
A Lewis basic site rich metal–organic framework featuring a hydrogen-bonded acetylene nano-trap for the efficient separation of C2H2/CO2†
Mengyue
Lu
a,
Zhiwei
Zhao
a,
Yuhao
Tang
a,
Yating
Wang
a,
Feifei
Zhang
*ab,
Jinping
Li
 ab and
Jiangfeng
Yang
*ab
ab and
Jiangfeng
Yang
*ab
aCollege of Chemistry and Chemical Engineering, Taiyuan University of Technology, Taiyuan 030024, Shanxi Province, China. E-mail: zhangfeifei0096@link.tyut.edu.cn; yangjiangfeng@tyut.edu.cn
bCollege of Chemistry and Chemical Engineering, Taiyuan University of Technology, Taiyuan 030024, Shanxi Province, China
First published on 14th January 2025
Abstract
The physical separation of C2H2 from CO2 on metal–organic frameworks (MOFs) has received a substantial amount of research interest due to its advantages of simplicity, security, and energy efficiency. However, the exploitation of ideal MOF adsorbents for C2H2/CO2 separation remains a challenging task due to their similar physical properties and molecular sizes. Herein, we report a unique C2H2 nano-trap constructed using accessible oxygen and nitrogen sites, which exhibits energetic favorability toward C2H2 molecules. This material exhibits a good acetylene capacity of 55.31 cm3 g−1 and high C2H2/CO2 selectivity of 7.0 under ambient conditions. We have combined in situ IR spectroscopy and in-depth theoretical calculations to unravel the synergistic interactions driven by the high density of accessible oxygen and nitrogen sites. Furthermore, dynamic breakthrough experiments confirmed the capability of TUTJ-201Ni for the separation of binary C2H2/CO2 mixtures. This study on Ni-based MOFs will enrich Lewis basic site rich MOFs for gas adsorption and separation applications.
Introduction
Acetylene (C2H2), which is a significant raw material used in chemical production, is of great importance in the petrochemical industry. In general, C2H2 is manufactured via the partial combustion of methane or cracking of hydrocarbons in which carbon dioxide (CO2) will inevitably be present.1,2 On the one hand, C2H2 and CO2 molecules have very similar molecular sizes with kinetic sizes of 3.32 × 3.34 × 5.7 Å (C2H2) and 3.18 × 3.33 × 5.36 Å (CO2), which leads to significant difficulty in their separation.3 In addition, the similar physical properties of C2H2 and CO2, with boiling points of 189.3 and 194.7 K, respectively, also hinder their separation.4 Consequently, there is a pressing need to develop efficient ways to purify C2H2 to better meet the demands for different domains. Currently, liquid absorption and cryogenic distillation processes are the two main techniques used to achieve the efficient purification of C2H2 from CO2. However, these processes are highly energy-intensive and often come with potential safety hazards.5 In this case, adsorption-based separation using porous solid adsorbents has attracted a great deal of research interest due to its lower cost and energy consumption.6 However, conventional adsorbents, such as activated carbon and zeolite, which were developed for the separation of C2H2 and CO2, exhibit relatively low separation selectivity and productivity.7Recently, metal–organic frameworks (MOFs), which are a new generation of high-capacity adsorbents, meet various separation needs and have exerted a huge amount of influence on gas separation and storage due to their high structural designability, tunable pore size, and adjustable internal surface properties.8–15 Many MOFs have been reported for the adsorption separation of C2H2/CO2 in order to fulfill this important task. From previous studies, an effective strategy to improve the C2H2/CO2 separation performance is the construction of strong C2H2 binding sites in the structure.16,17 Among them, C2H2 nano-traps with multiple adsorption sites can provide stronger binding interactions and high recognition capabilities.18–24 Niu et al.25 reported an ultra-strong acetylene nano-trap based on oppositely adjacent open metal sites in MOFs. Taking advantage of the designed C2H2 nano-trap, the anhydrous MOF26 demonstrated the strongest C2H2 binding capability with the largest Qst (79.1 kJ mol−1) and highest uptake at ultra-low pressure (<0.001 bar), as well as benchmark C2H2/CO2 selectivity at room temperature. Although the reported C2H2 nano-traps show a high separation performance, most of them tend to have high C2H2 binding energy, leading to a high desorption energy. Therefore, the construction of a new type of C2H2 nano-trap featuring both low heat of adsorption and high C2H2/CO2 selectivity is urgently needed for large-scale applications.
Accessible oxygen and nitrogen sites on the surface of MOFs have demonstrated the preferential binding of C2H2.27,28 However, previously reported C2H2 sorbents based on isolated oxygen or nitrogen binding sites are limited by their weak C–H⋯O and C–H⋯N interactions and often show low separation performance.29–34 Presumably, a C2H2 nano-trap based on accessible oxygen and nitrogen sites would improve the C2H2 adsorption capacity. More importantly, this new type of C2H2 nano-trap will exhibit excellent C2H2/CO2 separation performance under low C2H2 binding energy due to the weak recognition of CO2 by the oxygen and nitrogen sites. Upon consideration of these factors, we have prepared an ultra-microporous MOF (TUTJ-201Ni) taking advantage of C–H⋯O and C–H⋯N hydrogen bonding as nano-traps for C2H2 molecules, which features multiple accessible oxygen and nitrogen sites to increase the energetic favorability toward C2H2 molecules. The C2H2 and CO2 adsorption properties of TUTJ-201Ni have been characterized using isotherms, the heat of adsorption (Qst), and ideal adsorbed solution theory (IAST) selectivity. Breakthrough experiments were performed to evaluate its C2H2/CO2 separation performance under dynamic conditions. Moreover, theoretical calculations combined with in situ infrared (IR) spectroscopy were performed to further decipher the separation mechanism. Upon combining our experimental and computational results, TUTJ-201Ni can efficiently realize the separation of C2H2 and CO2, thus achieving the goal of enriching the C2H2 stream target.
Experimental section
Analysis of the gas adsorption and separation properties
C2H2 and CO2 single-component gas adsorption and desorption isotherms of the samples were measured at 273 and 298 K using a BSD-660 M analyzer (Beishide Instrument Technology (Beijing) Co., Ltd) applying the volumetric technique to precisely measure the amount of gas adsorbed.In situ IR spectroscopic analysis
The in situ IR spectra of adsorbed C2H2 and CO2 were obtained using a Bruker V70 instrument equipped with an MCT detector, a stainless steel high-temperature in situ IR cell, and a KBr window. The resolution of the IR spectrometer was set at 4 cm−1. For analysis, a specified amount of the adsorbent powder was placed in the sample cup of the high-temperature reaction cell and the powder scraped flat. The TUTJ-201Ni samples were activated at 423 K and 1 × 10−1 mbar for 6 h. During the activation process, the heating and cooling rates of the adsorbent were both 5 K min−1. After the adsorbent was cooled to the test temperature, the IR spectrum of the activated sample was recorded as the background spectrum. The tested gas was passed into the test system and the IR data were collected until the adsorbent was saturated.Breakthrough experiments
The breakthrough curves of TUTJ-201Ni were obtained at a mixed gas flow rate of 5–15 mL min−1. The prepared TUTJ-201Ni (1.2 g) particles were activated at 443 K and 1 × 10−1 mbar for 6 h and then placed in the adsorption column (∅ 4.0 mm × 100 mm) under anhydrous conditions. Before starting the test, the adsorption column was flushed with He gas at a flow rate of 15 mL min−1 and a temperature of 298 K, and the inlet gas was then switched to the test mixture at a total gas flow rate of 5–15 mL min−1. The spectra of the gas at the outlet of the adsorption column were recorded using an online mass spectrometer (HPR-20 EGA, Hiden, with a detection limit of 0.01%) to obtain the final breakthrough curves.Results and discussion
TUTJ-201Ni was synthesized based on our previously reported work. The framework is formed from the hydroxide groups bridging the regular octahedra of nickel atoms and organic linkers (Fig. 1). Each nickel metal center exhibits an octahedral coordination environment via its connection with four oxygen atoms and two nitrogen atoms, where three oxygen atoms and two nitrogen atoms are from five different organic linkers, whereas the remaining oxygen atoms originate from the bridging hydroxide group. TUTJ-201Ni exhibits rhombic-shaped one-dimensional channels ∼6.2 Å along the crystallographic b-axis and the pore walls are decorated by multiple oxygen and nitrogen sites.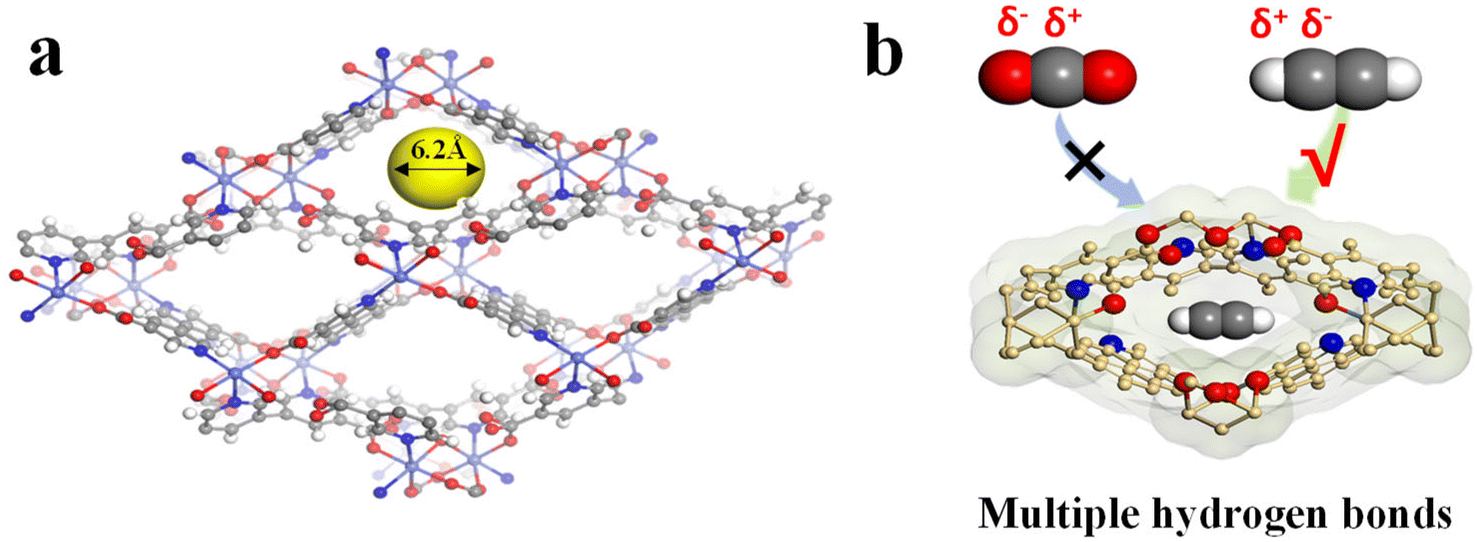 | ||
| Fig. 1 (a) 3D structures showing uniform one-dimensional channels. (b) Selective capture of the target molecule by the hydrogen-bonding nano-trap via host–guest multiple hydrogen-bonding interactions. | ||
Fig. 2a shows the adsorption behavior of TUTJ-201Ni for C2H2 and CO2 measured using the single-component gas adsorption isotherms obtained at 273 and 298 K, respectively. The C2H2 uptake capacity of TUTJ-201Ni was apparently higher than that of CO2. TUTJ-201Ni rapidly takes up C2H2, reaching a value of 36.1 cm3 g−1 at 298 K under 0.1 bar, but has a much lower CO2 uptake capacity of 14.2 cm3 g−1. As the pressure increased and reached 1.0 bar, the uptake of C2H2 reached as high as 55.3 cm3 g−1. In contrast, the CO2 uptake value was only 40.1 cm3 g−1. A comparison of the C2H2 and CO2 adsorption isotherms obtained for TUTJ-201Ni indicated the strong affinity for C2H2 over CO2. The C2H2 sorption amount observed at 298 K and 1.0 bar for TUTJ-201Ni was comparable to the values for some previously reported MOFs, including Cu@FAU (33.8 cm3 g−1),35 FJU-36 (52.2 cm3 g−1),36 ZNU-11 (45.5 cm3 g−1),37 NKMOF-1-Ni (61.0 cm3 g−1),38 Cu(bpy)NP (50.7 cm3 g−1),39 and NbU-7 (47.3 cm3 g−1)40 (Table S3†). The C2H2/CO2 uptake ratio reached up to 1.37 at 298 K and 1.0 bar, which is higher than the values previously reported for TIFSIX-2-Cu-I (1.05),41 MUF-17 (1.2),42 and Cu-AD-GA (1.28).43 In addition, successive adsorption/desorption cycles were measured at 298 K and a testing pressure of up to 1.0 bar without a regeneration process. TUTJ-201Ni exhibits excellent adsorption recyclability for C2H2 and CO2, as demonstrated by our cyclic C2H2 and CO2 adsorption measurements (Fig. 2b). To probe the porous nature of TUTJ-201Ni, we produced nitrogen adsorption and desorption isotherms of the samples at 77 K (Fig. S6†). The resulting N2 adsorption isotherms exhibit a typical type-I behavior, validating its inherent micro-porous structures.
 | ||
| Fig. 2 (a) C2H2 and CO2 adsorption isotherms obtained for TUTJ-201Ni at 273 and 298 K. (b) The recycling performance of TUTJ-201Ni obtained at 298 K and a testing pressure up to 1.0 bar without the regeneration process. (c) IAST selectivity of C2H2/CO2. (d) Qst curves calculated for C2H2 and CO2 sorption. (e) A comparison of the Qst value at zero coverage and IAST selectivity at 1 bar with other porous materials. | ||
To determine the separation performance of TUTJ-201Ni, the C2H2/CO2 selectivity was calculated using ideal adsorbed solution theory (IAST). Fig. 2c shows that the selectivity increases as the pressure decreases. At 298 K and 1.0 bar, the calculated C2H2/CO2 selectivity for the corresponding binary equimolar mixture is up to 7.0. This value is higher than those of most of the previously reported materials, including FJU-83 (4.8),44 CoV-bdc-ppy (2.7),45 Cu-AD-GA (2.6),43 and CAU-23 (3.8).46
The Qst values of C2H2 and CO2 were further calculated using the dual-site Langmuir–Freundlich model in order to evaluate the interactions formed between the adsorbents and gases (Fig. 2d). The Qst values of C2H2 were in the range of 28.86–34.66 kJ mol−1, which is notably higher than those of CO2 (27.46–29.84 kJ mol−1). The near zero coverage Qst values of C2H2 and CO2 are 34.66 and 29.84 kJ mol−1, respectively, which are in accordance with the above-mentioned gas adsorption results. The higher Qst observed for C2H2 over CO2 indicates the strong affinity of TUTJ-201Ni toward C2H2. The Qst value of C2H2 near zero coverage was lower than those of the previously reported top materials SIFSIX-DPA-Cu-I (46.53),47 ATC-Cu (79.1),25 UTSA-300a (57.6),48 and JXNU-18 (37.4)49 (Fig. 2e). From an industrial application perspective, the moderate adsorption enthalpy implies the potential to achieve regeneration under mild conditions, showing its promising potential for energy-friendly C2H2 purification from mixtures.
In order to better understand the ultra-high C2H2 storage density and separation performance of TUTJ-201Ni, Grand Canonical Monte Carlo (GCMC) simulations were performed to investigate the different binding sites of C2H2 and CO2. Fig. 3a and b show that the C2H2 and CO2 gas molecules both appear around the center of the rhombic pore, indicating that the pore centers were the most energetically favorable binding sites for both adsorbates. The density of C2H2 adsorbed in TUTJ-201Ni is obviously higher than that of CO2 and is in full agreement with their adsorption capacities. Subsequently, DFT calculations were carried out to directly visualize the locations of C2H2 and CO2 within TUTJ-201Ni. Simulations show that multiple binding sites existed. Fig. 3c and d show that the C2H2 molecule was preferentially adsorbed at the center of the MOF pore, where it formed three cooperative C–H⋯O interaction and one C–H⋯N hydrogen-bonding interaction with the nearby uncoordinated oxygen and nitrogen atoms with interaction distances ranging from 2.53 to 4.08 Å. The uncoordinated accessible oxygen and nitrogen sites can form preferential C2H2 binding interactions with the acidic H atoms via H-bonding and such interactions usually have moderate sorption heats according to the literature.50 Simulations show that multiple binding sites existed. In this case, plenty of carboxylate oxygen atoms were exposed on the pore walls. In contrast to C2H2, each CO2 molecule could only form four weak C–O⋯H interactions with distances of 3.31–4.45 Å. Such oxygen atoms can form hydrogen bonds with the acidic H atoms of C2H2 molecules but are unfavorable for adsorbing CO2 molecules with two electronegative O atoms due to the electrostatic repulsion interaction. Such subtle differences between C2H2 and CO2 can be maximized by the MOF bearing the Lewis base sites, thus enabling C2H2-selective C2H2/CO2 separation. The binding energies for the framework–C2H2 and framework–CO2 were calculated to be 34.66 and 29.84 kJ mol−1, which confirmed a stronger interaction formed between the framework and C2H2 molecules, and this order was consistent with our experimental data. In situ IR spectroscopy was carried out to probe further the interactions of C2H2 within TUTJ-201Ni. Fig. 4 shows the change in the stretching bands upon C2H2 loading. The adsorption bands at 3237 cm−1 that were assigned to ν(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H) gradually increased with dosing C2H2, indicating the interaction of C2H2 molecules with the framework via C–H⋯O and C–H⋯N. This weak interaction leads to changes in the intensity of the C–H peak in the in situ IR spectra. Similarly, ν(C
C–H) gradually increased with dosing C2H2, indicating the interaction of C2H2 molecules with the framework via C–H⋯O and C–H⋯N. This weak interaction leads to changes in the intensity of the C–H peak in the in situ IR spectra. Similarly, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) = 2333 cm−1 proves the presence of C2H2 and CO2 adsorbed in the MOF. Upon adsorption of C2H2 on TUTJ-201Ni, the intensity of the stretching band was significantly increased. This may be attributed to the high gas adsorption capacity of this material.
O) = 2333 cm−1 proves the presence of C2H2 and CO2 adsorbed in the MOF. Upon adsorption of C2H2 on TUTJ-201Ni, the intensity of the stretching band was significantly increased. This may be attributed to the high gas adsorption capacity of this material.
 | ||
| Fig. 3 (a) and (b) Density distributions of C2H2 and CO2 within TUTJ-201Ni at 298 K; the red dashed circles indicate the favorable adsorption sites. (c) and (d) The DFT-optimized adsorption configurations of C2H2 and CO2 in TUTJ-201Ni. | ||
 | ||
| Fig. 4 In situ IR spectra obtained for C2H2 (a) and CO2 (b) adsorbed on TUTJ-201Ni with different gas dosing durations at 298 K. | ||
Dynamic breakthrough experiments for an equimolar C2H2/CO2 mixture were carried out under different conditions in order to verify the adsorptive separation performance of binary mixtures under continuous flow conditions. Fig. 5a shows the dynamic breakthrough experiments carried out for an equimolar C2H2/CO2 mixture at a total flow rate of 5 mL min−1 under ambient conditions. CO2 first penetrated the adsorption bed and C2H2 was detected after a long period of time. The difference between the breakthrough times of the two gases reached 25.0 min, which offers a large operating window for C2H2 release during the pressure-swing adsorption process. Furthermore, five continuous cycling breakthrough experiments were performed on TUTJ-201Ni. No appreciable change in the retention time was observed, proving its good recyclability for C2H2/CO2 separation. After each cycle, the TUTJ-201Ni in the column can be readily regenerated within 30 min upon purging with He. To further achieve the target of practical applications, we also investigated the C2H2/CO2 separation performance at different gas flow rates of 10.0 and 15.0 mL min−1 (Fig. 5b). Although the breakthrough time decreased when the flow rate increased, the separation performance remained almost unchanged. Fig. 5c shows the breakthrough curves obtained under different temperatures and the results indicate that TUTJ-201Ni exhibited an excellent separation performance. In addition, the separation capacity of this material for high concentrations of C2H2 was also tested (Fig. 5d). When the C2H2/CO2 ratio was adjusted to 75/25 and 90/10, TUTJ-201Ni still exhibited a good separation effect. All of the breakthrough curves under various separation scenarios demonstrated that TUTJ-201Ni can effectively separate C2H2 and CO2.
 | ||
| Fig. 5 (a) Experimental breakthrough cycles obtained for C2H2/CO2 (50/50) separation on TUTJ-201Ni at 298 K and 1.0 bar. (b) Breakthrough curves obtained for TUTJ-201Ni with C2H2/CO2 (50/50) mixtures at 298 K and flow rates of 5, 10, and 15 mL min−1. (c) Experimental breakthrough curves obtained for TUTJ-201Ni during the separation of C2H2/CO2 (50/50) at 1.0 bar and different temperatures. (d) Experimental breakthrough curves obtained for 50/50, 75/25, and 90/10 mixtures at 298 K and 1.0 bar. | ||
Considering that the C2H2 raw stream is produced from the combustion of natural gas or cracking of hydrocarbons, the C2H2/CO2 separation process will be performed under harsh conditions in industrial applications. This requires adsorbent candidates with extremely high and long-term pH stability. Thus, we investigated the chemical stability of TUTJ-201Ni after treatment under different conditions. Fig. S3 and S4† show that TUTJ-201Ni exhibits no loss of crystallinity and no phase change after exposure to air for 1 week and different pH aqueous solutions using PXRD, indicating its good chemical stability.
Conclusions
In summary, we reported an oxygen and nitrogen based C2H2 nano-trap for the separation of C2H2 and CO2. The high C2H2 uptake capacity, remarkable C2H2/CO2 IAST selectivity, prominent experimental breakthrough performance, and theoretical calculation indicate that this C2H2 nano-trap is an ideal acetylene trapper for practical applications. Our work enriches the study of Lewis basic site rich MOFs with hydrogen-bonded acetylene nano-traps and provides an efficient functional porous adsorbent to address challenging gas separation applications.Author contributions
MYL: investigation, visualization, and writing original draft; ZWZ and YHT: investigation and visualization; YTW: visualization, validation and review; FFZ: in charge of conceptualization, data analysis, writing – review and editing. JPL and JFY: supervised and administered the project.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22408258, 22478272 and 22378287) and the Natural Science Foundation of Shanxi Province (No. 202303021222012).References
- Y. P. Li, Y. Wang, Y. Y. Xue, H. P. Li, Q. G. Zhai, S. N. Li, Y. C. Jiang, M. C. Hu and X. Bu, Angew. Chem., Int. Ed., 2019, 58, 13590–13595 CrossRef CAS PubMed.
- M. L. Foo, R. Matsuda, Y. Hijikata, R. Krishna, H. Sato, S. Horike, A. Hori, J. Duan, Y. Sato, Y. Kubota, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2016, 138, 3022–3030 CrossRef CAS PubMed.
- C. R. Reid and K. M. Thomas, J. Phys. Chem., 2001, 105, 10619–10629 CrossRef.
- S. C. Fan, Y. T. Li, Y. Wang, J. W. Wang, Y. Y. Xue, H. P. Li, S. N. Li and Q. G. Zhai, Inorg. Chem., 2021, 60, 18473–18482 CrossRef PubMed.
- W. Gong, H. Cui, Y. Xie, Y. Li, X. Tang, Y. Liu, Y. Cui and B. Chen, J. Am. Chem. Soc., 2021, 143, 14869–14876 CrossRef PubMed.
- B. Xing, S. Q. Yang, Q. Zhang and T. L. Hu, Dalton Trans., 2024, 53, 6993–6999 RSC.
- R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature, 2005, 436, 238–241 CrossRef PubMed.
- R. B. L. L. Li, R. Krishna, H. Li, S. Xiang, H. Wu, J. Li, W. Zhou and B. Chen, Science, 2018, 362, 443–446 CrossRef PubMed.
- X. Wang, H. Liu, Y. Li, X. Yang, F. Gao, X. Wang, Z. Kang, W. Fan and D. Sun, Coord. Chem. Rev., 2023, 482, 215093 CrossRef.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- C. B. Bai and D. Zhao, Science, 2020, 369, 372–373 CrossRef PubMed.
- R. B. Lin, S. Xiang, W. Zhou and B. Chen, Chem, 2020, 6, 337–363 Search PubMed.
- L. Zhou, P. Brantuas, A. Henrique, H. Reinsch, M. Wahiduzzaman, J. M. Greneche, A. E. Rodrigues, J. A. C. Silva, G. Maurin and C. Serre, Angew. Chem., Int. Ed., 2024, 63, e202320008 CrossRef.
- J. X. Wang, C. C. Liang, X. W. Gu, H. M. Wen, C. Jiang, B. Li, G. Qian and B. Chen, J. Mater. Chem. A, 2022, 10, 17878–17916 RSC.
- S. Q. Yang, T. L. Hu and B. Chen, Sci. China: Chem., 2023, 66, 2181–2203 CrossRef CAS.
- S. Q. Yang, R. Krishna, H. Chen, L. Li, L. Zhou, Y. F. An, F. Y. Zhang, Q. Zhang, Y. H. Zhang, W. Li, T. L. Hu and X. H. Bu, J. Am. Chem. Soc., 2023, 145, 13901–13911 CrossRef CAS PubMed.
- X. L. Cui, K. J. Chen, H. B. Xing, Q. W. Yang, R. Krishna, Z. B. Bao, H. Wu, W. Zhou, X. L. Dong, Y. Han, B. Li, Q. L. Ren, M. J. Zaworotko and B. L. Chen, Science, 2016, 353, 141–144 CrossRef CAS PubMed.
- L. Yang, X. Cui, Q. Yang, S. Qian, H. Wu, Z. Bao, Z. Zhang, Q. Ren, W. Zhou, B. Chen and H. Xing, Adv. Mater., 2018, 30, 1705374 CrossRef PubMed.
- M. Wriedt, J. P. Sculley, A. A. Yakovenko, Y. Ma, G. J. Halder, P. B. Balbuena and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 9804–9808 CrossRef CAS.
- O. T. Qazvini, R. Babarao, Z. L. Shi, Y. B. Zhang and S. G. Telfer, J. Am. Chem. Soc., 2019, 141, 5014–5020 CrossRef CAS.
- Z. Niu, X. Cui, T. Pham, P. C. Lan, H. Xing, K. A. Forrest, L. Wojtas, B. Space and S. Ma, Angew. Chem., Int. Ed., 2019, 58, 10138–10141 CrossRef CAS PubMed.
- H. G. Hao, Y. F. Zhao, D. M. Chen, J. M. Yu, K. Tan, S. Ma, Y. Chabal, Z. M. Zhang, J. M. Dou, Z. H. Xiao, G. Day, H. C. Zhou and T. B. Lu, Angew. Chem., Int. Ed., 2018, 57, 16067–16071 CrossRef CAS.
- X. Cui, Q. Yang, L. Yang, R. Krishna, Z. Zhang, Z. Bao, H. Wu, Q. Ren, W. Zhou, B. Chen and H. Xing, Adv. Mater., 2017, 29, 1606929 CrossRef.
- L. Yang, L. Yan, Y. Wang, Z. Liu, J. He, Q. Fu, D. Liu, X. Gu, P. Dai, L. Li and X. Zhao, Angew. Chem., Int. Ed., 2021, 60, 4570–4574 CrossRef CAS.
- Z. Niu, X. Cui, T. Pham, G. Verma, P. C. Lan, C. Shan, H. Xing, K. A. Forrest, S. Suepaul, B. Space, A. Nafady, A. M. Al-Enizi and S. Ma, Angew. Chem., Int. Ed., 2021, 60, 5283–5288 CrossRef CAS.
- Z. Niu, X. Cui, T. Pham, G. Verma, P. C. Lan, C. Shan, H. Xing, K. A. Forrest, S. Suepaul, B. Space, A. Nafady, A. M. Al-Enizi and S. Ma, Angew. Chem., Int. Ed., 2021, 60, 5283–5288 CrossRef CAS PubMed.
- Z. Zhang, S. B. Peh, Y. Wang, C. Kang, W. Fan and D. Zhao, Angew. Chem., Int. Ed., 2020, 59, 18927–18932 CrossRef.
- G. D. Wang, Y. Z. Li, W. F. Zhang, L. Hou, Y. Y. Wang and Z. Zhu, ACS Appl. Mater. Interfaces, 2021, 13, 58862–58870 CrossRef PubMed.
- H.-M. Wen, H. Wang, B. Li, Y. Cui, H. Wang, G. Qian and B. Chen, Inorg. Chem., 2016, 55, 7214–7218 CrossRef PubMed.
- Y. Zhang, X. Deng, X. Li, X. Liu, P. Zhang, L. Chen, Z. Yan, J. Wang and S. Deng, Sep. Purif. Technol., 2023, 316, 123751 CrossRef.
- C. Hao, H. Ren, H. Zhu, Y. Chi, W. Zhao, X. Liu and W. Guo, Sep. Purif. Technol., 2022, 290, 120804 CrossRef.
- C. Song, F. Zheng, Y. Liu, Q. Yang, Z. Zhang, Q. Ren and Z. Bao, Angew. Chem., Int. Ed., 2023, 62, e20231385 Search PubMed.
- P. Yan, J. Yang, D. Ma, S. Zhang, S. Zeng, L. Feng, S. Zhan and Q. Lin, Microporous Mesoporous Mater., 2020, 293, 109777 CrossRef.
- P. D. Zhang, X. Q. Wu, Q. Shuai, J. Yu, X. Zhang and J.-R. Li, ACS Mater. Lett., 2024, 6, 4632–4638 CrossRef.
- H. Li, C. Liu, C. Chen, Z. Di, D. Yuan, J. Pang, W. Wei, M. Wu and M. Hong, Angew. Chem., Int. Ed., 2021, 60, 7547–7552 CrossRef PubMed.
- L. Liu, Z. Yao, Y. Ye, L. Chen, Q. Lin, Y. Yang, Z. Zhang and S. Xiang, Inorg. Chem., 2018, 57, 12961–12968 CrossRef.
- Y. Han, Y. Jiang, J. Hu, L. Wang and Y. Zhang, Sep. Purif. Technol., 2024, 332, 125777 CrossRef.
- Y. L. Peng, T. Pham, P. Li, T. Wang, Y. Chen, K. J. Chen, K. A. Forrest, B. Space, P. Cheng, M. J. Zaworotko and Z. Zhang, Angew. Chem., Int. Ed., 2018, 57, 10971–10975 CrossRef PubMed.
- Y. Liu, J. H. Liu, H. T. Xiong, J. W. Chen, S. X. Chen, Z. L. Zeng, S. G. Deng and J. Wang, Nat. Commun., 2022, 13, 200 Search PubMed.
- J. Wu, Y. L. Wang, J. P. Xue, D. P. Wu and J. Li, Inorg. Chem., 2023, 62, 19997–20004 CrossRef CAS PubMed.
- K. J. Chen, H. S. Scott, D. G. Madden, T. Pham, A. Kumar, A. Bajpai, M. Lusi, K. A. Forrest, B. Space, J. J. Perry and M. J. Zaworotko, Chem, 2016, 1, 753–765 CAS.
- O. T. Qazvini, R. Babarao and S. G. Telfer, Chem. Mater., 2019, 31, 4919–4926 CrossRef CAS.
- Y. Zhang, W. Sun, B. Luan, J. Li, D. Luo, Y. Jiang, L. Wang and B. Chen, Angew. Chem., Int. Ed., 2023, 62, e202309925 CrossRef CAS.
- X. B. Xu, Y. Yang, Z. Yuan, F. Zheng, F. Xiang, Z. Bao, Z. Zhang and S. Xiang, Sep. Purif. Technol., 2023, 325, 124624 CrossRef.
- W. Wang, H. Yang, Y. Chen, X. Bu and P. Feng, J. Am. Chem. Soc., 2023, 145, 17551–17556 CrossRef CAS.
- Y. Ye, S. Xian, H. Cui, K. Tan, L. Gong, B. Liang, T. Pham, H. Pandey, R. Krishna, P. C. Lan, K. A. Forrest, B. Space, T. Thonhauser, J. Li and S. Ma, J. Am. Chem. Soc., 2021, 144, 1681–1689 CrossRef.
- J. J. You, H. P. Wang, T. T. Xiao, X. Y. Wu, L. Zhang and C. Z. Lu, Chem. Eng. J., 2023, 477, 147001 CrossRef CAS.
- R. B. Lin, L. Li, H. Wu, H. Arman, B. Li, R. G. Lin, W. Zhou and B. Chen, J. Am. Chem. Soc., 2017, 139, 8022–8028 CrossRef CAS PubMed.
- X. P. Fu, X. Y. Le, Y. H. Xiao, D. M. Zeng, K. A. Zhou, L. Huang, Y. L. Wang and Q. Y. Liu, Inorg. Chem., 2023, 62, 15031–15038 CrossRef CAS PubMed.
- M. Y. Gao, D. Sensharma, A. A. Bezrukov, Y. H. Andaloussi, S. Darwish, C. Deng, M. Vandichel, J. Zhang and M. J. Zaworotko, Small, 2023, 19, e2206945 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03411b |
| This journal is © The Royal Society of Chemistry 2025 |