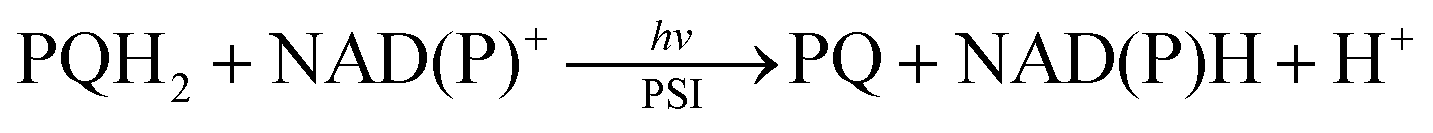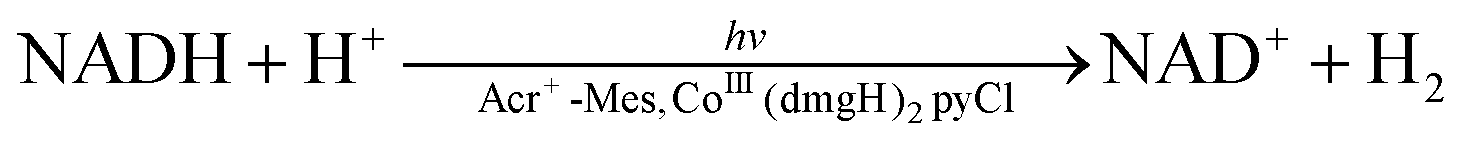DOI:
10.1039/D4CC06254J
(Feature Article)
Chem. Commun., 2025,
61, 3271-3282
Catalytic reduction of NAD(P)+ to NAD(P)H
Received
25th November 2024
, Accepted 14th January 2025
First published on 15th January 2025
Abstract
1,4-Dihydronicotinamide adenine dinucleotide (NADH) and its phosphate ester (NADPH) are essential cofactors required for all living cells, playing pivotal roles in multiple biological processes such as energy metabolism and biosynthesis. NADPH is produced during photosynthesis by the combination of photosystem II, where water is oxidised, and photosystem I, where NADP+ is reduced. This review focuses on catalytic NAD(P)+ (and its analogues) reduction to generate 1,4-NAD(P)H without formation of other regioisomers and the dimer. There are different ways for production of 1,4-NAD(P)H and its analogues. Firstly, electrocatalytic reduction of NAD(P)+ is discussed to clarify how the regioselective reduction of NAD(P)+ to 1,4-NAD(P)H is achieved with use of metal complex catalysts. The applied potential for the electrocatalytic reduction of NAD(P)+ to 1,4-NAD(P)H is much reduced by combination with the photocathode under photoirradiation. Then, mechanisms of hydrogenation of NAD(P)+ by H2 and transfer hydrogenation of NAD(P)+ by formate used as an electron and proton source to produce 1,4-NAD(P)H are discussed. Hydroquinone derivatives are also used as plastoquinol analogues, which act as hydride sources in a photosystem I model reaction, in which NAD(P)+ and its analogues are reduced by hydroquinone derivatives to form 1,4-NAD(P)H and its analogues using an NAD(P)+ reduction catalyst and a photoredox catalyst. The photosystem I model is then combined with a photosystem II model in which plastoquinone analogues are reduced to plastoquinol analogues by water to achieve the stoichiometry of photosynthesis, that is, photocatalytic reduction of NAD(P)+ by water.

Shunichi Fukuzumi
| Shunichi Fukuzumi received his BS degree in 1973 and PhD degree in 1978 from the Tokyo Institute of Technology. After working as a postdoctoral fellow (1978–1981) at Indiana University in the United States, he joined Osaka University as an assistant professor in the Department of Applied Chemistry in 1981. He was promoted to a Full Professor at Osaka University in 1994. His research interests are electron-transfer chemistry, redox catalysis, and artificial photosynthesis. He is a Distinguished Professor of Ewha Womans University and Professor Emeritus of Osaka University. |

Yong-Min Lee
| Yong-Min Lee received his PhD degree from Pusan National University, Korea (Major: Inorganic Chemistry), under the supervision of Professor Sung-Nak Choi in 1999. Then, he joined the Centro di Ricerca di Risonanze Magnetiche (CERM) at Università degli Studi di Firenze, Italy, as a postdoctoral fellow and researcher under the supervision of Professors Claudio Luchinat and Ivano Bertini (from 1999 to 2005). In 2006, he moved to Center for Biomimetic Systems at Ewha Womans University, as a Research Professor. Since 2009, he has been a Professor for Special Appointment at Ewha Womans University. |

Wonwoo Nam
| Wonwoo Nam earned his BS (Honors) degree in Chemistry from California State University, Los Angeles (1985), and his PhD degree in Inorganic Chemistry from UCLA (1990). After postdoctoral experience at UCLA, he became an Assistant Professor at Hong Ik University in 1991. He moved to Ewha Womans University in 1994, where he is presently a Distinguished Professor. His current research focuses on the mechanism of dioxygen activation and water oxidation in biomimetic and bioinorganic chemistry. |
1. Introduction
Cytochrome c oxidase requires 1,4-dihydonicotinamide adenine dinucleotide (NADH; see Fig. 1) as an e− and H+ source for the catalytic 4e−/4H+ reduction of dioxygen to water.1–6 Cytochrome P-450 enzyme also requires NADPH (Fig. 1) to reductively activate dioxygen for the catalytic oxygenation reactions.7–12 NADPH is produced by the regioselective NADP+ reduction by plastoquinol in photosystem I (PSI) via a ferredoxin-NADPH reductase (FNR), which ultimately reduces NADP+ into NADPH, whereas plastoquinol is formed by the photocatalytic reduction of plastoquinone by water in photosystem II (PSII) during photosynthesis.13–16 Overall NADPH is produced by the regioselective reduction of NADP+ using water as an e− and H+ source in photosynthesis [eqn (1)].13–16 Many industrially relevant enzymes depend on the cofactors NADH and NADPH, which are too expensive to be added in stoichiometric amounts.17–23 Therefore, extensive efforts have been made to develop efficient catalytic systems for NAD(P)H recycling with high activity without producing by-products such as the NAD(P) dimer and other regioisomers (1,2- and 1,6-dihydro forms).24–30
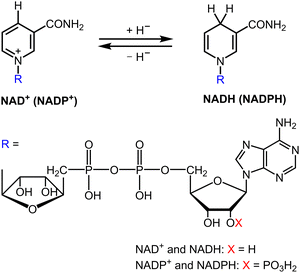 |
| | Fig. 1 Structural forms of NAD(P)+ and NAD(P)H. | |
This Feature Article has focused on catalytic production of 1,4-NAD(P)H by electrocatalytic reduction of NAD(P)+, hydrogenation of NAD(P)+ and photocatalytic reduction of NAD(P)+ to 1,4-NAD(P)H by electron donors including plastoquinol analogues with use of photoredox catalysts to mimic the molecular function of PSI.31 The catalytic mechanism is discussed to clarify how the regioselective NAD(P)+ reduction to the 1,4-dihydro form, NAD(P)H, is possible by the catalysts. Finally a PSII model, in which H2O is oxidized by a plastoquinone (PQ) analogue to produce O2 and a plastoquinol (PQH2) analogue,32,33 has been combined with the PSI model to achieve the stoichiometry of the photosynthesis, i.e., the photocatalytic reduction of NAD(P)+ by H2O to produce O2 and NAD(P)H [eqn (1)].31 Once NAD(P)H is produced by using H2O, combination of NAD(P)H dependent enzymes would make it possible to use H2O as a reductant for recycling NAD(P)H in a large number of industrial enzymatic reactions.
| | 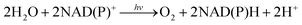 | (1) |
2. Electrocatalytic reduction of NAD(P)+
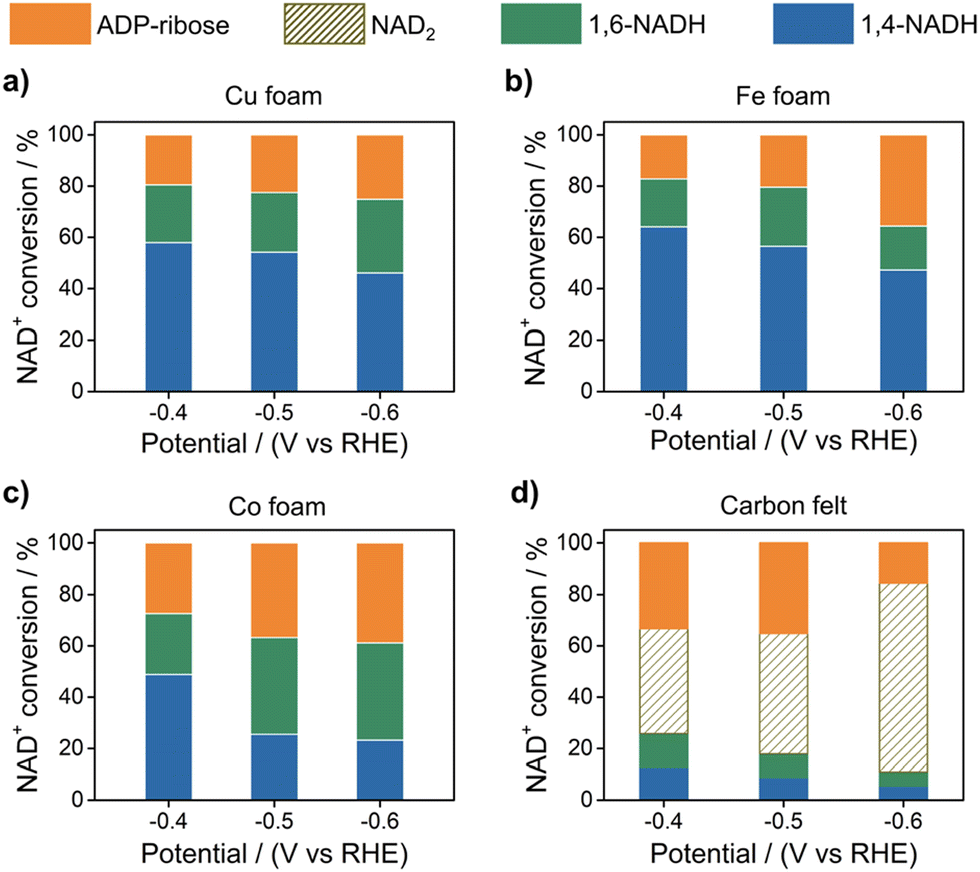
One method for nonenzymatic production of NAD(P)H is electrocatalytic reduction of NAD(P)+, although the production of NAD(P)H isomers remains a difficult issue.34–37 Electrocatalytic reduction of NAD+ to 1,4-NADH was reported with use of Cu, Fe, Co, and carbon electrodes.38Fig. 2 shows the reduction of NAD+ to different NADH isomers and the dimer when NAD+ is totally consumed after the electrocatalytic reduction at different applied potentials.38 The highest yield of 1,4-NADH using Cu, Fe, and Co electrodes was obtained as 58%, 64% and 49% at −0.4 V vs. RHE, respectively. In contrast to regioselective reduction of NAD+ at the metal electrodes, the yield of 1,4-NADH was much smaller (7.9%) at −0.4 V (vs. RHE) at the carbon electrode, which has a high proportion of the dimeric product (NAD2 > 40%). ADP-ribose was also produced probably by the fragmentation of NAD+.38
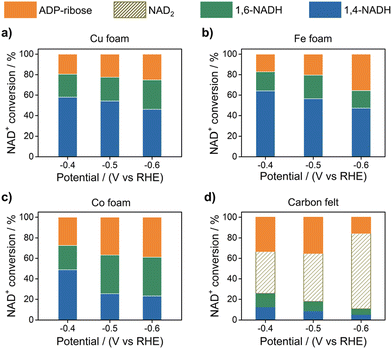 |
| | Fig. 2 Product yields obtained in the electrocatalytic reduction of NAD+ using (a) Cu, (b) Fe, (c) Co and (d) carbon electrodes depending on the applied potentials (pH 7; [phosphate buffer] = 0.10 M and [NAD+]ini = 1.0 mM). Reprinted with permission from ref. 38. Copyright 2022, Royal Society of Chemistry. | |
Because no NAD2 was produced in the electrocatalytic reduction of NAD+ with use of Cu, Fe, and Co electrodes (Fig. 2), it has been proposed that the surface-adsorbed hydrogen atom [*Had: asterisk (*) denotes the active site] is produced on Cu, Fe, and Co electrodes via proton-coupled electron transfer (step 1), followed by the reaction of *Had with NAD+ coupled with electron transfer (step 2) as shown in Scheme 1.38 The poor proton activation ability of the carbon electrode results in a low coverage of Had, when the electron-transfer reduction of NAD+ proceeds to produce NAD2 (step 2b in Scheme 1).38–41 The low selectivity to 1,4-NADH production in Fig. 2 should be improved for any application.
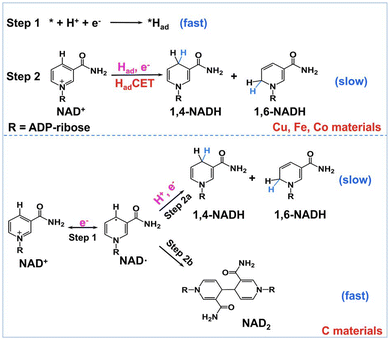 |
| | Scheme 1 Proposed NAD+ reduction mechanism at Had-rich Cu, Fe, and Co electrodes (top) and Had-poor C material electrodes (bottom). Reprinted with permission from ref. 38. Copyright 2022, Royal Society of Chemistry. | |
A cathode made from MWCNTs containing Ni nanoparticles (NP), Ni NP-MWCNTs (Scheme 2), was employed for the electrocatalytic reduction of NAD+ to produce 98% 1,4-NADH at a potential that is 700 mV more positive than that on carbon nanofibres (CNFs) and pure MWCNTs.42 The highly efficient production of 1,4-NADH on Ni NP-MWCNTs at low cathodic overpotentials results from the adsorption of activated hydrogen (Hads) on the NP-MWCNT electrode at low overpotentials, which facilitates the hydrogenation of NAD+ to produce 1,4-NADH.42
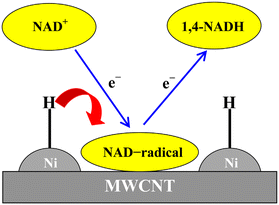 |
| | Scheme 2 The Ni NP-MWCNT electrode used for electrocatalytic production of 1,4-NADH. Reprinted with permission from ref. 42. Copyright 2017, John Wiley and Sons. | |
The incorporation of Ni nanoparticles (NPs) on TiO2 (Ni-TOTs) was reported to enhance the selective hydrogenation of NAD+ stabilized on TiO2 to produce high yield of the enzymatically active 1,4-NADH (93.8% at −0.9 V vs. Ag/AgCl).43 The Ti3+ and oxygen vacancies are suggested to play a crucial role in NAD+ adsorption, facilitating the selective hydrogenation by Ni-TOTs.43
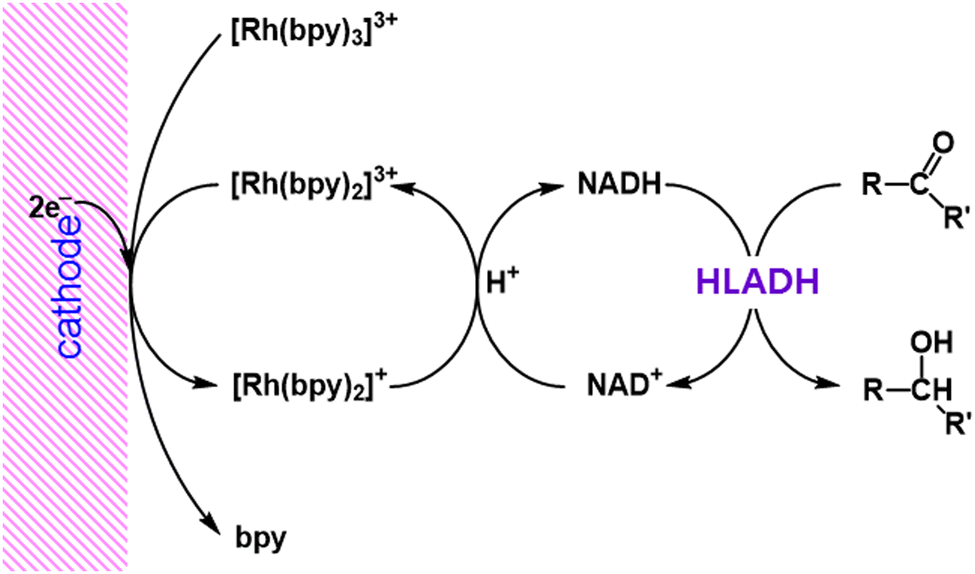
The regioselective electrocatalytic reduction of NAD+ to 1,4-NADH was achieved with use of [Rh(bpy)2]+ (bpy = 2,2′-bipyridine), which was produced by the electrochemical two-electron reduction of [Rh(bpy)3]3+, accompanied by release of a bpy ligand.44 The produced NADH is oxidised by cyclohexanone under catalysis of horse liver alcohol-dehydrogenase (HLADH) (Scheme 3).44
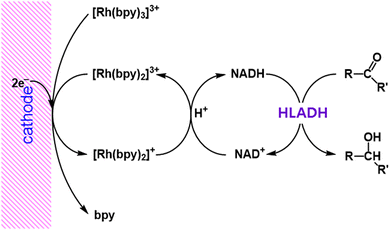 |
| | Scheme 3 Electrocatalytic production of NADH from NAD+ using [Rh(bpy)2]+.44 | |
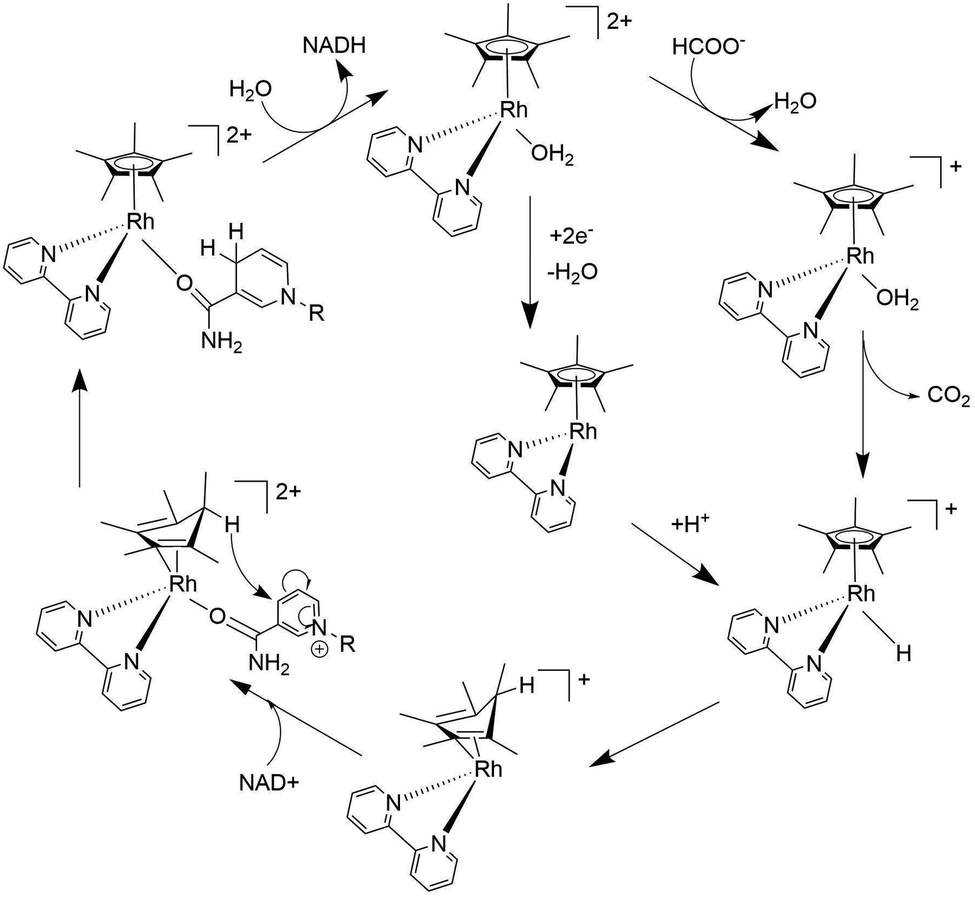
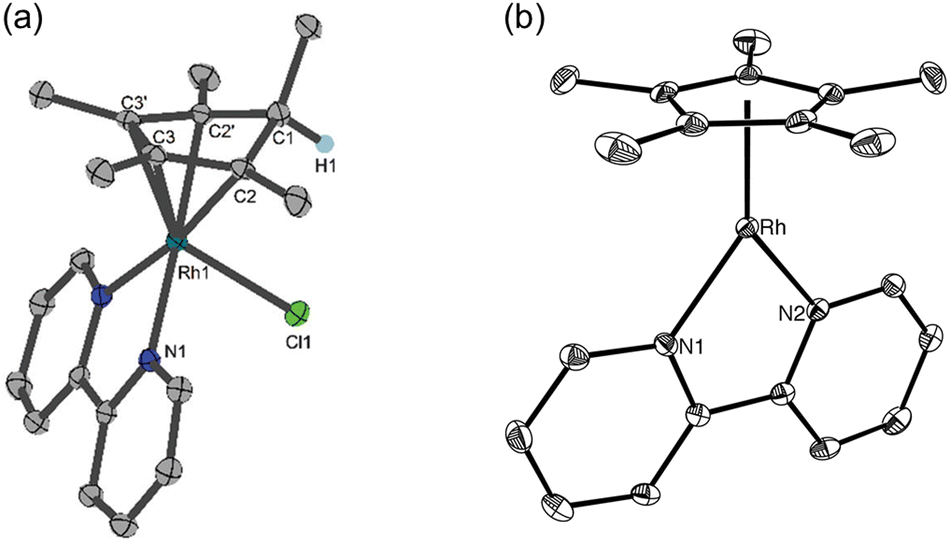
The aquo-(2,2′-bipyridine)(pentamethylcyclopentadienyl)-rhodium(III) complex ([Cp*Rh(bpy)(H2O)]2+) was also reported to act as an extremely effective redox catalyst for the electrocatalytic production of 1,4-NADH as well as for the chemical reduction of NAD(P)+ with formate as a hydride donor.45,46 The catalytic mechanism of the regioselective reduction of NAD+ with [Cp*Rh(bpy)(H2O)]2+ was proposed as shown in Scheme 4, where a Rh(III)-hydride complex ([Cp*Rh(bpy)H]+) was produced by the reaction of formate with [Cp*Rh(bpy)(H2O)]2+. [Cp*Rh(bpy)H]+ was converted to a Rh(I) complex with an η4-pentamethylcyclopentadiene ligand having a new C–H bond endo with respect to the metal centre ([(Cp*H)RhI(bpy)]+).28,47,48 The X-ray crystal structure of [(Cp*H)RhI(bpy)]+ is shown in Fig. 3a, where the C1–C2 distance (1.517(2) Å) is longer than the C2–C3 (1.440(3) Å) distance, confirming the diene structure.48 In sharp contrast, the crystal structure of the Cp*Rh(I) complex (Fig. 3b) shows only a 0.034 Å difference in the cyclopentadienyl C–C bonds.49 The coordination of the amide group of NAD+ to the Rh centre of [(Cp*H)Rh(bpy)]+ results in the regioselective reduction of NAD+ to 1,4-NADH, followed by the replacement of 1,4-NADH by H2O to regenerate [Cp*RhIII(bpy)(H2O)]2+ (Scheme 4).47,48
 |
| | Scheme 4 Proposed mechanism of catalytic hydrogenation of NAD+ by formate to 1,4-NADH with [Cp*Rh(bpy)(H2O)]2+. Reprinted with permission from ref. 48. Copyright 2016, Royal Society of Chemistry. | |
 |
| | Fig. 3 X-ray structures of (a) [(Cp*H)Rh(bpy)]+ and (b) [Cp*Rh(bpy)]+. Reprinted with permission from ref. 48. Copyright 2016, Royal Society of Chemistry. | |
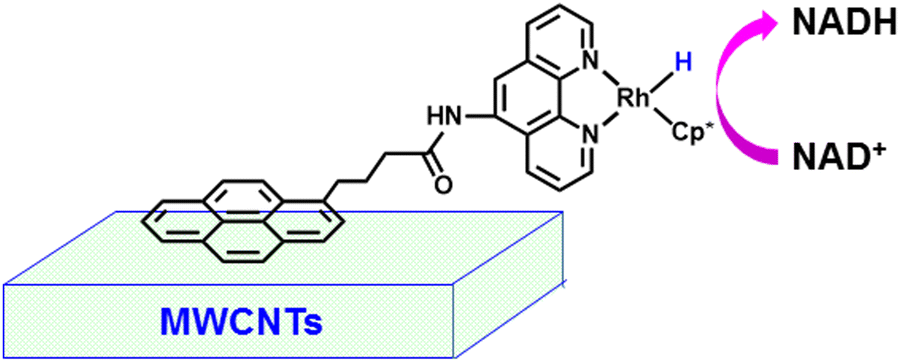
The immobilization of a rhodium complex with a pyrene-substituted phenanthroline ligand (pyr-Rh) was performed on multi-walled carbon nanotubes (MWCNTs) via π–π stacking to enhance the stability of the catalyst for the electrocatalytic production of NADH (Scheme 5).50 The pyr-Rh/MWCNT electrodes can also be applied to produce 1,4-NADH for enzymatic synthesis in which malate dehydrogenase (MDH) was incorporated for enzymatic reduction of oxaloacetic acid.50
 |
| | Scheme 5 Immobilization of the pyr-Rh complex onto MWCNTs. | |
The [Rh(Cp*)(bpy)Cl]+ complex was also immobilised on highly ordered three dimensional (3D) metal–organic framework (NU-1000) films at the zirconium nodes of NU-1000 (vide infra).51 The glassy carbon (GC) electrode was modified with carboxyl groups by electrochemical oxidation of 4-aminobenzoic acid (GC-COOH).51 An NU-1000 film was produced through the coordination by a Zr-oxo cluster (GC-Zr), followed by solvothermal synthesis (GC-NU1000).51 The NU-1000 film provided a 3D framework with regularly positioned nodes, where catalysts are loaded.51 The carboxyl-functionalized-Rh catalyst (Rh-COOH) was anchored onto the nodes of the film by solvent-assisted ligand incorporation (SALI) to prepare the Rh-immobilized electrode (GC-NU1000-Rh in Scheme 6).51 Electrocatalytic NAD+ reduction to 1,4-NADH was performed using GC-NU1000-Rh at an applied potential of −0.72 V in 30 mL of Tris buffer (pH 7.2) to afford a high TOF (∼1400 h−1) as well as a high faradaic efficiency (97%).51 This was coupled with an electroenzymatic conversion of pyruvate into L-lactate with a total TON of over 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 using L-lactate dehydrogenase.51
000 using L-lactate dehydrogenase.51
 |
| | Scheme 6 Electrocatalytic reduction of NAD+ to 1,4-NADH with use of a [Rh(Cp*)(bpy)Cl]+-functionalised NU-1000 film on a GC electrode (GC-NU1000-Rh). Reprinted with permission from ref. 51. Copyright 2022, American Chemical Society. | |
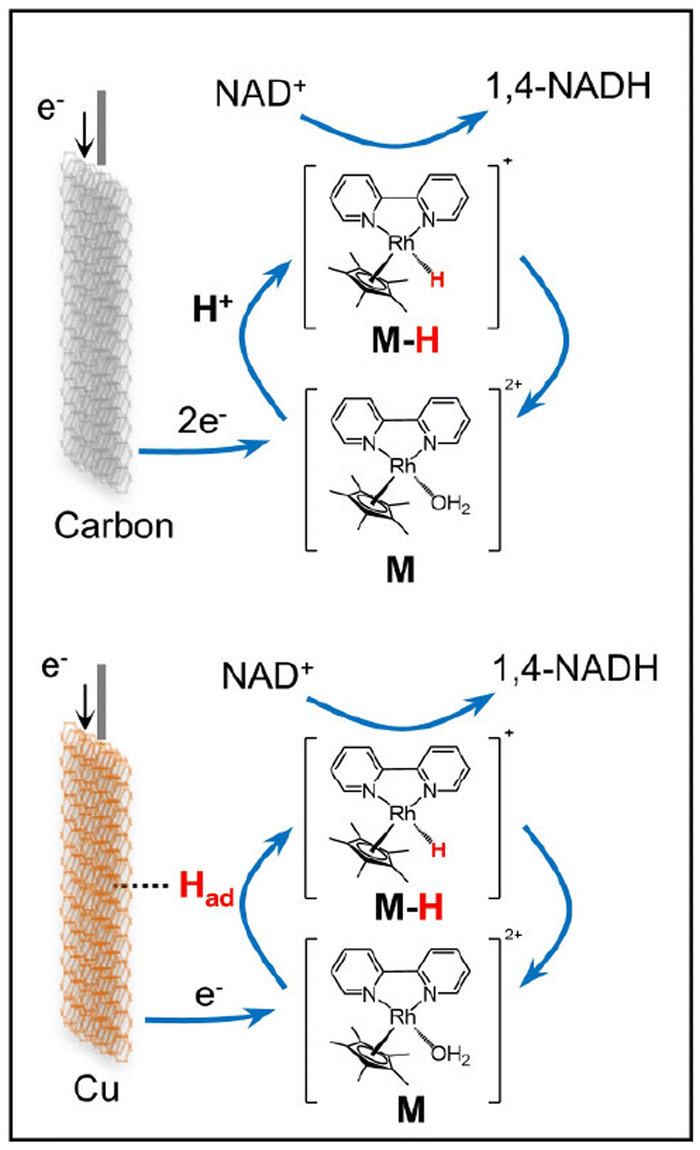
Prominent synergetic effects between various metal electrodes (Cu, Fe, Co, and Ni) and [Cp*Rh(bpy)(H2O)]Cl2 (or [Cp*Rh(phen)(H2O)]Cl2) in electrolytes were reported for electrocatalytic reduction of NAD+ to 1,4-NADH.52 For example, the normalized activity of the Cu-[Cp*Rh(bpy)(H2O)]Cl2 (Cu-M) system was 16 times higher than that of [Cp*Rh(bpy)(H2O)]Cl2 alone, whereas Cu alone showed trace catalytic activity at the same applied potential.52 The maximal selectivity of 1,4-NADH was enhanced from 63% for Cu alone to 95.3% for the coupled system.52 The Rh-hydride complex may be formed via adsorbed hydrogen (Had) on Cu and electron transfer, catalysing the NAD+ reduction to 1,4-NADH, whereas there is negligible Had on the surface of the carbon electrode (Scheme 7).52
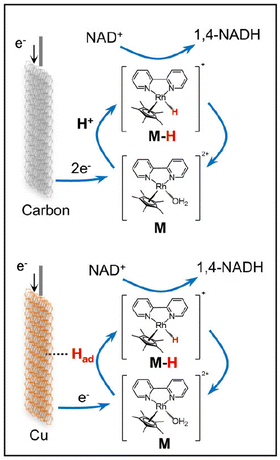 |
| | Scheme 7 Schematic description of the electrocatalytic NADH regeneration reaction with [Cp*Rh(bpy)(H2O)]Cl2 (M) alone and the Cu-M system. Reprinted with permission from ref. 52. Copyright 2024, American Chemical Society. | |
Ni sulfides (Ni3S2 and NiS2) were efficient for direct NAD+ reduction, but the selectivity to produce 1,4-NADH was not high.53 The coupled system of the Ni3S2 electrode and [Cp*Rh(bpy)(H2O)]Cl2 has enabled the NAD+ reduction to 1,4-NADH with both the highest activity and selectivity among previously reported results.53 In the direct NAD+ reduction on Ni3S2 in the absence of the Rh complex, the reaction proceeds via a hydride-transfer process or an Had-coupled electron transfer mechanism, suppressing the formation of NAD˙ and thereby preventing the production of byproduct NAD2 (Scheme 8a).53 However, the selectivity in 1,4-NADH is only 80% for Ni3S2.53 The low selectivity to produce 1,4-NADH is a common issue in the direct electrocatalytic reduction of NAD+ with heterogeneous catalysts.38–42,52–56 In the carbon-Rh electrode system, the NAD+ reduction proceeds via a hydride transfer from the Rh–hydride complex, but the carbon electrode is only a conductive electrode providing electrons to the Rh complex.54 A hydride transfer mechanism has been well established for the Rh-, Ru- and Ir-catalysed chemical reduction of NAD+ with formate as a reductant.57–68 In the Ni3S2-Rh system, Ni3S2 can transfer electrons and active hydrogen atoms to Rh efficiently via a concerted electron–proton transfer (CEPT) mechanism (Scheme 8b).53
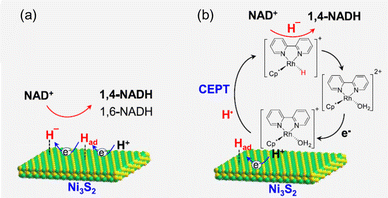 |
| | Scheme 8 Proposed mechanisms of NAD+ reduction into NADH for (a) Ni3S2 alone and (b) the Ni3S2-Rh system. Reprinted with permission from ref. 53. Copyright 2024, American Chemical Society. | |
3. Photoelectrocatalytic reduction of NAD(P)+
The photovoltage (Vph) of the p-type silicon nanowire (p-SiNW) photocathode composed of a buried n+p radial junction was enhanced for NADH production under solar light irradiation as shown in Scheme 9, where introduction of an n+ layer into p-SiNW results in an increase in the band bending at the n+/p interface to enhance the p-SiNW's Vph (435 mV).69 Electron transfer from the photoexcited electrons to [Cp*Rh(bpy)(H2O)]2+ (Mox mediator) afforded a benchmark onset potential (Eonset) of 0.393 V compared to the reversible hydrogen electrode (RHE) among reported SiNW-based photocathodes.69–72 Thus, the n+p-SiNW photocathode achieved Mred-mediated regioselective conversion of NAD+ to 1,4-NADH to afford a faradaic efficiency (FE) of 85% and a conversion rate of 1.6 μmol h−1 cm−1 at 0.2 V vs. RHE. The photoelectrocatalytic activity for NADH production remained for at least 12 h at the low cathodic potential.69
 |
| | Scheme 9 Reaction scheme of photocatalytic production of NADH at the photocathode driven by n+p-SiNWs. Reprinted with permission from ref. 69. Copyright 2023, American Chemical Society. | |
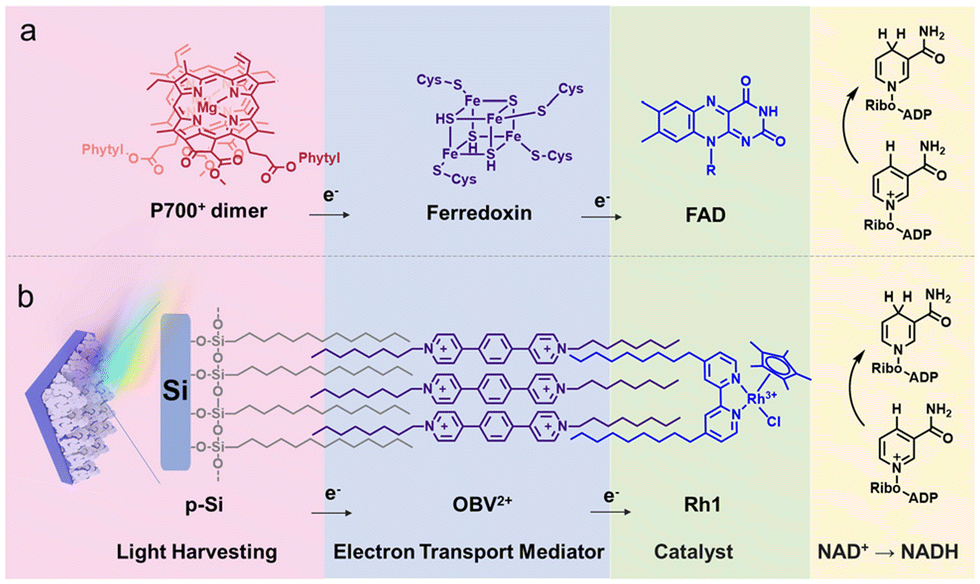
In PSI, photoinduced electron transfer from P700 in the thylakoid membrane to ferredoxin (Fd) occurs, followed by subsequent electron transfer to ferredoxin-NADP+ reductase (FNR), catalysing the solar-driven reduction of NADP+ to 1,4-NADPH (Fig. 4a).73,74 Inspired by the efficient photocatalytic function of PSI, an alkane-chain-substituted Rh1 complex ([Rh(Cp*)(bpy)Cl]+) was self-assembled on aliphatic chain-modified micro-pyramid p-type silicon array (p-Si) photocathodes (Fig. 4b).73 An electron-transfer mediator 4,4′-(1,4-phenylene)bis(1-octylpyridin-1-ium) (OBV2+) was employed to facilitate electron transfer from OBV2+ to the catalytic components. Rh1 and OBV2+ were assembled on the surface of SCA-modified p-Si via hydrophobic interactions to obtain electrode-supported “lipid-bilayer membrane” photocathodes.73 The incident photon-to-current efficiency (IPCE) of Rh1/OBV2+/SCA/p-Si for the photoelectrocatalytic reduction of NAD+ to 1,4-NADH was always higher than that of Rh1/SCA/p-Si over the entire spectral range.73 The IPCE of Rh1/OBV2+/SCA/p-Si at 665 nm was determined to be 3.2%, which was significantly larger than the IPCE of Rh1/SCA/p-Si (1.55%), because of the promotion of the photoinduced electron transfer and inhibition of interfacial charge recombination between the p-Si semiconductor and Rh1 catalyst.73 Thus, OBV2+ plays an important role as an electron mediator as compared with the photoelectrode without an electron mediator (Scheme 9).69,73
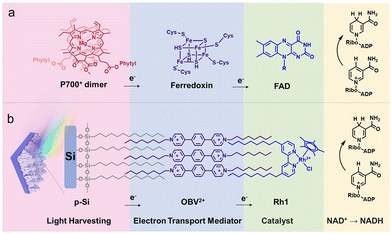 |
| | Fig. 4 (a) Electron flow from the PSI component to Fd and FAD. (b) Schematic of the photocathode with a lipid-bilayer membrane. Reprinted with permission from ref. 73. Copyright 2024, Royal Society of Chemistry. | |
4. Catalytic hydrogenation of NAD(P)+
Hydrogenation of NAD+ by H2 with [M2-OH2]0 occurred under basic conditions (e.g., pH = 8) to produce 1,4-NADH regioselectively.75 The yield of 1,4-NADH based on the initial mol% amount of NAD+ and the turnover number (TON) reached 97% and 9.3 (90 min), respectively.75 The turnover frequency (TOF) of H2 evolution from NADH increased with decreasing pH in the region between 4.1 and 7.0, which overlaps with the ratio of [M1-OH2]+, whereas the pH dependence of TOF for hydrogenation of NAD+ overlaps with the ratio of [M2-OH2]0.75 At pH 6.5, the TOF for the NADH formation was determined to be 36 h−1, whereas the TOF for the H2 evolution reached 52 h−1 at pH 4.1.75 Such pH dependence of TOF indicates that [M1-OH2]+ reacts with NADH to produce H2 and that [M2-OH2]0 reacts with H2 to reduce NAD+ to NADH, similar to the case of interconversion between HCOOH and H2 with use of the same Ir catalyst.76
The rate-determining step in the catalytic hydrogenation of NAD+ with hydrogen is the reaction of the Ir–H2O complex with H2 to form the Ir(III)-hydride complex, followed by fast hydride transfer to NAD+ to produce 1,4-NADH (the right-hand catalytic cycle in Scheme 10).75 Formation of the Ir(III)-hydride complex under normal pressure of hydrogen was detected by 1H-NMR, ESI mass, and UV-vis spectroscopies.75 This is the first demonstration of the interconversion between NADH and H2 using a water-soluble iridium-aqua complex, in which H2 is oxidized by NAD+ to produce H+ and NADH, whereas H2 and NAD+ are produced via the reduction of H+ by NADH, depending on pH, under normal pressure at room temperature.75
 |
| | Scheme 10 Proposed mechanism for catalytic interconversion between NADH and H2 with use of water soluble [C,N] cyclometalated Ir complexes (1 and 2) depending on pH. Reproduced with permission from ref. 75. Copyright 2011, American Chemical Society. | |
Enzyme-metal biohybrids composed of NAD+ reductase (NRase), biocatalytically synthesized small gold nanoparticles (Au NPs, <10 nm) and core–shell gold–platinum (Au@Pt) NPs were synthesized for tandem catalysis of hydrogenation of NAD+ with H2.77 As shown in Scheme 11a, electrons released in the oxidation of NADH with NRase were used for the reduction of metal salts (Mn+) to produce biohybrid Au NPs, Au core, and (Au@Pt) NPs.77 Au@Pt NPs prepared using NRase enzyme can re-donate electrons to NRase by utilizing its H2 oxidation properties at 1 bar and 25 °C, thereby selectively reducing NAD+ to the biologically active NADH cofactor (Scheme 11b).77 At [NRase] = 1.9 mg mL−1, 1,4-NADH was produced regioselectively, providing a novel route for continuous and H2-driven efficient NADH recycling.77 The H2-driven 1,4-NADH recycling was further coupled with alcohol dehydrogenase (YADH) for the reduction of enantioselective ketone.77 Use of H2 as a reductant for production of NADH has been previously explored using intact soluble hydrogenase enzymes78 and co-immobilised hydrogenase and NRase on carbon particles.79
 |
| | Scheme 11 (a) Formation of metal nanoparticles (MNPs) using NRase and NADH (or its analogue) as a reducing agent, while reducing Mn+ to M0. (b) Reduction of NAD+ to NADH by H2via H2 oxidation at the metal NP surface. Reprinted with permission from ref. 77. Copyright 2024, John Wiley and Sons. | |
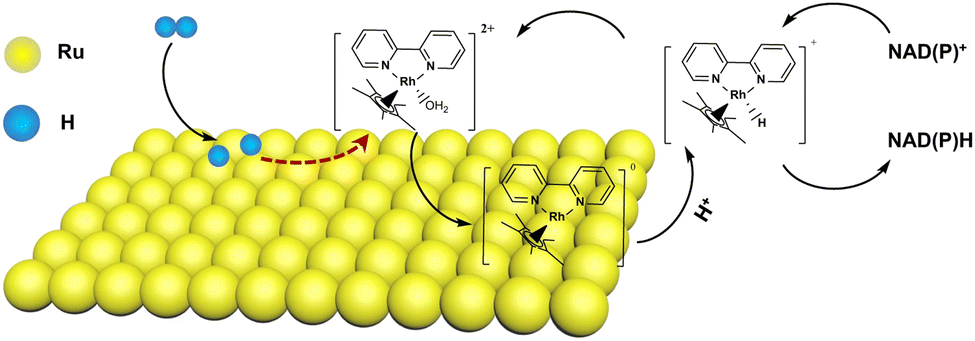
It has been reported that the simultaneous coupling of [Cp*Rh(bpy)(H2O)]2+ and supported Ru NPs enhanced both catalytic activity and regioselectivity for the hydrogenation of NAD(P)+, suggesting the efficient synergistic effect of the H2 dissociation ability of supported Ru NPs and the steric effect of [Cp*Rh(bpy)(H2O)]2+ in reducing NAD(P)+ to 1,4-NAD(P)H.80 NAD+ could be completely converted to 1,4-NADH at 25 °C and 1 bar H2 (>99% selectivity).80 NADPH with >95% yield was also successfully reproduced by use of this coupling system.80 The catalytic mechanism was proposed as shown in Fig. 5, where H2 was first dissociated into adsorbed hydrogen (H*) over Ru NPs. [Cp*Rh(bpy)(H2O)]2+ was reduced by H* to produce [Cp*Rh(I)(bpy)]0, which was deposited on the surface of Ru NPs (Fig. 5).80 [Cp*Rh(I)(bpy)]0 reacted with protons to produce [Cp*Rh(bpy)H]+.80 Then, hydride transfer from [Cp*Rh(bpy)H]+ to NAD(P)+ occurred via a ring-slipped mechanism,48 accompanied by regeneration of [Cp*Rh(bpy)(H2O)]2+.80 Nickel nanoparticles (NP) and [Cp*Rh(bpy)(H2O)]2+ were also integrated for H2-driven NAD(P)H regeneration through the immobilization of a Rh complex on a Ni/TiO2 surface via a bipyridine containing 3D porous organic polymer (POP).81
 |
| | Fig. 5 Coupling of [Cp*Rh(bpy)(H2O)]2+ and supported Ru NPs for NAD(P)+ hydrogenation. Reprinted with permission from ref. 80. Copyright 2022, Springer Nature. | |
Monocarbonyl diphosphine Ru(II) complexes were reported to be active for the regioselective reduction of NAD+ to 1,4-NADH by formate as the hydride source in aqueous media.82 The catalytic cycle is shown in Scheme 12, where the Ru(II) complexes react with formate to afford the hydride ruthenium complexes via β-hydride elimination accompanied by CO2 evolution.82 The hydride complexes hydrogenate NAD+ to produce 1,4-NADH, accompanied by regeneration of the Ru(II) aqua complex (Scheme 12).82 The Ru(II) aqua complex with the picolinamidate (pica) ligand was the most active, exhibiting a TOF of 6.08 h−1 in deuterated phosphate-buffered saline (PBS) at room temperature.82
 |
| | Scheme 12 Proposed catalytic pathway for the reduction of NAD+ with monocarbonyl diphosphine Ru(II) complexes in PBS. Reproduced with permission from ref. 82. Copyright 2023, American Chemical Society. | |
5. Photocatalytic reduction of NAD(P)+ by hydride donors
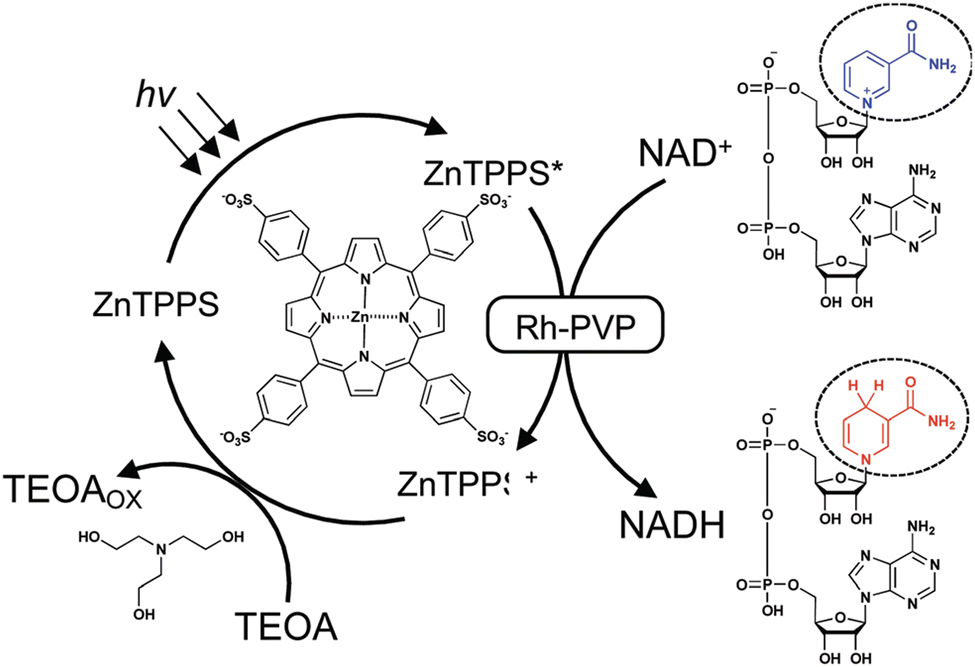
Rh nanoparticles dispersed in polyvinylpyrrolidone (Rh-PVP) were used for the visible-light-driven selective NADH production in the presence of a sacrificial electron donor, triethanolamine (TEOA), and a photosensitizer, zinc meso-5,10,15,20-tetrakis-(4-sulfonatophenyl)porphyrin (ZnTPPS), in the aqueous media, as shown in Scheme 13. It was confirmed that the reduction product of NAD+ with ZnTPPS and Rh-PVP was only 1,4-NADH.83 The photocatalytic reaction was started by electron transfer from ZnTPPS* to Rh-PVP by static quenching.83 NAD+ is adsorbed onto the surface of Rh-PVP by the carbonyl of the amide, similar to the Rh complex.47,54 The hydride species at the surface of Rh-PVP attacks the C4 position of nicotinamide and forms 1,4-NADH directly (Scheme 14a).83 Another possible mechanism is the interaction of NAD+ with an absorbed H atom on the surface of Rh-PVP coupled with an electron transfer (Scheme 14b).83 In any case, the hydride transfer is the key to avoid the radical intermediate and NAD dimer formation.83
 |
| | Scheme 13 Scheme of visible-light-driven NADH regeneration using the system composed of TEOA as an electron donor, ZnTPPS as a photosensitizer, Rh-PVP as a catalyst and NAD+. Reprinted with permission from ref. 83. Copyright 2021, Royal Society of Chemistry. | |
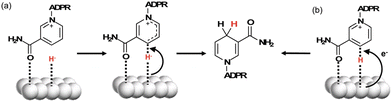 |
| | Scheme 14 Plausible mechanism of NADH regeneration using Rh-PVP by (a) H− transfer or (b) adsorbed H˙/electron transfer. Reprinted with permission from ref. 83. Copyright 2021, Royal Society of Chemistry. | |
When Rh-PVP was replaced by [Cp*Rh(bpy)(H2O)]2+, photocatalytic reduction of NAD+ by TEOA with ZnTPPS also occurred to produce 1,4-NADH selectively.84,85 ZnTPPS can be replaced by various photoredox catalysts such as Ir complexes (Scheme 15),86 Ru complexes,63,87 inorganic semiconductors,88 metal–organic frameworks (MOFs),89 conjugated porous polymers (CPPs)90 and quantum dots (QDs)91,92 for regioselective reduction of BNA+ (or NAD+) to 1,4-BNAH (or 1,4-NADH) with use of TEOA [or triethylamine (TEA)] as a hydride donor.
 |
| | Scheme 15 Photochemical production of 1,4-BNAH using a rhodium catalyst ([Cp*Rh(bpy)(H2O)]2+) with water soluble iridium(III) photosensitizers and TEOA for the regioselective BNA+ reduction to 1,4-BNAH by TEOA. Reprinted with permission from ref. 86. Copyright 2023, American Chemical Society. | |
6. A PSI functional model
In photosynthesis, PQH2 reduces NADP+ regioselectively to generate 1,4-NADPH via charge separation in the photosynthetic reaction centre (PRC) in PSI [eqn (2)].1 The first molecular model of PSI for NAD(P)H production has been reported by use of a hydroquinone derivative (X-QH2) as a PQH2 model compound, which regioselectively generates 1,4-NADH using a PRC model compound, 9-mesityl-10-methylacridinium ion (Acr+-Mes),93–96 and the NAD+ reduction catalyst, CoIII(dmgH)2pyCl,97–99 under visible light irradiation [eqn (3)].100| |  | (2) |
| | 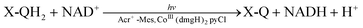 | (3) |
| | 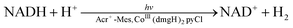 | (4) |
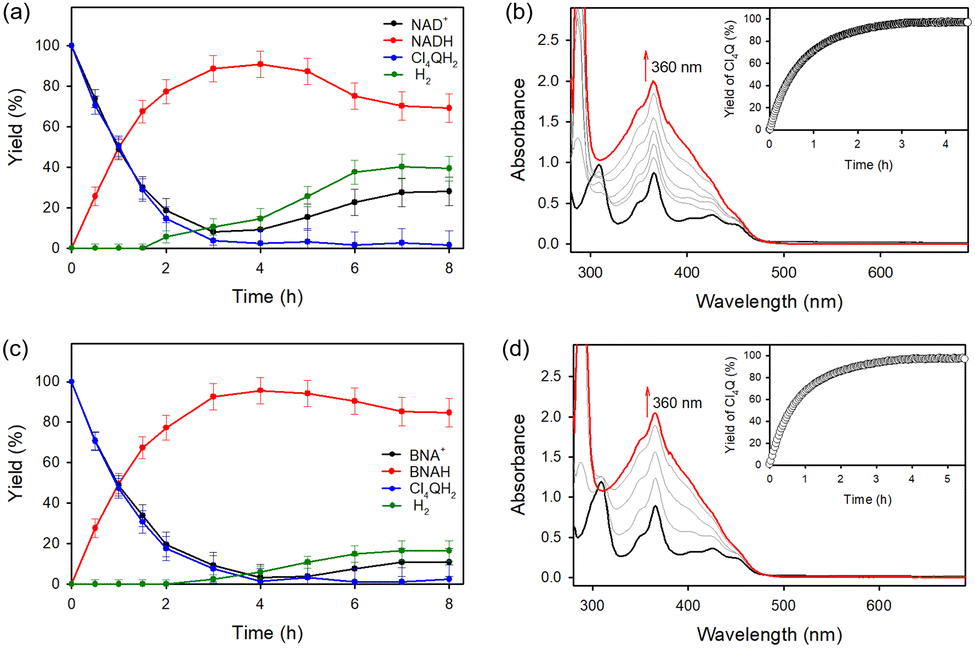
A quartz cell was used for the PSI model reaction [eqn (3)] in the two phases separated by a liquid membrane under visible light irradiation (Fig. 6).100 NADH was produced by the photocatalytic reduction of NAD+ by Cl4QH2 used as a PQH2 model compound, being detected by HPLC measurements. On the other hand, evolution of H2 was detected by GC (Fig. 7a).100 NAD+ is reduced regioselectively to 1,4-NADH, which is converted to H2 at the later stage of the NADH production (Fig. 7a) as given by eqn (4).100 The final yield for the conversion from NADH to H2 was almost 20%.100
 |
| | Fig. 6 A quartz cell employed for the PSI model reaction: photo-driven NAD+ reduction by X-QH2 in toluene (upper part) and a borate buffer/TFE mixed solution (v/v 19:1; lower part) containing X-QH2 (2.0 × 10−6 mol), NAD+ (4.0 × 10−6 mol), Acr+-Mes (1.2 × 10−6 mol) and CoIII(dmgH)2pyCl (1.5 × 10−6 mol) to produce 1,4-NADH under photoirradiation of Acr+-Mes in the aqueous/TFE phase. Reprinted with permission from ref. 100. Copyright 2024, American Chemical Society. | |
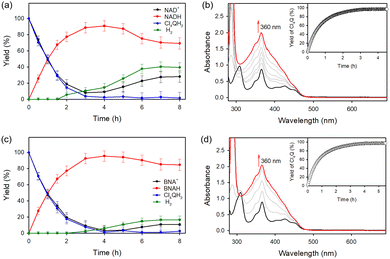 |
| | Fig. 7 (a) and (c) Time courses of a PSI model reaction: generation of (a) NADH and (c) BNAH in the photocatalytic reduction of (a) NAD+ (4.0 × 10−6 mol) and (c) BNA+ (4.0 × 10−6 mol), respectively, by Cl4QH2 (2.0 × 10−6 mol) with a PRC model compound (Acr+-Mes: 1.5 × 10−6 mol) and CoIII(dmgH)2pyCl (NAD+ reduction catalyst: 1.2 × 10−6 mol). (b) and (d) Visible absorption spectral changes in the photocatalytic reduction of (b) NAD+ (4.0 × 10−6 mol) and (d) BNA+ (4.0 × 10−6 mol) by Cl4QH2 (2.0 × 10−6 mol) with Acr+-Mes (1.5 × 10−6 mol) and CoIII(dmgH)2pyCl (1.2 × 10−6 mol) in a toluene/TFE/borate buffer (v/v/v 40:1:19; 3.0 mL) at 298 K. Insets show time profiles of the formation of Cl4Q. Reprinted with permission from ref. 100. Copyright 2024, American Chemical Society. | |
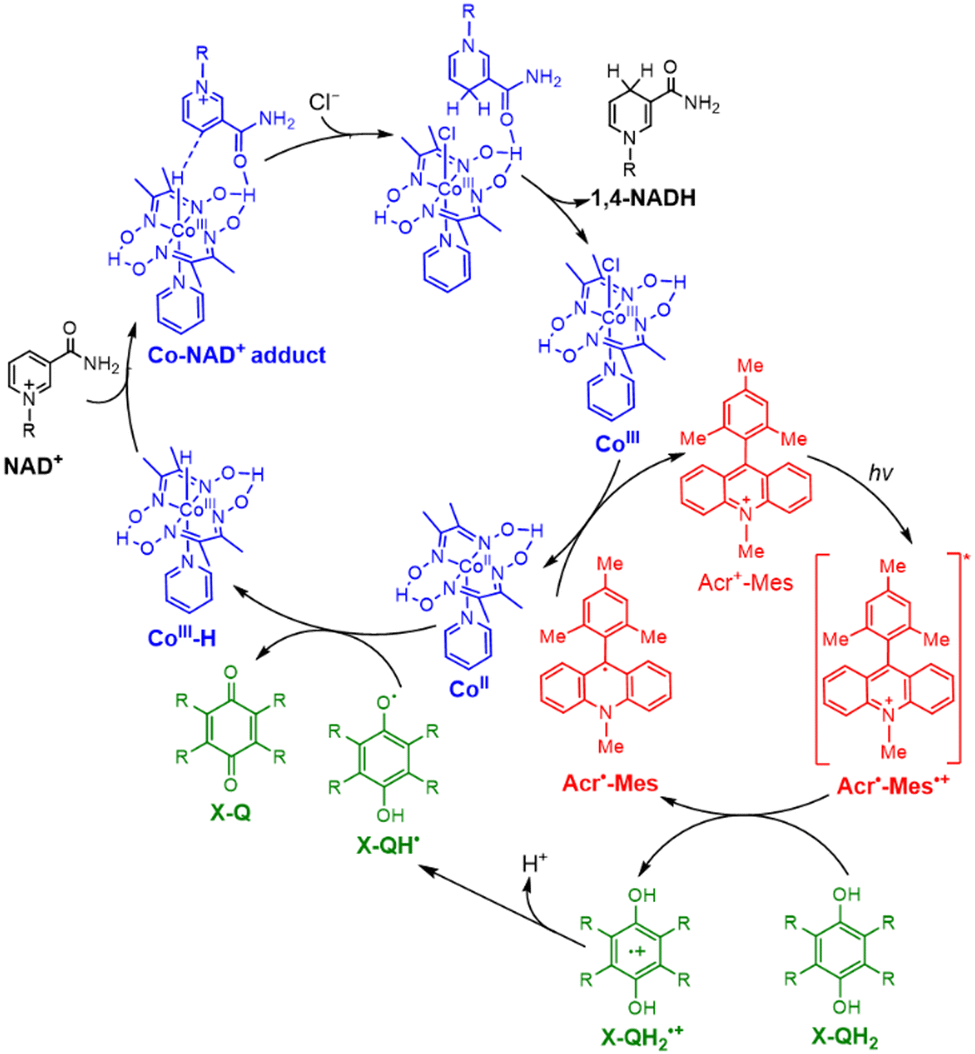
Cl4QH2 was converted to Cl4Q [eqn (3)] as indicated by UV-vis absorption spectral changes in Fig. 7b.100 When NAD+ was replaced by an NAD+ model compound (BNA+), 1,4-BNAH was also selectively produced (Fig. 7c), accompanied by formation of Cl4Q (Fig. 7d).100 At prolonged irradiation time, O2 is consumed by the photocatalytic oxidation of NADH or BNAH to produce H2O2. The photocatalytic mechanism of regioselective reduction of NAD+ by QH2 with a PRC model compound (Acr+-Mes) and an NAD+ reduction catalyst (CoIII(dmgH)2pyCl) (Scheme 16) is virtually the same as the case of photocatalytic H2 evolution from X-QH2 with Acr+-Mes and CoIII(dmgH)2pyCl.100 Firstly, photoexcitation of Acr+-Mes in the aqueous/TFE phase results in intramolecular electron transfer from the Mes moiety to the singlet excited state of the Acr+ moiety to produce the singlet electron-transfer (ET) state, followed by intersystem crossing to form the triplet ET state [3(Acr˙-Mes˙+)],93–95 which undergoes the ET oxidation of X-QH2 by the Mes˙+ moiety to produce X-QH2˙+ as well as the ET reduction of CoIII(dmgH)2pyCl by the Acr˙ moiety to produce [CoII(dmgH)2pyCl]−.100 X-QH2˙+ is deprotonated to produce the semiquinone radical (X-QH˙),100 followed by hydrogen atom transfer from X-QH˙ to [CoII(dmgH)2pyCl]− to produce X-Q and the Co(III)-hydride complex ([Co(H)(dmgH)2pyCl]−).100 Then, a H− transfer from the Co(III)-hydride complex to NAD+ proceeds via a six-membered ring transition state, where the hydride ion interacts with the C4-position of NAD+ to yield 1,4-NADH regioselectively.100 No other regioisomers, such as 1,2- and 1,6-NADH, were produced in the photocatalytic reduction of NAD+ by X-QH2 with Acr+-Mes and CoIII(dmgH)2pyCl (Scheme 16).100
 |
| | Scheme 16 Proposed mechanism of a PSI model reaction: the photocatalytic NAD+ reduction to 1,4-NADH by X-QH2 with a PRC model compound (Acr+-Mes) and a NAD+ reduction catalyst (CoIII(dmgH)2pyCl). Reprinted with permission from ref. 100. Copyright 2024, American Chemical Society. | |
7. Photocatalytic reduction of NAD(P)+ to NAD(P)H by water
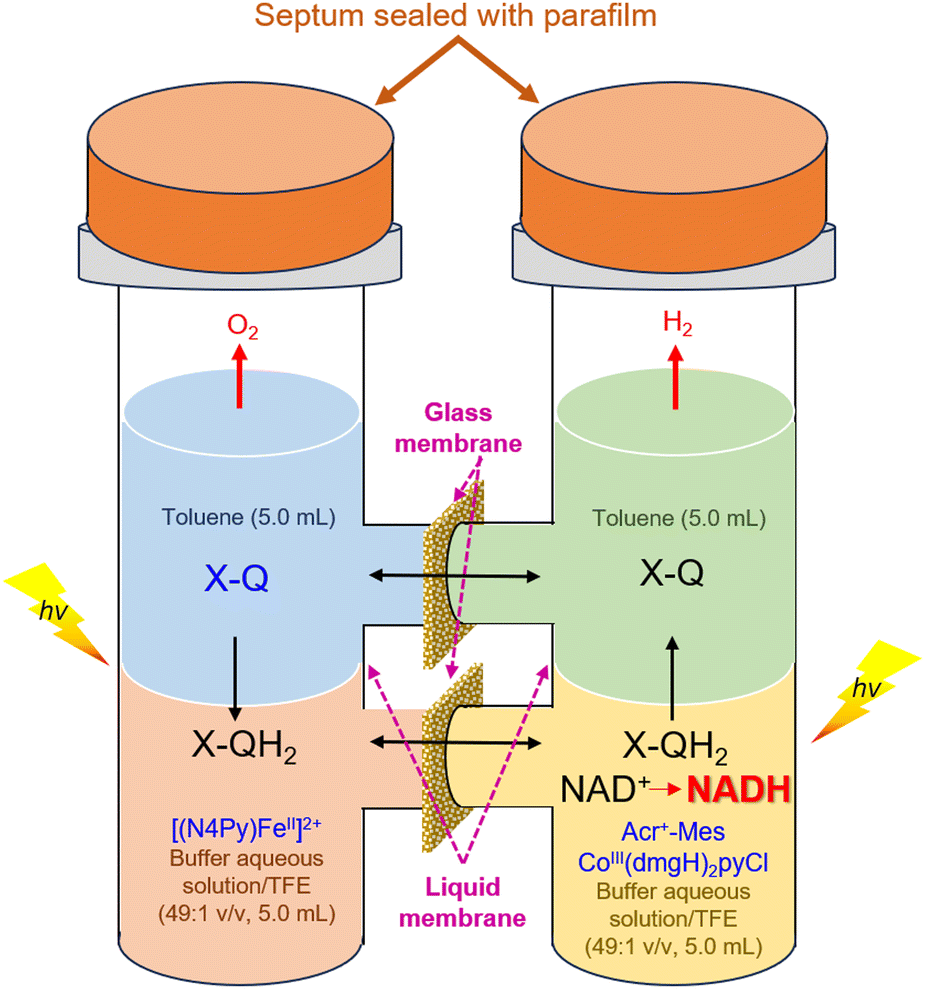
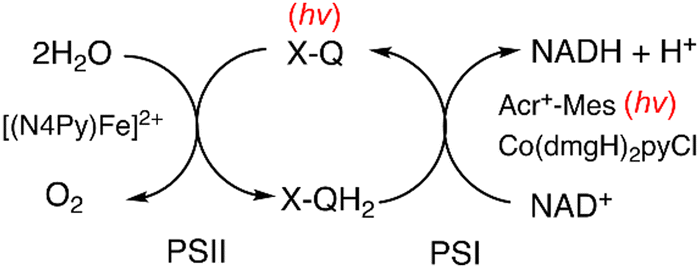
The same photocatalytic system for the water splitting has been used to achieve the reduction of NAD(P)+ to NAD(P)H by H2O, accompanied by oxidation of H2O to O2 as shown in Fig. 8.100,101 Photoirradiation of a mixed solution (two phase) of toluene, TFE and borate buffer (v/v/v 50:1:49; pH = 7.0) containing Cl4Q and [(N4Py)FeII]2+ in the left cell and a mixed solution (two phase) of toluene, TFE and borate buffer (v/v/v 50:1:49; pH = 7.0) containing NAD(P)+ [or an NAD+ model compound, 1-benzyl-3-carbamoylpyridinium cation (BNA+)], Acr+-Mes and CoIII(dmgH)2pyCl in the right cell resulted in regioselective formation of 1,4-NAD(P)H (or 1,4-BNAH) with almost 100% yield on the basis of the initial concentration of NAD(P)+ (or BNA+) used in the right cell together with production of O2 in the left cell.100 The TON for the production of NADH was 24, 16 and 12 on the basis of the initial concentrations of Cl4Q, CoIII(dmgH)2pyCl and Acr+-Mes, respectively.100 Thus, Cl4Q, CoIII(dmgH)2pyCl and Acr+-Mes act as combined catalysts for the overall photocatalytic NAD(P)+ reduction to 1,4-NAD(P)H by H2O.100 The photocatalytic NAD(P)+ reduction occurred similarly when Cl4QH2 was replaced by hydroquinone (QH2) and tetramethylhydroquinone (Me4QH2). Thus, a substituted p-benzoquinone (X-Q) generally acts as a plastoquinone (PQ) analogue, which is a redox catalyst in the photosynthesis (Scheme 17).100 The ratio of production of O2:NADH was 1:2, agreeing with the stoichiometry of photosynthesis [eqn (1)].100 Over long periods of light exposure, however, the concentration of O2 gradually decreased and the production of NADH ceased because H2 was produced by the reaction of NADH with H+ [eqn (4)].100
 |
| | Fig. 8 A photochemical O-type quartz tube employed for molecular photosynthesis: photocatalytic regioselective NAD+ reduction by H2O to produce 1,4-NADH by combining PSI and PSII model systems. Reprinted with permission from ref. 100. Copyright 2024, American Chemical Society. | |
 |
| | Scheme 17 Photocatalytic cycle for photocatalytic regioselective NAD+ reduction by H2O to produce 1,4-NADH and O2, achieved by combining PSI and PSII molecular models. Reprinted with permission from ref. 100. Copyright 2024, American Chemical Society. | |
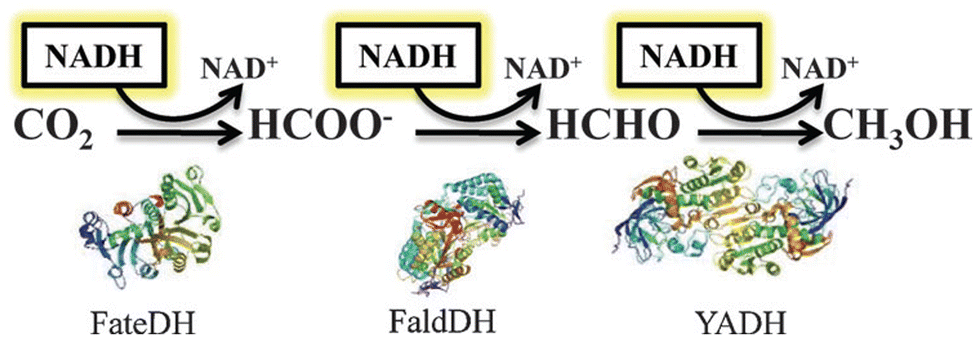
Once NADH is obtained by photocatalytic regioselective reduction of NAD+ by H2O (Scheme 17), NADH can reduce CO2 to methanol by using three dehydrogenases as shown in Scheme 18, where CO2 is reduced to formate by the catalysis of formate dehydrogenase (FateDH), then formate is further reduced to formaldehyde by the catalysis of formaldehyde dehydrogenase (FaldDH) and finally methanol is obtained by the catalysis of alcohol dehydrogenase (YADH).102 In the overall reaction, three equivalents of NADH are required to reduce CO2 to methanol.102 The optimized ratio of the three polyenzymes, i.e., FateDH, FaldH and YADH, was found to be 0.010, 0.15 and 0.75 g L−1 of commercially available enzymatic powder, respectively.102 Immobilization of the enzymes provides not only stabilisation and easier use, but also improved enzymatic activity.103–105
 |
| | Scheme 18 Reduction scheme of CO2 to methanol by three dehydrogenase enzymes (FateDH, FaldDH and YADH). Reprinted with permission from ref. 102. Copyright 2013, Royal Society of Chemistry. | |
8. Conclusion and perspectives
Electrocatalytic reduction of NAD(P)+ to 1,4-NAD(P)H has been achieved using Rh(III)–H complexes, which undergo hydride transfer to NAD(P)+ with the interaction of the amide group of NAD(P)+ with the metal centre to enable the regioselective reduction. The photoelectrocatalytic reduction of NAD(P)+ to 1,4-NAD(P)H has lowered the applied potential required for the NAD(P)+ reduction. The regioselective reduction of NAD(P)+ has also been achieved by hydrogenation of NAD(P)+ with H2, catalysed by metal complexes. Photocatalytic reduction of NAD(P)+ to 1,4-NADH can be made possible by combining photoredox catalysts and NAD(P)+ reduction catalysts in the presence of a sacrificial electron and proton donor such as TEOA and TEA. When hydroquinone derivatives were employed as plastoquinol analogues in a photosystem I molecular model system, photocatalytic NAD(P)+ reduction by hydroquinone derivatives occurred efficiently by using a simple photosynthetic reaction centre model, 9-mesityl-10-methylacridininum ion (Acr+-Mes), and an NAD(P)+ reduction catalyst, CoIII(dmgH)2pyCl, under photoirradiation to produce only 1,4-NAD(P)H and p-benzoquinone derivatives (plastoquinone analogues). This photosystem I model has been combined with a photosystem II model in which water is oxidized by plastoquinone analogues to achieve the stoichiometry of photosynthesis, i.e., photocatalytic reduction of NAD(P)+ to 1,4-NAD(P)H by water used as a reductant. Once NAD(P)H is produced by regioselective reduction of NAD(P)+ by H2O using solar energy, reduction of various substrates, including CO2 reduction, can occur through a combination of NAD(P)H dependent enzymes and PSI and PSII molecular models, providing a promising method for production of value-added products using water as a reductant and CO2 as a carbon source. The activity and stability of photoredox catalysts and NAD(P)+ reduction catalysts have yet to be improved for future practical applications.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the significant contributions of their collaborators and coworkers cited in the listed references and support from JSPS (23K04686 to S.F.) and NRF of Korea (NRF-2023R1A2C1007668 to Y.-M. L., NRF-2022H1D3A2A01045098 to S. F. and NRF-2021R1A3B1076539 to W. N.).
Notes and references
- S. A. Watson and G. P. McStay, Int. J. Mol. Sci., 2020, 21, 7254 CrossRef CAS PubMed.
- S. Yoshikawa and A. Shimada, Chem. Rev., 2015, 115, 1936–1989 CrossRef CAS PubMed.
- A. Shimada, T. Tsukihara and S. Yoshikawa, Front. Chem., 2023, 11, 1108190 CrossRef CAS PubMed.
- M. Wikström, K. Krab and V. Sharma, Chem. Rev., 2018, 118, 2469–2490 CrossRef PubMed.
- S. M. Adam, G. B. Wijeratne, P. J. Rogler, D. E. Diaz, D. A. Quist, J. J. Liu and K. D. Karlin, Chem. Rev., 2018, 118, 10840–11022 CrossRef CAS PubMed.
- S. Panda, H. Phan and K. D. Karlin, J. Inorg. Biochem., 2023, 249, 112367 CrossRef CAS PubMed.
-
G. C. Schröder, M. S. Smit and D. J. Opperman, Curr. Opin. Green Sustain. Chem., 2023, 39, 100734 Search PubMed.
- I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2277 CrossRef CAS PubMed.
- L. Zhang and Q. Wang, ChemBioChem, 2022, 23, e20210043 Search PubMed.
- J. He, X. Liu and C. Li, Molecules, 2024, 29, 2480 CrossRef CAS PubMed.
- S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949–1017 CrossRef CAS PubMed.
- F. P. Guengerich, Chem. Res. Toxicol., 2008, 21, 70–83 Search PubMed.
- D. Shevela, J. F. Kern, G. Govindjee and J. Messinger, Photosynth. Res., 2023, 156, 279–307 CrossRef CAS PubMed.
- N. Nelson and C. F. Yocum, Annu. Rev. Plant Biol., 2006, 57, 521–565 CrossRef CAS PubMed.
- M. M. Najafpour and S. I. Allakhverdiev, J. Photochem. Photobiol., B, 2015, 152, 173–175 CrossRef CAS PubMed.
- A. Stirbet, D. Lazár, Y. Guo and G. Govindjee, Ann. Bot., 2020, 126, 511–537 CrossRef CAS PubMed.
- R. Siedentop and K. Rosenthal, Int. J. Mol. Sci., 2022, 23, 3605 CrossRef CAS PubMed.
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC.
- S. Mordhorst and J. N. Andexer, Nat. Prod. Rep., 2020, 37, 1316–1333 RSC.
- R. A. Sheldon and D. Brady, ACS Sustainable Chem. Eng., 2021, 9, 8032–8052 CrossRef CAS.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- S. Huang, G. Chen and G. Ouyang, Chem. Soc. Rev., 2022, 51, 6824–6863 RSC.
- K.-Y. Wang, J. Zhang, Y.-C. Hsu, H. Lin, Z. Han, J. Pang, Z. Yang, R.-R. Liang, W. Shi and H.-C. Zhou, Chem. Rev., 2023, 123, 5347–5420 Search PubMed.
- H. Wu, C. Tian, X. Song, C. Liu, D. Yang and Z. Jiang, Green Chem., 2013, 15, 1773–1789 RSC.
- S. Fukuzumi, Y.-M. Lee and W. Nam, J. Inorg. Biochem., 2019, 199, 110777 CrossRef CAS PubMed.
- Y. S. Lee, R. Gerulskis and S. D. Minteer, Curr. Opin. Biotechnol, 2022, 73, 14–21 Search PubMed.
- G. Zhao, C. Yang, W. Meng and X. Huang, J. Mater. Chem. A, 2024, 12, 3209–3229 RSC.
- V. K. Sharma, J. M. Hutchison and A. M. Allgeier, ChemSusChem, 2022, 15, e202200888 CrossRef CAS PubMed.
- S. Immanuel, R. Sivasubramanian, R. Gul and M. A. Dar, Chem. – Asian J., 2020, 15, 4256–4270 CrossRef CAS PubMed.
- K. Bachosz, J. Zdarta, M. Bilal, A. S. Meyer and T. Jesionowski, Sci. Total Environ., 2023, 868, 161630 CrossRef CAS PubMed.
- S. Fukuzumi, Y.-M. Lee and W. Nam, iScience, 2024, 27, 110694 CrossRef CAS PubMed.
- Y. H. Hong, Y.-M. Lee, W. Nam and S. Fukuzumi, ACS Catal., 2023, 13, 308–341 CrossRef CAS.
- Y. H. Hong, J. Jung, T. Nakagawa, N. Sharma, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2019, 141, 6748–6754 Search PubMed.
- Y. Zhang and J. Liu, Curr. Opin. Electrochem., 2024, 46, 101506 CrossRef CAS.
- Y. Li, G. Liu, W. Kong, S. Zhang, Y. Bao, H. Zhao, L. Wang, L. Zhou and Y. Jiang, Green Chem. Eng., 2024, 5, 1–15 CrossRef.
- A. Weckbecker, H. Grçger and W. Hummel, Adv. Biochem. Eng. Biotechnol., 2010, 120, 195–242 CAS.
- S. Immanuel and R. Sivasubramanian, Mater. Sci. Eng. B, 2020, 114705 CrossRef CAS.
- F. Liu, C. Ding, S. Tian, S.-M. I. Lu, C. Feng, D. Tu, Y. Liu, W. Wang and C. Li, Chem. Sci., 2022, 13, 13361–13367 RSC.
- H. Jaegfeldt, Bioelectrochem. Bioenerg., 1981, 8, 355–370 CrossRef CAS.
- I. Ali, A. Gill and S. Omanovic, Chem. Eng. J., 2012, 188, 173–180 CrossRef CAS.
- I. Ali and S. Omanovic, Int. J. Electrochem. Sci., 2013, 8, 4283–4304 CrossRef CAS.
- I. Ali, N. Ullah, M. A. McArthur, S. Coulombe and S. Omanovic, Can. J. Chem. Eng., 2018, 96, 68–73 CrossRef CAS.
- N. H. A. Besisa, K.-S. Yoon, T. G. Noguchi, H. Kobayashi and M. Yamauchi, ACS Sustainable Chem. Eng., 2024, 12, 9874–9881 CrossRef CAS.
- R. Wienkamp and E. Steckhan, Angew. Chem., Int. Ed. Engl., 1982, 21, 782–783 CrossRef.
- R. Ruppert, S. Herrmann and E. Steckhan, Tetrahedron Lett., 1987, 28, 6583–6586 CrossRef CAS.
- E. Steckhan, S. Herrmann, R. Ruppert, E. Dietz, M. Frede and E. Spika, Organometallics, 1991, 10, 1568–1577 CrossRef CAS.
- H. C. Lo, O. Buriez, J. B. Kerr and R. H. Fish, Angew. Chem., Int. Ed., 1999, 38, 1429–1432 CrossRef CAS PubMed.
- C. L. Pitman, O. N. L. Finster and A. J. M. Miller, Chem. Commun., 2016, 52, 9105–9108 RSC.
- J. D. Blakemore, E. S. Hernandez, W. Sattler, B. M. Hunter, L. M. Henling, B. S. Brunschwig and H. B. Gray, Polyhedron, 2014, 84, 14–18 CrossRef CAS.
- B. Tan, D. P. Hickey, R. D. Milton, F. Giroud and S. D. Minteer, J. Electrochem. Soc., 2015, 162, H102–H107 CrossRef CAS.
- W. Li, C. Zhang, Z. Zheng, X. Zhang, L. Zhang and A. Kuhn, ACS Appl. Mater. Interfaces, 2022, 14, 46673–46681 CrossRef CAS PubMed.
- F. Liu, W. Shi, S. Tian, Y. Zhou, C. Feng, C. Ding and C. Li, J. Phys. Chem. C, 2024, 128, 5927–5933 CrossRef CAS.
- S. Tian, G. Long, P. Zhou, F. Liu, X. Zhang, C. Ding and C. Li, J. Am. Chem. Soc., 2024, 146, 15730–15739 CrossRef CAS PubMed.
- A. Azem, F. Man and S. Omanovic, J. Mol. Catal. A: Chem., 2004, 219, 283–299 CrossRef CAS.
- I. Ali, T. Khan and S. Omanovic, J. Mol. Catal. A: Chem., 2014, 387, 86–91 CrossRef.
- A. Damian, K. Maloo and S. Omanovic, Chem. Biochem. Eng. Q., 2007, 21, 21–32 CAS.
- V. Ganesan, D. Sivanesan and S. Yoon, Inorg. Chem., 2017, 56, 1366–1374 CrossRef CAS PubMed.
- M. Chrzanowska, A. Katafias and R. van Eldik, Inorg. Chem., 2020, 59, 14944–14953 CrossRef CAS PubMed.
- R. Ruppert, S. Herrmann and E. Steckhan, J. Chem. Soc., Chem. Commun., 1988, 1150–1151 RSC.
- V. Ganesan, J. J. Kim, J. Shin, K. Park and S. Yoon, Inorg. Chem., 2022, 61, 5683–5690 CrossRef CAS PubMed.
- F. Chen, J. J. Soldevila-Barreda, I. Romero-Canelón, J. P. C. Coverdale, J.-I. Song, G. J. Clarkson, J. Kasparkova, A. Habtemariam, V. Brabec, J. A. Wolny, V. Schünemann and P. J. Sadler, Dalton Trans., 2018, 47, 7178–7189 RSC.
- F. Chen, I. Romero-Canelón, J. J. Soldevila-Barreda, J.-I. Song, J. P. C. Coverdale, G. J. Clarkson, J. Kasparkova, A. Habtemariam, M. Wills, V. Brabec and P. J. Sadler, Organometallics, 2018, 37, 1555–1566 CrossRef CAS PubMed.
- Y. Matsubara, K. Koga, A. Kobayashi, H. Konno, K. Sakamoto, T. Morimoto and O. Ishitani, J. Am. Chem. Soc., 2010, 132, 10547–10552 CrossRef CAS PubMed.
- M. Chrzanowska, A. Katafias, R. van Eldik and J. Conradie, RSC Adv., 2022, 12, 21191–21202 RSC.
- M. Chrzanowska, A. Katafias and R. van Eldik, Front. Chem., 2023, 11, 1150164 CrossRef CAS PubMed.
- A. Bucci, S. Dunn, G. Bellachioma, G. M. Rodriguez, C. Zuccaccia, C. Nervi and A. Macchioni, ACS Catal., 2017, 7, 7788–7796 CrossRef CAS.
- L.-J. Zhao, C. Zhang, S. Zhang, X. Lv, J. Chen, X. Sun, H. Su, T. Murayama and C. Qi, Inorg. Chem., 2023, 62, 17577–17582 CrossRef CAS PubMed.
- L. Tensi, L. Rocchigiani, G. M. Rodriguez, E. Mosconi, C. Zuccaccia, F. De Angelis and A. Macchioni, Catal. Sci. Technol., 2023, 13, 6743–6750 RSC.
- E. Lineberry, J. Kim, J. Kim, I. Roh, J.-A. Lin and P. Yang, J. Am. Chem. Soc., 2023, 145, 19508–19512 CrossRef CAS PubMed.
- E. J. Son, J. W. Ko, S. K. Kuk, H. Choe, S. Lee, J. H. Kim, D. H. Nam, G. M. Ryu, Y. H. Kim and C. B. Park, Chem. Commun., 2016, 52, 9723–9726 RSC.
- S. H. Lee, G. M. Ryu, D. H. Nam, J. H. Kim and C. B. Park, ChemSusChem, 2014, 7, 3007–3011 CrossRef CAS PubMed.
- B. Zhang, S. Xu, D. He, R. Chen, Y. He, W. Fa, G. Li and D. Wang, J. Chem. Phys., 2020, 153, 064703 CrossRef CAS PubMed.
- M. Chen, F. Liu, Y. Wu, Y. Li, C. Liu, Z. Zhao, P. Zhang, Y. Zhao, L. Sun and F. Li, Chem. Commun., 2024, 60, 3319–3322 RSC.
- V. Massey, Biochem. Soc. Trans., 2000, 28, 283–296 CrossRef CAS PubMed.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 367–374 CrossRef CAS PubMed.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2021, 5, 7360–7367 RSC.
- L. B. F. Browne, T. Sudmeier, M. A. Landis, C. S. Allen and K. A. Vincent, Angew. Chem., Int. Ed., 2024, 63, e202404024 CrossRef CAS PubMed.
- R. Mertens, L. Greiner, E. C. D. van den Ban, H. B. C. M. Haaker and A. Liese, J. Mol. Catal. B, 2003, 24–25, 39–52 CrossRef CAS.
- H. A. Reeve, L. Lauterbach, O. Lenz and K. A. Vincent, Chem-CatChem, 2015, 7, 3480–3487 CrossRef CAS PubMed.
- M. Wang, Z. Zhao, C. Li, H. Li, J. Liu and Q. Yang, Nat. Commun., 2022, 13, 5699 CrossRef CAS PubMed.
- M. Wang, H. Dai and Q. Yang, Angew. Chem., Int. Ed., 2023, 62, e202309929 CrossRef CAS PubMed.
- D. Lovison, T. Berghausen, S. R. Thomas, J. Robson, M. Drees, C. Jandl, A. Pöthig, P. Mollik, D. P. Halter, W. Baratta and A. Casini, ACS Catal., 2023, 13, 10798–10823 CrossRef CAS.
- T. Katagiri and Y. Amao, New J. Chem., 2021, 45, 15748–15752 RSC.
- M. Takeuchi and Y. Amao, Chem. Lett., 2024, 53, upae014 CrossRef.
- M. Takeuchi and Y. Amao, Bull. Chem. Soc. Jpn., 2023, 96, 1206–1208 CrossRef CAS.
- M. R. Schreier, B. Pfund, D. M. Steffen and O. S. Wenger, Inorg. Chem., 2023, 62, 7636–7643 CrossRef CAS PubMed.
- Y. Matsubara and O. Ishitani, Coord. Chem. Rev., 2023, 477, 214955 CrossRef CAS.
- W. Lan, M. Wang, H. Dai and Q. Yang, Front. Chem. Sci. Eng., 2024, 18, 37 CrossRef CAS.
- Y. Chen, P. Li, J. Zhou, C. T. Buru, L. Đorđević, P. Li, X. Zhang, M. M. Cetin, J. F. Stoddart, S. I. Stupp, M. R. Wasielewski and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 1768–1773 CrossRef CAS PubMed.
- Y. Wang, X. Yue, H. Zhao, L. Ma, L. Zhou, Y. Liu, X. Zheng, Y. He, G. Liu and Y. Jiang, ChemSusChem, 2024, e202301868 CrossRef CAS PubMed.
- C.-H. Gao, S.-M. Zhang, F.-F. Feng, S.-S. Hu, Q.-F. Zhao and Y.-Z. Chen, J. Colloid Interface Sci., 2023, 652, 1043–1052 CrossRef CAS PubMed.
- I. N. Chakraborty, V. Jain, P. Roy, P. Kumar, C. P. Vinod and P. P. Pillai, ACS Catal., 2024, 14, 6740–6748 CrossRef CAS.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- T. Tsudaka, H. Kotani, K. Ohkubo, T. Nakagawa, N. V. Tkachenko, H. Lemmetyinen and S. Fukuzumi, Chem. – Eur. J., 2017, 23, 1306–1317 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed.
- K. Ohkubo, S. Matsumoto, H. Asahara and S. Fukuzumi, ACS Catal., 2024, 14, 2671–2684 CrossRef CAS.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- J. A. Kim, S. Kim, J. Lee, J.-O. Baeg and J. Kim, Inorg. Chem., 2012, 51, 8057–8063 CrossRef CAS PubMed.
- Y. H. Hong, Y.-M. Lee, W. Nam and S. Fukuzumi, Inorg. Chem., 2020, 59, 14838–14846 CrossRef CAS PubMed.
- Y. H. Hong, M. Nilajakar, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2024, 146, 5152–5161 CrossRef CAS PubMed.
- Y. H. Hong, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2022, 144, 695–700 CrossRef CAS PubMed.
- R. Cazelles, J. Drone, F. Fajula, O. Ersen, S. Moldovan and A. Galarneau, New J. Chem., 2013, 37, 3721–3730 RSC.
- C. Di Spiridione, M. Aresta and A. Dibenedetto, Adv. Energy Sustainability Res., 2024, 5, 2400081 CrossRef CAS.
- M. E. Aguirre, C. L. Ramírez and Y. Di Iorio, Chem. – Eur. J., 2023, 29, e202301113 CrossRef CAS PubMed.
- Y. Shu, W. Liang and J. Huang, Green Chem., 2023, 25, 4196–4221 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Yong-Min
Lee
*ab,
Yong-Min
Lee