
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sunflower oil-based thermosets via the Passerini three-component reaction†
Luis
Santos Correa
 a,
Silas
Leidenheimer
b and
Michael A. R.
Meier
*ab
a,
Silas
Leidenheimer
b and
Michael A. R.
Meier
*ab
aLaboratory of Applied Chemistry, Institute of Biological and Chemical Systems-Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology (KIT), 76344 Eggenstein-Leopoldshafen, Germany. E-mail: m.a.r.meier@kit.edu
bLaboratory of Applied Chemistry, Institute of Organic Chemistry (IOC), Karlsruhe Institute of Technology (KIT), Kaiserstraße 12, 76131 Karlsruhe, Germany
First published on 26th December 2024
Abstract
Tricarboxylic acids are molecules of interest for the synthesis of cross-linked polymers. Herein, a recently established route to a tricarboxylic acid from high oleic sunflower oil was utilized to synthesize renewable cross-linked polymers via the Passerini three-component reaction. Ten different polymers were synthesized by varying the diisocyanide and monoaldehyde components and subsequently characterized via infrared spectroscopy, swelling tests, thermogravimetric analysis, differential scanning calorimetry, and tensile tests. The characterization of the insoluble polymeric networks was complemented by the synthesis of model compounds to enable analysis in solution via nuclear magnetic resonance spectroscopy. Due to the fast curing of all polymers at room temperature, adhesive tests were performed to demonstrate their potential application as glues. Finally, one Passerini polymer was chemically reused by transesterification with methanol and catalytic amounts of sulfuric acid to establish a proof-of-concept of the circularity of these materials. Azelaic acid dimethyl ester, an industrially relevant compound used for the synthesis of polyamides, and a difunctional α-hydroxyamide were recovered in a yield of 75%. Repolymerization of the α-hydroxyamide to a polyurethane was performed to demonstrate a potential second life cycle of these compounds.
Introduction
The development and production of new polymeric materials from renewable resources are of paramount importance to reduce humanity's high dependency on depleting fossil resources. For instance, in 2022, a concerningly small share of less than 1% of the worldwide plastic production of >400 Mt was bio-based.1 Plant oils and fatty acids are the most important renewable feedstock of the chemical industry for polymer applications. In 2019, approximately 25% of the annual global production of 208 Mt vegetable oils was already used industrially for the production of biodiesel, oleochemicals, and other applications to substitute fossil-resources.2 In polymer chemistry, fatty acids are either functionalized at the carboxylic acid moiety by esterification or at their double bonds via oxidation, thiol–ene reaction, olefin metathesis or isomerizing functionalization to synthesize difunctional monomers that can be polymerized.3,4 Polyfunctional derivatives can be obtained from oils by functionalization of the triglyceride double bonds.5 Alternatively, cross-linked polymers can be synthesized from triglycerides by direct polymerization of the double bonds via acyclic triene metathesis polymerization or cationic/radical polymerization.3Besides the use of renewable feedstocks, chemical reactions must adhere as much as possible to the 12 principles of green chemistry, as established by Anastas and Warner in 1998.6,7 These principles act as guidelines to develop more sustainable processes and address topics such as waste production, atom economy, toxicity of the involved chemicals, the design of inherently safer chemistry, utilization of renewable resources, and the development of catalysts to reduce energy consumption.
Multicomponent reactions (MCRs) align with many of the 12 principles of green chemistry and are defined as reactions that combine at least three different starting compounds in a single one-pot reaction, in which most of the atoms of the starting materials are incorporated in the final product.8,9 Hence, waste generation is kept to a minimum due to the high atom economy of these reactions, and usually only benign by-products such as water, alcohols or amines are formed.10 The isocyanide based Passerini three-component reaction (P-3CR) was discovered in 1921 and describes the addition reaction between a carboxylic acid, an aldehyde, and an isocyanide to form an α-acyloxy amide.11,12 The high versatility of this reaction enables its use in step economic syntheses of several natural products and active pharmaceutical ingredients.12–14
Although the Passerini reaction is known to achieve near quantitative conversions at room temperature, its utilization as step-growth polymerization reaction was first reported 90 years after its discovery.15 Meier et al. reported polyesters with high molecular weights of up to 56 kDa that were obtained by a combination of a dicarboxylic acid, a dialdehyde, and a monoisocyanide at a reaction temperature of only 40 °C. Shortly after, several syntheses of new poly(ester-amide)s,16 polyamides,17 and polyesters18 were achieved by simple permutation of the starting materials’ functional groups. The multicomponent polymerization approach moreover easily allows the polymers to be grafted onto by the introduction of additional functional groups during the MCR step.17,18 So far, the synthesis of cross-linked polymers via isocyanide based MCRs has however been reported scarcely. Most studies focused on the synthesis of hydrogels from carboxylic acid group bearing polysaccharides such as alginic acid,19–22 hyaluronic acid,22,23 and carboxymethylcellulose22,24via the P-3CR or the Ugi four-component reaction. Furthermore, the polysaccharide chitosan was used as a polyamine component for the synthesis of Ugi-4CR based microgels.25 However, in all of these reports, the cross-linking was only performed with already polymeric starting materials. To the best of our knowledge, so far only one report by Xie et al. describes the synthesis of a cross-linked P-3CR polymer from terephthalaldehyde, 1,6-diisocyanohexane, and sebacic acid.26 However, due to the high molar excess of 50 mol% terephthalaldehyde, it is most likely that these materials exhibited a low degree of cross-linking and a high share of unreacted aldehyde groups. In addition, both the hydrogel approach and the polymer reported by Xie et al. used solvents during the polymerization, which is not desired as this generates additional waste.
Thus, we herein report for the first time the solvent-free synthesis of ten different P-3CR thermosets from a tricarboxylic acid, two diisocyanides, and five monoaldehydes. All components were by design chosen to be liquid to allow the facile preparation of solvent-free formulations. Moreover, isocyanide and aldehyde components were chosen to be at least in principle renewable in order to further increase the sustainability of these new materials. The used tricarboxylic acid is fully renewable and was synthesized from high oleic sunflower oil with a literature reported procedure.27
Experimental
Materials
All starting materials, solvents and reagents were purchased from chemical suppliers and used without further purification unless stated otherwise.Methods
Instruments
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 Da were used. System II (DMAc SEC): a PSS SECcurity2 SEC system based on Agilent Infinity 1260 II hardware was used for the measurements. The system was equipped with a refractive index detector. DMAc with 0.03 wt% lithium bromide was used at a flow rate of 1.0 ml min−1 and at 35 °C as the mobile phase. The analysis was performed on the following column system: Agilent Mixed-C column and Agilent Mixed-E column. For the calibration, polystyrene standards ranging from 370 to 2520000 Da were used. For sample preparation, 2.00 mg of analyte was dissolved in 1.50 ml of the solvent used in the respective system. All samples were filtered with a syringe filter prior to use, to avoid plugging of the injection setup or the column.
200 Da were used. System II (DMAc SEC): a PSS SECcurity2 SEC system based on Agilent Infinity 1260 II hardware was used for the measurements. The system was equipped with a refractive index detector. DMAc with 0.03 wt% lithium bromide was used at a flow rate of 1.0 ml min−1 and at 35 °C as the mobile phase. The analysis was performed on the following column system: Agilent Mixed-C column and Agilent Mixed-E column. For the calibration, polystyrene standards ranging from 370 to 2520000 Da were used. For sample preparation, 2.00 mg of analyte was dissolved in 1.50 ml of the solvent used in the respective system. All samples were filtered with a syringe filter prior to use, to avoid plugging of the injection setup or the column.
Oxidative cleavage of high oleic sunflower oil
In a 100 ml three-necked flask, high oleic sunflower oil (39.8 g, 45.0 mmol (based on the molecular weight of triolein (885.5 g mol−1)), 1.00 equiv.), ruthenium(III) acetylacetonate (359 mg, 900 μmol, 2.00 mol%) and pyridine-2,6-dicarboxylic acid (3.01 g, 18.0 mmol, 40.0 mol%) were dissolved in a mixture of tert-butanol (405 ml) and water (135 ml). The reaction mixture was stirred magnetically (400 rpm with a cross shaped stirring bar) at 80 °C for 24 h. After the reaction temperature reached 80 °C, hydrogen peroxide (35% aq. sol., 92.9 ml, 1.08 mol, 24.0 equiv.) was added slowly to the reaction solution using a syringe pump with a flow rate of 77.4 μl min−1 over a period of 20 h. After the entire reaction time passed, the reaction solution was diluted with water (200 ml) and tert-butanol was removed under reduced pressure to ensure easier phase separation during extraction. The aqueous phase was extracted with ethyl acetate (3 × 150 ml) and the combined organic layers were washed with saturated sodium chloride solution (3 × 100 ml), dried over sodium sulfate, filtered and the solvent was removed under reduced pressure. The reaction was conducted twice. Both reactions were combined and distilled in a Zinke apparatus in vacuo (100 °C, 0.1 mbar) to remove the cleavage product nonanoic acid (29.2 g, 185 mmol, 90%). The residue of the distillation was purified by flash column chromatography (cyclohexane/ethyl acetate, 4:1 + 1% formic acid, then 2:1 + 1% formic acid, and then 2:1 without formic acid) to obtain three fractions (fraction 1: residual nonanoic acid (2.50 g, 15.8 mmol, 8%), fraction 2: side product with low carboxylic acid content (18.0 g, 31%), fraction 3: sunflower oil-based triacid (20.8 g, 36%)) after the removal of the solvent and formic acid under reduced pressure. Analytical data for sunflower oil-based triacid: 1H NMR (400 MHz, DMSO-d6, ppm): δ = 11.95 (s, 3H), 5.18 (tt, J = 6.9, 3.7 Hz, 1H), 4.25 (dd, J = 12.0, 3.7 Hz, 2H), 4.12 (dd, J = 12.0, 6.5 Hz, 2H), 2.34–2.22 (m, 6H), 2.17 (t, J = 7.4 Hz, 6H), 1.49 (ddd, J = 14.6, 9.6, 5.5 Hz, 12H), 1.25 (d, J = 3.8 Hz, 18H), 0.89–0.78 (m, 0.17H); 13C NMR (126 MHz, DMSO-d6, ppm): δ = 174.5 (Cq, CO2H), 174.4 (Cq, CO2H), 172.5 (Cq, Cester), 172.2 (Cq, Cester), 68.8 (CH, Cglyceryl), 61.8 (CH2, Cglyceryl), 33.6 (CH2), 33.5 (CH2), 33.3 (CH2), 28.4 (CH2), 28.3 (CH2), 28.2 (CH2), 24.4 (CH2), 24.4 (CH2), 24.3 (CH2); IR (ATR, cm−1):  , 2929 (m), 2857 (w), 1738 (vs), 1703 (vs), 1456 (w), 1413 (w), 1378 (w), 1232 (m), 1160 (vs), 1133 (s), 1094 (m), 1033 (w), 938 (w), 728 (w); ESI-MS ([M − H]−, C30H49O12): calcd: 601.3230, found: 601.3229.
, 2929 (m), 2857 (w), 1738 (vs), 1703 (vs), 1456 (w), 1413 (w), 1378 (w), 1232 (m), 1160 (vs), 1133 (s), 1094 (m), 1033 (w), 938 (w), 728 (w); ESI-MS ([M − H]−, C30H49O12): calcd: 601.3230, found: 601.3229.
Quantitative 31P NMR spectroscopy
The amount of carboxylic acids and hydroxyl groups per mg of sample was determined by derivatization of the sunflower oil-based triacid sample using the phosphitylation agent 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (2-Cl-TMDP), according to a modified procedure of the one published by Kilpeläinen et al.29,30 First, the internal standard solution was prepared by dissolving endo-N-hydroxy-5-norbornene-2,3-dicarboximide (550 mg, 3.07 mmol) in pyridine (15 ml) and CDCl3 (10 ml). This resulted in a final concentration of 22 mg ml−1 or 123 mmol l−1. Then, under a standard atmosphere (no argon box), 28–32 mg of the analyte were weighed into a 10 ml screw-cap vial. CDCl3 (1 ml), pyridine (200 μl, 2.48 mmol) and the internal standard solution of endo-N-hydroxy-5-norbornene-2,3-dicarboximide (650 μl) were added and the mixture was agitated with a vortex mixer until the sample was fully dissolved (approximately 1 min). Finally, 2-Cl-TMDP (200 μl, 1.26 mmol) was added and the mixture was agitated for 1 min. Then, 1.0 ml of the solution was transferred to an NMR tube. 31P NMR measurements were performed immediately after the sample was prepared. The carboxylic acid and hydroxyl content was calculated according to eqn (S3) and (S4), ESI.†Synthesis of formamides and isocyanides
The syntheses of all formamides and isocyanides were conducted according to a literature reported procedure for the synthesis of aliphatic isocyanides.31 All syntheses and their analytical data are described in detail in the ESI.†General procedure for the synthesis of model compounds M1 to M5
Sunflower oil-based triacid (200 mg, 990 μmol, 1.00 equiv. of carboxylic acids), n-hexylisocyanide (165 mg, 1.49 mmol, 1.50 equiv. based on carboxylic acids), and the respective aldehyde (1.50 equiv. based on carboxylic acids) were weighed into a glass vial and the vial was sealed. Then, dichloromethane (1 ml) was added and the reaction mixture was stirred at room temperature for 3 days. The solvent was removed under reduced pressure and the crude product was purified by flash column chromatography (cyclohexane/ethyl acetate) to obtain the respective model compound. Exemplary analytical data for M4 (data of all other model compounds can be found in the ESI†): 1H NMR (400 MHz, CDCl3, ppm): δ = 6.10–5.89 (m, 3H, NH), 5.23 (p, J = 5.0 Hz, 1H, Hglyceryl), 5.13 (t, J = 6.0 Hz, 3H), 4.27 (dd, J = 11.9, 4.4 Hz, 2H, Hglyceryl), 4.12 (dd, J = 11.9, 5.9 Hz, 2H, Hglyceryl), 3.34–3.13 (m, 6H), 2.36 (t, J = 7.5 Hz, 6H, α-Hester), 2.29 (t, J = 7.4 Hz, 6H, α-Hester), 1.89–1.72 (m, 6H), 1.69–1.54 (m, 12H, β-Hester), 1.48 (p, J = 7.0 Hz, 6H), 1.34–1.00 (m, 72H), 0.95–0.78 (m, 18H); 13C NMR (101 MHz, CDCl3, ppm): δ = 173.2 (Cq, Cester), 172.8 (Cq, Cester), 172.5 (Cq, Cester), 169.9 (Cq, Camide), 74.1 (CH), 69.0 (CH, Cglyceryl), 62.2 (CH2, Cglyceryl), 39.3 (CH2), 34.3 (CH2, α-Cester), 34.2 (CH2, α-Cester), 34.0 (CH2, α-Cester), 32.0 (CH2), 31.9 (CH2), 31.5 (CH2), 29.6 (CH2), 29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 29.0 (CH2), 29.0 (CH2), 28.9 (CH2), 26.6 (CH2), 24.9 (CH2), 24.8 (CH2), 24.8 (CH2), 22.7 (CH2), 22.6 (CH2), 14.2 (CH3), 14.1 (CH3); IR (ATR, cm−1):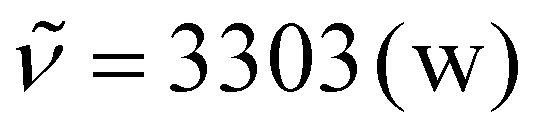 , 2952 (m), 2924 (vs), 2854 (s), 1741 (vs), 1656 (vs), 1537 (m), 1460 (m), 1418 (w), 1377 (w), 1299 (w), 1237 (m), 1159 (vs), 1132 (s), 1094 (s), 724 (w); ESI-MS ([M + H]+, C78H144N3O15): calcd: 1363.0592, found: 1363.0494.
, 2952 (m), 2924 (vs), 2854 (s), 1741 (vs), 1656 (vs), 1537 (m), 1460 (m), 1418 (w), 1377 (w), 1299 (w), 1237 (m), 1159 (vs), 1132 (s), 1094 (s), 724 (w); ESI-MS ([M + H]+, C78H144N3O15): calcd: 1363.0592, found: 1363.0494.
General procedure for Passerini three component polymerization
Sunflower oil-based triacid (600 mg, 1.00 equiv. of carboxylic acids) and the respective diisocyanide component (0.50 equiv. of carboxylic acids) were weighed into a 10 ml screw-cap vial and cooled to −20 °C with a sodium chloride ice cooling bath. The aldehyde component (1.00 equiv. of carboxylic acids) was freshly distilled and then weighed into the cooled vial. All compounds were mixed while cooling was maintained. The mixture was then poured into a poly(tetrafluoroethylene) form to produce bone shaped samples for tensile strength measurements (ESI chapter 2.5.5†). Polymers P1a to P3b were first cured at 5 °C for 30 minutes to prevent evaporation of the volatile aldehyde components, while polymers P4a to P5b were cured at room temperature for 30 minutes. All polymers were further cured at 50 °C for 24 h to guarantee full conversion of functional groups.Swelling tests for gel content determination
Each polymer sample (50–100 mg) was weighed and immersed in tetrahydrofuran for 3 days at room temperature. The sample was then removed from the solvent, dried at 70 °C and 10 mbar, and weighed again to determine the gel content (eqn (1)).Lap shear force adhesive tests
Test samples were prepared by cutting each substrate material (aluminum alloy, stainless-steel, wood, poly(methyl methacrylate), and glass) into rectangular pieces (ESI chapter 2.5.6†). For sample preparation, the respective monomers were premixed at 0 °C and then applied onto two rectangular pieces of the same substrate (gluing area of 2.25 cm2). The two surfaces were pressed together and cured at room temperature for 2 h. Afterwards, the two pieces were pulled apart in a tensile measurement to determine the ultimate tensile strength.Chemical recycling of the P-3CR polymer
Polymer P4a (3.00 g, 14.6 mmol ester groups, 1.00 equiv.) was suspended in methanol (120 ml, 2.96 mol, 203 equiv.) and sulfuric acid (96%, 286 mg, 2.91 mmol, 0.20 equiv.) was added. The reaction mixture was stirred for 5 h at 65 °C until P4a dissolved completely due to depolymerization. While cooling to room temperature, the diol compound 6 precipitated. The reaction volume was reduced to 60 ml and then filtered to isolate the diol (1.23 g, 2.69 mmol, 74%). The solvent methanol was removed from the filtrate and ethyl acetate (60 ml) was added. The organic phase was washed with water (3 × 40 ml) to remove glycerol and sulfuric acid, dried over sodium sulfate, and the solvent was removed under reduced pressure. The residue was then distilled in a Kugelrohr oven in vacuo (10 mbar, 150 °C) to isolate azelaic acid dimethyl ester (1.18 g, 5.45 mmol, 75%). Finally, the aqueous phase from the washing step was neutralized with sodium hydroxide (116 mg, 2.91 mmol, 0.20 equiv.) and distilled in a Kugelrohr oven to obtain glycerol (68 mg, 738 μmol, 30%). Analytical data for diol 6: Tm: 162 °C; 1H NMR (500 MHz, TFA-d, ppm): δ = 4.63 (dd, J = 8.7, 3.5 Hz, 2H), 3.44 (t, J = 7.3 Hz, 4H), 1.96–1.83 (m, 2H), 1.81–1.68 (m, 2H), 1.68–1.56 (m, 4H), 1.48–1.39 (m, 4H), 1.39 (s, 4H), 1.34–1.15 (m, 20H), 0.79 (t, J = 6.6 Hz, 6H); 13C NMR (126 MHz, TFA-d, ppm): δ = 180.7 (Cq, Camide), 73.8 (CH), 43.4 (CH2), 35.9 (CH2), 33.6 (CH2), 31.1 (CH2), 30.9 (CH2), 30.8 (CH2), 29.9 (CH2), 27.9 (CH2), 26.7 (CH2), 24.3 (CH2), 14.7 (CH3); IR (ATR, cm−1):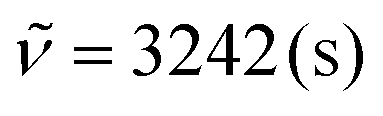 , 2953 (m), 2918 (vs), 2873 (w), 2849 (vs), 1638 (vs), 1622 (vs), 1543 (vs), 1468 (s), 1441 (w), 1373 (s), 1334 (m), 1327 (m), 1268 (w), 1176 (w), 1136 (w), 1082 (vs), 1034 (w), 802 (w), 722 (m), 704 (w), 679 (s), 552 (w), 480 (w), 466 (w); ESI-MS ([M + H]+, C26H53N2O4): calcd: 457.4000, found: 457.3996.
, 2953 (m), 2918 (vs), 2873 (w), 2849 (vs), 1638 (vs), 1622 (vs), 1543 (vs), 1468 (s), 1441 (w), 1373 (s), 1334 (m), 1327 (m), 1268 (w), 1176 (w), 1136 (w), 1082 (vs), 1034 (w), 802 (w), 722 (m), 704 (w), 679 (s), 552 (w), 480 (w), 466 (w); ESI-MS ([M + H]+, C26H53N2O4): calcd: 457.4000, found: 457.3996.
Repolymerization
Diol 6 (200 mg, 438 μmol, 1.00 equiv.) and hexamethylene diisocyanate (70.8 μl, 73.7 mg, 438 μmol, 1.00 equiv.) were added into a 1.5 ml screw-cap vial. The mixture was heated to 160 °C until a homogeneous solution formed and was then stirred for another 1 h. After cooling to room temperature, the reaction mixture was dissolved in hexafluoroisopropanol and precipitated into cold methanol. The precipitate was filtered, washed with methanol and dried at 70 °C and 10 mbar to obtain 125 mg of polymer (46%).Results and discussion
A literature known ruthenium catalyzed oxidative cleavage was used to synthesize the tricarboxylic acid glyceryl triazelate from high oleic sunflower oil (Scheme 1).32 The catalytic reaction was first reported by Behr et al. in 2013 for the oxidative cleavage of methyl oleate and was further optimized for the oxidative cleavage of high oleic sunflower oil (Scheme 1) by Meier et al.27 Its key features are the use of hydrogen peroxide as a greener oxidant, use of the benign solvents tert-butanol and water, and the tolerance towards ester groups as they remain uncleaved during this transformation. The successful isolation of the sunflower oil-based tricarboxylic acid was confirmed by 1H NMR, 13C NMR, and IR spectroscopy as well as mass spectrometry (ESI†). In the 1H NMR spectrum, a minor methyl proton integral is visible at 0.85 ppm (Fig. S3†). It was assumed that this signal corresponds to the formation of a dicarboxylic acid due to the presence of saturated fatty acid residues in the used sunflower oil, which are inert to the performed reaction. This is reasonable, since the used sunflower oil contained 7% saturated palmitic acid and stearic acid. The small integral of 0.17 corresponds to approximately 5% dicarboxylic acids. Hence, the isolated compound is practicably a tricarboxylic acid and will therefore be referred to as “triacid” in the following. The amount of carboxylic acid groups per gram of sample was determined quantitatively by 31P NMR spectroscopy according to a procedure introduced by Kilpeläinen et al.29,30 In this method, the carboxylic acid and hydroxyl groups of the sample are first transformed to their corresponding phosphite esters by reaction with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane and then quantified by 31P NMR spectroscopy using an internal standard. The determined carboxylic acid value of 4.95 μmol CO2H per mg of sample is almost identical to the carboxylic acid value of glyceryl triazelate (4.98 μmol CO2H per mg), again confirming the successful synthesis of the desired triacid. | ||
| Scheme 1 Ruthenium catalyzed oxidative cleavage of high oleic sunflower oil (DPA: pyridine-2,6-dicarboxylic acid and Ru(acac)3: ruthenium(III) acetylacetonate). | ||
Five model substances (M1–M5) were synthesized from the sunflower oil-based triacid, n-hexylisocyanide, and the five aldehydes acetaldehyde (1), butanal (2), hexanal (3), nonanal (4), and 2-ethylbutyraldehyde (5) to investigate the suitability of the triacid for Passerini three-component reactions and to obtain soluble compounds that allow full molecular characterization (Scheme 2). Each model compound was synthesized by stirring all three components for 3 days at room temperature. After purification by flash column chromatography, good yields between 50% and 70% were achieved. All model compounds were fully characterized by 1H NMR, 13C NMR and IR spectroscopy as well as mass spectrometry (ESI†). Exemplarily, the 1H NMR spectrum of compound M4 is depicted in Fig. 1. The characteristic signals of Passerini products such as amide protons at 6.03 ppm as well as CH protons of the newly formed α-acyloxy amide group at 5.13 ppm were detected.17,18 Moreover, the signals of the glyceryl ester moiety at 5.23 ppm and 4.12–4.27 ppm and those of the α-CH2 protons of ester groups at 2.29 ppm and 2.36 ppm were observed and they confirmed the successful isolation of the trifunctional Passerini product as a proof of concept.
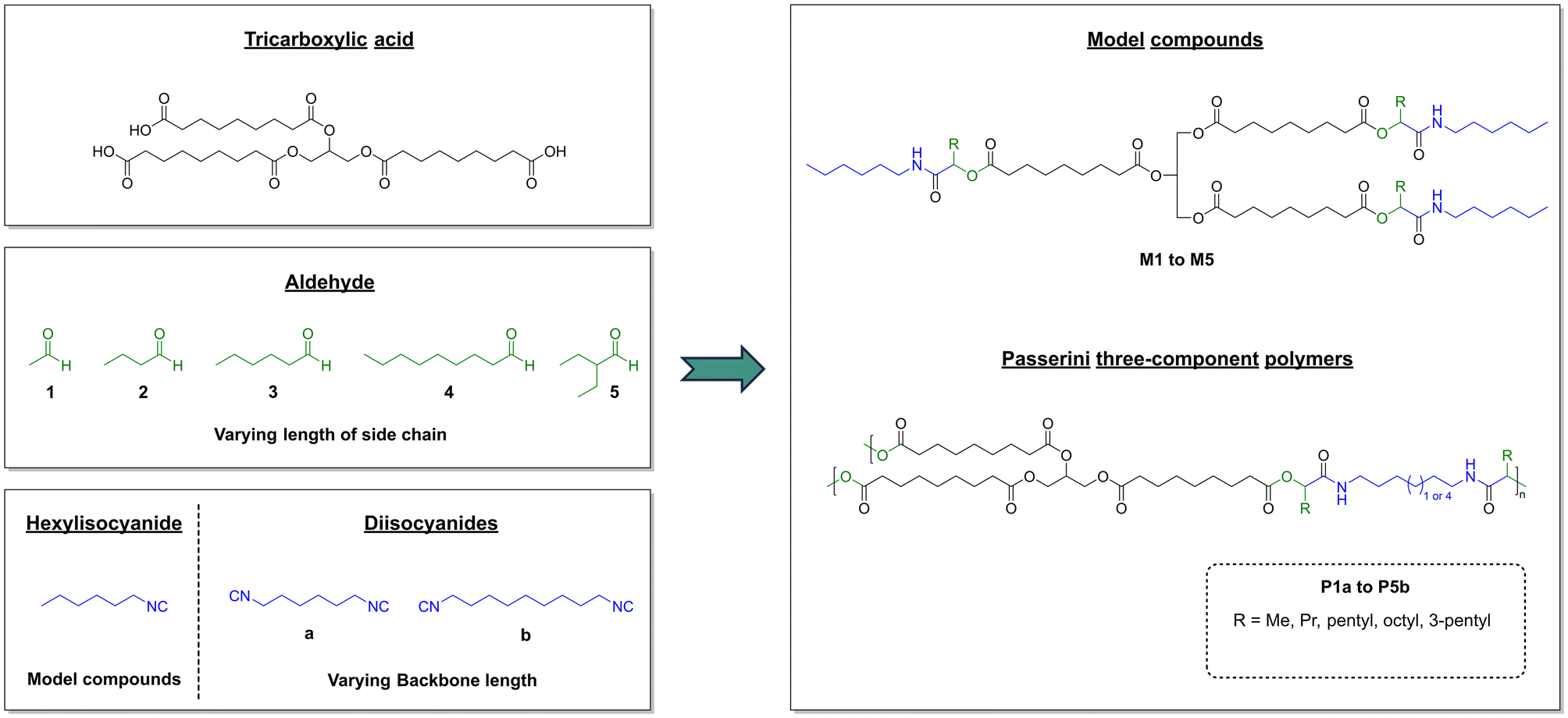 | ||
| Scheme 2 Synthesis of model compounds M1–M5 (top right) and polymers P1a–P5b (bottom right) via Passerini three-component reaction from the sunflower oil-based tricarboxylic acid, the five aldehydes acetaldehyde (1) butanal (2), hexanal (3), nonanal (4), and 2-ethylbutyraldehyde (5), and the three isocyanides n-hexylisocyanide, 1,6-diisocyanohexane (a) and 1,9-diisocyanononane (b). | ||
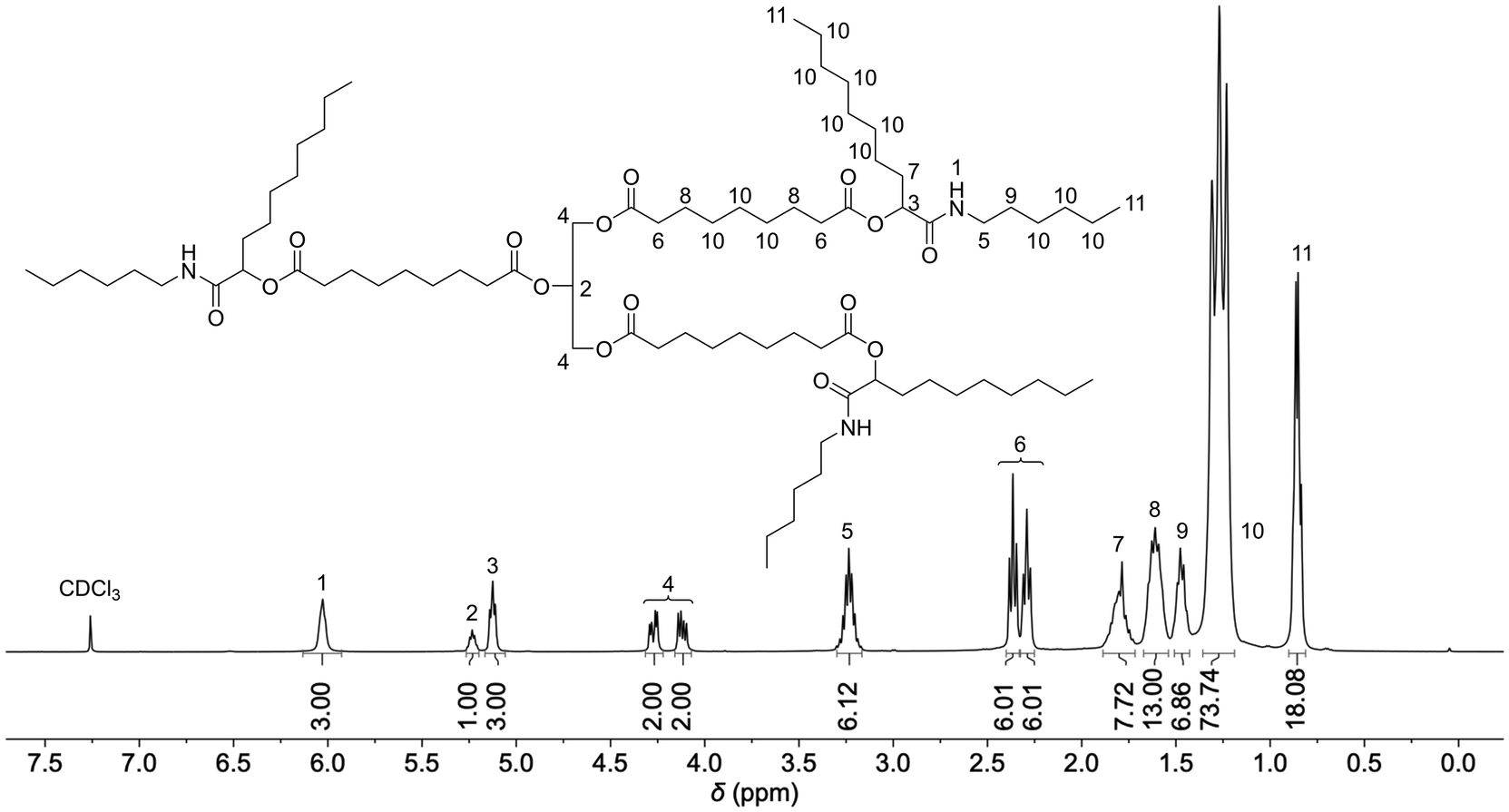 | ||
| Fig. 1 1H NMR spectrum of M4 in CDCl3. Integrals are referenced to protons 4 of the glyceryl moiety. | ||
The sunflower oil-based triacid was then reacted with aldehydes 1–5 and the two difunctional isocyanides 1,6-diisocyanohexane (a) and 1,9-diisocyanononane (b) for the synthesis of ten different cross-linked polymers via the Passerini three-component reaction (Scheme 2). All aldehyde and isocyanide components were chosen to be aliphatic and at least in principle renewable. For instance, 1,6-diisocyanohexane was synthesized from hexamethylenediamine, which can be produced from adiponitrile by hydrogenation.33 Adiponitrile can be synthesized from ammonia and adipic acid, which in turn can be produced biotechnologically from sugars.34–36 Another feasible route to hexamethylenediamine from adipic acid is the reduction of adipic acid ester to 1,6-hexanediol, which is then reacted with ammonia to yield the diamine.33 1,9-Diaminononane, the starting material for 1,9-diisocyanononane, can most likely be produced in the same way from azelaic acid, which is obtained industrially by oxidative cleavage of oleic acid.37,38 A biotechnological route to 1,9-diaminononane starting from oleic acid was recently reported by Park et al.39 All unbranched aldehydes occur in natural oils and can further be synthesized by dehydrogenation of the primary alcohols.40–42 2-Ethylbutyraldehyde is obtained by aldol condensation of butanal with acetaldehyde and subsequent hydrogenation.42
All polymerizations were performed in bulk, as all compounds were liquid at room temperature. First, the three components were mixed at −20 °C to guarantee a homogeneous mixture of all compounds before the polymerization started, as very rapid curing was observed in bulk. Then, the mixture was cast into a poly(tetrafluoroethylene) form to produce bone shaped samples for tensile strength measurements. Polymers P1a to P3b (please see Scheme 1 for compound labeling) were cured at 5 °C for 30 minutes to prevent the evaporation of the volatile aldehyde components, while polymers P4a to P5b were cured at room temperature for 30 minutes. All polymers were further cured at 50 °C for 24 h to guarantee full conversion of functional groups. For polymers P4a and P4b, a fast reaction was observed as the samples completely solidified after 5 minutes of curing at room temperature, while polymers P5a and P5b with the sterically more demanding 2-ethylbutyraldehyde took longer to solidify (10 minutes). It is therefore potentially possible to further increase the curing time by incorporating sterically more demanding and/or less reactive substrates into the polymer formulation. The reaction completeness was investigated qualitatively via infrared spectroscopy. The IR spectra of the sunflower-oil based triacid, nonanal (4), 1,6-diisocyanohexane (a), model compound M4, and polymer P4a are exemplarily depicted in Fig. 2. The characteristic IR signals of the starting materials, such as the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of carboxylic acids at 1703 cm−1, the COH combination band of aldehydes at 2823 cm−1 and 2714 cm−1, and the stretching vibration of isocyanides at 2146 cm−1 were neither visible in the IR spectra of the model compound M4 nor for polymer P4a. In the IR spectrum of M4, the characteristic signals of amide groups, such as the N–H stretching vibration (3305 cm−1), the CO stretching vibration (1655 cm−1), and N–H deformation vibration (1537 cm−1) were observed. The IR spectrum of polymer P4a was identical to that of M4, thereby confirming the successful P-3CR polymerization with near quantitative conversion of all functional groups.
O stretching vibration of carboxylic acids at 1703 cm−1, the COH combination band of aldehydes at 2823 cm−1 and 2714 cm−1, and the stretching vibration of isocyanides at 2146 cm−1 were neither visible in the IR spectra of the model compound M4 nor for polymer P4a. In the IR spectrum of M4, the characteristic signals of amide groups, such as the N–H stretching vibration (3305 cm−1), the CO stretching vibration (1655 cm−1), and N–H deformation vibration (1537 cm−1) were observed. The IR spectrum of polymer P4a was identical to that of M4, thereby confirming the successful P-3CR polymerization with near quantitative conversion of all functional groups.
 | ||
| Fig. 2 Stacked IR spectra of sunflower oil-based tricarboxylic acid, nonanal (4), 1,6-diisocyanohexane (a), model compound M4, and polymer P4a. Each IR spectrum is normalized to the strongest absorbance vibration. | ||
Swelling tests of all polymers were performed to further evaluate the degree of cross-linking. Each polymer was weighed and immersed in tetrahydrofuran for 3 days at room temperature to allow the complete solubilization of residual unreacted monomeric and oligomeric compounds inside the material. The polymer was then removed from the solvent, dried at 70 °C and 10 mbar, and weighed again. The gel content is the percentage of mass that is insoluble in THF and is therefore calculated by the division of the dried sample's mass m1 by the initial sample's mass m0 (eqn (1)).
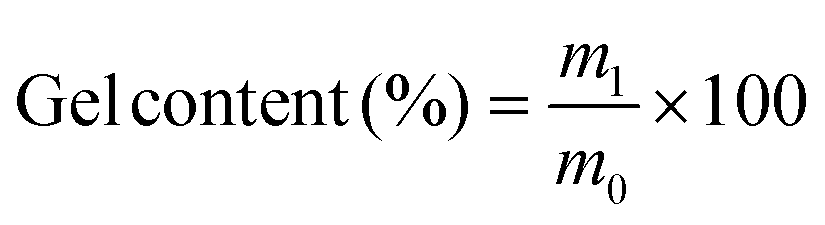 | (1) |
The determined values for the gel content of all polymers were between 96% and 99% (Table 1). Hence, both the results obtained from IR spectroscopy and the high gel contents from the swelling tests show that the P-3CR polymerization is suitable for thermosetting applications, as very high conversions and high degrees of cross-linking were achieved.
| Polymer | Gel content in THF/% | T d,5%/°C | T g/°C | Young's modulus/MPa | Ultimate tensile strength/MPa | Elongation at break/% |
|---|---|---|---|---|---|---|
| P1a | 98 ± 2 | 363 | 9 | 2.16 ± 0.41 | 1.64 ± 0.29 | 104 ± 8 |
| P1b | 96 ± 2 | 362 | 4 | 1.78 ± 0.40 | 1.34 ± 0.31 | 101 ± 2 |
| P2a | 96 ± 1 | 346 | 3 | 1.84 ± 0.10 | 1.45 ± 0.20 | 105 ± 10 |
| P2b | 98 ± 1 | 365 | 1 | 2.11 ± 0.08 | 1.68 ± 0.21 | 109 ± 13 |
| P3a | 97 ± 2 | 357 | −8 | 1.86 ± 0.08 | 1.24 ± 0.03 | 84 ± 4 |
| P3b | 96 ± 2 | 363 | −12 | 1.44 ± 0.09 | 0.92 ± 0.14 | 81 ± 16 |
| P4a | 97 ± 1 | 362 | −12 | 1.24 ± 0.02 | 0.85 ± 0.01 | 91 ± 1 |
| P4b | 97 ± 1 | 364 | −18 | 1.27 ± 0.04 | 0.87 ± 0.05 | 90 ± 6 |
| P5a | 98 ± 1 | 333 | 3 | 1.35 ± 0.04 | 2.20 ± 0.33 | 214 ± 26 |
| P5b | 99 ± 1 | 343 | 0 | 1.95 ± 0.05 | 1.98 ± 0.23 | 146 ± 14 |
The thermal stability of all materials was analyzed using thermogravimetric analysis (TGA). The obtained values for the temperature of 5 wt% loss (Td,5%) of polymers P1a–P5b ranged from 340 °C to 365 °C and were approximately 20 °C higher than the Td,5% values of model compounds M1–M5 (321 °C to 344 °C). The variation of aldehyde and isocyanide components had almost no influence on the thermal stability, as all polymers and all model compounds decomposed in a temperature range of 10 °C, respectively. Studies by Li et al. reported a Td,5% value of 335 °C for a P-3CR polymer synthesized from adipaldehyde, 1,6-diisocyanohexane, and undecanoic acid as well as Td,5% values ranging from 291 °C to 316 °C for polycaprolactone derivatives synthesized via P-3CR from 6-oxohexanoic acid and aliphatic isocyanides.17,18 For aliphatic poly(ester-amide)s, Td,5% values ranging from 267 °C to 290 °C were reported.43 A degradation temperature of 313 °C was reported for renewable isosorbide containing P-3CR polymers.44 The herein synthesized cross-linked polymers therefore exhibit an improved thermal stability compared to many linear P-3CR polymers.
Differential scanning calorimetry experiments revealed that all polymers showed glass transition temperatures (Tg) below room temperature (Table 1). Exemplarily, all DSC measurements of the polymer series a are depicted in Fig. 3.
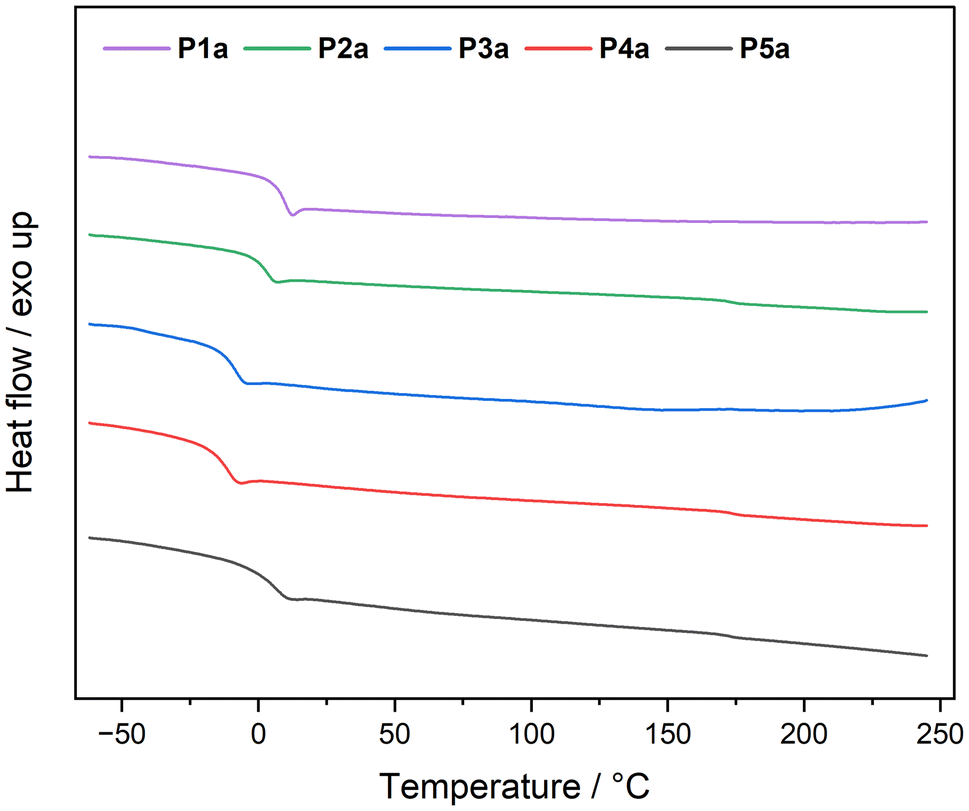 | ||
| Fig. 3 Differential scanning calorimetry measurements of polymer series a. | ||
The glass transition temperature decreased gradually with an increasing aldehyde side chain length from acetaldehyde (P1a) at 9 °C to nonanal (P4a) at −12 °C. The Tg of P5a was identical to that of P2a, presumably due to the similar steric hindrance of 2-ethylbutyraldehyde and butanal.
The same relationship between Tg and side chain length was observed for the polymer series b (Table 1 and Fig. S31†). Interestingly, every a polymer had a slightly higher Tg than the respective polymer from the b series. 1,9-Diisocyanononane (series b) is three carbon atoms longer than 1,6-diisocyanohexane (series a), resulting, on the one hand, in a lower density of amide groups per repeating unit and thus in less hydrogen bonding and on the other hand in a larger mesh size. Both factors contribute to a higher Tg in the a series.
Tensile strength measurements were performed to evaluate the mechanical properties of all samples. The manufactured samples and one exemplary measurement of P4a are depicted in Fig. 4a. All samples were flexible and could be bent by 180 °C without breaking. Moreover, linear elastic regions of at least 20% were observed for each measurement (Fig. S33–S42†). The determined Young's moduli are in the region on rubber-like materials and ranged from 1.24 MPa to 2.16 MPa (Table 1).
 | ||
| Fig. 4 Dog bone sample of polymer P4a with the associated tensile strength measurements (a) and adhesive test samples prepared from polymer P5a and different substrates (aluminum, stainless, wood, PMMA, and borosilicate glass) with the adhesive tests of stainless-steel after 2 h of curing at room temperature (b). | ||
Ultimate tensile strengths were in the range of 0.85 MPa to 2.20 MPa and the determined elongations at break ranged from 81% up to a maximum of 214%. It was observed that the Young's modulus decreases slightly but gradually with an increasing side chain length from P1a to P4b. Polymers P2a to P5b showed great reproducibility in the preparation of samples and had standard deviations between 1% and 5% for the Young's modulus and a maximum of 15% for the ultimate tensile strength. The high standard deviations of approximately 20% for P1a and P1b are attributed to the low boiling point of acetaldehyde and hence a more difficult reproducibility.
Due to the fast curing of all polymers at room temperature, a potential application as “three component glues” was imagined. For this proof of concept, polymer P5a was chosen since the sterically more demanding 2-ethylbutyraldehyde significantly increased the curing time and enabled a reproducible manufacturing of adhesive test samples. The substrates to be glued together were prepared by cutting aluminum alloy, stainless-steel, wood, poly(methyl methacrylate), and borosilicate glass into rectangular pieces. Polymer P5a was freshly prepared and applied onto two pieces of the same substrate. The two surfaces were then pressed together and cured at room temperature for 2 h. Afterwards, each substrate was measured in triplicate to determine the ultimate tensile strength. The prepared samples and the measurements performed with stainless-steel as the substrate are depicted in Fig. 4b.
The adhesion of P5a ranged from 0.94 MPa to 1.17 MPa for all surfaces except for stainless-steel, for which a stronger adhesion of 1.88 MPa was accomplished. In further experiments, wood surfaces were glued together and measured after 30 minutes, 60 minutes, and 120 minutes had passed (Fig. S46†). Already after 60 minutes of curing at room temperature, no further increase of the tensile strength could be detected, indicating a fully cured glue. These results prove that a multicomponent approach can be used to manufacture new thermosetting glues and that different substrates can be glued efficiently.
Finally, chemical degradation of polymer P4a was investigated to demonstrate a possible way to reutilize all components in a second life cycle after the materials reached their end of life. It was assumed that transesterification of P4a with methanol yields the three products glycerol, azelaic acid dimethyl ester, and the difunctional α-hydroxyamide 6 (Scheme 3).
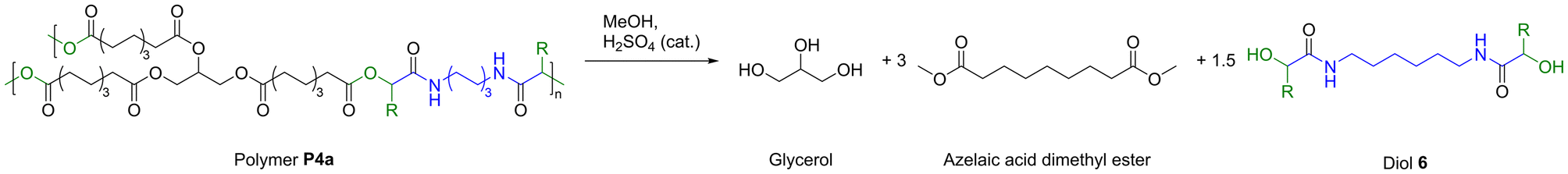 | ||
| Scheme 3 Chemical recycling of polymer P4a by transesterification with methanol and catalytic amounts of sulfuric acid. | ||
It should be noted that this approach of P-3CR and subsequent transesterification has already been utilized in the literature for the targeted synthesis of α-hydroxyamides.45,46 Hence, three grams of P4a were suspended in methanol and catalytic amounts of sulfuric acid were added. Intriguingly, the thermoset had completely dissolved after heating the reaction mixture to 65 °C for 5 h. Diol 6 precipitated as a white powder from the reaction mixture while cooling to room temperature and was thus easily isolated by filtration in a yield of 74% (1.26 g). The compound had a high melting point of 162 °C and was poorly soluble in common organic solvents due to the strong hydrogen bonding that originates from its amide groups. The molecular structure was confirmed by mass spectrometry and by NMR spectroscopy in deuterated trifluoroacetic acid (Fig. S49 and 50†). In the 1H NMR spectrum, the alpha amide proton signal is shifted downfield to 4.63 ppm due to the electron withdrawing effect of the hydroxy group. This was further confirmed by heteronuclear multiple bond correlation (HMBC) spectroscopy that showed correlation between this signal and the quaternary carbon atom of the amide at 180.7 ppm (Fig. S51†). Moreover, the characteristic signals of the amide functionality such as the CO stretching vibration at 1638 cm−1, the N–H stretching vibration at 3243 cm−1, and the N–H deformation vibration at 1543 cm−1 were observed in IR spectroscopy.
Methanol was then removed from the filtrate under reduced pressure. The residue that still contained a mixture of azelaic acid dimethyl ester and glycerol was extracted with ethyl acetate and washed with water to separate both compounds. Azelaic acid dimethyl ester was obtained in high purity by vacuum distillation of the organic phase in a yield of 75% (1.18 g) and glycerol was isolated by vacuum distillation of the aqueous phase in a yield of 30% (68 mg). The low glycerol yield is attributed to the small scale of recycling as 227 mg of glycerol corresponded to 100% yield. The chemical recycling of P4a was therefore successful with a simple transesterification approach. Noteworthy, azelaic acid dimethyl ester is an industrially relevant compound, which is used for cosmetic applications and the manufacture of polymers such as polyamides and polyesters.47–49 It was desired to repolymerize diol 6 with azelaic acid dimethyl ester to show that the recycled compounds can directly be incorporated into a new material. However, several polymerization efforts in bulk at 160 °C under 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) catalysis yielded only oligomeric compounds with an average molecular weight of 1900 Da (Fig. S54†). Unfortunately, also other catalysts (i.e. KOtBu or Sb2O3) did not lead to successful polymerization. The apparently low reactivity of 6 towards transesterification renders this diol with secondary hydroxyl groups impractical for poly(ester-amide) synthesis. Alternatively, it was discovered that diol 6 reacted readily with isocyanates in bulk. A reaction with hexamethylene diisocyanate yielded a poly(amide-urethane) with an average molecular weight of 17 kDa after one hour of reaction time at 160 °C (Fig. S55†). The polymer structure was further elucidated via NMR spectroscopy (Fig. S56 and 57†). In the 1H NMR spectrum, characteristic amide and urethane protons were visible at 6.89 ppm and 5.85 ppm, respectively. The corresponding quaternary carbon signals were observed at 171.5 ppm and 155.8 ppm in the 13C NMR spectrum. Although the use of isocyanates is rather unsustainable, we have demonstrated one possible way to reuse this difunctional α-hydroxyamide. Apart from this polymer application, α-hydroxyamides are found in pharmaceutical and natural products.50,51
Conclusions
A multicomponent polymerization approach for the synthesis of thermosets from a sunflower oil-derived tricarboxylic acid is presented. The triacid was produced from commercial sunflower oil via a ruthenium catalyzed oxidative cleavage, which uses hydrogen peroxide as a greener oxidant.27,32 As proof of concept for the multicomponent approach, five model compounds were synthesized via the Passerini three-component reaction (P-3CR) and fully characterized via1H NMR, 13C NMR, and IR spectroscopy and mass spectrometry. Afterwards, a set of ten P-3CR thermosets was synthesized from the triacid in combination with five monoaldehydes and two diisocyanides. The polymerization proceeds readily at room temperature and was performed without any additional solvents, rendering this process more sustainable. IR spectroscopy of the cured thermosets indicated a quantitative conversion of all aldehyde and isocyanide groups. Gel contents between 96% and 99% in THF point towards high degrees of cross-linking in all materials. Differential scanning calorimetry experiments revealed structure–property relationships, and it was possible to fine-tune the glass transition temperature in a range from −18 °C to 9 °C by variation of aldehyde and diisocyanide components. With increasing side chain length and increasing length of the diisocyanide, a gradual decrease of the Tg was observed. In tensile strength measurements, Young's moduli ranging from 1.24 MPa to 2.16 MPa, and elongations at break between 81% and 214% were determined. The presented system can be used as a “three component glue” since all compounds are liquid and no additional heating was required for curing. After 60 minutes of curing at room temperature, the adhesive exhibited ultimate tensile strengths between 0.94 MPa and 1.88 MPa on aluminum, steel, wood, PMMA, and glass surfaces. Finally, chemical reuse of polymer P4a was achieved by transesterification with methanol and catalytic amounts of sulfuric acid. The difunctional α-hydroxyamide 6 precipitated during recycling and was isolated in a good yield of 74%, while azelaic acid dimethyl ester was obtained by vacuum distillation in a yield of 75%. Eventually, a new polyurethane was synthesized from diol 6 and hexamethylene diisocyanate to exemplify a second life cycle of the recycled compound.Author contributions
Luis Santos Correa: conceptualization, data curation, formal analysis, investigation (synthesis and analytics), visualization, and writing – original draft; Silas Leidenheimer: investigation (synthesis); Michael A. R. Meier: conceptualization, resources, supervision, and writing – review & editing.Data availability
The data supporting this article have been included within the article and as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Dr Manfred Wilhelm and Dr Valerian Hirschberg for their support with tensile strength measurements and Alexander Jaks for manufacturing the required customized poly(tetrafluoroethylene) forms. Furthermore, the authors thank Prof. Dr Stefan Bräse, Prof. Dr Jörg Lahann, Prof. Dr Patrick Théato, and Birgit Huber for TGA measurements, Lara Hirsch for mass spectrometric measurements, Timo Sehn for fruitful discussions, and Severin Stalter for his help in the synthesis of a few compounds.References
- PlasticsEurope Deutschland e. V., Plastics – the fast Facts 2023, 2023, https://plasticseurope.org/knowledge-hub/plastics-the-fast-facts-2023/; access date: 07.11.2024.
- U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. R. Meier and J. O. Metzger, Angew. Chem., Int. Ed., 2021, 60, 20144–20165 CrossRef CAS PubMed.
- L. Montero de Espinosa and M. A. R. Meier, Eur. Polym. J., 2011, 47, 837–852 CrossRef CAS.
- S. Pandey, B. S. Rajput and S. H. Chikkali, Green Chem., 2021, 23, 4255–4295 RSC.
- J. C. Ronda, G. Lligadas, M. Galià and V. Cádiz, Eur. J. Lipid Sci. Technol., 2011, 113, 46–58 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- I. Ugi, A. Dömling and W. Hörl, Endeavour, 1994, 18, 115–122 CrossRef CAS.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
- M. Passerini, Gazz. Chem. Ital., 1921, 51, 126–129 CAS.
- A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef.
- L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, Chem. Sci., 2021, 12, 15445–15472 RSC.
- I. Akritopoulou-Zanze, Curr. Opin. Chem. Biol., 2008, 12, 324–331 CrossRef CAS PubMed.
- O. Kreye, T. Tóth and M. A. R. Meier, J. Am. Chem. Soc., 2011, 133, 1790–1792 CrossRef CAS PubMed.
- X.-X. Deng, L. Li, Z.-L. Li, A. Lv, F.-S. Du and Z.-C. Li, ACS Macro Lett., 2012, 1, 1300–1303 CrossRef CAS PubMed.
- Y.-Z. Wang, X.-X. Deng, L. Li, Z.-L. Li, F.-S. Du and Z.-C. Li, Polym. Chem., 2013, 4, 444–448 RSC.
- J. Zhang, M. Zhang, F.-S. Du and Z.-C. Li, Macromolecules, 2016, 49, 2592–2600 CrossRef CAS.
- S. König and I. Ugi, Z. Naturforsch., B, 1991, 46, 1261–1266 CrossRef.
- H. Bu, A.-L. Kjøniksen, K. D. Knudsen and B. Nyström, Biomacromolecules, 2004, 5, 1470–1479 CrossRef CAS PubMed.
- H. Bu, A.-L. Kjøniksen and B. Nyström, Eur. Polym. J., 2005, 41, 1708–1717 CrossRef CAS.
- A. E. J. de Nooy, D. Capitani, G. Masci and V. Crescenzi, Biomacromolecules, 2000, 1, 259–267 CrossRef CAS PubMed.
- V. Crescenzi, A. Francescangeli, D. Capitani, L. Mannina, D. Renier and D. Bellini, Carbohydr. Polym., 2003, 53, 311–316 CrossRef CAS.
- A. E. J. de Nooy, G. Masci and V. Crescenzi, Macromolecules, 1999, 32, 1318–1320 CrossRef CAS.
- N. Hauck, N. Seixas, S. P. Centeno, R. Schlüßler, G. Cojoc, P. Müller, J. Guck, D. Wöll, L. A. Wessjohann and J. Thiele, Polymers, 2018, 10, 1055 CrossRef PubMed.
- W. Lin, T. Sun, M. Zheng, Z. Xie, Y. Huang and X. Jing, RSC Adv., 2014, 4, 25114–25117 RSC.
- L. Santos Correa and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2023, 125, 2200171 CrossRef CAS.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
- A. W. T. King, J. Jalomäki, M. Granström, D. S. Argyropoulos, S. Heikkinen and I. Kilpeläinen, Anal. Methods, 2010, 2, 1499–1505 RSC.
- X. Meng, C. Crestini, H. Ben, N. Hao, Y. Pu, A. J. Ragauskas and D. S. Argyropoulos, Nat. Protoc., 2019, 14, 2627–2647 CrossRef CAS PubMed.
- K. A. Waibel, R. Nickisch, N. Möhl, R. Seim and M. A. R. Meier, Green Chem., 2020, 22, 933–941 RSC.
- A. Behr, N. Tenhumberg and A. Wintzer, RSC Adv., 2013, 3, 172–180 RSC.
- B. D. Herzog and R. A. Smiley, in Ullmann's Encyclopedia of Industrial Chemistry, 2012 Search PubMed.
- M. T. Musser, in Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed.
- J. Rios, J. Lebeau, T. Yang, S. Li and M. D. Lynch, Green Chem., 2021, 23, 3172–3190 RSC.
- R. A. Sheldon, J. Mol. Catal. A: Chem., 2016, 422, 3–12 CrossRef CAS.
- Emery Expands Ozonolysis Plant, Chem. Eng. News, 1959, 37, 25–26 Search PubMed.
- C. G. Goebel, A. C. Brown, H. F. Oehlschlaeger and R. P. Rolfes, Method of making azelaic acid, US 2813113, 1957.
- S.-Y. Hwang, J.-M. Woo, G. E. Choi, D.-K. Oh, J.-H. Seo and J.-B. Park, ACS Catal., 2024, 14, 4130–4138 CrossRef CAS.
- M. Eckert, G. Fleischmann, R. Jira, H. M. Bolt and K. Golka, in Ullmann's Encyclopedia of Industrial Chemistry, 2006 Search PubMed.
- D. K. Raff, in Ullmann's Encyclopedia of Industrial Chemistry, 2013 Search PubMed.
- C. Kohlpaintner, M. Schulte, J. Falbe, P. Lappe, J. Weber and G. D. Frey, in Ullmann's Encyclopedia of Industrial Chemistry, 2013 Search PubMed.
- Y.-T. Guo, C. Shi, T.-Y. Du, X.-Y. Cheng, F.-S. Du and Z.-C. Li, Macromolecules, 2022, 55, 4000–4010 CrossRef CAS.
- Z. Söyler, K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, Green Chem., 2018, 20, 214–224 RSC.
- R. M. Carrillo, A. G. Neo, L. López-García, S. Marcaccini and C. F. Marcos, Green Chem., 2006, 8, 787–789 RSC.
- M. Shiri, Z. Gholami-Koupaei, F. Bandehali-Naeini, M.-S. Tonekaboni, S. Soheil-Moghaddam, D. Ebrahimi, S. Karami and B. Notash, Synthesis, 2020, 3243–3252 CrossRef CAS.
- A. Fitton and K. L. Goa, Drugs, 1991, 41, 780–798 CrossRef CAS PubMed.
- A. Köckritz and A. Martin, Eur. J. Lipid Sci. Technol., 2011, 113, 83–91 CrossRef.
- B. Cornils and P. Lappe, in Ullmann's Encyclopedia of Industrial Chemistry, 2014, pp. 1–18 Search PubMed.
- H. A. Schenck, P. W. Lenkowski, I. Choudhury-Mukherjee, S.-H. Ko, J. P. Stables, M. K. Patel and M. L. Brown, Bioorg. Med. Chem., 2004, 12, 979–993 CrossRef CAS PubMed.
- M. R. Wood, K. M. Schirripa, J. J. Kim, S. D. Kuduk, R. K. Chang, C. N. Di Marco, R. M. DiPardo, B.-L. Wan, K. L. Murphy, R. W. Ransom, R. S. L. Chang, M. A. Holahan, J. J. Cook, W. Lemaire, S. D. Mosser, R. A. Bednar, C. Tang, T. Prueksaritanont, A. A. Wallace, Q. Mei, J. Yu, D. L. Bohn, F. C. Clayton, E. D. Adarayn, G. R. Sitko, Y. M. Leonard, R. M. Freidinger, D. J. Pettibone and M. G. Bock, Bioorg. Med. Chem. Lett., 2008, 18, 716–720 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical data of the synthesized compounds, details about the carboxylic acid value determination, and all tensile and adhesive test measurements. See DOI: https://doi.org/10.1039/d4py01358a |
| This journal is © The Royal Society of Chemistry 2025 |