
Asymmetric orbital hybridization at the MXene–VO2−x interface stabilizes oxygen vacancies for enhanced reversibility in aqueous zinc-ion batteries†
Yuan
Fang‡
a,
Chunhong
Qi‡
a,
Weichao
Bao
b,
Fangfang
Xu
 b,
Wei
Sun
*a,
Bin
Liu
c,
Xiqian
Yu
d,
Lianjun
Wang
a,
Wan
Jiang
a,
Pengpeng
Qiu
*a and
Wei
Luo
*a
b,
Wei
Sun
*a,
Bin
Liu
c,
Xiqian
Yu
d,
Lianjun
Wang
a,
Wan
Jiang
a,
Pengpeng
Qiu
*a and
Wei
Luo
*a
aState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China. E-mail: weisun@dhu.edu.cn; qiupengpeng@dhu.edu.cn; wluo@dhu.edu.cn
bState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Shanghai 200050, China
cSchool of Materials Science and Engineering, Shanghai University, Shanghai 200444, China
dBeijing Advanced Innovation Center for Materials, Genome Engineering, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China
First published on 14th November 2024
Abstract
Modulating the storage kinetics of Zn2+ through oxygen vacancy (Ov) manipulation represents a promising approach for developing cathode materials in aqueous rechargeable zinc-ion batteries (ZIBs). However, recent studies have shown that these Ovs can undergo migration and refilling during electrochemical cycling, leading to severe structural degradation and rapid capacity fading. Therefore, developing technologies to stabilize Ovs is critical for maximizing their efficiency, although it presents a significant challenge. Herein, we demonstrate a covalent heterostructure design that pushes the cycling performance of a vanadium dioxide (VO2) cathode to an unprecedented level. The rationale lies in the chemical growth of VO2 nanowall arrays on MXene nanosheets that leads to Ti–O–V asymmetric orbital hybridization (AOH) at the interface, which remarkably enhances the stability of Ovs on VO2. Due to this advanced cathode design, the prepared ZIBs exhibit highly reversible aqueous Zn2+ storage capacities and maintain a robust structure over 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 20 A g−1, without any significant capacity loss (1.4%). Detailed experimental and theoretical analyses indicate that Ti–O–V AOH facilitates a charge transfer pathway at the interface, allowing electrons to migrate from VO2 to the MXene surface, thereby stabilizing the Ovs both thermodynamically and kinetically. Our work offers an inspiring design principle for developing sustainable cathode materials for high-performance aqueous ZIBs and beyond, leveraging the synergistic effects of Ovs and interfacial orbital engineering.
000 cycles at 20 A g−1, without any significant capacity loss (1.4%). Detailed experimental and theoretical analyses indicate that Ti–O–V AOH facilitates a charge transfer pathway at the interface, allowing electrons to migrate from VO2 to the MXene surface, thereby stabilizing the Ovs both thermodynamically and kinetically. Our work offers an inspiring design principle for developing sustainable cathode materials for high-performance aqueous ZIBs and beyond, leveraging the synergistic effects of Ovs and interfacial orbital engineering.
Broader contextRecently, oxygen vacancy (Ov) engineering has been successfully applied in zinc ion batteries (ZIBs) to modulate the Zn2+ transport kinetics by inducing local lattice expansion. However, the Ovs on the cathode surface tend to capture oxygen from aqueous medium, leading to their refilling. Furthermore, the absence of oxygen atoms can create a local electric field, driving the migration of Ovs. These challenges can lead to severe structural degradation and rapid capacity fading of ZIBs. Therefore, it is urgent to develop stabilization technologies that maximize the utilization efficiency of Ovs. In this case, we designed a covalent heterostructure with asymmetric Ti–O–V orbital hybridization at the interface that greatly enhances the stability of Ovs both kinetically and thermodynamically. The accelerated migration of electrons from VO2 to the MXene surface is verified through detailed experimental and theoretical analyses, so the refill and migration of Ovs during Zn2+ shuttling are alleviated. Hence, the MXene–VO2−x cathode displayed a reversible specific capacity of 487.9 mA h g−1 at 0.2 A g−1 and a high capacity retention of 98.6% over 30000 cycles. This article draws noteworthy findings on the design, controllable synthesis, and characterization of the cathode, providing a clear direction for exploring the structure–performance relationship.
|
Introduction
Aqueous rechargeable batteries, assembled with water electrolytes, have recently emerged as a sustainable complement to Li-ion batteries due to their inherent safety, low cost, and environmentally friendly nature.1–5 Among them, Zn-ion batteries (ZIBs) are gaining momentum due to the distinctive properties of zinc anodes, including their large theoretical capacity (820 mA h g−1), low redox potential (−0.76 V vs. standard 2H+/H2), and excellent compatibility with water.6–9 However, a significant challenge in ZIBs lies in the development of cathode materials capable of reversibly intercalating high-polarity divalent Zn2+ to achieve both high capacity and long cycle life in full-cell applications. The manipulation of oxygen vacancies (Ovs) has been proven to be a powerful means to regulate the transport kinetics of Zn2+ by expanding the local lattice to the cathode without compromising the crystal integrity.10–12 However, recent investigations have revealed a significant challenge: Ovs on the cathode surface are prone to capturing oxygen from the aqueous medium, leading to their refilling and subsequent elimination during continuous electrochemical redox reactions.13,14 Additionally, the absence of oxygen atoms generates a local electric field, driving the migration of Ov, which inevitably decreases the battery performance (Fig. S1, ESI†).15,16 Despite these issues, the stability of Ovs has received limited attention in the design of efficient cathodes, and the exploration of reinforcing strategies remains sparse. Tuning the local electronic structure of Ovs by gradient refilling of different impurity atoms, such as nitrogen and sulfur, could substantially enhance the stability.14,17 However, this approach may also reduce the utilization efficiency of Ovs, which would negatively affect the cycle life of ZIBs.Recently, artificial heterostructures have garnered significant interest in materials science and condensed-matter physics. The interplay of spin, orbital, charge, and lattice degrees of freedom at the interface between two dissimilar phases or components can alter the atomic coordination environment compared to their bulk counterparts, leading to unique physicochemical properties.18,19 This suggests that interfacial heterostructure engineering could serve as a viable approach to modulate the electronic structure of Ovs, thereby influencing their behavior on the cathode materials. This study demonstrates the stabilization of Ovs on a vanadium dioxide (VO2) cathode through a covalent heterostructure design, which involves the uniform growth of VO2 nanowalls on the MXene nanosheets to induce interfacial Ti–O–V asymmetric orbital hybridization (AOH). Such a design is realized using a general hydrogen peroxide (H2O2)-assisted hydrothermal strategy. During the hydrothermal treatment, the surface of the MXene is gently oxidized by H2O2 molecules, exposing unterminated Ti–O bonds, which in turn facilitates interfacial orbital hybridization between the O 2p with V 3d. The resulting cathode delivers a remarkable reversible specific capacity of 487.9 mA h g−1 at 0.2 A g−1 and achieves an impressive capacity retention of 98.6% over 30000 cycles at 20 A g−1. Detailed experimental analysis and theoretical calculations suggest that the AOH of Ti–O–V at the interface promotes the migration of electrons from VO2 to the MXene, which aids in the stabilization of Ovs, leading to improved Zn2+ storage kinetics and structural integrity of the cathode. These results highlight the feasibility of combining orbital and vacancy engineering to construct high-performance cathodes for ZIBs.
Results and discussion
Material synthesis and structural evaluation
VO2 was chemically grown on MXene nanosheets to form a well-defined heterostructure with Ti–O–V AOH using a general H2O2-assisted hydrothermal approach followed by an oxidation–reduction phase transition process (Fig. S2, ESI†). The MXene nanosheets were first prepared by ultrasonically exfoliating accordion-like Ti3C2Tx MXenes. Then, the MXene nanosheets were dispersed into tetrahydrofuran (THF), followed by the addition of vanadyl acetylacetonate (VO(acac)2) as the V precursor and H2O2 as the nucleation agent. This mixture was hydrothermally treated to form uniform VO2–MXene–VO2 sandwich-like nanosheets (denoted as MXene–VO2). Finally, a high-temperature oxidation and reduction reaction was used to drive a repeated phase transition between VO2 and V2O5 to introduce Ovs in the MXene–VO2 heterostructure (denoted as MXene–VO2−x).Field-emission scanning electron microscopy (FESEM) images of the MXene–VO2 sample are shown in Fig. S3a and b (ESI†), revealing the uniform growth of contrasting VO2 nanowall arrays on the originally smooth surface of the MXene nanosheets (Fig. S4, ESI†). Additionally, the transmission electron microscopy (TEM), dark-field scanning TEM (STEM) images, and corresponding elemental mappings of MXene–VO2 further verify the homogenous distribution of polycrystalline VO2 nanowalls on the MXene nanosheets (Fig. S3c–e, ESI†). After phase transition, no significant change in the morphological structure of MXene–VO2−x is observed (Fig. S5, ESI†), implying a strong interaction (potentially orbital hybridization) between MXene and VO2. This interaction could protect the heterostructure from collapse at high temperatures, which is also indicated by the well-retained structure of MXene–V2O5 (the intermediate product formed during the phase transition process, Fig. S6, ESI†). The cross-sectional STEM images and corresponding elemental mappings of MXene–VO2−x (Fig. 1a and b) clearly demonstrate a thin MXene nanosheet enclosed by two VO2 nanowall layers. The high-resolution TEM (HRTEM) image reveals that the VO2 nanocrystals with a d-spacing of the (110) crystal plane (0.35 nm) grow vertically along the MXene nanosheet surface at the nanoscale (Fig. 1c). Atomic force microscopy (AFM) measurements on three selected areas indicate a thickness of approximately 27.5 nm for the VO2 nanowalls (Fig. S7, ESI†). To confirm the formation of Ovs in the MXene–VO2−x heterostructure, high-angle annular dark-field (HAADF)-STEM images were obtained. The lattice fringes of the (003) crystal planes of VO2 (d-spacing = 2.11 Å) can be observed with multiple lattice disorders (Fig. S8, ESI†) and evident atomic defects (Fig. 1d, highlighted by red circles). In contrast, the HRTEM and HAADF-STEM images of MXene–VO2 displayed in Fig. S9 (ESI†) show a smaller d-spacing of 2.06 Å and a completely ordered atomic arrangement. Notably, this synthetic strategy can be easily scaled up by employing a larger autoclave to produce more quantities of MXene–VO2−x (around 6 g) while maintaining the original quality (Fig. S10, ESI†), paving the way for potential practical production.
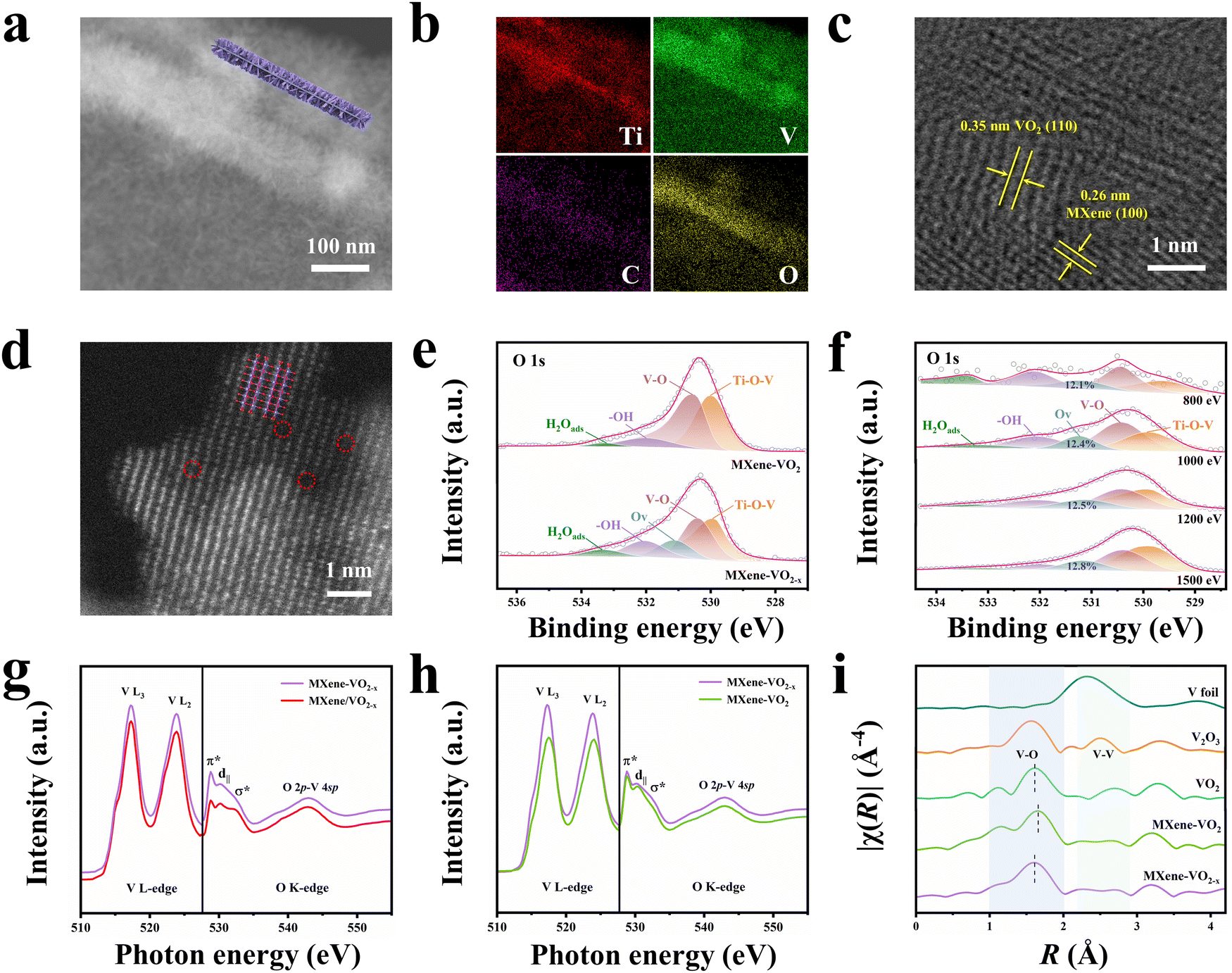 | ||
| Fig. 1 Morphology and structural characterization of MXene–VO2−x. (a) HAADF-STEM image of MXene–VO2−x (inset: Corresponding side-view model). (b) Elemental mapping showing the distributions of titanium, vanadium, carbon, and oxygen. (c) High-resolution TEM image of the heterointerface. (d) Atom-resolved HAADF-STEM image (inset: Crystal structure of VO2 (B)). (e) O 1s spectra of MXene–VO2 and MXene–VO2−x. (f) O 1s spectra of MXene–VO2−x recorded at incident photon energies of 800 to 1500 eV. (g) V L-edge and O K-edge sXAS spectra of MXene–VO2−x and MXene/VO2−x. (h) V L-edge and O K-edge sXAS spectra of MXene–VO2−x and MXene–VO2. (i) V K-edge k3-weighted χ(k)-function FT EXAFS spectra. | ||
X-ray photoelectron spectroscopy (XPS) was conducted to determine the elemental composition and chemical states of the prepared samples. The O 1s spectra for both MXene–VO2 and MXene–VO2−x (Fig. 1e) exhibit a Ti–O–V peak at 530.0 eV, suggesting the formation of AOH between MXene and VO2. The presence of a V5+ peak in the V 2p spectra further indicates the formation of Ti–O–V covalent bonds (Fig. S11b, ESI†). The O 1s spectrum of MXene–VO2−x shows an additional Ov peak compared to that of MXene–VO2, demonstrating the successful incorporation of Ovs into the VO2 structure. The depth-resolved synchrotron XPS profiles (Fig. 1f) reveal that the relative Ov peak area remains relatively constant at approximately 12.5% as the photon energy increases from 800 to 1500 eV, suggesting uniform distribution of Ovs in MXene–VO2−x. The presence of Ovs in the MXene–VO2−x heterostructure was also confirmed by the detection of a weak V4+ signal at a g-value of 1.96 in the electron paramagnetic resonance (EPR) analysis (Fig. S12a, ESI†). Both the C 1s and Ti 2p spectra show the presence of a Ti–C bond, which suggests that the MXene structure is well-preserved in the MXene–VO2−x heterostructure (Fig. S11c and d, ESI†). The X-ray diffraction (XRD) patterns of both MXene–VO2 and MXene–VO2−x (Fig. S12b, ESI†) show distinct diffraction peaks at 15.3°, 25.1°, 30.3°, 33.6°, 38.6°, 45.1°, and 48.7° that are well indexed to the (200), (110), (111), (310), (401), (−601), and (020) crystal planes of the monoclinic VO2 phase (PDF#81-2392). The MXene–VO2 composite exhibits an additional peak at about 9.5° that is ascribed to the (002) crystal plane of MXene. This peak is weaker and shifted to a larger angle compared to that of pure MXene nanosheets (8.3°), indicating the decrease in the interlayer spacing of the MXene caused by the close stacking of VO2. Magnified views of the XRD patterns (Fig. S12c and d, ESI†) show that the (110), (003), (−601), and (020) diffraction peaks of MXene–VO2−x are shifted to lower angles than those of MXene–VO2, which verify that the introduction of Ovs leads to the broadening of the lattice spacing.
Soft X-ray absorption spectroscopy (sXAS, Fig. 1g) was used to evaluate the detailed orbital information of MXene–VO2−x and MXene/VO2−x (prepared by physically mixing the MXene and VO2−x and further details are provided in the ESI†). The V–L edge regions of these samples show main peaks at around 517 and 524eV that are assigned to the electron excitations from V 2p3/2 to V 3d (V L3) and from V 2p1/2 to V 3d (V L2), respectively. MXene–VO2−x exhibits a higher L2/L3 ratio than MXene/VO2−x, primarily because the existence of Ti–O–V AOH results in an increased V oxidation state through electron transfer.20,21 Furthermore, the weaker d|| and σ* peaks in the O K-edge spectrum of MXene–VO2−x indicate a higher electron occupancy of the O 2p state in interfacial Ti–O–V bonds, which results from the electrons migrating away from the VO2 framework to MXenes. The sXAS spectra of MXene–VO2 and MXene–VO2−x were also compared, as shown in Fig. 1h. The V–L edge peak of MXene–VO2−x is shifted in the lower energy direction compared to that of MXene–VO2, implying that V is slightly reduced due to the generation of Ovs. Moreover, the substantially increased intensity of the σ* band in MXene–VO2−x is ascribed to the distorted local coordination of O atoms surrounding the vanadium atom.22,23 The atomic coordination structures of MXene–VO2−x and MXene–VO2 were examined by X-ray absorption fine structure (XAFS) measurements. As demonstrated by the X-ray absorption near-edge structure (XANES) spectra of the V K-edge (Fig. S13, ESI†), the main absorption edge of MXene–VO2 is close to that of the standard VO2 (B) sample. However, the V absorption edge position of MXene–VO2−x is shifted in the lower energy direction compared to that of MXene–VO2, implying that the vanadium ions are reduced due to the generation of Ovs. According to linear fitting with the standard samples, the average oxidation state of V in MXene–VO2−x (Fig. S14, ESI†) is approximately + 3.8, further revealing the presence of Ovs. The corresponding Fourier-transform extended X-ray absorption fine structure (FT EXAFS) spectra in Fig. 1i show a clear peak at ∼1.6 Å corresponding to the V–O scattering path in the first shell. Compared with the standard VO2 (B) sample, the V–O length of MXene–VO2 shifts to a higher value, which is possibly due to the Ti–O–V AOH. Moreover, the V–O peak of MXene–VO2−x is shifted to a slightly lower R value than that of MXene–VO2, indicating a reduction in the V–O bond length as a result of Ovs. According to the fitting results shown in Fig. S15 (ESI†), the coordination number of the V–O bond in MXene–VO2−x is lower than that in MXene–VO2 (4.7 vs. 6). The curve fitting parameters are reported in Table S1 (ESI†). The presence of a maximum peak at around 8 Å−1 in the wavelet transform (WT) of the EXAFS contour plots (Fig. S16, ESI†) is associated with the existence of the V–O bond. The observed shift in the k value is likely attributed to the structural defects in the MXene–VO2−x heterostructure.24 All these characterization results confirm the successful construction of an MXene–VO2−x heterostructure with Ti–O–V AOH at the interface and evenly distributed Ovs.
Origin of AOH in the MXene–VO2−x heterostructure
To understand the formation mechanism of AOH at the interface, several control experiments were carried out by adjusting the reaction conditions (Fig. S17, ESI†). The XRD pattern and TEM image of the sample prepared by hydrothermally reacting VO(acac)2 in the THF solvent (i.e., no MXene and no H2O2) suggest that this sample is amorphous. When MXene nanosheets are added, the surface of the MXene is barely coated with VO2, and only MXene peaks are detected in the XRD pattern. This implies that the nucleation and growth of VO2 do not occur on the surface of the MXene in a single THF solvent. When an equivalent amount of H2O is used instead of H2O2 during synthesis, the deposition of VO2 on the MXene nanosheets can also be achieved, but a very different morphological structure is formed. SEM, TEM, STEM, and corresponding elemental mapping images (Fig. S18, ESI†) clearly show that the deposited VO2 has a heterogeneous non-planar morphology on the MXene nanosheets. Moreover, the VO2 deposits are as large as 50 nm, sparsely distributed on the substrate, and isolated from each other. It should be noted that this structure exhibits high instability during the high-temperature phase transition process. Morphological characterization and XRD analysis of this sample after the high-temperature phase transition process, as shown in Fig. S19 and S20 (ESI†), indicate that the VO2−x nanoparticles are aggregated and the MXene nanosheets are completely oxidized to TiO2. This is possibly caused by the lack of robust orbital interaction at the heterointerface, as confirmed by XPS analysis (Fig. S21, ESI†). To further investigate the effect of H2O2 on the formation process, we varied both the amount of H2O2 and the hydrothermal reaction time during synthesis. With an increase in H2O2 concentration, the VO2 diffraction peak intensity is clearly strengthened and the VO2 nanowalls grow wider, longer, and denser (Fig. S22 and S23, ESI†). However, the synthesis performed with 9.1 vol% H2O2 leads to phase separation due to faster reaction kinetics between VO2 crystals. By varying the reaction time from 1 to 6 h with a fixed H2O2 dose, the thickness of the VO2 nanowalls on the MXene nanosheets can be tuned from 7.8 to 62.4 nm (Fig. S24 and S25, ESI†). Overall, these control experiments demonstrate that H2O2 plays an important role in controlling the chemical growth of VO2 nanowalls on the MXene surface to a desired thickness.On the basis of the structural analysis above, we first hypothesize that the H2O2 molecules employed in this work are more prone to concentrating the free V4+ near the MXene surface, thereby promoting heterogeneous nucleation instead of homogeneous nucleation occurring in the solution. This can be verified using the density functional theory (DFT) calculation of the adsorption energy of V4+ on three models including individual MXene, H2O-anchored MXene (MXene–H2O), and H2O2-anchored MXene (MXene–H2O2). As evident from Fig. 2a, the MXene–H2O2 surface exhibits a higher V4+ adsorption energy than the other two surfaces. Moreover, the classical nucleation theory suggests that strong metal–substrate bonding may result in smaller critical nucleate sizes, ultimately leading to the more uniform growth of the second phase.25–27 Given the oxidizing properties of H2O2, a potential explanation in this system is that the surface of the MXene nanosheets may undergo oxidation by H2O2, which generates a large number of unterminated Ti–O bonds. These bonds drive the orbital hybridization of the O 2p with V 3d orbitals (p–d hybridization) to form Ti–O–V AOH (Fig. 2b).
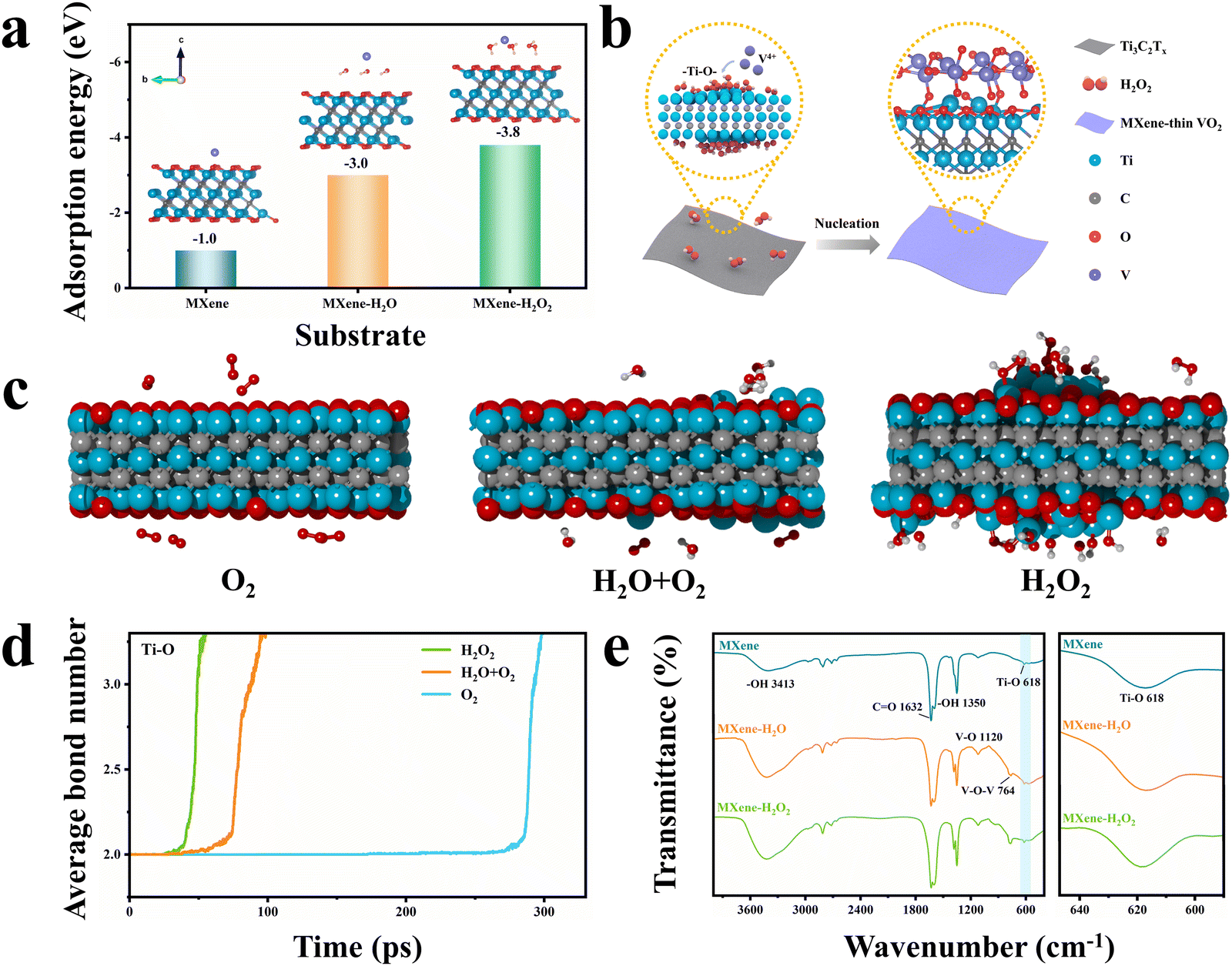 | ||
| Fig. 2 Formation mechanism of the MXene–VO2−x covalent heterostructure. (a) DFT calculation models for the adsorption of V4+ on the individual MXene, H2O-anchored MXene (MXene–H2O), and H2O2-anchored MXene (MXene–H2O2). (b) Schematic diagram of the predicted formation mechanism of the heterointerface. (c) MD simulated structure of MXene after oxidation in dry air (O2), wet air (H2O + O2), and H2O2 environments after 25 ps at a temperature of 1000 K (Ti: blue, C: gray, and O: red). (d) Change in the average bond number of Ti–O under dry air, wet air, and H2O2 environments. (e) FTIR spectra of samples under dry air, wet air, and H2O2 environments. | ||
To verify this proposed explanation, detailed theoretical and structural analyses were carried out using molecular dynamic (MD) simulations, TEM observations, XRD analysis, and Fourier transform infrared (FTIR) spectroscopy analysis. MD simulations were first performed to investigate the oxidation of MXene structures under three reaction conditions: dry air (200 O2 molecules), wet air (100 O2 and 100 H2O molecules), and H2O2 (200 H2O2 molecules) environments. Fig. 2c shows the structural models of the MXene under these three conditions at a simulation time of 25 ps, revealing that the MXene surface is more oxidized in the presence of H2O2 than under the other two atmospheres. Fig. S26 (ESI†) presents the evolution of the MXene structure with simulation times of 0, 50, 75 and 100 ps under three conditions at 1000 K. This result is in good agreement with the TEM analysis, as displayed in Fig. S27a–c (ESI†). The obtained TEM images clearly show that exposure to H2O2 roughens the MXene surface and causes the formation of some voids. The corresponding XRD patterns displayed in Fig. S27d (ESI†) reveal that the H2O2-treated MXene has a wider and lower (002) diffraction peak. To quantitatively evaluate the bond changes during the MXene oxidation process, the average bond numbers of the Ti–O, C–C, and Ti–C bonds at different MD simulation times were calculated (Fig. 2d and Fig. S28, ESI†). As the MD simulation time increases, there is a notable increase in the average number of C–C bonds, signifying their formation. Conversely, the average number of Ti–C bonds decreases, indicating the rupture of these bonds. In addition, the average number of Ti–O bonds rises with prolonged simulation time, attributed to the formation of additional functional Ti–O bonds resulting from the oxidation of the MXene. The MXene oxidation rate in the presence of H2O2 is higher than that in the dry and wet air environments, suggesting that H2O2-driven oxidation generates more Ti–O bonds. These MD results are in good agreement with the FTIR analysis (Fig. 2e), which confirms that the H2O2-treated MXene surface exhibits the largest number of Ti–O bonds. Therefore, it can be concluded that the growth of uniform VO2 nanowalls on the MXene nanosheets is due to the enhanced enrichment of V4+ close to the MXene surface and the generation of more Ti–O–V nuclei in the presence of H2O2.
To check the validity of the proposed mechanism, the adaptability of this approach for the growth of other metal oxides on the MXene surface was examined. Taking ZnO (MXene–ZnO) and MoO2 (Mxene–MoO2) as examples, TEM analysis demonstrates that these samples both exhibit sandwich structures that are similar to that of MXene–VO2 (Fig. S29 and S30, ESI†). The deconvoluted O 1s spectra of both MXene–ZnO and MXene–MoO2 (Fig. S31a and b, ESI†) show the presence of a Ti–O–M (Zn or Mo) peak, suggesting the formation of AOH between both metal oxides and the MXene substrate. Moreover, in addition to deposition on the MXene surface, the VO2 nanowalls can also be effectively grown onto the surfaces of graphene oxide (GO) and SiO2 (Fig. S32 and S33, ESI†) because these surfaces both have large amounts of unterminated functional oxygen groups. Growing VO2 nanowalls on the surfaces of carbon fibers (CFs) or carbon nanotubes (CNTs) would be appealing because these carbon materials exhibit high electrical conductivity and strong mechanical properties. However, the CF and CNT surfaces lack available oxygen functional groups, resulting in poor interaction between VO2 and these substrates. Thus, undesirable phase separation is observed and VO2 surface deposition does not occur (Fig. S34, ESI†). However, the strong acid treatment of these surfaces can lead to the uniform growth of VO2 nanowalls (Fig. S35 and S36, ESI†) with strong C–O–V AOH (Fig. S31c, ESI†). These results further corroborate the validity of the proposed mechanism.
Electrochemical performance measurements
To investigate the Zn2+ storage performance of the prepared cathodes, 2025 type coin cells were assembled using zinc foil anodes, a 3 M Zn(CF3SO3)2 aqueous electrolyte, and glass microfiber separators (see further details in the ESI†). The thickness of the VO2 nanowalls may affect the Ov stability. Therefore, the long-term cycling performance of MXene–VO2−x cathodes with various nanowall thicknesses (7.8, 27.5, and 62.4 nm) was measured at a current density of 1 A g−1 (Fig. S37, ESI†). The MXene–VO2−x cathode with a moderate thickness shows the most stable performance, which was then used to perform subsequent electrochemical measurements. As shown in Fig. 3a, cyclic voltammetry (CV) curves were obtained at a scan rate of 0.2 mV s−1 within the voltage window of 0.2 to 1.6 V (vs. Zn/Zn2+). Two pairs of redox peaks ascribed to the multiple insertion/extraction process of Zn2+ are observed for both the MXene–VO2−x and MXene/VO2−x cathodes. Through the comparison of the capacity contribution derived from two pair of redox peaks of MXene–VO2−x and MXene/VO2−x (Fig. S38, ESI†), the enhanced capacity of MXene–VO2−x primarily originates from the redox peak at a slow potential, attributed to the reversible reaction of V4+/V3+. Notably, the potential difference (ΔE) of the redox peaks of MXene–VO2−x is smaller than that of MXene/VO2−x, demonstrating weaker electrochemical polarization and more reliable reversibility of the MXene–VO2−x electrode reaction. The galvanostatic charge/discharge (GCD) curves of MXene–VO2−x and MXene/VO2−x were obtained at 0.2 A g−1 (Fig. 3b). The first and fourth GCD curves of MXene–VO2−x almost completely overlap, further suggesting the highly reversible redox behavior of this cathode. The MXene–VO2−x cathode delivers a notably enhanced and stable capacity of 487.2 mA h g−1, which is much higher than those of previously reported vanadium-based cathodes (Table S2, ESI†). Moreover, the Ragone plot shown in Fig. 3c reveals that the MXene–VO2−x cathode has specific energies of 683.2 W h kg−1 at 280 W kg−1 and 397.4 W h kg−1 at 28000 W kg−1, demonstrating the best performance among vanadium-based cathodes for aqueous ZIBs.28–36
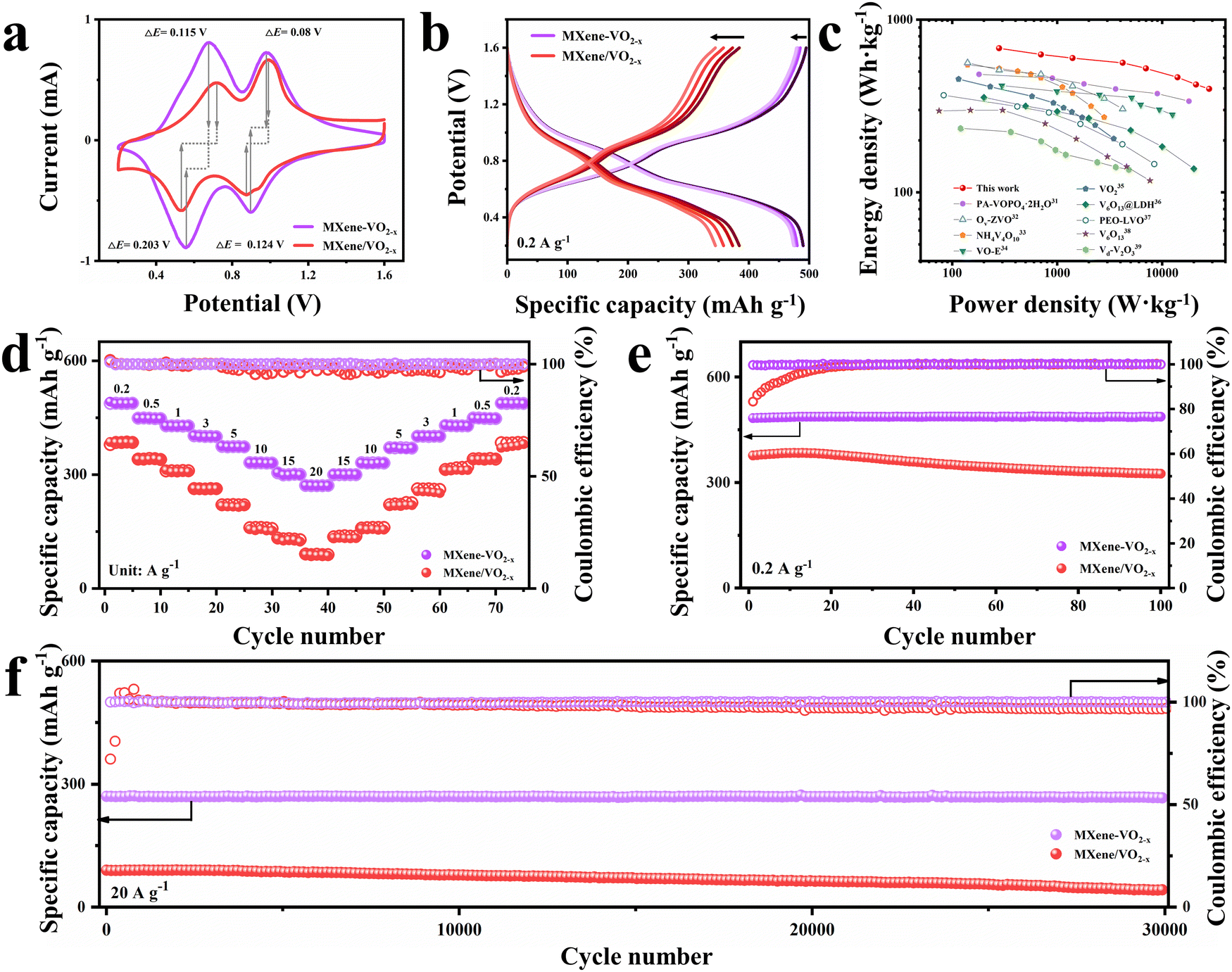 | ||
| Fig. 3 Electrochemical performance of aqueous zinc ion batteries. (a) CV curves of MXene–VO2−x and MXene/VO2−x at 0.2 mV s−1. (b) Charge/discharge curves of MXene–VO2−x and MXene/VO2−x at 0.2 A g−1. (c) Ragone plots of the MXene–VO2−x cathode compared with other vanadium-oxide-based electrode materials. (d) Rate performance of MXene–VO2−x and MXene/VO2−x cathodes. (e) Cycling performance of MXene–VO2−x and MXene/VO2−x cathodes at 0.2 A g−1. (f) Long-term cycling test of MXene–VO2−x and MXene/VO2−x cathodes at 20 A g−1. | ||
The ZIB assembled with MXene–VO2−x (Zn//MXene–VO2−x) shows superior rate performance as the current density increases from 0.2 to 20 A g−1 (Fig. 3d) compared to the ZIBs assembled with MXene/VO2−x and MXene–VO2 (Fig. S39, ESI†). Even at high rates of 10, 15, and 20 A g−1, the Zn//MXene–VO2−x battery still retains reversible capacities of 330.4, 300.7, and 270.5 mA h g−1, respectively. Upon returning to a low current density of 0.2 A g−1, the capacity of Zn//MXene–VO2−x recovers to 487.9 mA h g−1, indicating minimal capacity loss and highlighting the robust covalent heterostructure of MXene–VO2−x. Moreover, the GCD curves at different current densities (Fig. S40, ESI†) reveal the minimal impact on the specific capacity of the MXene–VO2−x cathode, suggesting a highly stable structural framework for Zn2+. The cycling performances of Zn//MXene–VO2−x and control batteries were also examined at both low (0.2 A g−1) and high (20 A g−1) current densities, as shown in Fig. 3e, f and Fig. S41 (ESI†). The control batteries exhibit similar cycling trends: the discharge capacity first increases during the initial cycles and then sharply declines in subsequent cycles. In contrast, the Zn//MXene–VO2−x battery demonstrates remarkable resilience, maintaining a capacity retention rate of ∼95.3% after enduring 100 cycles at 0.2 A g−1. Impressively, it still holds a capacity of 266.6 mA h g−1 even after enduring a staggering 30000 cycles at 20 A g−1, which is possibly attributed to the synergistic interplay of Ti–O–V AOH and Ov engineering. Besides, the scaled-up preparation of the MXene–VO2−x cathode shows a negligible effect on the cycling performance of the battery (Fig. S42, ESI†), indicating its immense potential for commercial applications. More impressively, the MXene–VO2−x cathode maintains its morphology and components effectively, regardless of whether it is subjected to long-term cycling at 0.2 A g−1 or 20 A g−1 (as evident in Fig. S43 and S44, ESI†). Inversely, the XRD pattern of the MXene/VO2−x cathode after large current cycles shows a new phase product of electrochemically inactive Zn3(OH)2V2O7·2H2O (PDF#50-0570) due to the instability of the electrode structure, thus leading to the attenuation of electrochemical capacity (Fig. S45, ESI†).
Detailed analysis of the aqueous Zn2+ storage mechanism
To analyze the charge storage behavior and calculate the pseudocapacitance contribution of the prepared cathodes, we performed CV tests at various scan rates. As the scan rate increases from 0.2 to 1.0 mV s−1, the MXene–VO2−x cathode maintains its CV peak shape and position effectively (Fig. 4a). In contrast, the MXene/VO2−x and MXene/VO2 cathodes experience significant polarization, as shown in Fig. S46 (ESI†), highlighting the MXene–VO2−x cathode's superior tolerance to higher scan rates and its excellent cycle reversibility. With the scan rate on the rise, the redox peaks of MXene–VO2−x show a sharp increase in current density, whereas the MXene/VO2−x and MXene/VO2 cathodes demonstrate a more subdued increase. To evaluate the relative contributions of capacitive and diffusion effects to the overall capacity, the relationship between the peak current (i) and scan rate (v) was analyzed: i = a·vb. When the value of b approaches 1.0, it indicates a significant pseudocapacitance contribution to the charge storage mechanism. The b values of peaks 1, 2, 3, and 4 of the MXene–VO2−x cathode are 0.941, 0.987, 0.905, and 0.930, respectively (Fig. 4b), suggesting that pseudocapacitance plays a primary role in determining the capacity of this cathode. For example, the capacitive contribution reaches up to 90.3% at a scan rate of 1.0 mV s−1 (Fig. S47, ESI†). As shown in Fig. 4c, the MXene/VO2−x cathode displays a larger capacitive contribution proportion than MXene/VO2, illustrating the enhanced pseudocapacitive charge storage mechanism after the introduction of Ov, which is consistent with the previous finding.10,37,38 The capacitive contribution of the MXene–VO2−x cathode is even higher than that of the MXene/VO2−x cathode at the evaluated scan rates, suggesting improved pseudocapacitive behavior and accelerated Zn2+ migration kinetics following the construction of AOH. The MXene–VO2−x cathode exhibits a higher Ds value (ion diffusion coefficient) than the MXene/VO2−x cathode in both the upper (Region I) and lower (Region II) plateaus of their galvanostatic intermittent titration (GITT) profiles (Fig. 4d), revealing the higher Zn2+ conductivity of MXene–VO2−x. Electrochemical impedance spectroscopy (EIS) measurements further indicate that the MXene–VO2−x cathode exhibits a lower charge transfer resistance than the MXene/VO2−x cathode (Fig. S48, ESI†). | ||
| Fig. 4 Electrochemical kinetics analyses and energy storage mechanism analyses of the MXene–VO2−x cathode. (a) CV curves at different scan rates. (b) Correspondinglog (peak current) versuslog (scan rate) plots at each redox peak. (c) Capacitive contribution to the capacity of MXene/VO2, MXene/VO2−x and MXene–VO2−x at different scan rates. (d) GITT curves and corresponding Zn2+ diffusion coefficients of MXene/VO2−x and MXene–VO2−x. (e) Ex situ sXAS at various states of discharge/charge. (f) O 1s spectra of MXene–VO2−x after discharging and charging. (g) V 2p3/2 spectra of MXene–VO2−x after discharging and charging. (h) In situ Raman spectra of the MXene–VO2−x electrode and corresponding charge/discharge profiles. | ||
The high stability and fast Zn2+ storage kinetics of the MXene–VO2−x cathode in ZIBs were also confirmed by a series of ex situ and in situ investigations. The ex situ sXAS spectra were recorded at various charge/discharge states (Fig. 4e). Upon full discharge of the battery to 0.2 V, the V L3-edge peak shifts to a lower energy. When the battery is charged back to 1.6 V, this peak gradually shifts back to a higher energy, reflecting the highly reversible redox activity and the stable crystalline structure of MXene–VO2−x. Meanwhile, the d|| and σ* hybridization peaks in the O K-edge also exhibit an evident reversible change in intensity, proving the reversible electron transfer at the interface during the electrochemical process. The Zn 2p XPS spectra of MXene–VO2−x were also obtained in the charged and discharged states (Fig. S49, ESI†). When discharged to 0.2 V, two strong Zn peaks are observed at 1021.2 and 1044.2 eV, corresponding to the insertion of Zn2+ into the MXene–VO2−x cathode. After the subsequent charging process, the intensity of Zn peaks decreases, suggesting the highly reversible extraction of Zn2+. The O 1s XPS spectra of the MXene–VO2−x cathode show that the content of Ti–O–V AOH remains similar at different voltages (Fig. 4f). In addition, the V 2p XPS spectra illustrate the reversible reduction and oxidation of V4+/V3+, confirming the high stability of Ovs during the electrochemical process (Fig. 4g). For comparison, the ex situ XPS measurement of the MXene/VO2−x cathode is shown in Fig. S50 (ESI†). The reduced Ov peak area in O 1s spectra indicates that the Ovs are refilled partly during electrochemical redox reactions, and this can also be demonstrated by the weaker V3+ signal in V 2p spectra after electrochemical cycling. The presence of the V5+ peak originates from the irreversible phase transition of the electrode structure to Zn3(OH)2V2O7·2H2O, which is consistent with the XRD pattern in Fig. S45 (ESI†). Ex situ XRD analysis of the MXene–VO2−x cathode was performed during the charge/discharge cycle within the potential window of 0.2–1.6 V (Fig. S51, ESI†). The (110) and (020) diffraction peaks of VO2 shift to a smaller degree during the discharge operation, indicating a typical in-plane expansion process. During the subsequent charge process, the peak positions recover gradually, illustrating good reversibility of the electrode reaction and the stabilization of the electrode structure. The corresponding TEM images of the MXene–VO2−x cathode at different voltages (Fig. S52 and S53, ESI†) verify that the heterostructure is well-retained during the electrochemical process. In the online recorded Raman spectra (Fig. 4h), the peak at ∼124 cm−1 corresponds to the skeleton bending vibration of layered VO2,39 whereas the two peaks at ∼448 and ∼490 cm−1 are attributed to the bending and stretching vibrations of V–O, respectively.40 The reversible change of these peaks during the Zn2+ insertion/extraction process confirms the structural stability of the MXene–VO2−x cathode. All these experimental results demonstrate that the synergistic combination of AOH and Ov can efficiently boost the electrochemical performance of VO2 cathodes.
Theoretical calculation verification
In order to elucidate the chemical origin of the remarkable storage capacity and stability of the MXene–VO2−x cathode, we performed DFT calculations to gain deeper insights into the electronic interactions between AOH and Ovs. An Ov is formed by removing an oxygen atom between two vanadium atoms, resulting in two distinct configurations: equatorial and apical Ovs (Fig. S54, ESI†). Through the calculation of formation energy (Ef) of Ovs, Ovs at the apical sites were found to exhibit a significantly lower Ef value (2.9 eV) than Ovs at the equatorial sites (4.18 eV), suggesting that apical Ovs are more likely to be formed than equatorial Ovs. Therefore, the following calculations are based on the formation of apical Ovs in the VO2 crystal structure. Owing to the strong orbital coupling, charge transfer would occur at the interface. Analyses of the sliced electron localization function (ELF, Fig. 5a) and Bader charge (Fig. S55, ESI†) reveal that the layer of VO2 adjacent to the interface donates 0.87 |e| through the Ti–O–V AOH to the MXene surface. Following the introduction of Ovs in VO2, the framework transfers an increased number of electrons (1.72 |e|) to the MXene, highlighting a strong electronic interaction between AOH and Ovs. The electron migration along Ti–O–V AOH is attributed to the difference in the band centers of V-3d, O-2p and Ti-3d (Fig. 5b).41,42 Note that the electron transfer from VO2 to the MXene via AOH creates a charge transfer pathway for O atoms in VO2, which aids in the release of electrons from these atoms and promotes the formation of Ovs.43,44 The DFT calculation further confirms that the construction of Ti–O–V AOH significantly lowers the formation energy (Ef) of Ovs, regardless of the number of slabs, suggesting that the Ovs exhibit enhanced chemical stability within the MXene–VO2−x framework (Fig. 5c and Fig. S56, S57, ESI†).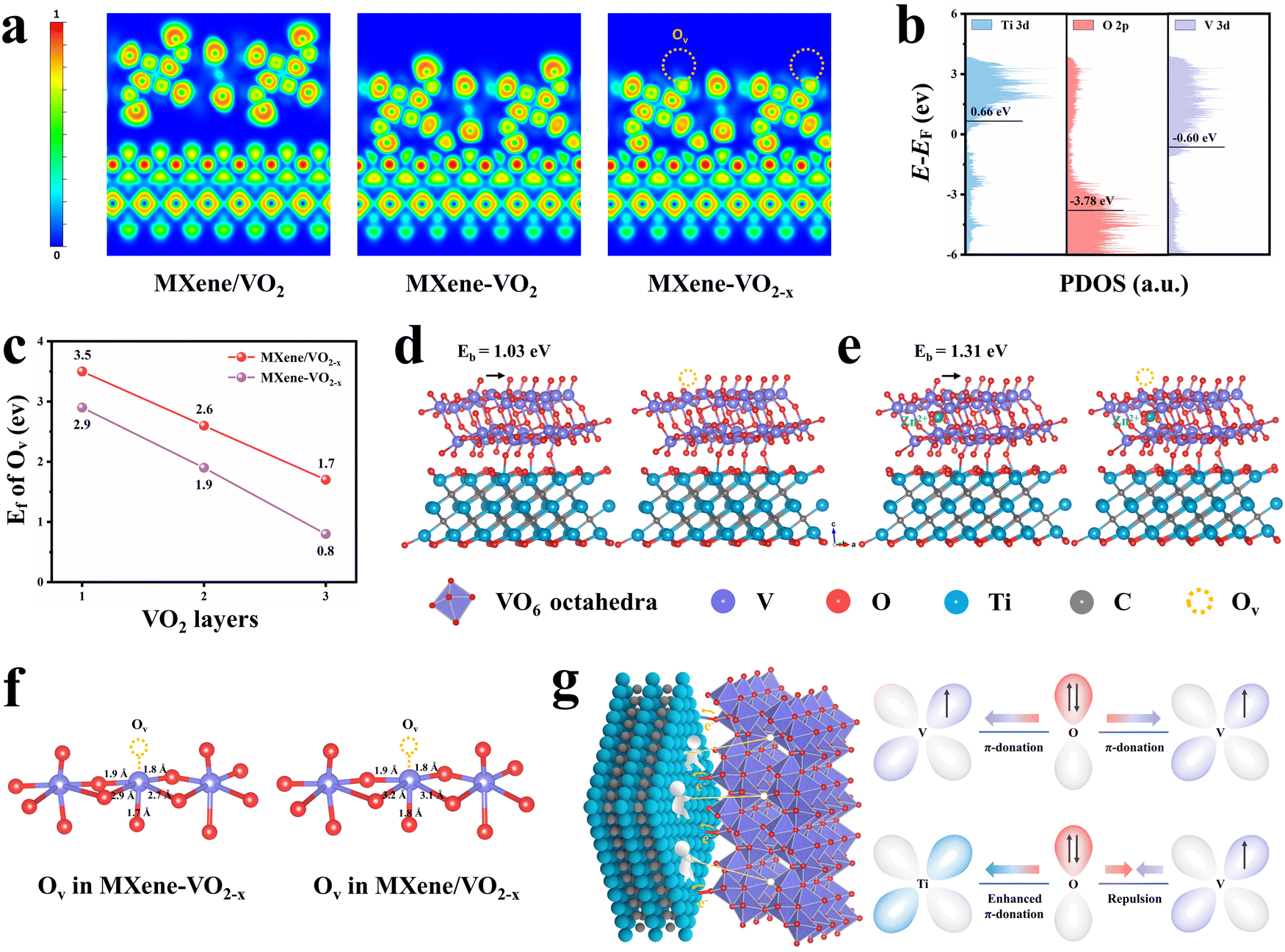 | ||
| Fig. 5 Theoretical verification for stabilizing Ovs on the VO2 cathode. (a) Sliced ELF of MXene/VO2, MXene–VO2, and MXene–VO2−x. The isovalues of 1 and 0 indicate complete electron localization and complete electron delocalization, respectively. (b) Projected density of states (PDOS) of Ti-3d, O-2p, and V-3d bands, with the black bar showing the band centers. (c) Calculated Ef values of Ovs at MXene/VO2−x and MXene–VO2−x as a function of VO2 layer number. (d) Calculated models for the migration of oxygen atoms to neighboring sites on MXene–VO2−x in the absence of intercalated Zn2+. (e) Calculated models for the migration of oxygen atoms to neighboring sites on MXene–VO2−x in the presence of intercalated Zn2+. (f) Optimized geometric structure near the Ovs of MXene–VO2−x and MXene/VO2−x. Measured bond distances are indicated. (g) Schematic illustration showing the mechanism of Ti–O–V AOH in the MXene–VO2−x cathode. | ||
Interestingly, the transfer of charges through Ti–O–V AOH also significantly influences the kinetic behaviors of Ovs. Fig. 5d shows the model of O migrating to the neighbor vacant site without the intercalation of Zn2+. The calculated migration energy barrier (Eb) of Ovs on the surface of MXene–VO2−x is 1.03 eV, slightly larger than that on MXene/VO2−x (0.95 eV, Fig. S58, ESI†), suggesting that Ovs are less prone to migration in the former case. This trend becomes even more evident when Zn2+ are intercalated into the framework, as seen in the comparison of 1.31 vs. 0.73 eV. This implies that the formation of AOH significantly stabilizes the Ovs during electrochemical cycling (Fig. 5e and Fig. S59, ESI†), ascribed to the improved geometric stability of the VO2−x frameworks after the migration away from electrons. Specifically, the electrostatic repulsion between the adjacent V atoms and connected O atoms can be weakened through Ti–O–V AOH, resulting in a shortened V–O bond length surrounding Ovs (Fig. 5f). This phenomenon can be further elucidated by examining the electronic configurations of Ti–O–V AOH. Unlike the V–O–V orbital coupling in bulk VO2, the substitution of V with Ti atoms disrupts charge symmetry. The unoccupied orbitals of Ti4+ can engage in π-donation with bridging O,45,46 while the electron repulsion between V4+ and O2− on the opposite side further enhances this interaction, leading to increased orbital coupling and strong electron transfer from VO2 to MXene (Fig. 5g). Consequently, the electrostatic interaction between the VO2−x framework and intercalated Zn2+ is significantly diminished, which can be verified through the zeta potential measurement (Fig. S60a, ESI†). The adsorption energies of Zn2+ at different sites on the cathode materials were also analyzed through DFT calculations, showing the lowest Zn2+ adsorption energy on the MXene–VO2−x cathode (Fig. S60b and S61, ESI†). The energy barriers of Zn2+ migration were calculated to further assess the dynamic performance of Zn2+ during the discharge/charge process (Fig. S62 and S63, ESI†). The calculated Zn2+ diffusion barrier in MXene–VO2−x is much lower than that of individual MXene and VO2, which may be attributed to the fast interfacial charge transfer of MXene–VO2−x. This gives rise to the smallest migration energy barriers for Zn2+ (∼0.15 eV), thereby facilitating rapid Zn2+ storage kinetics and enhancing the structural stability of the MXene–VO2−x cathode during the discharge/charge process.
Conclusions
Through detailed experimental and theoretical analyses, we demonstrated a rational covalent heterostructure design to stabilize Ovs on VO2 model cathodes for use in ZIBs. The cathode was achieved through a facile H2O2-assisted hydrothermal strategy, where H2O2 was crucial in facilitating the uniform deposition of VO2 nanowall arrays onto the MXene surface to form tight interfacial Ti–O–V AOH. DFT calculations suggested that these Ti–O–V AOH at the interface significantly influence the stabilization of Ovs by migrating electrons from the VO2 framework to the MXene surface. Thus, the refilling and migration of Ovs during Zn2+ shuttling were inhibited. Benefitting from this design, the prepared MXene–VO2−x cathode displayed superb Zn2+ storage performance, with a reversible specific capacity of 487.9 mA h g−1 at 0.2 A g−1 and a high capacity retention of 98.6% over 30000 cycles. This study offers important insight into the role of covalent heterostructures in improving the utilization efficiency of Ovs and provides guidance for the development of future battery cathode materials for specific application requirements.
Author contributions
W. L. and P. P. Q. conceived the project. Y. F. synthesized the samples and performed the structural characterization. W. S. carried out the electrochemical tests. C. H. Q performed the theoretical calculations. P. P. Q, W. S. and Y. F. wrote the manuscript. All authors analyzed the results and commented on the manuscript.Data availability
All the original data can be obtained from the corresponding authors upon reasonable request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was sponsored by the National Natural Science Foundation of China (52225204, 52173233, 52202085, and 52002059), the Innovation Program of Shanghai Municipal Education Commission (2021-01-07-00-03-E00109), the Natural Science Foundation of Shanghai (23ZR1479200), “Shuguang Program” Supported by the Shanghai Education Development Foundation and Shanghai Municipal Education Commission (20SG33), the Fundamental Research Funds for the Central Universities (2232024Y-01) and the DHU Distinguished Young Professor Program (LZA2022001 and LZB2023002). We are thankful for the support provided for using the BL14W1 beamline at the Shanghai Synchrotron Radiation Facility.References
- N. Zhang, X. Y. Chen, M. Yu, Z. Q. Niu, F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2020, 49, 4203–4219 RSC.
- Y. Zhang, F. Wan, S. Huang, S. Wang, Z. Q. Niu and J. Chen, Nat. Commun., 2020, 11, 2199 CrossRef CAS PubMed.
- R. F. Service, Science, 2021, 372, 890–891 CrossRef CAS.
- X. X. Jia, C. F. Liu, Z. G. Neale, J. H. Yang and G. Z. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS.
- D. J. Dong, T. R. Wang, Y. Sun, J. Fan and Y.-C. Lu, Nat. Sustainability, 2023, 6, 1474–1484 CrossRef.
- F. Wan and Z. Q. Niu, Angew. Chem., Int. Ed., 2019, 58, 16358–16367 CrossRef CAS.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- D. L. Han, C. J. Cui, K. Y. Zhang, Z. X. Wang, J. C. Gao, Y. Guo, Z. C. Zhang, S. C. Wu, L. C. Yin, Z. Weng, F. Y. Kang and Q.-H. Yang, Nat. Sustainability, 2022, 5, 205–213 CrossRef.
- L. L. Wang, K.-W. Huang, J. T. Chen and J. R. Zheng, Sci. Adv., 2019, 5, eaax 4279 CrossRef CAS.
- H.-S. Kim, J. B. Cook, H. Lin, J. S. Ko, S. H. Tolbert, V. Ozolins and B. Dunn, Nat. Mater., 2017, 16, 454–460 CrossRef CAS.
- C. Guo, S. J. Yi, R. Si, B. J. Xi, X. G. An, J. Liu, J. F. Li and S. L. Xiong, Adv. Energy Mater., 2022, 12, 2202039 CrossRef CAS.
- Y. Q. Zhang, L. Tao, C. Xie, D. D. Wang, Y. Q. Zou, R. Chen, Y. Y. Wang, C. K. Jia and S. Y. Wang, Adv. Mater., 2020, 32, 1905923 CrossRef CAS.
- Z. H. Xiao, Y. Wang, Y.-C. Huang, Z. X. Wei, C.-L. Dong, J. M. Ma, S. H. Shen, Y. F. Li and S. Y. Wang, Energy Environ. Sci., 2017, 10, 2563–2569 RSC.
- M. Wang, G. Y. Zhao, X. M. Bai, W. J. Yu, C. H. Zhao, Z. R. Gao, P. B. Lyu, Z. Y. Chen and N. Q. Zhang, Adv. Energy Mater., 2023, 13, 2301730 CrossRef CAS.
- P. F. Yan, J. M. Zheng, Z.-K. Tang, A. Devaraj, G. Y. Chen, K. Amine, J.-G. Zhang, L.-M. Liu and C. M. Wang, Nat. Nanotechnol., 2019, 14, 602–608 CrossRef CAS PubMed.
- C. H. Zhao, B. Jiang, Y. Huang, X. Sun, M. Wang, Y. Zhang and N. Q. Zhang, Energy Environ. Sci., 2023, 16, 5490–5499 RSC.
- L. Z. Zhuang, Y. Jia, H. L. Liu, Z. H. Li, M. R. Li, L. Z. Zhang, X. Wang, D. J. Yang, Z. H. Zhu and X. D. Yao, Angew. Chem., Int. Ed., 2020, 59, 14664–14670 CrossRef CAS PubMed.
- N. B. Aetukuri, A. X. Gray, M. Drouard, M. Cossale, L. Gao, A. H. Reid, R. Kukreja, H. Ohldag, C. A. Jenkins, E. Arenholz, K. P. Roche, H. A. Dürr, M. G. Samant and S. S. P. Parkin, Nat. Phys., 2013, 9, 661–666 Search PubMed.
- Z. L. Liao, E. Skoropata, J. W. Freeland, E.-J. Guo, R. Desautels, X. Gao, C. Sohn, A. Rastogi, T. Z. Ward, T. Zou, T. Charlton, M. R. Fitzsimmons and H. N. Lee, Nat. Commun., 2019, 10, 589 Search PubMed.
- Z.-Y. Yu, Y. Duan, J.-D. Liu, Y. Chen, X.-K. Liu, W. Liu, T. Ma, Y. Li, X.-S. Zheng, T. Yao, M.-R. Gao, J.-F. Zhu, B.-J. Ye and S.-H. Yu, Nat. Commun., 2019, 10, 2799 Search PubMed.
- Y. Duan, X.-L. Zhang, F.-Y. Gao, Y. Kong, Y. Duan, X.-T. Yang, X.-X. Yu, Y.-R. Wang, S. Qin, Z. Chen, R. Wu, P.-P. Yang, X.-S. Zheng, J.-F. Zhu, M.-R. Gao, T.-B. Lu, Z.-Y. Yu and S.-H. Yu, Angew. Chem., Int. Ed., 2023, 62, e202217275 CrossRef CAS PubMed.
- L. H. Yeo, A. Srivastava, M. A. Majidi, R. Sutarto, F. He, S. M. Poh, C. Diao, X. Yu, M. Motapothula, S. Saha, S. Ojha, D. Kanjilal, P. E. Trevisanutto, M. B. H. Breese, T. Venkatesan and A. Rusydi, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 081112 CrossRef.
- J. L. Wang, C.-S. Hsu, T.-S. Wu, T.-S. Chan, N.-T. Suen, J.-F. Lee and H. M. Chen, Nat. Commun., 2023, 14, 6576 CrossRef CAS PubMed.
- X. D. Yang, B. Ouyang, P. Q. Shen, Y. Q. Sun, Y. S. Yang, Y. N. Gao, E. J. Kan, C. C. Li, K. Xu and Y. Xie, J. Am. Chem. Soc., 2022, 144, 11138–11147 CrossRef CAS.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS.
- K.-J. Wu, E. C. M. Tse, C. X. Shang and Z. X. Guo, Prog. Mater. Sci., 2022, 123, 100821 CrossRef CAS.
- G. L. Zhan, Z.-F. Cai, K. Strutyński, L. H. Yu, N. Herrmann, M. Martínez-Abadía, M. Melle-Franco, A. Mateo-Alonso and S. D. Feyter, Nature, 2022, 603, 835–840 CrossRef CAS.
- L. F. Hu, Z. Y. Wu, C. J. Lu, F. Ye, Q. Liu and Z. M. Sun, Energy Environ. Sci., 2021, 14, 4095–4106 RSC.
- J.-J. Ye, P.-H. Li, H.-R. Zhang, Z.-Y. Song, T. J. Fan, W. Q. Zhang, J. Tian, T. Huang, Y. T. Qian, Z. G. Hou, N. Shpigel, L.-F. Chen and S. X. Dou, Adv. Funct. Mater., 2023, 33, 2305659 Search PubMed.
- Y. Xu, G. L. Fan, P. X. Sun, Y. Guo, Y. Y. Wang, X. J. Gu, L. M. Wu and L. Yu, Angew. Chem., Int. Ed., 2023, 62, e202303529 CrossRef CAS PubMed.
- Z. H. Wang, Y. Song, J. Wang, Y. L. Lin, J. M. Meng, W. B. Cui and X.-X. Liu, Angew. Chem., Int. Ed., 2023, 62, e202216290 CrossRef CAS PubMed.
- Z. Y. Cao, L. P. Wang, H. Zhang, X. Zhang, J. W. Liao, J. C. Dong, J. Y. Shi, P. Y. Zhuang, Y. D. Cao, M. X. Ye, J. F. Shen and P. M. Ajayan, Adv. Funct. Mater., 2020, 30, 2000472 CrossRef CAS.
- Y. H. Dai, C. Y. Zhang, J. W. Li, X. Gao, P. Hu, C. M. Ye, H. Z. He, J. X. Zhu, W. Zhang, R. W. Chen, W. Zong, F. Guo, I. P. Parkin, D. J. L. Brett, P. R. Shearing, L. Q. Mai and G. J. He, Adv. Mater., 2024, 36, 2310645 CrossRef CAS.
- M. Liao, J. W. Wang, L. Ye, H. Sun, Y. Z. Wen, C. Wang, X. M. Sun, B. J. Wang and H. S. Peng, Angew. Chem., Int. Ed., 2020, 59, 2273–2278 CrossRef CAS PubMed.
- M. H. Wu, C. Shi, J. W. Yang, Y. Zong, Y. Chen, Z. G. Ren, Y. X. Zhao, Z. Li, W. Zhang, L. Y. Wang, X. L. Huang, W. Wen, X. L. Li, X. Ning, X. C. Ren and D. M. Zhu, Adv. Mater., 2024, 36, 2310434 CrossRef CAS.
- K. F. Zhu, S. Q. Wei, H. W. Shou, F. R. Shen, S. M. Chen, P. J. Zhang, C. D. Wang, Y. Y. Cao, X. Guo, M. Luo, H. J. Zhang, B. J. Ye, X. J. Wu, L. H. He and L. Song, Nat. Commun., 2021, 12, 6878 CrossRef CAS PubMed.
- D. Z. Wu, Y. H. Kang, F. Wang, J. Yang, Y. Q. Xu, Y. C. Zhuang, J. Y. Wu, J. Zeng, Y. Yang and J. B. Zhao, Adv. Energy Mater., 2023, 13, 2301145 CrossRef CAS.
- Z. Y. Peng, Y. T. Huang, A. G. Bannov, S. L. Li, L. Tang, L. C. Tan and Y. W. Chen, Energy Environ. Sci., 2024, 17, 3384–3395 RSC.
- J. T. Huang, H. P. Liang, Y. Tang, B. G. Lu, J. Zhou and S. Q. Lian, Adv. Energy Mater., 2022, 12, 2201434 CrossRef CAS.
- J. W. Li, K. McColl, X. K. Lu, S. Sathasivam, H. B. Dong, L. Q. Kang, Z. N. Li, S. Zhao, A. G. Kafizas, R. Wang, D. J. L. Brett, P. R. Shearing, F. Corà, G. J. He, C. J. Carmalt and I. P. Parkin, Adv. Energy Mater., 2020, 10, 2000058 CrossRef CAS.
- J. M. Tian, Y. Rao, W. H. Shi, J. W. Yang, W. J. Ning, H. Y. Li, Y. G. Yao, H. S. Zhou and S. H. Guo, Angew. Chem., Int. Ed., 2023, 62, e202310894 CrossRef CAS.
- R. L. Liu, C. Wang, Y. Yan, R. Z. Wang and G. Chen, ACS Catal., 2024, 14, 3955–3965 CrossRef CAS.
- T.-N. Ye, S.-W. Park, Y. F. Lu, J. Li, M. Sasase, M. Kitano, T. Tada and H. Hosono, Nature, 2020, 583, 391–395 CrossRef CAS PubMed.
- S. Z. Li, G. Wang, H. F. Lv, Z. J. Lin, J. S. Liang, X. Liu, Y.-G. Wang, Y. H. Huang, G. X. Wang and Q. Li, J. Am. Chem. Soc., 2024, 146, 17659–17668 CrossRef CAS PubMed.
- S. Y. Yao, S. Y. Wang, Y. M. Liu, Z. S. Hou, J. R. Wang, X. Y. Gao, Y. F. Sun, W. J. Fu, K. Q. Nie, J. Z. Xie, Z. Y. Yang and Y.-M. Yan, J. Am. Chem. Soc., 2023, 145, 26699–26710 CrossRef CAS.
- H. J. Huang, W. Geng, X. Z. Wu, Y. Y. Zhang, L. Xie, T. Ma and C. Cheng, Angew. Chem., Int. Ed., 2024, 63, e202310811 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04466e |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2025 |