
On peptide bond formation from protonated glycine dimers in the gas phase: computational insight into the role of protonation†
Léo
Lavy
 a,
Denis
Comte
ac,
Florent
Calvo
b,
Bernadette
Farizon
a,
Michel
Farizon
*a and
Tilmann D.
Märk
c
a,
Denis
Comte
ac,
Florent
Calvo
b,
Bernadette
Farizon
a,
Michel
Farizon
*a and
Tilmann D.
Märk
c
aUniversité Claude Bernard Lyon 1, CNRS/IN2P3, UMR 5822, Institut de Physique des 2 Infinis de Lyon, F-69622 Villeurbanne, France. E-mail: m.farizon@ip2i.in2p3.fr
bUniversité Grenoble Alpes, CNRS, LIPhy, 38000 Grenoble, France
cInstitut für Ionenphysik und Angewandte Physik, Universität Innsbruck, A-6020 Innsbruck, Austria
First published on 6th January 2025
Abstract
Peptide bond formation from the pure protonated glycine dimer, H+(Gly)2, and from the mixed protonated glycine–diglycine dimer, H+Gly2(Gly), was recently found experimentally to occur in gas-phase experiments in the absence of any catalyst and especially under anhydrous conditions [J. Phys. Chem. A, 2023, 127, 775]. In this contribution we further examine the conditions of such unimolecular reactions by means of density-functional theory calculations at the DFT/M06 2X/6-311G++(2df,p) level, focusing in particular on the role played by the protonation site. Two pathways, stepwise and concerted, are identified for the pure protonated dimer, and six pathways are examined for the mixed dimer. The lowest-energy barriers for peptide bond formation are generally found when the reaction occurs precisely at the protonation site. In contrast, the highest barrier is obtained when the dipeptide is protonated away from the reaction site, in which case the peptide bond is formed similarly as with two neutral glycine molecules as the reaction partners. Protonated glycine monomers can also be hydrogen-bonded with the dipeptide, leading to energy barriers that lie inbetween those extreme cases.
Introduction
The production of primordial peptides continues to be one of the highly important topics associated with the origin of life.1 Conversion of amino acids into peptides is essential for our understanding of the origin of life in the Universe, including the key formation of biomolecules under prebiotic conditions. Significant prebiotic synthesis of polyatomic compounds could take place in interstellar media, energetically driven by cosmic-ray ionization, ultraviolet irradiation and electric discharge.2 In this context, peptide bond formation in the gas phase is relevant in astrochemistry where ion–molecule processes are expected to play a crucial role in the synthesis of relatively complex molecules in the interstellar medium.3,4Several laboratory works have been carried out to address the possible formation of peptide bonds in the gas phase.5–12 In particular, Wincel et al.5 showed that ion–molecule reactions involving the methionine and glutamic amino acids may promote the synthesis of protonated dipeptides and their subsequent elongation towards polypeptides. Photoexcitation of proton-bound peptide complexes by Lee et al.6 led to water elimination and the formation of longer amino-acid chains, suggesting that proton-bound dimers are long-lived intermediates along the pathway toward polypeptides. By experimentally producing cluster ions of amino acids, Singh et al.7 concluded that water elimination associated with the formation of a new peptide covalent bond marked the initial step towards the formation of the primordial peptides. Peptide elongation in the gas phase has been observed in the laboratory for β-alanine,8 glycine,9 serine10,11 and triptophan.12
The possible presence of amino acids in the interstellar medium lead to an extensive radioastronomical search for the simplest amino acid, glycine. Glycine has been detected in the Murchison meteorite13 while diglycine was reported in both the Yamato-791198 and the Murchison meteorites.14 The observation of glycine in the coma of the comet 67P/Churyumov–Gerasimenko by the ROSINA mass spectrometer during the Rosetta mission15 strongly suggests that glycine could be present in celestial bodies. A precursor of glycine was also detected surrounding a Sun-like young star16,17 while characteristic glycine bands have been detected in hot corrinos.18 The current state-of-the-art in spectroscopic techniques could enable the conclusive detection of glycine in protoplanetary systems19 in the near future.
The harsh radiation conditions experienced in vast regions of Space, especially cosmic rays and UV photons,20,21 could provide the energy needed to trigger ion–molecule chemical reactions, such as the ones needed to grow peptides.22 It was recently shown that diglycine could be formed by exciting a protonated glycine dimer H+(Gly)2 in a single high-velocity atomic collision.9 Here the new peptide bond was produced even in the absence of any solid substrate or available solvent. In the same study, diglycine and glycine monomers were also found to associate abundantly into the mixed protonated dimer complex H+Gly2(Gly).9 Upon exposure to the same 8 keV argon atoms, protonated triglycine was observed, confirming that the experimental conditions are appropriate for peptide elongation beyond the initial dipeptide. To further address the possible role of water in peptide bond formation, the measurements were repeated with or without additional water in the molecular beams. Rather surprisingly, no mixed cluster ions containing both glycine and water molecules were observed in the presence of water in the beam, similar mass spectra distribution patterns being observed as in the anhydrous case. Such a demixing behaviour is in striking contrast with earlier experiments carried on the same experimental device on other molecules such as pyridine or methanol.23,24
From the theoretical perspective, the uncatalyzed peptide bond formation in the gas phase has been studied by Jensen et al.25 who considered the reaction between two glycine molecules. These authors addressed the mechanism of the reaction and its concerted or stepwise nature, the barrier heights involved in the process, the effects of entropy or the molecular and electronic details at the transition state. Redondo et al.22 performed a thorough computational study of the possible ion–molecule processes that could lead to the formation of a peptide bond in the gas phase, also involving the glycine amino acids but with one protonated reaction partner. In this work too, concerted and stepwise reactions were found to compete to produce the protonated dipeptide under isolated conditions, and it was concluded that peptide elongation could take place under interstellar conditions if glycine is indeed present in the first place.
Under atmospheric conditions, Gale et al.26 theoretically investigated water-mediated peptide bond formation and predicted that glycine should persist in the atmosphere in abundant hydrated form. Recently, Harold et al.27 provided a plausible pathway for peptide growth by modelling the early stages of polyglycine elongation with two and three glycines, under conditions pertaining to the prebiotic atmosphere. By examining different pathways and associated transition states, these authors concluded that the formation of diglycine and triglycine in atmospheric water nanoclusters in the prebiotic atmosphere is kinetically favoured through N-to-C pathways, the transition state being further stabilized by the addition of water molecules.
The aforementioned experimental results,9 together with the seminal computational work by Redondo and coworkers,22 highlight the importance of protonation on the ability of the isolated amino acids to react and form a new peptide bond once exposed to appropriate radiative excitation. In the present contribution, we examine in further details the underlying reaction pathways leading to the formation of the diglycine and triglycine products from the homo (glycine–glycine) and hetero or mixed (glycine–diglycine) dimers, in protonated form. The various protonation sites among the reaction partners enable different reaction pathways, strongly related to the concerted and stepwise mechanisms identified in ref. 22. Here we build upon this computational observation to guide our search of new pathways for the more complex case involving the mixed glycine–diglycine dimer.
The paper is organized as follows. In the next section, we briefly describe the quantum chemistry methodology employed to characterize the energy minima, transition states, and reaction pathways connecting the isolated amino acids and mixed dimers to the diglycine and triglycine peptides, respectively. The role of protonation is then discussed first in general by comparing to the unprotonated case, then more specifically on the important role played by the protonation site on the likely reaction pathway and associated barrier. We finally summarize and conclude by suggesting future research directions to extend the present effort.
Methods
Density-functional theory (DFT) was used as our main computational approach to determine the reaction pathways for pure and mixed glycine dimers in bare and protonated forms, and characterize all the relevant stationary points, namely endpoints, intermediate minima, and connecting transition states. As we are dealing with isolated molecules in the gas phase, the temperature was kept to 0 K, and energies were only corrected with zero-point contributions evaluated at the harmonic level of theory, with empirical anharmonic corrections under the form of a simple scaling factor.More precisely, the M06-2X28 exchange–correlation functional was chosen owing to its good performance for describing non-covalent interactions,29 together with the 6-311G++(2df,p) basis set which includes polarization functions and diffuse orbitals, allowing an accurate description of hydrogen-bonded and cationic systems.30 All stationary points were checked from the number of strictly positive normal mode frequencies, and the zero-point harmonic energy was scaled by the factor 0.9663 appropriate for the chosen method.31 For each transition state identified, intrinsic reaction coordinates (IRC) calculations were successfully carried out to determine the two local minima connected by this transition state. The transition state configurations were initially explored at a lower level of theory, namely Hartree–Fock with the 6-21G basis set, before being refined at the higher level DFT/M06-2X/6-311G++(2df,p). Similarly, the IRC calculations were started at this low level of theory optimizing the minima at higher level. The entire conformer space has not been explored in detail in the reaction paths. Connections between successive minima in the reaction pathways were not established by IRC, as we focus mainly on peptide bond formation and the related water elimination process, which are the rate-limiting steps in the reaction.32,33 Our search also produced additional transition states associated with proton transfer or internal rotations, usually involving relatively low energy barriers, thus easily accessible and not affecting those rate-limiting steps. In particular, the role of the terminal nitrogen will be particularly scrutinized as it is the most stable protonation site found for both the glycine and the diglycine reagents, in agreement with the higher basicity of primary amines relative to carboxylic acids (see ref. 22) and references therein. The geometrical structure of all relevant stationary points discussed below are provided as ESI.†
Results and discussion
In line with earlier studies,22 we explored stepwise and concerted mechanisms for peptide bond formation. In all the mechanisms discussed below, the proton is always located on the terminal nitrogen. Fig. 1 provides a schematic overview of the three scenarios that arise when these constraints are considered. Note that, unlike the stepwise mechanism, the concerted mechanism can occur under two distinct forms: one where the proton is present at the reaction site and another one where it is absent. The stepwise mechanism is characterized by the formation of an intermediate complex. The color-coding indicated by the arrows in the figure will be used consistently throughout the article. | ||
| Fig. 1 Schematic representation of the three types of reactions studied in this work: stepwise mechanism (blue/pink arrows), concerted without H+ (green arrow), and concerted with H+ (red arrows). | ||
Fig. 2 shows the two main reaction pathways associated with peptide bond formation between two glycine molecules, one of them being protonated, with characteristic energies as obtained from our DFT calculations. All energies are given relative to the dimer in its lowest-energy conformer, and the evaporation energies corresponding to placing the monomers at infinite separation are also reported, with the structures of neutral34,35 and protonated36 glycine being taken from the appropriate literature. The two pathways represented in this figure both involve water elimination and can be described as concerted or stepwise, in accordance with the earlier analysis of Redondo and coworkers.22
 | ||
| Fig. 2 Reaction pathways leading to the formation of protonated diglycine from neutral and protonated glycine monomers, as computed at the DFT/M06-2X/6311G++(2df,p) level of theory at 0 K. The reported energies are corrected for zero-point energy in the harmonic approximation, frequencies being scaled by the factor 0.9663, and given relative to the ground state energy of the H+(Gly)2 dimer ion. The vertical black arrows highlight the evaporation energies needed to dissociate the system into infinitely separated H+Gly and Gly fragments. The structures associated with all stationary states are schematically shown next to their energy level, green arrows pointing at the specific protonation site. | ||
IRC calculations were performed on both sides of the transition states, producing starting points for the minima that were subsequently reoptimized using the DFT method. The mechanism associated with the lowest barrier (TS′ 1 at 1.82 eV) is concerted and involves a reaction between the protonated amine group (–NH3+) and the carboxylic acid group (–COOH). This transition state leads to the formation of the dipeptide with the excess proton on the amide group (1.49 eV).
The second mechanism has a higher barrier (TS′ 2 at 2.00 eV) is stepwise and has the neutral amine group react with the carboxylic acid group to form an intermediate complex. Water elimination takes place subsequently from this complex, leading to a protonated diglycine conformer that is more stable (0.71 eV) that the previous structure originating from the concerted process. Both mechanisms are associated with barriers that lie higher than the evaporation energy required to separate the two reaction partners (1.32 eV), confirming that they need to be significantly activated by external excitation to be even observable.
The two reaction pathways differ in terms of the proton involvement. For the concerted mechanism, the excess proton is a direct actor involved in the formation of the peptide bond, while in the stepwise mechanism it remains as a relatively distant spectator. During the second step of the stepwise mechanism, the proton is transferred and this helps in the elimination of a water molecule, consistently with the lowest barrier found in our calculations. These two transition states were already investigated by Redondo et al.22 and the energies reported in Fig. 2 are in good agreement with their results, our TS for the concerted mechanisms being higher by 0.17 eV due to differences in the underlying theory. A better agreement was reached by conducting more accurate single point CCSD(T) calculations on top of the DFT structures, still employing the 6-311++G(2df,p) basis set, eventually reducing the error to less than 10 meV.7 The slight differences in the quantum chemical method also naturally produce minor deformations in the structures of the minima.
The reaction pathways leading to peptide elongation from the mixed protonated glycine dimer H+Gly2(Gly) were approached similarly as in the homodimer case, inspired by the results previously discussed. In particular, we examined the role of the protonation site on the reactants, namely by assuming that either the glycine or the diglycine partners were carrying the excess proton, and further imposing the protonation site to be at or away from the peptide bond reaction site.
Fig. 3 shows the most significant concerted pathways identified when the proton is initially localized on the dipeptide, (H+Gly2)(Gly). Here again, all energies are given relative to the ground state energies of the dimer in its lowest-energy conformers (see Fig. S1 of the electronic ESI,† for a comparison between different conformers), and the evaporation energies corresponding to placing the monomers at infinite separation are also reported.
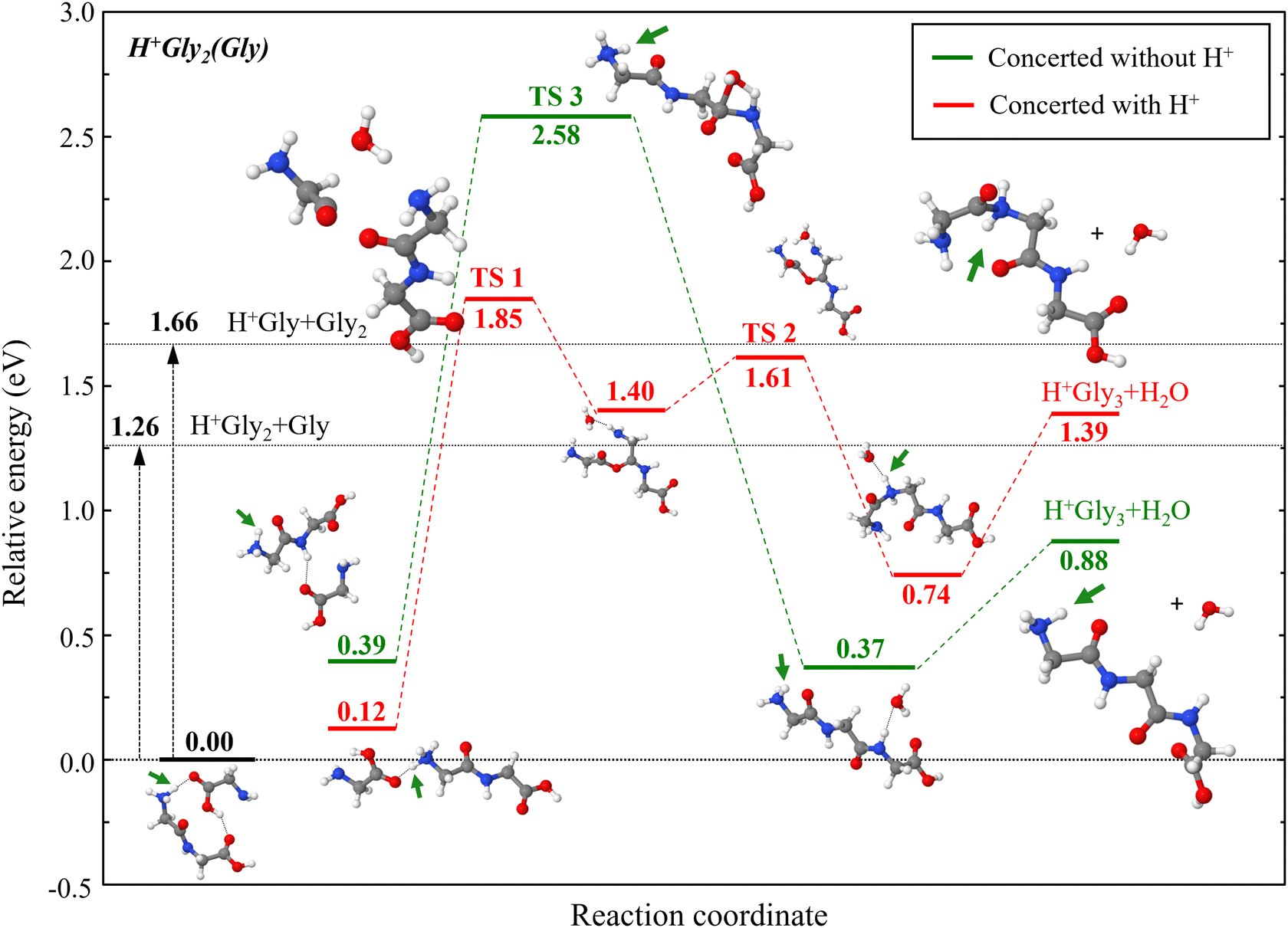 | ||
| Fig. 3 Concerted reaction pathways leading to the formation of protonated triglycine from mixed dimers of glycine with protonated diglycine, as computed at the DFT/M06-2X/6311G++(2df,p) level of theory at 0 K. The reaction in which the protonation site is on the newly formed peptide bond is shown in red, the other reaction not directly involving the excess proton being depicted in green. The reported energies are corrected for zero-point energy in the harmonic approximation, frequencies being scaled by the factor 0.9663, and given relative to the ground state energy of the H+Gly2(Gly) dimer ion. The vertical black arrows highlight the evaporation energies needed to dissociate the system into infinitely separated fragments. The structures associated with all stationary states are schematically shown next to their energy level, green arrows pointing at the specific protonation site. | ||
One particular pathway, involving as its rate-determining step a transition state denoted as TS 1 in Fig. 3, directly involves the excess proton in the newly formed peptide bond and therefore resembles TS′ 1 obtained for the homodimer. The IRC calculations reveal an intermediate minimum where a water molecule connects the two nitrogens, as well as a second transition state (TS 2) that is lower in energy than TS 1. The triglycine product formed by this pathway is protonated on the nitrogen of the peptide bond.
In the second pathway shown in Fig. 3, the excess proton is not involved in the newly formed peptide bond, its transition state (TS 3) being unsurprisingly similar to TS′ 2 of the homoglycine case (see Fig. 2). However, unlike TS′ 2, the water molecule is readily formed in TS 3, in a concerted way according to Redondo et al.22 The tripeptide thus formed is protonated on the terminal amine group. Strikingly, the energy difference in the rate-limiting transition states (1.85 eV vs. 2.58 eV) correlate with the reaction site involving the excess proton either directly, or as a distant spectator, respectively. The protonated tripeptides formed along the two pathways also differ from one another, a more stable conformer (0.88 eV vs. 1.39 eV) being obtained if the proton is a spectator. In addition, it is important to notice that, for the mixed system too, the two energy barriers for the formation of the tripeptide again lie above the evaporation energy corresponding to the infinite separation between the glycine and diglycine partners, independently of the protonation site, confirming the need for a significant excitation energy to trigger such reactions.
Fig. 4 shows the concerted pathways obtained when the proton is initially localized on the glycine monomer, (Gly2)(H+Gly). The path associated with the transition state denoted as TS 5 has the excess proton bound to the reaction site, very similarly as TS 1 and TS′ 1. The triglycine conformer thus formed keeps the proton close to the peptide bond. Its energy is higher than that determined for the pathway rate-limited by TS 4 for which the excess proton lies away from the reaction site of the newly formed peptide bond, and remains so along the entire path. In this case, the triglycine obtained is protonated at the nitrogen terminus. For this system, the barrier for peptide bond formation is lower when the excess proton lies away from the reactive site, albeit by a reduced magnitude (TS 4 at 2.14 eV vs. TS 5 at 2.27 eV). This contrasts with the results discussed in Fig. 3 assuming the dipeptide initially carries the excess proton. This is because the proton on the amine group of glycine can form a hydrogen bond while the carboxylic group forms the peptide bond.
 | ||
| Fig. 4 Same as Fig. 3, with the glycine monomer carrying the excess proton instead of diglycine, in the glycine–diglycine heterodimer reaction. | ||
The four reaction pathways presented so far for the protonated mixed glycine–diglycine system all fall into the concerted mechanism category, according to the terminology followed by Redondo et al.22 Two other transition states, TS 6 and TS 8, were obtained by rotating the glycine carboxylic group by 180° in TS 4 and TS 3 respectively, and are found to be part of stepwise mechanisms similar to TS′ 2 of the homodimer case (see Fig. 2). They are presented in Fig. 5. These two stepwise mechanisms only differ by which reactant is protonated. Starting from protonated diglycine, two transition states are found, very close in energy but the highest of which denoted as TS 9 lying 2.36 eV above the common reference. This pathway leads to a triglycine conformer protonated on a carboxylic acid oxygen. Protonating instead the glycine monomer, the reaction threshold given by TS 6 is lower (1.98 eV) owing to the formation of a hydrogen bond already identified in TS 4. Triglycine is also protonated on the carboxylic acid oxygen with this pathway. In the two stepwise mechanisms, and despite not being directly involved in the peptide bond formed from (Gly2)(H+Gly), the protonation site in the reactant significantly influences the energy of the transition state, thus the accessibility of the tripeptide product. These stepwise mechanisms have a lower activation energy than their concerted counterparts.
 | ||
| Fig. 5 Stepwise reaction pathways leading to the formation of protonated triglycine from mixed dimers of glycine with protonated diglycine (blue) and protonated glycine with diglycine (pink), as computed at the DFT/M06-2X/6311G++(2df,p) level of theory at 0 K. The reported energies are corrected for zero-point energy in the harmonic approximation, frequencies being scaled by the factor 0.9663, and given relative to the ground state energy of the H+Gly2(Gly) dimer ion. The vertical black arrows highlight the evaporation energies needed to dissociate the system into infinitely separated fragments. The structures associated with all stationary states are schematically shown next to their energy level, green arrows pointing at the specific protonation site. | ||
Fig. 6 finally summarizes and simplifies all pathways leading to protonated triglycine from mixed glycine–diglycine dimers, only keeping the initial protonated mixed dimer, the limiting step transition state, and specific triglycine conformers resulting from each corresponding pathway. These six reactions can be sorted into three main groups depending on where does the excess proton lie on the separated reaction partners, highlighted in three distinct colours in Fig. 6. We thus distinguish one first group with the excess proton lying far away from the reaction site (TS 3 and TS 9), another group with the proton on the glycine monomer (TS 4 and TS 6), and finally a group with the proton at the site of the newly formed peptide bond (TS 1 and TS 5). The latter group is similar to the transition state obtained for the reaction between glycine and protonated glycine (TS′ 1). While the transition states in these groups generally correlate with lower energies as the excess proton moves away from the reaction site, one minor exception should be noted with TS 5 at 2.27 eV involving the protonated glycine monomer. Keeping in mind that in all cases the evaporation energies of 1.26 and 1.66 eV leading to the dissociation into the separated monomers are lower than the chemical reaction barriers, it is clear that peptide elongation can only occur with a sufficiently strong excitation, consistently with the experimental observations of ref. 9.
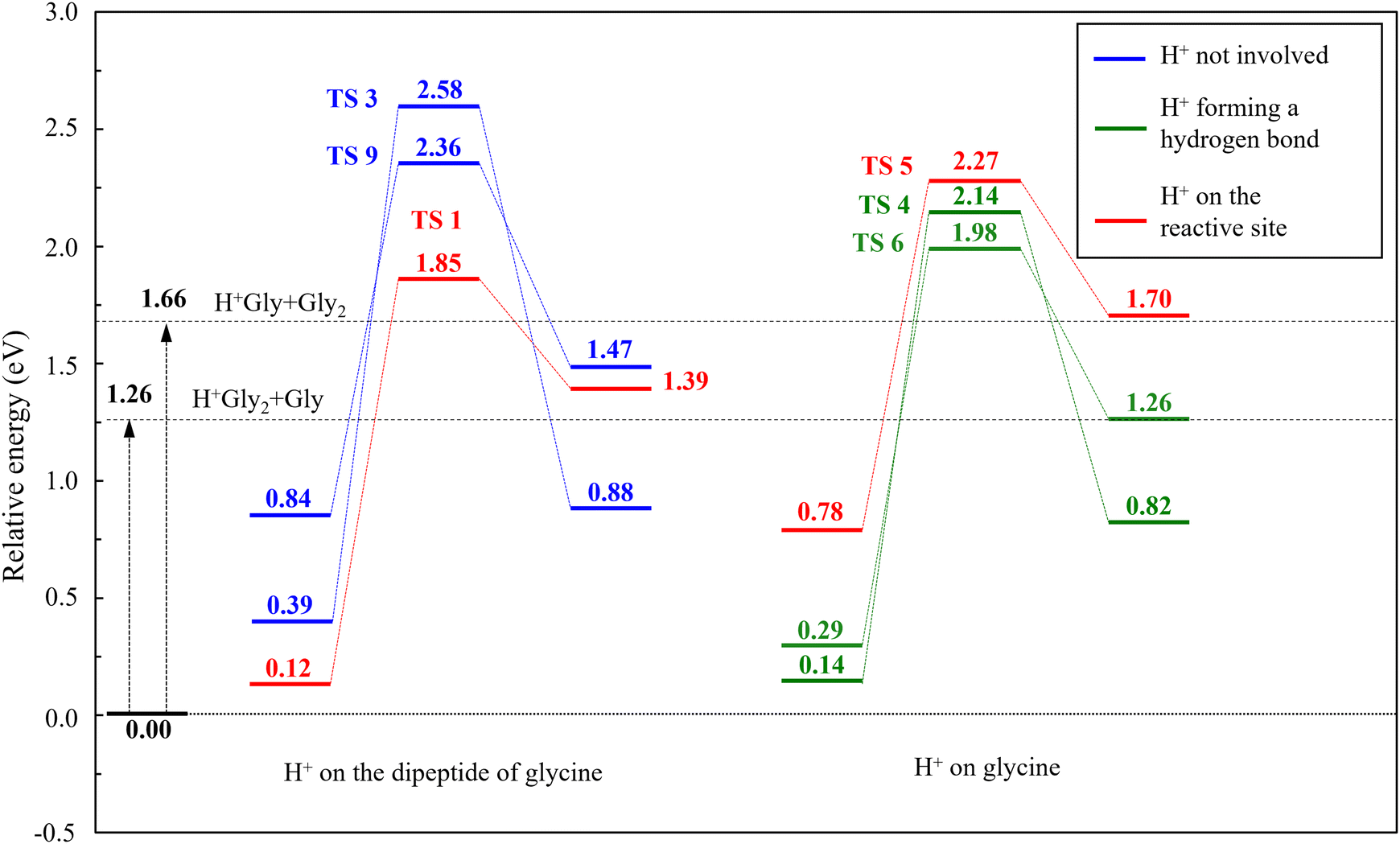 | ||
| Fig. 6 Simplified reaction profiles summarizing the paths leading to protonated triglycine from the mixed dimer H+Gly2(Gly) in the cases where diglycine (left) or the glycine monomer (right) carries the excess proton. Only the energies of the initial, rate-determining transition and final states are shown. | ||
Conclusions
Different reaction pathways for the peptide bond formation from homo and mixed protonated dimers of glycine were theoretically determined and characterized using density-functional theory. By extending earlier seminal work by Redondo and coworkers22 on the simple glycine case to the mixed dimer system with a diglycine partner, the influence of the protonation site of the reactants on the transition state energies could be investigated in broader details. Our results notably show that the diversity of reaction pathways is greater in the mixed glycine–diglycine dimer, H+Gly2(Gly), compared to the homodimer H+(Gly)2.If the dipeptide is protonated away from the reaction site, peptide elongation is a rather energy-demanding process similar to that found for the neutral reaction between two glycine amino acids. Conversely, if the excess proton is bound to the reaction site leading to a new peptide bond, the resulting energy barrier is significantly reduced. Intermediate cases involving protonated glycine can also lead to transition states in which the excess proton is not on the reactive site, but still actively participates to the reaction by stabilizing the dimer through a hydrogen bond.
The present computational work indicates how protonation in general, and the specific protonation site in particular, can both influence the energy barriers for peptide elongation. Low-energy pathways such as intramolecular proton transfer were not investigated but are also expected to take place, likely playing a role on the protonation site. The pathways presented here outline the reaction process and come as support for the experimentally observed production of protonated triglycine peptides from collision-induced excitation of mixed diglycine–glycine dimers.9 In particular, as was emphasized in our calculated reaction pathways, the formation of a new peptide bond competes with the evaporation into the infinitely separated partners, a process which is barrierless and thermodynamically much favoured. The excess energy provided by the collision in the experiment is therefore needed to trigger the reaction kinetically. In this respect, it would be interesting to investigate the dynamical effects arising from such processes using in particular velocity map imaging, as partly explored on the eliminated water molecules in our earlier experimental work.9 From the computational perspective, additional insight could be provided by going beyond the static, quantum chemical picture pursued here by considering explicitly dynamical effects, e.g. by performing ab initio molecular dynamics simulations such as those pioneered by Saitta and coworkers37 or, more recently, by Sangiovanni et al.38
Author contributions
Léo Lavy: investigation, formal analysis, visualization, writing – review & editing. Denis Comte: investigation, formal analysis, writing – review & editing. Florent Calvo: conceptualization, writing – review & editing, Bernadette Farizon: conceptualization, investigation, supervision, writing – review & editing. Michel Farizon: conceptualization, data curation, investigation, supervision, writing – review & editing, Tilmann Märk: conceptualization, supervision, writing – review & editing.Data availability
A part of data supporting this article have been included as part of the ESI.† All the other data that led to the present findings are available upon request to the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the LABEX Lyon Institute of Origins (ANR-10-LABX-0066) within the program “Investissements d′Avenir” (ANR-11-IDEX-0007) of the French government operated by the National Research Agency (ANR) and to the University of Innsbruck [Leopold-Franzens-Universität Innsbruck (LFU)] for their financial support. They acknowledge the Institut de Physique des 2 Infinis de Lyon (IP2I) for technical support, particularly Raphaël Fillol and Peter Calabria. Calculations were supported by the Centre de Calcul de l′Institut National de Physique Nucléaire et de Physique des Particules (CCIN2P3).Notes and references
- M. Frenkel-Pinter, M. Samanta, G. Ashkenasy and L. J. Leman, Chem. Rev., 2020, 120, 4707 CrossRef CAS.
- C. R. Arumainayagam, E. Herbst, A. N. Heays, E. Mullikin, M. Farrah and M. G. Mavros, in Prebiotic Photochemistry: From Urey–Miller-like Experiments to Recent Findings, ed. F. Saija and G. Cassone, The Royal Society of Chemistry, 2021, pp. 9–36 Search PubMed.
- C. Chyba and C. Sagan, Nature, 1992, 355, 6356 CrossRef.
- P. Redondo, C. Barrientos and A. Largo, Astrophys. J., 2014, 793, 32 CrossRef.
- H. Wincel, R. H. Fokkens and N. M. M. Nibbering, Rapid Commun. Mass Spectrom., 2000, 14, 135 CrossRef CAS.
- S. Lee, S. J. Valentine, J. P. Reilly and D. E. Clemmer, J. Am. Chem. Soc., 2011, 133, 15834 CrossRef CAS PubMed.
- A. Singh, S. Kaur, J. Kaur and P. Singh, Rapid Commun. Mass Spectrom., 2014, 28, 2019 CrossRef CAS PubMed.
- P. Rousseau, et al. , Nat. Commun., 2020, 11, 1 CrossRef.
- D. Comte, et al. , J. Phys. Chem. A, 2023, 127, 3 CrossRef.
- M. Nihamkin, A. Isaak, A. Albeck, Y. Mastai and Y. Toker, J. Phys. Chem. Lett., 2020, 11, 23 CrossRef PubMed.
- O. Licht, et al. , Angew. Chem., 2023, 62, 15 Search PubMed.
- L. Tiefenthaler, J. Kočišek and P. Scheier, Eur. Phys. J. D, 2020, 74, 5 CrossRef.
- K. Kvenvolden, et al. , Nature, 1970, 228, 5275 CrossRef.
- A. Shimoyama and R. Ogasawara, Origins Life Evol. Biospheres, 2002, 32, 2 Search PubMed.
- K. Altwegg, et al. , Sci. Adv., 2016, 2, 5 Search PubMed.
- C. Favre, et al. , Astrophys. J., 2018, 862, 1 CrossRef.
- F. Hoyle and N. C. Wickramasinghe, Astrophys. Space Sci., 1999, 268, 1/3 Search PubMed.
- P. Gorai, et al. , Astrophys. J., 2020, 895, 2 CrossRef.
- M. N. Drozdovskaya, E. F. van Dishoeck, M. Rubin, J. K. Jørgensen and K. Altwegg, Mon. Not. R. Astron. Soc., 2019, 490, 1 CrossRef.
- E. Herbst, Chem. Soc. Rev., 2001, 30, 168 RSC.
- E. Herbst and E. F. Van Dishoeck, Annu. Rev. Astron. Astrophys., 2009, 47, 1 CrossRef.
- P. Redondo, H. Martínez, Á. Cimas, C. Barrientos and A. Largo, Phys. Chem. Chem. Phys., 2013, 15, 31 RSC.
- L. Feketeová, et al. , Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 45 CrossRef.
- T. Salbaing, et al. , J. Chem. Phys., 2024, 160, 094301 CrossRef CAS PubMed.
- J. H. Jensen, K. K. Baldridge and M. S. Gordon, J. Phys. Chem. A, 1992, 96, 8340 CrossRef CAS.
- A. G. Gale, T. T. Odbadrakh, B. T. Ball and G. C. Shields, J. Phys. Chem. A, 2020, 124, 4150 CrossRef CAS PubMed.
- S. E. Harold, S. L. Warf and G. C. Shields, Phys. Chem. Chem. Phys., 2023, 25, 28517 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 1–3 Search PubMed.
- E. G. Hohenstein, S. T. Chill and C. D. Sherrill, J. Chem. Theory Comput., 2008, 4, 12 CrossRef.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 3 Search PubMed.
- J. P. Merrick, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 45 CrossRef PubMed.
- B. Paizs, I. P. Csonka, G. Lendvay and S. Suhai, Rapid Commun. Mass Spectrom., 2001, 15, 8 CrossRef.
- P. B. Armentrout, A. L. Heaton and S. J. Ye, J. Phys. Chem. A, 2011, 115, 41 CrossRef PubMed.
- A. G. Csaszar, J. Am. Chem. Soc., 1992, 114, 24 CrossRef.
- P. D. Godfrey and R. D. Brown, J. Am. Chem. Soc., 1995, 117, 2019 CrossRef CAS.
- M. Noguera, L. Rodríguez-Santiago, M. Sodupe and J. Bertran, J. Mol. Struct., 2001, 537, 307 CrossRef CAS.
- A. M. Saitta and F. Saija, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13768 CrossRef CAS PubMed.
- D. G. Sangiovanni, et al. , Phys. Chem. Chem. Phys., 2018, 20, 17751 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Conformers of the protonated dimer of dipeptide and glycine (Fig. S1) and Cartesian coordinates for the M06-2X structures of H+Gly2(Gly) pathways in Å (Table SI). See DOI: https://doi.org/10.1039/d4cp04437a |
| This journal is © the Owner Societies 2025 |