
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural contributions of Zn in enhancing CO2 hydrogenation to methanol over ZnxZrOy catalysts†
Zinat Zanganeh ,
Max Bols,
Parviz Yazdani,
Hilde Poelman and
Mark Saeys*
,
Max Bols,
Parviz Yazdani,
Hilde Poelman and
Mark Saeys*
Laboratory for Chemical Technology, Ghent University, Technologiepark 125, B-9052 Ghent, Belgium. E-mail: mark.saeys@ugent.be
First published on 12th December 2024
Abstract
Single-reactor CO2 conversion to light olefins via methanol is currently obstructed by the incompatible reaction conditions for the CO2 to methanol and methanol to olefin steps. The conventional Cu/ZnO–Al2O3 CO2 hydrogenation catalysts produce excessive CO and rapidly deactivate at the high temperatures preferred for methanol to olefins with zeolite or SAPO catalysts. ZnxZrOy catalysts are a promising alternative to Cu/ZnO–Al2O3. We studied ZnxZrOy with varying Zn doping levels, using XRD, XPS, H2-TPR, CO2-TPD, N2-physisorption, DRIFT, and Raman spectroscopy, along with CO2 conversion and methanol selectivity measurements, to examine structure-performance relationships in CO2 hydrogenation to methanol. The interplay between dopant concentration, calcination temperature, and crystal structure dictates the catalyst's phase composition, which correlates with catalytic performance. The pristine ZrO2 is a mixture of tetragonal and monoclinic phases. At Zn/Zr = 0.01, the tetragonal phase is dominant, while for Zn/Zr = 0.07–0.28, the cubic phase is obtained. Above Zn/Zr = 0.28, phase separation of ZnO occurs. For CO2 hydrogenation to methanol, a Zn/Zr = 0.07–0.28 performs best. Zinc addition increases catalyst surface area, pore volume, basicity, and reducibility. XPS analysis reveals zinc enrichment near the surface and the formation of Zr–O–Zn species upon Zn incorporation into ZrO2. A clear correlation between Zn content and catalyst activity is generally absent, but this relationship becomes evident in cubic-phase materials. At least in part, the relevance of zinc doping for CO2 to methanol lies in its ability to distort the structure of zirconia, creating a cubic phase, with implications for selectivity that correlate with the adsorption of CO2 and H2.
1. Introduction
Catalytic hydrogenation of CO2 to methanol (CTM) is an exothermic reaction facing slow reaction kinetics at low temperatures and low, thermodynamically limited selectivity at higher temperatures. The predominant methanol production route converts methane-derived syngas to methanol at 200–300 °C over a benchmark Cu/ZnO–Al2O3 catalyst through a CO2 intermediate, involving water–gas-shift, with additional CO2 feeding to match the net reaction stoichiometry.1 To circumvent the thermodynamic limitation and enable higher temperature conditions, a tandem process is considered with in situ methanol to olefins (MTO) conversion over a Brønsted acid catalyst, regularly a zeolite or SAPO material.2,3 The CTM-MTO tandem offers a direct selective route to C2–C3 olefins4 as an alternative to Fischer–Tropsch synthesis with limited selectivity to light olefins.5 The MTO reaction is optimized above 380 °C. This is a mismatch with the lower temperatures required for existing CTM catalysts, which are selective to CO instead of methanol at these high temperatures.6 Several alternative CTM catalysts are considered. Zinc-doped zirconium oxide (ZnxZrOy) is particularly interesting for its improved CO2 to methanol conversions and selectivity at higher temperatures, achieving 85% methanol selectivity at 320 °C.7 Other metal dopants in zirconia have also been explored, yielding promising catalysts with diverging specifications.6,8–10Most studies attribute zinc's promotional effect to its incorporation in the active sites interacting with CO2 and H2.7,11 H2-TPR measurements demonstrate that Zn addition enhances reducibility, increasing H2 consumption and shifting it to lower temperatures.12 A lowered take-off temperature for H2–D2 isotope scrambling, from 250 to 147 °C, is reported in Zn-doped versus undoped ZrO2.13 DFT calculations support this finding, indicating a low-barrier heterolytic H2 dissociation on ZnxZrOy with the formation of Zn–H.13 Zn addition may affect both surface oxygen concentration and its chemical nature. Counteracting the beneficial effect of surface oxygen in H2 activation for CTM conversion,14 oxygen vacancies have been reported as active sites for CO2 activation in this process. Through EPR and XPS measurements, several types of oxygen vacancies and their concentrations have been associated with coordinatively unsaturated Zr atoms (CUS-Zr). These surface species were correlated to CO2 adsorption and reverse water gas shift (RWGS) activity.15 However, various forms of CO2 adsorption on the ZrO2 surface are described.16–20 Among these, the insertion of CO2 into Zr–OH is associated with oxygen vacancies and CUS-Zr on the surface.17,21,22 Besides CO2 and H2 activation, combining activated CO2 and H2 on the surface to form intermediates towards methanol is required. DFT has suggested that Zn facilitates the hydrogenation of surface Zr-bound intermediates through the proximity of Zn–H surface species.13 Differences in surface intermediates and weaker CO2 adsorption have been observed in DRIFTS on ZnxZrOy compared to ZrO2.13,23 It has been suggested that formate's C–O bond cleavage is easier on asymmetric Zn–O–Zr sites than on symmetric Zr–O–Zr sites, facilitating the formation of C–H bonds and methanol synthesis.13,24
Numerous additional explanations for the Zn promotional effect are found in literature, which are not necessarily mutually exclusive. XRD results show that Zn addition contracts the ZrO2 crystal lattice, forms a solid solution, and alters ZrO2 crystal phases.12,23,25 The incorporation of ZnO nanoparticles into the ZrO2 matrix is pointed out by EXAFS and correlates with the formation of Zn–O–Zr species.11 Some researchers point at the ZnO–ZrO2 interface as the dominant active site in CO2 hydrogenation.11,23,26 Other researchers, however, hypothesize that the growth of ZnOx clusters during the reaction improves methanol synthesis by enhancing H2 dissociation.27 ZrO2 inherently contains weak Zr4+–O2− Lewis acid–base (LAB) pairs,28 whose reactivity in heterolytic H2 dissociation is influenced by the crystal phase, exposed facets, and the orientation of LAB pair sites.29,30 The introduction of Zn distorts the ZrO2 lattice, leading to changes in both bulk and surface properties.
Zn can influence the concentration, nature, and proximity of Zr–OH, surface oxygen, and CUS-Zr in various ways while also introducing Zn–H, Zn–O–Zr, Zn–O–Zn, and ZnOx motifs. Conclusive evidence for their relative contributions to CTM activity and selectivity is lacking. To offer another viewpoint to this puzzle, this study investigates the association between Zn doping and the ZrO2 crystal phase and its subsequent impact on CTM. The aspect of the crystal phase has been largely overlooked in the Zn promotional effect, in contrast to other promotor metals (Y, Mg, Ca,…).31,32 Most studies report ZrO2 transitioning to the tetragonal phase after Zn addition.11,13,22,26,33,34 Notably, a recent study emphasized the influence of the ZrO2 crystal phase on methanol synthesis, showing that the tetragonal phase achieved higher methanol selectivity and space–time yield compared to the monoclinic phase. This was attributed to well-dispersed ZnO clusters on tetragonal ZrO2, particularly under catalytic conditions.26 However, doping ZrO2 with Zn has been reported by several authors to induce a crystal structure alteration, stabilizing the cubic phase either in the bulk via coprecipitation or near the surface through impregnation.7,23 Despite these observations, the direct relationship between the cubic phase of doped ZrO2 and CTM catalysis has so far not been investigated.35–37
We prepared ZnxZrOy through coprecipitation, with Zn/Zr molar ratios ranging from 0.01 to 0.56. Zn0.19ZrOy samples are calcined at various temperatures. We measure surface area, pore volume, temperature-programmed reducibility, CO2 uptake, XRD, Raman, DRIFT and XPS spectra, and CTM catalysis to correlate structure and performance. The data indicate that the ‘one fits all’ active-site or surface structure explanations suggested in the literature are likely incomplete descriptions of the Zn promotion of ZrOx for CTM.
2. Experimental
2.1. Catalyst synthesis
ZnxZrOy samples were synthesized through coprecipitation, using nitrate precursors of zinc and zirconium, and sodium carbonate as the precipitating agent. Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, Sigma-Aldrich, purity ≥ 99%) was dissolved in a variable ratio into a 0.11 M solution of zirconium oxynitrate (ZrO(NO3)2·5H2O, Sigma-Aldrich, purity ≥ 98%) under stirring at 65 °C. Subsequently, a 0.30 M sodium carbonate solution (Na2CO3, Sigma-Aldrich, purity ≥ 99%) was added dropwise as the precipitating agent until the pH reached 9. The resulting suspension was aged for one hour at 65 °C. The precipitate was washed with deionized water until a pH of 7 was obtained. After filtration, the obtained material was dried overnight at 80 °C and then calcined for 3 hours at 500 °C under static air (heating ramp of 3 °C min−1). The dried Zn0.19ZrOy was also calcined at 550, 700, and 800 °C using the same heating ramp. The catalysts are denoted as ZnxZrOy-T, where x indicates the Zn/Zr molar ratio and T is the calcination temperature. The ZnO reference sample was synthesized using the same procedure by precipitating zinc nitrate hexahydrate with sodium carbonate, followed by aging, filtration, washing, and drying, but it was calcined at 350 °C.2.2. Catalyst characterization
X-ray diffraction (XRD) measurements of the calcined catalysts were conducted on a Siemens Diffractometer Kristalloflex D5000, applying Cu Kα radiation (λ = 0.154 nm). The powder diffraction pattern was recorded in a 2θ range of 20–70° with a step size of 0.025°. The data were analysed employing DIFFRAC.EVA V5.2 software. Raman spectra of the powdered samples were recorded on a Raman spectrograph using a KAISER optical system coupled with a 532 nm laser source. The textural properties of the as-prepared catalysts were analysed by nitrogen adsorption/desorption isotherms at −196 °C in a Micrometrics Tristar-II 3020 instrument. Before the measurements, 400 mg of the calcined samples (150–250 μm) were degassed at 350 °C for 6 hours using a Micromeritics Smart Prep device. The data were analysed using MicroActive software version 5.00. The specific surface area was determined using the Brunauer–Emmett–Teller (BET) method, utilizing nitrogen adsorption data at relative pressures between 0.05 and 0.30 to estimate surface area based on multilayer adsorption theory. The pore volume and pore size were calculated using the Barrett–Joyner–Halenda (BJH) method, derived from the Kelvin equation, relating the changes in the adsorbed or desorbed volume at different pressures to the pore radius, assuming cylindrical pore geometry. SEM-EDX measurements were performed using a JEOL JSM-5400 equipped with an INCAx X-ray detector. The measurements were carried out at energies between 5 to 20 kV.The temperature-programmed experiments were conducted in a Micromeritics AutoChem 2920 equipped with a thermal conductivity detector and mass spectrometer. The temperature of the catalyst bed was measured with a K-type thermocouple touching the sample. Typically, 100 mg of the calcined samples (150–250 μm) were loaded into a quartz U-tube and pretreated under 60 Nml Ar min−1 at 500 °C for 1 h at atmospheric pressure. The temperature-programmed reduction (H2-TPR) profile was recorded by heating the sample from 50 to 750 °C at a rate of 10 °C min−1, while exposing it to a 5% H2/Ar stream (60 Nml min−1) and maintaining the temperature at 750 °C for 10 minutes. At the same time, a mass spectrometer measured the formed water during the process. The CO2 adsorption capacity of the as-prepared samples was studied by CO2 temperature-programmed desorption (CO2-TPD). CO2 adsorption was carried out under 60 Nml min−1 CO2 at 50 °C for 2 hours. After evacuating the physically adsorbed CO2 for 2 hours at 50 °C, the temperature-programmed desorption (TPD) profile was recorded by heating the sample from 50 to 500 °C at 10 °C min−1 under 60 Nml min−1 helium. The desorbed CO2 was quantified by integrating the area under the deconvoluted desorption profile.
The surface chemical properties of samples were studied using X-ray photoelectron spectroscopy (XPS) with an SSI S-probe equipped with a monochromatic Al Kα source (hν = 1486.6 eV). Data acquisition was carried out under ultra-high vacuum conditions (9 × 10−7 Pa). Survey scans were measured at a constant pass energy of 140.8 eV. High-resolution scans were recorded at 90.15 eV for Zr 3d, Zn 2p, O 1s, and C 1s core level for ZrO2, Zn0.07ZrOy-500, Zn0.19ZrOy-500, Zn0.19ZrOy-700, and Zn0.19ZrOy-800. All binding energies were calibrated using the C 1s peak of adventitious carbon at a binding energy of 284.8 eV.
Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS) measurements were conducted at 10 bar using a Harrick flow reactor equipped with ZnSe windows mounted in a Praying Mantis accessory within a Bruker Tensor 27 FTIR. To clarify the effect of Zn doping on ZrO2, we selected ZrO2, Zn0.19ZrOy, and Zn0.56ZrOy for our DRIFT study. 30 mg of samples were diluted with 100 mg KBr and placed in the sample holder cup of a high-temperature cell. After pretreatment at 400 °C for 1 hour under an Ar atmosphere, background IR spectra were collected under Ar. The gas mixture was then introduced into the cell via mass flow controllers, and each spectrum was recorded at a resolution of 4 cm−1, with an average of 32 scans per spectrum. In the CO2 adsorption experiment, CO2 was adsorbed by flowing 2 ml min−1 CO2 in 16 ml min−1 Ar for 40 minutes, followed by the removal of weakly adsorbed species under Ar. For the methanol adsorption experiment, 2.3 ml min−1 of methanol with 130 ml min−1 Ar was introduced for 30 minutes at 200 °C. After methanol adsorption, physically adsorbed species were removed in an 130 ml min−1 Ar flow, and the spectra of the remaining adsorbed methanol were collected.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2:Ar = 3:1:1 at constant pressure and flowrate (45 bar and GHSV = 21000 Nml gcat−1 h−1). The reaction temperature was incrementally increased from 300 to 400 °C in intervals of 25 °C and held for 2 hours at each temperature to establish steady-state conditions. The stability of the Zn0.19ZrOy-500 catalyst was evaluated by varying the reaction temperature and subsequently returning to the initial temperature, allowing for assessment of its performance consistency and resilience under elevated temperatures. The outlet gas line was maintained at 150 °C to prevent condensation of the reaction products. The reaction effluent was analysed online using a TRACE-1310 GC analyser equipped with Rt-Ubond (FID), Hayesep N and ShinCarbon-ST (1st TCD), and RT-QBond and RT-MolSieve 5A (2nd TCD) columns. The fulfilment of the Wheeler–Weisz and Carberry criteria confirmed the absence of transport limitations. The catalysts' performance was evaluated by calculating CO2 conversion (X(CO2)), methanol selectivity (S(CH3OH)), and methanol space–time yield (STY(CH3OH)) as follows:
:CO2:Ar = 3:1:1 at constant pressure and flowrate (45 bar and GHSV = 21000 Nml gcat−1 h−1). The reaction temperature was incrementally increased from 300 to 400 °C in intervals of 25 °C and held for 2 hours at each temperature to establish steady-state conditions. The stability of the Zn0.19ZrOy-500 catalyst was evaluated by varying the reaction temperature and subsequently returning to the initial temperature, allowing for assessment of its performance consistency and resilience under elevated temperatures. The outlet gas line was maintained at 150 °C to prevent condensation of the reaction products. The reaction effluent was analysed online using a TRACE-1310 GC analyser equipped with Rt-Ubond (FID), Hayesep N and ShinCarbon-ST (1st TCD), and RT-QBond and RT-MolSieve 5A (2nd TCD) columns. The fulfilment of the Wheeler–Weisz and Carberry criteria confirmed the absence of transport limitations. The catalysts' performance was evaluated by calculating CO2 conversion (X(CO2)), methanol selectivity (S(CH3OH)), and methanol space–time yield (STY(CH3OH)) as follows:
 | (1) |
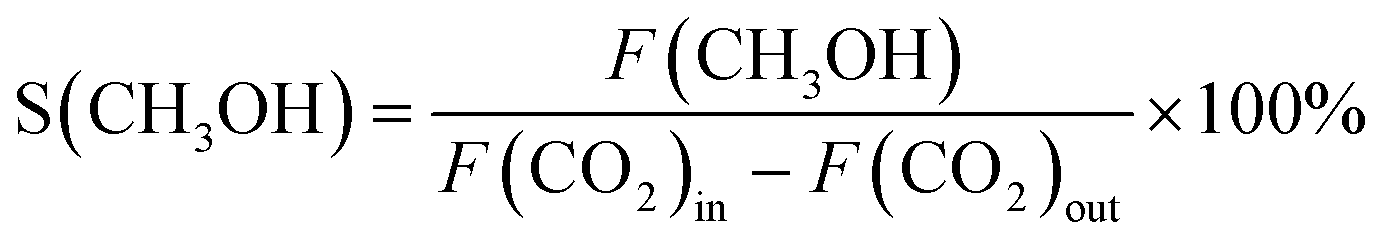 | (2) |
 | (3) |
3. Results and analysis
3.1. Structure and general properties of the ZnxZrOy catalysts
The preparation of the ZnxZrOy precipitate follows a commonly used coprecipitation recipe comparable to other studies on CTM.7,13 Seven different Zn/Zr molar ratios were used to prepare the ZrO2, Zn0.01ZrOy, Zn0.07ZrOy, Zn0.19ZrOy, Zn0.28ZrOy, Zn0.44ZrOy and Zn0.56ZrOy precursors. After calcination at 500 °C, their physical properties and those of the ZnO sample as an extra reference were measured (Table 1). As reported in the literature, Zn0.19ZrOy performs best in CTM conversion and selectivity at 350 °C.7 This sample is, therefore, besides the standard 500 °C calcination temperature, also calcined at 550, 700, and 800 °C. Different levels of Zn addition and calcination temperatures are expected to change the main crystal phase of the bulk and distribute Zn differently between bulk and surface.38 The bulk phase correlates to surface properties that underpin heterogeneous catalysis through exposed crystal planes and surface energy, including porosity, surface area, and texture.| Sample | Crystal phasea | Surface areab (m2 g−1) | Pore volume × 10−3c (cm3 g−1) | Avg. Pore sizec (Å) | n(H2)d (μmol g−1) | n(CO2)e (μmol g−1) | Zn/Zr (molar ratio) | XPS (mol%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nominal composition | EDXf | XPS | Zr | O | Zn | |||||||
| a Identified in XRD (m: monoclinic, t: tetragonal, and c: cubic phase).b Determined using the BET method.c Determined from BJH adsorption isotherm.d Measured by integrating the peaks in H2-TPR between 50–700 °C.e Measured by integrating the peaks in CO2-TPD between 50–500 °C.f Calculated from EDX results at 20 kV electron beam energy. | ||||||||||||
| ZnO | h-ZnO | 31.5 | 230 | 316 | — | 1.6 | — | — | — | — | — | — |
| ZrO2 | (m + t) ZrO2 | 21.9 | 26 | 45 | 20.3 | 14.0 | — | — | — | 24.8 | 75.2 | — |
| Zn0.01ZrOy-500 | t-Phase | 24.5 | 28 | 40 | 28.5 | 16.2 | 0.01 | 0.01 | 0.01 | 41.0 | 58.4 | 0.6 |
| Zn0.07ZrOy-500 | c-Phase | 35.9 | 47 | 48 | 37.4 | 23.3 | 0.07 | 0.05 | 0.18 | 26.0 | 69.3 | 4.7 |
| Zn0.19ZrOy-500 | c-Phase | 31.2 | 50 | 58 | 44.6 | 26.3 | 0.19 | 0.13 | 0.22 | 23.1 | 71.8 | 5.1 |
| Zn0.28ZrOy-500 | c-Phase | 22.8 | 46 | 54 | 41.4 | 37.5 | 0.28 | 0.20 | 0.30 | 30.7 | 60.3 | 9.0 |
| Zn0.44ZrOy-500 | c-Phase | 17.5 | 32 | 66 | 72.5 | 5.3 | 0.44 | 0.48 | 0.59 | 28.1 | 55.2 | 16.7 |
| +h-ZnO | ||||||||||||
| Zn0.56ZrOy-500 | c-Phase | 12.9 | 32 | 91 | 116.8 | 3.8 | 0.56 | 0.66 | 0.76 | 25.1 | 55.8 | 19.1 |
| +h-ZnO | ||||||||||||
| Zn0.19ZrOy-550 | c-Phase | 34.0 | 55 | 58 | 41.0 | 26.9 | 0.19 | 0.12 | 0.29 | 24.5 | 68.4 | 7.1 |
| Zn0.19ZrOy-700 | c-Phase | 9.2 | 35 | 154 | 27.3 | 37.9 | 0.19 | 0.11 | 0.43 | 20.4 | 70.8 | 8.8 |
| Zn0.19ZrOy-800 | (m + t) phases | 1.1 | 9 | 322 | 23.3 | 2.6 | 0.19 | 0.10 | 0.24 | 23.0 | 71.6 | 5.5 |
| +h-ZnO | ||||||||||||
Elemental analysis using scanning electron microscopy equipped with energy-dispersive X-ray spectroscopy (SEM-EDX) confirmed the successful Zn addition into ZrO2 (Tables 1 and S1†). In XRD (Fig. 1), no ZnO crystals were detected in the samples with Zn/Zr ratios of 0.01–0.19, indicating that Zn is either finely distributed within these samples, present as small-sized crystallites (<3 nm), or is amorphous. On samples with Zn/Zr = 0.44–0.56, a hexagonal ZnO phase is visible beside the main ZrO2 phase. Calcination of Zn0.19ZrOy at 800 °C also causes ZnO phase separation and ZrO2 phase transition to monoclinic and tetragonal.
 | ||
| Fig. 1 (a) XRD patterns of ZnxZrOy-500 catalysts, (b) enlarged XRD patterns of ZnxZrOy-500 catalysts in the 2θ range of 29.5–31.0°, and (c) XRD patterns of Zn0.19ZrOy-T calcined at 500, 550, 700, and 800 °C (m-ZrO2: PDF card 81.1314, t-ZrO2: PDF card 50-1089, c-ZrO2: PDF card 65-0461, and h-ZnO: PDF card 05-0664). | ||
The higher surface Zn/Zr ratio detected in XPS (Table 1), compared to the nominal elemental composition, suggests an enrichment of Zn near the surface,12,23 implying the formation of an imperfect mixed metal oxide with a higher concentration of Zn in the skin layer than in the bulk; increasing the calcination temperature to 700 °C results in a higher concentration of Zn near the surface. Upon calcination at 800 °C, however, the Zn surface excess decreases (Fig. S2†), correlating with the appearance of ZnO crystals in XRD due to sintering.
Three main ZrO2 phases are found in the XRD spectra: tetragonal, monoclinic, and cubic (Fig. 1a). In agreement with earlier reports, pristine ZrO2 contains monoclinic and tetragonal phases.7 A small addition of Zn (Zn/Zr = 0.01) results in a partial phase transformation from monoclinic to tetragonal, while larger amounts of Zn lead to the appearance of a cubic phase. From Zn/Zr = 0.07 upwards, the lattice also contracts with increasing Zn content, indicated by the shift of the diffraction peak near 30° to higher angles (Fig. 1b). This shift is consistent with the displacement of Zr (0.82 Å) by the smaller Zn cation (0.74 Å)15 within the crystal lattice. A Zn/Zr molar ratio ≥ 0.07 leads to the formation of the cubic phase, as evidenced by the presence of symmetrical single peaks instead of double peaks at 2θ = 35.5°, 50.9° and 60.5°. ZnO remains undetectable up to Zn/Zr = 0.19. Excess Zn in the Zn0.28ZrOy-500 sample, surpassing the solid solution limit (Zn/Zr = 0.24),23 alters the crystalline structure (Fig. 1b). The alteration induced by high zinc concentration broadens the peaks, shrinks the unit cell due to inhomogeneously strained crystallites, and shifts the diffraction peaks to higher angles.7,39,40 The broadening peaks may indicate the presence of multiple phases within the material.7 In Zn0.44ZrOy-500 and Zn0.56ZrOy-500, the excess ZnO gives rise to measurable diffractions, in addition to the cubic ZrO2 diffractions.
Calcining the Zn0.19ZrOy sample at higher temperatures (Fig. 1c) maintains the cubic phase at least up to 700 °C. At 800 °C, ZnO phase separation and cubic phase transformation to tetragonal and monoclinic phases occur. The samples' Raman spectra (Fig. 2 and ESI† section 2 – Raman spectroscopy) confirm the same crystal phases as those measured by XRD.
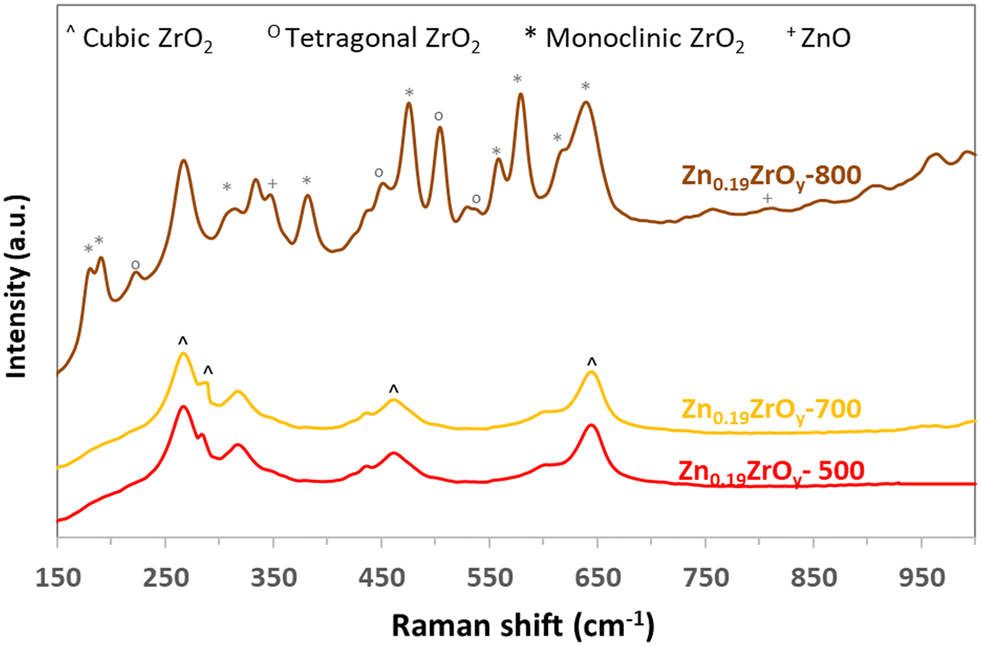 | ||
| Fig. 2 Raman spectra of Zn0.19ZrOy-T calcined at 500, 700, and 800 °C. | ||
Table 1 and Fig. S1† present the nitrogen adsorption/desorption results, all indicating mesoporous materials. Up to Zn/Zr = 0.28, surface area and pore volume increase with Zn loading. Beyond this, surface area decreases. The Zn0.44ZrOy-500 and Zn0.56ZrOy-500 isotherms resemble a bimodal mesoporous material, combining ZrO2 and ZnO isotherms (Fig. S1a†). The region corresponding to P/P0 > 0.87 implies the presence of larger mesopores attributed to the formation of ZnO particles, consistent with the XRD results. The other hysteresis region, within the range 0.45 < P/P0 < 0.87, represents the smaller mesopores of the mixed oxide. Elevating the calcination temperature to 700 °C and 800 °C alters the pore geometry, resulting in partially blocked pores (Fig. S1b†) and reduced surface area. The pore size distribution in Fig. S1c and d† illustrates changes in pore size resulting from zinc addition and the influence of calcination temperature on pore size distribution. The graphs show that pore enlargement occurs with calcination temperature exceeding 550 °C.
The Zr 3d and Zn 2p XPS spectra are shown in Fig. 3. The ZrO2 profile can be fitted with three Gaussian-shaped doublets, each exhibiting a spin–orbital splitting of 2.4 eV, typical for zirconia. The bands centred at 180.9 and 182.3 eV are assigned to the monoclinic and tetragonal phases of ZrO2, respectively.41 The shift of the bands upon adding Zn indicates an alteration in the electronic and structural properties of Zr species in ZnxZrOy.42 The high energy band centred at 182.6 eV corresponds to bulk Zr in cubic ZrO2, indicating the phase transformation from tetragonal to cubic.43,44 The low energy band at 181.3 eV is assigned to Zr in Zn–O–Zr motifs.9,23,38,42,45,46 Zinc incorporation generates new surface Zr species in lower oxidation states, with a 179.9 eV centred XPS band, especially in the Zn0.19ZrOy-500 sample. Elevating the calcination temperature to 700 °C leads to a decrease in the concentration of Zr species in lower oxidation states, Fig. 3a and Table S2.†
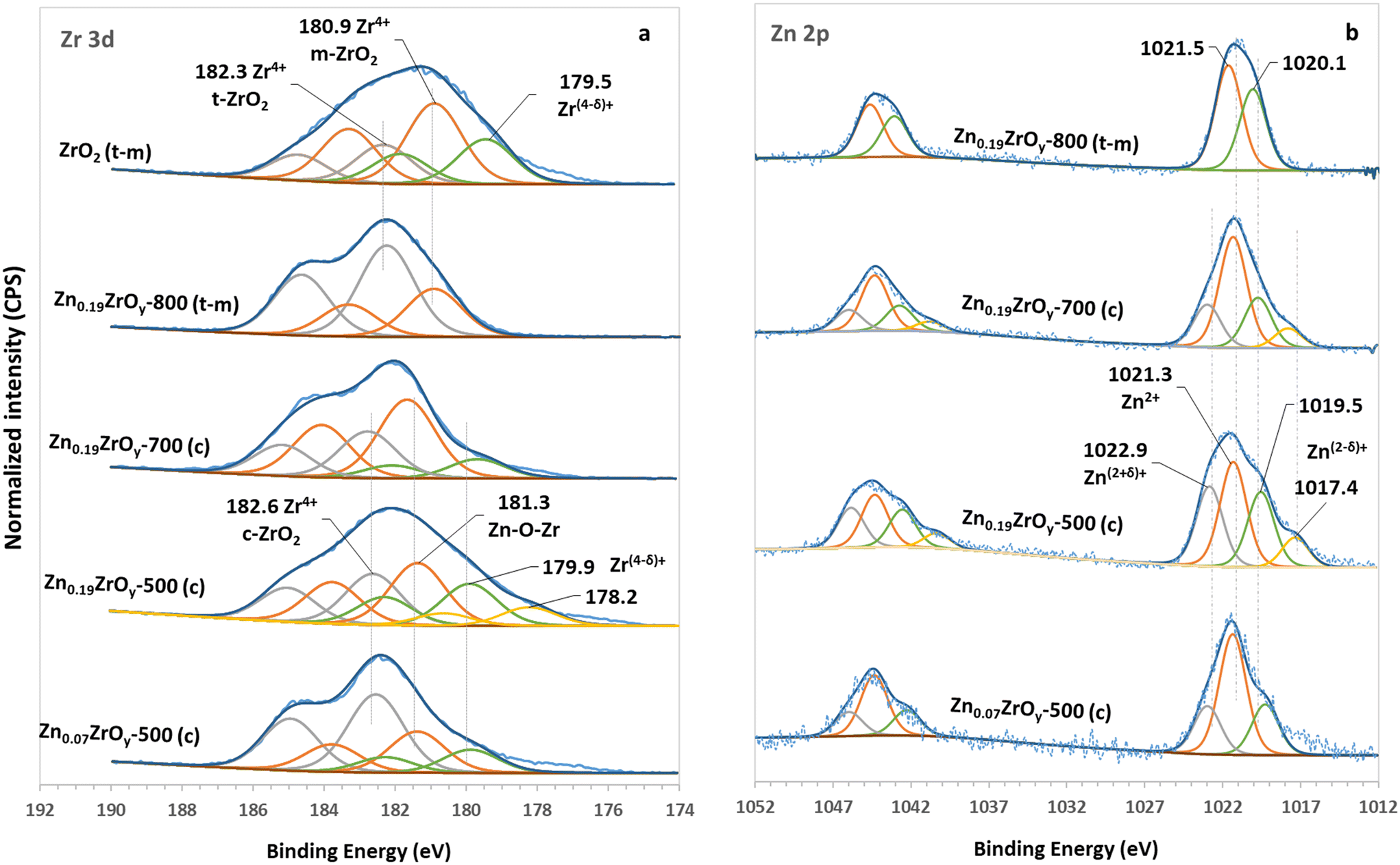 | ||
| Fig. 3 XPS spectra of (a) Zr 3d in ZrO2, Zn0.07ZrOy-500, Zn0.19ZrOy-500, Zn0.19ZrOy-700, and Zn0.19ZrOy-800 and (b) Zn 2p in Zn0.07ZrOy-500, Zn0.19ZrOy-500, Zn0.19ZrOy-700, and Zn0.19ZrOy-800. | ||
Further increasing the calcination temperature to 800 °C shifts the Zr 3d5/2 bands to 180.9 and 182.3 eV, resembling the Zr 3d doublets observed in the pure ZrO2 spectrum. This shift is attributed to the phase transformation of zirconia and the formation of monoclinic and tetragonal crystals at 800 °C, consistent with the XPS spectra observed for pure ZrO2.
The Zn 2p spectra of the samples are shown in Fig. 3b. The binding energies of Zn 2p3/2 and Zn 2p1/2 in pure hexagonal ZnO centre at 1021.0 and 1044.1 eV, respectively.42 Metallic Zn has a similar binding energy to ZnO, but isolated metallic Zn is not expected in these samples. The presence of Zn species at higher binding energy (1022.6 eV) in ZnxZrOy originates from the different coordination structures, suggesting that the Zn species are in contact with the oxygen of ZrO2, forming Zn–O–Zr bonds.46 This coordination environment, with higher electronegativity than the oxygen ligand in bulk ZnO, contributes to the observed shift in binding energy38 and suggests a charge transfer between O2−, Zr4+, and Zn2+.45 These observations point to the possible incorporation of Zn into the ZrO2 lattice, forming a mixed oxide. The Zn–O–Zr species disappear after calcination above 700 °C due to the reduced contact between Zn and ZrO2 related to the decreased surface area38 and the formation of ZnO particles. Zn 2p3/2 and Zn 2p1/2 peaks become broader in Zn0.19ZrOy-500, originating from the partially charged Zn species. Increasing the calcination temperature to 700 °C reduces the 1017.4 eV band associated with Zn(2−δ)+ species, and this peak disappears entirely when the sample is calcined at 800 °C. The high-intensity peak centred at 1020.1 eV in Zn0.19ZrOy-800 is likely linked to the phase-separated ZnO species at the interface of ZrO2.
The reducibility of the catalysts with H2 is directly relevant to the activation of H2 required for the CO2 hydrogenation catalysis.7 Fig. 4 shows H2 temperature-programmed reduction profiles. The reduction of ZrO2 is challenging and requires high temperatures.13 Increasing the Zn/Zr ratio up to 0.19 progressively shifts the reduction to lower temperatures and increases the reducibility (quantified in Table 1), revealing the presence of more reducible oxygen species on the surface, which aligns with the observation of low binding energies in Zr and Zn XPS. In the Zn0.44ZrOy-500 and Zn0.56ZrOy-500 samples, the exposed ZnO particles result in noticeable H2 consumption compared to the Zn0.01ZrOy-500 sample. The Zn0.28ZrOy-500 reduction profile shifts to lower temperatures, likely due to the formation of nano-sized ZnO particles with facilitated reduction compared to bulk ZnO. Increasing the calcination temperature gradually shifts the reduction to lower temperatures. In the Zn0.19ZrOy-700 sample, the low-temperature reduction can be attributed to the enrichment of the surface with Zn, as seen in XPS. In the Zn0.19ZrOy-800 sample, ZnO phase separation lowers the initial reduction temperatures.
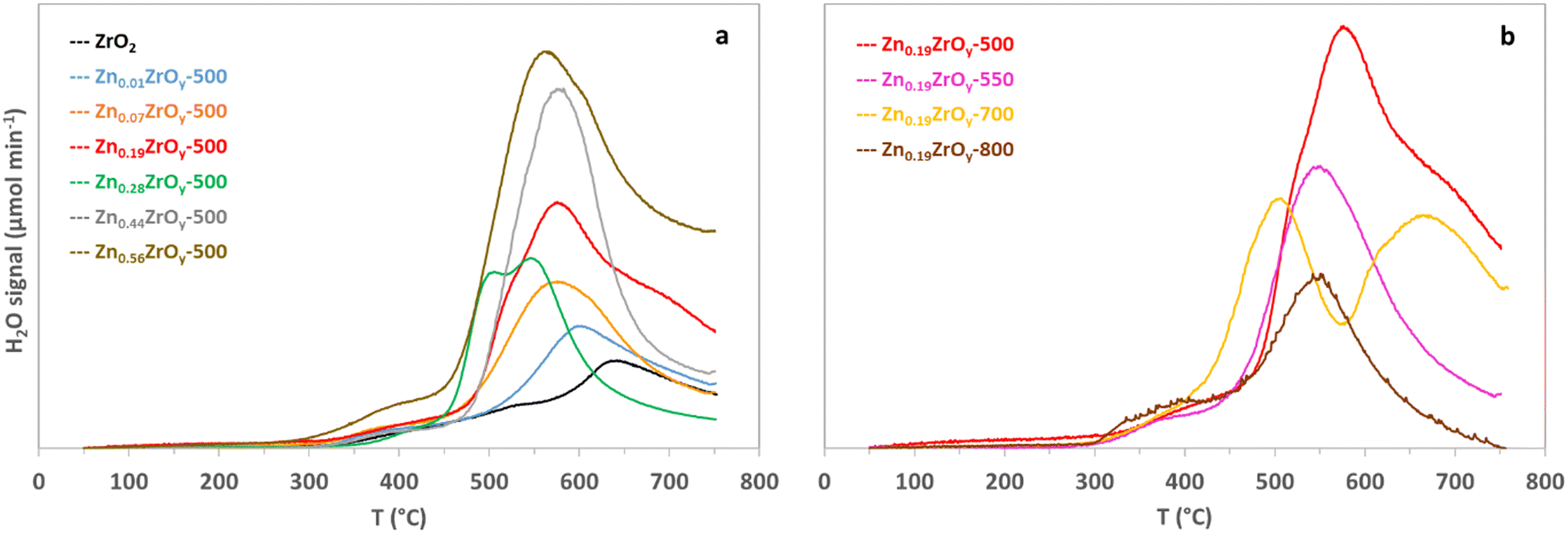 | ||
| Fig. 4 Temperature-programmed H2 reduction of (a) ZnxZrOy-500 catalysts and (b) Zn0.19ZrOy-T calcined at 500, 550, 700, and 800 °C. | ||
CO2 activation is generally linked to the acid–base properties of the zirconia catalyst surface, with basic surface OH sites forming bicarbonate and acid–base Lewis pairs forming monodentate and bidentate carbonate species.47,48 The changes in ZrO2 basicity upon Zn incorporation and the impact of calcination temperature were assessed using temperature-programmed CO2 desorption (CO2-TPD, Fig. 5). Deconvolution of the profiles (Fig. S4†) reveals three desorption features in each sample. According to their peak desorption temperatures, these correspond to weak (100 °C), medium (150–190 °C), and strong (>200 °C) basic sites. The medium and strong basic sites have been linked to chemisorbed CO2 on Zn–O–Zr sites or surface oxygen vacancies.8 CO2 adsorption gradually increases with Zn loading from 14.0 μmol g−1 for ZrO2 to 37.5 μmol g−1 for Zn0.28ZrOy-500, and then declines sharply (Table 1). Introducing zinc in a Zn/Zr ratio of 0.01 leads to a 17% increase in total CO2 adsorption without notable alterations in the CO2 desorption trend (Fig. 5a). This correlates with the transition of monoclinic zirconia to the tetragonal phase, characterized by higher basicity compared to the monoclinic phase,49 and results in a marginal enhancement in catalyst activity. In the ZrO2 catalyst comprising monoclinic and tetragonal phases, strong surface CO2 adsorption sites are present. The addition of Zn, however, reduces the strength of these strong basic sites, likely due to the phase transformation from monoclinic, which contains stronger adsorption sites,50 to tetragonal and cubic phases. Weaker basic sites are reported to facilitate CO2 activation and its conversion to formate.51 Increasing the Zn/Zr ratio up to 0.28 enhances the number of basic sites. The CO2 desorption profiles of Zn0.07ZrOy-500 and Zn0.19ZrOy-500 exhibit remarkable similarity. The strong basic sites account for half of the total basicity, and the introduction of Zn shifts the temperature of the strong basic sites to lower temperatures (Table S3†). A higher calcination temperature shifts the strong basic sites to slightly higher temperatures (Fig. 5b). At 700 °C, the temperature at which the catalyst is on the verge of phase transformation, the basicity increases to 37.9 μmol g−1. With a further increase in the calcination temperature to 800 °C, the basicity decreases sharply, primarily attributable to limited surface area (see Table 1) and the accumulation of ZnO crystals on the surface, which have low CO2 adsorption capacity.
 | ||
| Fig. 5 Temperature-programmed CO2 desorption of (a) ZnxZrOy-500 catalysts and (b) Zn0.19ZrOy-T calcined at 500, 550, 700, and 800 °C. | ||
3.2. DRIFTS
Understanding the adsorption of CO2 and methanol on ZrO2 and ZnxZrOy mixed oxides is helpful to unravel the effect of Zn on surface interactions and catalytic behaviour. To this end, we studied the formation of surface species from CO2 and methanol adsorption, followed by evacuation under argon. | ||
| Fig. 6 DRIFT spectra of (a) CO2 adsorption at 325 °C and 10 bar on ZrO2, Zn0.19ZrOy-500, and Zn0.56ZrOy-500 after 40 minutes under 2 ml min−1 CO2 and 16 ml min−1 Ar and (b) after 40 minutes evacuation at 325 °C and 10 bar under 16 ml min−1 Ar (m: monodentate, b: bidentate, wi: weakly interacting, and p: polydentate species). | ||
On Zn0.19ZrOy-500, CO2 initially adsorbs as monodentate carbonates (m-CO32−) species and weakly interacting bicarbonates (wi-HCO3−). With time, again bidentate carbonates (b-CO32−) emerge, but now accompanied by the appearance of polydentate carbonates (p-CO32−), Fig. 6a and S5.† Following evacuation under Ar (Fig. 6b), the weakly interacting bicarbonates (wi-HCO3−) disappear, the monodentate carbonates (m-CO32−) decrease, the polydentate carbonates (p-CO32−) remain stable, and the DRIFTS features of the bidentate carbonates shift to lower wavenumbers, indicating changes in their binding strength. The formation of bidentate carbonates at different wavelengths, 1578 cm−1 and 1564 cm−1, is reported to correspond to two slightly different structural arrangements without further specification.52
The addition of Zn alters the surface sites, as observed in DRIFTS, and this coincides with changes in CO2 adsorption behaviour, as observed in CO2-TPD (Fig. 5). A commonly stated explanation is an alteration of surface OH groups,13,17,52,53 however the underlying drivers remain unclear. A possible explanation is an altered surface OH distribution on different ZrOx phases. The monoclinic phase is described to be rich in surface OH groups,17,52,54 while the cubic phase exposes facets with less surface OH.55 XRD analysis (Fig. 1) indicates a shift towards the cubic phase upon Zn incorporation, but the correspondence to the amount of surface bicarbonates in DRIFTS and CO2-TPD is not straightforward. Additionally, XPS data (Fig. 3) confirm the formation of Zn–O–Zr sites on the surface upon Zn addition. These Zn–O–Zr sites provide new adsorption sites,13 possibly supporting the polydentate adsorption of carbonate, characterized by peaks at 1400–1480 cm−1.17,54
Limited CO2 adsorption on Zn0.56ZrOy-500 is in line with the formation of ZnO covering the surface, as confirmed by XRD and N2 physisorption. This is consistent with the limited CO2 adsorption capability of ZnO, as evidenced by the CO2-TPD results (Fig. 5).
 | ||
| Fig. 7 DRIFT spectra of methanol adsorption at 200 °C and 10 bar on ZrO2, Zn0.19ZrOy-500, and Zn0.56ZrOy-500 (the spectra were recorded after evacuation of physically adsorbed species under a flow of 130 ml min−1 Ar). | ||
Dissociative methanol adsorption is observed on all tested samples, but on Zn0.56ZrOy-500, these peaks shift to higher wavenumbers, indicating further changes in the surface's electronic environment due to increased Zn content and ZnO formation. The peak intensity at 1160 cm−1 on ZrO2 also decreases and shifts to 1150 cm−1 upon Zn addition, reflecting alterations in the adsorption sites.
3.3. Performance test
The performance for CO2 hydrogenation was evaluated at 45 bar and 300–400 °C for all synthesized catalysts. Methanol and CO were the main products. Trace amounts of methane and dimethyl ether were also detected in the reactor effluent. Methane formation occurs only at elevated temperatures, above 350 °C, reaching a maximum formation of 0.25% of the methanol over Zn0.56ZrOy-500 at 400 °C. DME is produced through the sequential dehydration of methanol on acid sites, reaching a maximum formation of 3.11% of the methanol over Zn0.07ZrOy-500 at 375 °C. The CTM performance of Zn0.19ZrOy-500 at 350 °C was tested by increasing the reaction temperature and then returning to 350 °C. The catalyst maintained stable methanol selectivity and STY throughout temperature fluctuations, demonstrating its robustness and ability to perform well under changing thermal conditions (Fig. S6†).Fig. 8a illustrates the temperature dependence of CO2 conversion and methanol selectivity. Zinc introduction into zirconia increases both CO2 conversion and methanol selectivity across the temperature range. A Zn/Zr ratio of 0.01 has a negligible impact on the catalyst's CTM activity compared to pure ZrO2. This doping level was also insufficient to stabilize the cubic phase. Samples containing Zn/Zr = 0.07–0.19 exhibit the highest CO2 conversion among the tested samples. For reaction temperatures up to 350 °C, the selectivity to methanol maximally increases to more than threefold that of the Zn/Zr = 0.01 catalyst for the Zn/Zr = 0.19 catalyst, with the major increase with Zn doping occurring between Zn/Zr = 0.01 and 0.07. However, selectivity sharply declines above 350 °C, especially for the Zn/Zr ≥ 0.07 catalysts. The decline in selectivity with increasing conversion, induced by decreasing the flow rate (Fig. 8c), for Zn0.19ZrOy-500, suggests sequential reactions of methanol, Scheme 1.
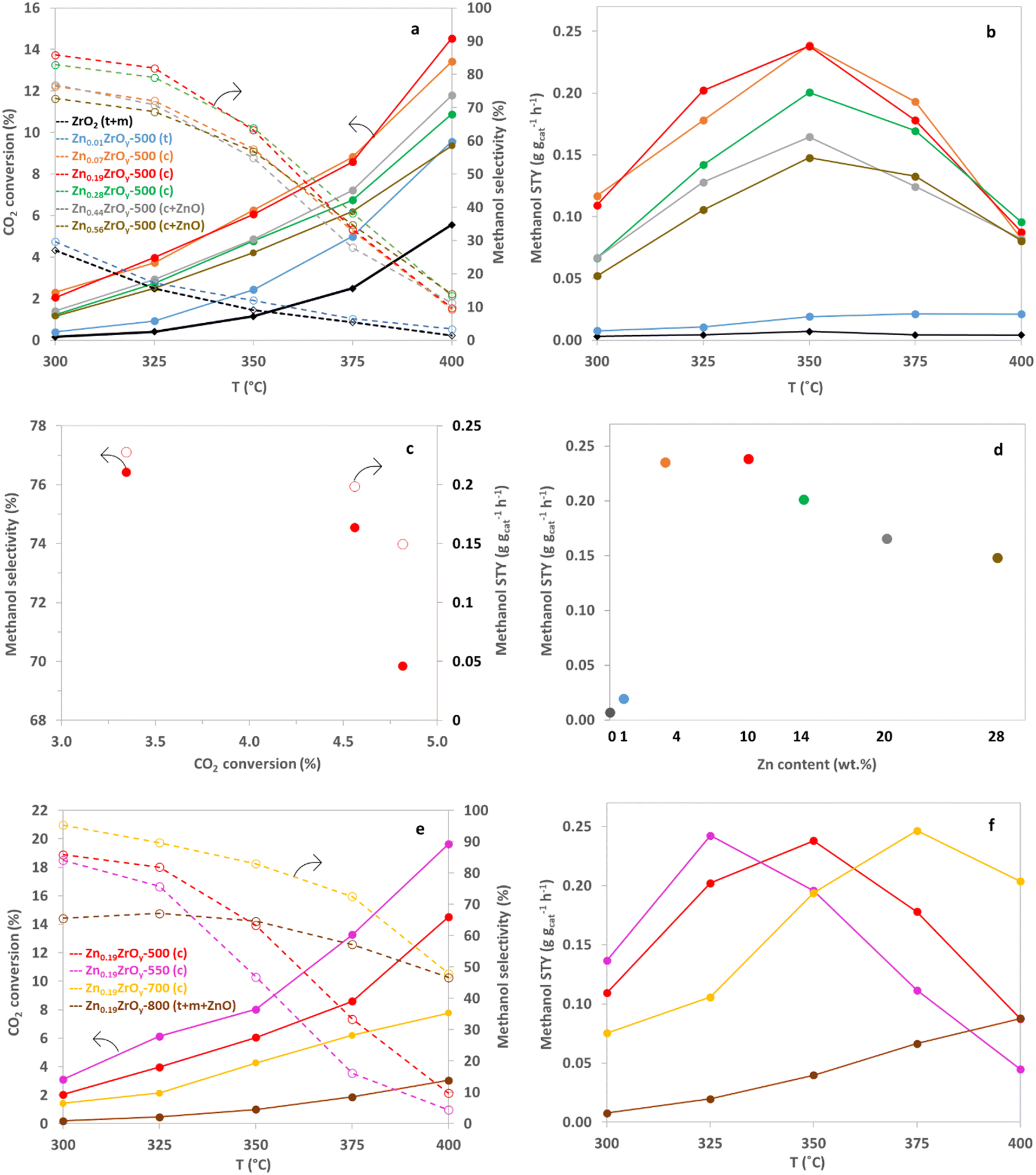 | ||
| Fig. 8 Effect of temperature on (a) CO2 conversion and methanol selectivity and (b) methanol STY of ZnxZrOy-500 catalysts, at 45 bar, H2:CO2:Ar = 3:1:1, and GHSV = 21000 Nml gcat−1 h−1. (c) Methanol selectivity versus CO2 conversion on Zn0.19ZrOy-500 at 325 °C, 45 bar, H2:CO2:Ar = 3:1:1, and GHSV = 31500, 21000, and 15750 Nml gcat−1 h−1. (d) Effect of Zn loading on methanol STY at 350 °C, 45 bar, H2:CO2:Ar = 3:1:1, and GHSV = 21000 Nml gcat−1 h−1. (e) Effect of temperature on CO2 conversion and methanol selectivity and (f) methanol STY of Zn0.19ZrOy-T catalysts, at 45 bar, H2:CO2:Ar = 3:1:1, and GHSV = 21000 Nml gcat−1 h−1 (full lines: CO2 conversion-dotted lines: methanol selectivity). | ||
 | ||
| Scheme 1 CO2 hydrogenation to methanol and CO and sequential reaction of methanol. | ||
For all catalysts with a Zn/Zr > 0.01, the methanol space–time yield (STY) peaks at 350 °C. The maximum methanol STY, 0.24 g gcat−1 h−1, is achieved with Zn/Zr = 0.07, for which a dominant cubic phase was found. This high STY is maintained up to Zn/Zr = 0.19, after which it gradually decreases with further increases in Zn/Zr ratios (Fig. 8b), accompanied by the formation of hexagonal ZnO. Although overall and surface Zn/Zr ratios vary, the catalytic activity is similar for Zn0.07ZrOy-500 and Zn0.19ZrOy-500 (Fig. 8d). The decrease in catalyst activity at higher Zn loadings up to Zn/Zr = 0.56 (in agreement with the literature),7 suggests that the quantity of zinc does not necessarily lead to a rise in the number of active sites. Instead, it tends to augment the abundance of other sites, such as ZnO, with lower activity levels.
These results align with literature, where CO2 hydrogenation at 320–350 °C, 20–50 bar, GHSV = 24000 ml gcat−1 h−1, and H2:CO2 = 3–4 gives a methanol selectivity ranging from 74 to 86%, with methanol formation rates of 0.2–0.3 g gcat−1 h−1.13,22,33,34,59,60 An exception was observed for 13% ZnO–ZrO2, which achieved a higher methanol formation rate of 0.5 g gcat−1 h−1 under the same conditions.7 Operating at lower GHSV values of 4000–10800 ml gcat−1 h−1 did not enhance performance, with the methanol formation rate dropping to 0.15–0.18 g gcat−1 h−1.8,61 An overview of CTM catalysis in literature is included in the ESI† (Table S4, ESI† section 7).
We selected Zn0.19ZrOy-500 as the best-performing catalyst and conducted catalytic tests on samples calcined at 550, 700, and 800 °C (Fig. 8e). Within the examined temperature range, conversion is highest for Zn0.19ZrOy-550 but declines for higher calcination temperatures. Methanol selectivity becomes less temperature-dependent when using catalysts calcined at 700 and 800 °C. Fig. 8f illustrates the methanol space–time yield plotted against temperature. Higher calcination temperatures shift the point of maximum STY to a higher temperature.
4. Discussion
4.1. Preparation parameters for different zirconia phases
Through varying degrees of zinc doping and different calcination temperatures, we obtained ZnxZrOy materials of monoclinic, tetragonal, and cubic polymorphs, as demonstrated in earlier literature.62 Monoclinic zirconia is stable at low temperatures, transitioning to the tetragonal phase at around 1170 °C, and then into the cubic phase at 2370–2680 °C.63 The stability of the tetragonal or cubic phase depends on grain size and chemical composition. Particles larger than 30 nm are most stable as monoclinic ZrO2, those smaller than 14 nm as tetragonal ZrO2, and particles smaller than 6 nm as the metastable cubic ZrO2 phase.43,64–66 The tetragonal or cubic zirconia can additionally be stabilized by doping an aliovalent cation such as Zn2+ into zirconia,16,67–69 by adding stabilizing oxides,70 or by adjusting the calcination temperature.63 Doped cations replace some zirconium ions in the crystal lattice, altering its electronic structure and introducing lattice strains that hinder oxygen atoms' movement and inhibit phase transitions from cubic to other crystalline phases.43,71–73 With increasing Zn doping, the dominant ZrO2 crystal phase alters from monoclinic to tetragonal and then to cubic. At calcination temperatures above 800 °C, the kinetic barriers preserving the metastable cubic phase are surpassed,74,75 leading to the transition of the material into tetragonal and monoclinic phases.In this study, beyond Zn/Zr = 0.28, ZnO phase separation happens, observed as hexagonal ZnO. Synthesizing catalysts with a Zn/Zr ratio up to 0.28 increased surface area, pore volume, and basicity. However, the Zn/Zr ratio exceeding 0.44 resulted in bimodal mesoporous materials, indicating the formation of ZnO particles and lower surface area, leading to inferior catalytic performance.
4.2. Surface property correlations and CTM catalysis
Adding zinc at a Zn/Zr ratio of 0.01 has a negligible impact on the catalyst's activity in methanol synthesis and is insufficient to stabilize the cubic phase. With Zn/Zr = 0.07 and 0.19, enhanced methanol STY, 34 times higher than the unpromoted ZrO2 catalyst, is observed at 350 °C and this coincides with the phase transition to cubic phase. With Zn/Zr ratios of 0.44–0.56, again lower activity is obtained (Fig. S8†) which coincides with zinc phase separation forming hexagonal ZnO. This aligns with existing literature,23 indicating other types of Zn sites, such as ZnO, with lower activity levels are formed at such high Zn/Zr ratios.This study aims to assess the structure–function relationship by which the ZnxZrOy crystal phase influences CTM catalysis. The investigated samples yield a large variety of surface physical and chemical properties. From the correlation plot in Fig. 9a, it is clear that many variables correlate strongly among the assessed samples. When focusing exclusively on the pure cubic-phase samples, however, the correlations are very different, highlighting distinct behaviour within this subset. Moreover, the correlation plot indicates a strong positive correlation between cubic phase and selectivity, space–time yield, and CO2 conversion in CTM at 350 °C. The other phases on the other hand show a negative correlation. The addition of Zn to ZrO2 enables and enhances CO2 conversion and methanol selectivity, as shown in Fig. 9b. Most interestingly, while a general trade-off between conversion and selectivity is observed among the tested samples, those with a pure cubic phase achieve the highest selectivity for methanol at a given conversion at 350 °C. Although ZrO2 and Zn0.19ZrOy-800 convert CO2 to a similar extent and exhibit comparable crystal phases, the presence of Zn in Zn0.19ZrOy-800 promotes selective methanol production in the reaction.
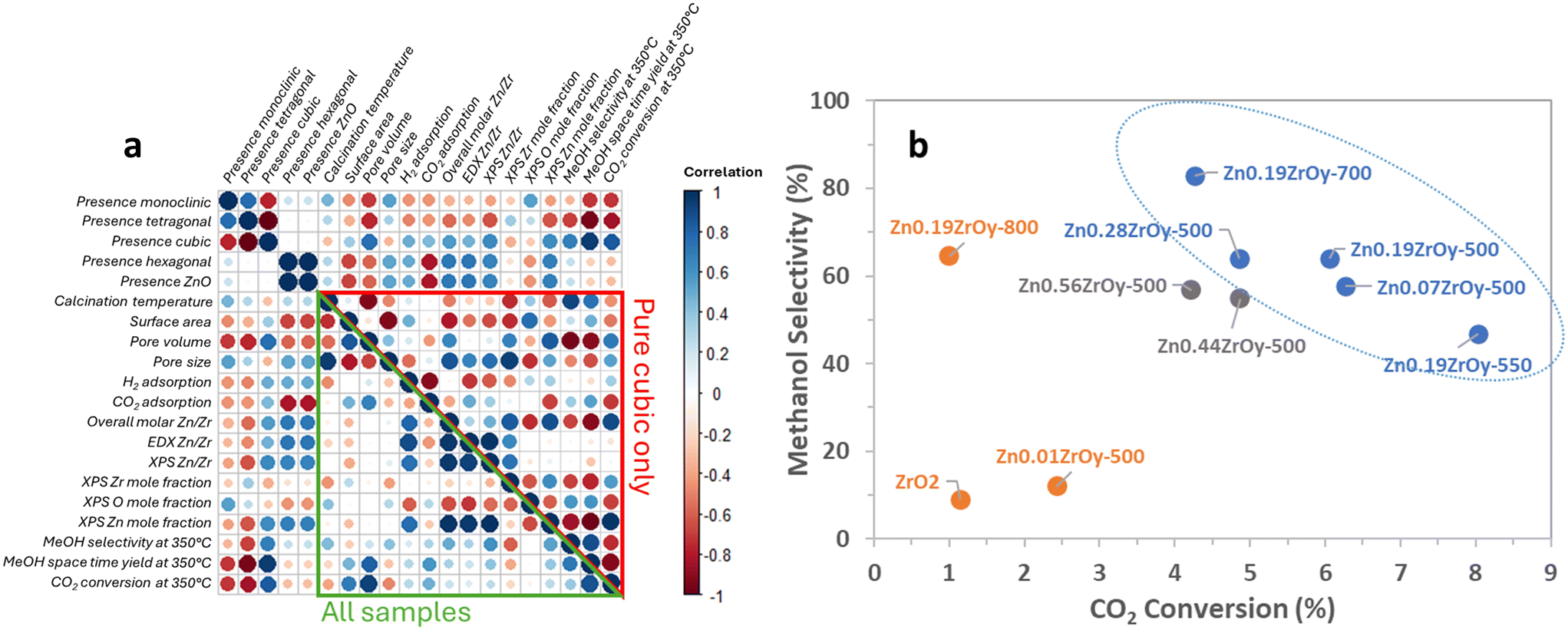 | ||
| Fig. 9 (a) Surface property correlations and CTM catalysis within all samples and (b) methanol selectivity versus CO2 conversion at 350 °C, 45 bar, H2:CO2:Ar = 3:1:1, and GHSV = 21000 Nml gcat−1 h−1 (blue colour: pure cubic-phase, grey colour: cubic-phase + h-ZnO, and orange colour: the rest of the samples). | ||
The cubic materials are also those with the highest pore volume and surface area, smallest pore size, and highest surface Zn. We therefore additionally look at surface properties in relation to pore volume, surface area, or surface zinc concentration as these metrics more suitably pinpoint desirable properties of the surface structure on a molecular level. Individual two-dimensional plots are shown in Fig. 10 and 11, and ESI† section 9 – parameters correlation where it can also be seen that samples with a pure cubic phase occupy distinct regions in the plots (shown in blue).
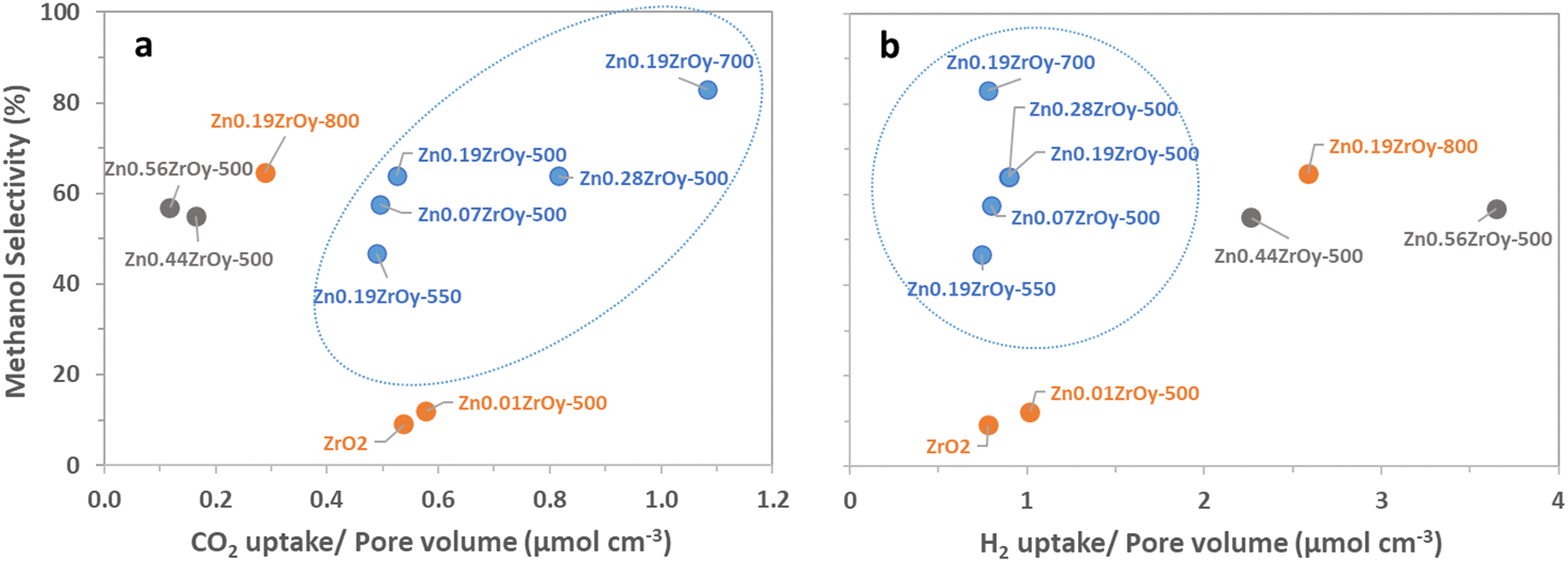 | ||
| Fig. 10 Effect of pore-volume normalized (a) CO2 uptake and (b) H2 uptake on methanol selectivity at 350 °C, 45 bar, H2:CO2:Ar = 3:1:1, and GHSV = 21000 Nml gcat−1 h−1 (blue colour: pure cubic-phase, grey colour: cubic-phase + h-ZnO, and orange colour: the rest of the samples). | ||
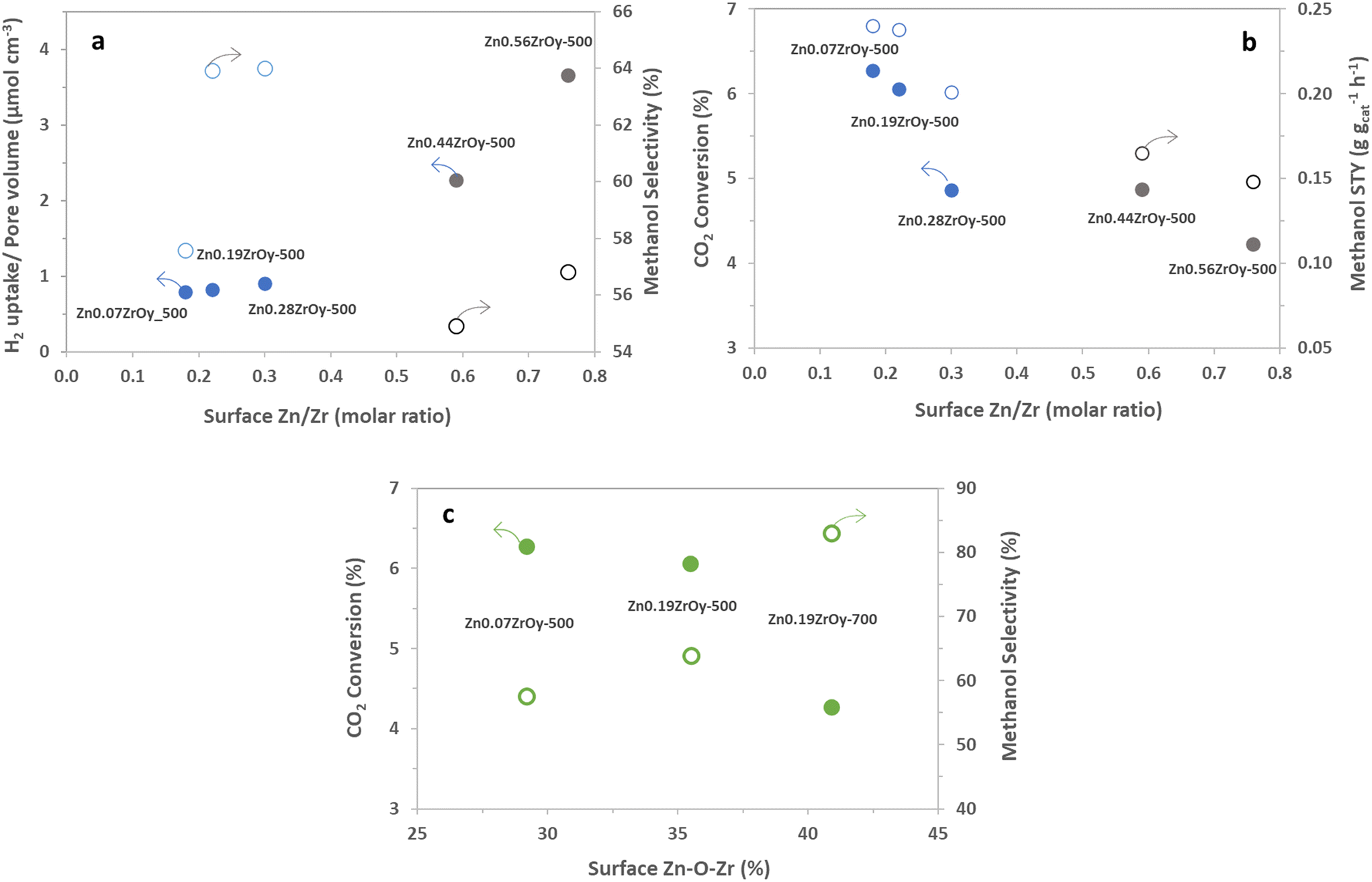 | ||
| Fig. 11 For a sub-selection of the measured samples, all with a dominant cubic phase; effect of (XPS) surface Zn/Zr ratio on (a) pore volume normalized H2 uptake (solid points) and methanol selectivity (empty points), and (b) CO2 conversion (solid points) and methanol STY (empty points), (blue colour: pure cubic-phase and grey colour: cubic-phase + h-ZnO) and (c) effect of (XPS) surface Zn–O–Zr species on CO2 conversion (solid points) and methanol selectivity (empty points), at 350 °C, 45 bar, H2:CO2:Ar = 3:1:1, and GHSV = 21000 Nml gcat−1 h−1. | ||
For all samples, there is a clear positive correlation between specific surface area and conversion and for samples containing cubic phase, there is a clear negative correlation between selectivity and pore volume (Fig. S7†) in which the cubic-phase-free samples do not fit. From Fig. 9b and S8b,† while conversion correlates strongly positively with pore volume (and thus negatively with selectivity, as is logically expected) for all samples, the pure cubic samples still demonstrate high selectivity at high pore volumes. This may, in part, be attributed to their unique combining of high CO2 adsorption per pore volume and selectivity as well as their low H2 uptake per pore volume (Fig. 10). From the available data, we cannot draw full conclusions on what sets the cubic phase samples apart from the others in this regard, but the observed beneficial effect of lower H2 uptake is surprising. It counterintuitively seems to favour selectivity to the more reduced methanol over CO, potentially by limiting further methanol decomposition to CO, while hydrogen activation and spillover are commonly seen as the rate-determining step (RDS) in methanol synthesis.74,75 The existence of the cubic phase may confound solid-phase diffusion during synthesis, which could enhance favourable zinc speciation at the surface. While surface zinc is essential, CTM catalysis appears to be more advantageous with cubic phase catalysts for reasons not fully addressed by current data. Coupled with literature, linking CO2 adsorption to oxygen vacancy sites17,21,22 suggests that surface zinc indeed facilitates CO2 adsorption. However, the cubic phase seems to provide a specific combination of oxygen vacancies and surface zinc, promoting CO formation only to a limited extent compared to other catalysts.15
The role of the crystal phase in methanol synthesis was highlighted in a recent study examining monoclinic versus tetragonal phases. The tetragonal phase achieved significantly higher methanol selectivity (81% versus 39%) and STY (0.04 versus 0.02 g gcat−1 h−1) compared to the monoclinic phase at 320 °C, using a ZnxZrOy catalyst.26 In the present study, methanol STY reached 0.20 g gcat−1 h−1 on the cubic phase sample, with comparable selectivity, underscoring the importance of stabilizing the optimal crystal phase. Clearly, certain crystal phases, in particular the cubic one, enhance the performance of ZnxZrOy catalyst in CTM. In addition to the Zn-induced phase change observed in XRD (Fig. 1), DRIFTS indicates that Zn influences surface basicity, which also affects CTM catalysis by means of CO2 and methanol chemisorption (Fig. 5–7). Hence, Zn contributes to CTM catalysis by inducing structural changes in two ways. Disentangling the latter requires further study of the quantitative structure–function relations.
It is informative to further analyse surface Zn's electronic properties for the samples containing cubic phase ZnxZrOy, and their association with CTM catalysis (Fig. 11). For the cubic samples calcined at 500 °C, particularly in the ZnO phase-separated samples (Zn0.44ZrOy and Zn0.56ZrOy), a positive correlation is observed between the increased surface Zn/Zr concentration and the pore-volume normalized H2 uptake (Fig. 11a). This suggests a role for surface Zn (or ZnO) in steering selectivity through the surface density of adsorbed H2. In Fig. 11a, methanol selectivity peaks at a surface Zn/Zr ratio of 0.2–0.4. However, further increasing the surface Zn/Zr ratio and the formation of ZnO particles negatively affects methanol selectivity. Fig. 11b confirms that CO2 conversion and methanol STY are optimal within a narrow range of surface Zn/Zr ratios. More specifically, the presence of Zr–O–Zn bonds correlates positively with methanol selectivity and negatively with CO2 conversion, (Fig. 11c), suggesting that H2 activation over Zn–O–Zr sites is not rate controlling, since a higher Zn–O–Zr surface fraction yields a lower conversion. This implies that the electronic properties of the surface atoms are crucial in determining the catalyst's behaviour, especially when the crystal phase is cubic. This electronic interaction is presumably more important than the physical characteristics of the pores, as it directly affects the reactivity and selectivity of the catalysts. This is supported by efforts to increase surface area using the evaporation-induced self-assembly method (EISA) to achieve 96 m2 g−1 compared to 48 m2 g−1 with the coprecipitation method, which resulted in only a minor increase in CO2 conversion, from 4% to 5.5% at 320 °C.33
5. Conclusions
This study explored the influence of Zn on the structure of ZnxZrOy and its catalytic performance in CO2 hydrogenation to methanol. Zinc incorporation into the zirconia lattice induces a phase transformation from monoclinic and tetragonal to mainly tetragonal phase in Zn0.01ZrOy-500. The cubic phase becomes dominant from a Zn/Zr ratio of 0.07. Catalysts with a Zn/Zr ratio of 0.07 and 0.19 demonstrate enhanced methanol formation. The cubic phase remains stable up to the calcination temperature of 700 °C, above which a transition to tetragonal and monoclinic phases occurs. The decreased catalytic activity of such samples can be attributed to the formation of agglomerated ZnO particles and the phase transformation.An optimal zinc content for CTM catalysis facilitates the formation of a mixed oxide, resulting in the formation of the cubic phase and enhances CO2 adsorption sites. Elevating the calcination temperature further indicates the significance of the cubic zirconia crystal phase. Within the pure cubic-phase samples, the surface Zn/Zr ratio and the concentration of Zr–O–Zn species correlate positively with CO2 adsorption capacity and methanol selectivity. Insufficient zinc content fails to stabilize the cubic phase, while excessive zinc content leads to ZnO phase separation. Both are detrimental to catalyst performance. Although the surface Zn/Zr ratio shows a positive correlation with pore volume-normalized H2 uptake, increased H2 uptake does not necessarily translate to improved CTM performance. Rather, a narrow range of surface Zn/Zr is advantageous, enhancing both methanol selectivity and CO2 conversion.
Further analysis of the catalysts at 350 °C reveals the underlying drivers for CO2 conversion to methanol, indicating that moderate Zn doping combined with a pure cubic phase induces the most effective CTM catalysts regarding methanol space–time yield (STY). Because a variety of catalytically relevant surface properties vary simultaneously, pinpointing the exact reason for this observation on an active site level remains challenging, both from our collected data and existing literature. A possible explanation lies in the changed concentration and distribution of surface basicity on the zirconia surface. Nevertheless, we identify informative correlations for CTM catalysis and can conclude that a ‘one-fits-all’ explanation of the role of zinc in promoting ZrOx catalysts for CTM is inadequate. It is an example of the commonly encountered complexity in catalytic structure–function relationships.
Data availability
The processed data supporting this article (N2 physisorption, XPS, SEM-EDX, CO2-TPD, DRIFTS and catalyst stability test) have been included as part of the ESI.† Raw data are available on request from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was financially supported by C2O (Light-assisted and thermal heterogeneous catalysis for the direct conversion of CO2 to olefins) Flanders Industry Innovation Moonshot (AIO SBO 2021 000 701).References
- G. Pacchioni, From CO2 to Methanol on Cu/ZnO/Al2O3 Industrial Catalyst. What Do We Know about the Active Phase and the Reaction Mechanism?, ACS Catal., 2024, 14, 2730–2745 CrossRef CAS.
- S. Chen, J. Wang, Z. Feng, Y. Jiang, H. Hu, Y. Qu, S. Tang, Z. Li, J. Liu, J. Wang and C. Li, Hydrogenation of CO2 to Light Olefins over ZnZrOx/SSZ-13, Angew. Chem., Int. Ed., 2024, 63, 1–7 Search PubMed.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, Highly Selective Conversion of Carbon Dioxide to Lower Olefins, ACS Catal., 2017, 7, 8544–8548 CrossRef CAS.
- M. Flores-Granobles and M. Saeys, Quantitative analysis of CO2 emissions reduction potential of alternative light olefins production processes, Green Chem., 2023, 25, 6459–6471 RSC.
- W. Zhang, S. Wang, S. Guo, Z. Qin, M. Dong, J. Wang and W. Fan, Effective conversion of CO2 into light olefins along with generation of low amounts of CO, J. Catal., 2022, 413, 923–933 CrossRef CAS.
- P. Zhang, L. Ma, F. Meng, L. Wang, R. Zhang, G. Yang and Z. Li, Boosting CO2 hydrogenation performance for light olefin synthesis over GaZrOx combined with SAPO-34, Appl. Catal., B, 2022, 305, 121042 CrossRef CAS.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol, Sci. Adv., 2017, 3, 1701290 CrossRef PubMed.
- W. Zhang, S. Wang, S. Guo, Z. Qin, M. Dong, J. Wang and W. Fan, Effective conversion of CO2 into light olefins over a bifunctional catalyst consisting of La-modified ZnZrOx oxide and acidic zeolite, Catal. Sci. Technol., 2022, 12, 2566–2577 RSC.
- W.-H. Feng, M.-M. Yu, L.-J. Wang, Y.-T. Miao, M. Shakouri, J. Ran, Y. Hu, Z. Li, R. Huang, Y.-L. Lu, D. Gao and J.-F. Wu, Insights into Bimetallic Oxide Synergy during Carbon Dioxide Hydrogenation to Methanol and Dimethyl Ether over GaZrOx Oxide Catalysts, ACS Catal., 2021, 11, 4704–4711 CrossRef CAS.
- L. Zhang, B. Geng, P. Wang, H. Kang, H. Xiao, J. Jia and H. Wu, Highly efficient ZnCeZrOx/SAPO-34 catalyst for the direct conversion of CO2 into light olefins under mild reaction conditions, Appl. Catal., A, 2023, 657, 119141 CrossRef CAS.
- D. Salusso, E. Borfecchia and S. Bordiga, Combining X-ray Diffraction and X-ray Absorption Spectroscopy to Unveil Zn Local Environment in Zn-Doped ZrO2 Catalysts, J. Phys. Chem. C, 2021, 125, 22249–22261 CrossRef CAS.
- L. D. R. Silva-Calpa, P. C. Zonetti, C. P. Rodrigues, O. C. Alves, L. G. Appel and R. R. de Avillez, The ZnxZr1−xO2−y solid solution on m-ZrO2: Creating O vacancies and improving the m-ZrO2 redox properties, J. Mol. Catal. A: Chem., 2016, 425, 166–173 CrossRef CAS.
- Z. Feng, C. Tang, P. Zhang, K. Li, G. Li, J. Wang, Z. Feng and C. Li, Asymmetric Sites on the ZnZrOx Catalyst for Promoting Formate Formation and Transformation in CO2 Hydrogenation, J. Am. Chem. Soc., 2023, 145, 12663–12672 CrossRef CAS PubMed.
- K. Lee, M. P. Dickieson, M. Jung, Y. Yang and N. Yan, Structure Sensitivity of ZnZrOx Catalysts in CO2 Hydrogenation to Methanol: Significance of Surface Oxygen Content and Synthesis Strategy, ACS Catal., 2024, 14, 3074–3089 CrossRef CAS.
- L. H. Chagas, P. C. Zonetti, C. R. V. Matheus, C. R. K. Rabello, O. C. Alves and L. G. Appel, The Role of the Oxygen Vacancies in the Synthesis of 1, 3-Butadiene from Ethanol, ChemCatChem, 2019, 11, 5625–5632 CrossRef CAS.
- E.-M. Köck, M. Kogler, T. Bielz, B. Klötzer and S. Penner, In Situ FT-IR Spectroscopic Study of CO2 and CO Adsorption on Y2O3, ZrO2, and Yttria-Stabilized ZrO2, J. Phys. Chem. C, 2013, 117, 17666–17673 CrossRef PubMed.
- K. Pokrovski, K. T. Jung and A. T. Bell, Investigation of CO and CO2 Adsorption on Tetragonal and Monoclinic Zirconia, Langmuir, 2001, 17, 4297–4303 CrossRef CAS.
- T. J. Keskitalo, M. K. Veringa Niemelä and A. O. I. Krause, Modeling of the Adsorption and Desorption of CO2 on Cu/ZrO2 and ZrO2 Catalysts, Langmuir, 2007, 23, 7612–7619 CrossRef CAS PubMed.
- J. Kondo, H. Abe, Y. Sakata, K.-I. Maruya, K. Domen and T. Onishi, Infrared studies of adsorbed species of H2, CO and CO2 over ZrO2, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 511–519 RSC.
- F. Sha, S. Tang, C. Tang, Z. Feng, J. Wang and C. Li, The role of surface hydroxyls on ZnZrOx solid solution catalyst in CO2 hydrogenation to methanol, Chin. J. Catal., 2023, 45, 162–173 CrossRef CAS.
- M. D. Rhodes and A. T. Bell, The effects of zirconia morphology on methanol synthesis from CO and H2 over Cu/ZrO2 catalysts Part I. Steady-state studies, J. Catal., 2005, 233, 198–209 CrossRef CAS.
- P. Ticali, D. Salusso, R. Ahmad, C. Ahoba-Sam, A. Ramirez, G. Shterk, K. A. Lomachenko, E. Borfecchia, S. Morandi, L. Cavallo, J. Gascon, S. Bordiga and U. Olsbye, CO2 hydrogenation to methanol and hydrocarbons over bifunctional Zn-doped ZrO2/zeolite catalysts, Catal. Sci. Technol., 2021, 11, 1249–1268 RSC.
- S. Tada, N. Ochiai, H. Kinoshita, M. Yoshida, N. Shimada, T. Joutsuka, M. Nishijima, T. Honma, N. Yamauchi, Y. Kobayashi and K. Iyoki, Active Sites on ZnZrOx Solid Solution Catalysts for CO2-to-Methanol Hydrogenation, ACS Catal., 2022, 12, 7748–7759 CrossRef CAS.
- Y. Kim, T. S. B. Trung, S. Yang, S. Kim and H. Lee, Mechanism of the Surface Hydrogen Induced Conversion of CO2 to Methanol at Cu(111) Step Sites, ACS Catal., 2016, 6, 1037–1044 CrossRef CAS.
- E. A. Redekop, T. Cordero-Lanzac, D. Salusso, A. Pokle, S. Oien-Odegaard, M. F. Sunding, S. Diplas, C. Negri, E. Borfecchia, S. Bordiga and U. Olsbye, Zn Redistribution and Volatility in ZnZrOx Catalysts for CO2 Hydrogenation, Chem. Mater., 2023, 35, 10434–10445 CrossRef CAS PubMed.
- X. Zhang, X. Yu, R. G. Mendes, P. Matvija, A. E. M. Melcherts, C. Sun, X. Ye, B. M. Weckhuysen and M. Monai, Highly Dispersed ZnO Sites in a ZnO/ZrO2 Catalyst Promote Carbon Dioxide-to-Methanol Conversion, Angew. Chem., Int. Ed., 2024, 202416899 Search PubMed.
- X. Zhang, G. Zhang, X. Zhou, Z. Wang, Y. Liu, J. Zhu, C. Song and X. Guo, Identification of the Active Sites on ZnO/ZrO2 for CO2 Hydrogenation to CO and Methanol, Ind. Eng. Chem. Res., 2023, 62, 21173–21181 CrossRef CAS.
- K. Tanabe and T. Yamaguchi, Acid-base bifunctional catalysis by ZrO2 and its mixed oxides, Catal. Today, 1994, 20, 185–197 CrossRef CAS.
- K. Tanabe, Surface and catalytic properties of ZrO2, Mater. Chem. Phys., 1985, 13, 347–364 CrossRef CAS.
- N. R. Jaegers, V. Danghyan, J. Shangguan, C. Lizandara-Oueyo, P. Deshlahra and E. Iglesia, Heterolytic C–H Activation Routes in Catalytic Dehydrogenation of Light Alkanes on Lewis Acid–Base Pairs at ZrO2 Surfaces, J. Am. Chem. Soc., 2024, 146, 25710–25726 CrossRef CAS PubMed.
- S. Kim, C. A. Jhaveri and E. Sasmaz, Impact of Yttria-Stabilized Zirconia on Direct CO2 Hydrogenation to Light Olefins over a Tandem Catalyst Composed of In2O3/YSZ and SAPO-34, Energy Fuels, 2023, 37, 7361–7371 CrossRef CAS.
- S. G. Giniyatova, A. L. Kozlovskiy, R. I. Shakirzyanov, N. O. Volodina, D. I. Shlimas and D. B. Borgekov, Structural, Dielectric, and Mechanical Properties of High-Content Cubic Zirconia Ceramics Obtained via Solid-State Synthesis, Appl. Sci., 2023, 13, 10989 CrossRef CAS.
- Z. Han, C. Tang, F. Sha, S. Tang, J. Wang and C. Li, CO2 hydrogenation to methanol on ZnO-ZrO2 solid solution catalysts with ordered mesoporous structure, J. Catal., 2021, 396, 242–250 CrossRef CAS.
- K. Lee, U. Anjum, T. P. Araújo, C. Mondelli, Q. He, S. Furukawa, J. Pérez-Ramírez, S. M. Kozlov and N. Yan, Atomic Pd-promoted ZnZrOx solid solution catalyst for CO2 hydrogenation to methanol, Appl. Catal., B, 2022, 304, 120994 CrossRef CAS.
- M. T. Nikolajsen, J.-C. Grivel, A. Gaur, L. P. Hansen, L. Baumgarten, N. C. Schjødt, U. V. Mentzel, J.-D. Grunwaldt, J. Sehested, J. M. Christensen and M. Høj, Surface ZnOx on zirconia is highly active for high temperature methanol synthesis, J. Catal., 2024, 431, 115389 CrossRef CAS.
- F. C. F. Marcos, F. M. Cavalcanti, D. D. Petrolini, L. Lin, L. E. Betancourt, S. D. Senanayake, J. A. Rodriguez, J. M. Assaf, R. Giudici and E. M. Assaf, Effect of operating parameters on H2/CO2 conversion to methanol over Cu-Zn oxide supported on ZrO2 polymorph catalysts: Characterization and kinetics, Chem. Eng. J., 2022, 427, 130947 CrossRef CAS.
- T. Witoon, J. Chalorngtham, P. Dumrongbunditkul, M. Chareonpanich and J. Limtrakul, CO2 hydrogenation to methanol over Cu/ZrO2 catalysts: Effects of zirconia phases, Chem. Eng. J., 2016, 293, 327–336 CrossRef CAS.
- C. Temvuttirojn, Y. Poo-Arporn, N. Chanlek, C. K. Cheng, C. C. Chong, J. Limtrakul and T. Witoon, Role of Calcination Temperatures of ZrO2 Support on Methanol Synthesis from CO2 Hydrogenation at High Reaction Temperatures over ZnOx/ZrO2 Catalysts, Ind. Eng. Chem. Res., 2020, 59, 5525–5535 CrossRef CAS.
- T. Yamamoto and A. Kurimoto, Ga Ion-doped ZrO2 Catalyst Characterized by XRD, XAFS, and 2-Butanol Decomposition, Anal. Sci., 2020, 36, 41–46 CrossRef CAS PubMed.
- K. Anandan, K. Rajesh, K. Gayathri, S. Vinoth Sharma, S. G. Mohammed Hussain and V. Rajendran, Effects of rare earth, transition and post transition metal ions on structural and optical properties and photocatalytic activities of zirconia (ZrO2) nanoparticles synthesized via the facile precipitation process, Phys. E, 2020, 124, 114342 CrossRef CAS.
- P. Lackner, Z. Zou, S. Mayr, U. Diebold and M. Schmid, Using photoelectron spectroscopy to observe oxygen spillover to zirconia, Phys. Chem. Chem. Phys., 2019, 21, 17613–17620 RSC.
- S. Han, D. Zhao, T. Otroshchenko, H. Lund, U. Bentrup, V. A. Kondratenko, N. Rockstroh, S. Bartling, D. E. Doronkin, J.-D. Grunwaldt, U. Rodemerck, D. Linke, M. Gao, G. Jiang and E. V. Kondratenko, Elucidating the Nature of Active Sites and Fundamentals for their Creation in Zn-Containing ZrO2–Based Catalysts for Nonoxidative Propane Dehydrogenation, ACS Catal., 2020, 10, 8933–8949 CrossRef CAS.
- Y. Maithani, J. A. Khan, B. R. Mehta and J. P. Singh, Cubic phase optimization and influence of post-annealing on microstructure, optical, wetting, and nanomechanical properties of zirconia thin films, Ceram. Int., 2023, 49, 1048–1060 CrossRef CAS.
- K. Samson, M. Śliwa, R. P. Socha, K. Góra-Marek, D. Mucha, D. Rutkowska-Zbik, J. F. Paul, M. Ruggiero-Mikołajczyk, R. Grabowski and J. Słoczyński, Influence of ZrO2 Structure and Copper Electronic State on Activity of Cu/ZrO2 Catalysts in Methanol Synthesis from CO2, ACS Catal., 2014, 4, 3730–3741 CrossRef CAS.
- J. Ding, Z. Li, W. Xiong, Y. Zhang, A. Ye and W. Huang, Structural evolution and catalytic performance in CO2 hydrogenation reaction of ZnO-ZrO2 composite oxides, Appl. Surf. Sci., 2022, 587, 152884 CrossRef CAS.
- Z. Zhang, Y. Huang, H. Ma, W. Qian, H. Zhang and W. Ying, Syngas-to-olefins over MOF-derived ZnZrOx and SAPO-34 bifunctional catalysts, Catal. Commun., 2021, 152, 106292 CrossRef CAS.
- D. Wierzbicki, R. Baran, R. Dębek, M. Motak, M. E. Gálvez, T. Grzybek, P. Da Costa and P. Glatzel, Examination of the influence of La promotion on Ni state in hydrotalcite-derived catalysts under CO2 methanation reaction conditions: Operando X-ray absorption and emission spectroscopy investigation, Appl. Catal., B, 2018, 232, 409–419 CrossRef CAS.
- L. J. I. Coleman, W. Epling, R. R. Hudgins and E. Croiset, Ni/Mg-Al mixed oxide catalyst for the steam reforming of ethanol, Appl. Catal., A, 2009, 363, 52–63 CrossRef CAS.
- Z.-Y. Ma, C. Yang, W. Wei, W.-H. Li and Y.-H. Sun, Surface properties and CO adsorption on zirconia polymorphs, J. Mol. Catal. A: Chem., 2005, 227, 119–124 CrossRef CAS.
- K. Taek Jung, Y. G. Shul and A. T. Bell, The Preparation and Surface Characterization of Zirconia Polymorphs, Korean J. Chem. Eng., 2001, 18, 992–999 CrossRef.
- Z. Zhang, L. Zhang, M. J. Hülsey and N. Yan, Zirconia phase effect in Pd/ZrO2 catalyzed CO2 hydrogenation into formate, Mol. Catal., 2019, 475, 110461 CrossRef CAS.
- B. Bachiller-Baeza, I. Rodriguez-Ramos and A. Guerrero-Ruiz, Interaction of Carbon Dioxide with the Surface of Zirconia Polymorphs, Langmuir, 1998, 14, 3556–3564 CrossRef CAS.
- N. H. M. D. Dostagir, C. R. Tomuschat, K. Oshiro, M. Gao, J. Hasegawa, A. Fukuoka and A. Shrotri, Mitigating the Poisoning Effect of Formate during CO2 Hydrogenation to Methanol over Co-Containing Dual-Atom Oxide Catalysts, JACS Au, 2024, 4(3), 1048–1058 CrossRef CAS PubMed.
- F. C. F. Marcos, J. M. Assaf, R. Giudici and E. M. Assaf, Surface interaction of CO2/H2 mixture on mesoporous ZrO2: Effect of crystalline polymorph phases, Appl, Surf. Sci., 2019, 496, 143671 CrossRef CAS.
- S. N. Basahel, T. T. Ali, M. Mokhtar and K. Narasimharao, Influence of crystal structure of nanosized ZrO2 on photocatalytic degradation of methyl orange, Nanoscale Res. Lett., 2015, 10, 73 CrossRef PubMed.
- L. E. Briand, W. E. Farneth and I. E. Wachs, Quantitative determination of the number of active surface sites and the turnover frequencies for methanol oxidation over metal oxide catalysts I. Fundamentals of the methanol chemisorption technique and application to monolayer supported molybdenum oxide catalysts, Catal. Today, 2000, 62, 219–229 CrossRef CAS.
- L. J. Burcham, L. E. Briand and I. E. Wachs, Quantification of Active Sites for the Determination of Methanol Oxidation Turn-over Frequencies Using Methanol Chemisorption and in Situ Infrared Techniques. 2. Bulk Metal Oxide Catalysts, Langmuir, 2001, 17, 6175–6184 CrossRef CAS.
- M. Badlani and I. E. Wachs, Methanol: A “Smart” Chemical Probe Molecule, Catal. Lett., 2001, 75, 137–149 CrossRef CAS.
- T. P. Araújo, J. Morales-Vidal, T. Zou, M. Agrachev, S. P. O. Willi, R. N. Grass, G. Jeschke, S. Mitchell, N. López and J. Pérez-Ramírez, Design of Flame-Made ZnZrOx Catalysts for Sustainable Methanol Synthesis from CO2, Adv. Energy Mater., 2023, 13, 2204122 CrossRef.
- Q. Ren, K. Yang, F. Liu, M. Yao, J. Ma, S. Geng and J. Cao, Role of the structure and morphology of zirconia in ZnO/ZrO2 catalyst for CO2 hydrogenation to methanol, Mol. Catal., 2023, 547, 113280–113280 CrossRef CAS.
- D. Xu, X. Hong and G. Liu, Highly dispersed metal doping to ZnZr oxide catalyst for CO2 hydrogenation to methanol: Insight into hydrogen spillover, J. Catal., 2021, 393, 207–214 CrossRef CAS.
- S. Tsunekawa, S. Ito, Y. Kawazoe and J.-T. Wang, Critical Size of the Phase Transition from Cubic to Tetragonal in Pure Zirconia Nanoparticles, Nano Lett., 2003, 3, 871–875 CrossRef CAS.
- T. Chraska, A. H. King and C. C. Berndt, On the size-dependent phase transformation in nanoparticulate zirconia, Mater. Sci. Eng., A, 2000, 286, 169–178 CrossRef.
- Y. L. Zhang, X. J. Jin, Y. H. Rong, T. Y. Hsu, D. Y. Jiang and J. L. Shi, The size dependence of structural stability in nano-sized ZrO2 particles, Mater. Sci. Eng., A, 2006, 438–440, 399–402 CrossRef.
- S. Jayakumar, P. V. Ananthapadmanabhan, T. K. Thiyagarajan, K. Perumal, S. C. Mishra, G. Suresh, L. T. Su and A. I. Y. Tok, Nanosize stabilization of cubic and tetragonal phases in reactive plasma synthesized zirconia powders, Mater. Chem. Phys., 2013, 140, 176–182 CrossRef CAS.
- A. Bumajdad, A. Abdel Nazeer, F. Al Sagheer, S. Nahar and M. I. Zaki, Controlled Synthesis of ZrO2 Nanoparticles with Tailored Size, Morphology and Crystal Phases via Organic/Inorganic Hybrid Films, Sci. Rep., 2018, 8, 22088 Search PubMed.
- S. Shukla and S. Seal, Mechanisms of room temperature metastable tetragonal phase stabilisation in zirconia, Int. Mater. Rev., 2005, 50, 45–64 CrossRef CAS.
- L. M. G. Rojas, C. A. Huerta-Aguilar, E. Navarrete, E. Llobet and P. Thangarasu, Enhancement of the CO2 Sensing/Capture through High Cationic Charge in M-ZrO2 (Li+, Mg2+, or Co3+): Experimental and Theoretical Studies, ACS Appl. Mater. Interfaces, 2023, 15, 25952–25965 CrossRef PubMed.
- A. O. Zhigachev, V. V. Rodaev, D. V. Zhigacheva, N. V. Lyskov and M. A. Shchukina, Doping of scandia-stabilized zirconia electrolytes for intermediate-temperature solid oxide fuel cell: A review, Ceram. Int., 2021, 47, 32490–32504 CrossRef CAS.
- A. K. Nikumbh and P. V. Adhyapak, Formation characterization and rheological properties of zirconia and ceria-stabilized zirconia, Nat. Sci., 2010, 2, 694–706 CAS.
- C. Colbea, D. Avram, B. Cojocaru, R. Negrea, C. Ghica, V. G. Kessler, G. A. Seisenbaeva, V. Parvulescu and C. Tiseanu, Full Tetragonal Phase Stabilization in ZrO2 Nanoparticles Using Wet Impregnation: Interplay of Host Structure, Dopant Concentration and Sensitivity of Characterization Technique, Nanomaterials, 2018, 8, 988–988 CrossRef PubMed.
- M. Raza, D. Cornil, S. Lucas, R. Snyders, S. Konstantinidis and J. Cornil, Oxygen vacancy stabilized zirconia (OVSZ); a joint experimental and theoretical study, Scr. Mater., 2016, 124, 26–29 CrossRef CAS.
- W. Kim, M. Choi and K. Yong, Generation of oxygen vacancies in ZnO nanorods/films and their effects on gas sensing properties, Sens. Actuators, B, 2015, 209, 989–996 CrossRef CAS.
- K. Bocam, C. Anunmana and T. Eiampongpaiboon, Grain size, crystalline phase and fracture toughness of the monolithic zirconia, J. Adv. Prosthodont., 2022, 14, 285–293 CrossRef PubMed.
- N. C. Horti, M. D. Kamatagi, S. K. Nataraj, M. N. Wari and S. R. Inamdar, Structural and optical properties of zirconium oxide (ZrO2) nanoparticles: effect of calcination temperature, Nano Express, 2020, 1, 01002 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01175a |
| This journal is © The Royal Society of Chemistry 2025 |