
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comprehensive coordination chemistry of iminophosphonamides
Bhupendra Goswami
 *a and
Peter W. Roesky
*bc
*a and
Peter W. Roesky
*bc
aDepartment of Chemistry, School of Chemical Sciences and Pharmacy, Central University of Rajasthan, Ajmer 305817, Rajasthan, India. E-mail: bhupendra.goswami@curaj.ac.in
bInstitute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
cInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Kaiserstr. 12, 76131, Germany
First published on 9th April 2025
Abstract
The iminophosphonamide [R2P(NR′)2]− ligand is an analog of phosphate (R2PO2−), replacing its oxygen atoms with two amide groups. This review aims to provide the first comprehensive report on coordination chemistry dealing with iminophosphonamide ligands, focusing on the s-block, p-block, transition, and f-block metals. In particular, this monoanionic ligand's coordination mode and reactivity with the metal centers are discussed. The last section of the review is dedicated to the reported chiral iminophosphonamine ligands and corresponding metal complexes.
 Bhupendra Goswami | Bhupendra Goswami obtained his integrated BS-MS dual degree from the Indian Institute of Science Education and Research (IISER) Mohali, India, in 2017. He joined as a doctoral student at the Karlsruhe Institute of Technology, Germany, under the supervision of Prof. Peter W. Roesky and was awarded Dr rer. nat. in 2020. Dr Goswami worked as a postdoc researcher with Prof. Dominik Munz at the University of Saarland, Germany (2021–2022). Subsequently, he joined the Central University of Rajasthan, as an Assistant Professor at the Department of Chemistry in 2022. His current area of research is synthetic inorganic chemistry, coordination chemistry of transition metals and their photoluminescence. |
 Peter W. Roesky | Peter W. Roesky obtained his diploma in 1992 from the University of Würzburg and his doctoral degree from the Technical University of Munich (with Prof. W. A. Herrmann) in 1994. He was a postdoc with Prof. T. J. Marks at Northwestern University (1995–1996). In 1999 he completed his Habilitation at the University of Karlsruhe. As a Full Professor, he joined the faculty of the Freie Universität Berlin in 2001. In 2008, he became a Full Professor of inorganic functional materials at the Karlsruhe Institute of Technology (KIT). From 2013 to 2015, he served as Dean of the Faculty of Chemistry and Biosciences at KIT. In 2019, he received a JSPS Invitational Fellowship for Research in Japan and in 2020, a Reinhart Koselleck Project of the German Science Foundation. In 2022, he was given the Frank H. Spedding Award for research in Rare Earth Science and in 2024, the Marianne-Baudler-Award of the German Chemical Society (GDCh). Currently, he is the chair of the Wöhler Association for Inorganic Chemistry of the GDCh. His current research interest revolves around the synthetic inorganic and organometallic chemistry of s-block metals, silicon, group 15, gold, and lanthanides. |
1 Introduction
In coordination chemistry, tailor-made ligand systems are crucial for creating metal complexes with specific properties.1 Monoanionic chelating ligands such as β-diketiminates,2 aminotroponimates,3 bis(phosphinimino)methanides,4,5 amidinates,6–8 and closely related guanidinates8–12 are a well-established class of ligand systems. Of these N-donor ligands, amidinates belong to the NXN ligand framework with a carbon atom at the center (i.e., X = C). The coordinated complexes containing an NXN motif are typically referred to as non-metallocene derivatives. Upon replacing the central atom X, many N–X–N-type bidentate chelating ligands can be created, and such ligands are thought of as hetero analogs of the allylic ligand.The boraamidinate, sulfinamidinate, triazenide, and iminophosphonamide systems are examples of N–X–N ligands, where X = BR,13,14 SR,15–18 N,19,20–23 and PR2,24–26 respectively (Fig. 1). As expected, electronic tuning of the ligand backbone occurs upon alteration of the central atom X in the NXN ligand. It is reasonable to assume that these heteroatomic ligand systems have a similar coordination chemistry. Nonetheless, the ligand system's closure inspection indicates that the corresponding bond lengths and bite angles vary, which significantly affects how the corresponding complexes behave chemically. In contrast to the other NXN ligands, the iminophosphonamides (X = PR2) have the longest X–N bond (1.60 Å), which result into a comparatively wider bite angle in their metal complexes.26 Furthermore, it has been experimentally observed that some iminophosphonamides can exist in a zwitterionic form, denoted as N−–P+–N−, where the negative charge is localized on the nitrogen atoms of the iminophosphonamide ligand. This zwitterionic form is a key difference in the electronic properties between iminophosphonamides and closely related amidinates.27
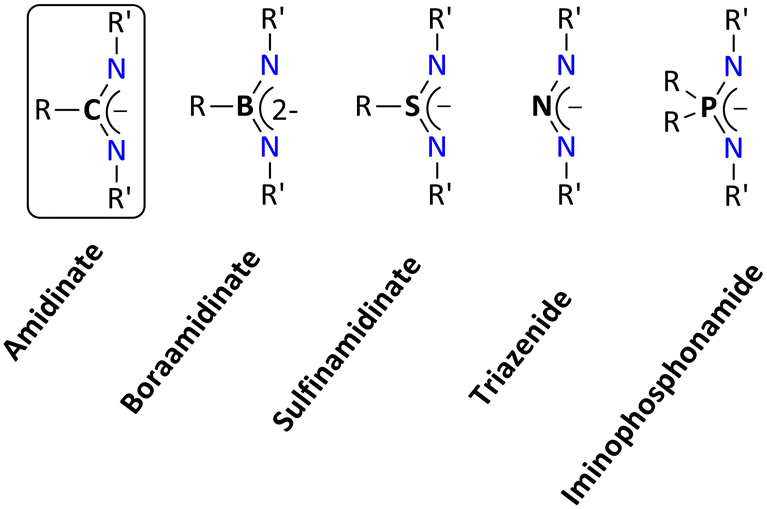 | ||
| Fig. 1 Different derivatives of the amidinate ligand system. | ||
Additionally, the highest occupied molecular orbital (HOMO) of iminophosphonamide ligands can exhibit either C2v or Cs symmetry, whereas the HOMO of amidinate ligands demonstrates only C2v symmetry. The presence of Cs symmetry in iminophosphonamide ligands allows for effective in-plane donation of π-electron density from the nitrogen center to the dxz orbital of the metal center. In contrast, the C2v symmetric HOMO of amidinates results in only lateral donation, leading to significant puckering of the metallacycles.27
Furthermore, the presence of a phosphorus center in the ligand backbone makes it easy to track the reaction progress via 31P NMR spectroscopy. Moreover, the central R2P unit of the iminophosphonamide offers a higher steric demand in comparison to the other NXN ligand frameworks. By changing the substituents (R, R′) in the iminophosphonamide ligand, the steric factor can be varied.
The following three types of bonding modes can be offered by the iminophosphonamide ligand, which depends upon the type of the metal atoms (Fig. 2).
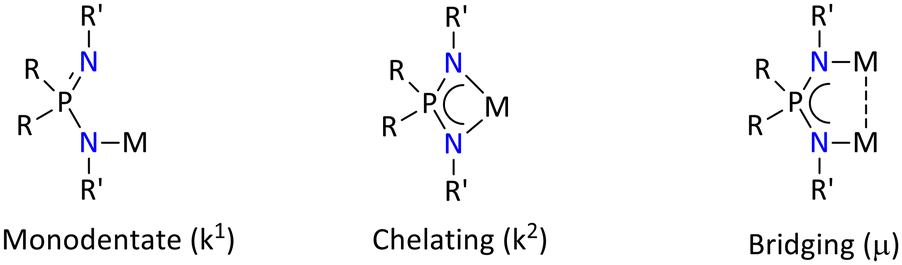 | ||
| Fig. 2 General coordination modes observed in the iminophosphonamide ligand. | ||
Generally, the following two major preparative approaches can be used to synthesize the iminophosphonamide ligand (Scheme 1):
(1) Kirsanov condensation: in this approach, a bromo phosphonium salt, [R2PXX′]+Br− (X and X′ = Cl or Br), is generated by oxidizing tertiary phosphine, R2PX (X = Cl and Br) with bromine. In the second step, the produced bromo phosphonium salt can react with primary amines (R′NH2) to form a diamino phosphonium salt, which is then transformed into the equivalent iminophosphonamine with the aid of a strong base.28,29
(2) Staudinger reaction: the Staudinger reaction for the synthesis of iminophosphonamine is more versatile. In this preparative approach, a reaction is carried out between a stoichiometric quantity of an organic azide (R′N3) and phosphonamine (R2PNHR′) or phosphine (R2PH), which immediately releases N2 and yields the iminophosphonamine ligand.30–32
 | ||
| Scheme 1 General synthetic methods for iminophosphonamine (B = base).33 | ||
To avoid any potential hazards associated with the disubstituted phosphanes and azides in the Staudinger reaction, Kirsanov condensation could be employed as an alternative synthetic method.
Based on the above-mentioned synthetic methods, various iminophosphonamine ligands have been reported in the literature. Using these ligands, the literature presents a variety of iminophosphonamide metal complexes, ranging from main group, over transition metals to rare earth metals. The synthesized metal complexes can be formed either by direct deprotonation (Scheme 2a) or through salt metathesis reactions (Scheme 2b).
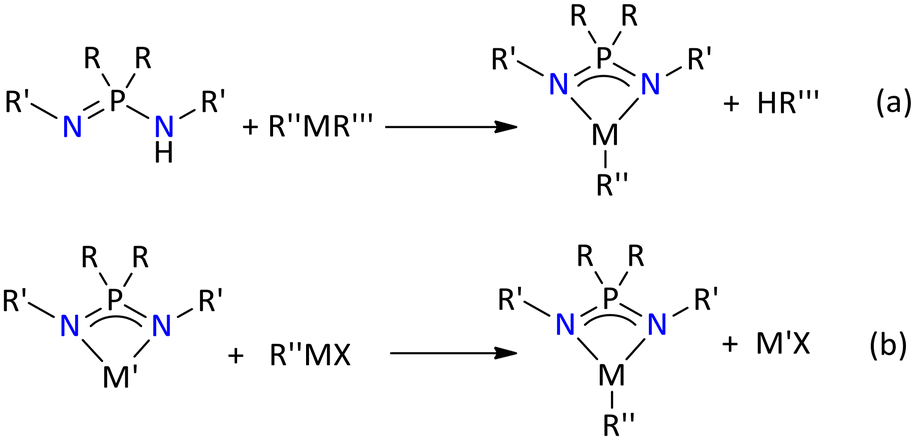 | ||
| Scheme 2 Common chemical reactions involved in the synthesis of iminophosphonamide metal complexes, by (a) direct deprotonation or (b) through salt metathesis reactions. | ||
To the best of our knowledge, reviews dealing with the coordination chemistry of most of the above-mentioned NXN ligand systems are known with the exception of iminophosphonamide ligands (Fig. 1).13,34–36 Some parts of the review have already been published in the dissertation of one of the authors.37 This review will give a comprehensive account of the coordination chemistry dealing with the iminophosphonamide ligand, particularly coordination modes and reactivity with the s-block, p-block, transition, and rare earth metal elements. This also includes the developments in the field of enantiopure iminophosphonamine ligands so far.
2. Achiral complexes of iminophosphonamides
2.1 s-Block metal complexes
The s-block metal complexes of the iminophosphonamine ligand are important since these metal complexes serve as reagents to transfer the ligand to other metal centers.The alkali metal complexes of the ligand are commonly synthesized by deprotonation of the iminophosphonamines with alkali metal precursors such as n-BuLi, NaH, [Na{N(SiMe3)2}], KH, [K{N(SiMe3)2}], [Rb{N(SiMe3)2}], and [Cs{N(SiMe3)2}] or direct deprotonation of elemental metals such as rubidium and caesium. The literature represents alkali metal complexes of various NPN motifs such as {Ph2P(2,6-iPr2C6H3N)2}H, {Ph2P(2,4,6-Me3C6H2N)2}H,38,39 {Ph2P(2,6-iPr2C6H3NH)P(Ph2)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(C6H2-2,4,6-Me3))},40 and {Ph2PN(SiMe3)2}H.41
N(C6H2-2,4,6-Me3))},40 and {Ph2PN(SiMe3)2}H.41
Upon closely looking at the structures of alkali metal complexes of the various ligands, it is observed that depending on the ionic radius of the metals and N-substituents, the aggregation state of the complexes differs. For example, the solid state of the potassium salt [K{Ph2P(2,6-iPr2C6H3N)2}]n is polymeric, as depicted in Scheme 3.38
 | ||
| Scheme 3 Syntheses of Li, Na and K complexes of {Ph2P(ArN)2} (Ar = 2,6-iPr2C6H3 and 2,4,6-Me3C6H2).38 | ||
The K⋯C contact length in the polymeric potassium complex lies in the range of 3.100–3.468 Å. The 31P{1H} NMR spectrum of the polymeric potassium complex exhibits only one signal at δ = −17.5 ppm, suggesting the symmetric coordination of the ligand in the solution. 31P{1H} of the polymeric potassium is slightly upfield shifted compared to its starting ligand, δ = −16.7 ppm. In contrast, the deprotonation of the {Ph2P(2,6-iPr2C6H3N)2}H ligand with n-BuLi and Na(SiMe3)2 resulted in the formation of dimeric complexes [Li{Ph2P(2,6-iPr2C6H3N)2}]2 and [Na{Ph2P(2,6-iPr2C6H3N)2}]2, respectively. However, upon recrystallization, the later mother-liquor of the dimeric lithium complex [Li{Ph2P(2,6-iPr2C6H3N)2}]2 resulted in the formation of unusual crystals of [Ph2P(2,6-iPr2C6H3N){2,6-iPr2C6H3N(Ph2P)(N–NN-2,6-iPr2C6H3)}Li2] and [{2,6-iPr2C6H3N(Ph2P)(N–NN-2,6-iPr2C6H3)}2Li2]. The formation of these phosphazide products was attributed to the incomplete N2 evolution during the Staudinger reaction of ligand formation (Scheme 4).
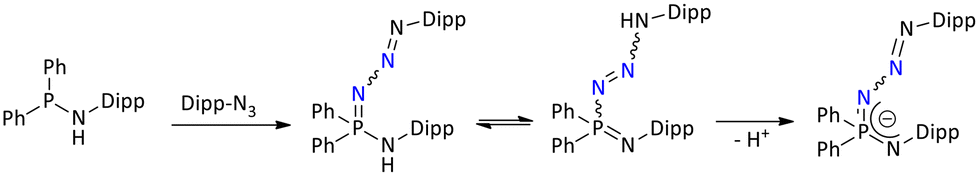 | ||
| Scheme 4 Formation of phosphazide through incomplete N2 evolution during the Staudinger reaction.38 | ||
Additionally, the THF adducts [Li{Ph2P(2,6-iPr2C6H3N)2}(thf)] and [Li{Ph2P(2,6-iPr2C6H3N)2}(thf)2] were isolated either by performing the reaction in n-hexane/THF or by the addition of THF to the dimeric [Li{Ph2P(2,6-iPr2C6H3N)2}]2 complex. The dimeric molecular structure of the sodium corresponding complex exhibited Na⋯η6-Dipp–arene interaction with the second ligand having an average Na⋯C centroid distance of 2.50 Å.
Moreover, the deprotonation of {Ph2P(2,4,6-Me3C6H2N)2}H with n-BuLi in n-hexane resulted in the formation of a dimeric complex, [Li2{Ph2P(2,4,6-Me3C6H2N)2}2]. The treatment of the {Ph2P(2,4,6-Me3C6H2N)2}H ligand with Na{N(SiMe3)2} in toluene resulted in the formation of a homoleptic sodium complex, [Na2{Ph2P(2,4,6-Me3C6H2N)2}2], and the molecular structure of this homoleptic complex was undetermined. However, the THF adduct of the sodium complex could be identified by recrystallizing the product in THF. Additionally, the potassium complex crystallized in polymeric form [K{Ph2P(2,4,6-Me3C6H2N)2}]n similar to [K{Ph2P(2,6-iPr2C6H3N)2}]n.
Furthermore, when the ligand {Ph2P(Me3SiN)2}H was deprotonated with n-BuLi, NaH, KH, Rb, and Cs, the coordination pattern in the series from Li to Cs complexes exhibits a non-uniform trend (Scheme 5).41 The molecular structures of the lithium and potassium complexes exhibit monomeric forms, [{Ph2P(NSiMe3)2}Li(thf)2] and [{Ph2P(NSiMe3)2}K(thf)4], where the central metal ions are coordinated by two and four THF molecules, respectively. However, the sodium complex nonuniformly crystalized as highly reactive sodium/sodiumate ion pair [{Ph2P(NSiMe3)2}2Na][Na(thf)6]. In this ion paired sodium complex, the metal ions exit in two different coordination states, one sodium is octahedrally coordinated by six THF molecules, whereas the other ion is tetrahedrally coordinated by four nitrogen atoms of the iminophosphonamide ligand. Conversely, Rb and Cs metal complexes are found in dimeric [{Ph2P(Me3SiN)2Rb(thf)}2] and [{Ph2P(Me3SiN)2Cs}2] forms.
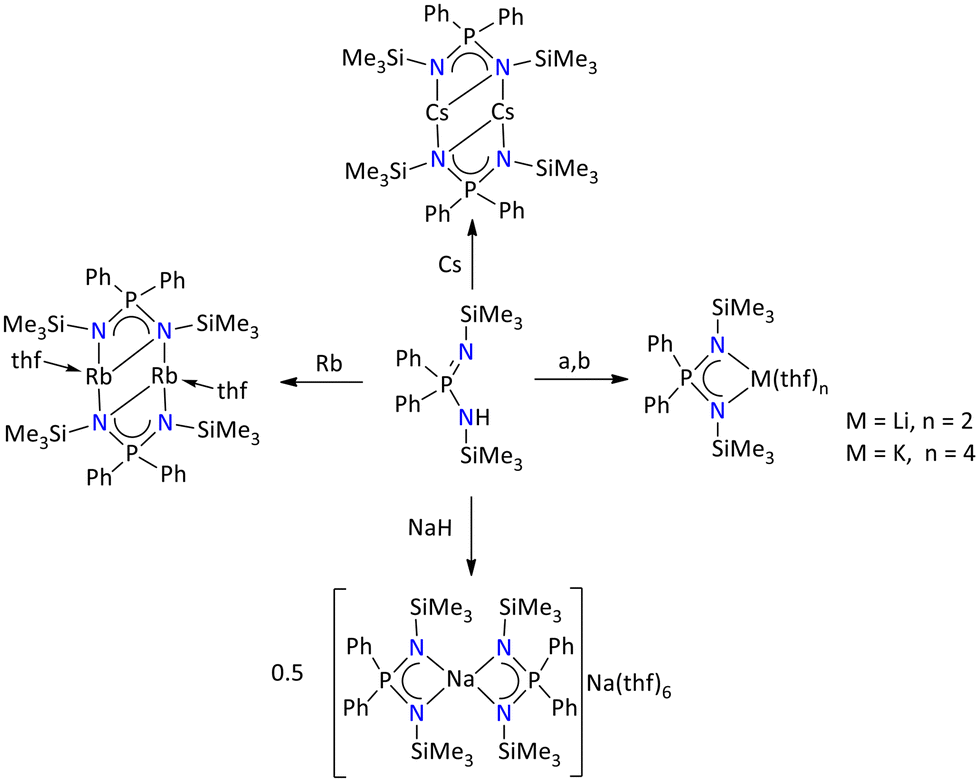 | ||
| Scheme 5 Iminophosphonamide complexes of alkali metals: (a) n-BuLi and (b) KH.41 | ||
Iminophosphonamide alkaline earth metal complexes are generally synthesized by deprotonating the relevant iminophosphonamines with alkaline earth metal amides, [M{N(SiMe3)2}2].
The alkaline earth metals (M = Be, Mg, Ca, Sr, and Ba), as opposed to alkali metal complexes of the {Ph2P(Me3SiN)2}− ligand, show a consistent coordination pattern (Scheme 6).42 The alkaline earth metal complexes exist in a mononuclear form with additional coordination of THF molecules, [{Ph2P(NSiMe3)2}2M(thf)m], depending on the ionic radius of central metals.
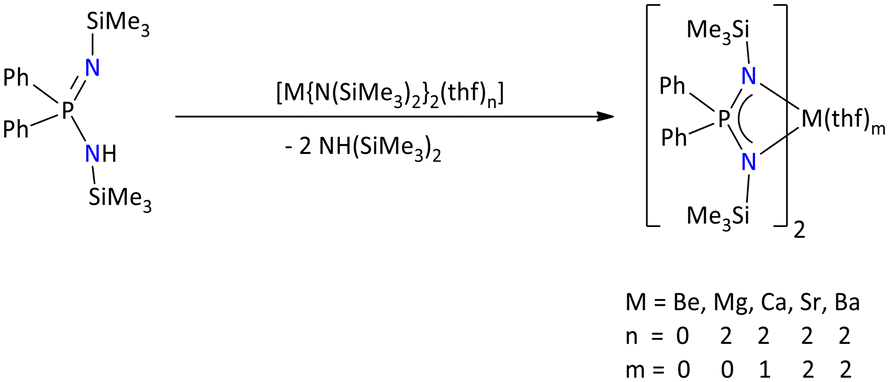 | ||
| Scheme 6 Iminophosphonamide complexes of alkaline earth metals.42 | ||
Furthermore, Stasch has incorporated sterically protected iminophosphonamide ligands such as {Ph2P(2,6-iPr2C6H3N)2}− to stabilize the low-valent [Mg]+1 complexes.43
In this regard, the iminophosphonamide ligand was first deprotonated by MeMgX (X = Br and I), which resulted in the formation of [{Ph2P(2,6-iPr2C6H3N)2}MgX(Et2O)]. To form an ether-free Mg(II) complex, the ether adduct of the iminophosphonamide [{Ph2P(2,6-iPr2C6H3N)2}MgX(Et2O)] complex was heated in toluene under reduced pressure, which afforded [{Ph2P(2,6-iPr2C6H3N)2}2Mg2(μ-X)2]. One electron reduction of the halide bridged magnesium complex was performed using [{(Mesnacnac)Mg}2], which resulted in the low valent magnesium [{Ph2P(2,6-iPr2C6H3N)2}2Mg2] complex (Scheme 7). The stability of the low valent magnesium species could be attributed to the P(V) center and steric and kinetic factors of the iminophosphonamide ligand. The Mg(I) species was characterized by multinuclear NMR spectroscopy and single-crystal X-ray diffraction. The observed Mg–Mg bond distance, 2.8445(9) Å, in the complex is also in the range of previous reports.43–46 Moreover, when two equivalents of tert-butyl isocyanate reacted with Mg(I) species, it resulted in the formation of colorless C–C-coupled product [{Ph2P(2,6-iPr2C6H3N)2Mg}2(NtBuCO)2]. This product has a bridging tert-butyl isocyanate fragment that coordinates the Mg2+ centers via N, N′ and O, O′ coordination modes.
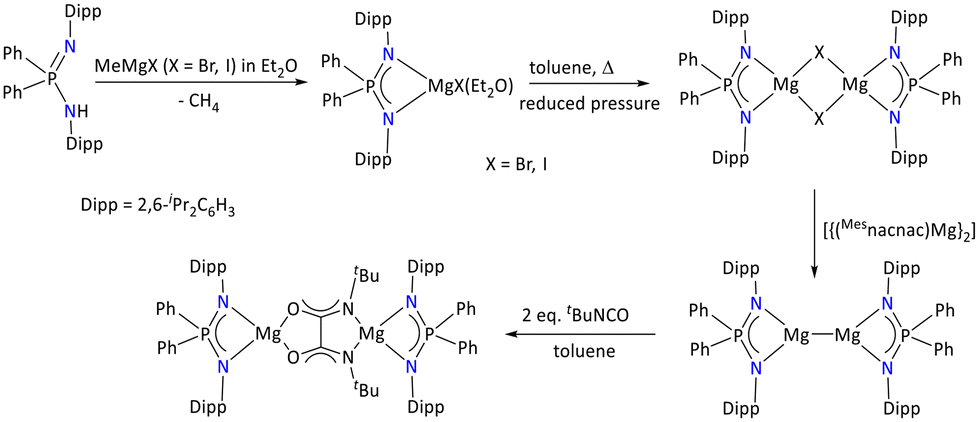 | ||
| Scheme 7 Low-valent magnesium iminophosphonamide complexes and their reactivity.43 | ||
This dimeric magnesium complex, [{Ph2P(2,6-iPr2C6H3N)2}2Mg2] was also used to examine the mechanisms of anionic ligand exchange and fullerene reduction.47
In 2001, Stalke's group altered the coordination ability of the {Ph2P(Me3SiN)2}− iminophosphonamide ligand, by replacing the phenyl substituents of phosphorous with pyridyl moieties; thus, a novel Janus head {Py2P(Me3SiN)2}− ligand was reported.48 The ligand was synthesized by the reaction of di(2-pyridyl)phosphane with 2 equiv. of trimethylsilylazide via the Staudinger method. The NPN coordination pocket in the {Py2P(Me3SiN)2}− ligand and two distant pyridyl substituents indicate that the Janus head ligand exhibits two possible chelating coordination pockets.
At room temperature, the protonated Janus-type ligand reacts with [M{N(SiMe3)2}2] (M = Sr or Ba) furnishing [{Py2P(NSiMe3)2}2Sr(thf)] and [{Py2P(NSiMe3)2}2Ba(4,4′-bipy)]n complexes. The 4,4′-bipyridine ligands help to reveal a zigzag polymeric arrangement in the solid-state structure of the barium complex (Scheme 8).48
 | ||
| Scheme 8 Janus head iminophosphonamide-based alkaline earth metal complexes.48 | ||
2.2. p-Block: group 13 and 14 compounds of the iminophosphonamides
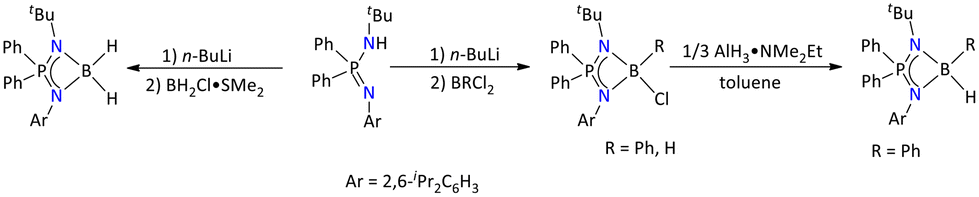 | ||
| Scheme 9 Syntheses of iminophosphonamide-based borane complexes.49 | ||
The formation of all these complexes was confirmed by multinuclear NMR and IR spectroscopy and HRMS, and the molecular structures of all these complexes were determined by single-crystal X-ray diffraction. The molecular structures suggested that the boron center adopts tetrahedral geometry for all these compounds.
The aluminum complexes of the iminophosphonamide {Ph2P(Me3SiN)2}− were reported by H. Roesky. These complexes were obtained either by a base elimination reaction between the iminophosphonamine {Ph2P(Me3SiN)2}H and trialkyl/triamide precursors of group 13 or by salt metathesis processes involving group 13 metal halides and in situ produced lithium iminophosphonamide (Scheme 10).50,51 The tetracoordinated dimethyl complexes [{Ph2P(NSiMe3)2}Me2M] (M = Al, Ga, or In) exhibited distinct melting points. By determining the matching cryoscopic molecular weight, the monomeric character of these metal complexes was verified.50 The aluminum monohydride complex, which was synthesized by the reaction of [Ph2P(NSiMe3)2H] and AlH3·NMe3, adopted a distorted trigonal bipyramidal geometry. The P–N bond lengths for each of these complexes are consistent with previously reported single and double bond lengths.52 Few group 13 metal complexes based on distinct iminophosphonamide backbones such as {Ph2P(2,6-iPr2C6H3N)(NtBu)}−26 and {PH(tBu)(2,6-iPr2C6H3N)2}−53 have been structurally characterized.
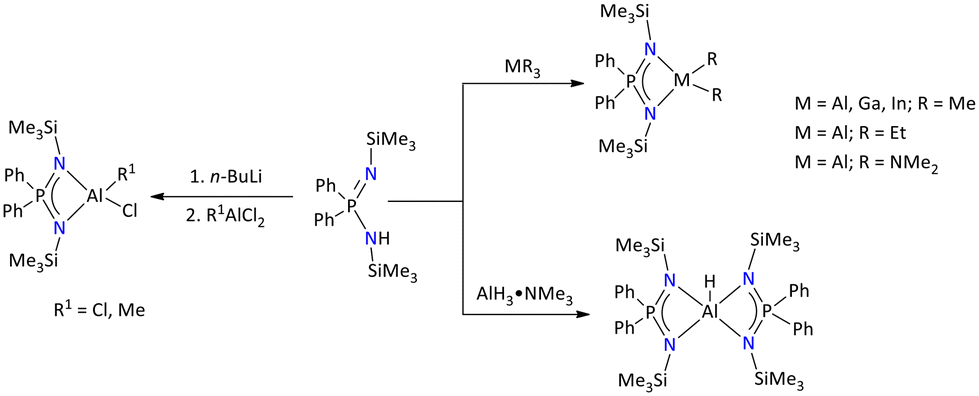 | ||
| Scheme 10 Synthesis of group 13 iminophosphonamide complexes.51 | ||
In this context, the deprotonation of the iminophosphonamine ligand {Ph2P(2,6-iPr2C6H3N)(NtBuH)}26 with AlH3·NMe2H in an equivalent ratio resulted in the formation of dimeric aluminum complex [{Ph2P(2,6-iPr2C6H3N)(NtBu)}AlH(μ-H)2AlH{Ph2P(2,6-iPr2C6H3N)(NtBu)}] based on the solid-state structure (Scheme 11). The molecular structure revealed that each aluminum center adopted a distorted trigonal bipyramidal geometry.
 | ||
| Scheme 11 Group 13 metal complexes substituted by the bulky iminophosphonamide ligand.26 | ||
The terminal hydride, one of the bridging hydrides and one of the nitrogen atoms of the iminophosphonamide ligand adopted the equatorial plane, whereas the remaining bridging hydride and another nitrogen of the ligand occupied the axial plane with the N–Al–H bond angle of 159.50(2)°. The aluminum hydride signals were assigned to a broad singlet at δ = 5.20 ppm in the 1H NMR spectrum. Further deprotonation of the ligand {Ph2P(2,6-iPr2C6H3N)(NtBuH)} with AlMe3 furnished facile formation of [{Ph2P(2,6-iPr2C6H3N)(NtBu)}AlMe2]. The Al–Me resonance appeared as a singlet at δ = 0.12 ppm in the 1H NMR spectrum of the complex. Furthermore, the 31P{1H} NMR spectrum of the complex showed a single peak at δ = 30.23 ppm. Additionally, treatment of [{Ph2P(2,6-iPr2C6H3N)(NtBu)}AlMe2] with B(C6F5)3 resulted in the formation of cationic aluminum species [{Ph2P(2,6-iPr2C6H3N)(NtBu)}AlMe]+[B(C6F5)3Me]−, which was characterized by multinuclear NMR spectroscopy. It was observed that the cationic species slowly converts into neutral tetracoordinated aluminum species via Me and C6F5 exchange. Additionally, the salt metathesis reaction between the lithium salt generated in situ and ECl3 (E = B, Al, and Ga) furnished the corresponding metal dichloride complexes, [{Ph2P(2,6-iPr2C6H3N)(NtBu)}ECl2] (E = B, Al, and Ga). The identity of these complexes was confirmed by multinuclear NMR and IR spectroscopy, HRMS and X-ray diffraction techniques.
Vrána et al. investigated the reactivity of bis(organoamino)phosphanes PhP(NHR)(NHR′) (R = R′ = tBu; R = tBu, R′ = 2,6-iPr2C6H3; R = R′ = Ph) and tBuP(NHDip)2 with Al(III) compounds (Scheme 12).53 In this regard, the reaction of PhP(NHtBu)2 and PhP(NHtBu)(2,6-iPr2C6H3NH) with AlMe3 resulted in the formation of [{PhP(NHR)(NR′)}AlMe2] (R = R′ = tBu; R = tBu, R′ = 2,6-iPr2C6H3) complexes via methane elimination. Furthermore, upon heating, these complexes showed H atom migration from nitrogen to the phosphorous center, which resulted in [{PhHP(NR)(NR′)}AlMe2] (R = R′ = tBu; R = tBu, R′ = 2,6-iPr2C6H3) complexes. In contrast to this, when PhP(NHPh)2 was reacted with AlMe3, it resulted in the formation of an intermediate adduct, [Ph(H)P(NHPh)(NPh)AlMe3], which was converted into [Ph(H)P(NPh)2AlMe2] due to the first hydrogen atom migration followed by methane elimination. Furthermore, hydrogen atom migration was observed when the bis(organoamino)phosphanes {R′′P(NHR)(NHR′)} (R′′ = Ph, R = R′ = tBu; R′′ = Ph, R = tBu, R′ = 2,6-iPr2C6H3 and R′′ = tBu, R = R′ = 2,6-iPr2C6H3) were partially deprotonated by n-BuLi, whereupon the reaction with AlCl3 resulted in [{R′′HP(NR)(NR′)}AlCl2] (R′′ = Ph, R = R′ = tBu; R′′ = Ph, R = tBu, R′ = 2,6-iPr2C6H3 and R′′ = tBu, R = R′ = 2,6-iPr2C6H3).
 | ||
| Scheme 12 Reactivity study of Al(III) compounds with bis(organoamino)phosphanes, PhP(NHR)(NHR′).53 | ||
Apart from this, Nakata's group has demonstrated nucleophilic substitution reactions of [{Ph2P(NtBu)2}AlCl2] using various nucleophiles (Nu) such as N(SiMe3)2−, {Fe(CO)2Cp}− and Cp− that resulted in the formation of an aluminum(III) monochloride complex bearing a nucleophile, [{Ph2P(NtBu)2}AlCl(Nu)] (Scheme 13).54 The identity of all these aluminum(III) mono- and di-chloride complexes was confirmed by various spectroscopic techniques and the molecular structures were identified by single-crystal X-ray diffraction.
 | ||
| Scheme 13 Syntheses of the aluminum(III) monochloride complex bearing a nucleophile, [{Ph2P(NtBu)2}AlCl(Nu)].54 | ||
Interestingly, by taking advantage of the sterically protected iminophosphonamide {Ph2P(2,6-iPr2C6H3N)2}− ligand, Stasch's group reported monovalent group 13 complexes [{Ph2P(2,6-iPr2C6H3N)2}E] (Ga(I), In(I), and Tl(I)) (Scheme 14).55
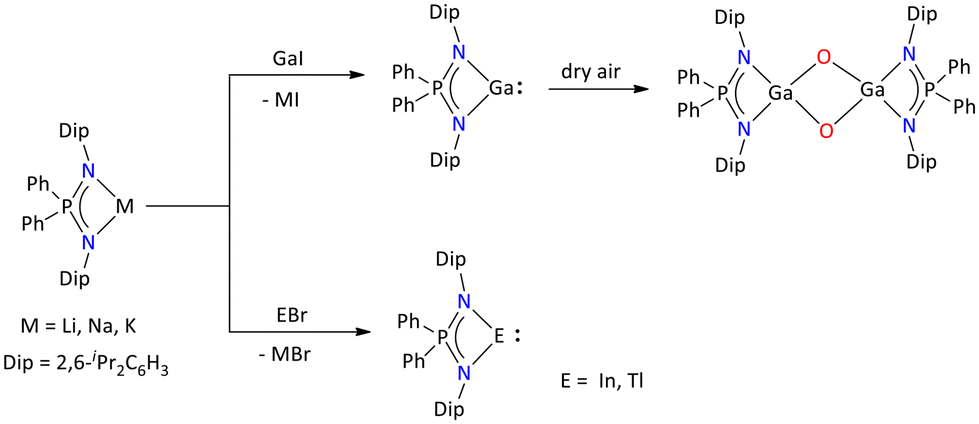 | ||
| Scheme 14 Syntheses of the monovalent heavier group 13 iminophosphonamide complexes.55 | ||
Other than the monomeric form for Ga(I), a weakly bonded dimer was obtained, which was structurally characterized. The Ga(I) complex upon exposure to dry air affords an oxidized Ga(III) complex, [{Ph2P(2,6-iPr2C6H3N)2}Ga(μ2-O)2Ga{Ph2P(2,6-iPr2C6H3N)2}].
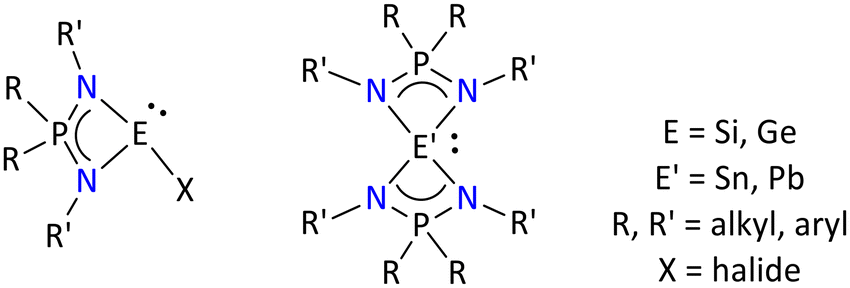 | ||
| Fig. 3 Literature-known tetrylenes based on iminophosphonamides.65–70 | ||
These examples include the structurally characterized germylene [Ph2P(NtBu)(2,6-iPr2C6H3N)GeX] (where X = Cl, OtBu, OTf), [(RO)2P(NSiMe3)2]GeCl, [{(RO)(Me3SiO)P(NSiMe3)2} GeCl], (R = iPr) and stannylene [{Ph2P(NSiMe3)2}2Sn] and [{(Ph)(R)P(NtBu)2}SnX] (where R = CH2Ph or 8-C9H6N; X = Cl or N(SiMe3)2) compounds.
Recently, an iminophosphonamide variant (i.e. [{Ph2P(NtBu)2}SiCl]) of the amidinate [{PhC(NtBu)2}SiCl] has been reported by Nakata's group.71 The study suggested that the chlorosilylene supported by iminophosphonamide has stronger σ-donor ability in comparison to common N-heterocyclic carbenes (NHCs) and other NHSis. Further when the synthesized silylene reacted with ½ equivalent of [RhCl(cod)]2, it led to the formation of a chlorosilylene-Rh(I) complex, [RhCl(cod){Ph2P(NtBu)2SiCl}]. The 29Si{1H} NMR spectrum of the complex exhibited a doublet at δ = 27.3 ppm, which is due to the coupling of 29Si with 103Rh and 31P (1JSi–Rh = 114.5 Hz; 2JSi–P = 13.4 Hz). Further, the cationic tris(silylene)-Rh(I) complex, [RhCl{Ph2P(NtBu)2SiCl}3]+, and the 14-electron Y-shaped Rh(I) complex, [RhCl{Ph2P(NtBu)2SiCl}2], were produced by the reactions of {Ph2P(NtBu)2SiCl} with [RhCl(cod)]2 in 1/4 and 1/6 equivalents, respectively. Additionally, treating the {Ph2P(NtBu)2SiCl} with [Pd(PPh3)4] resulted in the formation of a homoleptic Pd(0) complex, [Pd{Ph2P(NtBu)2SiCl}3]. Interestingly, when the chlorosilylene was treated with [PdMe2(tmeda)], it afforded a μ3-silylene-bridged tetranuclear palladium complex, with different oxidation states of the palladium centre (Scheme 15).72 The XPS analysis of the cluster suggested the presence of mixed oxidation states, Pd(0) and Pd(II), in the complex.
 | ||
| Scheme 15 Syntheses of an iminophosphonamide monochloride silylene and its reactivity.71,72 | ||
Further, Nakata's group has demonstrated that when the salt metathesis reaction was performed between chlorosilylene and KN(SiMe3)2 or K[CpFe(CO)2], the formation of silaimine instead of expected silylene was seen (Scheme 16). It was observed that the conversion of silaimine occurred via ring opening rearrangement of sterically hindered four-membered silylene. The DFT study suggest that combination of steric hinderance and the electronic factor of the substituents is the main reason for this intramolecular P–N bond cleavage of the intermediate silylene.73
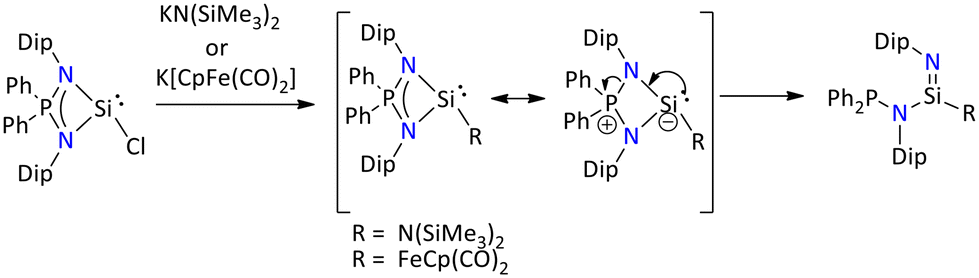 | ||
| Scheme 16 Generation of silaimine via intramolecular P–N bond cleavage of iminophosphonamido silylene.73 | ||
It is also demonstrated that the synthesis of [{Ph2P(NtBu)2}SiX] (X = Br and I) by heavier halo substitution of the chlorosilylene could be achieved via a halogen exchange reaction between the chlorosilylene [{Ph2P(NtBu)2}SiCl] and alkali halides (Scheme 17). The resulting Si–X bonds of [{Ph2P(NtBu)2}SiX] (X = Br, I) are considerably longer than expected possibly due to repulsion between the Si and the halogen lone pairs.74 Further, the oxidative addition of these heavier halo silylene with elemental selenium afforded a colorless product of bromo-and iodo-silylene selone. Moreover, the reaction of chlorosilylene with Fe(CO)5 in THF resulted in the formation of silylene–tetracarbonyliron(0), [Fe(CO)4{Ph2P(NtBu)2SiCl}], which is characterized by various spectroscopic techniques, and the molecular structure was determined by single-crystal X-ray diffraction.75
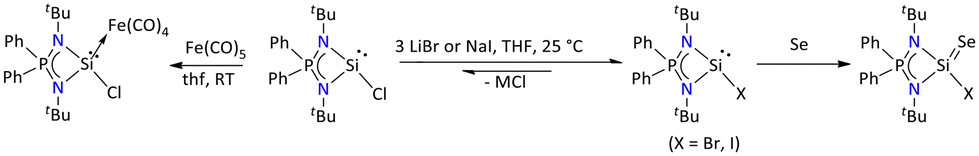 | ||
| Scheme 17 Reactivity of [{Ph2P(NtBu)2}SiCl].74,75 | ||
Furthermore, other than Si(II), Nakata's group explored the iminophosphonamide ligand with its heavier analogs such as Ge(II), Sn(II), and Pb(II) (Schemes 18 and 20).76–78 The salt elimination reaction between the lithium salt of the iminophosphonamide ligand and GeCl2·dioxane gave the three-coordinated chlorogermylenes of the type {Ph2P(NR)2GeCl} (R = tBu; R = Dipp = 2,6-iPr2C6H3).78 When {Ph2P(NtBu)2GeCl} was reacted with ½ equivalent of [Rh(cod)Cl]2 the formation of a chlorogermylene-Rh(I) complex, [RhCl(cod){Ph2P(NtBu)2GeCl}], was seen. In contrast to this, when {Ph2P(NDipp)2GeCl} was treated with ½ equivalent of [Rh(cod)Cl]2 in the presence of CO, it resulted in the formation of a five-membered germarhodacycle, [{Ph2P(NDipp)2}Rh(CO)2GeCl2] (Scheme 18).
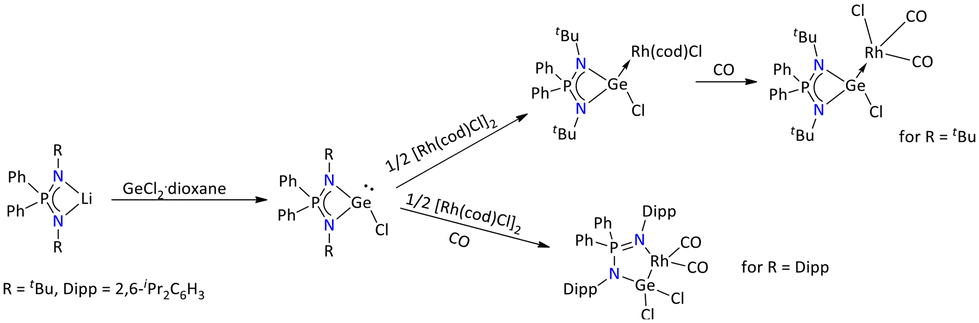 | ||
| Scheme 18 Syntheses of variably iminophosphonamide-substituted chlorogermylene and their Rh(I) complexes.78 | ||
The application of a germylene-based ligand was also demonstrated by using the chlorogermylene-Rh(I) complex [RhCl(cod){Ph2P(NtBu)2GeCl}] as a catalyst to hydrosilylate and hydroborate diphenylacetylene.
The lithium salt of the ligand, [Li{Ph2P(2,6-iPr2C6H3N)(NtBu)}], generated in situ was treated with one equivalent of GeCl2·dioxane to give the germylene monochloride [{Ph2P(2,6-iPr2C6H3N)(NtBu)}GeCl].65 Its reactivity was further investigated towards generating adducts with transition metals, oxidation, and salt metathesis processes (Scheme 19).
 | ||
| Scheme 19 Synthesis and reactivity of an iminophosphonamide-stabilized germylene monochloride.65 | ||
More recently, Driess and coworkers have reported the synthesis of a new chelating bis(iminophosphonamido-germylene)xanthene ligand, {Ph2P(NtBu)2Ge(II)(Xant)Ge(II)(NtBu)2PPh2}, (Xant = 9,9-dimethyl-xanthene-4,5-diyl).79 The redox noninnocent character of the ligand was used to stabilize monoatomic, two-coordinate Bi(I) and Bi(II) species. The nucleophilic nature of the Bi(I) species was investigated through redox reactions with AgOTf and MeOTf, leading to the formation of Bi(III) species.79
Moreover, [{Ph2P(NtBu)2}SnCl], synthesized by Nakata's group, was employed as a precatalyst for the hydroboration of carbonyls and imines. The result of the DFT calculations proposed that the hydroboration of carbonyl involves [{Ph2P(NtBu)2}SnH] as an intermediate, which proceeded through hydride transfer to carbonyl groups and eventually resulted in the boronate esters.76
Apart from this, Nakata's group has reported various low-valent Pb(II) species ligated by the iminophosphonamide {Ph2P(NtBu)2}−. The general salt elimination reaction of [{Ph2P(NtBu)2}Li] with PbCl2 furnished the chloroplumbylene {Ph2P(NtBu)2PbCl}. Further substitution of the chloride with {N(SiMe3)2}− resulted in the formation of aminoplumbylene [Ph2P(NtBu)2PbN(SiMe3)2]. A mono cationic plumbyliumylidene [Ph2P(NtBu)2Pb:]+ was generated through the chloride abstraction of [Ph2P(NtBu)2PbCl] with NaB(C6F5)4 (Scheme 20). The cationic [Ph2P(NtBu)2Pb:]+[B(C6F5)4]− plumbyliumylidene served as an efficient catalyst for the hydroboration of benzophenone and benzaldehyde.77
 | ||
| Scheme 20 Iminophosphonamide-stabilized plumbylenes and plumbyliumylidenes.77 | ||
Moreover, tetracoordinated [{Ph2P(NSiMe3)2}2E] (E = Sn and Pb) complexes of Sn(II) and Pb(II) have been reported by salt metathesis route (Scheme 21).67 The 31P{1H} NMR spectrum of [{Ph2P(NSiMe3)2}2E] showed a single resonance at δ = 25.4 ppm, whereas the 29Si NMR spectrum showed a doublet at δ = −5.2 ppm, with a coupling constant of 2JSi–P = 8 Hz. The tetrylenes exist in a distorted trigonal bipyramidal geometry, with the lone pair occupying the equatorial position. The molecular structure of the stanylene and plumbylene showed that both the four membered metalylene (i.e., [{Ph2P(NSiMe3)2}2M], M = Sn and Pb) planes are twisted with each other possibly due to the presence of lone pair and steric crowding from SiMe3 groups.
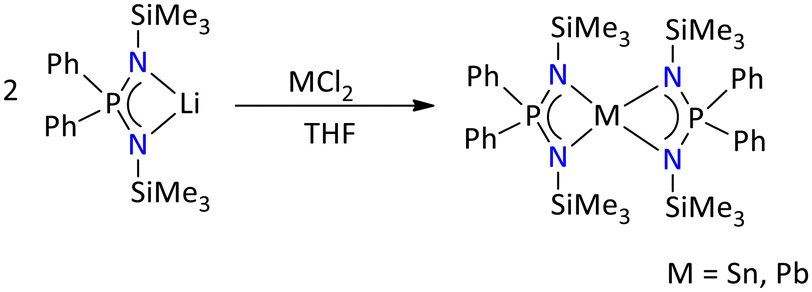 | ||
| Scheme 21 Synthesis of iminophosphonamide-stabilized homoleptic stanylene and plumbylene.67 | ||
2.3 Transition metal complexes of the iminophosphonamide ligands
Numerous studies have been conducted on amidinate (NCN)-stabilized transition metal complexes, and their catalytic applications in a range of organic transformations have been documented.80,81 The transition metal chemistry of the iminophosphonamide (NPN) ligand is still in its infancy compared to amidinate ligands. However, some iminophosphonamide transition metal complexes have been used as catalysts in recent decades, for example in alkene polymerizations,82,83 cyclopropanation reactions,84,85 and olefin oligomerizations.86 | ||
| Scheme 22 Group 4 (M = Ti and Zr) iminophosphonamide complexes as efficient polymerization catalysts.87 | ||
Also the bis(iminophosphonamide) complexes, [{Ph2P(NR)2}2MCl2] (R = p-tolyl, benzyl and M = Ti, Zr) were investigated towards ethylene polymerization. The result of catalysis suggested 50 to 100 times higher catalytic activity of the [{Ph2P(NR)2}2ZrCl2] (R = p-tolyl, benzyl) complex than its Ti analogue.
Collins’ group also reported a range of group 4 metal complexes coordinated by bis(iminophosphonamide). Piano-stool-type iminophosphonamide compounds were formed. In this regard, the amine elimination reaction between variably substituted iminophosphonamine, {Ph2P(NR)2}H (R = p-tolyl, benzyl, SiMe3, C6F6, 3,5-(CF3)2Ph) and CpZr(NMe2)3 followed by metathesis with excess Me2NH·HCl resulted in the formation of [Cp{Ph2P(NR)2}ZrCl2] (R = p-tolyl, benzyl, SiMe3, C6F5, 3,5-(CF3)2Ph). During the amine elimination, the intermediate [Cp{Ph2P(NC6F5)2}Zr(NMe2)2] rearranged in a novel terminal difluoride complex, [Cp{Ph2P(NC6F4NMe2)2}ZrF2], as described in Scheme 23.
 | ||
| Scheme 23 Piano-stool-type iminophosphonamide complexes of zirconium.88,89 | ||
Furthermore, when [Cp{Ph2P(NR)2}ZrCl2] (R = p-tolyl, benzyl, SiMe3, C6F6, 3,5-(CF3)2Ph) was reacted with MeMgBr or MeLi, it furnished the corresponding dialkyl complex.88,89
It was observed that the bis(iminophosphonamide)-ligated zirconium complexes, [{Ph2P(NR)2}2ZrX2] (R = p-tolyl, benzyl, C6F5; X = Cl and Me) and piano-stool-type iminophosphonamide-ligated zirconium complexes, [Cp{Ph2P(NR)2}ZrX2] (R = p-tolyl, benzyl, C6F5; X = Cl and Me), showed good catalytic activity towards ethylene polymerization in the presence of activators such as [Ph3C][B(C6F5)4] or PMAO.
The iminophosphonamide complexes of titanium and zirconium of the form [{Ph2P(NtBu)(NR)}2MCl2] (M = Ti, Zr and R = Ph, SiMe3) were synthesized by reacting the corresponding iminophosphonamide ligand and MCl4 (M = Ti and Zr) (Scheme 24).90
 | ||
| Scheme 24 Synthesis of Ti and Zr complexes of iminophosphonamides.90 | ||
The ethylene polymerization using these complexes as catalysts did not give a satisfactory result, which was anticipated due to steric crowding around the metal center.
Furthermore, Manners’ group reported perhalogenated bis(iminophosphonamide) complexes, [{Cl2P(NSiMe3)2}MCl3]·THF (M = Ti, Zr and Hf) and [{Cl2P(NSiMe3)2}NbCl4], by the reaction of {Cl2PN(SiMe3)2(NSiMe3)} with corresponding metal halides in dichloromethane. The formation of these complexes was anticipated as the [3 + 1] cyclocondensation of the ligand and metal center, resulting in the elimination of Me3SiCl (Scheme 25). Interestingly, a mixture of product formation was observed when 1.5 eq. of the ligand {Cl2PN(SiMe3)2(NSiMe3)} was treated with NbCl5. The reaction mixture exhibited many signals between δ = 14.5 and 6.5 ppm in the 31P{1H} NMR spectrum. Finally, the product was characterized by single-crystal X-ray diffraction, which showed the formation of unusual heterocycles [Cl3Nb{N(SiMe3)PCl2N}2Cl2Nb{N(SiMe3)PCl2N(SiMe3)}] and [Cl2Nb{N(SiMe3)PCl2N(SiMe3)}]2-μ-Cl(NPCl2N); the later complex was considered as the first example of Nb complex containing the six-membered ClNbNPNNb ring.91
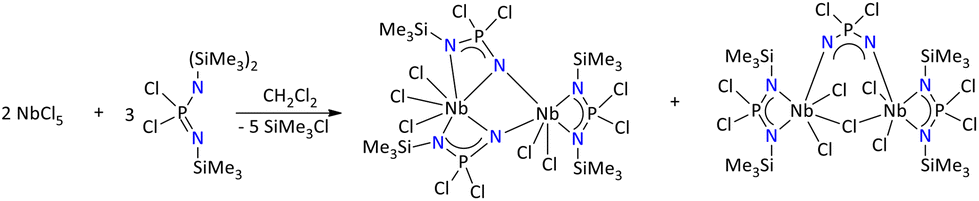 | ||
| Scheme 25 Synthesis of unusual heterocycles of niobium stabilized by iminophosphonamide.91 | ||
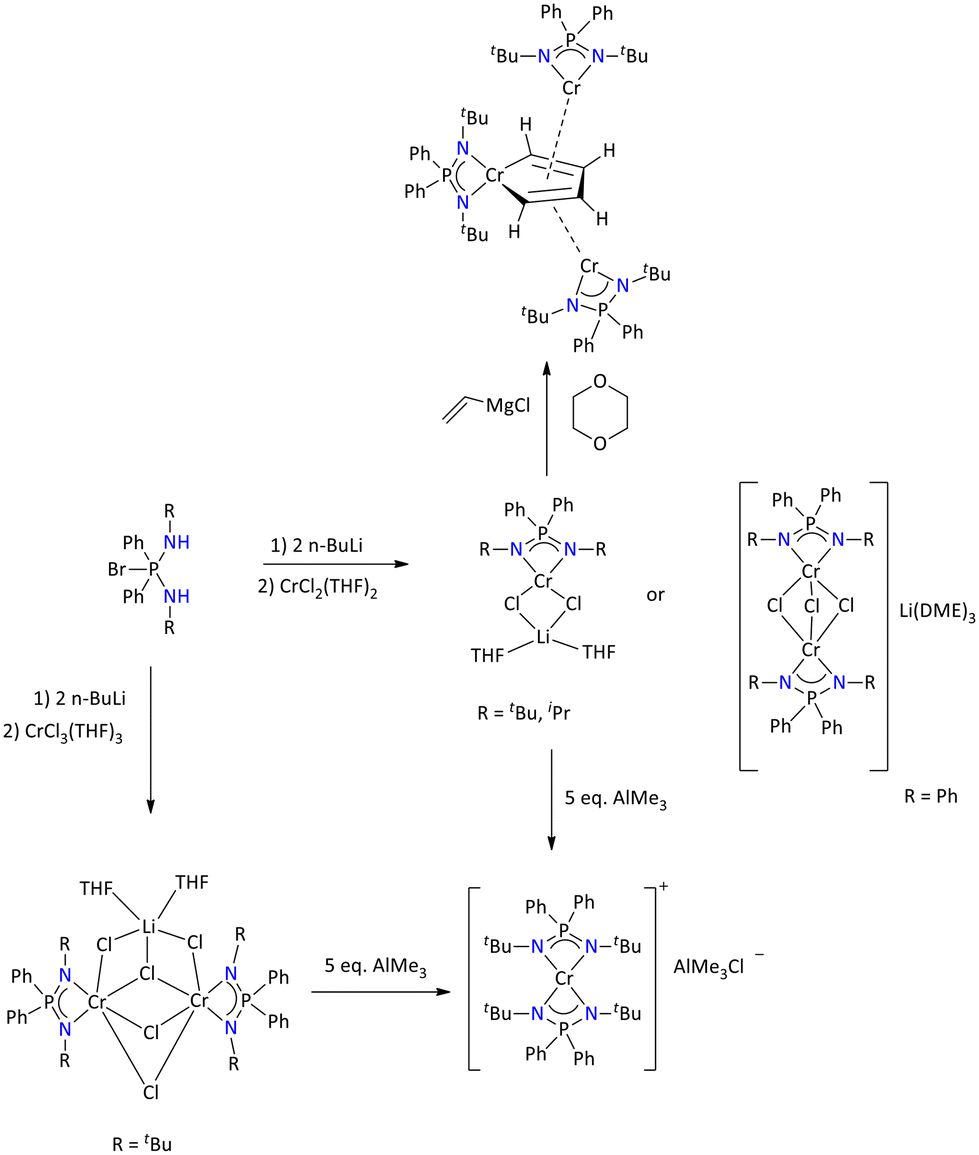 | ||
| Scheme 26 Chromium complexes of iminophosphonamide as active catalysts for ethylene oligomerization.86,92 | ||
Furthermore, treatment of the divalent chromium complex [{PPh2N(tBu)2}Cr(μ-Cl)2Li(THF)2] with the Grignard reagent CH2CH-MgCl resulted in the formation of the trinuclear chromium complex [π-{Ph2P(NtBu)}2Cr]2{μ,μ′,η4,η4′-C4H4}[σ-{Ph2P(NtBu)}2Cr]. The trinuclear complex was also structurally characterized by single-crystal X-ray diffraction. The molecular structure of the complex suggests that the one chromium center possessing iminophosphonamide, (i.e., [{Ph2P(NtBu)2}Cr]), is part of a planar metalla cyclopentadiene ring, whereas the other chromium centers are situated perpendicular of the metalla cycle with π bonds. This complex served as an efficient and selective catalyst for ethylene trimerization.92
The Fe(III) complex of the iminophosphonamide [{Ph2PN(Ph)2}3Fe] was synthesized by treating the iron(III) silylamide [Fe{N(SiMe3)2}3] with the phosphinohydrazine {NPh(Ph2P)NHPh} in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 proportion, respectively (Scheme 27). During the deprotonation reaction of the phosphinohydrazines, it rearranged and converted into the iminophosphonamide {Ph2P(NPh)2}. The synthesized iron(III) complex was isolated as fine yellow crystals that were stable towards the air. The complexes displayed strong absorptions at 1300 cm−1 and 1270 cm−1 in their IR spectra, which were attributed to the stretching vibrations of the N–P–N ligand backbone.93
:3 proportion, respectively (Scheme 27). During the deprotonation reaction of the phosphinohydrazines, it rearranged and converted into the iminophosphonamide {Ph2P(NPh)2}. The synthesized iron(III) complex was isolated as fine yellow crystals that were stable towards the air. The complexes displayed strong absorptions at 1300 cm−1 and 1270 cm−1 in their IR spectra, which were attributed to the stretching vibrations of the N–P–N ligand backbone.93
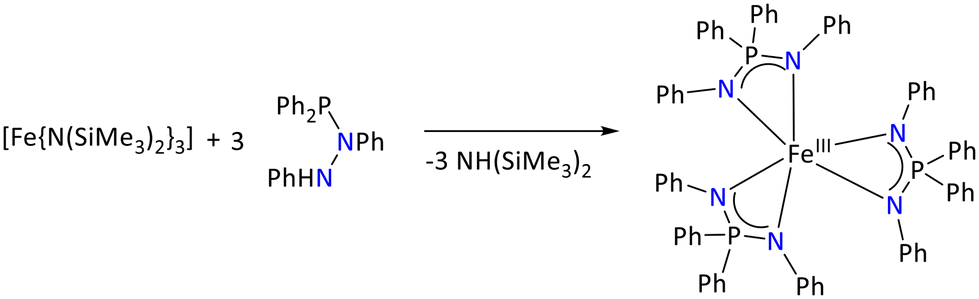 | ||
| Scheme 27 Synthesis of the homoleptic Fe(III) iminophosphonamide complex.93 | ||
The 16 VE and 18 VE half sandwich complexes of ruthenium containing [(η6-C6Me6){R2P(NR′)2}RuCl] and [(η6-C6Me6){R2P(NR′)2}Ru]+[BF4]− (R = Ph, Et and R′ = p-Tol, Me) were synthesized by reacting the appropriate precursor of variably substituted iminophosphonamides with [(C6Me6)RuCl2]2 followed by halide abstraction, respectively (Scheme 28).27,94–96 The centroid (Ru–C6Me6) distance falls between 1.662(4) Å and 1.675(3) Å, which is a common range for neutral half-sandwich ruthenium complexes. The Ru–Cl bond in the range of (2.437–2.445 Å) of the 18 VE [(η6-C6Me6){R2P(NR′)2}RuCl] complexes is elongated, and dissociates even in an apolar solvent, forming the corresponding 16 VE [(η6-C6Me6){R2P(NR′)2}Ru]+[Cl]− cationic complex that can be readily separated as salts with weakly-coordinating anions such as BF4−.
 | ||
| Scheme 28 Synthesis of the half-sandwich Ru(II) iminophosphonamide complex.27 | ||
The stability of the 16 VE cation of the ruthenium complex arises from the sufficient π-donation from the iminophosphonamide ligand. The characterization of these complexes was carried out by NMR spectroscopy, X-ray diffraction, and elemental analysis. The molecular structure of the 16 VE cationic ruthenium complex suggested the two-legged piano-stool-type geometry with the iminophosphonamide ligand perpendicular to the C6H6 ring. These complexes also served as efficient catalysts in the ROMP polymerization of norbornene.
Furthermore, Poli's group investigated the ruthenium complex of the iminophosphonamide, [(η6-Cym){Ph2P(N-pTol)2}RuCl], as a precatalyst for ketone transfer hydrogenation by isopropanol, which revealed the hydridic ruthenium complex, [(η6-Cym){Ph2P(N-pTol)2}RuH], as the active catalyst in a basic medium (Scheme 29).97 This ruthenium hydride complex is stabilized by dihydrogen bonding in the isopropanol. The transfer hydrogenation mechanism was investigated by NMR spectroscopy and DFT calculations.
 | ||
| Scheme 29 Generation of ruthenium hydride complex stabilized by iminophosphonamide.97 | ||
Moreover, Poli and Kalsin's group reported a variety of ruthenium arene complexes, [(Arene)RuCl{R2P(NR′)2}] bearing various substituents on iminophosphonamide ligands and a variety of arene rings (Fig. 4).94
 | ||
| Fig. 4 Variety of ruthenium iminophosphonamide complexes.94,95 | ||
The general synthesis of these complexes is shown in Scheme 30. These complexes were characterized by multinuclear NMR spectroscopy and X-ray diffraction analysis. A comparative investigation of these complexes as precatalysts was carried out towards acetophenone transfer hydrogenation in basic and base-free isopropanol.
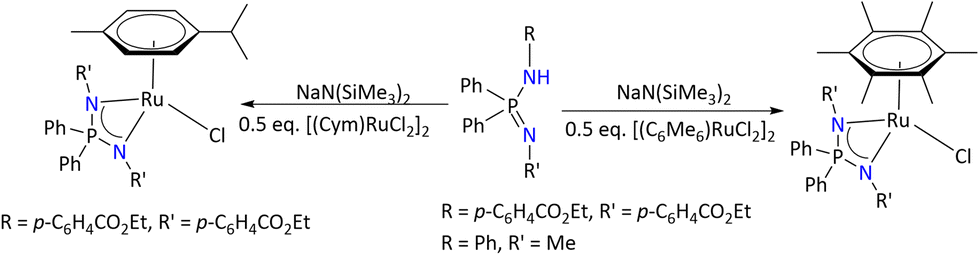 | ||
| Scheme 30 Syntheses of the piano-stool-type Ru(II) iminophosphonamide complex.94 | ||
The study revealed two distinct reaction pathways depending on the basicity of the N-substituents. In the case of [(Arene)RuCl{R2P(NR′)2}] (R′ = aryl), the active catalyst was observed only once the neutral hydride [(Arene)RuH{R2P(NR′)2}] is generated in basic isopropanol, whereas when one of the nitrogen substituents is Me (i.e., R′ = Me) in [(Arene)RuCl{R2P(NR′)2}], it readily catalyzed the reaction without base (Scheme 31).
 | ||
| Scheme 31 Possible reaction pathways for acetophenone hydrogenation-catalyzed Ru(II) iminophosphonamide.94 | ||
In the literature, the number of structurally characterized Co(II) complexes is limited to a few iminophosphonamide backbones such as {Ph2P(p-tBu-C6H4N)2} and {Ph2P(2,4,6-Me3C6H2N)2}.40,93 The synthesis of the later homoleptic cobalt complex is described below (Scheme 32).40 The molecular structure of the homoleptic [{Ph2P(p-tBu-C6H4N)2}2Co] and [{Ph2P(2,4,6-Me3C6H2N)2}2Co] is isostructural, and suggests that the central Co(II) atom adopts a distorted tetrahedral geometry with dihedral angles of 82.7(2)° and 88.01°, respectively, between two average CoN2P planes.
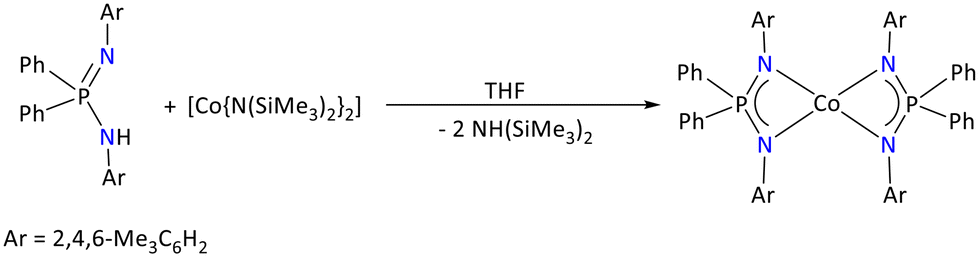 | ||
| Scheme 32 Synthesis of the homoleptic Co(II) iminophosphonamide complex.40 | ||
The four Co–N bond distances of these complexes are also in agreement with each other [{Ph2P(p-tBu-C6H4N)2}2Co] (1.999–2.025 Å) and [{Ph2P(2,4,6-Me3C6H2N)2}2Co] (2.015(1)–2.045(1) Å).
More recently, Kalsin's group has reported novel examples of structurally characterized half-sandwiched 18 VE and 16 VE rhodium iminophosphonamide complexes.98 These complexes, [Cp*RhX{(Ph2P(N–R)(N–R′)}] (Cp* = C5Me5; X = Cl; X = PF6; R, R′ = p-Tol, p-Tol; p-Tol, Me; and Me, Me) coordinated with either symmetric or non-symmetric iminophosphonamides (Scheme 33). Most of the half-sandwich rhodium complexes were synthesized in a similar way to the analogous ruthenium complexes.27,98 Furthermore, the reactivities of the cationic 16 VE rhodium complexes [(η5-C5Me5){Ph2P(N-p-Tol)(NR)}Ru]+[PF6]− (R = p-Tol and Me) were studied towards the reaction with pyridine and CO. The result indicated that due to the low coordination enthalpy of the pyridine adduct, it is stable only at low temperatures. In contrast when CO was reacted with the cationic complex, it resulted in the formation of a CO-inserted product.
 | ||
| Scheme 33 Synthesis of the cationic half-sandwich rhodium complex and its reactivity.98 | ||
Additionally, the fulvene complexes [(η4-C5Me4CH2){Ph2P(N-p-Tol)(NR)}Rh] (R = p-Tol and Me) were generated by the reaction of [(η5-C5Me5){Ph2P(N-p-Tol)(NR)}RuCl] (R = p-Tol and Me) with NaN(SiMe3)2. Further the reaction of [(η4-C5Me4CH2){Ph2P(N-p-Tol)(NR)}Rh] (R = p-Tol) with isoprene was carried out, which resulted in the formation of [(η5:η1-C5Me4(CH2)C(Me)CHCH2)Rh{Ph2P(N-p-Tol)2}], which was considered as the first example of 1,4-metallacycloaddition of diene to the fulvene complexes [(η4-C5Me4CH2){Ph2P(N-p-Tol)(NR)}Rh] (Scheme 34).
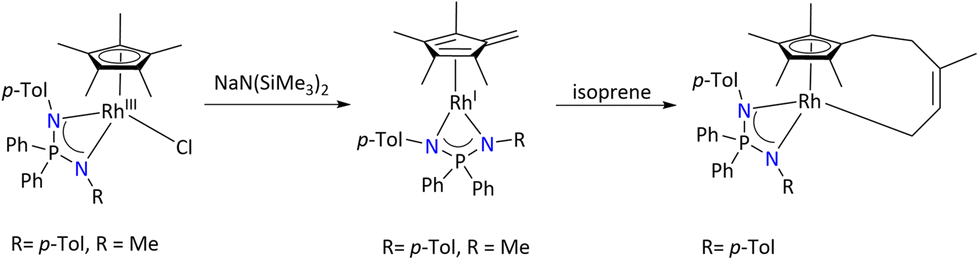 | ||
| Scheme 34 Synthesis of the fulvene rhodium complex and its reactivity with diene.98 | ||
 | ||
| Scheme 35 Syntheses of nickel and palladium iminophosphonamide complexes.82,99 | ||
During the formation of these compounds, bis(η3-allyl) Ni/Pd complexes underwent an unanticipated allyl rearrangement due to the iminophosphonamide ligand, which resulted in the production of a σ-bonded π-allyl group. These compounds acted as an active catalyst for the polymerization of ethylene at 70 °C and 50 bar.
Later in 1985, Fink's group reported that the combination of the iminophosphonamide ligand and [Ni(COD)2] or [Ni(η3-C3H5)2] is a capable system for the polymerization of α-olefins into chains with variable methyl branching at low temperatures.99
Collins’ group has synthesized a variety of Ni iminophosphonamide complexes, as described in Scheme 36.100
 | ||
| Scheme 36 Syntheses of discrete iminophosphonamide Ni(II) complexes.40,100 | ||
[(η3-C3H5){Ph2P(NR)2}Ni] (R = SiMe3 and p-Tol) was synthesized by the reaction of in situ generated salt of the differently substituted {Ph2P(NR)2} (R = SiMe3, 2,4,6-Me3C6H2, p-Tol) ligands with [(η3-C3H5)NiBr]2. These complexes are stable in the open air for a short time. The NMR spectroscopy of these complexes revealed the non-fluxional behavior of the π-allyl group; however, through {Ph2P(NR)2} ligand reorientation, the chemical exchange could be observed. The molecular structure of these [(η3-C3H5){Ph2P(NR)2}Ni] complexes revealed the long Ni⋯N bond distance in the range of 1.941(1)–1.973(2) Å having an acute NPN bite angle with the ligand. When [(η3-C3H5){Ph2P(NR)2}Ni] was employed as a catalyst in the polymerization of ethylene, it resulted in no active catalysis. In search of an active catalyst, [{Ph2P(NSiMe3)2}NiPh(PPh3)] was prepared from [NiPh(PPh3)2Br] and in situ generated {Ph2P(NSiMe3)2}− ligand. The complex is active for 1-hexene isomerization without any activator, and produces R-methyl styrene when reacting with propene. However, it was observed that in the presence of suitable phosphine scavengers, [{Ph2P(NSiMe3)2}NiPh(PPh3)] can polymerize ethylene. Furthermore, all these complexes are active towards the dimerization of ethylene in the presence of polymethylaluminoxane (PMAO).
Various homoleptic Ni(II) complexes such as [{iPr2P(NMe)2}2Ni], [{Ph2P(2,4,6-Me3-C6H2N)2}2Ni], and [{Ph2P(NPh)2}2Ni] have also been reported and structurally characterized.40,101 In this regard, Singh's group reported the synthesis of homoleptic [{Ph2P(2,4,6-Me3-C6H2N)2}2Ni] by in situ deprotonation of the corresponding iminophosphonamine ligand followed by treatment with [(PPh3)2NiCl2] (Scheme 36). The characterization of the Ni(II) complex was further carried out by NMR spectroscopy and single-crystal X-ray diffraction. The 1H NMR spectrum of the complex showed chemically non-equivalent protons of the o-methyl protons of the mesityl rings, which was attributed to the agostic interaction of the o-methyl protons with the Ni(II) centers. Furthermore, the 31P{1H} NMR spectrum of the Ni(II) complex exhibited single resonance at −16.83 ppm, which is significantly upfield shifted as compared to the corresponding ligand (36 ppm). The non-equivalence nature of the o-methyl protons of the mesityl rings and agnostic interaction was further confirmed by the molecular structure of the Ni(II) complex having a Ni⋯H distance of 2.673(1) Å.
Wang's group has reported various nickel iminophosphonamide complexes having a carbene substituent at the side arm.102 These complexes efficiently catalyzed cross-coupling reactions of Grignard reagents with aryl chlorides or fluorides (Scheme 37).
 | ||
| Scheme 37 Cross coupling using carbine-based Ni(II) iminophosphonamide complexes.102 | ||
Zeise's salt analogue of the iminophosphonamide [{(CH3)2P(NCH3)2}PtCl(C2H4)] was reported by Nahrstedt's group, by treating the lithium salt of the corresponding iminophosphonamine with [KPtCl3(C2H4)] in 2/1 molar ratio, respectively (Scheme 38).103 The formation of the metal complex was confirmed and characterized by multinuclear NMR spectroscopy.
 | ||
| Scheme 38 Synthesis of Zeise's analogue iminophosphonamide ligand.103 | ||
Ustynyuk and Kalsin's group reported various palladium and platinum complexes of the iminophosphonamide (Scheme 39).96,104 The structural analysis of these complexes was carried out by single-crystal X-ray diffraction. The results suggested an intramolecular weak C–H⋯Pd interaction in [{Ph2P(p-iPr-C6H4N)2}Pd(PPh3)Cl] and an intermolecular weak C–H⋯Pd interaction in homoleptic [{Ph2P(p-iPr-C6H4N)2}Pd] complexes. It was observed that these weak interactions influenced the MNPN metallacycle planarity. The synthesized palladium complexes [{Ph2P(p-iPr-C6H4N)2}Pd(PPh3)Cl] and [{Ph2P(p-COOEt-C6H4N)2}Pd(η3-allyl)] were also tested as catalysts in the Tsuji–Trost allylation and the Suzuki–Miyaura cross-coupling reactions.
 | ||
| Scheme 39 Syntheses of palladium and platinum iminophosphonamide complexes.96,104 | ||
More recently, Stassner's group reported the cyclometalated NHC Pt(II) complexes of iminophosphonamide (Scheme 40).105 These complexes were synthesized by first treating the NHC salt with Ag2O, which resulted in the formation of silver carbene complexes. Further transmetalation of the silver(I) carbene complex with [Pt(COD)Cl2] led to the formation of chloro bridged platinum complexes [{C∧C*}(μ-Cl)Pt]2 (C∧C* = cyclometalated N-heterocyclic carbene). Finally, a salt metathesis reaction with the in situ prepared lithium salt of the iminophosphonamide and chloro bridged platinum complexes yielded the expected platinum iminophosphonamide complexes [{Ph2P(NPh)2}(C∧C*)Pt] after purification by neutral column chromatography.
 | ||
| Scheme 40 Synthesis of platinum iminophosphonamide complexes.105 | ||
The formation of complexes was confirmed by various techniques such as multinuclear NMR spectroscopy, elemental analysis, single-crystal X-ray diffraction and mass spectrometry as well. The 195Pt NMR spectrum of the iminophosphonamide complex showed a signal at −3510 ppm (R = H) and −3483 ppm (R = 5,6-OC6H4), which is comparatively downfield shifted compared to the chloro bridged platinum complexes (−4050 ppm) in deuterated dimethyl sulfoxide.
The molecular structure of the complex suggested that the nitrogen atoms of the ligand coordinated to the metal center with 79.37(7)° NPtN bite angle (R = H). Furthermore, the NPtN and NPN planes are almost uniplanar with a slight deviation of 2.73°. The photophysical study of these complexes exhibited that depending on the NHC substituents, the emissive states of the Pt(II) complexes vary. The study of these complexes was examined by cyclic voltammetry (CV) and differential pulse voltammetry (DPV), and the results were supported by density functional theory (DFT).
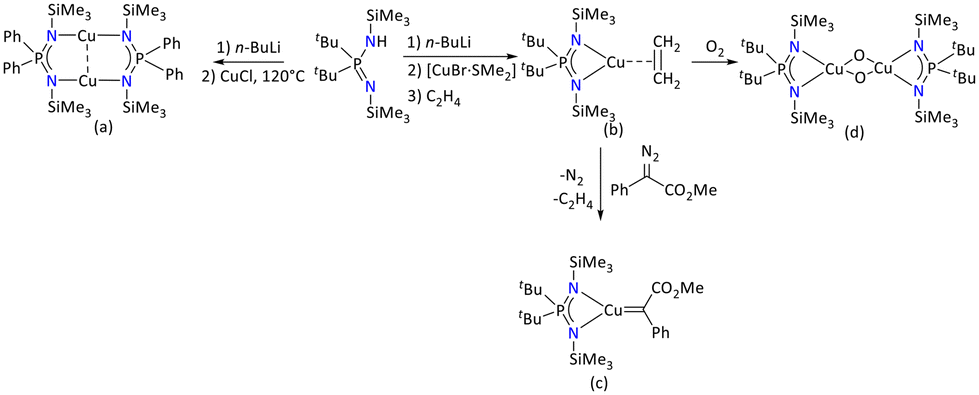 | ||
| Scheme 41 Iminophosphonamide-supported (a) copper(I) binuclear copper complex, (b) olefin-copper(I) complex, (c) copper(I) carbene and (d) bis(μ-oxo) copper(III) species.106–108 | ||
Only one structurally characterized example of a Cu(II) iminophosphonamide complex has been reported and was accessed by the deprotonation of {Ph2P(2,4,6-Me3C6H2N)2}H with [Cu{N(SiMe3)2}2], resulting in the formation of the mononuclear homoleptic complex [{Ph2P(2,4,6-Me3C6H2N)2}2Cu] (Scheme 42).40
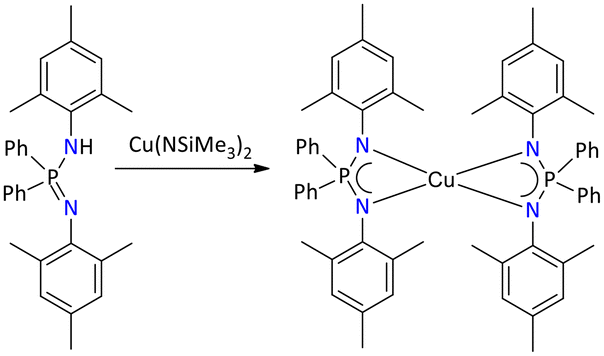 | ||
| Scheme 42 Synthesis of iminophosphonamide-supported homoleptic copper(II) complex.40 | ||
Bhoomishankar's group reported the silver complexes of pyridyl substituted iminophosphonamide.110,111 In this regard, treatment of [P(NHpy)4]Cl with AgClO4 led to the formation of a pentanuclear Ag(I) complex, [Ag5{P(Npy)2(NHpy)2}2][ClO4]3. Alternatively, this complex can also be synthesized by treating [P(NHpy)4]Cl with a base first. This resulted in the formation of a neutral iminophosphonamine, P(Npy)(NHpy)3 ligand. Subsequent reaction with AgClO4 also formed the pentanuclear Ag(I) complex [Ag5{P(Npy)2(NHpy)2}2][ClO4]3 (Scheme 43). The five-fold positive charge due to five Ag(I) ions in the cluster is stabilized by two anionic iminophosphonamide ligands and three perchlorate anions. Both ligands functioned as hexadentate donor ligands. The molecular structure of the metal complex revealed that the intermetallic Ag⋯Ag interaction lies in the range from 2.9647(7) Å to 3.1772(12) Å and is comparable with the literature.112 Additionally another Ag(I) complex, [Ag5{P(Npy)2(NHpy)2}2][OTf]3, was synthesized by reacting [P(NHpy)4]Cl with the more reactive silver triflate Ag(OTf). In both compounds, the same cation, [Ag5{P(Npy)2(NHpy)2}2]+, suggested the thermodynamic stability of the pentanuclear Ag(I) complex with different counter anions.
 | ||
| Scheme 43 Synthesis of silver(I) iminophosphonamide complexes.110,111 | ||
To the best of our knowledge, divalent zinc complexes have only been obtained by using {Ph2P(2,6-iPr2C6H3N)2}39 and {Py2P(Me3SiN)2}48,113 as non-chiral ligands. In this regard, Stasch's group has demonstrated that a monovalent zinc complex could be stabilized by the sterically demanding {Ph2P(2,6-iPr2C6H3N)2} ligand (Scheme 44). As shown, the heteroleptic zinc complex [{Ph2P(2,6-iPr2C6H3N)2}Zn(μ-X)2Zn{Ph2P(2,6-iPr2C6H3N)2}] can be obtained either by salt metathesis between the potassium salt of the ligand and ZnBr2 or by reacting the pre-synthesized heteroleptic complex [{Ph2P(2,6-iPr2C6H3N)2}ZnR] with I2. Further, the single electron reduction of [{Ph2P(2,6-iPr2C6H3N)2}Zn(μ-X)2Zn{Ph2P(2,6-iPr2C6H3N)2}] by [{(Mesnacnac)Mg}2] afforded the monovalent zinc complex [{Ph2P(2,6-iPr2C6H3N)2}Zn]2. The intermetallic Zn–Zn distance is 2.3132(9) Å, which is consistent with previously known dimeric Zn(I) species.
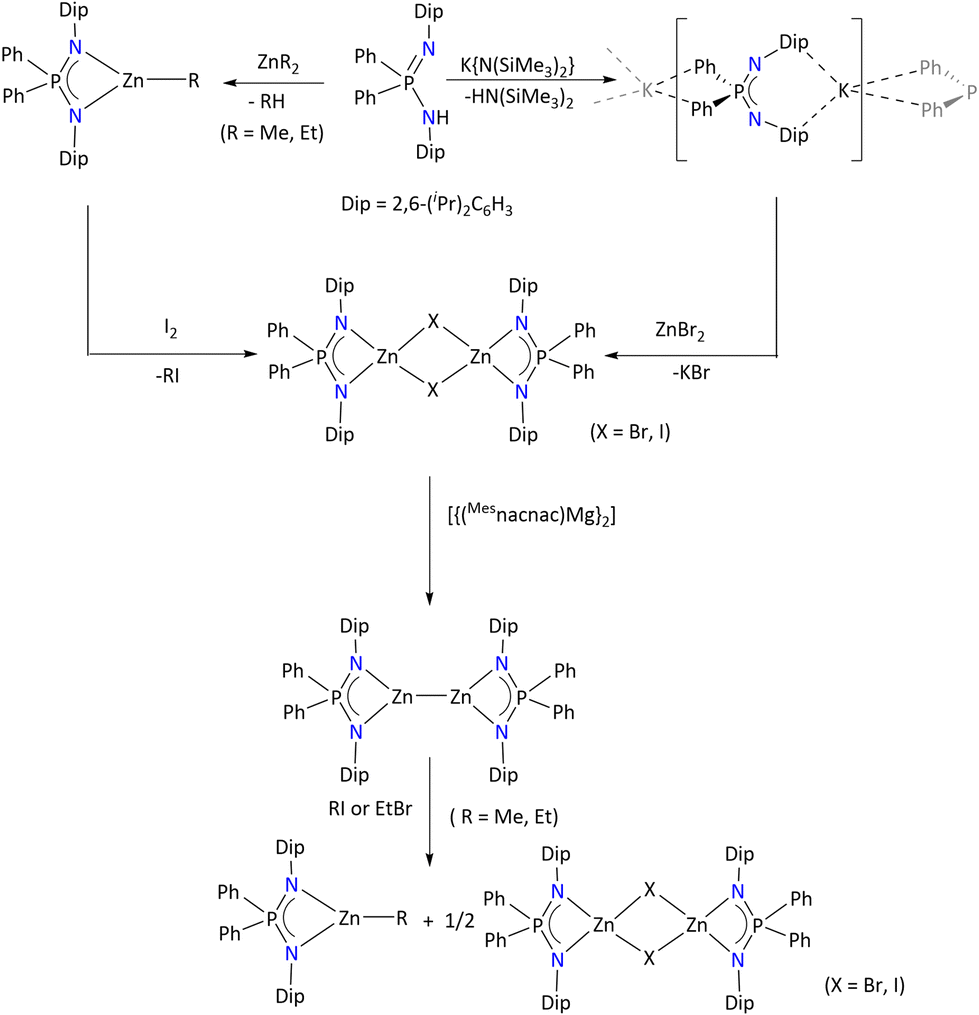 | ||
| Scheme 44 Syntheses of Zn(I) and Zn(II) iminophosphonamide complexes. | ||
The 1H NMR spectrum of [{Ph2P(2,6-iPr2C6H3N)2}Zn]2 shows the absence of a [{Ph2P(2,6-iPr2C6H3N)2}ZnH] species, which further support the presence of a Zn–Zn bond in the complex. Moreover, the reactivity of this Zn(I) complex was explored towards small molecules such as RI (R = Me and Et) or EtBr, which resulted in the umpolung of the alkyl halides.
2.4 f-Block metal complexes based on iminophosphonamides
Compared to the well-researched amidinate chemistry of rare earth metals, there has been relatively little investigation into the related coordination chemistry with iminophosphonamides. Most contributions since the 1990s have come from the field's pioneers, Edelmann and Schumann.39,114–116 In these early reports, ligands consisting of silyl, aryl, and alkyl-substituted N-centers of various iminophosphonamide complexes have been generally used in rare earth metal chemistry (Fig. 5).117 | ||
| Fig. 5 Iminophosphonamide ligands used in rare earth metal complexes.117 | ||
The early reports of lanthanide iminophosphonamide metal complexes are based on silyl-substituent (I, Fig. 5). In particular, Edelmann's group reported and structurally characterized the Sm(III) and Yb(II) complexes [{Ph2P(NSiMe3)2}2Sm(μ-I2)Li(thf)2] and [{Ph2P(NSiMe3)2}2Yb(thf)2] bearing the same iminophosphonamide ligand in 1991 (Scheme 45).116 Furthermore, Edelmann's group reported in 1995, a series of [{Ph2P(NSiMe3)2}2Ln-COT] (COT = cyclooctatetraene) (Ln = Ce, Pr, Nd, and Sm) complexes bearing the same ligand (Scheme 45).115 These complexes were obtained by salt metathesis of [{Ph2P(NSiMe3)2}Li] and [Ln(μ-Cl)(thf)2COT]2.
 | ||
| Scheme 45 Syntheses of silyl-substituted iminophosphonamide complexes of lanthanides.115,116 | ||
Most of the alkyl and aryl substituted iminophosphonamide lanthanide complexes were used as catalysts for the polymerization of olefins.118–121 In this regard, in 2010, Cui and co-workers demonstrated that selective isoprene polymerization could be achieved when different lanthanide metal complexes were employed as catalysts (Scheme 46). The bis(alkyl)iminophosphonamide complex of lutetium, [Ph2P{(2-pyridyl-N)(NC6H4Me2)}(thf)Lu(CH2SiMe3)2], was synthesized by the deprotonation reaction of the iminophosphonamine Ph2P{(2-pyridyl-NH)(NC6H4Me2)} with [Lu(CH2SiMe3)3(thf)2]. Furthermore, when the Ph2P{(2-pyridyl-NH)(NC6H4Me2)} ligand was reacted with [Nd(BH4)3(thf)3], an unprecedented reaction occurred leading to the formation of a bis(borohydride) neodymium complex. The synthesized lutetium complex [Ph2P{(2-pyridyl-N)(NC6H4Me2)}(thf)Lu(CH2SiMe3)2] in combination with proper cocatalysts ([PhMe2NH][B(C6F5)4] and AliBu3) afforded the 3,4 selectivity, whereas the neodymium borohydride complex selectively afforded the trans-1,4 product in the presence of ([PhMe2NH][B(C6F5)4] and MgnBu2).120
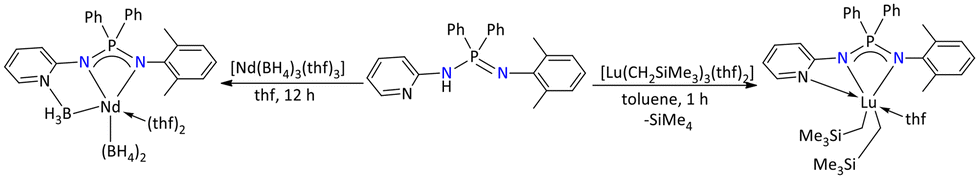 | ||
| Scheme 46 Synthesis of selective polymerization catalysts based on iminophosphonamide.120 | ||
Cui's group in 2014, also reported the dialkyl complexes of lutetium ligated with variably substituted iminophosphonamide of type II (Fig. 5 and Scheme 47). The structural determination of these complexes was carried out by various spectroscopic techniques including single-crystal X-ray diffraction.
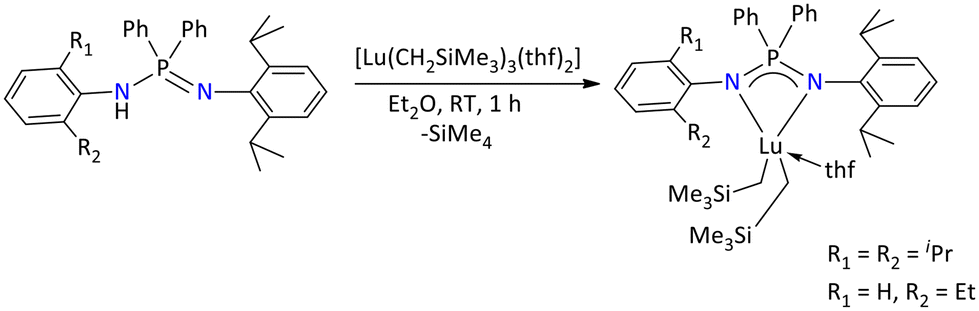 | ||
| Scheme 47 Synthesis of dialkyl lutetium complexes of iminophosphonamides.118 | ||
These complexes were tested as catalysts in the polymerization of isoprene. The result indicated that all these complexes appeared as effective 3,4 regioselective catalysts. Depending on the ligand substituent, the syndio- and iso-3,4 selectivity of the isoprene polymerization was observed, which was supported by DFT calculations.118
Furthermore, a series of bis(alkyl) complexes [{Ph2P(2,4,6-Me3C6H2N)(NAr)}Ln(CH2SiMe3)2(thf)2] (Ln = Lu, Y, Sc, and Er; Ar = Ph and 2-pyridyl) were generated and structurally characterized.119 These complexes were tested for isoprene polymerization in the presence of [Ph3C][B(C6F5)4] and AliBu3.
Research on the catalytic polymerization of isoprene by Cui revealed that the substituents on the nitrogen atoms and the atomic radii of the corresponding central metal atoms determine the catalytic activity and selectivity.122 Furthermore, it was investigated how the molecular architectures of the complexes and their catalytic capabilities correlate.
The study on regio- and stereoselective isoprene polymerization was further carried out using dialkyl lanthanide complexes of iminophosphonamide [{Ph2P(2,6-iPrC6H3N)2}Ln(R)2(thf)] (Ln = Sc, Lu, Y, and Er; R = CH2SiMe3 and for Ln = La and Nd; R = CH2-p-Tol) as catalysts in the presence of activator systems [Ph3C][B(C6F5)4]/AliBu3 and [Ph3C][B(C6F5)4]/AlEt3, respectively. The study found that the lanthanide complexes, [{Ph2P(2,6-iPrC6H3N)2}Ln(CH2SiMe3)2(thf)] (Ln = Sc, Lu, Y, and Er) with a smaller ionic size in the presence of [Ph3C][B(C6F5)4]/AliBu3 tend to favor a high 3,4-regio and stereoselectivity for the polymerization of isoprene (up to 98.1%) While the 4-methylbenzyl neodymium and lanthanum complexes [{Ph2P(2,6-iPrC6H3N)2}Ln(CH2-p-Tol)2(thf)] (Ln = La and Nd) in the presence of [Ph3C][B(C6F5)4]/AlEt3 demonstrated efficient catalytic activity in the trans-1,4-selective polymerization of isoprene (Scheme 48).123,124
 | ||
| Scheme 48 Regio- and stereoselective isoprene polymerization using iminophosphonamide-stabilized lanthanide complexes.123 | ||
Of the three above-described NPN ligand backbone types, Sundermeyer's group initially introduced the {Ph2P(NtBu)2} ligand (Fig. 5, III) into the rare earth metal chemistry in 2016.117 For instance, dialkyl or monoalkyl substituted rare earth metal (Sc and Y) complexes of {Ph2P(NtBu)2} can be synthesized by reacting the iminophosphonamine with [Ln(CH2SiMe3)3(thf)3] in an appropriate ratio (Scheme 49).
 | ||
| Scheme 49 Syntheses of mono- and dialkyl lanthanide complexes.117 | ||
Alternatively, {Ph2P(NtBu)2}H was reacted with ScCl3 and MeLi to give [{Ph2P(NtBu)2}2Sc(Me)thf] (Scheme 49).
Moreover, the mono-alkyl substituted complexes are reactive towards the abstraction of acidic –CH protons (Scheme 50).117 In this regard, an in situ prepared yttrium mono alkyl [{Ph2P(NtBu)2}2Y(CH2SiMe3)] complex when treated with phenylacetylene resulted in the formation of a highly air- and moisture-sensitive colorless yttrium alkyne complex. The formation of the alkynyl complex was confirmed by multinuclear NMR spectroscopy. The 31P{1H} NMR spectra of the yttrium alkynyl [{Ph2P(NtBu)2}2Y(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)] complex exhibited a single resonance at δ = 17.8 ppm upfield shifted compared to the monoalkyl yttrium complex (δ = 19.5 ppm). Furthermore, the reaction of [{Ph2P(NtBu)2}2Y(CH2SiMe3)] with [PhNHMe2][B(C6F5)4] afforded the cationic yttrium complex [{Ph2P(NtBu)2}2Y]+[B(C6F5)4]−. The formation of the cationic complex was confirmed by NMR spectroscopy, which displayed signals corresponding to PhNMe2 and SiMe4. The other characteristic signals in the multinuclear NMR spectra were also consistent with the product formation.117
CPh)] complex exhibited a single resonance at δ = 17.8 ppm upfield shifted compared to the monoalkyl yttrium complex (δ = 19.5 ppm). Furthermore, the reaction of [{Ph2P(NtBu)2}2Y(CH2SiMe3)] with [PhNHMe2][B(C6F5)4] afforded the cationic yttrium complex [{Ph2P(NtBu)2}2Y]+[B(C6F5)4]−. The formation of the cationic complex was confirmed by NMR spectroscopy, which displayed signals corresponding to PhNMe2 and SiMe4. The other characteristic signals in the multinuclear NMR spectra were also consistent with the product formation.117
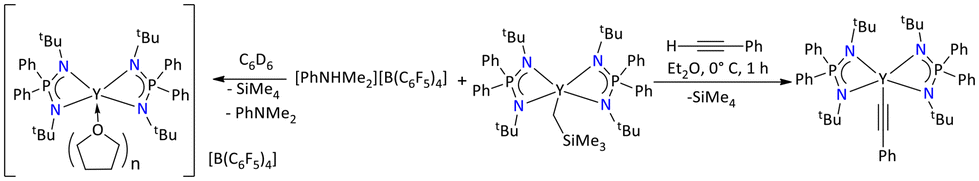 | ||
| Scheme 50 Syntheses of cationic and alkynyl yttrium complexes.117 | ||
More recently, Konchenko's group reported a variety of tris-iminophosphonamide lanthanide complexes utilizing chromophore 2-(phen-2′-yl)-1,3-benzothiazole (Pbt) substituents at the nitrogen centers of the iminophosphonamine, Ph2P(HNPbt)(NPbt).125
The corresponding iminophosphonamide lanthanide complexes were synthesized by a salt metathesis between LnCl3 and in situ generated K{Ph2P(NPbt)2} regardless of the stoichiometry of the precursors. In all cases, [Ln{Ph2P(NPbt)2}2]+ (Ln = Y, Sm, Gd, and Dy), where [Ln{Ph2P(NPbt)2}2]+ exists as cationic fragment and [Ph2P(NPbt)2]− as its counter anion, were isolated (Scheme 51). Furthermore, the photoluminescence of Ph2P(HNPbt)(NPbt) showed two emission bands in the visible range, suggesting the relationship between the color of luminescence and the excitation wavelength. In contrast, only a single band was observed when the photoluminescence was measured in solution. This peculiar observation is conditioned to the specific effect of packing in the solid state of the ligand. Furthermore, the potassium salt K{Ph2P(NPbt)2}, [Gd{Ph2P(NPbt)2}2][Ph2P(NPbt)2] and [Dy{Ph2P(NPbt)2}2][Ph2P(NPbt)2], when excited, showed bright emission bands of different colors depending on the metal center, both in solution and in the solid state.
 | ||
| Scheme 51 Synthesis of luminescent lanthanide complexes.125 | ||
More recently, Konchenko's group reported the yttrium complex using a nonsymmetric benzothiazole-functionalized iminophosphonamide ligand. The yttrium complex was synthesized by in situ deprotonation of iminophosphonamine with potassium hydride followed by a reaction of yttrium trichlorides in THF (Scheme 52). The molecular structure of the protonated ligand and yttrium complex showed that there is intermolecular π-stacking between the 2-(phen-2′-yl)-1,3-benzothiazole (Pbt) fragments.126 Edelmann's group reported both lanthanide and actinide iminophosphonamide metal complexes (M = Nd, Pr, Th, and U) by reacting [{Ph2P(NSiMe3)2}Li(thf)2]2 with anhydrous chlorides of lanthanide and actinides (Scheme 53).114 The formation of these metal complexes was accessed through multinuclear NMR spectroscopy, elemental analysis, and mass spectrometry. The 31P{1H} NMR spectrum of the thorium complex, [{Ph2P(NSiMe3)2}2ThCl2], and the dioxouranium complex, [{Ph2P(NSiMe3)2}2UO2], displayed a single resonance in the positive region at δ = 17.4 ppm and δ = 42.9 ppm, respectively, while the rest of the metal complexes exhibited a single peak in the negative region ranging from −231.3 ppm to −139.6 ppm.
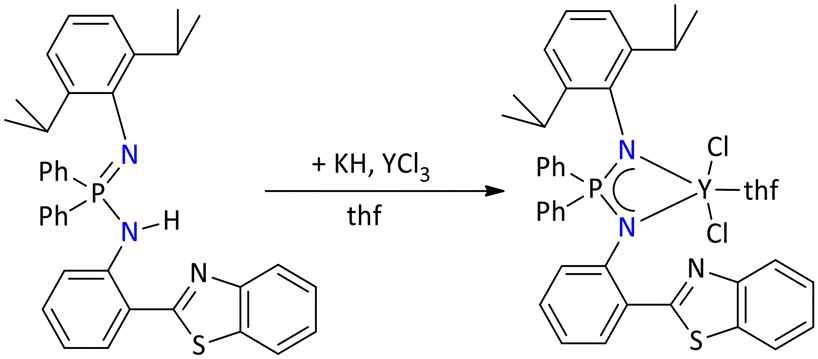 | ||
| Scheme 52 Synthesis of a luminescent yttrium complex.126 | ||
 | ||
| Scheme 53 Syntheses of lanthanide and actinide iminophosphonamide complexes.114 | ||
3 Enantiopure iminophosphonamide metal complexes
As discussed in the above sections, the coordination chemistry dealing with nonchiral iminophosphonamide is well explored. In contrast to this, few reports of chiral iminophosphonamide are known in the literature.In this context, Hill's group reported the DACH (DACH = trans-1,2-diaminocyclohexane) bis(iminophosphonamide)-bridged ligands in 2007.121 The DACH substituted ligands were synthesized by deprotonating the trans-1,2-diaminocyclohexane with 2 eq. of n-BuLi in toluene, first. Subsequent reaction with 2 eq. of Ph2PCl resulted in an exothermic reaction due to precipitation of LiCl. Further reaction with the appropriate azide afforded rac-[trans-1,2-C6H12{HNP(Ph2)N(Ar)}2] (Scheme 54). The 1H NMR spectrum of the ligands exhibited the expected signals including the NH resonances; additionally, the purity was confirmed by the appearance of single signals in their corresponding 31P{1H} NMR spectrum. This variably substituted rac-[trans-1,2-C6H12{HNP(Ph2)N(Ar)}2] iminophosphonamine at one of the nitrogen atoms was treated with 1 eq. of triethyl aluminum. This led to the formation of the monometallic aluminum complex rac-[trans-1,2-C6H12{NP(Ph2)N(Ar)}2AlEt] having a N4 chelate, whereas when the ligand was treated with 2 eq. of AlEt3 it furnished the bimetallic aluminum complex having N2 chelates.
 | ||
| Scheme 54 Synthetic route for rac-[trans-1,2-C6H12{HNP(Ph2)N(Ar)}2].121 | ||
Similarly, when the ligands rac-[trans-1,2-C6H12{HNP(Ph2)N(Ar)}2] were reacted with [M(NR2)3] (M = Y and Sc; R = SiMe3 and SiHMe2), it resulted in the formation of the N4 chelates of M = Y and Sc (Scheme 55). The bimetallic aluminum complexes rac-[trans-1,2-C6H12{NP(Ph2)N(Ar)}2(AlEt2)2] (Ar = 2,4,6-Me3C6H2, 2,6-Me2C6H3) displayed the single resonances in the 31P{1H} NMR spectrum in a very narrow range (i.e., 34 ppm to 37 ppm), suggesting the symmetric coordination environment around the metal centers.
 | ||
| Scheme 55 Syntheses of metal (M = Al, Sc, and Y) complexes of rac-[trans-1,2-C6H12{HNP(Ph2)N(Ar)}2].121 | ||
In contrast, the 31P{1H} monometallic aluminum complex rac-[trans-1,2-C6H12{NP(Ph2)N(Ar)}2AlEt] (Ar = 2,4,6-Me3C6H2 and 2,6-Me2C6H3) exhibited two peaks with an equal integral ratio based on integration. These signals were assigned by the Karplus relationship with the axial cyclohexane protons.
Further, these monometallic and bimetallic complexes were employed as catalysts for methylmethacrylate (MMA) polymerization at room temperature. The results showed that both mono metallic rac-[trans-1,2-C6H12{NP(Ph2)N(Ar)}2AlEt] (Ar = 2,4,6-Me3C6H2 and 2,6-Me2C6H3) and bimetallic aluminum complexes, rac-[trans-1,2-C6H12{NP(Ph2)N(Ar)AlEt2}2] (Ar = 2,4,6-Me3C6H2 and 2,6-Me2C6H3), require three-component Gibson initiators,127 (L2AlR/MAD/Ni(acac)2) (MAD = bis(2,6-di-tert-butyl-4-methylphenoxide)aluminum methyl) for active polymerization to yield a syndiotactic (>80%) pattern with broad polydispersity indices (∼2). In contrast, efficient catalysis with a mostly isotactic (>80%) pattern close to monodisperse was observed when the yttrium complexes, rac-[trans-1,2-C6H12{NP(Ph2)N(2,4,6-Me3C6H2)}2Y(NR2)] (R = SiMe3 and SiHMe2), were employed as catalysts.
Later in 2017, Guan's group has reported a variety of functionalized chiral iminophosphonamines, where the chiral centers were present at one nitrogen atom of the diamines (i.e., (1R,2R)-diaminocyclohexane or (1R,2R)-diphenylethylenediamine) (Fig. 6).128
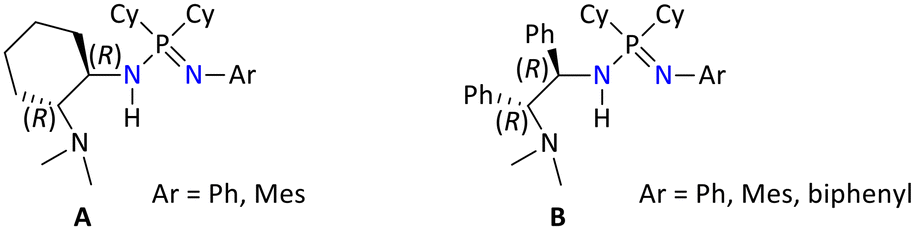 | ||
| Fig. 6 Variably substituted chiral iminophosphonamines. | ||
Further metal complexation of these ligands was accessed by treating the ligands with metal benzyl compounds of alkaline earth and rare earth metals (Scheme 56).
 | ||
| Scheme 56 Syntheses of alkaline and rare earth metal complexes of chiral iminophosphonamines.128 | ||
The 1H NMR spectra of the metal complexes confirmed complete deprotonation of the ligand. Further, the synthesized metal alkyl complexes bearing chiral iminophosphonamines were investigated as catalysts in the cross-dehydrogenative coupling of amines with prochiral silanes. The best catalytic activity was observed with the yttrium complex bearing a 1,2-diphenyl ethylenediamine moiety compared to the one with a 1,2-diamino cyclohexane moiety. This catalyst was further subjected to broad substrates of amine and prochiral silane, which showed excellent yield of the silylamines (Table 1).128 All of the isolated silylamines were found to have a negative specific optical rotation.
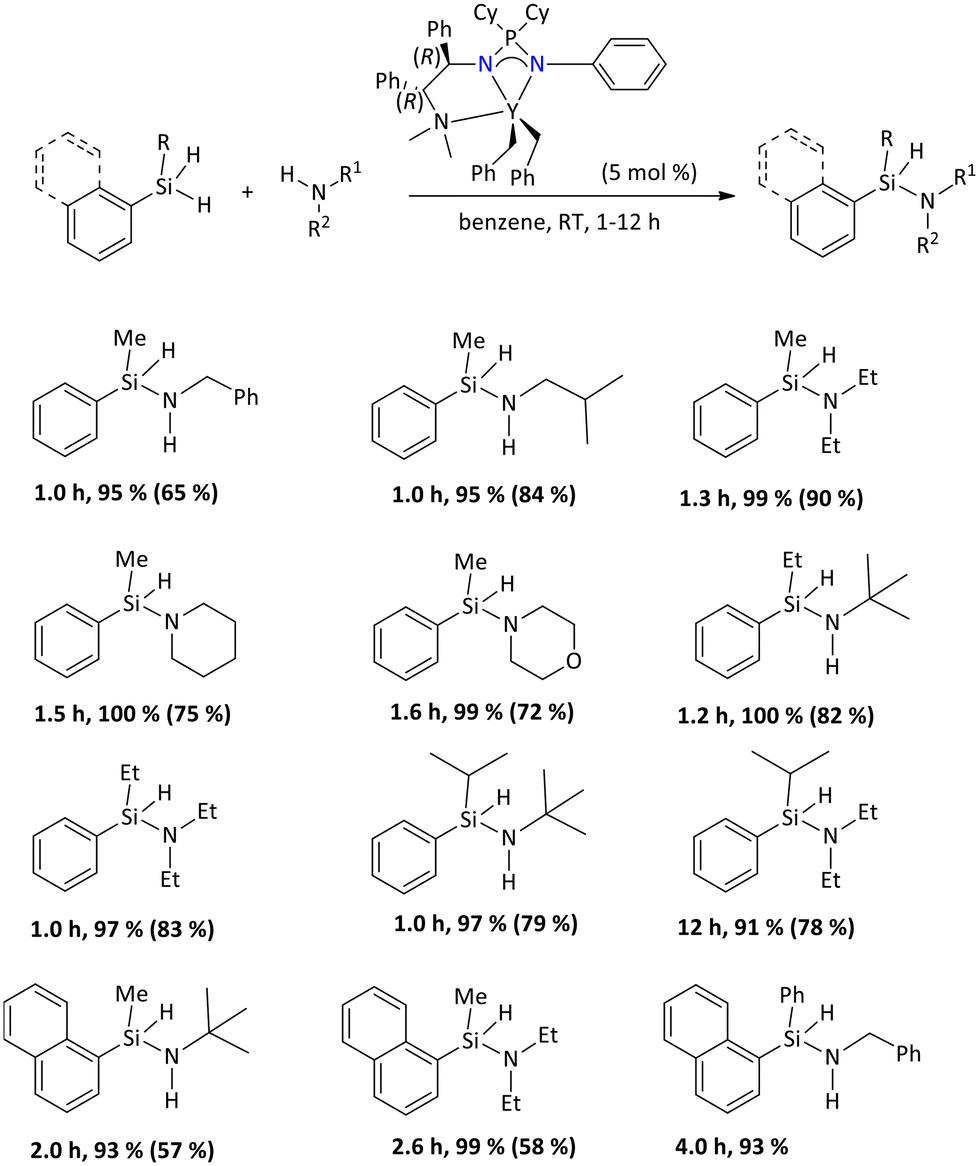 |
Due to the high moisture sensitivity of the isolated silylamines, the conventional chiral HPLC method to determine ee was unsuccessful. Therefore, to assess the ee of silylamines, the stable derivative of silylamines was isolated. In this regard, the silylamine-boron derivative was synthesized by direct deprotonation/boronation in a single pot (Scheme 57). The resulting chiral silylamine boron derivative was analyzed using chiral HPLC, which exhibited the best catalytic activity having 71% isolated yield and 23% ee when the yttrium complex bearing 1,2-diphenyl ethylenediamine at one end and biphenyl substituent at another nitrogen end of the iminophosphonamide was employed as the catalyst.
 | ||
| Scheme 57 Asymmetric chiral silamine catalysis using the yttrium benzyl complex.128 | ||
Further in 2019, Rufanov's group reported various C2-symmetric bis(iminophosphonamide) ligands.33 These ligands were synthesized by reacting the (R)-1,1′-binaphthyl-2,2′-diamine {(R)-Binam} with Ph2PCl with Et3N in DCM, resulting in the formation of (R)-N,N′-bis(diphenylphosphanyl)-2,2′-diamino-1,1′-binaphthyl [(R)-Binam-P]. Further, the Staudinger reaction was performed between the (R)-Binam-P and R′-N3 (R′ = tBu, Dip, and 1-Ad), which resulted in a series of C2-symmetric bis(iminophosphonamide) ligands [(R)-Binam(Ph2PN(H)R′)2] (R′ = tBu, Ad and Dip) (Scheme 58). The ligands were characterized by multinuclear NMR (1H, 31P, 13C) spectroscopy, EI-MS and elemental analysis, whereas the molecular structure of [(R)-Binam(Ph2PN(H)tBu)2] was determined by growing suitable crystals for X-ray diffraction in a concentrated solution of [(R)-Binam(Ph2PN(H)tBu)2] in DMSO. The ligand crystallized with 2 molecules of DMSO, forming N–H⋯O hydrogen bonds with the DMSO molecules.
 | ||
| Scheme 58 Synthesis of C2-symmetric bis(iminophosphonamide) ligands.33 | ||
To the best of our knowledge, the coordination chemistry of the [(R)-Binam(Ph2PN(H)R′)2] is yet unknown.
In contrast to the above-described chiral iminophosphonamines, where chirality was close to one nitrogen atom, the chirality centers close to both nitrogen atoms of the iminophosphonamines were largely uncommon. In this regard, P. Roesky's group reported in 2019 that the [Ph2P{N(R)CH(CH3)Ph}2] ligand has chiral centers at both ends of the ligand.129 The ligand was synthesized by performing a Staudinger reaction between (R)-α-methylbenzyl azide and HN(R-CHMePh)(PPh2). The constitution of the ligand was confirmed by NMR spectroscopy, IR spectroscopy, and elemental analysis, and its molecular structure was accessed by single-crystal X-ray diffraction.
When the ligand [Ph2P{HN(R)CH(CH3)Ph}{N(R)CH(CH3)Ph}] was deprotonated with appropriate alkali metal bases, it resulted in the formation of dimeric metal complexes, [Ph2P{N(R)CH(CH3)Ph}2M2] [M = Li, Na, K, and Rb]. Interestingly, when the ligand was treated with cesium precursors, it formed dimeric [Ph2P{N(R)CH(CH3)Ph}2Cs2] and 1D-polymeric [Ph2P{N(R)CH(CH3)Ph}Cs]n species in 1/1 ratio (Scheme 59). Complete deprotonation of the ligand was confirmed by the disappearance of the NH signal in the 1H NMR spectrum of the metal complexes. The 31P{1H} NMR spectra of all these metal complexes also exhibited single resonances in the range from δ = 12.6 to δ = 20.6 ppm, which is downfield shifted as compared to the ligand [Ph2P{HN(R)CH(CH3)Ph}{N(R)CH(CH3)Ph}] (δ = 2.7 ppm). Moreover, the molecular structures of these metal complexes were confirmed by X-ray diffraction, which revealed that the cesium complex adopts both dimeric and polymeric forms, whereas the rest of the metal complexes exist in dimeric forms. The photoluminescence (PL) investigations at variable temperatures of the ligand [Ph2P{N(R)CH(CH3)Ph}2] and all its alkali metal complexes [Ph2P{N(R)CH(CH3)Ph}M]2 [M = Li, Na, K, Rb, and Cs] revealed that the ligand is fluorescent in nature at all temperatures. In contrast, the alkali metal complexes showed phosphorescence at low temperatures (<100 K) and thermally activated delayed fluorescence (TADF) above ∼150 K with a PL quantum efficiency of up to 36% for [Ph2P{N(R)CH(CH3)Ph}2Na2].
 | ||
| Scheme 59 Syntheses of alkali metal complexes of [Ph2P{N(R)CH(CH3)Ph}].129 | ||
DFT calculations revealed the small S1/T1 energy gap for all the alkali metal complexes; further, the effective intersystem crossing S1/T1 energy states was due to the dimeric forms of these metal complexes.129
To understand the effect of the ligand substituent on the photophysical properties of the metal complexes, symmetrically [Ph2P{N(R)CH(CH3)Ph}2], [Ph2P{N(R)CH(CH3)naph}2] (naph = naphthyl)- and non-symmetrically [Ph2P{NDipp}{N(R)CH(CH3)Ph}] (Dipp = 2,6-iPr2C6H3)-substituted chiral iminophosphonamines were synthesized. The new symmetric ligand [Ph2P{N(R)CH(CH3)naph}2] and the non-symmetric [Ph2P{NDipp}{N(R)CH(CH3)Ph}] ligand were obtained by reacting the iminophosphorane with corresponding azides.130
Performing transamination between the [Ca{N(SiMe3)2}(thf)2] and above-mentioned symmetric and non-symmetric ligands afforded the homoleptic calcium complexes, [{Ph2P{N(R)CH(CH3)Ph}2}2Ca], [{Ph2P{N(R)CH(CH3)naph}2}2Ca] and [{Ph2P(NDipp){N(R)CH(CH3)Ph}}2Ca] (Scheme 60). The PL study of these metal complexes in solid state at various temperatures exhibited blue green PL. The emission of [{Ph2P{N(R)CH(CH3)naph}2}2Ca] was assigned to the fluorescence of naphthyl rings, whereas the emission of complexes [{Ph2P{N(R)CH(CH3)Ph}2}2Ca] and [{Ph2P(NDipp){N(R)CH(CH3)Ph}}2Ca] exhibited phosphorescence at low temperatures and TADF at elevated temperatures. Different PL lifetimes suggested that the ligand substituents played a vital role in the PL of these calcium complexes. Moreover, all these homoleptic calcium complexes showed excellent catalytic activity in the hydroboration of ketones at room temperature.130 Further the coordination chemistry of the [Ph2P{N(R)CH(CH3)Ph}2] ligand was explored with group 13 metal centers.131 In this regard, when the ligand was treated with LiAlH4, it resulted in the formation of [Ph2P{N(R)CH(CH3)Ph}2AlH]. Treating the lithium salt of the ligand [Ph2P{N(R)CH(CH3)Ph}Li]2 with MCl3 (M = Al and Ga) resulted in the formation of corresponding monochloride complexes [Ph2P{N(R)CH(CH3)Ph}2MCl] (M = Al and Ga). Moreover, the halide abstraction of the monochloride complexes afforded corresponding tetracoordinated cationic complexes [{Ph2P{N(R)CH(CH3)Ph}2}2Al]+[GaCl4]− and [{Ph2P{N(R)CH(CH3)Ph}2}2Ga]+[AlCl4]− (Fig. 7).
 | ||
| Scheme 60 Syntheses of chiral homoleptic calcium complexes.130 | ||
 | ||
| Fig. 7 Monometallic aluminum and gallium complexes of the [Ph2P{N(R)CH(CH3)Ph}2] ligand.131 | ||
All these complexes were thoroughly characterized by multinuclear NMR and IR spectroscopy, elemental analysis, and X-ray diffraction.131
The effect of substituent variation in the iminophosphonamide metal complexes on photoluminescence (PL) was further investigated with d10 configured metal centers. In this regard, various Cu(I) and Zn(II) metal complexes of iminophosphonamides were synthesized (Scheme 61). The binuclear Cu(I) complexes [Ph2P{(NR)(NR′)}2Cu2] (R = R′ = (R)CH(CH3)Ph and R = R′ = (R)CH(CH3)naph) were synthesized by a salt metathesis reaction between CuCl and lithium and the potassium salt of the iminophosphonamides [Ph2P{(NR)(NR′)}2M2] (M = Li, for R = R′ = (R)CH(CH3)Ph and M = K for R = R′ = (R)CH(CH3)naph). Similar the one-pot reaction of CuCl, K(NSiMe3)2 and the iminophosphonamines {Ph2P{(NHR)(NR′)} (R = (R)CH(CH3)Ph; R′ = 2,6-iPr2C6H4 and R = R′ = 2,6-iPr2C6H4) in equivalent ratios resulted in the formation of corresponding binuclear Cu(I) complexes [Ph2P{(NR)(NR′)}2Cu2] (R = (R)CH(CH3)Ph; R′ = 2,6-iPr2C6H4 and R = R′ = 2,6-iPr2C6H4). Furthermore, when the deprotonation of the iminophosphonamine {Ph2P(NHR)(NR′)} (R = R′ = (R)CH(CH3)Ph; and R = (R)CH(CH3)Ph; R′ = 2,6-iPr2C6H4) was performed with ZnR2 (R = Me and Ph), it resulted in the formation of corresponding homoleptic complexes, [Ph2P{(NR)(NR′)}2Zn] (R = R′ = (R)CH(CH3)Ph and R = (R)CH(CH3)Ph; R′ = 2,6-iPr2C6H4). However, the deprotonation reaction between Ph2P{(2,6-iPr2C6H4N)2}H and ZnPh2 resulted in the formation of a heteroleptic complex, [Ph2P{(2,6-iPr2C6H4N)2}ZnPh], due to the steric crowding from the Dipp (Dipp = 2,6-iPr2C6H4) group. The formation of all these complexes was proven by various analytical techniques such as single-crystal X-ray diffraction, multinuclear NMR spectroscopy, elemental analysis and IR spectroscopy. The solid-state PL measurement of these metal complexes in the temperature interval 5–295 K revealed that depending on the ligand substituents, the PL ranged from prompt fluorescence to TADF to long-lived phosphorescence. Additionally, step-scan FTIR spectroscopy was performed for [{Ph2P(2,6-iPr2C6H4N)2}Cu]2 to characterize the excited triplet state on the microscopic time scale.132
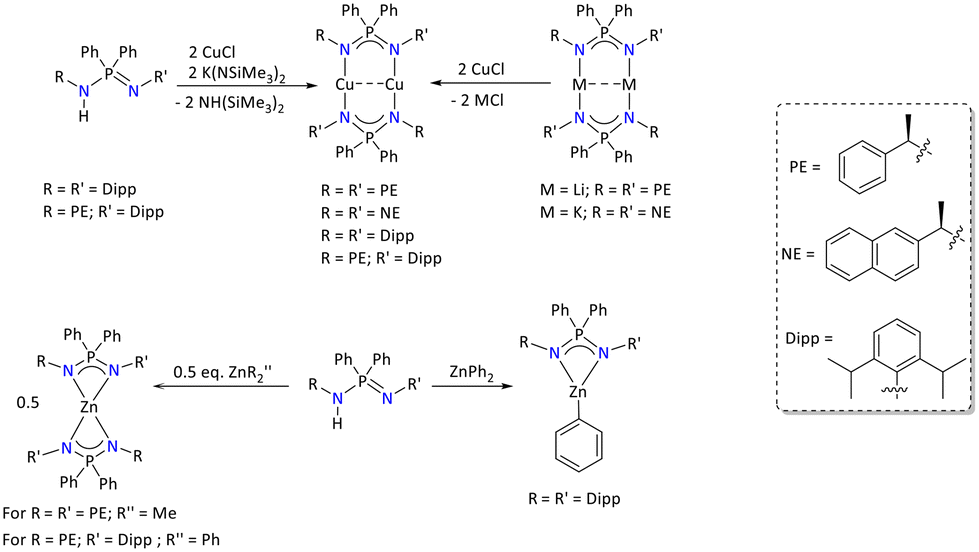 | ||
| Scheme 61 Syntheses of the selected Cu(I) and Zn(II) iminophosphonamide complexes for photoluminescence study.132 | ||
Moreover, photoluminescence studies were further extended to the lanthanide metal complexes. Therefore, chiral homoleptic lanthanide complexes [{Ph2P{N(R)CH(CH3)Ph}2}3Ln] (Ln = Y, La, Sm, Tb, and Lu) were synthesized by performing a salt metathesis reaction between [{Ph2P{N(R)CH(CH3)Ph}2}K]2 and LnCl3 in 1.5/1 molar ratio (Scheme 62).133 The Sm(III) complex exhibited both ligand- and metal-centered PL depending on the excitation wavelength. For the PL of the Tb(III) complex a quantum yield of 15% in the solid state was observed.
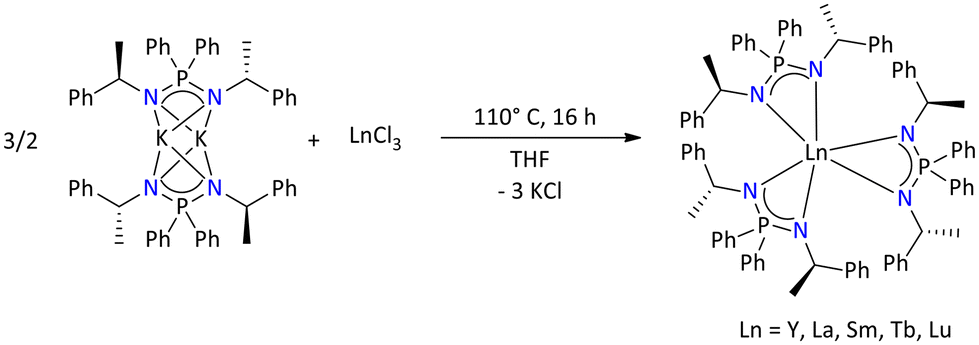 | ||
| Scheme 62 Syntheses of chiral homoleptic lanthanide complexes.133 | ||
Very recently, the group 4 (i.e., Zr and Hf) metal complexes of chiral iminophosphonamide ligands [Ph2P{N(R)CH(CH3)Ph}2] and [Ph2P{NDipp}{N(R)CH(CH3)Ph}] (Dipp = 2,6-iPr2C6H3) have been reported.134 These metal complexes were synthesized by adopting the amine elimination method between [M(NMe2)4] (M = Zr and Hf) and [Ph2P{HN(R)CH(CH3)Ph}2] and [Ph2P{HNDipp}{N(R)CH(CH3)Ph}] in equimolar ratios (Scheme 63). The synthesized metal complexes were extensively characterized using several spectroscopic and analytical techniques. The structural determination of the complexes was carried out by using single-crystal X-ray diffraction.
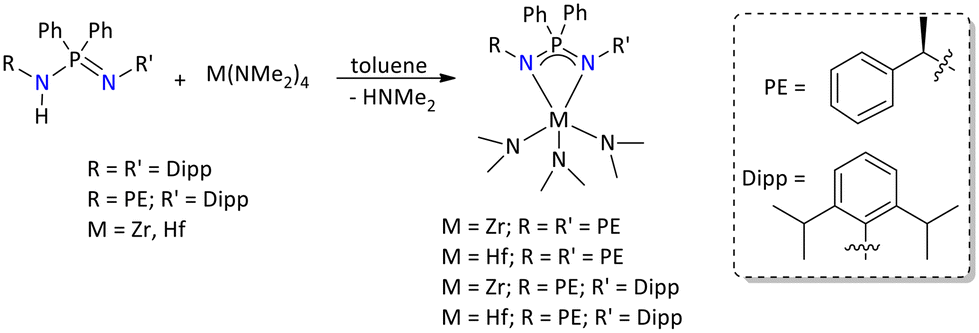 | ||
| Scheme 63 Syntheses of chiral zirconium and hafnium metal complexes.134 | ||
Furthermore, the catalytic efficiency of the zirconium complex of the symmetric ligand [Ph2P{(NR)(NR′)}Zr(NMe2)3] (R = R′ = (R)CH(CH3)Ph) and the hafnium complex of the unsymmetrical ligand [Ph2P{(NR)(NR′)}Hf(NMe2)3] (R = (R)CH(CH3)Ph and R′ = Dipp) was explored towards intramolecular hydroamination and hydroboration reactions, respectively.
4. Conclusions
In conclusion, we present an account of coordination compounds of nonchiral iminophosphonamides with s-block, p-block, transition, and f-block metals. The broad application of this ligand in wide areas of coordination chemistry is caused by various advantages: (i) The substituents on the nitrogen atoms can be varied by a wide range of functional groups including enantiomeric pure substituents. (ii) Due to the stepwise synthesis of iminophosphonamides, asymmetric substitution patterns can easily be realized. This allows for, e.g., a convenient access to bridged bis(iminophosphonamides), which may act as chelating ligands. (iii) The steric demand of the iminophosphonamide can be adjusted due to the flexible variation of the substituents on the nitrogen atoms. (iv) By having a phosphorous atom in the backbone of the ligand, the reaction can be monitored in situ by 31P NMR spectroscopy. This makes iminophosphonamide superior to other N,N′-chelating ligands such as amidinates.The review demonstrate that the iminophosphonamides differ more significantly from the amidinates than their formal similarity would suggest. However, the chemistry of iminophosphonamides in comparison to amidinates is still significant less developed. With this review, we hope to inspire chemists considering iminophosphonamides as suitable ligands for their research.
Author contributions
Both the authors did the literature research and wrote parts of the manuscript. PWR originated the idea and supervised the work.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
BG thanks the Central University of Rajasthan for support. PWR was supported by the German Research Foundation (DFG) through grant no. 546228048 (RO 2008/25-1).References
- T. Kottke and D. Stalke, Design of Self-Adapting N-Heteroaromatic Substituted Claw Ligands as E−/M+(E = P-Block Element, M = Main-Group Metal) Charge Spacers, Chem. Ber., 1997, 130, 1365–1374 CrossRef CAS.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, The Chemistry of β-Diketiminatometal Complexes, Chem. Rev., 2002, 102, 3031–3066 CrossRef CAS PubMed.
- P. W. Roesky, The co-ordination chemistry of aminotroponiminates, Chem. Soc. Rev., 2000, 29, 335–345 RSC.
- A. Jain, H. Karmakar, P. W. Roesky and T. K. Panda, Role of Bis(phosphinimino)methanides as Universal Ligands in the Coordination Sphere of Metals across the Periodic Table, Chem. Rev., 2023, 123, 13323–13373 Search PubMed.
- T. K. Panda and P. W. Roesky, Main-group and transition-metal complexes of bis(phosphinimino)methanides, Chem. Soc. Rev., 2009, 38, 2782–2804 RSC.
- P. C. Junk and M. L. Cole, Alkali-metal bis(aryl)formamidinates: a study of coordinative versatility, Chem. Commun., 2007, 1579–1590 RSC.
- F. T. Edelmann, Lanthanide amidinates and guanidinates: from laboratory curiosities to efficient homogeneous catalysts and precursors for rare-earth oxide thin films, Chem. Soc. Rev., 2009, 38, 2253–2268 RSC.
- S. T. Barry, Amidinates, guanidinates and iminopyrrolidinates: Understanding precursor thermolysis to design a better ligand, Coord. Chem. Rev., 2013, 257, 3192–3201 CrossRef CAS.
- A. A. Trifonov, Guanidinate and amidopyridinate rare-earth complexes: Towards highly reactive alkyl and hydrido species, Coord. Chem. Rev., 2010, 254, 1327–1347 CrossRef CAS.
- T. Chlupatý and A. Růžička, Hybrid amidinates and guanidinates of main group metals, Coord. Chem. Rev., 2016, 314, 103–113 CrossRef.
- P. J. Bailey and S. Pace, The coordination chemistry of guanidines and guanidinates, Coord. Chem. Rev., 2001, 214, 91–141 CrossRef CAS.
- F. T. Edelmann, Lanthanide amidinates and guanidinates in catalysis and materials science: a continuing success story, Chem. Soc. Rev., 2012, 41, 7657–7672 RSC.
- C. Fedorchuk, M. Copsey and T. Chivers, The coordination chemistry of boraamidinate ligands, Coord. Chem. Rev., 2007, 251, 897–924 CrossRef CAS.
- S. Harder, Syntheses and structures of bora-amidinate lanthanide(III) complexes, Dalton Trans., 2010, 39, 6677–6681 RSC.
- M. M. Meinholz, S. K. Pandey, S. M. Deuerlein and D. Stalke, Access to new Janus head ligands: linking sulfur diimides and phosphanes for hemilabile tripodal scorpionates, Dalton Trans., 2011, 40, 1662–1671 RSC.
- F. Pauer, J. Rocha and D. Stalke, Synthesis and crystal structure of bis(12-crown-4)lithium bis[N,N′-bis(trimethylsilyl)benzenesulphinamidino]lithiate(1−); the first observation of three different lithium-7 environments in high-resolution solid-state NMR spectroscopy, J. Chem. Soc., Chem. Commun., 1991, 1477–1479 RSC.
- R. Fleischer, B. Walfort, A. Gbureck, P. Scholz, W. Kiefer and D. Stalke, Raman Spectroscopic Investigation and Coordination Behavior of the Polyimido SVI Anions [RS(NR)3]− and [S(NR)4]2−, Chem. – Eur. J., 1998, 4, 2266–2274 CrossRef CAS.
- F. T. Edelmann, F. Knösel, F. Pauer, D. Stalke and W. Bauer, Solid-state and solution structures of three lithiumsulfinimidamides: Identification of two distinct structural types, J. Organomet. Chem., 1992, 438, 1–10 CrossRef CAS.
- S.-O. Hauber, F. Lissner, G. B. Deacon and M. Niemeyer, Stabilization of Aryl–Calcium, –Strontium, and –Barium Compounds by Designed Steric and π-Bonding Encapsulation, Angew. Chem., Int. Ed., 2005, 44, 5871–5875 CrossRef CAS PubMed.
- N. Nimitsiriwat, V. C. Gibson, E. L. Marshall, P. Takolpuckdee, A. K. Tomov, A. J. P. White, D. J. Williams, M. R. J. Elsegood and S. H. Dale, Mono- versus Bis-chelate Formation in Triazenide and Amidinate Complexes of Magnesium and Zinc, Inorg. Chem., 2007, 46, 9988–9997 CrossRef CAS PubMed.
- S.-O. Hauber and M. Niemeyer, Stabilization of Unsolvated Europium and Ytterbium Pentafluorophenyls by π-Bonding Encapsulation through a Sterically Crowded Triazenido Ligand, Inorg. Chem., 2005, 44, 8644–8646 CrossRef CAS PubMed.
- S. G. Alexander, M. L. Cole, C. M. Forsyth, S. K. Furfari and K. Konstas, Bulky triazenide complexes of alumino- and gallohydrides, Dalton Trans., 2009, 2326–2336 RSC.
- C. GuhaRoy, R. J. Butcher and S. Bhattacharya, Rhodium complexes of 1,3-diaryltriazenes: Usual coordination, N–H bond activation and, N–N and C–N bond cleavage, J. Organomet. Chem., 2008, 693, 3923–3931 Search PubMed.
- A. Steiner, S. Zacchini and P. I. Richards, From neutral iminophosphoranes to multianionic phosphazenates. The coordination chemistry of imino–aza-P(V) ligands, Coord. Chem. Rev., 2002, 227, 193–216 Search PubMed.
- M. Witt and H. W. Roesky, Transition and Main Group Metals in Cyclic Phosphazanes and Phosphazenes, Chem. Rev., 1994, 94, 1163–1181 CrossRef CAS.
- B. Prashanth and S. Singh, A new bulky iminophosphonamide as an N,N′-chelating ligand: synthesis and structural characterization of heteroleptic group 13 element complexes, Dalton Trans., 2014, 43, 16880–16888 RSC.
- T. Y. A. Peganova, I. S. Sinopalnikova, A. S. Peregudov, I. V. Fedyanin, A. Demonceau, N. A. Ustynyuk and A. M. Kalsin, The half-sandwich 18- and 16-electron arene ruthenium iminophosphonamide complexes, Dalton Trans., 2016, 45, 17030–17041 RSC.
- T. A. Peganova and A. M. Kalsin, Synthesis of Nonsymmetric Iminophosphonamines by Kirsanov Condensation, Synthesis, 2020, 433–440 CAS.
- O. J. Scherer and G. Schieder, Metallorganische Aminophosphine und Phosphinimine, Chem. Ber., 1968, 101, 4184–4198 CrossRef CAS.
- H. Staudinger and J. Meyer, Über neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine, Helv. Chim. Acta, 1919, 2, 635–646 CrossRef CAS.
- Y. G. Gololobov and L. F. Kasukhin, Recent advances in the staudinger reaction, Tetrahedron, 1992, 48, 1353–1406 CrossRef CAS.
- L. P. Spencer, R. Altwer, P. Wei, L. Gelmini, J. Gauld and D. W. Stephan, Pyridine− and Imidazole−Phosphinimine Bidentate Ligand Complexes: Considerations for Ethylene Oligomerization Catalysts, Organometallics, 2003, 22, 3841–3854 Search PubMed.
- K. A. Rufanov, I. Y. Titov, A. R. Petrov, K. Harms and J. Sundermeyer, Synthesis of Binam-P Derived C2-Symmetric bis-Iminophosphonamide Ligands. Molecular Structure of [(R)-Binam(Ph2PN(H)tBu)2], Z. Anorg. Allg. Chem., 2019, 645, 559–563 Search PubMed.
- J. Barker and M. Kilner, The coordination chemistry of the amidine ligand, Coord. Chem. Rev., 1994, 133, 219–300 CrossRef CAS.
- F. T. Edelmann, N-silylated benzamidines: versatile building blocks in main group and coordination chemistry, Coord. Chem. Rev., 1994, 137, 403–481 CrossRef.
- M. R. Gyton, A. R. Leverett, M. L. Cole and A. I. McKay, Bulky bis(aryl)triazenides: just aspiring amidinates? A structural and spectroscopic study, Dalton Trans., 2020, 49, 5653–5661 RSC.
- B. Goswami, Enantiopure Iminophosphonamide Complexes: Synthesis, Photoluminescence and Catalysis, Cuvillier Verlag, 2020 Search PubMed.
- A. L. Hawley and A. Stasch, Structural Diversity in Sterically Demanding Diiminophosphinato Alkali Metal Complexes, Eur. J. Inorg. Chem., 2015, 258–270 CrossRef CAS.
- A. Stasch, Synthesis, Structure, and Reactivity of a Dimeric Zinc(I) Compound Stabilized by a Sterically Demanding Diiminophosphinate Ligand, Chem. – Eur. J., 2012, 18, 15105–15112 CrossRef CAS PubMed.
- B. Prashanth and S. Singh, Bulky iminophosphonamines for N–P–N coordination: Synthesis and structural characterization of lithium iminophosphonamides and homoleptic bis-chelates of Co(II), Ni(II) and Cu(II), J. Chem. Sci., 2015, 127, 315–325 Search PubMed.
- A. Steiner and D. Stalke, Unexpected coordination of aminoiminophosphoranate ligands with alkali metals, Inorg. Chem., 1993, 32, 1977–1981 CrossRef CAS.
- R. Fleischer and D. Stalke, Syntheses and Structures of [(THF)nM{(NSiMe3)2PPh2}2] Complexes (M = Be, Mg, Ca, Sr, Ba; n = 0–2): Deviation of Alkaline Earth Metal Cations from the Plane of an Anionic Ligand, Inorg. Chem., 1997, 36, 2413–2419 CrossRef CAS PubMed.
- A. Stasch, Synthesis of a Dimeric Magnesium(I) Compound by an MgI/MgII Redox Reaction, Angew. Chem., Int. Ed., 2014, 53, 10200–10203 CrossRef CAS PubMed.
- C. Jones and A. Stasch, in Alkaline-Earth Metal Compounds: Oddities and Applications, ed. S. Harder, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 73–101 Search PubMed.
- A. Stasch and C. Jones, Stable dimeric magnesium(I) compounds: from chemical landmarks to versatile reagents, Dalton Trans., 2011, 40, 5659–5672 RSC.
- M. Westerhausen, Molecular Magnesium(I) Compounds: From Curiosity to Kudos, Angew. Chem., Int. Ed., 2008, 47, 2185–2187 Search PubMed.
- S. R. Lawrence, D. B. Cordes, A. M. Z. Slawin and A. Stasch, Mechanistic insights of anionic ligand exchange and fullerene reduction with magnesium(I) compounds, Dalton Trans., 2019, 48, 16936–16942 Search PubMed.
- S. Wingerter, M. Pfeiffer, A. Murso, C. Lustig, T. Stey, V. Chandrasekhar and D. Stalke, Phosphorus-Based Ambidentate Chelating Ligands: Pyridyl-N− and Imido-N−Metal Coordination in the Py2P(NSiMe3)2 Anion, J. Am. Chem. Soc., 2001, 123, 1381–1388 CrossRef CAS.
- B. Prashanth, N. K. Srungavruksham and S. Singh, Mononuclear Neutral Boron Hydrides Affordable as [N,N′] Chelates of Iminophosphonamides, ChemistrySelect, 2016, 1, 3601–3606 CrossRef CAS.
- H. Schmidbaur, K. Schwirten and H.-H. Pickel, Kleine anorganische Ringe, II. Ein cyclisches Alumophosphazan und seine Gallium- und Indiumanalogen, Chem. Ber., 1969, 102, 564–567 CrossRef CAS.
- B. Nekoueishahraki, H. W. Roesky, G. Schwab, D. Stern and D. Stalke, Synthesis and Structural Characterization of Aluminum Iminophosphonamide Complexes, Inorg. Chem., 2009, 48, 9174–9179 CrossRef CAS PubMed.
- H. R. Allcock, in Phosphorus-nitrogen Compounds, ed. H. R. Allcock, Academic Press, 1972, pp. 385–395 Search PubMed.
- J. Vrána, R. Jambor, A. Růžička, M. Alonso, F. De Proft and L. Dostál, Reactivity of Bis(organoamino)phosphanes with Aluminum(III) Compounds: Straightforward Access to Diiminophosphinates by Means of Hydrogen-Atom Migration – An Experimental and Theoretical Study, Eur. J. Inorg. Chem., 2014, 2014, 5193–5203 Search PubMed.
- K. Nakaya, A. Ishii and N. Nakata, Aluminum(III) di- and monochlorides incorporating an N,N’-chelating iminophosphonamide ligand: synthesis and structures, Mendeleev Commun., 2022, 32, 71–73 CrossRef CAS.
- A. L. Hawley, C. A. Ohlin, L. Fohlmeister and A. Stasch, Heavier Group 13 Metal(I) Heterocycles Stabilized by Sterically Demanding Diiminophosphinates: A Structurally Characterized Monomer–Dimer Pair For Gallium, Chem. – Eur. J., 2017, 23, 447–455 CrossRef CAS PubMed.
- D. H. Harris and M. F. Lappert, Monomeric, volatile bivalent amides of group IV elements, M(NR12)2 and M(NR1R2)2(M=Ge, Sn, or Pb; R1=Me3Si, R2=Me3C), J. Chem. Soc., Chem. Commun., 1974, 895–896 RSC.
- P. J. Davidson, D. H. Harris and M. F. Lappert, Subvalent Group 4B metal alkyls and amides. Part I. The synthesis and physical properties of kinetically stable bis[bis(trimethysilyl)methyl]-germanium(II), -tin(II), and -lead(II), J. Chem. Soc., Dalton Trans., 1976, 2268–2274 Search PubMed.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Stable Heavier Carbene Analogues, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed.
- W.-P. Leung, K.-W. Kan and K.-H. Chong, Reactions of some organogermanium(II) chlorides, Coord. Chem. Rev., 2007, 251, 2253–2265 CrossRef CAS.
- O. Kühl, N-heterocyclic germylenes and related compounds, Coord. Chem. Rev., 2004, 248, 411–427 CrossRef.
- C.-W. So, H. W. Roesky, J. Magull and R. B. Oswald, Synthesis and Characterization of [PhC(NtBu)2]SiCl: A Stable Monomeric Chlorosilylene, Angew. Chem. Int. Ed., 2006, 45, 3948–3950 CrossRef CAS PubMed.
- S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, High Yield Access to Silylene RSiCl (R = PhC(NtBu)2) and Its Reactivity toward Alkyne: Synthesis of Stable Disilacyclobutene, J. Am. Chem. Soc., 2010, 132, 1123–1126 CrossRef CAS PubMed.
- S. Nagendran, S. S. Sen, H. W. Roesky, D. Koley, H. Grubmüller, A. Pal and R. Herbst-Irmer, RGe(I)Ge(I)R Compound (R = PhC(NtBu)2) with a Ge−Ge Single Bond and a Comparison with the Gauche Conformation of Hydrazine, Organometallics, 2008, 27, 5459–5463 CrossRef CAS.
- L. Álvarez-Rodríguez, J. A. Cabeza, P. García-Álvarez and D. Polo, The transition-metal chemistry of amidinatosilylenes, -germylenes and -stannylenes, Coord. Chem. Rev., 2015, 300, 1–28 CrossRef.
- B. Prashanth and S. Singh, Concise access to iminophosphonamide stabilized heteroleptic germylenes: chemical reactivity and structural investigation, Dalton Trans., 2016, 45, 6079–6087 RSC.
- O. A. Brusylovets, O. V. Vinichenko, A. I. Brusilovets, T. Lis, E. Bonnefille, S. Mazières and C. Couret, New substituents for the stabilization of low-coordinate germanium species: Use in access to diazogermylenes, Polyhedron, 2010, 29, 3269–3276 CrossRef CAS.
- U. Kilimann, M. Noltemeyer and F. T. Edelmann, Viergliedrige anorganische ringsysteme des zweiwertigen Zinns und Bleis: Synthese und struktur von chelatstabilisierten Stannylenen und Plumbylenen, J. Organomet. Chem., 1993, 443, 33–42 CrossRef.
- S. R. Foley, C. Bensimon and D. S. Richeson, Facile Formation of Rare Terminal Chalcogenido Germanium Complexes with Alkylamidinates as Supporting Ligands, J. Am. Chem. Soc., 1997, 119, 10359–10363 CrossRef CAS.
- E. Bonnefille, S. Mazières, N. Saffon and C. Couret, Reactivity of a germa-alkyne: Evidence for a germanone intermediate in the hydrolysis and alcoholysis processes, J. Organomet. Chem., 2009, 694, 2246–2251 CrossRef CAS.
- E. Carl and D. Stalke, Germanium(II) and Tin(II) Halide Complexes Containing the Triimido Sulfur(VI) Phosphanyl Ligand, Eur. J. Inorg. Chem., 2015, 2015, 2052–2056 CrossRef CAS.
- S. Takahashi, J. Sekiguchi, A. Ishii and N. Nakata, An Iminophosphonamido-Chlorosilylene as a Strong σ-Donating NHSi Ligand: Synthesis and Coordination Chemistry, Angew. Chem., Int. Ed., 2021, 60, 4055–4059 CrossRef CAS PubMed.
- J. Sekiguchi, Y. Kazama, A. Ishii and N. Nakata, Synthesis of a homoleptic tris(silylene)–palladium(0) complex and a silylyne-bridged tetranuclear palladium cluster, Chem. Commun., 2023, 59, 9844–9847 RSC.
- S. Takahashi, A. Ishii and N. Nakata, Formation of silaimines from a sterically demanding iminophosphonamido chlorosilylene via intramolecular N–P bond cleavage, Chem. Commun., 2021, 57, 6728–6731 Search PubMed.
- S. Takahashi, J. Sekiguchi, K. Nakaya, A. Ishii and N. Nakata, Halogen-Exchange Reactions of Iminophosphonamido-Chlorosilylenes with Alkali Halides: Convenient Synthesis of Heavier Halosilylenes, Inorg. Chem., 2022, 61, 7266–7273 Search PubMed.
- S. Takahashi, K. Nakaya, A. Ishii and N. Nakata, [N,N′-Di-tert-butyl-P,P-diphenylphosphinimidic Amidato-κN,κN′]chlorosilicon-κSi-tetracarbonyliron, Molbank, 2022, 2022, M1433 Search PubMed.
- K. Nakaya, S. Takahashi, A. Ishii, K. Boonpalit, P. Surawatanawong and N. Nakata, Hydroboration of carbonyls and imines by an iminophosphonamido tin(II) precatalyst, Dalton Trans., 2021, 50, 14810–14819 RSC.
- K. Nakaya, S. Takahashi, A. Ishii and N. Nakata, Iminophosphonamido-Supported Plumbylenes and Plumbyliumylidenes: Synthesis and Properties, Inorg. Chem., 2022, 61, 15510–15519 CrossRef CAS PubMed.
- S. Takahashi, S. Kamiyama, A. Ishii and N. Nakata, Syntheses of Iminophosphomamido Chlorogermylenes and Their Complexation with a Rhodium(I) Complex, Chem. – Asian J., 2024, 19, e202300968 CrossRef CAS PubMed.
- J. Xu, S. Pan, S. Yao, C. Lorent, C. Teutloff, Z. Zhang, J. Fan, A. Molino, K. B. Krause, J. Schmidt, R. Bittl, C. Limberg, L. Zhao, G. Frenking and M. Driess, Stabilizing Monoatomic Two-Coordinate Bismuth(I) and Bismuth(II) Using a Redox Noninnocent Bis(germylene) Ligand, J. Am. Chem. Soc., 2024, 146, 6025–6036 CrossRef CAS PubMed.
- S. Collins, Polymerization catalysis with transition metal amidinate and related complexes, Coord. Chem. Rev., 2011, 255, 118–138 CrossRef CAS.
- H. Nagashima, H. Kondo, T. Hayashida, Y. Yamaguchi, M. Gondo, S. Masuda, K. Miyazaki, K. Matsubara and K. Kirchner, Chemistry of coordinatively unsaturated organoruthenium amidinates as entry to homogeneous catalysis, Coord. Chem. Rev., 2003, 245, 177–190 CrossRef CAS.
- W. Keim, R. Appel, A. Storeck, C. Krüger and R. Goddard, Novel Nickel- and Palladium-Complexes with Aminobis(imino)phosphorane Ligands for the Polymerization of Ethylene, Angew. Chem., Int. Ed. Engl., 1981, 20, 116–117 CrossRef.
- A. Dworak, W. Walach and B. Trzebicka, Cationic polymerization of glycidol. Polymer structure and polymerization mechanism, Macromol. Chem. Phys., 1995, 196, 1963–1970 CrossRef CAS.
- B. F. Straub, F. Rominger and P. Hofmann, A Fluxional α-Carbonyl Diazoalkane Complex of Copper Relevant to Catalytic Cyclopropanation, Organometallics, 2000, 19, 4305–4309 CrossRef CAS.
- B. F. Straub and P. Hofmann, Copper(I) Carbenes: The Synthesis of Active Intermediates in Copper-Catalyzed Cyclopropanation, Angew. Chem., Int. Ed., 2001, 40, 1288–1290 CrossRef CAS PubMed.
- K. Albahily, S. Licciulli, S. Gambarotta, I. Korobkov, R. Chevalier, K. Schuhen and R. Duchateau, Highly Active Ethylene Oligomerization Catalysts, Organometallics, 2011, 30, 3346–3352 Search PubMed.
- R. Vollmerhaus, P. Shao, N. J. Taylor and S. Collins, Synthesis of and Ethylene Polymerization Using Iminophosphonamide Complexes of Group 4, Organometallics, 1999, 18, 2731–2733 CrossRef CAS.
- R. Vollmerhaus, R. Tomaszewski, P. Shao, N. J. Taylor, K. J. Wiacek, S. P. Lewis, A. Al-Humydi and S. Collins, Synthesis and Structure of Group 4 Iminophosphonamide Complexes, Organometallics, 2005, 24, 494–507 Search PubMed.
- R. Tomaszewski, R. Vollmerhaus, A. Al-Humydi, Q. Wang, N. J. Taylor and S. Collins, Ethylene and 1-hexene polymerization using zirconium iminophosphonamide complexes, Can. J. Chem., 2006, 84, 214–224 CrossRef CAS.
- C. Qi and S. Zhang, Titanium and zirconium complexes with aminoiminophosphorane ligands, Appl. Organomet. Chem., 2006, 20, 70–73 CrossRef CAS.
- E. Rivard, A. R. McWilliams, A. J. Lough and I. Manners, Synthesis and characterization of perhalogenated diazaphosphametalletidines containing transition metals from group 4 and 5, J. Chem. Soc., Dalton Trans., 2002, 2173–2179 RSC.
- K. Albahily, V. Fomitcheva, S. Gambarotta, I. Korobkov, M. Murugesu and S. I. Gorelsky, Preparation and Characterization of a Reduced Chromium Complex via Vinyl Oxidative Coupling: Formation of a Self-Activating Catalyst for Selective Ethylene Trimerization, J. Am. Chem. Soc., 2011, 133, 6380–6387 Search PubMed.
- Y. V. Fedotova, A. N. Kornev, V. V. Sushev, Y. A. Kursky, T. G. Mushtina, N. P. Makarenko, G. K. Fukin, G. A. Abakumov, L. N. Zakharov and A. L. Rheingold, Phosphinohydrazines and phosphinohydrazides M(–N(R)–N(R)–PPh2)n of some transition and main group metals: synthesis and characterization: Rearrangement of Ph2P–NR–NR– ligands into aminoiminophosphorane, RNPPh2−NR–, and related chemistry, J. Organomet. Chem., 2004, 689, 3060–3074 CAS.
- A. M. Kalsin, T. A. Peganova, I. S. Sinopalnikova, I. V. Fedyanin, N. V. Belkova, E. Deydier and R. Poli, Mechanistic diversity in acetophenone transfer hydrogenation catalyzed by ruthenium iminophosphonamide complexes, Dalton Trans., 2020, 49, 1473–1484 RSC.
- I. S. Sinopalnikova, T. Y. A. Peganova, V. V. Novikov, I. V. Fedyanin, O. A. Filippov, N. V. Belkova, E. S. Shubina, R. Poli and A. M. Kalsin, Coordinatively Labile 18-Electron Arene Ruthenium Iminophosphonamide Complexes, Chem. – Eur. J., 2017, 23, 15424–15435 CrossRef CAS PubMed.
- T. Y. A. Peganova, A. V. Valyaeva, A. M. Kalsin, P. V. Petrovskii, A. O. Borissova, K. A. Lyssenko and N. A. Ustynyuk, Synthesis of Aminoiminophosphoranate Complexes of Palladium and Platinum and X-ray Diffractional Investigation of the Weak C−H⋯Pd Interactions Affecting the Geometry of the PdNPN Metallacycles, Organometallics, 2009, 28, 3021–3028 CrossRef CAS.
- I. S. Sinopalnikova, T. A. Peganova, N. V. Belkova, E. Deydier, J.-C. Daran, E. S. Shubina, A. M. Kalsin and R. Poli, Ruthenium p-Cymene Iminophosphonamide Complexes: Activation under Basic Conditions and Transfer Hydrogenation Catalysis, Eur. J. Inorg. Chem., 2018, 2018, 2285–2299 CrossRef CAS.
- R. I. Nekrasov, T. Y. A. Peganova, I. V. Fedyanin, E. I. Gutsul, O. A. Filippov, N. V. Belkova and A. M. Kalsin, Versatile Reactivity of Half-Sandwich Rhodium(III) Iminophosphonamide Complexes, Inorg. Chem., 2022, 61, 16081–16092 CrossRef CAS PubMed.
- V. M. Möhring and G. Fink, Novel Polymerization of α-Olefins with the Catalyst System Nickel/Aminobis(imino)phosphorane, Angew. Chem., Int. Ed. Engl., 1985, 24, 1001–1003 CrossRef.
- R. L. Stapleton, J. Chai, N. J. Taylor and S. Collins, Ethylene Polymerization Using Discrete Nickel(II) Iminophosphonamide Complexes, Organometallics, 2006, 25, 2514–2524 CrossRef CAS.
- R. Boese, M. Düppmann, W. Kuchen and W. Peters, Chelatkomplexe LM/n von Übergangsmetallen mit Iminophosphin-säureamidato-Liganden R2P(NR′)2− (= L), Z. Anorg. Allg. Chem., 1998, 624, 837–845 CrossRef CAS.
- W.-J. Guo and Z.-X. Wang, Cross-Coupling of ArX with ArMgBr Catalyzed by N-Heterocyclic Carbene-Based Nickel Complexes, Org. Chem., 2013, 78, 1054–1061 CrossRef CAS PubMed.
- O. J. Scherer and A. Nahrstedt, Synthesis of a Zeise Salt Derivative with a Phosphorus Nitrogen Ylide as Chelate Ligand, Angew. Chem., Int. Ed. Engl., 1979, 18, 234–235 CrossRef.
- T. Y. A. Peganova, O. A. Filippov, N. V. Belkova, I. V. Fedyanin and A. M. Kalsin, The Origin of the MNXN Metallacycle Flexibility in the Chelate Iminophosphonamide and Amidinate Transition Metal Complexes, Eur. J. Inorg. Chem., 2018, 2018, 5098–5107 CrossRef CAS.
- S. Stipurin, F. Wurl and T. Strassner, C∧C* Platinum(II) Complexes with PtXPX Metallacycle Forming (X = N and S) Auxiliary Ligands: Synthesis, Crystal Structures, and Properties, Organometallics, 2022, 41, 313–320 CrossRef CAS.
- B. F. Straub, F. Eisenträger and P. Hofmann, A remarkably stable copper(I) ethylene complex: synthesis, spectroscopy and structure, Chem. Commun., 1999, 2507–2508 RSC.
- B. F. Straub, F. Rominger and P. Hofmann, A neutral dicopper(II) bis(μ-oxo) complex from a copper(I) ethylene iminophosphanamide and O, Chem. Commun., 2000, 1611–1612 RSC.
- H. Ackermann, O. Bock, U. Müller and K. Dehnicke, Synthese und Kristallstruktur des Aminoimino-Phosphinat–Kupfer(I)-Komplexes [Cu(Me3SiNP(Ph)2NSiMe3)]2, Z. Anorg. Allg. Chem., 2000, 626, 1854–1856 CrossRef CAS.
- H. L. Hermann, G. Boche and P. Schwerdtfeger, Metallophilic Interactions in Closed-Shell Copper(I) Compounds—A Theoretical Study, Chem. – Eur. J., 2001, 7, 5333–5342 CrossRef CAS PubMed.
- A. K. Gupta, F. A. S. Chipem and R. Boomishankar, A 2-pyridyl (py) attached phosphine imine [P(Npy)(NHpy)3] and an imido phosphinate ion [P(Npy)2(NHpy)2]− in its Ag(i) complex, Dalton Trans., 2012, 41, 1848–1853 RSC.
- A. K. Gupta, A. K. Srivastava and R. Boomishankar, A tetrakis(amido)phosphonium cation containing 2-pyridyl (2Py) substituents, [P(NH2Py)4]+ and its reactivity studies with Ag(I) salts, J. Chem. Sci., 2015, 127, 619–626 CrossRef CAS.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier Science, 2012 Search PubMed.
- F. Baier, Z. Fei, H. Gornitzka, A. Murso, S. Neufeld, M. Pfeiffer, I. Rüdenauer, A. Steiner, T. Stey and D. Stalke, Phosphane- and phosphorane Janus Head ligands in metal coordination, J. Organomet. Chem., 2002, 661, 111–127 CrossRef CAS.
- A. Recknagel, M. Witt and F. T. Edelmann, Synthese von Metallacyclophosphazenen mit Praseodym, Neodym, Uran und Thorium, J. Organomet. Chem., 1989, 371, C40–C44 CrossRef CAS.
- H. Schumann, J. Winterfeld, H. Hemling, F. E. Hahn, P. Reich, K.-W. Brzezinka, F. T. Edelmann, U. Kilimann, M. Schäfer and R. Herbst-Irmer, Metallorganische Verbindungen der Lanthanoide, 89. Cyclooctatetraenyl-Komplexe der frühen Übergangsmetalle und Lanthanoide, 6. (Cyclooctatetraenyl)[N,N′-bis(trimethylsilyl)benzamidinato]- und -[diphenylbis(trimethylsilylimido)phosphinato]-Komplexe der Seltenen Erden; Röntgenstrukturanalyse von (C8H8)Tm[PhC(NSiMe3)2](THF), (C8H8)Lu[μ4-MeOC6H4C(NSiMe3)2](THF) und (C8H8)Nd[Ph2P(NSiMe3)2](THF), Chem. Ber., 1995, 128, 395–404 CrossRef CAS.
- A. Recknagel, A. Steiner, M. Noltemeyer, S. Brooker, D. Stalke and F. T. Edelmann, Diiminophosphinate des Lithiums, Samariums und Ytterbiums: Molekülstrukturen von Li[Ph2P(NSiMe3)2](THF)2 und [Ph2P(NSiMe3)2]2Sm(μ-I)2Li(THF)2, J. Organomet. Chem., 1991, 414, 327–335 CrossRef CAS.
- K. A. Rufanov, N. K. Pruß and J. Sundermeyer, Simple entry into N-tert-butyl-iminophosphonamide rare-earth metal alkyl and chlorido complexes, Dalton Trans., 2016, 45, 1525–1538 RSC.
- B. Liu, L. Li, G. Sun, J. Liu, M. Wang, S. Li and D. Cui, 3,4-Polymerization of Isoprene by Using NSN- and NPN-Ligated Rare Earth Metal Precursors: Switching of Stereo Selectivity and Mechanism, Macromolecules, 2014, 47, 4971–4978 CrossRef CAS.
- S. Li, D. Cui, D. Li and Z. Hou, Highly 3,4-Selective Polymerization of Isoprene with NPN Ligand Stabilized Rare-Earth Metal Bis(alkyl)s. Structures and Performances, Organometallics, 2009, 28, 4814–4822 CrossRef CAS.
- Y. Yang, K. Lv, L. Wang, Y. Wang and D. Cui, Isoprene polymerization with aminopyridinato ligand supported rare-earth metal complexes. Switching of the regio- and stereoselectivity, Chem. Commun., 2010, 46, 6150–6152 RSC.
- S. A. Ahmed, M. S. Hill, P. B. Hitchcock, S. M. Mansell and O. St John, DACH-Bridged (DACH = trans-1,2-Diaminocyclohexane) Bis(iminophosphonamide) Derivatives of Groups 3 and 13 and Their Use in the Enantiomorphic Polymerization of Methyl Methacrylate, Organometallics, 2007, 26, 538–549 CrossRef CAS.
- S. Li, W. Miao, T. Tang, W. Dong, X. Zhang and D. Cui, New Rare Earth Metal Bis(alkyl)s Bearing an Iminophosphonamido Ligand. Synthesis and Catalysis toward Highly 3,4-Selective Polymerization of Isoprene, Organometallics, 2008, 27, 718–725 CrossRef CAS.
- B. Liu, G. Sun, S. Li, D. Liu and D. Cui, Isoprene Polymerization with Iminophosphonamide Rare-Earth-Metal Alkyl Complexes: Influence of Metal Size on the Regio- and Stereoselectivity, Organometallics, 2015, 34, 4063–4068 CrossRef CAS.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- D. K. Sinitsa, E. K. Pylova, O. A. Mironova, D. A. Bashirov, A. A. Ryadun, T. S. Sukhikh and S. N. Konchenko, Lanthanide complexes with a new luminescent iminophosphonamide ligand bearing phenylbenzothiazole substituents, Dalton Trans., 2024, 53, 2181–2192 RSC.
- D. K. Sinitsa, T. S. Sukhikh, N. A. Pushkarevsky and S. N. Konchenko, New Asymmetric Iminophosphonamide Ligand and Its Yttrium Complex: Synthesis and Crystal Structure, J. Struct. Chem., 2024, 65, 666–675 CrossRef CAS.
- P. A. Cameron, V. C. Gibson and D. J. Irvine, Nickel-Catalyzed Generation of Schiff Base Aluminum Enolate Initiators for Controlled Methacrylate Polymerization, Angew. Chem., Int. Ed., 2000, 39, 2141–2144 CrossRef CAS PubMed.
- N. Li and B.-T. Guan, Yttrium–Benzyl Complexes Bearing Chiral Iminophosphonamide Ligands: Synthesis and Application in Catalytic Asymmetric Amine-Silane Dehydrocoupling Reactions, Adv. Synth. Catal., 2017, 359, 3526–3531 CrossRef CAS.
- T. J. Feuerstein, B. Goswami, P. Rauthe, R. Köppe, S. Lebedkin, M. M. Kappes and P. W. Roesky, Alkali metal complexes of an enantiopure iminophosphonamide ligand with bright delayed fluorescence, Chem. Sci., 2019, 10, 4742–4749 RSC.
- B. Goswami, T. J. Feuerstein, R. Yadav, R. Köppe, S. Lebedkin, M. M. Kappes and P. W. Roesky, Enantiopure Calcium Iminophosphonamide Complexes: Synthesis, Photoluminescence, and Catalysis, Chem. – Eur. J., 2021, 27, 4401–4411 CrossRef CAS PubMed.
- B. Goswami, R. Yadav, C. Schoo and P. W. Roesky, Neutral and cationic enantiopure group 13 iminophosphonamide complexes, Dalton Trans., 2020, 49, 675–681 RSC.
- B. Goswami, T. J. Feuerstein, R. Yadav, S. Lebedkin, P. J. Boden, S. T. Steiger, G. Niedner-Schatteburg, M. Gerhards, M. M. Kappes and P. W. Roesky, Thermally Activated Delayed Fluorescence and Phosphorescence Quenching in Iminophosphonamide Copper and Zinc Complexes, Chem. – Eur. J., 2021, 27, 15110–15119 CrossRef PubMed.
- B. Goswami, X. Sun, T. J. Feuerstein, M. T. Gamer and P. W. Roesky, Homoleptic Enantiopure Lanthanide Complexes: Synthesis, Structure, and Characterization, Organometallics, 2023, 42, 1317–1323 CrossRef CAS.
- B. Goswami, R. Yadav, H. Joshi and P. Roy, Zirconium and Hafnium Complexes of Enantiopure Iminophosphonamides, Eur. J. Inorg. Chem., 2025, e202400627 CrossRef CAS.
| This journal is © the Partner Organisations 2025 |