
Electrochemical protonation/deprotonation of TiNb2O7 in protic ionic liquids†
Masahiro
Shimizu
 *,
Takuya
Kawai
,
Tomonori
Ichikawa
and
Susumu
Arai
*,
Takuya
Kawai
,
Tomonori
Ichikawa
and
Susumu
Arai
Department of Materials Chemistry, Faculty of Engineering, Shinshu University, 4-17-1 Wakasato, Nagano, 380-8553, Japan. E-mail: shimizu@shinshu-u.ac.jp; Fax: +81-26-269-5627; Tel: +81-26-269-5627
First published on 18th February 2025
Abstract
In recent years, there has been growing interest in rechargeable batteries utilizing protons or hydronium ions as charge carriers, driven by the rapid ionic conduction enabled by the proton-specific Grötthuss mechanism. However, the use of acidic aqueous electrolytes introduces side reactions, such as irreversible hydrogen evolution and the dissolution of active materials into the electrolyte, which are influenced by the reaction potential of the active materials. These challenges complicate the identification and development of active materials. While some combinations of Brønsted acids and bases may potentially compromise the advantages of the Grötthuss mechanism, this study successfully demonstrated the electrochemical protonation of TiNb2O7 using protic ionic liquids as electrolytes. Acetic acid (AcOH) and 1,1,1-trifluoro-N-((trifluoromethyl)sulfonyl)methanesulfonamide (HTFSA) were employed as Brønsted acids, while 1-methylimidazole (Im) and 1,8-diazabicyclo[5.4.0]-7-undecene (DBU) were utilized as Brønsted bases. Irreversible hydrogen evolution was dominant in an aqueous buffer solution consisting of citric acid and trisodium citrate. The AcOH/DBU system showed negligible charge/discharge capacities within the cut-off potential range of −1.5 to +0.25 V. In contrast, AcOH/Im and HTFSA/Im systems exhibited reversible capacities of 61 and 55 mA h g−1, respectively, during the first cycle. However, their Coulomb efficiencies were significantly low below 20%. Meanwhile, HTFSA/DBU, despite a lower reversible capacity of 40 mA h g−1 (corresponding to H0.5TiNb2O7), achieved a Coulomb efficiency exceeding 90%. Notably, it maintained an average Coulomb efficiency of 96% over 50 cycles without any capacity degradation.
Introduction
With the growing significance of storage batteries, recent years have seen active research on not only divalent (Zn2+, Mg2+)1–7 and trivalent (Al3+)8–10 metal cations but also anions, such as fluoride ions (F−),11–13 as carrier ions. On the other hand, there are not enough studies on proton/hydronium ions as charge carriers. If the fast ionic conduction specific to proton based on the Grötthuss mechanism can be utilized, rapid charge–discharge performance can be offered. However, in the case of acidic aqueous solutions, which are typical proton conductors, irreversible hydrogen or oxygen evolution occurs dominantly depending on reaction potentials of active materials, and there is a concern that proton-insertion/deinsertion into/from host materials do not proceed. From this point of view, buffer solutions14–18 are used and molecular crowding electrolytes19,20 are being actively considered. The electrolytes also contribute to suppressing the dissolution of active materials into acidic aqueous solutions. Veronica et al. succeeded in suppressing the dissolution of Ti3+ from H2Ti3O7 and improving the reversible capacity by using phosphate buffer as the electrolyte.21 The suppression of Ti3+ dissolution (generation of surface pitting) associated proton adsorption is also evident from SEM observation after electrochemical tests. On the other hand, as an attempt to adjust the reaction potential based on the crystal structure, we have focused on rutile TiO2.22 The rutile TiO2 synthesized by the hydrothermal method allows electrochemical protonation at a higher potential compared to anatase TiO2, thereby mitigating irreversible hydrogen evolution and improving cycle performance. As outlined above, the complexity of developing active materials for proton rechargeable batteries arises from side reactions such as the dissolution of active materials and irreversible hydrogen evolution. These issues make it inherently challenging to determine whether the synthesized samples can electrochemically accommodate protons.Since these undesirable side reactions predominantly occur in electrolytes using water as the solvent, this study focused on proton conduction in non-aqueous solvents, specifically exploring protic ionic liquids23,24 as electrolyte candidates. Protic ionic liquids are formed through proton transfer from Brønsted acid to Brønsted base.25 Angel et al. classified them into good and poor categories based on their ΔpKa, with a threshold of 10.26,27 Good ionic liquids exhibit high boiling points and ionic conductivities comparable to those of aprotic ionic liquids, whereas poor ionic liquids are characterized by lower boiling points and reduced ionic conductivities due to incomplete proton transfer. Protic ionic liquids have been extensively studied for their potential applications in electrochemical devices, serving as proton conductors in a new class of fuel cells28–33 and as alternatives to alkaline aqueous electrolytes in nickel-metal hydride batteries.34 N. Chaabene and J. Monnier were the first to demonstrate the phase transition of AB5-type alloys (A: rare earth, B: 3d late transition metal) into the hydride phase using the protic ionic liquid consisting of pyrrolidine (Pyrr) and acetic acid (AcOH). Recently, a pseudo-protic ionic liquid exhibiting proton conductivity facilitated by a Grötthuss-like mechanism, as proposed by Umebayashi et al.,35–38 has been reported to enhance the electrochemical protonation of VO2.39 Unlike the aqueous electrolyte (12 M H3PO4 aq.), the pseudo-protonic ionic liquid (AcOH/C1Im = 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) demonstrated remarkable cycling stability, retaining 75% of reversible capacity even after 200 cycles.
:1) demonstrated remarkable cycling stability, retaining 75% of reversible capacity even after 200 cycles.
In this study, we report electrochemical protonation/deprotonation of TiNb2O7 in protic ionic liquids. Wadsley–Roth phase TiNb2O7 with a crystallographic shear structure is a promising candidate for energy storage of Li ions.40–43 We hypothesized that TiNb2O7 could accommodate protons inside the structure and subsequently investigated its electrochemical behavior. To suppress undesirable side reactions, protic ionic liquids were employed as electrolytes. This study examines the correlation between the ΔpKa of the Brønsted acid/base, the activation energy, the protonation potential, and the reversible capacity.
Experimental section
Preparation and characterization of protic ionic liquids as electrolytes
Protic ionic liquids were prepared by mixing of the following combinations of Brønsted acid/base (Fig. 1a) in an Ar-filled glovebox: acetic acid (AcOH; FUJIFILM Wako Pure Chemical Corporation, 99%), 1,1,1-trifluoro-N-((trifluoromethyl)sulfonyl)methanesulfonamide (HTFSA; Kanto Chemical Co., Inc., 98%), 1-methylimidazole (Im; Sigma-Aldrich, 99%), and 1,8-diazabicyclo[5.4.0]-7-undecene (DBU; Sigma-Aldrich, 99%). Thermogravimetric analysis was conducted using a TG-DTA instrument (Thermo Plus TG8120, Rigaku) from room temperature to 500 °C under flowing N2 at a rate of 200 mL min−1. Differential scanning calorimetry (DSC) was performed with a calorimeter (Thermo Plus EVO2, Rigaku) using a sealed aluminum ampoule at a cooling/heating rate of 10 °C min−1. Ionic conductivity was evaluated by an electrochemical impedance analyzer (SP-300; Bio-Logic Science Instruments) using an airtight four-probe cell. | ||
| Fig. 1 (a) Chemical structures of Brønsted acid and base used in this study. (b) Thermogravimetric traces, (c) differential scanning calorimetry curves, and (d) ionic conductivities of (AcOH)x(Im)1−x, (HTFSA)x(Im)1−x, (AcOH)x(DBU)1−x, and (HTFSA)x(DBU)1−x (x = 0.1, 0.2, 0.3, 0.4, 0.5). | ||
Synthesis of TiNb2O7 and electrochemical tests
TiNb2O7 was synthesized by the hydrothermal methods, as detailed below.42 Niobium pentachloride (NbCl5; Sigma-Aldrich, 99%) was dissolved in ethanol and mixed with titanium (IV) oxysulfate solution (15 wt% TiOSO4 in dilute sulfuric acid; Sigma-Aldrich, 99.99%) in a stoichiometric ratio. Ammonia solution (28–30% purity; Kanto Chemical Co., Inc.) was then added dropwise until the pH reached 8. The mixture was transferred to a hydrothermal synthesis vessel and heat-treated at 160 °C for 5 h. The resulting precipitates were washed with deionized water and dried under vacuum to obtain amorphous nano-sized precursors. The target sample was synthesized by heating the precursors at 1000 °C for 30 min in air.Electrochemical protonation/deprotonation of the TiNb2O7 was studied using slurry-based composite electrodes as the working electrode. 80 wt% TiNb2O7 powder, 10 wt% acetylene black as the conductive additive, and 10 wt% polyvinylidene (PVDF) as the binder with N-methyl-2-pyrrolidone (NMP) were mixed and uniformly casted onto a Ti current collector (thickness: 18 μm) using a doctor blade, then dried under vacuum at 120 °C for 12 h. Three-electrode cells were composed of the working electrode, an activated carbon electrode as the counter electrode, and an Ag/AgCl wire as the reference electrode immersed in a saturated NaCl aqueous solution. The loading mass of the active material and activated carbon were 1.0 and 4.5 mg cm−2, respectively.
Results and discussion
Typical acidic aqueous solutions as proton/hydronium-ion conducting electrolytes have high conductivity, while low thermal stability is a concern. Fig. 1b shows thermogravimetric curves for mixtures of Brønsted acid (AcOH, HTFSA) and Brønsted base (Im, DBU) under an N2 atmosphere. When the Brønsted acid is AcOH, there is no change in thermal stability at any concentration ratio regardless of the Brønsted base. Considering the respective boiling points, the weight loss observed in these systems using AcOH as the conjugate acid is evaporation rather than decomposition. In contrast, the use of HTFSA dramatically enhanced the thermal resistance with decomposition temperatures (10 wt% loss) of 375 and 435 °C for Im and DBU, respectively, at a molar ratio of 5:5. This is attributed to stabilization as a result of proton transfer from HTFSA to Im or DBU, respectively, through ionization.44–46 Based on the results of thermogravimetric analysis, the system with AcOH as the Brønsted acid is classified as a pseudo-protic ionic liquid,36 while the HTFSA system is a protic ionic liquid.44 For x = 0.4 or less in the case of using HTFSA, the thermal resistance was improved with increasing fraction of HTFSA, though not as much as for equimolar ratios. This suggests partial ionization associated with proton transfer. In each of the four electrolytes, the ΔpKa are 2.4 for AcOH/Im, 8.7 for AcOH/DBU, 17.1 for HTFSA/Im, and 23.4 for HTFSA/DBU.25,26,36,45 Watanabe et al. reported that the relationship between ΔpKa and thermal stability of mixtures of DBU as the Brønsted bases and various conjugated acids, and found that mixtures with ΔpKa > 20 showed thermal stability similar to aprotic ionic liquids, which supports the above-mentioned results.45 Meanwhile, the systems of HTFSA as the Brønsted acid are not a liquid phase at room temperature at molar ratio of 5:5 regardless of the Brønsted base (Fig. 1c). In the molar ratio of 4:6 (Brønsted acid/base), with all samples showing the liquid phase at room temperature, their ionic conductivities are 2.83 for AcOH/Im, 0.04 for AcOH/DBU, 4.66 for HTFSA/Im, and 0.50 mS cm−1 for HTFSA/DBU, respectively (Fig. 1d and Table S1, ESI†).
XRD patterns (Fig. 2a) show that TiNb2O7 synthesized by the hydrothermal method is obtained as a single phase without any impurities. Compared to the sample prepared by the calcination (∼1 μm), the hydrothermal treatment provides smaller particle sizes of 200 nm as a primary particle (Fig. 2b). In our previous studies on rutile TiO2,22 we observed that the extent of protonation decreases with increasing particle size, with fine particles exhibiting a greater protonation capacity. Building on this prior knowledge, we selected fine TiNb2O7 synthesized via the hydrothermal method as the target material for this study. Indeed, we confirmed that the TiNb2O7 particles prepared by the solid-state synthesis showed only negligible charge–discharge capacity (Fig. S1, ESI†).
 | ||
| Fig. 2 (a) XRD patterns and (b) SEM images of TiNb2O7 synthesized by different methods for comparison, the pattern and sem image of TiNb2O7 prepared by calcination is also shown. | ||
To explore the electrochemical protonation behavior of TiNb2O7 at room temperature, electrolytes with a molar ratio of 4:6 in which proton transfer occurs to be ionized partially were used (Fig. S2 and S3, ESI†). Note that AcOH/Im with 4:6 molar ratio is classified as pseudo protic ionic liquid as reported by Umebayashi et al. In the Raman spectra of the mixture, dissociated AcO− was not detected in the AcOH/Im system, but in the AcOH/DBU system.36 Comparing the cathodic limit of 1 mol dm−3 (M) AcOH on a Pt foil, the reductive decomposition potentials for all electrolytes were extended (Fig. 3a). Replacing imidazole with DBU in the electrolyte significantly improved the resistance to reduction. Interestingly, the anodic limits are different for systems with the same TFSA anion, which probably reflects the strength of the interaction between the TFSA anions and the protonated cations. In the pseudo protic ionic liquid of AcOH/Im, a reduction current below −0.75 V and a corresponding response on the oxidation side were observed, suggesting proton insertion/deinsertion into/from TiNb2O7 (Fig. 3b). On the other hand, the presence of AcO− on the Raman spectrum of AcOH/DBU even in the molar ratio of 4:6 (Fig. S3, ESI†) indicates that DBU is protonated, but the redox reaction hardly proceeded. This is because the stability of the protonated cation is higher in DBU than in Im, and protons are less likely to be transferred to TiNb2O7. When HTFSA was used as the Brønsted acid, the reversibility was improved from 33% to 69% by using DBU as Brønsted base compared to Im. However, the reduction/oxidation responses associated with proton-insertion/deinsertion were decreased, and the onset potential of protonation was also shifted to the negative side. These findings highlight that the combination of Brønsted acids/bases is a highly important factor in regulating reaction potentials. As an overall trend, as the cathodic limit on the electrolytes becomes more negative, the protonation potential for the TiNb2O7 also shifted in a more negative direction. Fig. 3c displays Nyquist plots of the TiNb2O7 electrodes in the electrolyte of HTFSA/DBU. One distinct semicircle was recognized below −1.35 V, and the dimension of the semicircle became smaller at −1.40 V, which is assigned to charge transfer resistance (Rct) involved with protonation. The lack of semicircle due to interfacial resistance, as observed in a graphite electrode used in Li-ion batteries, suggests that the solid electrolyte interphase (SEI) is either not present or has negligible resistance in the HTFSA/DBU system. The temperature dependence of Rct was measured from 298 to 323 K to examine the kinetics of protonation. The inverse of the charge transfer resistances as a function of reciprocal temperature obeyed the following Arrhenius equation (Fig. 3d):47
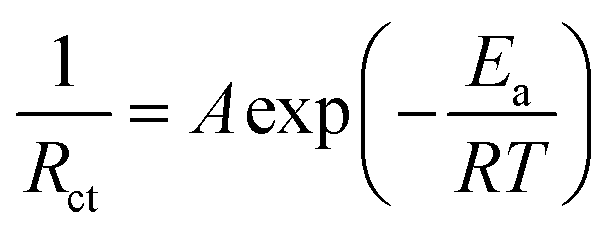 | (1) |
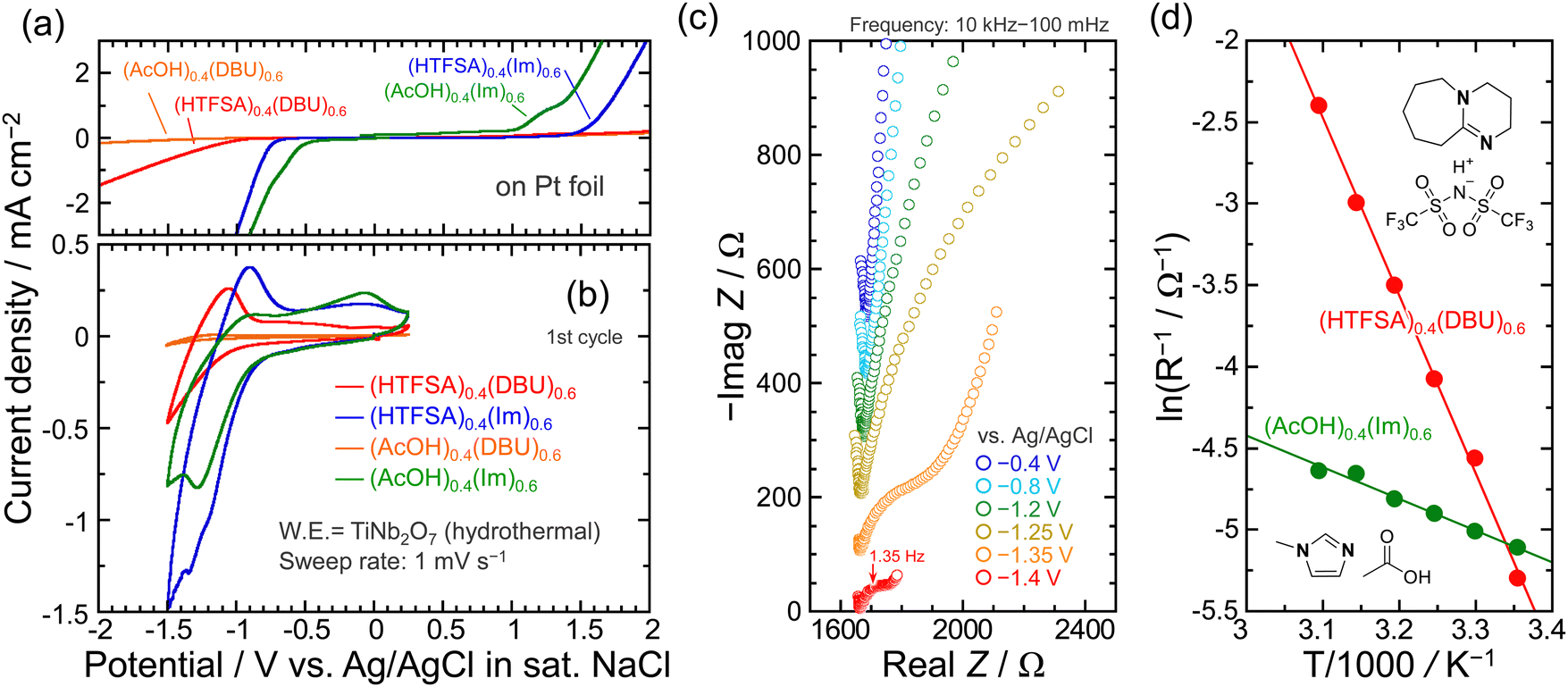 | ||
| Fig. 3 (a) Linear sweep voltammograms of a pt foil and (b) cyclic voltammograms of TiNb2O7 (hydrothermal) electrodes in protic ionic liquids (electrolytes) at a sweep rate of 1 mV s−1. (c) Nyquist plots of the TiNb2O7 electrodes. (d) Temperature dependence of the inverse of charge transfer resistances (1/Rct). In the nyquist plots, each spectrum is shifted by 100 Ω toward −Z-axis for clarity. Activation energies were estimated from the slopes. | ||
Fig. 4a exhibits galvanostatic charge−discharge profiles of the TiNb2O7 electrodes at a current density of 387 mA g−1 in the potential range between −1.5 and +0.25 V vs. Ag/AgCl. At this stage, we have not yet determined the theoretical capacity of TiNb2O7. The current density of 387 mA g−1 was chosen based on this theoretical capacity when TiNb2O7 is used as the negative electrode in Li-ion batteries.40–43 When the aqueous-based buffer solution consisting of 1 M citric acid and 1 M trisodium citrate was applied, the potential did not reach the lower cut-off limit and remained at ca. 1 V. After the charge capacity reached 400 mA h g−1, the switch to discharge process resulted in a reversible capacity of 35 mA h g−1. Most of the charge capacity is attributed to the consumption for hydrogen evolution as a side reaction. As inferred from the results of the CV measurements, the AcOH/DBU system provided little charge/discharge capacity. The stagnation of potential was also observed in AcOH/Im and HTFSA/Im and should be due to the decomposition of electrolytes. Considering the activation energy for protonation of TiNb2O7 in the AcOH/DBU system, it should exhibit a charge–discharge capacity comparable to that of HTFSA/Im. However, it shows almost no electrochemical activity. One possible explanation is the formation of a surface layer on TiNb2O7 due to the decomposition of the electrolyte. However, at this stage, we have not yet identified the underlying cause of the uniquely inactive charge–discharge behavior of AcOH/DBU. Nevertheless, AcOH/Im and HTFSA/Im showed 61 and 55 mA h g−1, respectively at the first cycle (Fig. 4b). After 50 cycles, the deprotonation capacities are 32 and 65 mA h g−1 for AcOH/Im and HTFSA/Im, respectively. In both systems, the Coulomb efficiency was notably low, indicating the accumulation of electrolyte-decomposition products on the surface of TiNb2O7. The differences in the capacity retention observed after 50 cycles are presumed to originate from the composition of the surface layers; however, this aspect was not further examined in this study. On the other hand, at the first cycle, HTFSA/DBU yielded a relatively smaller reversible capacity of 40 mA h g−1 compared to the two mentioned above but demonstrated a higher Coulomb efficiency of 90%. Furthermore, HTFSA/DBU exhibited stable cycling performance up to 50 cycles without any capacity loss, maintaining an average Coulomb efficiency of 96%. Based on the obtained reversible capacity, the stored protons are 0.5 moles per TiNb2O7. We attempted to store protons by setting the lower cut-off potential to the negative side of −1.7 V (Fig. S4, ESI†). The reversible capacity at the first cycle increased to 64 mA h g−1, while the Coulomb efficiency decreased to 17%. For the electrolytes except AcOH/DBU, the protonation potential shifted to the negative side as the ΔpKa is larger. The combination of Brønsted acid and Brønsted base, defined by their ΔpKa, serves as a key factor in tuning the protonation potential of the active material. The obtained results should greatly contribute to the design of electrolytes for proton rechargeable batteries. Some combinations of Brønsted acid/base lose the advantage of fast proton conduction by Grötthuss mechanism, the use of protic ionic liquids allows proton storage in active materials that could not function in aqueous electrolytes due to dominant hydrogen evolution.
 | ||
| Fig. 4 (a) Galvanostatic charge/discharge (protonation/deprotonation) curves and (b) dependence of reversible capacities and coulombic efficiencies on cycle numbers for TiNb2O7 (hydrothermal) electrodes at a current density of 387 mA g−1 in the cut-off potential between −1.5 and +0.25 V vs. Ag/AgCl in sat. NaCl. For comparison, the electrochemical behavior in the aqueous buffer solution of 1 M citric acid and 1 M trisodium citrate is also shown. | ||
Conclusions
This study demonstrated the electrochemical protonation/deprotonation of TiNb2O7 in protic ionic liquids. The relationship between ΔpKa and the activation energy for protonation of TiNb2O7 was examined using four different combinations for Brønsted acid/base. AC impedance measurements revealed an activation energy of 16 kJ mol−1 in the AcOH/Im system, which exhibited the smallest ΔpKa (2.4), whereas the activation energy increased to 90 kJ mol−1 in the HTFSA/DBU system (23.4). These results suggest that a larger ΔpKa stabilizes the protonated Brønsted base, thereby hindering proton transfer to the active material. The HTFSA/DBU system achieved a high Coulombic efficiency of 90% and a deprotonation capacity of 40 mA h g−1 during the first cycle. In contrast, the aqueous-based buffer solution composed of 1 M citric acid and 1 M trisodium citrate showed dominant irreversible hydrogen evolution during charging. Moreover, the HTFSA/DBU system maintained an average Coulombic efficiency exceeding 96%, with no observable capacity loss over 50 cycles.Data availability
The data that support the findings of this study are openly available in SOAR-RD at https://soar-rd.shinshu-u.ac.jp/search/detail.html?systemId=gCkVbUkh&lang=ja&st=researcher, ref. 1.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (B) (24K01601) from the Japan Society for the Promotion of Science (JSPS).References
- S. Inoguchi, A. Kitada, K. Fukami and K. Murase, J. Electrochem. Soc., 2020, 167, 162511 CrossRef CAS.
- M. Shimizu, K. Hirahara and S. Arai, Phys. Chem. Chem. Phys., 2019, 21, 7045–7052 RSC.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., 2012, 51, 933–935 CrossRef CAS PubMed.
- A. Mitelman, M. D. Levi, E. Lancry, E. Levi and D. Aurbach, Chem. Commun., 2007, 4212–4214 RSC.
- T. Mandai and M. Watanabe, J. Mater. Chem. A, 2023, 11, 9755–9761 RSC.
- T. Hatakeyama, N. L. Okamoto, K. Shimokawa, H. Li, A. Nakao, Y. Uchimoto, H. Tanimura, T. Kawaguchi and T. Ichitsubo, Phys. Chem. Chem. Phys., 2019, 21, 23749–23757 RSC.
- K. Shimokawa, T. Atsumi, N. L. Okamoto, T. Kawaguchi, S. Imashuku, K. Wagatsuma, M. Nakayama, K. Kanamura and T. Ichitsubo, Adv. Mater., 2021, 33, e2007539 CrossRef PubMed.
- Y. Fang, K. Yoshii, X. Jiang, X.-G. Sun, T. Tsuda, N. Mehio and S. Dai, Electrochim. Acta, 2015, 160, 82–88 CrossRef CAS.
- M. C. Lin, M. Gong, B. Lu, Y. Wu, D. Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B. J. Hwang and H. Dai, Nature, 2015, 520, 325–328 CrossRef PubMed.
- T. Mandai and P. Johansson, J. Phys. Chem. C, 2016, 120, 21285–21292 CrossRef CAS.
- T. Yamamoto, K. Matsumoto, R. Hagiwara and T. Nohira, ACS Appl. Energy Mater., 2019, 2, 6153–6157 CrossRef CAS.
- A. Inoo, J. Inamoto and Y. Matsuo, ACS Appl. Mater. Interfaces, 2022, 14, 56678–56684 CrossRef CAS PubMed.
- Y. Ito, C. Lee, Y. Miyahara, S. Yamazaki, T. Yamada, K. Hiraga, T. Abe and K. Miyazaki, Chem. Mater., 2022, 34, 8711–8718 CrossRef CAS.
- M. Mateos, N. Makivic, Y. S. Kim, B. Limoges and V. Balland, Adv. Energy Mater., 2020, 10, 2000332 CrossRef CAS.
- N. Makivić, J.-Y. Cho, K. D. Harris, J.-M. Tarascon, B. Limoges and V. Balland, Chem. Mater., 2021, 33, 3436–3448 CrossRef.
- Y.-S. Kim, S. Kriegel, K. D. Harris, C. Costentin, B. Limoges and V. Balland, J. Phys. Chem. C, 2017, 121, 10325–10335 CrossRef CAS.
- Y. S. Kim, K. D. Harris, B. Limoges and V. Balland, Chem. Sci., 2019, 10, 8752–8763 RSC.
- S. Saeed, J. Fortunato, K. Ganeshan, A. C. T. Duin and V. Augustyn, ChemElectroChem, 2021, 8, 4371–4379 CrossRef CAS.
- S. Wu, J. Chen, Z. Su, H. Guo, T. Zhao, C. Jia, J. Stansby, J. Tang, A. Rawal, Y. Fang, J. Ho and C. Zhao, Small, 2022, 18, e2202992 CrossRef PubMed.
- K. Kawai, S. H. Jang, Y. Igarashi, K. Yazawa, K. Gotoh, J. Kikkawa, A. Yamada, Y. Tateyama and M. Okubo, Angew. Chem., 2024, e202410971 Search PubMed.
- J. Fortunato, Y. K. Shin, M. A. Spencer, A. C. T. van Duin and V. Augustyn, J. Phys. Chem. C, 2023, 127, 11810–11821 CrossRef CAS.
- M. Shimizu, D. Nishida, A. Kikuchi and S. Arai, J. Phys. Chem. C, 2023, 127, 17677–17684 CrossRef CAS.
- H. Ohno and M. Yoshizawa, Solid State Ionics, 2002, 154–155, 303–309 CrossRef CAS.
- M. Yoshizawa, W. Ogihara and H. Ohno, Electrochem. Solid-State Lett., 2001, 4, E25–E27 CrossRef CAS.
- M. Yoshizawa, W. Xu and C. A. Angell, J. Am. Chem. Soc., 2003, 125, 15411–15419 CrossRef CAS PubMed.
- W. Xu and C. A. Angell, Science, 2003, 302, 422–425 CrossRef CAS PubMed.
- J.-P. Belieres and C. A. Angell, J. Phys. Chem. B, 2007, 111, 4926–4937 CrossRef CAS PubMed.
- M. Yoshizawa and H. Ohno, Chem. Commun., 2004, 1828–1829 RSC.
- M. A. Susan, A. Noda, S. Mitsushima and M. Watanabe, Chem. Commun., 2003, 938–939 RSC.
- A. Noda, M. A. B. H. Susan, K. Kudo, S. Mitsushima, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2003, 107, 4024–4033 CrossRef CAS.
- U. A. Rana, M. Forsyth, D. R. MacFarlane and J. M. Pringle, Electrochim. Acta, 2012, 84, 213–222 CrossRef CAS.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- O. Danyliv and A. Martinelli, J. Phys. Chem. C, 2019, 123, 14813–14824 CrossRef CAS.
- N. Chaabene, J. Zhang, M. Turmine, E. Kurchavova, V. Vivier, F. Cuevas, M. Mateos, M. Latroche and J. Monnier, J. Power Sources, 2023, 574, 233176 CrossRef CAS.
- R. Kanzaki, H. Doi, X. Song, S. Hara, S. Ishiguro and Y. Umebayashi, J. Phys. Chem. B, 2012, 116, 14146–14152 CrossRef CAS PubMed.
- H. Doi, X. Song, B. Minofar, R. Kanzaki, T. Takamuku and Y. Umebayashi, Chem. – Eur. J., 2013, 19, 11522–11526 CrossRef CAS PubMed.
- H. Watanabe, T. Umecky, N. Arai, A. Nazet, T. Takamuku, K. R. Harris, Y. Kameda, R. Buchner and Y. Umebayashi, J. Phys. Chem. B, 2019, 123, 6244–6252 CrossRef CAS PubMed.
- H. Watanabe, N. Arai, H. Jihae, Y. Kawana and Y. Umebayashi, J. Mol. Liq., 2022, 352, 118705 CrossRef CAS.
- S. Park, S.-I. Nishimura, A. Kitada and A. Yamada, ACS Appl. Energy Mater., 2024, 7, 4347–4352 CrossRef CAS.
- J.-T. Han, Y.-H. Huang and J. B. Goodenough, Chem. Mater., 2011, 23, 2027–2029 CrossRef CAS.
- K. Ise, S. Morimoto, Y. Harada and N. Takami, Solid State Ionics, 2018, 320, 7–15 CrossRef CAS.
- N. Takami, K. Ise, Y. Harada, T. Iwasaki, T. Kishi and K. Hoshina, J. Power Sources, 2018, 396, 429–436 CrossRef CAS.
- K. J. Griffith, I. D. Seymour, M. A. Hope, M. M. Butala, L. K. Lamontagne, M. B. Preefer, C. P. Kocer, G. Henkelman, A. J. Morris, M. J. Cliffe, S. E. Dutton and C. P. Grey, J. Am. Chem. Soc., 2019, 141, 16706–16725 CrossRef CAS PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 208, 206–237 CrossRef PubMed.
- M. S. Miran, H. Kinoshita, T. Yasuda, M. A. Susan and M. Watanabe, Phys. Chem. Chem. Phys., 2012, 14, 5178–5186 RSC.
- M. S. Miran, H. Kinoshita, T. Yasuda, M. A. Susan and M. Watanabe, Chem. Commun., 2011, 47, 12676–12678 RSC.
- M. Shimizu, T. Koya, A. Nakahigashi, N. Urakami, T. Yamakami and S. Arai, J. Phys. Chem. C, 2020, 124, 13008–13016 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Raman spectra of protic ionic liquids; galvanostatic charge/discharge profiles of TiNb2O7 (PDF). See DOI: https://doi.org/10.1039/d4cp04651j |
| This journal is © the Owner Societies 2025 |