
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a series of rare-earth-based multi-anion chalcogenide iodides RE3Si2SexS8−xI (RE = La, Ce, Pr, and Nd) using the flux-assisted boron–chalcogen mixture method†
Gopabandhu
Panigrahi
,
Habiba Binte
Kashem
,
Gregory
Morrison
 and
Hans-Conrad
zur Loye
*
and
Hans-Conrad
zur Loye
*
Department of Chemistry and Biochemistry, University of South Carolina, Columbia, SC 29208, United States. E-mail: zurloye@mailbox.sc.edu
First published on 12th March 2025
Abstract
Single crystals and polycrystalline powders of rare earth mixed chalcogenide iodides La3Si2Se1.21S6.79I, Ce3Si2Se1.39S6.61I, Pr3Si2Se1.22S6.78I, and Nd3Si2Se1.18S6.82I were prepared using the reactive flux-assisted boron–chalcogen mixture (BCM) method at 850 °C. All compounds crystallized in the monoclinic crystal system, space group C2/c (space group number 15). The series adopts the La3Si2O8Cl structure type, containing isolated SiQ4 tetrahedra connected by REQ8 (RE = La, Ce, Pr and Nd) polyhedra; this arrangement creates tunnels that are filled by I atoms. The partial substitution of S by Se was carried out to modulate the optical properties. Phase pure samples and uniform solid solutions were obtained for all compositions as determined using powder X-ray diffraction patterns. Polycrystalline powders were used for physical property measurements, including magnetic susceptibility and UV-Vis diffuse reflectance. The solid-state UV-Vis data for the polycrystalline La3Si2Se1.21S6.79I, Ce3Si2Se1.39S6.61I, and Pr3Si2Se1.22S6.78I samples revealed band gaps of Eg = 2.5(1), 2.2(1), and 2.3(1) eV, typical of semiconductors. Magnetic measurements indicated that Ce3Si2Se1.39S6.61I and Nd3Si2Se1.18S6.82I exhibit paramagnetic behavior with slightly negative Weiss constants θ = −25 and −38. The photoluminescence spectrum of Ce3Si2Se1.39S6.61I exhibits a broad emission band around ∼493 nm.
Introduction
Rare-earth chalcogenides exhibit diverse structural chemistry and display a broad range of physical properties, primarily due to their 4f electrons and, at times, due to their stability in higher coordination environments. This combination of factors leads to a variety of intriguing physical behaviors, making rare-earth chalcogenides promising candidates for different applications, including use as phosphors, magnetic materials, magneto-optical devices, and optical materials for various applications.1–5 The physical properties of rare-earth chalcogenides can be further altered by modifying their compositions, particularly through the incorporation of mixed cations or mixed anions.The introduction of more than one type of anion into metal chalcogenides can significantly impact both the structure and the properties of the resulting materials. This enhancement often leads to novel characteristics that are unattainable in materials with only a single anion or in mixed-cation compounds,6 where examples include transparent conductors, inorganic pigments, luminescent materials, cathode materials in batteries, thermoelectric materials, and even superconductors.7–14 To date, however, mixed-anion compounds that incorporate heavier chalcogenides alongside halides remain relatively rare. In the quaternary system RE-Tt-Q-X (where RE = rare-earth metals, Tt = group 14 elements Si, Ge, or Sn, Q = chalcogenides S, Se, or Te, and X = halides Cl, Br, or I), only a few quaternary phases have been reported.15–20 Similarly, compounds that specifically explore mixed chalcogenide–halide compositions are rare. One notable system where this has been studied is Ce3Si2(S1−ySey)8I,21 in which sulfur (S) is partially or fully substituted by selenium (Se), resulting in three different compositions. This appears to be the only system in which this has been reported. The three distinct compositions each exhibit a striking change in the luminescence emission as sulfur is progressively replaced by selenium. This color tunability, driven by anion substitution, illustrates the type of optical control that can be achieved in such mixed-anion systems, pointing to their potential for use in advanced optical materials.
Motivated by this, we extended our research to explore a broader range of lanthanide-based compounds that incorporate mixed chalcogens in similar RE3Si2S8X-type structures. Our focus was to further understand and expand the scope of Ce3Si2(S1−ySey)8I by synthesizing different mixed chalcogenide materials, denoted as RE3Si2SexS8−xI, where RE = La, Ce, Pr and Nd, and sulfur is partially substituted by selenium. The synthesis of these mixed-anion compounds typically involves solid-state methods starting with rare earth metals that can lead to materials with tailored properties.22 In contrast, our synthesis used rare-earth oxide precursors and sodium iodide (NaI) as a reactive flux.23,24 The use of NaI as a flux in the reaction promotes the incorporation of the iodide anion into the final structure, facilitating the formation of chalcogenide-iodide compounds. Herein we describe their crystal structures and physical properties including magnetic behavior, UV-Vis absorption, photoluminescence, and thermal stability.
Experimental section
RE2O3 (RE = La and Nd) (99.9%, Alfa Aesar), CeO2 (99.9%, Alfa Aesar), Pr6O11 (99.9%, Alfa Aesar), sulfur powder (99.5% Fisher Scientific), selenium powder (99.999% Fisher Scientific), boron (crystalline 100 mesh, 99.9%, Beantown Chemicals), and SiO2 (amorphous powder, 99.9%, Alfa Aesar). The NaI used for synthesis was stored in a drying oven set to 260 °C to maintain the anhydrous state of the salts.The single crystals of La3Si2Se1.21S6.79I and Nd3 Si2Se1.18S6.82I were synthesized by the addition of 50 mg of RE2O3 (RE = La and Nd), 5 mg of SiO2, 40 mg of S, 10 mg of Se, 20 mg of B and 250 mg of NaI into heavily carbon coated fused silica tubes (12 mm OD and 10 mm ID). For the single crystal synthesis of Ce3Si2Se1.39S6.61I and Pr3Si2Se1.22S6.78I, 50 mg of CeO2 or 50 mg of Pr6O11 were used as the rare earth source. The carbon coated fused silica tubes were evacuated to 10−4 torr and flame sealed using a methane/oxygen torch. The sealed fused silica tubes were placed into a programmable muffle furnace set to heat to 850 °C in 20 h, dwell at this temperature for 20 h, and cool to 550 °C in 20 h, at which point the furnace was shut off and allowed to cool down to room temperature.
The polycrystalline sample of RE3Si2SexS8−xI synthesized by the addition of 150 mg of RE2O3, 150 mg of CeO2, or 150 mg of Pr6O11, 15 mg of SiO2, 120 mg of S, 30 mg of Se, 60 mg of B and 700 mg of NaI into a heavily carbon coated fused silica tube (12 mm OD and 10 mm ID). The heating profile for the synthesis of polycrystalline materials followed the same heating profile explained above.
The resulting products exhibited stability to air and moisture. They were washed three times with methanol and sonicated in methanol to remove any remaining flux. The phase purity of the compounds was confirmed by powder X-ray diffraction analysis and these samples were used for physical property measurements (Fig. 1).
 | ||
| Fig. 1 Optical microscopy images of (a) La3Si2Se1.21S6.79I, (b) Ce3Si2Se1.39S6.61I, (c) Pr3Si2Se1.22S6.78I and (d) Nd3 Si2Se1.18S6.82I. | ||
Caution: Boron sulfides or boron selenides that might form during the syntheses described above are prone to moisture sensitivity, leading to the generation of H 2 S/H2Se gas upon contact with water or moisture. Therefore, all reaction procedures must be conducted within fume hoods, with strict adherence to safety protocols.
Single crystal X-ray diffraction (SCXRD)
X-ray intensity data from crystals of RE3Si2SexS8−xI were collected at 300(2) K using a Bruker D8 QUEST diffractometer equipped with a PHOTON-II area detector and an Incoatec microfocus source (Mo Kα radiation, λ = 0.71073 Å).25 The working voltage and operating currents were 50 kV and 1.4 mA, respectively, throughout the experiment. The detector to crystal distance of 40 mm was fixed, and an exposure time of 10 s per frame was used for data collection. The raw area detector data frames were reduced and corrected for absorption effects using the SAINT+ and SADABS programs.26 Final unit cell parameters were determined by least-squares refinement over a large number (>9000) of reflections taken from the data set. An initial structural model was obtained using SHELXT.27 Subsequent difference Fourier calculations and full-matrix least-squares refinement against F2 were performed using SHELXL-2018.28All RE3Si2SexS8−xI compounds crystallize in the monoclinic crystal system in the space group C2/c. The space group C2/c was chosen based on the Laue symmetry, intensity statistics, and systematic absences.
The asymmetric unit consists of two lanthanum atoms (RE = La, Ce, Pr and Nd), one silicon atom, one iodine and four mixed sulfur/selenium atomic positions. All atoms are located on the Wyckoff site 8f, except for RE2 and I, which are located on a site 4e. The mixed S/Se sites were identified from the behavior of the displacement parameters, which became abnormally small if refined as 100% sulfur or abnormally large if refined as 100% Se. Atomic positions were standardized using the program STRUCTURE TIDY. No additional symmetry was detected by the ADDSYM program in PLATON.29,30 Trial refinements of the site occupancy parameters then confirmed this, with each site refining to greater than 100% S or to less than 100% Se. For the final model, each site was constrained to 100% total occupancy with the S/Se ratio allowed to vary. Distinct sulfur and selenium positions could not be resolved. All atoms were refined with anisotropic displacement parameters. The largest residual electron density peak and hole in the final difference map, site occupancy, crystallographic and refinement data pertinent to all title compounds are provided in Table 1 and Table S1,† and interatomic distances are listed in Table S2.†
| Chemical formula | La3Si2Se1.21S6.79I | Ce3Si2Se1.39S6.61I | Pr3Si2Se1.22S6.78I | Nd3Si2Se1.18S6.82I |
|---|---|---|---|---|
| Formula weight | 913.16 | 925.11 | 919.63 | 927.50 |
| Crystal system, space group | Monoclinic, C2/c | Monoclinic, C2/c | Monoclinic, C2/c | Monoclinic, C2/c |
| Temperature (K) | 298 | 298 | 297 | 300 |
| a, b, c (Å) | 16.1720(3), 7.9517(1), 10.9848(2) | 16.0737(3), 7.8940(1), 10.9299(2) | 15.9823(4), 7.8496(1), 10.8814(2) | 15.9313(4), 7.8058(2), 10.8413(2) |
| β (°) | 97.702(1) | 97.598(1) | 97.698(1) | 97.768(1) |
| V (Å3) | 1399.85(4) | 1374.67(4) | 1352.82(5) | 1335.82(5) |
| Z | 4 | 4 | 4 | 4 |
| μ (mm−1) | 15.49 | 16.83 | 17.38 | 18.21 |
| Crystal size (mm) | 0.07 × 0.05 × 0.04 | 0.07 × 0.04 × 0.03 | 0.07 × 0.02 × 0.02 | 0.13 × 0.03 × 0.02 |
| Absorption correction | Multi-scan (“SADABS” Sheldrick, 2016/2) | Multi-scan (“SADABS” Sheldrick, 2016/2) | Multi-scan (“SADABS” Sheldrick, 2016/2) | Multi-scan (“SADABS” Sheldrick, 2016/2) |
| T min, Tmax | 0.390, 0.495 | 0.360, 0.495 | 0.342, 0.495 | 0.261, 0.495 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 665, 2135, 2061 665, 2135, 2061 |
38987, 2099, 2010 |
35512, 2055, 2030 |
46262, 2422, 2387 |
| R int | 0.031 | 0.028 | 0.026 | 0.034 |
| (sin θ/I)max (Å−1) | 0.714 | 0.714 | 0.714 | 0.758 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.010, 0.042, 1.47 | 0.010, 0.022, 1.08 | 0.008, 0.019, 1.26 | 0.011, 0.024, 1.22 |
| Δρmax, Δρmin (e Å−3) | 0.99, −1.13 | 1.09, −0.58 | 0.62, −0.38 | 1.49, −0.50 |
Powder X-ray diffraction (PXRD)
Powder X-ray diffraction (PXRD) data were obtained using finely ground powder samples of La3Si2Se1.21S6.79I, Ce3Si2Se1.39S6.61I, Pr3Si2Se1.22S6.78I and Nd3Si2Se1.18S6.82I prepared using the flux assisted BCM synthesis method. PXRD data were collected on a Bruker D2 PHASER diffractometer using Cu-Kα radiation (λ = 1.5418 Å) over the 2θ range of 5–65° with a step size of 0.02°.Energy-dispersive X-ray spectroscopy (EDS)
EDS data were collected on single crystals by mounting them directly onto a SEM stub using conducting carbon tape. Qualitative elemental analyses were performed utilizing a Tescan Vega-3 SEM instrument, which was fitted with a Thermo EDS attachment. The SEM operated in a low-vacuum mode with an accelerating voltage of 15–20 kV and an accumulation time of 30 seconds. The summarized EDS results can be found in the ESI (Table S3†).UV-visible spectroscopy
The UV-visible reflectance spectra of polycrystalline samples of La3Si2Se1.21S6.79I, Ce3Si2Se1.39S6.61I, and Pr3Si2Se1.22S6.78I were recorded using a PerkinElmer lambda 35 UV-Vis spectrophotometer. The spectrophotometer was operated in the diffuse reflectance mode and was equipped with an integrating sphere. Reflectance data were converted internally to absorbance via the Kubelka–Munk function. Spectra were recorded over the 200–900 nm range at room temperature under ambient conditions on the powder sample.31Thermogravimetric analysis (TGA)
TGA was performed on polycrystalline powder samples of Ce3Si2Se1.39S6.61I using an SDT Q600 thermogravimetric analyzer. An alumina pan was used as the sample holder. The samples were heated from room temperature to 500 °C at a constant rate of 5 °C min−1 in nitrogen environments with a controlled flow rate of 100 mL min−1. After TGA, the resulting powders were further analyzed by powder X-ray diffraction (PXRD) to identify their phases.Photoluminescence
Photoluminescence data of Ce3Si2Se1.39S6.61I were collected using a HORIBA Scientific microspectrometer, equipped with a HORIBA iHR320 imaging spectrograph and a Synapse CCD detector. A confocal 375 nm diode laser was used as the excitation source. Data acquisition was performed using LabSpec 6 software over the 400–800 nm range, utilizing a 10.0 mW laser power and a 10× UV objective.Results and discussion
We successfully synthesized four new compounds in the RE3Si2SexS8−xI (RE = La, Ce, Pr, and Nd) series by using the flux-assisted boron–chalcogen mixture (BCM) method. The synthesis process involved the use of RE2O3 (La or Nd), CeO2 and Pr6O11, and SiO2 oxide reagents as rare earth and silicon sources, respectively. Boron played a crucial role in this reaction by acting as a reducing agent to remove oxygen from the oxide reagents, which has been explained extensively in earlier work.1,22,32,33 This step was essential to ensure that the rare earth and silicon elements were available in their reduced forms, enabling them to react with elemental sulfur and selenium to form the desired chalcogenide compounds. In our experiments, an excess of NaI flux was employed to promote the formation of both single crystals and polycrystalline materials and also as a source of I.After synthesis, the reaction products were thoroughly washed with methanol to dissolve the residual NaI flux and eliminate any impurities. To verify the composition, we conducted energy dispersive spectroscopy (EDS) analyses. These analyses confirmed the presence of all expected elements in the crystal (Table S3†) and no extraneous elements were detected. The phase-purity of the samples was confirmed by PXRD analysis (Fig. 2 and Fig. S1–S3†), and these compounds were used for further characterization. Additionally, our observations revealed that all synthesized phases remained stable under ambient conditions. The thermal stability of the Ce3Si2Se1.39S6.61I polycrystalline powder samples was investigated through thermogravimetric analysis (TGA) conducted under a nitrogen atmosphere, with the temperature increasing progressively up to 500 °C (Fig. S4†). The TGA results indicated minimal weight loss over the examined temperature range, suggesting the stability of the material under an N2 atmosphere. To verify the findings and assess any potential structural changes induced by heating, powder X-ray diffraction (PXRD) analysis was performed on the samples after the TGA measurements. The PXRD patterns of the post-TGA samples revealed no observable changes, confirming that the phase is quite stable, and no oxidation or decomposition happened during the TGA experiment (Fig. S5†).
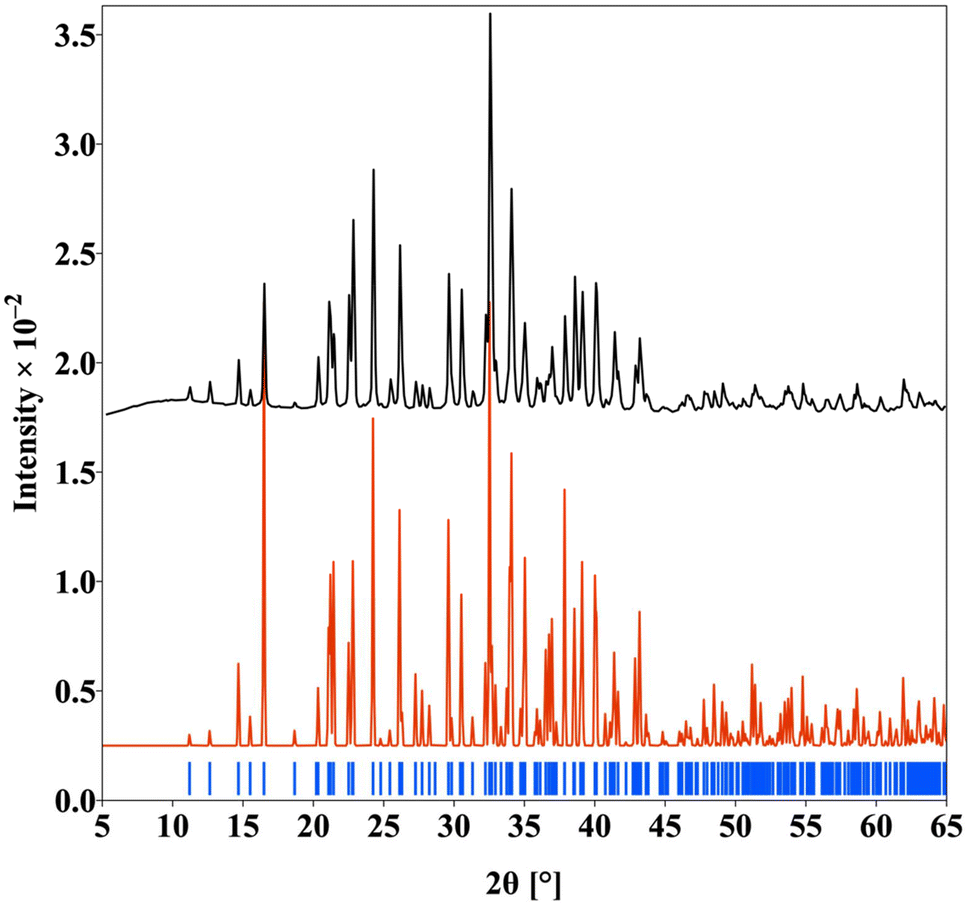 | ||
| Fig. 2 The PXRD pattern of the polycrystalline Nd3Si2Se1.18S6.82I compound (simulated pattern (red), experimental pattern (black) and Bragg position (blue)). | ||
Crystal structure
The compounds RE3Si2SexS8−xI crystallize in the monoclinic C2/c space group, sharing an isostructural relationship with the previously reported La3Si2O8Cl compound. The monoclinic crystal structure of the RE3Si2SexS8−xI compounds is also consistent with other known compounds such as RE3Si2O8Cl, RE3Si2O8X (X = Cl, Br, and I), RE3Ge2S8I, and RE3(Ge1−xSix)2S8I.16,18,34,35 The unit cell contains eight independent atomic sites, with four mixed chalcogenide sites (8f) being occupied by both S and Se. For the mixed-chalcogen RE3Si2SexS8−xI compounds, structural refinements provided evidence for site preferences of S and Se. Only one of the four chalcogen sites (labeled Q1 in Fig. 3d) contained a significant amount of Se (35%), while the remaining three sites are predominantly occupied by S with only ∼10% Se for sites Q2 and Q3 and less than ∼5% for site Q4. The details of the site occupancies of S and Se in RE3Si2SexS8−xI are provided in Table S1.† The Q atoms, specifically Q1, Q2, and Q3, which exhibit a higher concentration of selenium (Se), are strategically positioned around the channel within the unit cell structure where they can interact with the iodine in the center of the channel, as illustrated in Fig. 5. It is likely that the softer and more covalent selenium prefers interactions with the soft and covalent iodine, resulting in higher selenium occupancies in Q1, Q2, and Q3, vs. Q4.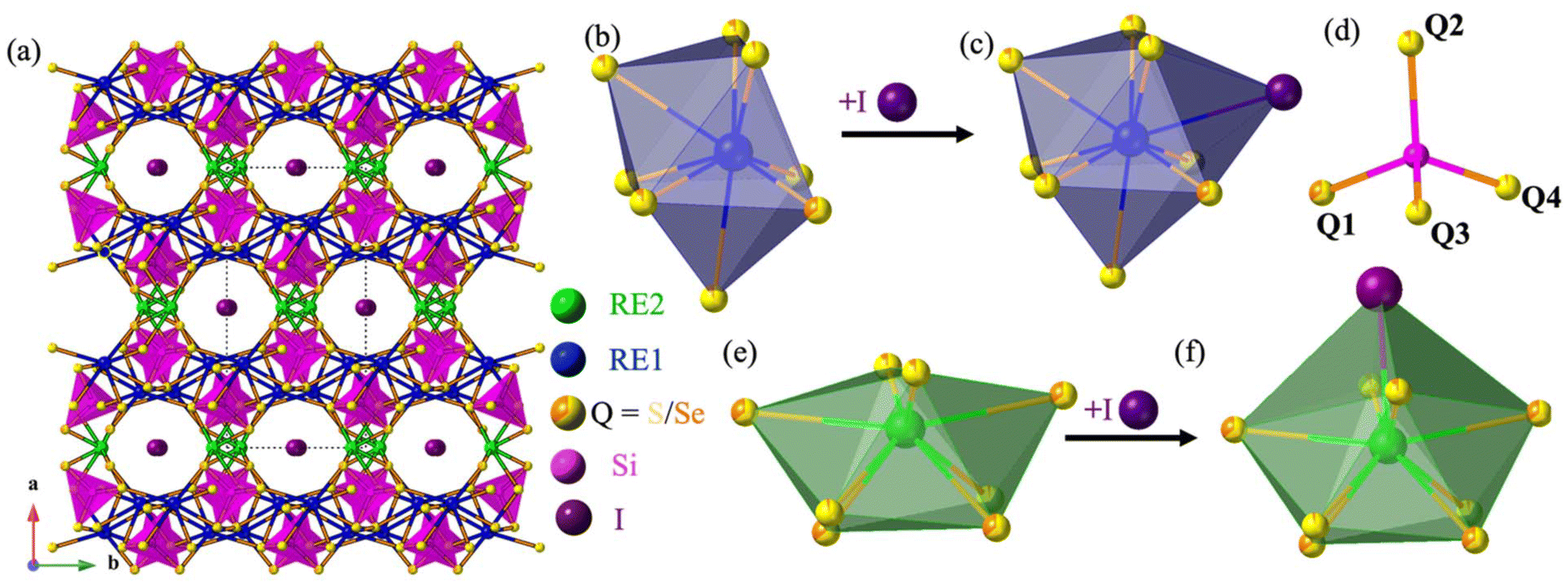 | ||
| Fig. 3 The (a) unit cell view of RE3Si2SexS8−xI compounds, (b) RE1Q8 bicapped trigonal prism, (c) RE2Q8 bicapped trigonal prism, (d) SiQ4 tetrahedron, (e) RE2Q8 bicapped trigonal prism, and (f) RE1Q8I tricapped trigonal prism. | ||
The three-dimensional tunnel structure of RE3Si2SexS8−xI along the c-direction is shown in Fig. 3a. It consists of isolated SiQ4 tetrahedra that are connected by REQ8 (RE = La, Ce, Pr and Nd) polyhedra; this arrangement creates tunnels that are filled by I atoms (Fig. 3b, d and e). Both RE1 and RE2 form bicapped trigonal prisms with eight neighboring chalcogens, which extend to a tricapped trigonal prismatic structure once the additional long bond to I is included (Fig. 3c and f). The complete 3D structure contains two different layers 2∞[RE1SiQ4] and 1∞[RE2Q6] connected via edge sharing (Fig. 4c and d). The 2∞[RE1SiQ4] layers are constructed by zigzagging ribbons of face-sharing 1∞[RE1Q5] polyhedra that extend along the b-axis (Fig. 4a and b); SiQ4 tetrahedra are located above and below the 1∞[RE1Q5] layers. The resulting 3D structure consisting of these two layers displays tunnels along the c-axis that are filled by iodine (Fig. 3a).35
 | ||
| Fig. 4 (a) 2D 1∞[RE1Q5] layers along the b-axis, (b) 2D 1∞[RE1Q5] ribbons along the a-axis, (c) 2∞[RE1SiQ4] layers viewed along the b-direction, and (d) total 3D view of 3∞[RE3Si2SexS8−x]1+, with RE2Q8 bicapped trigonal prisms bridging between 2∞[RE1SiQ4]1− layers. | ||
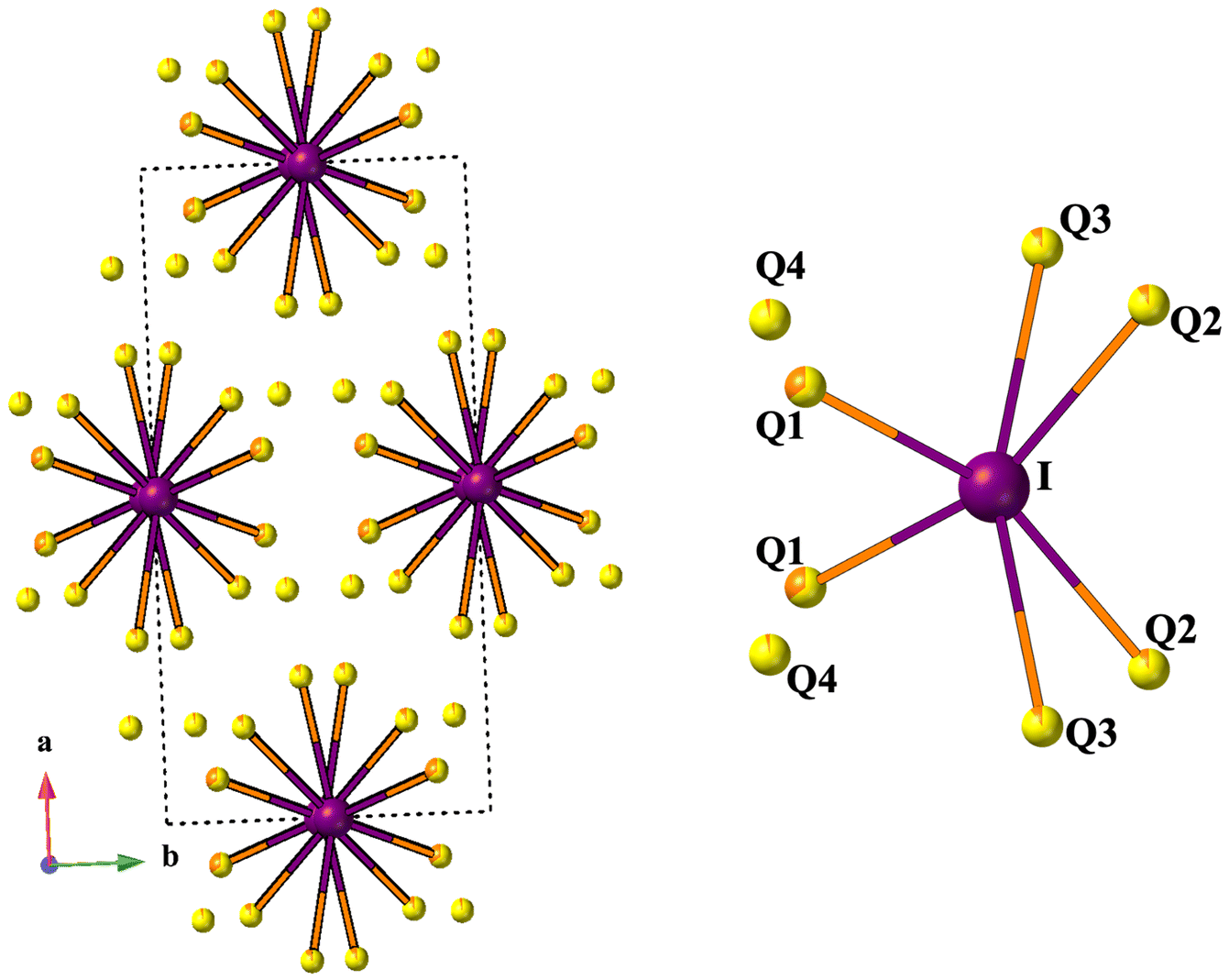 | ||
| Fig. 5 The coordination environment of I with Q atoms (Q = S/Se). Iodine is purple and Q is S/Se yellow/orange. | ||
The RE–Q bonds within the bicapped trigonal prisms of the RE3Si2SexS8−xI compositions exhibit bond lengths ranging from 2.9420(5) Å to 3.167(1) Å and the Si–Q bonds within these tetrahedra range from 2.1157(8) Å to 2.259(1) Å. These bond lengths are consistent with those found in related chalcogenide compounds, suggesting that the RE–Q and Si–Q bonding interactions in RE3Si2SexS8−xI are typical of this class of materials.36–41 The minimum RE⋯RE distance decreases from La (dLa–La = 4.5034(5) Å) to Nd (dNd–Nd = 4.4065(5) Å), presumably due to the lanthanide contraction. The lanthanide contraction also may cause the shrinkage of the cavity size where the minimum I⋯I distance also decreases from 5.4966(4) Å to 5.4067(4) Å. The RE–I bonds range from RE1–I of 3.4773(5) Å to 3.4488(5) Å and RE2–I of 3.3333(5) Å to 3.2496(5) Å.
Magnetic properties
The temperature-dependent magnetization of polycrystalline samples of Ce3Si2Se1.39S6.61I and Nd3Si2Se1.18S6.82I was measured over the temperature range of 2 K–300 K. The magnetic susceptibility (χ) was found to follow the Curie law upon cooling down to 2 K with no evidence of long-range magnetic ordering for Nd3Si2Se1.18S6.82I; however, an antiferromagnetic transition (TN = 6 K) was observed for the Ce3Si2Se1.39S6.61I compound. The magnetic susceptibility vs. temperature plots of Ce3Si2Se1.39S6.61I and Nd3Si2Se1.18S6.82I are shown in Fig. 6a and b. The M vs. H plots of Ce3Si2Se1.39S6.61I and Nd3Si2Se1.18S6.82I are shown in Fig. 6c and d which are collected over an applied magnetic field of 7 T to −7 and 5 T to −5 T. The inverse susceptibility data were fitted to the Curie law using the high temperature region (200–300 K) to calculate the Curie (C) and Weiss (θ) constants (see the inset of Fig. 6a and b). | ||
| Fig. 6 Molar susceptibility and inverse molar susceptibility vs. temperature plots for polycrystalline (a) Ce3Si2Se1.39S6.61I and (b) Nd3Si2Se1.18S6.82I measured in an applied field of 1000 Oe; magnetization as a function of applied field plots for polycrystalline (c) Ce3Si2Se1.39S6.61I and (d) Nd3Si2Se1.18S6.82I measured at 2 K. | ||
The effective magnetic moments determined from the linear fit are 4.61μB and 6.57μB per formula unit, for the Ce and Nd compositions, which are in good agreement with the theoretical moments expected for Ce3+ and Nd3+ ions, with values of 4.39μB and 6.27μB, respectively. The negative Weiss constants (θ = −25 and −38) observed for the Ce3Si2Se1.39S6.61I and Nd3Si2Se1.18S6.82I compounds indicate antiferromagnetic interactions between f-electrons on neighboring rare-earth ions, likely due to the intervening sulfur and selenium ions. The minimum distance between neighboring Nd atoms observed is 4.4065(5) Å. No difference between the field-cooled (FC) and zero field-cooled (ZFC) data was observed; the FC data are shown in Fig. 6 and the ZFC data are shown in Fig. S6.† The M vs. H plot measured at 2 K begins to saturate at higher magnetic fields as expected, Fig. 6c and d.
UV-visible diffuse reflectance spectroscopy
The optical properties of the quaternary compounds La3Si2Se1.21S6.79I, Ce3Si2Se1.39S6.61I, and Pr3Si2Se1.22S6.78I were examined using polycrystalline powder samples. The Kubelka–Munk equation α/S = (1 − R)2/2R was used (where α is the absorption coefficient, R is the reflectance, and S is the scattering coefficient) to analyze the optical absorption data, as shown in Fig. 7. The absorption data reveal band gaps of 2.5(1) eV, 2.2(1) eV and 2.3(1) eV for the polycrystalline La3Si2Se1.21S6.79I, Ce3Si2Se1.39S6.61I, and Pr3Si2Se1.22S6.78I samples, consistent with the semiconducting nature of the composition. This band gap value of Ce3Si2Se1.39S6.61I is lower than that observed for the related compound Ce3Si2S8I, and Ce3Si2S6Se1I which have reported band gaps of 2.78 eV and 2.39 eV, respectively, and higher than that of Ce3Si2Se8I (1.92 eV).21 The observed reduction in the band gap may be attributed to the substitution of selenium (Se) for sulfur (S) in the lattice. Selenium, being less electronegative and having a higher atomic radius than sulfur, can contribute to a decrease in the overall band gap by enhancing the covalency in the bonding. Consequently, the introduction of Se into Ce3Si2Se1.39S6.61I is the likely reason for the narrower band gap in Ce3Si2Se1.39S6.61I relative to Ce3Si2S8I.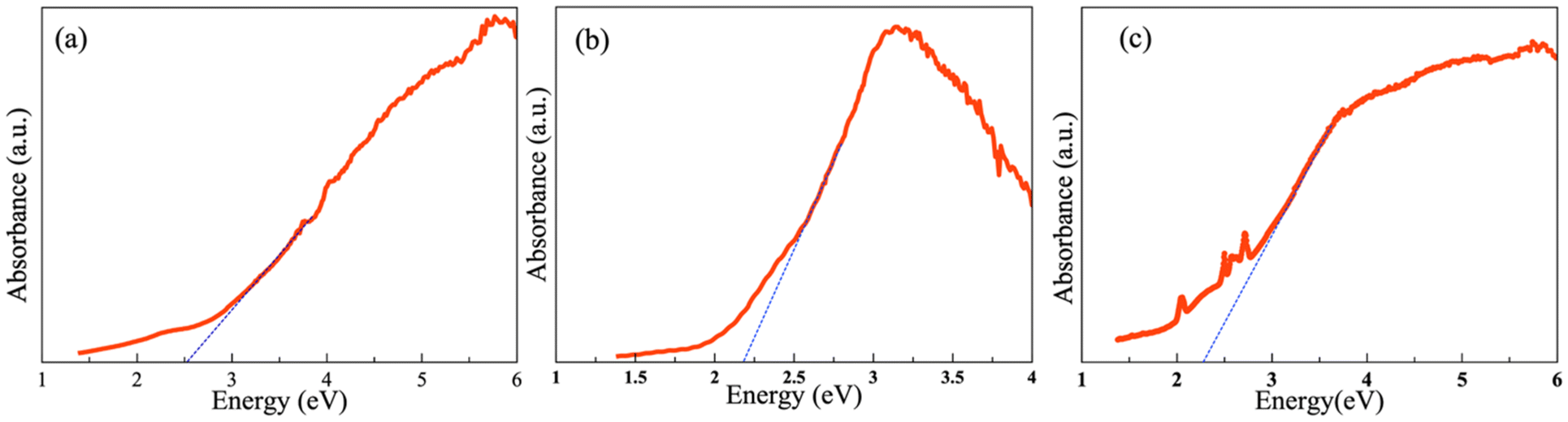 | ||
| Fig. 7 Optical absorption plot of polycrystalline (a) La3Si2Se1.21S6.79I, (b) Ce3Si2Se1.39S6.61I, and (c) Pr3Si2Se1.22S6.78I. | ||
Photoluminescence
Photoluminescence spectra were recorded using a large (2.4 mm × 0.8 mm × 0.6 mm) yellow crystal of Ce3Si2Se1.39S6.61I. The spectra were recorded using a 375 nm ultraviolet diode laser as the excitation source. The spectra revealed a broad emission band centered around ∼493 nm, which is attributed to the 5d–4f electronic transition of the Ce3+ ions (Fig. 8). This emission, observed in the blue-green region, is similar to that reported for the related compound Ce3Si2S8I which shows strong luminescence in the blue region at ∼462 nm. The slight redshift in emission observed for Ce3Si2Se1.39S6.61I can likely be attributed to differences in the local environment around the Ce atom, possibly due to the influence of the selenium atoms. The photoluminescence in this compound thus appears to be sensitive to the local coordination environment around the Ce atom. The little asymmetric shape of the emission peak may arise from the presence of two distinct types of emissive centers, which is consistent with the existence of two rare-earth (RE) sites within the structure. These RE sites experience slightly different local environments due to the inherent disorder of the surrounding sulfur (S) and selenium (Se) atoms. Variations in the local coordination geometry and ligand field strength at each site can lead to differences in emission energy, resulting in overlapping of emission bands. This overlap can cause spectral broadening and asymmetry in the photoluminescence peak. We also investigated its scintillation ability by illuminating a powder sample of Ce3Si2Se1.39S6.61I with Cu X-rays. The green scintillation that results confirms the ability of this material to scintillate. (Fig. S7†). These observations suggest that selective substitution of chalcogen atoms in this family of compounds can be used to fine-tune the photoluminescence emission behavior.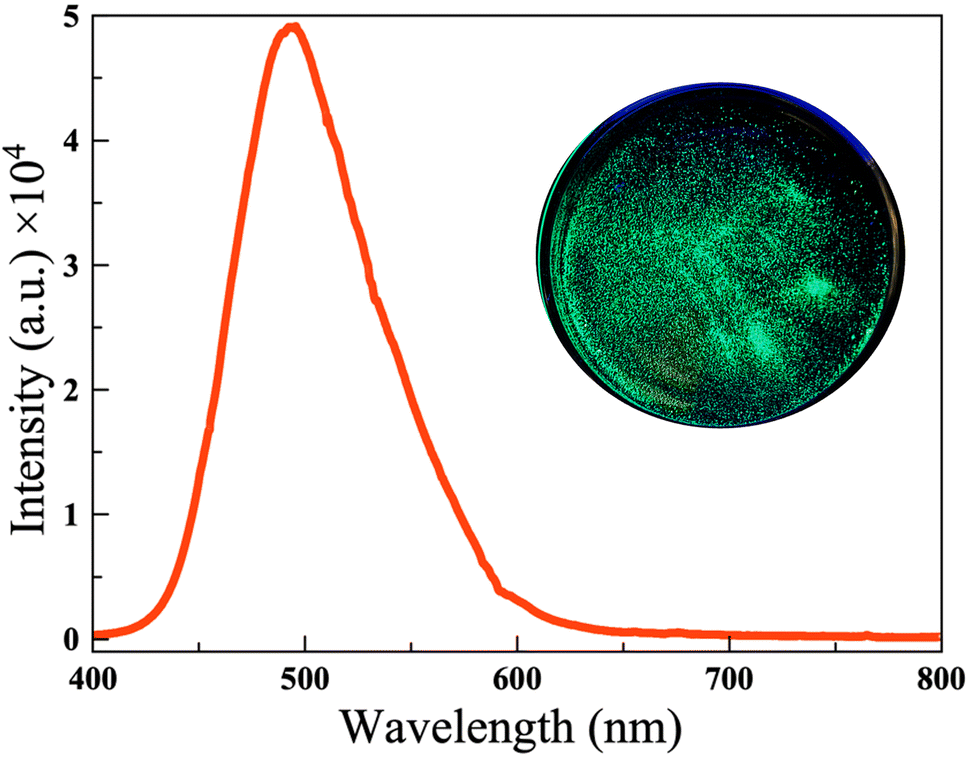 | ||
| Fig. 8 Emission spectra of the Ce3Si2Se1.39S6.61I single crystal (the inset figure shows the green color of the sample under exposure to UV light). | ||
Conclusion
The rare-earth mixed chalcogenide iodides La3Si2Se1.21S6.79I, Ce3Si2Se1.39S6.61I, Pr3Si2Se1.22S6.78I, and Nd3Si2Se1.18S6.82I were successfully synthesized using a flux-assisted BCM method, yielding phase-pure samples that crystallize in the monoclinic C2/c space group. These compounds exhibit a three-dimensional tunnel structure resembling the La3Si2O8Cl type, with isolated SiQ4 tetrahedra connected to rare-earth polyhedra to generate a tunnel structure in which the tunnels are occupied by iodide ions. Partial substitution of S with Se impacts the optical properties of these materials, as demonstrated by UV-visible spectroscopy and photoluminescence studies. Specifically, Ce3Si2Se1.39S6.61I has a band gap of 2.2(1) eV classifying it as a semiconductor. It also exhibits luminescence behavior with a broad emission band around 493 nm. The materials appear to be paramagnetic, as exemplified by Nd3Si2Se1.18S6.82I, which exhibits paramagnetic behavior with a negative Weiss constant. These findings highlight the potential of these mixed chalcogenide iodides for optical applications, where the optical properties can be influenced by the substitution of Se for S.Data availability
The CCDC 2388413–2388416† entries encompass the supplementary crystallographic data associated with this paper. Raw data for other measurements are available upon request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge the support from the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering, under award DE-SC0018739.References
- G. Panigrahi, A. A. Berseneva, G. Morrison, A. A. King, R. L. Conner, L. G. Jacobsohn and H.-C. zur Loye, Inorg. Chem., 2024, 63, 12849–12857 CrossRef CAS PubMed.
- S.-P. Lee, C.-H. Huang, T.-S. Chan and T.-M. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 7260–7267 CrossRef CAS PubMed.
- K. Mitchell and J. A. Ibers, Chem. Rev., 2002, 102, 1929–1952 Search PubMed.
- H.-J. Zhao, P.-F. Liu and L.-M. Wu, Dalton Trans., 2021, 50, 2075–2082 RSC.
- G. B. Jin, E. S. Choi, R. P. Guertin, C. H. Booth and T. E. Albrecht-Schmitt, Chem. Mater., 2011, 23, 1306–1314 CrossRef CAS.
- A. Tassanov, H. Lee, Y. Xia and J. M. Hodges, J. Am. Chem. Soc., 2024, 146, 32627–32639 Search PubMed.
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 9, 772 CrossRef PubMed.
- J. K. Harada, N. Charles, K. R. Poeppelmeier and J. M. Rondinelli, Adv. Mater., 2019, 31, 1805295 CrossRef PubMed.
- H. Kageyama, T. Yajima, Y. Tsujimoto, T. Yamamoto, C. Tassel and Y. Kobayashi, Bull. Chem. Soc. Jpn., 2019, 92, 1349–1357 Search PubMed.
- Y.-Y. Li, W.-J. Wang, H. Wang, H. Lin and L.-M. Wu, Cryst. Growth Des., 2019, 19, 4172–4192 CrossRef CAS.
- K. Maeda, F. Takeiri, G. Kobayashi, S. Matsuishi, H. Ogino, S. Ida, T. Mori, Y. Uchimoto, S. Tanabe, T. Hasegawa, N. Imanaka and H. Kageyama, Bull. Chem. Soc. Jpn., 2022, 95, 26–37 CrossRef CAS.
- N. Recham, J.-N. Chotard, L. Dupont, C. Delacourt, W. Walker, M. Armand and J.-M. Tarascon, Nat. Mater., 2010, 9, 68–74 Search PubMed.
- M. Jansen and H. P. Letschert, Nature, 2000, 404, 980–982 Search PubMed.
- Y. Kamihara, T. Watanabe, M. Hirano and H. Hosono, J. Am. Chem. Soc., 2008, 130, 3296–3297 CrossRef CAS PubMed.
- J.-R. Xiao, S.-H. Yang, F. Feng, H.-G. Xue and S.-P. Guo, Coord. Chem. Rev., 2017, 347, 23–47 CrossRef CAS.
- D. Mumbaraddi, A. K. Iyer, E. Üzer, V. Mishra, A. O. Oliynyk, T. Nilges and A. Mar, J. Solid State Chem., 2019, 274, 162–167 CrossRef CAS.
- G. Gauthier, S. Kawasaki, S. Jobic, P. Macaudiere, R. Brec and J. Rouxel, J. Alloys Compd., 1998, 275–277, 46–49 CrossRef CAS.
- R. Riccardi, D. Gout, G. Gauthier, F. Guillen, S. Jobic, A. Garcia, D. Huguenin, P. Macaudière, C. Fouassier and R. Brec, J. Solid State Chem., 1999, 147, 259–268 CrossRef CAS.
- S. T. Hatscher and W. Urland, Z. Anorg. Allg. Chem., 2001, 627, 2198–2200 CAS.
- S. T. Hatscher and W. Urland, Acta Crystallogr., Sect. E:Struct. Rep. Online, 2002, 58, i100–i102 Search PubMed.
- D. Mumbaraddi, V. Mishra, M. Jomaa, X. Liu, A. Karmakar, S. Thirupurasanthiran, V. K. Michaelis, A. P. Grosvenor, A. Meldrum and A. Mar, Chem. Mater., 2023, 35, 6039–6049 CAS.
- L. S. Breton, V. V. Klepov and H.-C. zur Loye, J. Am. Chem. Soc., 2020, 142, 14365–14373 Search PubMed.
- R. L. Gitzendanner and F. J. DiSalvo, Inorg. Chem., 1996, 35, 2623–2626 CAS.
- L. Jiang, L. Zhang, X. Jiang, G. Lv and Y. Su, J. Alloys Compd., 2024, 970, 172544 CAS.
- APEX3 Version 2019.1-0 and SAINT+ Version 8.40A, Bruker Nano, Inc., Madison, WI, USA, 2019 Search PubMed.
- SADABS-2016/2: L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 Search PubMed.
- (a) SHELXT: G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 Search PubMed; (b) SHELXL: G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, ShelXle: A Qt Graphical User Interface for SHELXL, J. Appl. Crystallogr., 2011, 44(6), 1281–1284 Search PubMed.
- L. M. Gelato and E. Parthé, J. Appl. Crystallogr., 1987, 20, 139–143 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CAS.
- P. Kubelka, Z. Tech. Phys., 1931, 12, 593–601 Search PubMed.
- A. A. Berseneva, V. V. Klepov, K. Pal, K. Seeley, D. Koury, J. Schaeperkoetter, J. T. Wright, S. T. Misture, M. G. Kanatzidis, C. Wolverton, A. V. Gelis and H.-C. zur Loye, J. Am. Chem. Soc., 2022, 144, 13773–13786 Search PubMed.
- S.-H. Lin, J.-G. Mao, G.-C. Guo and J.-S. Huang, J. Alloys Compd., 1997, 252, L8–L11 Search PubMed.
- P. Gravereau, B. Es-Sakhi and C. Fouassier, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 1884–1887 CrossRef.
- G. Gauthier, S. Kawasaki, S. Jobic, P. Macaudière, R. Brec and J. Rouxel, J. Mater. Chem., 1998, 8, 179–186 RSC.
- L. D. Gulay, O. S. Lychmanyuk, I. D. Olekseyuk, M. Daszkiewicz, J. Stępień-Damm and A. Pietraszko, J. Alloys Compd., 2007, 431, 185–190 CrossRef CAS.
- A. A. King, L. S. Breton, G. Morrison, M. D. Smith, M. Liang, P. S. Halasyamani and H.-C. zur Loye, Inorg. Chem., 2023, 62, 7446–7452 CrossRef CAS PubMed.
- J. He, Z. Wang, X. Zhang, Y. Cheng, Y. Gong, X. Lai, C. Zheng, J. Lin and F. Huang, RSC Adv., 2015, 5, 52629–52635 Search PubMed.
- W. Xing, N. Wang, Y. Guo, Z. Li, J. Tang, K. Kang, W. Yin, Z. Lin, J. Yao and B. Kang, Dalton Trans., 2019, 48, 17620–17625 RSC.
- G. Li, Y. Chu, J. Li and Z. Zhou, Dalton Trans., 2020, 49, 1975–1980 RSC.
- M. Usman, M. D. Smith, G. Morrison, V. V. Klepov, W. Zhang, P. S. Halasyamani and H.-C. zur Loye, Inorg. Chem., 2019, 58, 8541–8550 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: EDS tabulated data, PXRD patterns, ZFC magnetic susceptibility plots, scintillation image, and tables of atomic parameters and bond lengths. CCDC 2388413–2388416. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03506b |
| This journal is © The Royal Society of Chemistry 2025 |