubMed.
- F. Basuli, U. J. Kilgore, X. Hu, K. Meyer, M. Pink, J. C. Huffman and D. J. Mindiola, Cationic and Neutral Four-Coordinate Alkylidene Complexes of Vanadium(IV) Containing Short V=C Bonds, Angew. Chem., Int. Ed., 2004, 43, 3156–3159 CrossRef CAS PubMed.
- F. Basuli, B. C. Bailey, D. Brown, J. Tomaszewski, J. C. Huffman, M.-H. Baik and D. J. Mindiola, Terminal vanadium-neopentylidyne complexes and intramolecular cross-metathesis reactions to generate azametalacyclohexatrienes, J. Am. Chem. Soc., 2004, 126, 10506–10507 CrossRef CAS PubMed.
- B. C. Bailey, H. Fan, E. W. Baum, J. C. Huffman, M.-H. Baik and D. J. Mindiola, Intermolecular C-H bond activation promoted by a titanium alkylidyne, J. Am. Chem. Soc., 2005, 127, 16016–16017 CrossRef CAS PubMed.
- J. T. Lyon and L. Andrews, Electron Deficient Carbon−Titanium Triple Bonds: Formation of Triplet XC÷TiX3 Methylidyne Complexes, Inorg. Chem., 2006, 45, 9858–9863 CrossRef CAS PubMed.
- B. C. Bailey, J. C. Huffman and D. J. Mindiola, Intermolecular Activation of C−X (X = H, O, F) Bonds by a Ti≡CtBu Linkage, J. Am. Chem. Soc., 2007, 129, 5302–5303 CrossRef CAS PubMed.
- B. C. Bailey, H. Fan, J. C. Huffman, M.-H. Baik and D. J. Mindiola, Room Temperature Ring-Opening Metathesis of Pyridines by a Transient Ti≡C Linkage, J. Am. Chem. Soc., 2006, 128, 6798–6799 CrossRef CAS PubMed.
- B. C. Bailey, H. Fan, J. C. Huffman, M.-H. Baik and D. J. Mindiola, Intermolecular C−H Bond Activation Reactions Promoted by Transient Titanium Alkylidynes. Synthesis, Reactivity, Kinetic, and Theoretical Studies of the Ti≡C Linkage, J. Am. Chem. Soc., 2007, 129, 8781–8793 CrossRef CAS PubMed.
- D. Adhikari, F. Basuli, J. H. Orlando, X. F. Gao, J. C. Huffman, M. Pink and D. J. Mindiola, Zwitterionic and Cationic Titanium and Vanadium Complexes Having Terminal M-C Multiple Bonds. The Role of the β-Diketiminate Ligand in Formation of Charge-Separated Species, Organometallics, 2009, 28, 4115–4125 CrossRef CAS.
- A. R. Fout, J. Scott, D. L. Miller, B. C. Bailey, M. Pink and D. J. Mindiola, Dehydrofluorination of Hydrofluorocarbons by Titanium Alkylidynes via Sequential C-H/C-F Bond Activation Reactions. A Synthetic, Structural, and Mechanistic Study of 1,2-CH Bond Addition and β-Fluoride Elimination, Organometallics, 2009, 28, 331–347 CrossRef CAS.
- J. Scott and D. J. Mindiola, A tribute to Frederick Nye Tebbe. Lewis acid stabilized alkylidyne, alkylidene, and imides of 3d early transition metals, Dalton Trans., 2009, 8463–8472 RSC.
- B. C. Bailey, A. R. Fout, H. Fan, J. Tomaszewski, J. C. Huffman and D. J. Mindiola, An Alkylidyne Analogue of Tebbe's Reagent: Trapping Reactions of a Titanium Neopentylidyne by Incomplete and Complete 1,2-Additions, Angew. Chem., Int. Ed., 2007, 46, 8246–8249 CrossRef CAS PubMed.
- J. A. Flores, V. N. Cavaliere, D. Buck, B. Pintér, G. Chen, M. G. Crestani, M.-H. Baik and D. J. Mindiola, Methane activation and exchange by titanium-carbon multiple bonds, Chem. Sci., 2011, 2, 1457–1462 RSC.
- V. N. Cavaliere, M. G. Crestani, B. Pinter, M. Pink, C. H. Chen, M.-H. Baik and D. J. Mindiola, Room temperature dehydrogenation of ethane to ethylene, J. Am. Chem. Soc., 2011, 133, 10700–10703 CrossRef CAS PubMed.
- A. K. Hickey, M. G. Crestani, A. R. Fout, X. Gao, C. H. Chen and D. J. Mindiola, Dehydrogenation of hydrocarbons with metal-carbon multiple bonds and trapping of a titanium(II) intermediate, Dalton Trans., 2014, 43, 9834–9837 RSC.
- M. Kamitani, B. Pinter, K. Searles, M. G. Crestani, A. Hickey, B. C. Manor, P. J. Carroll and D. J. Mindiola, Phosphinoalkylidene and -alkylidyne Complexes of Titanium: Intermolecular C–H Bond Activation and Dehydrogenation Reactions, J. Am. Chem. Soc., 2015, 137, 11872–11875 CrossRef CAS PubMed.
- P. Zatsepin, E. Lee, J. Gu, M. R. Gau, P. J. Carroll, M.-H. Baik and D. J. Mindiola, Tebbe-like and Phosphonioalkylidene and -alkylidyne Complexes of Scandium, J. Am. Chem. Soc., 2020, 142, 10143–10152 CrossRef CAS PubMed.
- P. Deng, X. Shi, X. Gong and J. Cheng, Trinuclear scandium methylidyne complexes stabilized by pentamethylcyclopentadienyl ligands, Chem. Commun., 2021, 57, 6436–6439 RSC.
- S. Hernandez, D. S. Belov, V. Krivovicheva, S. Senthil and K. V. Bukhryakov, Decreasing the Bond Order between Vanadium and Oxo Ligand to Form 3d Schrock Carbynes, J. Am. Chem. Soc., 2024, 146, 18905–18909 CrossRef CAS PubMed.
- M. G. Jafari, J. B. Russell, H. Lee, B. Pudasaini, D. Pal, Z. Miao, M. R. Gau, P. J. Carroll, B. S. Sumerlin, A. S. Veige, M.-H. Baik and D. J. Mindiola, Vanadium Alkylidyne Initiated Cyclic Polymer Synthesis: The Importance of a Deprotiovanadacyclobutadiene Moiety, J. Am. Chem. Soc., 2024, 146, 2997–3009 CrossRef CAS PubMed.
- O. Theilmann, M. Ruhmann, A. Villinger, A. Schulz, W. W. Seidel, K. Kaleta, T. Beweries, P. Arndt and U. Rosenthal, [Cp2TiIII(NCy)2C-TiIIICp2]: A Transient Titanocene Carbene Complex?, Angew. Chem., Int. Ed., 2010, 49, 9282–9285 CrossRef CAS PubMed.
- M. Haehnel, S. Hansen, A. Spannenberg, P. Arndt, T. Beweries and U. Rosenthal, Highly Strained Heterometallacycles of Group 4 Metallocenes with Bis(diphenylphosphino)amide Ligands, Chem.–Eur. J., 2012, 18, 10546–10553 CrossRef CAS PubMed.
- M. Haehnel, M. Ruhmann, O. Theilmann, S. Roy, T. Beweries, P. Arndt, A. Spannenberg, A. Villinger, E. D. Jemmis, A. Schulz and U. Rosenthal, Reactions of Titanocene Bis(trimethylsilyl)acetylene Complexes with Carbodiimides: An Experimental and Theoretical Study of Complexation versus C–N Bond Activation, J. Am. Chem. Soc., 2012, 134, 15979–15991 CrossRef CAS PubMed.
- K. Kaleta, M. Ruhmann, O. Theilmann, S. Roy, T. Beweries, P. Arndt, A. Villinger, E. D. Jemmis, A. Schulz and U. Rosenthal, Experimental and Theoretical Studies of Unusual Four-Membered Metallacycles from Reactions of Group 4 Metallocene Bis(trimethylsilyl)acetylene Complexes with the Sulfurdiimide Me3SiN=S=NSiMe3, Eur. J. Inorg. Chem., 2012, 2012, 611–617 CrossRef CAS.
- L. Becker, V. V. Burlakov, P. Arndt, A. Spannenberg, W. Baumann, H. Jiao and U. Rosenthal, Reactions of Group 4 Metallocene Complexes with Mono- and Diphenylacetonitrile: Formation of Unusual Four- and Six-Membered Metallacycles, Chem.–Eur. J., 2013, 19, 4230–4237 CrossRef CAS PubMed.
- T. Beweries, M. Haehnel and U. Rosenthal, Recent advances in the chemistry of heterometallacycles of group 4 metallocenes, Catal. Sci. Technol., 2013, 3, 18–28 RSC.
- M. Haehnel, S. Hansen, K. Schubert, P. Arndt, A. Spannenberg, H. Jiao and U. Rosenthal, Synthesis, Characterization and Reactivity of Group 4 Metallocene Bis(diphenylphosphino)acetylene Complexes—A Reactivity and Bonding Study, J. Am. Chem. Soc., 2013, 135, 17556–17565 CrossRef CAS PubMed.
- M. Haehnel, J. B. Priebe, J. C. H. Yim, A. Spannenberg, A. Brückner, L. L. Schafer and U. Rosenthal, Four-Membered Heterometallacyclic d0 and d1 Complexes of Group 4 Metallocenes with Amidato Ligands, Chem.–Eur. J., 2014, 20, 7752–7758 CrossRef CAS PubMed.
- E. P. Beaumier, C. P. Gordon, R. P. Harkins, M. E. McGreal, X. Wen, C. Copéret, J. D. Goodpaster and I. A. Tonks, Cp2Ti(κ2-tBuNCNtBu): A Complex with an Unusual κ2 Coordination Mode of a Heterocumulene Featuring a Free Carbene, J. Am. Chem. Soc., 2020, 142, 8006–8018 CrossRef CAS PubMed.
- U. Rosenthal, Update for Reactions of Group 4 Metallocene Bis(trimethylsilyl)acetylene Complexes: A Never-Ending Story?, Organometallics, 2020, 39, 4403–4414 CrossRef CAS.
- L. Pauling, The nature of the chemical bond. IV. The energy of single bonds and the relative electronegativity of atoms, J. Am. Chem. Soc., 1932, 54, 3570–3582 CrossRef CAS.
- A. L. Allred, Electronegativity values from thermochemical data, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef CAS.
- R. L. Gdula, M. J. A. Johnson and N. W. Ockwig, Nitrogen-Atom Exchange Mediated by Nitrido Complexes of Molybdenum, Inorg. Chem., 2005, 44, 9140–9142 CrossRef CAS PubMed.
- P. G. Hayes, W. E. Piers and M. Parvez, Cationic Organoscandium β-Diketiminato Chemistry: Arene Exchange Kinetics in Solvent Separated Ion Pairs, J. Am. Chem. Soc., 2003, 125, 5622–5623 CrossRef CAS PubMed.
- C. H. Suresh and G. Frenking, 1,3-Metal–Carbon Bonding and Alkyne Metathesis: DFT Investigations on Model Complexes of Group 4, 5, and 6 Transition Metals, Organometallics, 2012, 31, 7171–7180 CrossRef CAS.
- G. Erker, R. Lecht, R. Schlund, K. Angermund and C. Krüger, Vanadium Carbene Complexes by Reaction of (Diene)metallocenes with [CpV(CO)4], Angew. Chem., Int. Ed. Engl., 1987, 26, 666–668 CrossRef.
- B. Hessen, A. Meetsma and J. H. Teuben, Alkylidene complex of vanadium: synthesis and structure of cyclopentadienyl[bis(dimethylphosphino)ethane](neopentylidene)vanadium(III), J. Am. Chem. Soc., 1989, 111, 5977–5978 CrossRef CAS.
- B. Hessen, A. Meetsma, F. Van Bolhuis, J. H. Teuben, G. Helgesson and S. Jagner, Chemistry of carbon monoxide free monocyclopentadienylvanadium(I) alkene and alkyne complexes, Organometallics, 1990, 9, 1925–1936 CrossRef CAS.
- M. Berlekamp, G. Erker and J. L. Petersen, Structure of a metallacyclic metaloxycarbene vanadium complex prepared from CpV(CO)4, J. Organomet. Chem., 1993, 458, 97–103 CrossRef CAS.
- B. Hessen, J. K. F. Buijink, A. Meetsma, J. H. Teuben, G. Helgesson, M. Haakansson, S. Jagner and A. L. Spek, a-, b-, and d-Hydrogen abstraction in the thermolysis of paramagnetic vanadium(III) dialkyl complexes, Organometallics, 1993, 12, 2268–2276 CrossRef CAS.
- J.-K. F. Buijink, J. H. Teuben, H. Kooijman and A. L. Spek, Synthesis, Molecular Structure, and Reactivity of a Half-Sandwich Vanadium(III) Imido Complex: The First Vanadium(V) Alkylidene, Organometallics, 1994, 13, 2922–2924 CrossRef CAS.
- M. Grehl, M. Berlekamp and G. Erker, A Nine-Membered Metallacyclic Metalloxycarbene Vanadium Complex, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 1772–1774 CrossRef.
- M. Moore, S. Gambarotta, G. Yap, L. M. Liable-Sands and A. L. Rheingold, Formation of a vanadium(V) bicyclic carbene–amide complex via insertion of alkyne into a V–C bond, Chem. Commun., 1997, 643–644 RSC.
- J. Yamada, M. Fujiki and K. Nomura, A Vanadium(V) Alkylidene Complex Exhibiting Remarkable Catalytic Activity for Ring-Opening Metathesis Polymerization (ROMP), Organometallics, 2005, 24, 2248–2250 CrossRef CAS.
- U. J. Kilgore, C. A. Sengelaub, M. Pink, A. R. Fout and D. J. Mindiola, A Transient VIII–Alkylidene Complex: Oxidation Chemistry Including the Activation of N2 to Afford a Highly Porous Honeycomb-Like Framework, Angew. Chem., Int. Ed., 2008, 47, 3769–3772 CrossRef CAS PubMed.
- G. Liu, X. Lu, M. Gagliardo, D. J. Beetstra, A. Meetsma and B. Hessen, Vanadium (β-(Dimethylamino)ethyl)cyclopentadienyl Complexes with Diphenylacetylene Ligands, Organometallics, 2008, 27, 2316–2320 CrossRef CAS.
- W. Zhang and K. Nomura, Facile Synthesis of (Imido)vanadium(V)−Alkyl, Alkylidene Complexes Containing an N-Heterocyclic Carbene Ligand from Their Trialkyl Analogues, Organometallics, 2008, 27, 6400–6402 CrossRef CAS.
- U. J. Kilgore, H. Fan, M. Pink, E. Urnezius, J. D. Protasiewicz and D. J. Mindiola, Phosphinidene group-transfer with a phospha-Wittig reagent: a new entry to transition metal phosphorus multiple bonds, Chem. Commun., 2009, 4521–4523 RSC.
- S. Zhang, M. Tamm and K. Nomura, 1,2-C−H Activation of Benzene Promoted by (Arylimido)vanadium(V)-Alkylidene Complexes: Isolation of the Alkylidene, Benzyne Complexes, Organometallics, 2011, 30, 2712–2720 CrossRef CAS.
- K. Nomura, K. Suzuki, S. Katao and Y. Matsumoto, Ring-Opening Polymerization of THF by Aryloxo-Modified (Imido)vanadium(V)-alkyl Complexes and Ring-Opening Metathesis Polymerization by Highly Active V(CHSiMe3)(NAd)(OC6F5)(PMe3)2, Organometallics, 2012, 31, 5114–5120 CrossRef CAS.
- K. Hatagami and K. Nomura, Synthesis of (Adamantylmido)vanadium(V)-Alkyl, Alkylidene Complex Trapped with PMe3: Reactions of the Alkylidene Complexes with Phenols, Organometallics, 2014, 33, 6585–6592 CrossRef CAS.
- K. Nomura, B. K. Bahuleyan, K. Tsutsumi and A. Igarashi, Synthesis of (Imido)vanadium(V) Alkyl and Alkylidene Complexes Containing Imidazolidin-2-iminato Ligands: Effect of Imido Ligand on ROMP and 1,2-C–H Bond Activation of Benzene, Organometallics, 2014, 33, 6682–6691 CrossRef CAS.
- S. Chaimongkolkunasin and K. Nomura, (Arylimido)Vanadium(V)-Alkylidenes Containing Chlorinated Phenoxy Ligands: Thermally Robust, Highly Active Catalyst in Ring-Opening Metathesis Polymerization of Cyclic Olefins, Organometallics, 2018, 37, 2064–2074 CrossRef CAS.
- D. Rehder, Vanadium NMR of organovanadium complexes, Coord. Chem. Rev., 2008, 252, 2209–2223 CrossRef CAS.
- P. G. Hayes, W. E. Piers and M. Parvez, Synthesis, Structure, and Ion Pair Dynamics of β-Diketiminato-Supported Organoscandium Contact Ion Pairs, Organometallics, 2005, 24, 1173–1183 CrossRef CAS.
- V. Iaroshenko, Organophosphorus Chemistry: From Molecules to Applications, John Wiley & Sons, 2019 Search PubMed.
- S. S. Batsanov, Van der Waals Radii of Elements, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- M. R. Churchill and J. W. Ziller, Crystal structure of a deprotonated tungstenacyclobutadiene complex, (η5-C5H5)W[C3(CMe3)2]Cl; characterization of an η3-RCCCR ligand, J. Organomet. Chem., 1985, 281, 237–248 CrossRef CAS.
- J. H. Freudenberger and R. R. Schrock, Multiple metal-carbon bonds. 42. Formation of tungstenacyclobutadiene complexes containing a proton in the ring and their conversion to deprotio tungstenacyclobutadiene complexes, Organometallics, 1986, 5, 1411–1417 CrossRef CAS.
- J. Heppekausen, R. Stade, A. Kondoh, G. Seidel, R. Goddard and A. Furstner, Optimized synthesis, structural investigations, ligand tuning and synthetic evaluation of silyloxy-based alkyne metathesis catalysts, Chem.–Eur. J., 2012, 18, 10281–10299 CrossRef CAS PubMed.
- R. Lhermet and A. Fürstner, Cross-Metathesis of Terminal Alkynes, Chem.–Eur. J., 2014, 20, 13188–13193 CrossRef CAS PubMed.
- L. G. McCullough, M. L. Listemann, R. R. Schrock, M. R. Churchill and J. W. Ziller, Multiple metal-carbon bonds. 34. Why terminal alkynes cannot be metathesized. Preparation and crystal structure of a deprotonated tungstacyclobutadiene complex, W(η5-C5H5)[C3(CMe3)2]Cl, J. Am. Chem. Soc., 1983, 105, 6729–6730 CrossRef CAS.
- X. Shi, S. Li, M. Reiß, A. Spannenberg, T. Holtrichter-Rößmann, F. Reiß and T. Beweries, 1-Zirconacyclobuta-2,3-dienes: synthesis of organometallic analogs of elusive 1,2-cyclobutadiene, unprecedented intramolecular C–H activation, and reactivity studies, Chem. Sci., 2021, 12, 16074–16084 RSC.
- X. Shi, S. Li, A. Spannenberg, F. Reiß and T. Beweries, Selective 1,2-insertion of carbodiimides and substrate-divergent silyl group migration at 1-metallacyclobuta-2,3-dienes, Inorg. Chem. Front., 2023, 10, 3584–3594 RSC.
- S. Li, M. Schroder, A. Prudlik, X. Shi, A. Spannenberg, J. Rabeah, R. Francke, B. Corzilius, F. Reiss and T. Beweries, A General Concept for the Electronic and Steric Modification of 1-Metallacyclobuta-2,3-dienes: A Case Study of Group 4 Metallocene Complexes, Chem.–Eur. J., 2024, 30, e202400708 CrossRef CAS PubMed.
- J. B. Russell, D. Konar, T. M. Keller, M. R. Gau, P. J. Carroll, J. Telser, D. W. Lester, A. S. Veige, B. S. Sumerlin and D. J. Mindiola, Metallacyclobuta-(2,3)-diene: A Bidentate Ligand for Stream-line Synthesis of First Row Transition Metal Catalysts for Cyclic Polymerization of Phenylacetylene, Angew. Chem., Int. Ed., 2024, 63, e202318956 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- E. Van Lenthe and E. J. Baerends, Optimized Slater-type basis sets for the elements 1–118, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- R. G. Parr, S. R. Gadre and L. J. Bartolotti, Local density functional theory of atoms and molecules, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 2522–2526 CrossRef CAS PubMed.
- R. F. Nalewajski, J. Mrozek and A. Michalak, Two-electron valence indices from the Kohn-Sham orbitals, Int. J. Quantum Chem., 1997, 61, 589–601 CrossRef CAS.
- I. Mayer, Charge, bond order and valence in the AB initio SCF theory, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS.
- C. H. Suresh and M.-H. Baik, α, β-(C–C–C) Agostic bonds in transition metal based olefin metathesis catalyses, Dalton Trans., 2005, 2982–2984 RSC.
- J. I. Rodríguez, R. F. Bader, P. W. Ayers, C. Michel, A. W. Götz and C. Bo, A high performance grid-based algorithm for computing QTAIM properties, Chem. Phys. Lett., 2009, 472, 149–152 CrossRef.
- J. I. Rodríguez, An efficient method for computing the QTAIM topology of a scalar field: The electron density case, J. Comput. Chem., 2013, 34, 681–686 CrossRef PubMed.
- A. D. Becke and K. E. Edgecombe, A simple measure of electron localization in atomic and molecular systems, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, spectral data, crystallographic and computational studies. CCDC 2379015 and 2379016. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05884d |
| This journal is © The Royal Society of Chemistry 2024 |