
SiO2 assisted Cu0–Cu+–NH2 composite interfaces for efficient CO2 electroreduction to C2+ products†
Zi-Yang
Zhang‡
a,
Hao
Tian‡
a,
Han
Jiao
a,
Xin
Wang
a,
Lei
Bian
a,
Yuan
Liu
 a,
Nithima
Khaorapapong
b,
Yusuke
Yamauchi
cde and
Zhong-Li
Wang
*a
a,
Nithima
Khaorapapong
b,
Yusuke
Yamauchi
cde and
Zhong-Li
Wang
*a
aTianjin Key Laboratory of Applied Catalysis Science & Technology, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China. E-mail: wang.zhongli@tju.edu.cn
bMaterials Chemistry Research Center, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen 40002, Thailand
cAustralian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, Brisbane, QLD 4072, Australia
dDepartment of Materials Process Engineering, Graduate School of Engineering, Nagoya University, Nagoya, 464–8603 Japan
eDepartment of Chemical and Biomolecular Engineering, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul, 03722 South Korea
First published on 13th December 2023
Abstract
The electrochemical CO2 reduction reaction (CO2RR) for high value-added multi-carbon product (C2+) production over copper oxide-based catalysts is an important way to realize the carbon cycle. However, developing effective reaction interfaces and microenvironments to improve the Faraday efficiency (FE) and current density of C2+ remains a major challenge. Herein, we construct Cu0–Cu+–NH2 composite interfaces with the assistance of SiO2. Using Cu2O nanoparticles as a model catalyst, a layer of porous SiO2 is first coated on the surface of the particles, and then, a silane coupling agent containing –NH2 is bonded on the surface of SiO2. The strong interaction between SiO2 and Cu2O at the interface induces the oxidation effect of low valent Cu, and even under the CO2RR, part of Cu+ is reduced to Cu0 and part of Cu+ still maintains positive valence, forming the interface of Cu0–Cu+. SiO2 also acts as a bridge between copper species and –NH2 to create a Cu catalyst–NH2 group interface. With the help of the synergistic effect of the composite interfaces, the optimized Cu2O@SiO2–NH2 catalyst achieves a FE of 81.2% for C2+ products with a current density of 292 mA cm−2 at −1.7 V versus a reversible hydrogen electrode. In situ Raman and attenuate total reflectance-infrared absorption spectroscopy spectra show that the interaction between surface –NH2 and CO2 molecules enhances the adsorption and activation process of CO2 and promotes the formation of CO intermediates (*CO). On the Cu0–Cu+ interface, the C–C coupling process between *CO is accelerated, and the two interfaces synergistically promote the generation of C2+ products. This work provides a new strategy for constructing composite interfaces to improve the CO2RR to C2+ products.
1. Introduction
The excessive emission of CO2 breaks the balance of the natural carbon cycle;1 therefore, it is urgent to develop new technologies for CO2 recycling and utilization to realize the sustainable development of resources and energy, and mitigate global warming. The electrochemical CO2 reduction reaction (CO2RR) coupled with renewable electric energy can not only synthesize high-value-added products from CO2 but also realize energy storage at the same time, which is a promising way of CO2 utilization.2,3 Up to now, it has been reported that at least 16 distinct products are formed from the CO2RR, such as CO, CH4, HCOOH, C2H4, C2H5OH, C3H7OH, and so on.4,5 Among the CO2RR products, C2+ can be used as a chemical raw material and fuel, leading to wide application.1,6 Consequently, researchers have made significant endeavors in the synthesis of C2+ through the CO2RR. However, CO2 is a linear and inert molecule with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond energy of up to 750 kJ mol−1.7 Thus, achieving CO activation under milder conditions requires the use of appropriate catalysts. Currently reported catalysts for CO2RR synthesis of C2+ products are mainly Cu-based catalysts, because they have a moderate adsorption strength for C1 intermediates in the CO2RR process, which is neither too strong for desorption nor too weak for further adsorption activation for subsequent reactions, to promote the dimerization between C1 intermediates and generate C2+ products.1,8
O bond energy of up to 750 kJ mol−1.7 Thus, achieving CO activation under milder conditions requires the use of appropriate catalysts. Currently reported catalysts for CO2RR synthesis of C2+ products are mainly Cu-based catalysts, because they have a moderate adsorption strength for C1 intermediates in the CO2RR process, which is neither too strong for desorption nor too weak for further adsorption activation for subsequent reactions, to promote the dimerization between C1 intermediates and generate C2+ products.1,8
Among Cu-based catalysts, the oxides of Cu and their derivatives are an important type of catalyst with high reactivity.9–11 During the reduction and reconstruction process, abundant metal-oxide (Cu0–Cuδ+) interfaces are generated for Cu oxides, which significantly enhance the activity of the catalysts and improve the rate of C–C coupling.12 Therefore, their electronic structure13 and morphology14,15 have been widely studied to improve the activity of the CO2RR to C2+ products. Especially, recent studies have found that the adsorption strength of the CO intermediate (*CO) on Cuδ+ species (0 < δ < 2) is stronger than that on Cu0 species at the metal-oxide interface,16–18 which is beneficial to increase the concentration of *CO on the catalyst surface and promote the *CO dimerization step. Therefore, it is important to improve the stability of Cuδ+ species under reduction conditions to produce C2+ products. For example, Zhou et al.16 reported a B-doped Cu catalyst to regulate the local electronic structure of Cu and improve the stability of positive valence Cu. Consequently, the adsorption and dimerization of *CO can be controlled by adjusting the average oxidation valence of Cu, which makes the Cu(B) catalyst achieve 79% faradaic efficiency (FE) of C2 in the CO2RR process. In addition, Yan et al.19 reported a hexagonal boron nitride (h-BN) modified Cu2O catalyst, where the strong electron interaction between the two components of Cu2O and BN makes the electrons on Cu2O transfer to BN to strengthen the Cu–O bond, thus stabilizing the Cu+ species during the CO2RR. Upon applying the Cu2O-BN catalyst for the CO2RR process, the ratio of C2H4/CO increased by 1.62 times compared with that of the Cu2O catalyst. Similarly, Zang et al.20 designed a carbon-coated CuOx (CuOx@C) catalyst and the carbon layer on the catalyst surface effectively stabilized Cu+ species, thereby facilitating the C–C coupling process. In the CO2RR process, the FE of ethanol reached 46%, and the partial current density reached 166 mA cm−2. A series of studies have proven that the interfaces between Cu+ and Cu0 regions could promote C2+ production. Additionally, organic molecules containing amino groups (–NH2) are often used as surface modifiers to regulate the surface properties of catalysts, thereby altering the reaction microenvironment during the CO2RR. Li et al.21 constructed a molecular–catalyst interface by modifying a layer of an N-aryl-dihydropyridine-based oligomer on the surface of the Cu catalyst, which made the catalyst exhibit excellent ethylene selectivity with an FE of 72% at −0.83 V versus the reversible hydrogen electrode (vs. RHE; the same potential scale is used in the following discussion unless otherwise specified) during the CO2RR. Similarly, Chen et al.22 incorporated polyamine on Cu electrodes. Due to the strong binding force between the CO2/CO molecule and –NH2, the polyamine–Cu interface formed a microenvironment with locally high concentrations of CO2 and CO, which accelerates the formation of ethylene and the FE reached 72% at −0.97 V. According to the above discussion, the Cu0–Cu+ reaction interface and –NH2 induced molecular–catalyst interface both can promote the formation of C2+ products in the CO2RR.
The widely suggested CO2-to-C2+ conversion mechanisms show that the promoted CO2 activation, the faster formation and adsorption of the C1 intermediate, and the accelerated C–C coupling process are essential to increase the selectivity of C2+ products. However, developing a reaction interface that simultaneously meets the requirements of three aspects still faces great challenges. In this work, we construct a Cu0–Cu+–NH2 composite reaction interface with the assistance of SiO2, which includes both the Cu-based Cu0–Cu+ interface and the Cu catalyst–NH2 group interface. We have fully utilized two characteristics of amorphous SiO2: firstly, it can uniformly coat inorganic nanoparticles, and secondly, it can bond with silane coupling agents to introduce organic functional groups. Using Cu2O nanoparticles as a model catalyst, a layer of porous SiO2 is first coated on the surface of the particles (named Cu2O@SiO2). Under the conditions of electrochemical reduction, part of Cu+ is reduced to Cu0, and part of Cu+ maintains positive valence under the strong interaction of SiO2, forming the interface of Cu0–Cu+. Then, a silane coupling agent containing –NH2 is bonded on the surface of SiO2 (named Cu2O@SiO2–NH2), and the SiO2 coating acts as a bridge between copper species and –NH2 to form a Cu catalyst–NH2 group interface. With the help of the synergistic effect of the Cu0–Cu+–NH2 composite interfaces, the optimized Cu2O@SiO2–NH2 catalyst achieves a selectivity of 81.2% for C2+ products at a current density of 292 mA cm−2 at −1.7 V (without iR compensation). In situ Raman and attenuate total reflectance-infrared absorption spectroscopy (ATR-IRAS) spectra show that the interaction between surface –NH2 and CO2 molecules enhances the adsorption and activation process of CO2 and promotes the formation of *CO, which increases the local concentration of surface *CO. On the Cu0–Cu+ interface, the C–C coupling process between *CO is accelerated, and the two interfaces synergistically promote the generation of C2+ products. This work provides a new strategy for constructing composite interfaces to promote the CO2RR to C2+ product conversion under high current density.
2. Experimental section
2.1 Materials
All reagents involved in this study were commercially available and used without further purification: CuCl2·2H2O (AR, JiangTian), NaOH (AR, MACKLIN), Na2CO3 (AR, JiangTian), KCl (AR, JiangTian), ethyl orthosilicate (GC, Aladdin), aminopropyl triethoxysilane (AR, HEOWNS), ascorbic acid (AR, JiangTian), n-octane (AR, MERYER), L-arginine (AR, JiangTian), polyvinylpyrrolidone (MW = 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, MACKLIN). All aqueous solutions were prepared with deionized water. A proton exchange membrane (Nafion 211, DuPont), Nafion (5 wt%, Sigma-Aldrich), isopropanol (>99%, Aladdin), carbon NPs (50 nm, Sigma-Aldrich), and graphite (Aladdin) were obtained.
000, MACKLIN). All aqueous solutions were prepared with deionized water. A proton exchange membrane (Nafion 211, DuPont), Nafion (5 wt%, Sigma-Aldrich), isopropanol (>99%, Aladdin), carbon NPs (50 nm, Sigma-Aldrich), and graphite (Aladdin) were obtained.
2.2 Catalyst preparation
Cu2O dodecahedron nanoparticles were synthesized by a method reported in the literature.23 1.11 g PVP was dissolved into deionized water, and then 10 mL of 0.1 mol L−1 CuCl2·2H2O was also added into the above solution dropwise, under stirring and heating to 55 °C. Then 10 mL of 2 mol L−1 NaOH solution was added. After stirring at 55 °C for 30 min, 10 mL of 1 mol L−1 ascorbic acid solution was added to the mixture and continued to stir at 55 °C for 3 h. Then, the precipitation was centrifuged and washed in deionized water and anhydrous ethanol 3 times to obtain Cu2O dodecahedron nanoparticles.Then the Cu2O nanoparticles were modified with SiO2 and –NH2.24 0.73 g n-octane and 0.014 g L-arginine were added into 15 mL deionized water and stirred vigorously at 60 °C. n-Octane was used as the solvent to prevent excessive polymerization of SiO2, and to control the formation process of SiO2 on the surface of Cu2O. After that, 25 μL ethyl orthosilicate (TEOS) was added to the mixture and stirred at 60 °C for 4 h. Then 0.2 g Cu2O dodecahedron nanoparticles were added into the mixture and stirred at room temperature for 20 h. The precipitation was centrifuged and washed three times with deionized water and anhydrous ethanol to obtain Cu2O@SiO2 nanoparticles. The obtained Cu2O@SiO2 nanoparticles were evenly dispersed into 25 mL anhydrous ethanol, and then 25 μL aminopropyl triethoxysilane (APTES) was added, and stirred at room temperature for 24 h. The precipitation was centrifuged, washed three times with anhydrous ethanol and deionized water, and dried by vacuum at room temperature for 12 h. Cu2O@SiO2–NH2 nanoparticles were obtained. For the control sample of Cu2O@SiO2, the amount of TEOS was increased to 50 μL during the SiO2 coating process to maintain a coating thickness similar to that of the Cu2O@SiO2–NH2 sample.
2.3 Catalyst characterization
Scanning electron microscopy (SEM) images were obtained on a Hitachi S-4800 field emission scanning electron microscopy. Transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) were conducted on a JEOL F200 transmission electron microscope with an acceleration voltage of 200 kV. The samples were dispersed in ethanol and ultrasonically treated for 20 min, and then the samples were added dropwise onto a Mo grid ultrathin carbon film. X-ray diffraction was conducted on a Bruker D8 Focus X-ray diffractometer using Cu Kα radiation (λ = 0.15418 nm). The tube voltage and current were 40 kV and 40 mA, respectively. The diffraction angle of samples was recorded from 20° to 80° (2θ) with a scan rate of 5° min−1. N2 physisorption analysis was conducted at −196 °C using a Tristar 3000 Micromeritics instrument. XPS was conducted on a Thermo-Fisher Scientistic K-Alpha+ instrument. The X-ray radiation source was Al Kα (hν = 1486.6 eV) with an X-ray power of 150 W and the spot size was 400 μm. The pass energy was 50 eV. The XPS was calibrated with a C 1s binding energy of 284.8 eV. X-ray absorption fine structure (XAFS) measurements were performed to probe the valence state and the bonding information of Cu species on a TableXAFS-500 X-ray absorption fine structure spectrometer. The CO2 adsorption experiment was carried out on a BEL SORP-max at 298 K.2.4 Electrode preparation
We first prepared a conductive gas-diffusion layer by sputtering the Cu layer on a PTFE film, copper target (99.999%). To prepare the catalyst ink, 10 mg of the pre-catalyst and 2 mg of carbon were dispersed in a mixture of 1 mL propanol and 30 μL of 5 wt% Nafion solution (Sigma-Aldrich); 10 mg carbon was dispersed in a mixture of 1 mL of propanol and 50 μL of 5 wt% Nafion solution; 10 mg graphite was dispersed in a mixture of 1 mL of propanol and 70 μL of 5 wt% Nafion solution, and then they were sonicated for at least 1 hour. The ink was airbrushed onto a 2 × 3 cm2 Cu/PTFE substrate with a loading of ∼1 mg cm−2; the carbon–graphite mixture was sprayed on the catalyst surface in turn. The PTFE-based gas diffusion electrode could enhance CO2 gas mass transfer through hydrophobic PTFE channels and the carbon powder and graphite powder with certain hydrophobicity on the surface of the catalyst could tailor the wettability of the electrolyte and prevent the catalyst from being flooded by aqueous electrolyte. A stainless-steel mesh was used as the anode. Before the reaction, the steel (1.5 × 2 cm2) was sonicated in acetone and deionized water for 30 min, respectively, and then dried by nitrogen purging for further use.2.5 Electrochemical CO2 reduction measurement
The CO2RR activity of the catalyst was investigated by performing electrolysis in a flow-cell configuration using 1 M KCl as the cathodic and 2 M KOH as the anodic electrolyte. Cl− anions can specifically adsorb on the inner Helmholtz plane (IHP) of the catalyst surface, which not only confines CO2 and facilitates electron transfer from the electrode to CO2via the Xad–C bond but also improves the *CO adsorption for favorable C–C coupling.25 Moreover, the formation of OH− during the CO2RR in the KCl electrolyte leads to a local alkaline environment, and the use of high pH can greatly accelerate the production rates for C2+ products.26 The flow cell consists of a gas diffusion layer as the working electrode (0.5 cm2), a proton exchange membrane (Nafion 211), a stainless-steel mesh (1500 mesh) as the counter electrode, and Ag/AgCl (saturated KCl) as the reference electrode. An electrochemical workstation (CHI660, Chenhua, Shanghai) was used to perform the CO2RR test. CO2 was supplied into gas chambers with a constant rate of 10 mL min−1 by using a mass-flow controller, and the outlet gas flow rate was also recorded by the flow controller. The cathodic electrolyte (30 mL) was circulated through the electrolyte chambers under a constant flow (5 mL min−1) via a peristaltic pump. The anodic electrolyte was circulated through the anodic chamber by a gas–liquid mixed flow pump. Reactions were tested via chronoamperometry for 30 min at different applied potentials from −1.1 V to −1.9 V. Gas products were analyzed via online GC (Shimadzu 2010) with a Thermal conductivity detector (TCD) (column: Agilent Carbon Plot (30 m × 0.32 mm × 3 μm)). The FE of gas products was calculated based on the following:where Cproduct is the concentration of the gas-phase products (mol L−1), νCO2 is the flow rate of CO2, t is the reaction time, e is the number of transferred electrons for the product, F the Faraday constant 96
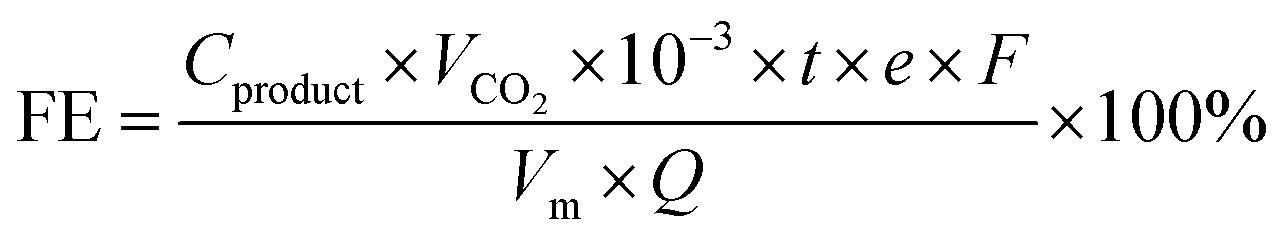 485 C mol−1, Vm is the gas mole volume, and Q is the total quantity of electric charge.
485 C mol−1, Vm is the gas mole volume, and Q is the total quantity of electric charge.
The liquid products were determined by H NMR (JEOL JNM ECZ600R 600 MHz), in which 300 μL electrolyte was mixed with 300 μL D2O and 10 μL diluent dimethyl sulfoxide (DMSO), wherein DMSO served as an internal standard. The concentration of liquid products was calculated based on the following:
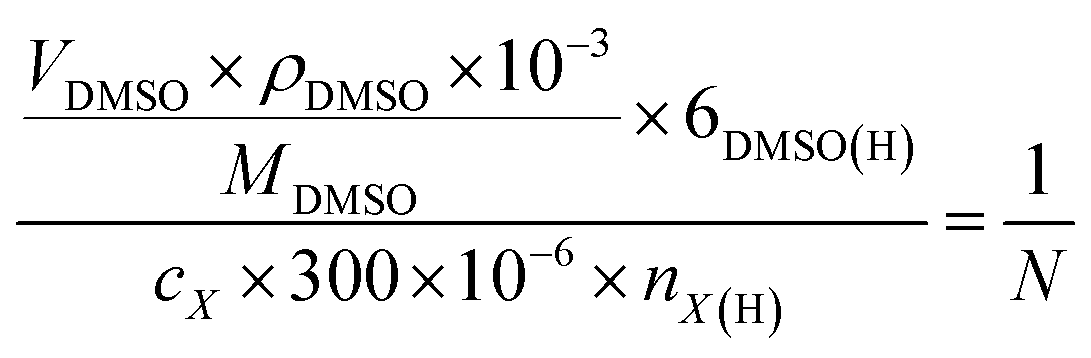
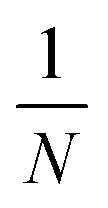 is the ratio of the product peak area to DMSO area in the nuclear magnetic spectrum.
is the ratio of the product peak area to DMSO area in the nuclear magnetic spectrum.
The FE of liquid products was computed from:
| FE = e × F × n/Q |
Potentials were referenced to the RHE based on the following:
| ERHE = E (vs. Ag/AgCl) + 0.197 V + 0.059 × pH |
2.6 In situ Raman test
In situ Raman experiments were conducted by Confocal Raman Microscopy (Horiba) with a 785 nm near-infrared laser in a homemade electrolyzer cell. A platinum electrode and Ag/AgCl electrode were used as the counter and reference electrodes, respectively. The catalyst for the cathode was sprayed on carbon paper (0.5 × 1 cm2) using ionomer solution as a binder. The electrolyte (1 M KCl) was saturated with CO2 solution and CO2 was continued to inject at a flow rate of 5 mL min−1. A long focal length lens (Leica, 50×) was used for focusing and collecting the incident and scattered laser light. Electrolysis at different cathodic potentials was performed for 10 min before signal collection.2.7 In situ ATR-IRAS measurement
In situ ATR-IRAS experiments were performed on a Nicolet iS20 spectrometer equipped with a HgCdTe (MCT/A) detector and a VeeMax III (PIKE Technologies) accessory in a homemade single-cell electrolyzer. A platinum electrode and Ag/AgCl electrode were used as the counter and reference electrodes. A fixed-angle Si prism (60°) coated with a catalyst embedded into the bottom of the cell served as the working electrode. Before testing, the detector was cooled with liquid nitrogen for at least 30 min to maintain a stable signal. Electrolysis at different cathodic potentials was carried out for 3 min with chronoamperometry by spectrum collection (32 scans, 4 cm−1 resolution). All spectra were subtracted from the background.3. Results and discussion
3.1 Synthesis of catalysts and characterization analysis
As shown in Fig. 1, the synthesis of Cu2O@SiO2–NH2 can be divided into three steps. Firstly, Cu2O dodecahedron nanoparticles were synthesized by a precipitation method. Second, the Cu2O NPs were mixed with TEOS uniformly, and the SiO2 formed during the hydrolysis of TEOS is uniformly covered on the surface of Cu2O NPs to form Cu2O@SiO2 samples. Finally, the Cu2O@SiO2 nanoparticles and ATPES were evenly dispersed with anhydrous ethanol to obtain Cu2O@SiO2–NH2.24 The detailed procedure is discussed in the Experimental section.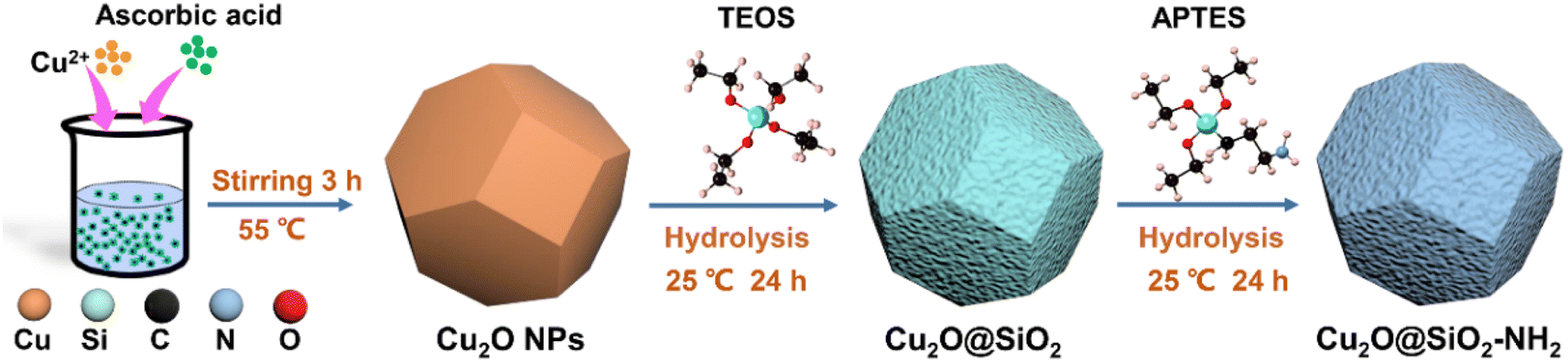 | ||
| Fig. 1 The schematic illustration of the Cu2O@SiO2–NH2 synthesis process. | ||
The morphology of the catalyst was characterized by SEM. As shown in Fig. 2a–c and S1,† the Cu2O nanoparticles exhibit a regular dodecahedral shape. After SiO2 coating, the surface of the nanoparticles becomes rough, indicating the successful coating of SiO2, and there may be a porous structure (Fig. 2b and S1b†). In the subsequent modification process of –NH2, the whole particle still maintained a complete dodecahedral structure. The results show that the SiO2 coating and –NH2 modification have no significant effect on the overall structure of Cu2O nanoparticles (Fig. 2a–c and S1†). The structure of Cu2O@SiO2–NH2 and control samples were characterized by TEM. As shown in Fig. S2,† the Cu2O nanoparticle exhibits a smooth surface in the TEM image, and the high-resolution TEM (HR-TEM) image shows that there is a lattice fringe d = 0.213 nm on the catalyst surface, which is assigned to the Cu2O (200) surface (Fig. S2b†). The Cu and O elements overlap with the STEM image in EDX element mapping (Fig. S2c†). In the TEM images of the Cu2O@SiO2 sample (Fig. S3a–c†), a lattice fringe of d = 0.246 nm can be seen in the inner layer, which belongs to the Cu2O (111) crystal plane, and an amorphous SiO2 coating with a thickness of about 10 nm located at the outer layer can be seen, which construct the obvious Cu2O–SiO2 interface. At the same time, the STEM and EDX element distribution maps further confirm the formation of the coating structure (Fig. S3d and e†). For the Cu2O@SiO2–NH2 sample, a uniform coating layer with a thickness of about 13 nm can be seen on the surface of Cu2O nanoparticles (Fig. 2a–f). By comparison with Cu2O@SiO2, it can be preliminarily confirmed that the Cu2O@SiO2 sample surface has been successfully modified with –NH2. The lattice fringe of d = 0.302 nm of the Cu2O (110) facet in close contact with the SiO2–NH2 coating layer in HR-TEM images (Fig. 2g) can confirm the formation of a Cu2O–SiO2–NH2 interface. Furthermore, the EDX elemental maps show that the Si and N elements are mainly distributed on the surface of Cu2O nanoparticles, and the overlay image of element distribution further proves the formation of Cu2O@SiO2–NH2. The surface pore distribution on the surface of Cu2O and Cu2O@SiO2–NH2 catalysts was characterized by using N2 isothermal adsorption–desorption curves. As illustrated in Fig. S4a,† the surface of Cu2O exhibits almost no pore structure. In contrast, the N2 isothermal adsorption–desorption curve of the Cu2O@SiO2–NH2 catalyst reveals a gradual uptake of nitrogen gas with a hysteresis loop, indicating the presence of irregular pores on its surface. At the same time, combined with the pore size distribution diagram in Fig. S4b,† it shows that there are mesoporous pores with an average pore size of 3.32 nm distributed on the Cu2O@SiO2–NH2 catalyst surface. And its specific surface area increases from 6 m2 g−1 of Cu2O to 40 m2 g−1 of Cu2O@SiO2–NH2.
 | ||
| Fig. 2 The SEM images of (a) Cu2O, (b) Cu2O@SiO2, and (c) Cu2O@SiO2–NH2; and the structural characterization of Cu2O@SiO2–NH2 (d and e) TEM images, (f) HR-TEM image and (g) EDX mapping. | ||
X-ray diffraction (XRD) was conducted to analyze the chemical compositions of Cu2O@SiO2–NH2 and the control samples. As shown in Fig. 3a and S5,† five obvious diffraction peaks in the XRD pattern of Cu2O are located at 2θ = 29°, 36°, 42.5°, 62° and 74°, which belong to the (110), (111), (200), (220) and (311) crystal planes of Cu2O, respectively. However, the position of the diffraction peaks of Cu2O dodecahedron nanoparticles have no obvious change after SiO2 coating and –NH2 modification, indicating that the process of SiO2 coating and –NH2 modification have no significant effect on the crystal phase structure of Cu2O nanoparticles. The surface chemical state of the samples was characterized by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 3b, for the Cu 2p XPS spectrum, there are only two peaks at the binding energies of 932.3 and 952.0 eV for the Cu2O catalyst, which are ascribed to Cu+ 2p3/2 and 2p1/2, respectively.27 Meanwhile for Cu2O@SiO2 and Cu2O–SiO2–NH2 catalysts, a satellite peak at 943 eV appears in the Cu 2p XPS spectra, indicating the presence of Cu2+ species.28 Meanwhile, the asymmetric peaks of Cu 2p can be deconvoluted into two sets of peaks. The first group of peaks with binding energies of 932.3 and 952.0 eV are attributed to 2p3/2 and 2p1/2 of Cu+ species. The second set of peaks at 934.6 and 954.3 eV belong to 2p3/2 and 2p1/2 of Cu2+ species.29 Compared with Cu2O, both Cu2+ and Cu+ species exist on the surface of Cu2O@SiO2 and Cu2O@SiO2–NH2, indicating strong interaction between SiO2 and Cu species. This result is further confirmed by the Cu LMM X-ray induced Auger transition spectra (XAES) (Fig. 3c), where the Cu LMM XAES of Cu2O sample shows a symmetrical peak at 916.8 eV, which belongs to Cu+ species, while the Cu LMM XAES of Cu2O@SiO2 and Cu2O@SiO2–NH2 show an asymmetric peak with a wider full width at half maximum (FWHM), indicating the coexistence of Cu+ and Cu2+ species at 916.8 and 917.9 eV.30 As is well known, XRD is the analysis of the entire bulk phase, while XPS is surface analysis. No CuO phase is observed in the XRD pattern (Fig. 3a), indicating a low content of Cu2+. XPS spectra (Fig. 3b) show a clear Cu2+ peak, indicating that Cu2+ is mainly present on the surface, while for Cu2O@SiO2–NH2 and Cu2O@SiO2 catalysts, it is at the interface between Cu2O and SiO2. The production of Cu2+ indicates that the strong interaction between SiO2 and Cu2O at the interface induces the oxidation effect of low valent Cu, which will affect the reduction of Cu2O in the CO2RR.31 Moreover, XPS analysis was also performed on the Cu2O@ sample that was treated under the same conditions without TEOS and APTES, and there is no significant difference in Cu 2p XPS between the Cu2O@ and Cu2O samples, which indicates that the treatment process did not oxidize Cu+ to Cu2+. Therefore, it can be confirmed that the strong interaction between SiO2 and Cu species at the Cu2O–SiO2 interface promotes the formation of Cu2+ species. In addition, the chemical state of the –NH2 group on the surface was characterized by N 1s XPS. As shown in Fig. 3d, the spectrum of N 1s XPS can be divided into two peaks located at the binding energies of 400 and 403.2 eV, respectively. According to a previous report, N 1s XPS at low binding energy belongs to the –NH2 group, while the peak at high binding energy belongs to –NH3+.32 This means that part of the –NH2 group on Cu2O@SiO2–NH2 has been protonated, which may increase the local pH on the catalyst surface.22
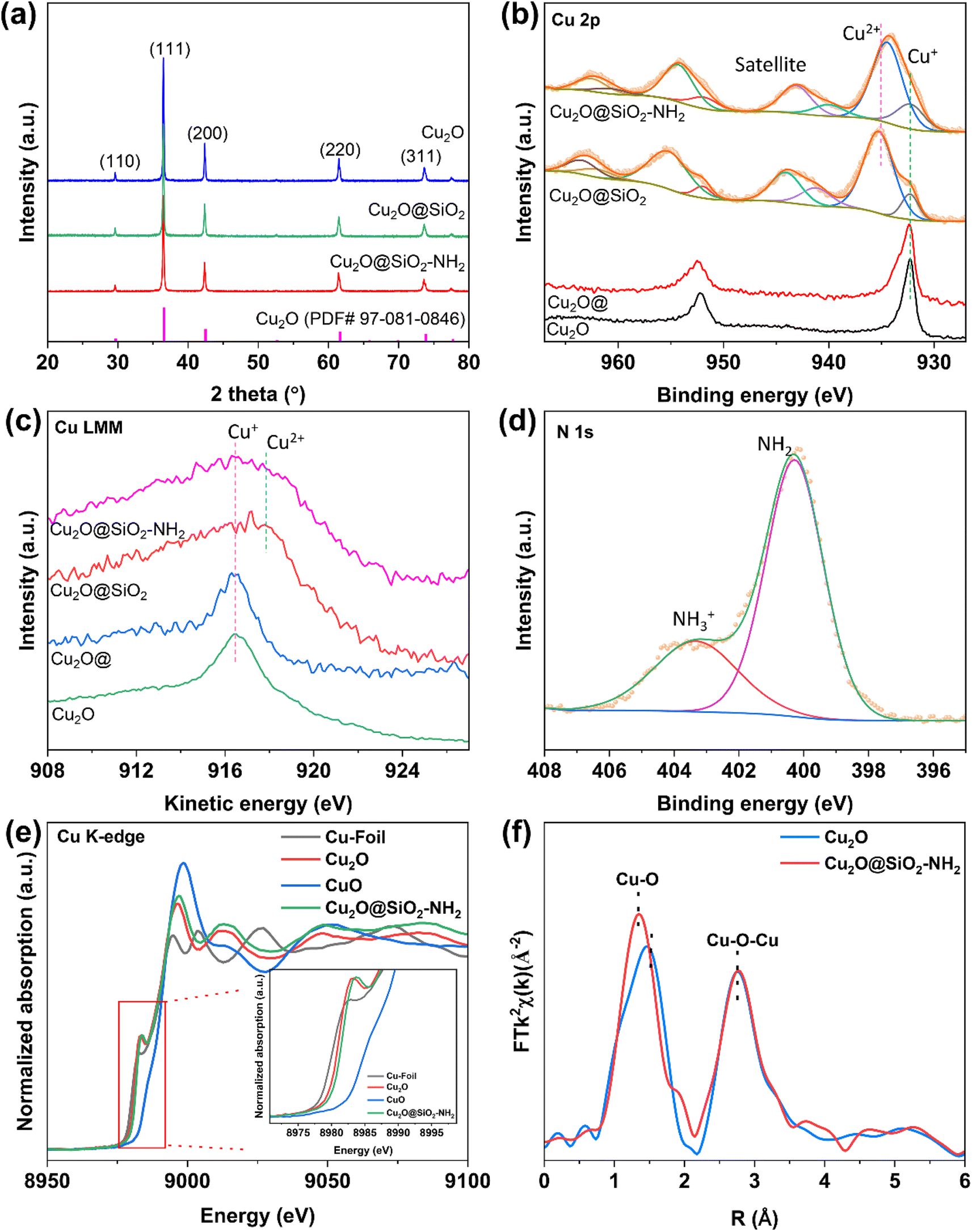 | ||
| Fig. 3 Chemical composition characterization of Cu2O, Cu2O@SiO2 and Cu2O@SiO2–NH2. (a) XRD patterns, (b) Cu 2p XPS spectra, (c) Cu LMM XAES spectra, (d) N 1s XPS spectrum, (e) XANES spectra and (f) Fourier transformed EXAFS spectra of the Cu K-edge. | ||
To explore more detailed structural information, the samples are further investigated by X-ray absorption fine structure spectroscopy (XAFS). Fig. 3e shows the X-ray absorption near-edge structure (XANES) spectra of Cu. In the Cu K-edge spectra, the pre-edge peak at 8987 eV is attributed to the dipole-forbidden 1s to 3d electron transition, which represents the fingerprint of Cu2+. Moreover, the absorption edge of the curve located between those for Cu2O and CuO shows the averaged valence state of Cu species in Cu2O@SiO2–NH2 between +1 and +2, which is consistent with XPS data. Moreover, the extended X-ray absorption fine structure (EXAFS) spectra of the Cu K-edge show that compared to pure Cu2O, the Cu2O@SiO2–NH2 catalyst exhibits a shorter Cu–O bond length in the first shell, indicating that SiO2 enhances the bonding between Cu2+/Cu+ and O (Fig. 3f). To demonstrate the interaction between –NH2 groups and CO2, the CO2 adsorption capacity of the catalyst was tested. As shown in Fig. S6,† compared to Cu2O@SiO2, Cu2O@SiO2–NH2 exhibits stronger CO2 adsorption, indicating that the modification of –NH2 significantly enhances the CO2 adsorption capacity, which will play an important role in promoting the activity of the CO2RR.
3.2 Electrochemical CO2 reduction performance
The catalytic performance of Cu2O@SiO2–NH2 for the CO2RR was evaluated in a flow-cell with 1 M KCl cathode electrolyte (Fig. S7†). The gas and liquid phase products were detected by online gas chromatography (GC) and 1H nuclear magnetic resonance (1H NMR), respectively, (Fig. S8 and S9†). Fig. 4a shows the linear sweep voltammetry (LSV) curves of Cu2O@SiO2–NH2 and the control samples. The cathode current density increases sharply after CO2 gas is introduced, corresponding to the catalytic CO2RR. When CO2 is replaced by N2, the cathodic current of Cu2O@SiO2–NH2 decreases significantly, which indirectly indicates the low HER activity of Cu2O@SiO2–NH2. Under a N2 atmosphere, the HER current density of the Cu2O@SiO2–NH2 catalyst is relatively low compared to Cu2O. Another possible reason is that the modification of SiO2 and –NH2 groups hinders the diffusion of H2O molecules to the catalyst surface, while the CO2 atmosphere eliminates this obstacle and accelerates the adsorption and diffusion of H2O and CO2. The current density of the CO2RR on Cu2O@SiO2–NH2 is significantly higher than that on Cu2O and Cu2O@SiO2, suggesting that Cu2O@SiO2–NH2 has a strong CO2RR activity. Due to the exposure of the Cu2O catalyst to the electrolyte solution, its HER current density under the N2 atmosphere is slightly higher than that of Cu2O@SiO2 and Cu2O@SiO2–NH2, indicating that the HER is more likely to occur in the Cu2O catalyst.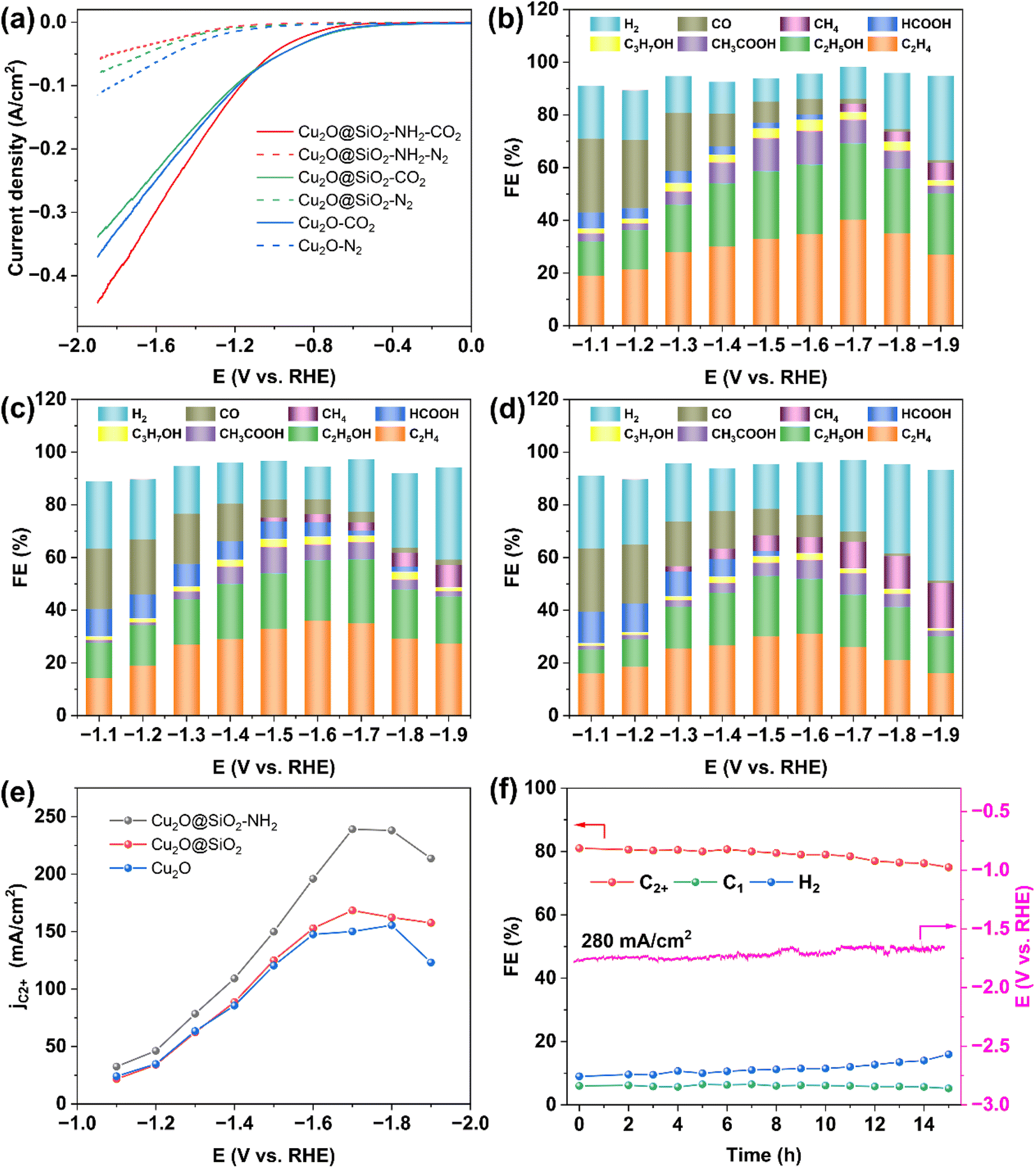 | ||
| Fig. 4 CO2RR performance on Cu2O@SiO2–NH2, Cu2O@SiO2 and Cu2O catalysts. (a) LSV curves toward the CO2RR; product distributions and corresponding faradaic efficiencies produced by Cu2O@SiO2–NH2 (b), Cu2O@SiO2 (c), and Cu2O (d), (e) the partial current density of C2+ products, and (f) stability test of Cu2O@SiO2–NH2 at 280 mA cm−2 in 1 M KCl electrolyte. | ||
The product distribution of the CO2RR over Cu2O@SiO2–NH2 and the control samples in the cathode potential range of −1.1 to −1.9 V as shown in Fig. 4b–d. The total FE of C2+ products exhibits a volcanic trend relative to the cathodic potential change over Cu2O@SiO2–NH2 (Fig. 4b), and the total current density increases from 80 to 375 mA cm−2 (Fig. S10†). Under a current density of 292 mA cm−2 at −1.7 V, the FE of C2+ products reaches the highest value of 81.2% (including C2H4: 40.2%, C2H5OH: 29%, CH3COOH: 9%, and C3H7OH: 3%), which has a higher activity than most reported catalysts (Table S1†). The FE of H2 and C1 is only 12% and 6%, respectively. In comparison, the FE of C2+ is 68.3% (C2H4: 35%, C2H5OH: 24.3%, CH3COOH: 6.5%, and C3H7OH: 2.5%) over Cu2O@SiO2 at −1.7 V (Fig. 4c). Meanwhile for Cu2O dodecahedron nanoparticles, the C2+ FE of Cu2O is only 61.7% (C2H4: 31%, C2H5OH: 21%, CH3COOH: 7%, and C3H7OH: 2.7%) at the optimum potential of −1.6 V (Fig. 4d). As a result, the ratio of C2+/C1 increases significantly from 4 in Cu2O to 13.5 in Cu2O@SiO2–NH2 (Fig. S11a†). In addition, the partial current densities of C2+ products of different samples increase from 147 mA cm−2 of Cu2O to 168 mA cm−2 of Cu2O@SiO2 during the CO2RR process, and then it further increases to 237 mA cm−2 over Cu2O@SiO2–NH2 (Fig. 4e and S11b–d†). These results indicate that the Cu2O@SiO2–NH2 catalyst has a high selectivity of C2+ products in the process of the CO2RR.
By comparison, it can be found that when Cu2O nanoparticles are coated with SiO2, the HER is inhibited and the selectivity of C2+ products increases in the full cathodic potential range. This indicates that the SiO2 coating layer helps to promote the C–C coupling process. According to the Cu 2p XPS results, the Cu species on the surface will maintain a high valence state after coating with SiO2, thus improving the adsorption strength of *CO on Cu species and increasing the surface coverage of *CO,33 followed by enhanced C–C coupling. After modifying the surface of the Cu2O@SiO2 sample with the –NH2 group, the CO selectivity increases at low cathodic potential, indicating that the CO2RR process is accelerated. According to previous reports, the interaction between nucleophilic N in –NH2 and electrophilic C in CO2 molecules can enrich CO2, thus increasing the local CO2 concentration on the catalyst surface,22,34,35 and increasing the conversion rate of CO2 to CO. Therefore, the reaction environment with a high local concentration of *CO can be provided at higher cathodic potential. Meanwhile, according to the N 1s XPS results (Fig. 3d), part of the –NH2 group on the catalyst surface interacts with H2O to form protonated –NH3+ and release OH− at the same time, leading to a higher local pH value of the reaction micro-environment,22 which is beneficial to promote the dimerization process of the C1 intermediate.36 Therefore, the positively charged Cu species coupled with the reaction microenvironment with sufficient CO supply and a high local pH value created by the surface –NH2 group significantly enhance the formation of C2+ products from the CO2RR and increase the FE of C2+ to 81.2% (Fig. 4b). However, in the Cu2O catalyst without SiO2 coating, the FE of CH4 increases at high potential (Fig. 4d). According to a previous report, the CO2RR is conducive to the formation of the C1 product over the bulk Cu catalyst in neutral electrolyte.37,38 Therefore, it can be inferred that Cu2O is rapidly reduced to form Cu in the reaction process, which leads to the formation of CH4 and promotes the HER process at the same time, while the SiO2 coating promotes the formation of C2+ by stabilizing the positively charged Cu species in the catalyst.31 To eliminate the influence of the Cu/PTFE substrate on the analysis results, the CO2RR performance of the Cu/PTFE substrate was tested under the same conditions. It is found that the CO2RR to C2+ product conversion over Cu/PTFE shows a maximum FE of 26.2% at −1.5 V, and the FE of H2 reaches 58% with a total current density of only 67 mA cm−2 (Fig. S12†). The partial current density of C2+ products over Cu/PTFE is only 17 mA cm−2, compared to 237 mA cm−2 over Cu2O@SiO2–NH2, which shows that the main activity source of the CO2RR to C2+ products is originated from the target catalyst, and the Cu/PTFE substrate has little effect on the activity of the catalysts. The pH value of the electrolyte was also tested during the CO2RR. Due to the generation of OH− at the cathode and the reaction of some OH− with CO2 to generate CO32−, the pH increased from 6.82 at 0 V to 10.11 at −1.9 V (Fig. S13†), and the local alkaline environment may facilitate C–C coupling.2 In addition to the excellent FE of C2+, Cu2O@SiO2–NH2 also exhibits high stability at high current densities. The FE of C2+ from the CO2RR over Cu2O@SiO2–NH2 does not decrease significantly for 15 h with 280 mA cm−2 current density in 1 M KCl electrolyte and remains above 75% (Fig. 4f).
3.3 Characterization of the samples after the activity test
Considering the reconstruction phenomenon of the oxidation state Cu-based catalyst during the CO2RR, it is necessary to further explore the activity source of Cu2O@SiO2–NH2 and the control samples, so the structure and composition of the samples after the activity test were characterized (the samples after the test were stored in a vacuum). As shown in TEM images of Fig. S14–S16,† the morphologies of Cu2O@SiO2–NH2 and Cu2O@SiO2 catalysts have no obvious change after the CO2RR, except that the thickness of the SiO2–NH2 layer slightly decreases, indicating that the coating layer is partially dissolved during the reaction. It can be explained by the fact that the formation of OH− during the CO2RR increases the pH of the solution, and SiO2 reacts with OH− leading to a decrease in the coating layer thickness. Meanwhile, the HR-TEM images of Cu2O@SiO2–NH2 and Cu2O@SiO2 samples (Fig. S14c, d and S15c, d†) show lattice fringes of d = 0.302, 0.213 and 0.209 nm, which belong to the (110) and (200) of Cu2O and Cu (111) crystal facets, respectively. This indicates that the oxidation state Cu on the catalyst surface is partially reduced, forming the Cu–Cu2O interface; therefore, the metal-oxide interface effect can effectively improve the formation rate of C2+ from the CO2RR.20 At the same time, the elements overlap each other in the EDX mapping of Cu2O@SiO2–NH2 and Cu2O@SiO2 samples, which proves that the Cu2O@SiO2–NH2 catalyst still maintains a complete coating structure after the CO2RR (Fig. S14e, f and S15e†). In contrast, the morphology of the Cu2O catalyst after the CO2RR is significantly changed (Fig. S16a and b†), and only the lattice fringe of the Cu (111) facet (d = 0.2086 nm) can be observed in the HR-TEM image (Fig. S16c†). This indicates that Cu2O nanoparticles are completely reduced to Cu during the CO2RR. Notably, irregular holes can be observed on the surface of Cu2O nanoparticles (Fig. S16d†), which further indicates that Cu2O nanoparticles are reduced, leading to a change in the morphology of nanoparticles. The EDX mapping also confirmed the morphology change (Fig. S16e†), where the Cu element is not evenly distributed. By comparing the TEM images of Cu2O@SiO2–NH2, Cu2O@SiO2 and Cu2O catalysts, the results indicate that SiO2 coating can stabilize the morphology of Cu2O nanoparticles, inhibit the reduction of oxidized Cu in the catalyst, and form a stable metal-oxide interface in the reduction process, which accelerates the C–C coupling step during the CO2RR.XRD characterization was carried out to analyze the chemical composition of the catalyst after the CO2RR. As shown in Fig. 5a, there is only one diffraction peak at 2θ = 43.2° in the XRD pattern of the Cu2O catalyst, which is attributed to metal Cu (PDF# 97-004-3493), indicating that Cu2O is completely reduced during the CO2RR. There are two sets of diffraction peaks in the XRD patterns of Cu2O@SiO2–NH2 and Cu2O–SiO2 catalysts, which are located at 2θ = 36.4°, 42.5° and 2θ = 43.2°. They belong to the Cu2O (PDF# 97-018-0846) and Cu phases, respectively. The XRD results show that Cu2O nanoparticles coated with SiO2 inhibit the reduction of Cu+ species and form a metal-oxide interface in the CO2RR process. Combined with TEM characterization results, it is further demonstrated that SiO2 coating is beneficial to stabilize the oxidized copper in Cu2O nanoparticles during the CO2RR, due to the strong interaction between SiO2 and Cu2O species. At the same time, the Cu 2p XPS spectrum of Cu2O after the CO2RR (Fig. 5b) shows only two peaks located at 932.0 and 951.8 eV, belonging to Cu0 species.39 Meanwhile for Cu 2p XPS of Cu2O@SiO2 and Cu2O@SiO2–NH2, there is weak satellite peaks at 942–944 eV and acromial peaks at 934.7 eV, indicating the presence of a small amount of Cu2+ on the surface.40 We further analyzed the Cu LMM XAES spectra of Cu2O@SiO2–NH2 and control samples after the CO2RR. As shown in Fig. 5c, there is only one peak located at 918.1 eV in the Cu LMM XAES of the Cu2O catalyst, which belongs to Cu0 species, suggesting that all Cu+ species are reduced to Cu0 species. In contrast, the Cu LMM XAES of Cu2O@SiO2 and Cu2O@SiO2–NH2 catalysts can be fitted into two peaks at 916.2 and 918.1 eV, belonging to the Cu+ and Cu0 species, respectively.19,41 Compared with the Cu 2p XPS before the CO2RR (Fig. 3b), the surface of the Cu2O@SiO2–NH2 catalyst is mainly Cu2+, and after the CO2RR, it is mainly mixed Cu+ and Cu0, indicating that SiO2 can stabilize part of Cu+ and form Cu+–Cu0 interfaces. The results are consistent with TEM and XRD images. Meanwhile, the Si 2p XPS also can be detected in Cu2O@SiO2–NH2 and Cu2O@SiO2 catalysts, which confirms the presence of SiO2 on the catalyst surface after the CO2RR (Fig. S17†). Moreover, the N 1s XPS spectrum shows the retention of –NH2 groups on the surface and the presence of groups in two states of –NH2 and –NH3+, indicating that the surface –NH2 continued to play a role in the enrichment of CO2 during the reaction process and accelerated the conversion rate of CO2 (Fig. 5d).
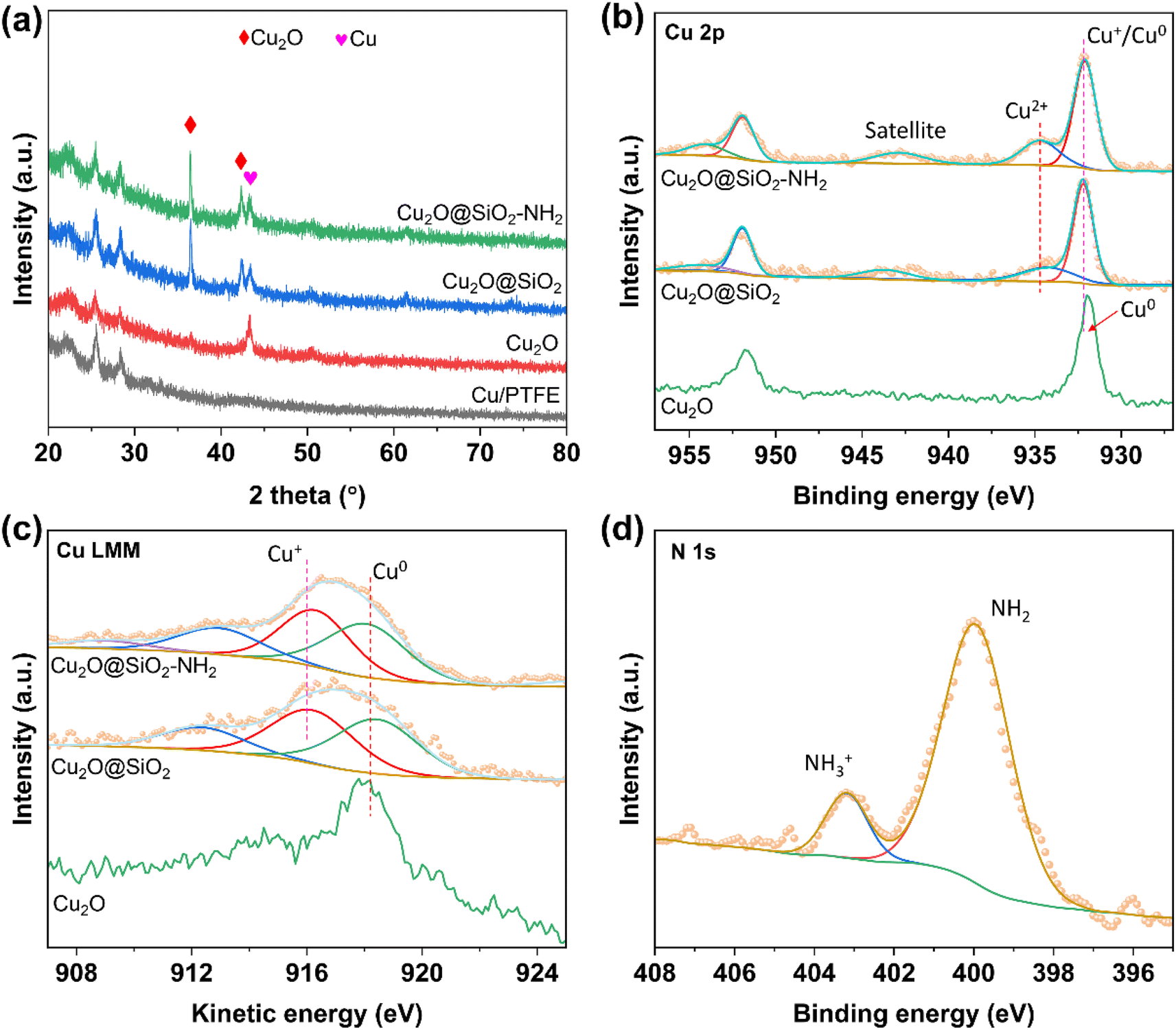 | ||
| Fig. 5 Chemical composition characterizations of Cu2O, Cu2O@SiO2 and Cu2O@SiO2–NH2 after the CO2RR. (a) XRD patterns, (b) Cu 2p XPS spectra, (c) Cu LMM XAES spectra and (d) N 1s XPS spectrum. | ||
3.4 Reaction mechanism study
In situ Raman experiments were performed to study the valence changes of Cu species of Cu2O@SiO2–NH2 and control samples, and to detect the key intermediates in the CO2RR process. As shown in Fig. 6, under open-circuit potential conditions, the two Raman peaks at 145 and 213 cm−1 belong to the Cu2O phase.13 When the cathodic potential is applied, the peaks of the Cu2O phase still exist in the Raman spectrum of Cu2O@SiO2–NH2, indicating that the Cuδ+ species can be well preserved in the CO2RR process. Similarly, this phenomenon has also been observed on the Cu2O@SiO2 catalyst, where Cu2O species can exist stably with the increase of cathodic potential. In contrast, for the Cu2O catalyst, the two Raman peaks at 145 and 213 cm−1 only can be observed under the open-circuit potential conditions. However, when a cathodic potential of −1.1 V is applied, the two peaks disappear, suggesting that the Cu2O on the catalyst surface is completely reduced to metal Cu. The above analysis results imply that the strong interaction between SiO2 and Cu2O can improve the stability of Cuδ+ species on the catalyst surface. This is consistent with the results of TEM and XRD of samples after the CO2RR. More importantly, in the Raman spectrum of Cu2O@SiO2–NH2, an obvious peak at 529 cm−1 is observed, which is related to the chemisorption of CO2 on the copper surface (*CO2ad),42,43 and it further enhances with the increase of cathodic potential applied. This shows that the catalyst has strong CO2 adsorption and activation ability (Fig. 6a). In contrast, the peak intensity of *CO2ad at 529 cm−1 on Cu2O@SiO2 and Cu2O catalysts decreases sharply (Fig. 6b and c), indicating that *CO2ad is less adsorbed on the surface. Therefore, this result shows that –NH2 enhances the ability of the catalyst to adsorb and activate CO2. As mentioned above, the –NH2 group can enrich CO2 molecules, thus improving the CO2 coverage on the catalyst surface.22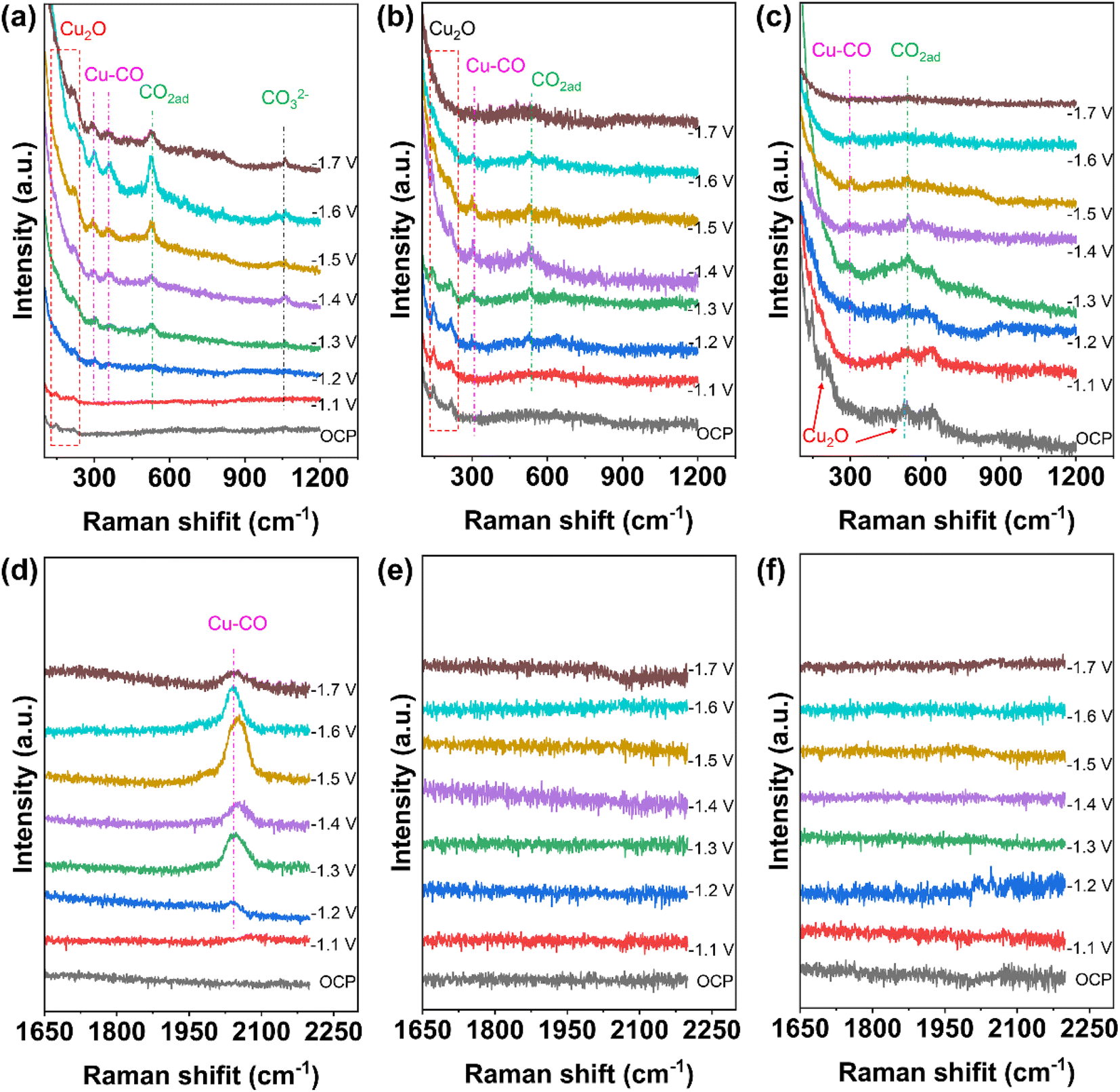 | ||
| Fig. 6 In situ Raman spectra of the catalysts under CO2RR conditions. (a and d) Cu2O@SiO2–NH2, (b and e) Cu2O@SiO2, and (c and f) Cu2O. | ||
Moreover, the peaks at 297 and 373 cm−1 are attributed to the rotation and stretching vibrations of *CO on Cu (Cu–CO) in the Raman spectra,12,44 which indicate the formation of CO and the adsorption of *CO on the catalyst surface (Fig. 6a–c). At the same time, the most obvious peak at 2000–2080 cm−1 is associated with *COa (Fig. 6d).27 It is worth noting that the adsorption peak of *CO first increases and then decreases with the increase of cathodic potential, indicating that the coverage of *CO on the catalyst surface increases first with the cathodic potential, which promotes the C–C coupling step. However, after the C–C coupling reaction, the *CO adsorbed on the surface will be consumed, thus weakening the adsorption peak of *CO.44 In contrast, the Cu–CO Raman signal on Cu2O@SiO2 is weaker (Fig. 6b and e), indicating the low surface coverage of *CO. Moreover, the Cu–CO Raman signal is the weakest on the Cu2O catalyst (Fig. 6c and f). The results of in situ Raman show that the interaction between –NH2 and CO2 creates a high local concentration of the CO2 microenvironment at the Cu0–Cu+–NH2 composite interfaces, which accelerates the activation of CO2 and generation of CO, and enhances the adsorption of *CO.45 At the same time, the Cu0/Cu+ synergistic effect at the interface may promote the C–C coupling step. However, the Cu0–Cu+ interface without –NH2 on the Cu2O@SiO2 surface leads to a low local concentration of the CO2 microenvironment, which suppresses the CO generation rate, followed by a slow C–C coupling step. The lack of a SiO2 coating layer on the Cu2O catalyst leads to the quick reduction of Cu2O to metal Cu, thus reducing the adsorption strength of CO.
In situ attenuated total reflectance-infrared absorption spectroscopy (ATR-IRAS) was carried out under CO2RR conditions to monitor the adsorption intermediates and get insight into the CO2RR mechanism more precisely. As shown in Fig. 7, when the applied cathodic potential is higher than −1.1 V, new infrared (IR) absorption peaks begin to appear in the ATR-IRAS of all catalysts. The most obvious peak appears at 1650 cm−1 in the ATR-IRAS of Cu2O@SiO2–NH2 and Cu2O@SiO2 (Fig. 7a, b), which is related to the IR absorption peak of H2O.46 The intensity of the IR absorption peak increases with the increase of cathodic potential, indicating that the process of H2O adsorption and activation is accelerated at high cathodic potential. In contrast, on the surface of the Cu2O catalyst, the intensity of this absorption peak decreases sharply. This suggests that the surface of Cu2O@SiO2 can promote the adsorption and activation of H2O. This can be explained by the fact that the metal-oxide interface (Cu0–Cu+) can regulate the dissociation process of H2O, resulting in high coverage of *H species on the catalyst surface.47,48 The presence of a metal-oxide interface is also confirmed by the XRD and TEM results. From the CO2RR equations in Table S2,† it can be seen that the CO2RR must involve the participation of active *H, such as the transfer of 12 protons and 12 electrons to generate ethylene and ethanol, where the protons obtain electrons and become active *H. Therefore, the peak intensity of the H2O peak represents the ability to activate H2O to produce active *H, which is partially involved in the CO2RR and partially in the HER. For catalysts with high CO2RR activity, most of the active *H is involved in the CO2RR. In contrast, for catalysts with poor CO2RR activity, most of the active *H is involved in the HER. From Table S2,† it can also be seen that the number of electrons transferred is directly proportional to the number of protons consumed, which means that the number of active *H is directly proportional to the current density. In Fig. S11,† the current density of C2+ in the CO2RR process of the Cu2O@SiO2–NH2 catalyst is 237 mA cm−2, and the current density of H2 is only 35.32 mA cm−2, while for the Cu2O catalyst, the current density of C2+ is only 147 mA cm−2, but the current density of H2 is 76.55 mA cm−2. From this result, it can be seen that more active *H is generated in the Cu2O@SiO2–NH2 catalyst, and most of it is involved in the CO2RR. Although the HER current density of Cu2O is high, the total current density is low, resulting in less active *H, which is consistent with the H2O peak intensity in the ATR-IRAS spectra.
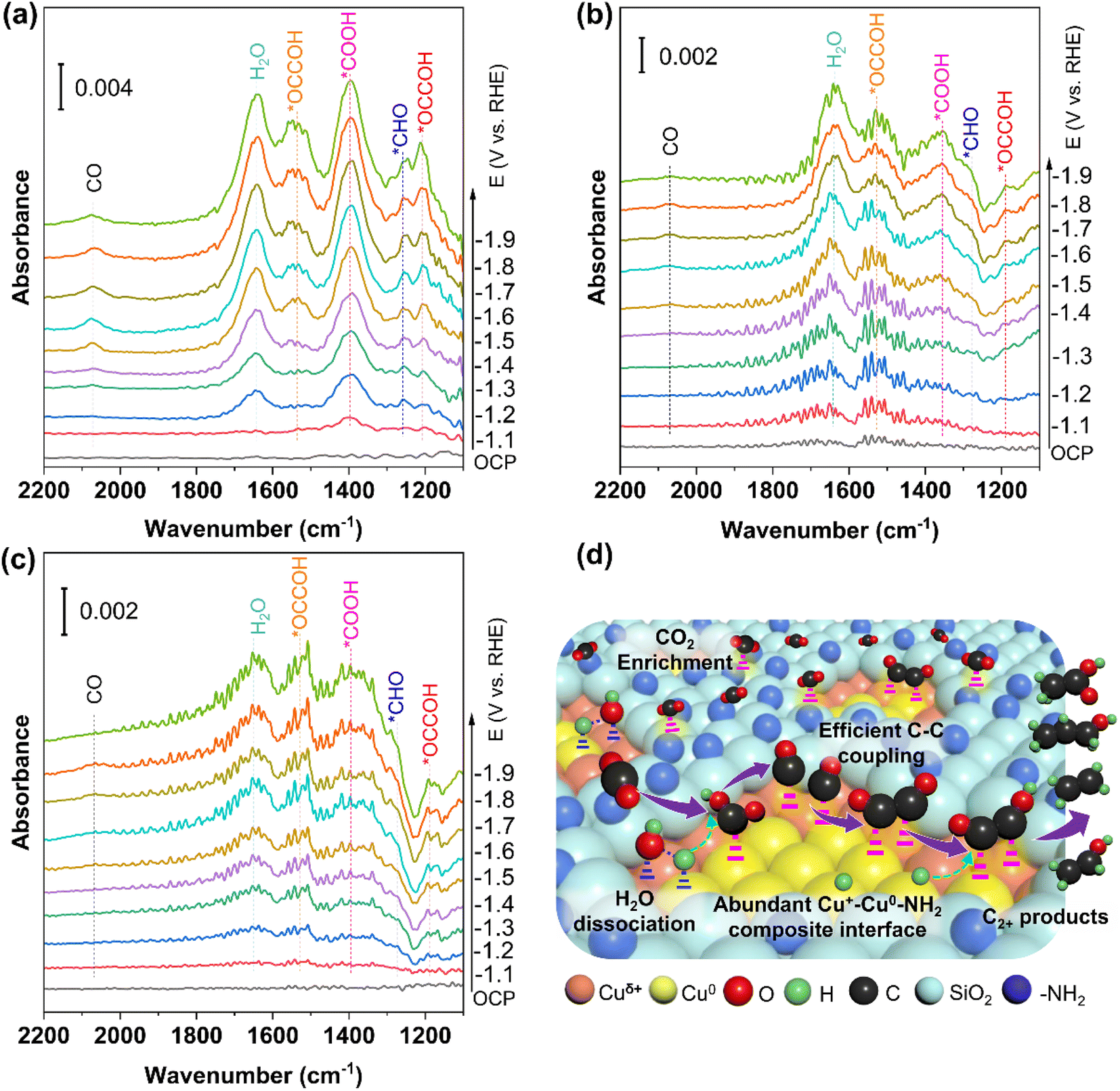 | ||
| Fig. 7 In situ ATR-IRAS spectra of catalysts under CO2RR conditions and the reaction mechanism. (a) Cu2O@SiO2–NH2, (b) Cu2O@SiO2, and (c) Cu2O and (d) reaction mechanism of the CO2RR to C2+ product formation over the Cu2O@SiO2–NH2 catalyst. | ||
In addition to the H2O absorption peak, the other strong peak located at 1390 cm−1 belongs to the *COOH species,49 implying that the Cu2O@SiO2–NH2 catalyst has strong adsorption and activation ability for CO2 molecules. The simultaneous adsorption and activation of H2O and CO2 molecules indicate a good coupling between H2O dissociation and CO2 reduction. The activated *CO2 species and surface *Had species promote the formation of *CO species, which can be confirmed by the *CO absorption peak appearing around 2070 cm−1 in the ATR-IRAS spectrum.46 There is an obvious *CO IR absorption peak on the Cu2O@SiO2–NH2 surface, indicating the high coverage of *CO on the Cu2O@SiO2–NH2 surface, which promotes the formation of C2+ products (Fig. 7a). In contrast, the ATR-IRAS of Cu2O@SiO2 shows a weak *CO IR absorption peak, which corresponds to a low CO coverage (Fig. 7b). However, no obvious IR absorption peak of *CO was found on the surface of Cu2O, indicating weak *CO adsorption capacity on the surface, because Cu2O was reduced to Cu0, followed by low *CO coverage (Fig. 7c). This finding proves that the presence of –NH2 can improve the adsorption and activation of CO2 on the catalyst surface, and the Cu+ species can promote the adsorption of CO, thus improving the surface coverage of *CO.
In addition, the peaks of ATR-IRAS at 1260 cm−1, 1205 cm−1, and 1530 cm−1 are related to the intermediate species of *CHO and *OCCOH on the catalyst, respectively (Fig. 7).49 The absorption peak intensity of *OCCOH increases with the cathodic potential, which is consistent with the enhancement trend of the C2+ product formation rate with the change of the cathodic potential. Compared with Cu2O@SiO2 and Cu2O catalysts, the IR absorption peak of the C2 intermediate on Cu2O@SiO2–NH2 is more intense, indicating that the C2 intermediate has a stable adsorption structure on the catalyst surface, which is conducive to promoting C–C coupling, followed by higher C2+ product selectivity. Therefore, on the surface of Cu2O@SiO2–NH2, the enhancement of the CO2 adsorption activation process improves the surface coverage of *CO, thus accelerating the C–C coupling process. The high coverage of *Had species can enhance the protonation process of C2+ intermediates and desorption of products, thus promoting the efficient formation of C2+ products from the CO2RR process.50,51
According to the above analysis results, the CO2RR mechanism on the surface of Cu2O@SiO2–NH2 can be reasonably proposed (Fig. 7d). Firstly, CO2 reaches the three-phase reaction interface through the gas diffusion layer, and then is enriched by –NH2 at the Cu0–Cu+–NH2 composite interfaces, forming a local microenvironment with a high concentration of CO2.34 CO2 molecules diffused through the porous SiO2 coating layer to reach the Cu0–Cu+ interface, and adsorbed and activated at the active site to form *CO2ad. At the same time, the H2O molecules are activated by the Cu0–Cu+ interface to increase the concentration of *H, which couples with CO2 activation to form *COOH, and promotes the formation of *CO. Then the Cu+ enhances the adsorption of *CO, which facilitate the C–C coupling process at the Cuδ+/Cu0 interface. The C2 intermediate is protonated by *H on the catalyst surface, which promotes product desorption and thus accelerates the formation of C2+ products. This mechanism is consistent with the theoretical calculation results reported in the literature. For example, the electron density around the C atoms in *CO on the Cu2O–Cu interface is higher than that on Cu2O, which reduces the reaction energy barrier of C–C coupling to form *OCCO. Meanwhile, the energy barrier of H2O dissociation on Cu, Cu2O, and Cu2O–Cu is 2.33, 2.15, and 1.64 eV, respectively; therefore, the faster H2O dissociation and promoted C–C coupling contribute to accelerated C2+ product formation from the CO2RR on the Cu2O–Cu interface.48
4. Conclusion
In this work, dodecahedral Cu2O@SiO2–NH2 nanoparticles have been synthesized by hydrolysis of TEOS and APTES on the surface of Cu2O particles for the CO2RR to C2+ products. A series of characterization results show electronic interaction between Cu2O and SiO2–NH2, and the Cu0–Cu+–NH2 composite interface is formed in the CO2RR process. With the help of the interface effect, the FE of C2+ products reaches 81.3% with a current density of 290 mA cm−2, and is stable for 15 h without significant degradation of activity. Combined with XPS and in situ Raman spectroscopy, the high activity and stability originate from the interaction between SiO2 and Cu2O, which stabilizes the positively charged Cu species and creates a stable Cu0–Cu+ interface under CO2RR conditions. Meanwhile, the surface –NH2 can enrich CO2 and promote the adsorption and activation of CO2 on the catalyst surface, which accelerates the formation of *CO; the –NH2 protonation process increases the local pH, which creates a reaction microenvironment that facilitates C–C coupling at the Cu0–Cu+–NH2 composite interface, and improves the efficiency of the CO2RR to C2+. This research provides a new idea for the surface modification of Cu-based catalysts and the design of an efficient Cu0–Cu+–NH2 composite interface.Author contributions
Conceptualization: Z.-L. W.; methodology: Z.-Y. Z. and H. T.; software: H. T.; validation: Z.-Y. Z. and H. J.; formal analysis: X. W., L. B., and Y. L.; investigation: Y. L. and N. K.; resources: Z.-L. W.; data curation: all authors; writing – original draft: all authors; writing – review & editing: all authors; visualization: Z.-Y. Z. and Z.-L. W.; supervision: Y. Y. and Z.-L. W.; project administration: Y. Y. and Z.-L. W.; funding acquisition: N. K., Y. Y., and Z.-L. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (NSFC) [grant numbers 22075201 and 21962014] and the National Key Research and Development Program of China [2022YFB4101800]. N. K. acknowledges the Office of the Ministry of Higher Education, Science, Research and Innovation (MHESRI) under the Reinventing University System/Visiting Professor Program 2023 for partial support. This work used the Queensland node of the NCRIS-enabled Australian National Fabrication Facility (ANFF).References
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef PubMed
.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wanga and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC
- Y. Lei, Z. Wang, A. Bao, X. Tang, X. Huang, H. Yi, S. Zhao, T. Sun, J. Wang and F. Gao, Chem. Eng. J., 2023, 453, 139663 CrossRef CAS
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC
- M. Fang, M. Wang, Z. Wang, Z. Zhang, H. Zhou, L. Dai, Y. Zhu and L. Jiang, J. Am. Chem. Soc., 2023, 145, 11323–11332 CrossRef CAS PubMed
- Z.-Y. Zhang, H. Tian, L. Bian, S.-Z. Liu, Y. Liu and Z.-L. Wang, J. Energy Chem., 2023, 83, 90–97 CrossRef CAS
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS PubMed
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed
- P.-P. Yang, X.-L. Zhang, P. Liu, D. J. Kelly, Z.-Z. Niu, Y. Kong, L. Shi, Y.-R. Zheng, M.-H. Fan, H.-J. Wang and M.-R. Gao, J. Am. Chem. Soc., 2023, 145, 8714–8725 CAS
- H. Li, Y. Jiang, X. Li, K. Davey, Y. Zheng, Y. Jiao and S.-Z. Qiao, J. Am. Chem. Soc., 2023, 145, 14335–14344 CrossRef CAS PubMed
- D. Zhong, D. Cheng, Q. Fang, Y. Liu, J. Li and Q. Zhao, Chem. Eng. J., 2023, 470, 143907 CrossRef CAS
- J. Zhang, Y. Wang, Z. Li, S. Xia, R. Cai, L. Ma, T. Zhang, J. Ackley, S. Yang, Y. Wu and J. Wu, Adv. Sci., 2022, 9, 2200454 CrossRef CAS PubMed
- S. Mu, H. Lu, Q. Wu, L. Li, R. Zhao, C. Long and C. Cui, Nat. Commun., 2022, 13, 3694 CrossRef CAS PubMed
- Y. Jiang, X. Wang, D. Duan, C. He, J. Ma, W. Zhang, H. Liu, R. Long, Z. Li, T. Kong, X. J. Loh, L. Song, E. Ye and Y. Xiong, Adv. Sci., 2022, 9, 2105292 CrossRef CAS PubMed
- Y. Zhou, Y. Liang, J. Fu, K. Liu, Q. Chen, X. Wang, H. Li, L. Zhu, J. Hu, H. Pan, M. Miyauchi, L. Jiang, E. Cortes and M. Liu, Nano Lett., 2022, 22, 1963–1970 CrossRef CAS PubMed
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS
- K. Hadjiivanov and H. Knözinger, Phys. Chem. Chem. Phys., 2001, 3, 1132–1137 RSC
- Y. Zhou, Y. Yao, R. Zhao, X. Wang, Z. Fu, D. Wang, H. Wang, L. Zhao, W. Ni, Z. Yang and Y.-M. Yan, Angew. Chem., Int. Ed., 2022, 61, e202205832 CrossRef CAS PubMed
- Y. Zang, T. Liu, P. Wei, H. Li, Q. Wang, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2022, 134, e202209629 CrossRef
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CrossRef CAS
- Z.-Z. Wu, X.-L. Zhang, Z.-Z. Niu, F.-Y. Gao, P.-P. Yang, L.-P. Chi, L. Shi, W.-S. Wei, R. Liu, Z. Chen, S. Hu, X. Zheng and M.-R. Gao, J. Am. Chem. Soc., 2022, 144, 259–269 CrossRef CAS PubMed
- L. Li, W. Gu, J. Liu, S. Yan and Z. P. Xu, Nano Res., 2015, 8, 682–694 CrossRef CAS
- P.-P. Yang, X.-L. Zhang, P. Liu, D. J. Kelly, Z.-Z. Niu, Y. Kong, L. Shi, Y.-R. Zheng, M.-H. Fan, H.-J. Wang and M.-R. Gao, J. Am. Chem. Soc., 2023, 145, 8714–8725 CAS
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed
- G.-Y. Duan, X.-Q. Li, G.-R. Ding, L.-J. Han, B.-H. Xu and S.-J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202110657 CrossRef CAS PubMed
- X. Zhou, J. Shan, L. Chen, B. Y. Xia, T. Ling, J. Duan, Y. Jiao, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2022, 144, 2079–2084 CrossRef CAS PubMed
- C. Azenha, C. Mateos-Pedrero, M. Alvarez-Guerra, A. Irabien and A. Mendes, Chem. Eng. J., 2022, 445, 136575 CrossRef CAS
- H.-Q. Liang, S. Zhao, X.-M. Hu, M. Ceccato, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2021, 11, 958–966 CrossRef CAS
- J. Li, A. Ozden, M. Wan, Y. Hu, F. Li, Y. Wang, R. R. Zamani, D. Ren, Z. Wang, Y. Xu, D.-H. Nam, J. Wicks, B. Chen, X. Wang, M. Luo, M. Graetzel, F. Che, E. H. Sargent and D. Sinton, Nat. Commun., 2021, 12, 2808 CrossRef CAS PubMed
- X. Fang, S. Wu, Y. Wu, W. Yang, Y. Li, J. He, P. Hong, M. Nie, C. Xie, Z. Wu, K. Zhang, L. Kong and J. Liu, Appl. Surf. Sci., 2020, 518, 146226 CrossRef CAS
- D. Zeng, C. Li, W. Wang, L. Zhang, Y. Zhang, J. Wang, L. Zhang, X. Zhou and W. Wang, Chem. Eng. J., 2023, 461, 142133 CrossRef CAS
- Y. Zhao, X. Zu, R. Chen, X. Li, Y. Jiang, Z. Wang, S. Wang, Y. Wu, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2022, 144, 10446–10454 CrossRef CAS PubMed
- W. Li, Z. Yin, Z. Gao, G. Wang, Z. Li, F. Wei, X. Wei, H. Peng, X. Hu, L. Xiao, J. Lu and L. Zhuang, Nat. Energy, 2022, 7, 835–843 CrossRef CAS
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. de Arquer, A. Kiani, J. P. Edwards, P. D. Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed
- H. Xiao, T. Cheng and W. A. Goddard III, J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS PubMed
- Q. Fan, X. Zhang, X. Ge, L. Bai, D. He, Y. Qu, C. Kong, J. Bi, D. Ding, Y. Cao, X. Duan, J. Wang, J. Yang and Y. Wu, Adv. Energy Mater., 2021, 11, 2170140 CrossRef
- Y. Jiang, C. Choi, S. Hong, S. Chu, T.-S. Wu, Y.-L. Soo, L. Hao, Y. Jung and Z. Sun, Cell Rep. Phy. Sci., 2021, 2, 100356 CrossRef CAS
- L. Xu, X. Ma, L. Wu, X. Tan, X. Song, Q. Zhu, C. Chen, Q. Qian, Z. Liu, X. Sun, S. Liu and B. Han, Angew. Chem., Int. Ed., 2022, 61, e202210375 CrossRef CAS PubMed
- C. Chen, X. Yan, Y. Wu, S. Liu, X. Zhang, X. Sun, Q. Zhu, H. Wu and B. Han, Angew. Chem., Int. Ed., 2022, 61, e202202607 CrossRef CAS PubMed
- X. Yan, C. Chen, Y. Wu, S. Liu, Y. Chen, R. Feng, J. Zhang and B. Han, Chem. Sci., 2021, 12, 6638–6645 RSC
- K. Yao, J. Li, H. Wang, R. Lu, X. Yang, M. Luo, N. Wang, Z. Wang, C. Liu, T. Jing, S. Chen, E. Cortes, S. A. Maier, S. Zhang, T. Li, Y. Yu, Y. Liu, X. Kang and H. Liang, J. Am. Chem. Soc., 2022, 144, 14005–14011 CrossRef CAS PubMed
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z. J. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 15344–15347 CrossRef CAS PubMed
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S.-Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS PubMed
- C.-T. Dinh, A. Jain, F. P. G. de Arquer, P. De Luna, J. Li, N. Wang, X. Zheng, J. Cai, B. Z. Gregory, O. Voznyy, B. Zhang, M. Liu, D. Sinton, E. J. Crumlin and E. H. Sargent, Nat. Energy, 2019, 4, 107–114 CrossRef CAS
- S. Wang, D. Wang, B. Tian, X. Gao, L. Han, Y. Zhong, S. Song, Z. Wang, Y. Li, J. Gui, M. G. Sendeku, Y. Zhang, Y. Kuang and X. Sun, Sci. China Mater., 2023, 66, 1801–1809 CrossRef CAS
- M. Zheng, P. Wang, X. Zhi, K. Yang, Y. Jiao, J. Duan, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2022, 144, 14936–14944 CrossRef CAS PubMed
- H. Zhang, J. Gao, D. Raciti and A. S. Hall, Nat. Catal., 2023, 6, 807–817 CrossRef CAS
- J. Feng, L. Zhang, S. Liu, L. Xu, X. Ma, X. Tan, L. Wu, Q. Qian, T. Wu, J. Zhang, X. Sun and B. Han, Nat. Commun., 2023, 14, 4615 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05652j |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |