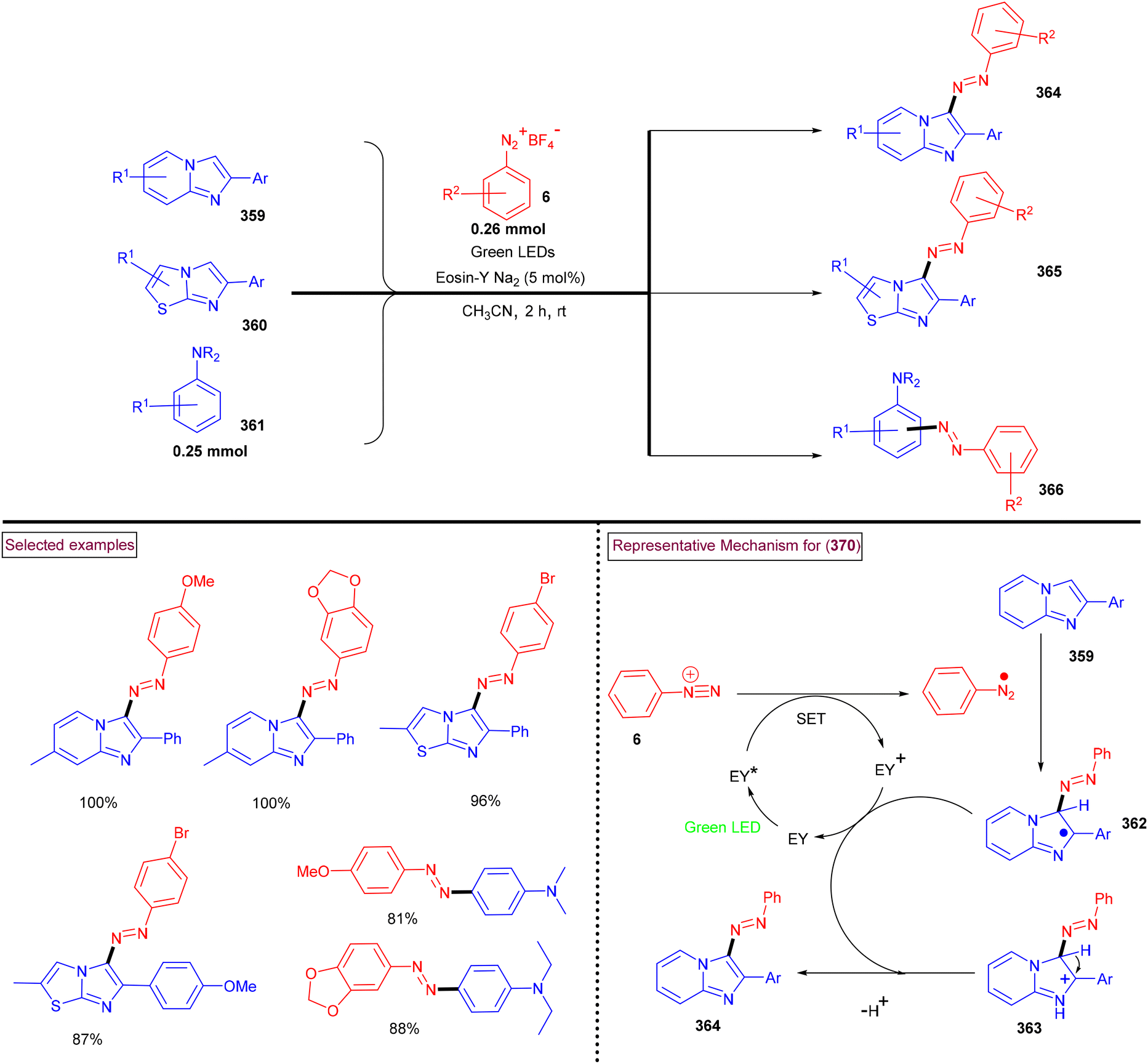
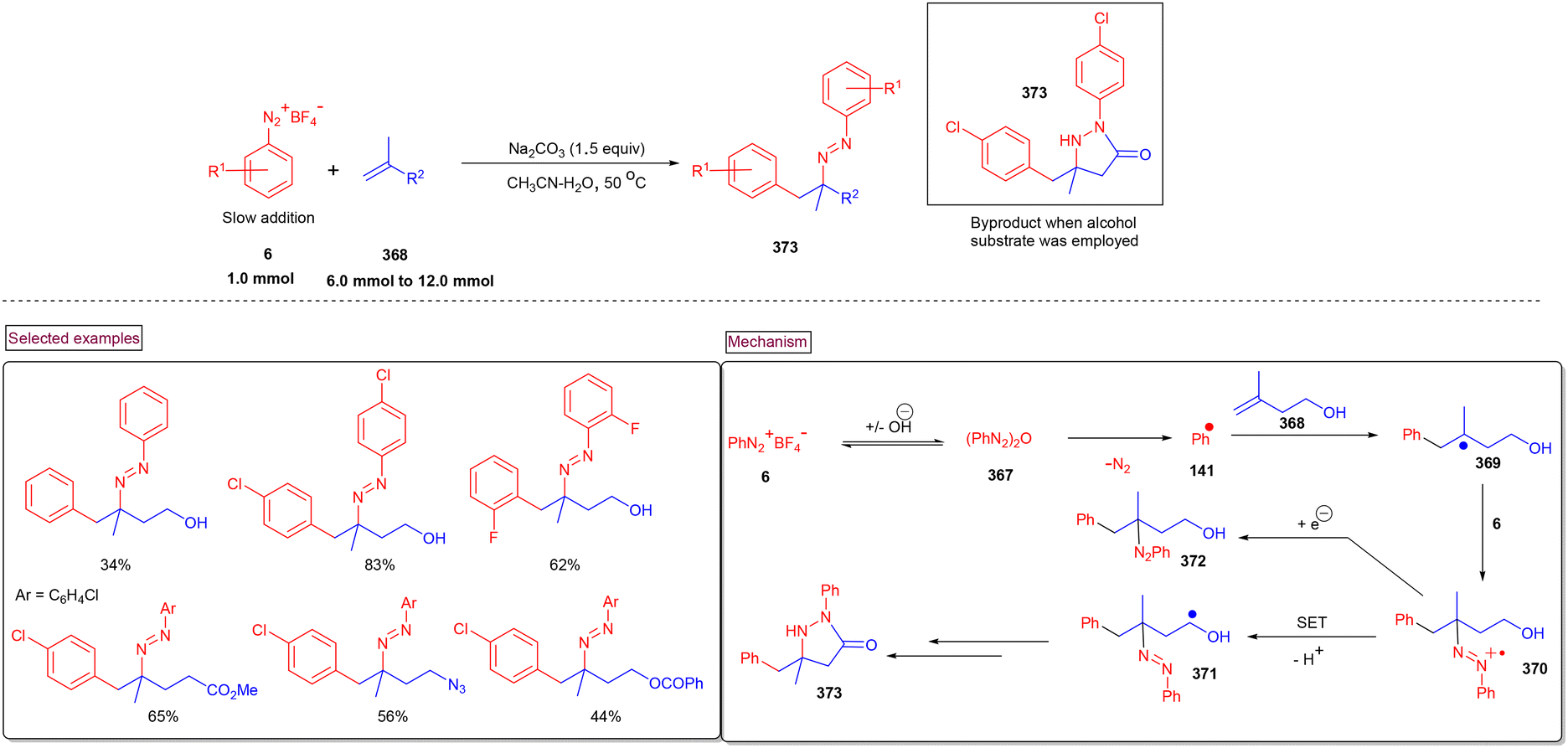
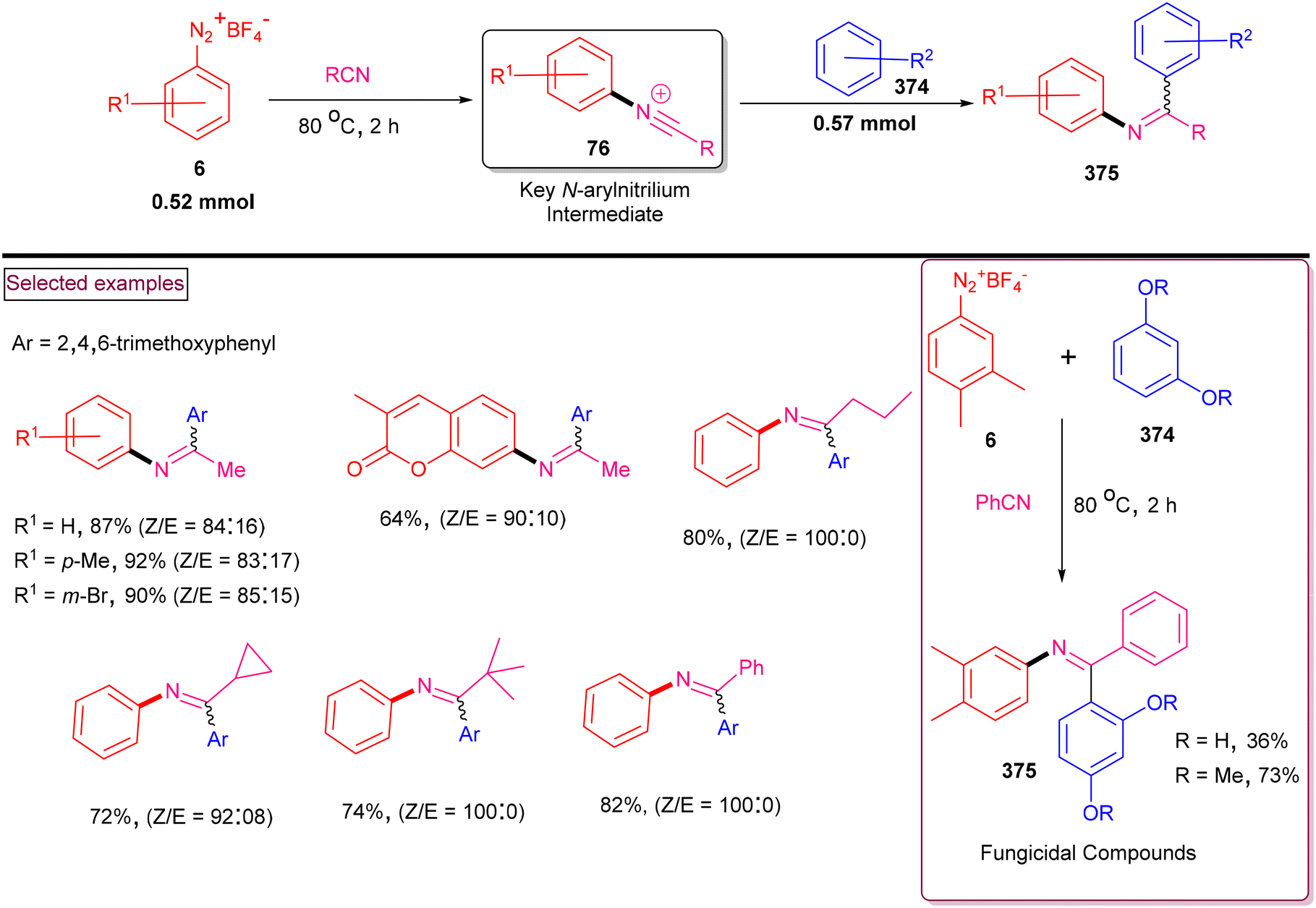
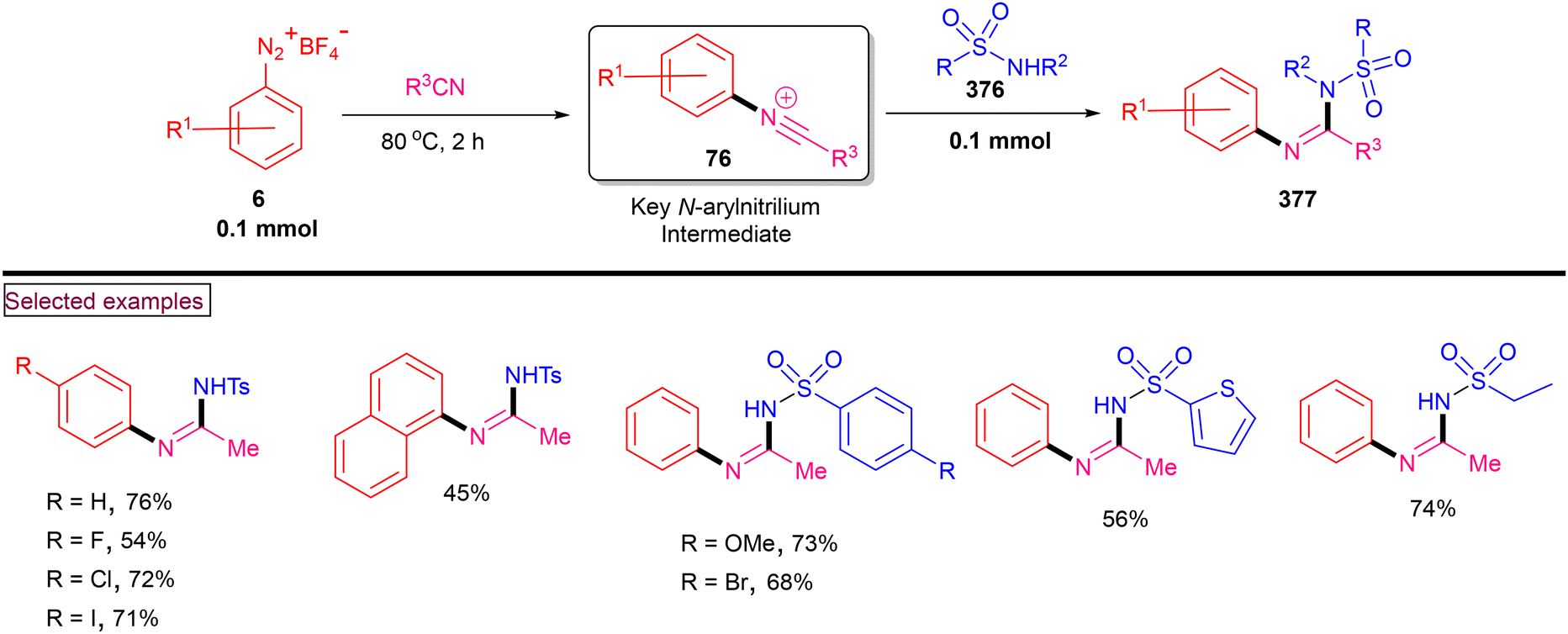
DOI:
10.1039/D4QO01776E
(Review Article)
Org. Chem. Front., 2025,
12, 256-327
Transition-metal-free approaches to access N-heterocycles and valuable intermediates from aryldiazonium salts
Received
23rd September 2024
, Accepted 11th November 2024
First published on 13th November 2024
Abstract
The discovery of novel and efficient synthetic methods to achieve valuable N-fused heterocycles has been an important research area in organic chemistry for several decades. Owing to the importance of N-fused heterocycles in medicinal chemistry, materials sciences, and organic synthesis, numerous efforts have been devoted to designing robust methods for the construction of N-heterocyclic molecules. Aryldiazonium salts are a privileged and readily accessible class of reagents/synthetic building blocks used in a diverse range of disciplines in chemical sciences. Aryldiazonium salts based on conventional C–C bond construction strategies have been well established. Alternatively, owing to their high electrophilic capacity, aryldiazonium salts have been broadly utilized in the efficient, atom-economic and rapid assembly of structurally unique nitrogen-containing heterocycles via the formation of C–N bonds in recent years. Herein, we review a diverse range of transition-metal-free synthetic methodologies using aryldiazonium salts as key synthetic precursors to afford highly useful N-heterocyclic frameworks over the past nine years (2015 to date). In addition, the application of these salts to prepare synthetically useful intermediates and precursors is also discussed. These N-heterocyclic motifs and intermediates are highly prevalent in a variety of natural products, agrochemicals and material applications.
1. Introduction
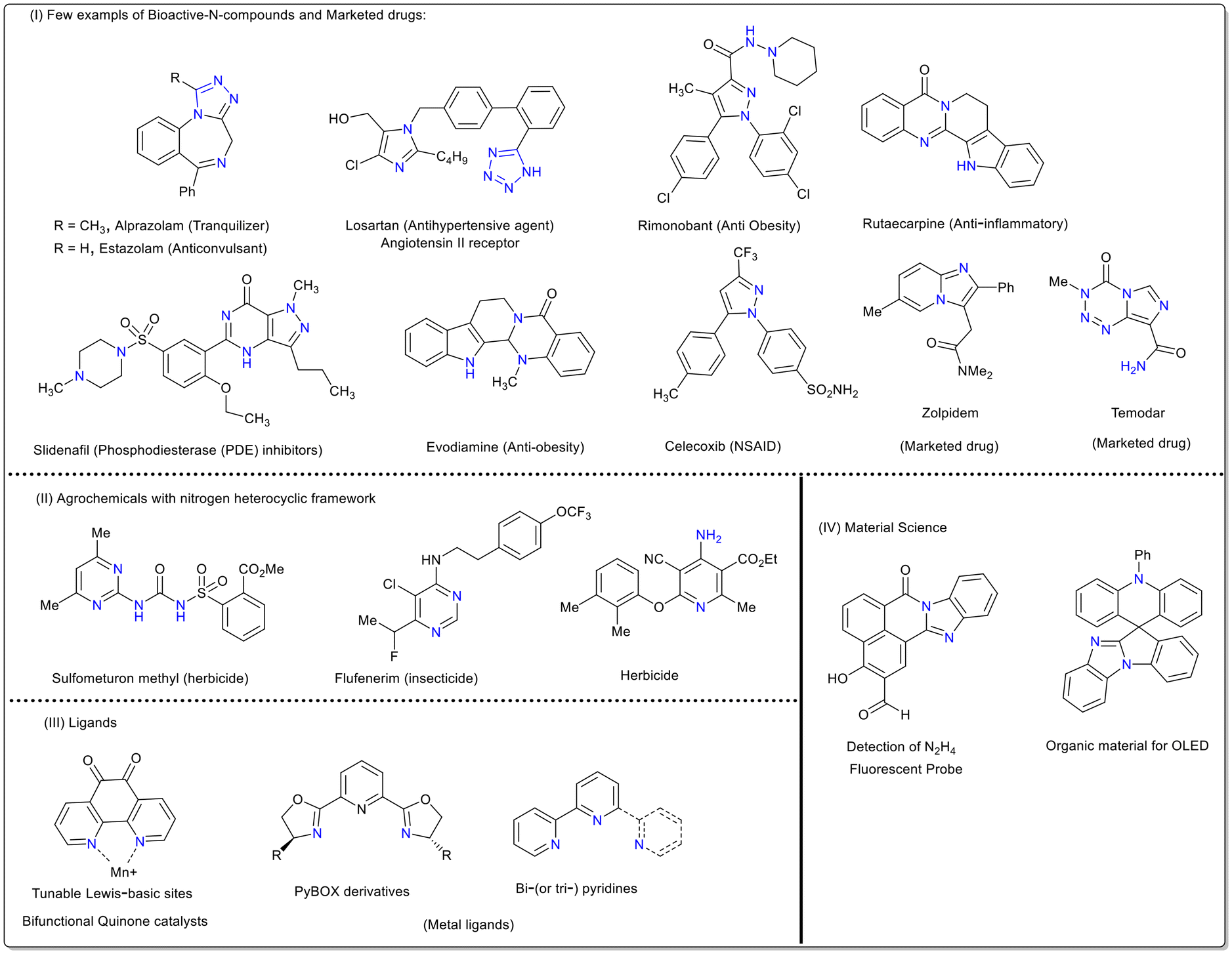
N-Based heterocycles represent an important and widespread class of molecules receiving special recognition in drug discovery.1–5 As supported by big data analysis, the synthesis of N-heterocycles is one of the most common reaction strategies in the organic/medicinal chemistry field.6,7 These heterocyclic units are key structural backbones of several natural products, dyes and functional molecules.8–10 Moreover, 60% of US-FDA approved drugs possess nitrogen frameworks in their structure.11–14 These drugs showcase a broad-spectrum therapeutic profile with antifungal, antibacterial, anticancer, anticonvulsant, antimalarial and anti-inflammatory activities (Fig. 1).15–21 In addition, they play a vital role in various biochemical processes involving RNA, DNA and co-enzymes.22–24 Owing to their importance, extensive investigations have been undertaken to design numerous synthetic methods for the efficient construction of N-heterocycles over the past few decades.25–29 Over the years, transition-metal-catalysed strategies have been proven to be remarkable tools, producing a diverse range of nitrogen heterocycles through unique reaction pathways.30a,b In particular, aryldiazonium salts are widely used in combination with transition-metal-catalysed conditions to afford nitrogen-rich scaffolds.30c–g Although metal-catalysed construction of N-heterocycles is dominant in the literature, the reaction conditions are generally air- and moisture-sensitive as well as toxic and require costly ligands. Their threshold value in pharmaceutically important end products is also crucial. Alternatively, conceptually distinct metal-free systems have also been developed to afford these ubiquitous N-heterocyclic skeletons via a sustainable and atom-economic fashion. This review focuses on the preparation, representative examples, mechanistic explanation and post synthetic modifications achieved using aryldiazonium salts exclusively under transition metal-free conditions. Most of the discussed nitrogen skeletons such as tetrazoles, triazoles, indazoles, pyrazoles, phenanthridines, quinolines, and quinazolines are frequently found in a diverse range of biologically important compounds. Overall, this review will provide synthetic insights with a comprehensive and updated guide towards the synthesis of N-heterocycles and key intermediates in organic chemistry.
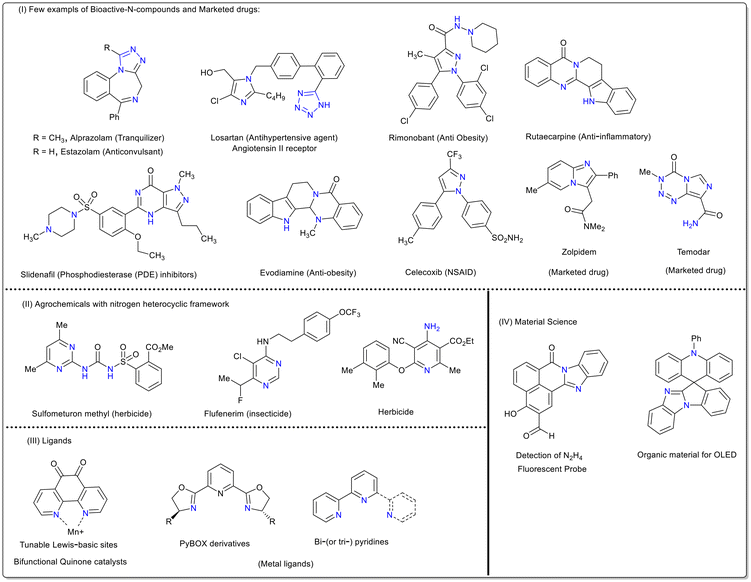 |
| | Fig. 1 Illustrative examples of nitrogen-rich biologically active molecules. | |
2. Diverse reactivity profiles of aryldiazonium salts
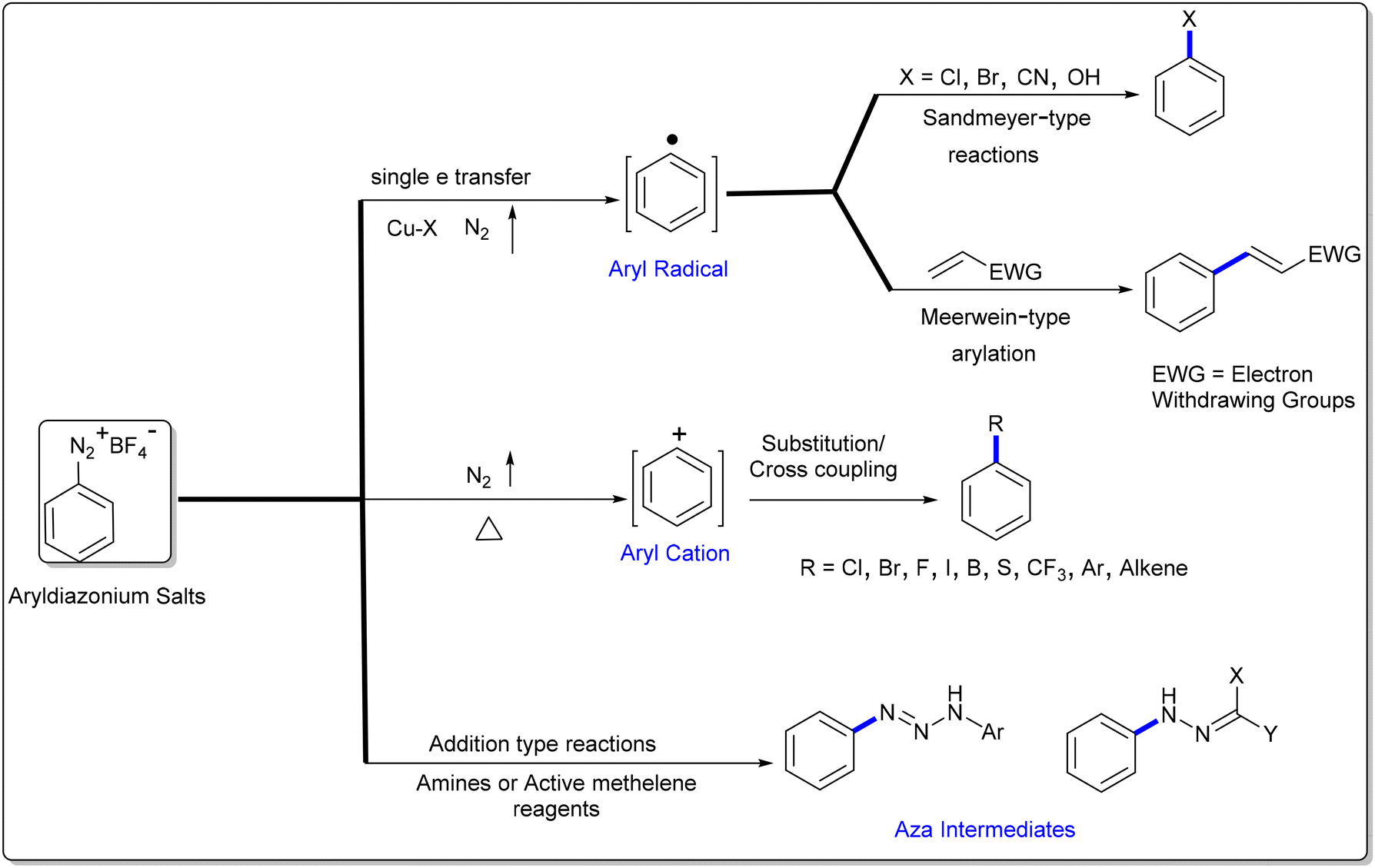
Aryldiazonium salts are readily available and versatile reagents widely utilized in a diverse range of organic transformations, azo dyes and materials.31–34 Typically, aryldiazonium salts are represented as Ar–N2+X−, where X is a non-nucleophilic inorganic or organic anion (X = halides, BF4, PF6, and OTf). The large-scale production of aryldiazonium salts is challenging due their explosive nature; however, this challenge be bypassed via continuous flow technology,35,36in situ diazotization and by modulating the counter-ion to improve the stability.37 Since the discovery of diazonium salts (1858, Griess),38 these species have played a key role in organic synthesis owing to their multifaceted reactivity (Scheme 1). Due to the intrinsic electrophilicity associated with the ability to lose N2, aryldiazonium salts are mostly utilized as aryl precursors in substitution-type conversions, as exemplified by various renowned organic named reactions such as Sandmeyer-type reactions of C–X bond construction (X = Cl, Br or CN, 1884),39 Balz–Schiemann reaction (C–F, 1927),40 Gomberg–Bachmann (C–C, 1924),41 Heck–Matsuda reaction (arylation of olefines),42,43 Pschorr reaction (intramolecular arylation, 1896),44 and Meerwein arylation (1939).45 In addition, C–S, C–P, C–B and C–CF3 bond-forming reactions from aryldiazonium salts have also received significant attention.46 Recently, aryldiazonium salts in combination with visible-light mediated photo redox catalysis have offered various elegant methods for the construction of aryl C–C, C–Sn and C–Se bonds.47–51 Alternatively, nucleophilic addition-type transformations of aryldiazonium salts are also known, where the nitrogen is retained. In this sub-group, the aryldiazonium ion acts as a reactive nitrogen electrophile (amidine-mediated N–N bond formation, Japp–Klingemann reaction)52–70 and radical acceptor in radical-mediated addition reactions.71–74 Furthermore, aryldiazonium salts are also known to act as dual synthons such as aryl precursors and radical acceptors.75
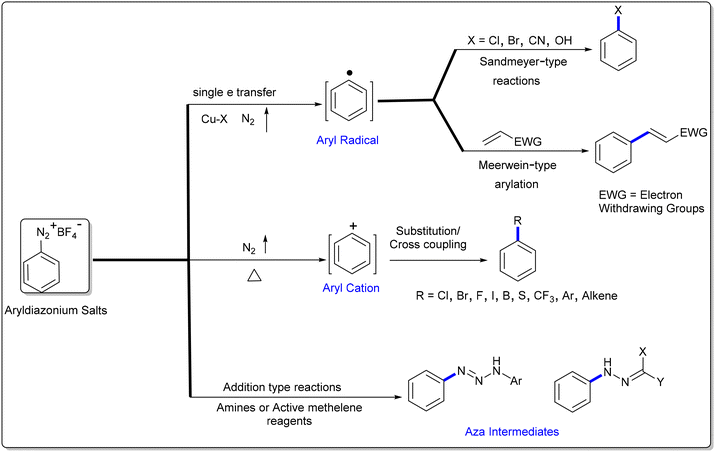 |
| | Scheme 1 Multifaceted reactivity profile of aryldiazonium salts. | |
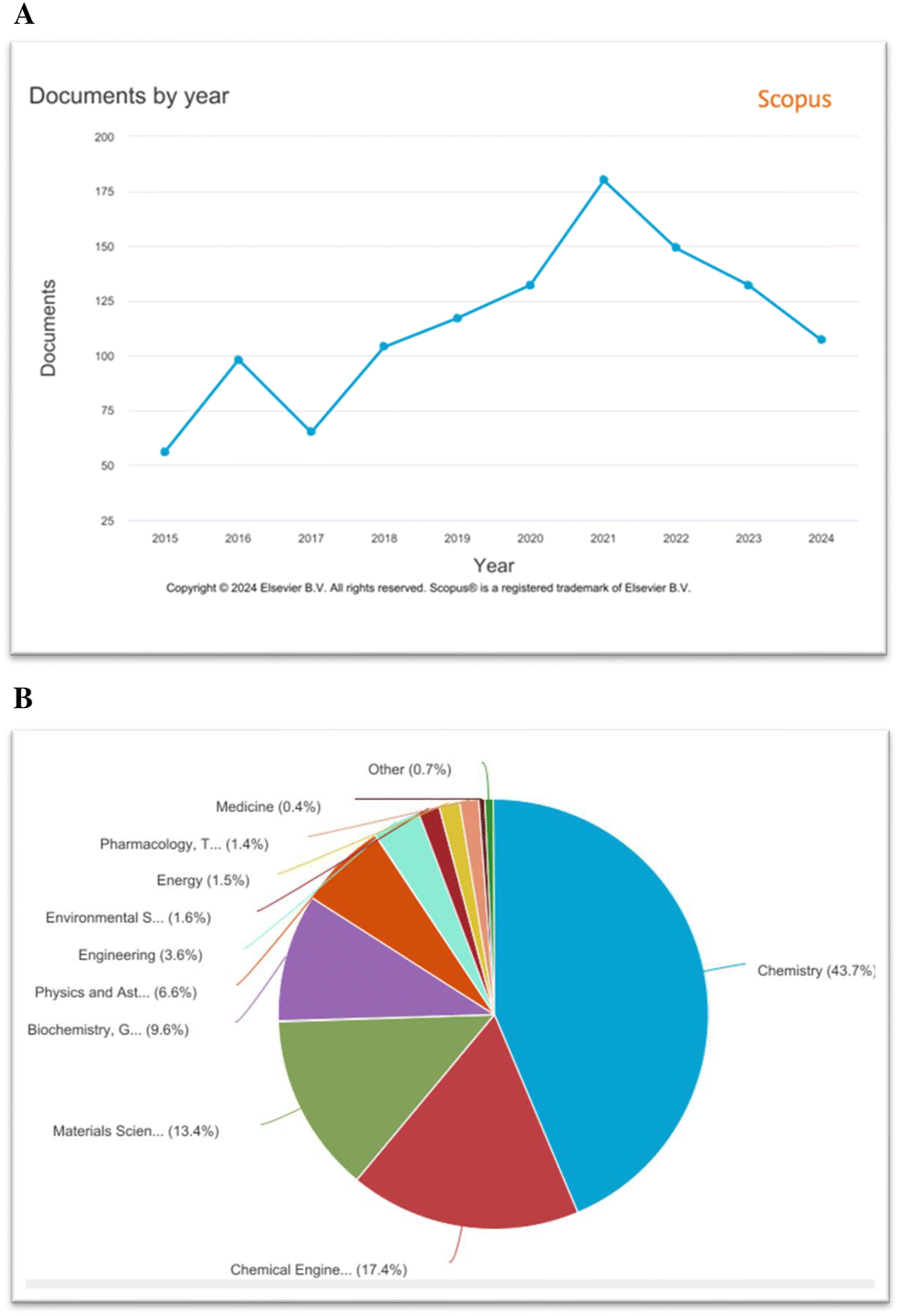
Transition metal-catalysed transformations of aryldiazonium salts were well-established after the discovery that Pd(0)-complexes catalysed the direct arylation of olefines by aryldiazonium salts (Matsuda, 1977).43 After this report, aryldiazonium salts have been widely utilized in various cross-coupling reactions as an arylating partner.76 In addition, a range of novel functional group conversions and methodologies has been developed based on the aryldiazonium moiety.31 However, despite these enrapturing advancements, the eco-friendly development of synthetic protocols with notable atom-/step economy, higher yields and broad functional group tolerance is necessary. In this pursuit, a renaissance has been witnessed in the aryldiazonium salt-based transition metal-free synthesis of N-rich heterocycles including cascade/tandem approaches, multi-component strategies, cycloadditions, visible light-induced and methods.77–80 As shown in Fig. 2A, according to our Scopus search results, the aryldiazonium-based construction of nitrogen heterocycles is an active research theme in chemistry. In addition, aryldiazonium salts are mainly utilized in chemistry (43.7%), followed by materials science and chemical engineering (Fig. 2B).
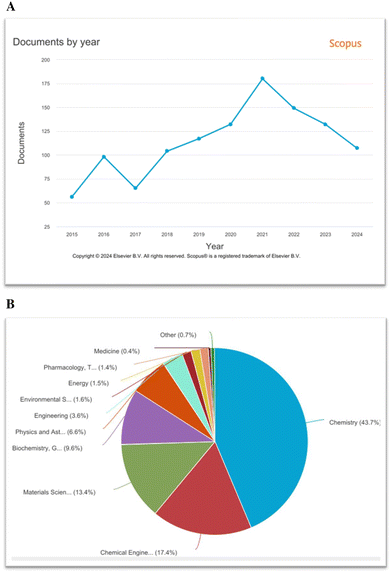 |
| | Fig. 2 (A) Scopus results for the keyword search “aryldiazonium salts and nitrogen heterocycles” for the years 2015–2024. (B) Documents categorized on the basis of subject area using Scopus. | |
3. Transition metal-free approaches based on aryldiazonium salts
Due to our continued interest in the construction of N-heterocycles, we aim to provide an easy comprehension and insights into the exploration of aryldiazonium chemistry under transition-metal-free conditions. The synthetic methodologies and applications published from 2015 to date are compiled. For convenience, this account is organized based on the type of N-heterocycle and the proposed pathways are discussed with representative examples where appropriate. In addition, some of the key intermediates and/or substrates prepared from aryldiazonium salts under transition metal-free conditions are also emphasized. Overall, the goal of this report is not only to present the synthetic advancements of aryldiazonium chemistry, but also provide inspiration and insights for future developments in this field.
3.1. Tetrazole derivatives
Tetrazoles are an important class of N-rich heterocyclic scaffolds in medicinal chemistry and drug discovery. These ubiquitous derivatives possess broad spectrum biological activities such as anti-hypertensive (Losartan, Valsartan, Azosemide, and Candesartan),81 anti-HIV (Etravirine),82 antibiotic (Cefamandole and Tedizolid),83 antiallergic,84 anti-convulsant (Corazolum),85 anti-Alzheimer's (Tomelukast),86 antifungal (TAK-456)87 and antihistaminic drugs (Tazanoplast).88 To date, 23 USFDA approved drugs contain tetrazole substituents in their structures. Several distinctive approaches including multi-component reactions and [3 + 2] cycloadditions are discussed in this section.
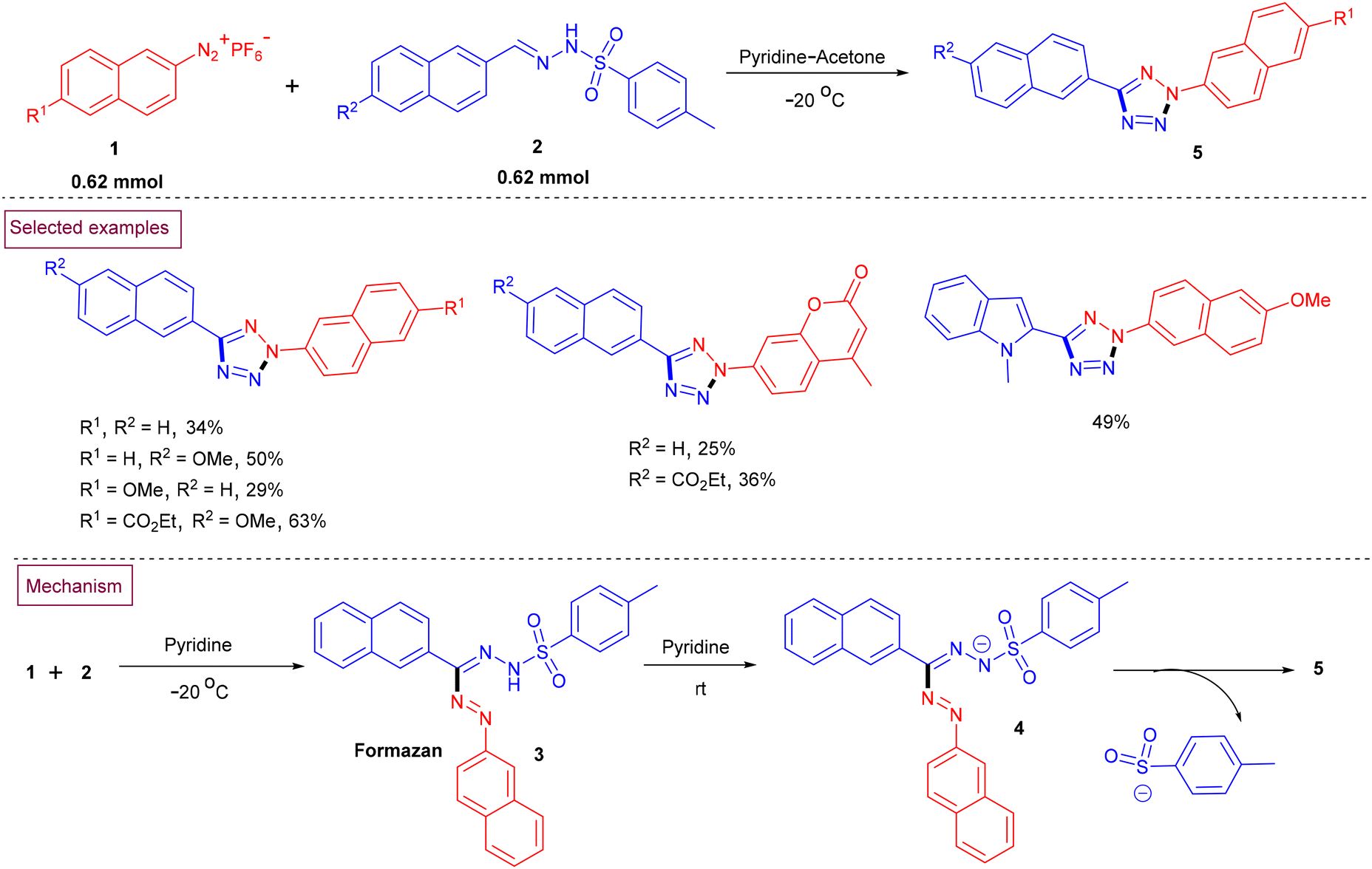
A reaction between arylsulfonyl hydrazones (2) and aryldiazonium salts (1) to prepare a series of 2,5-difunctionalized binaphthyl, naphthyl and biphenyl tetrazole derivatives (5) in acetone/pyridine medium was reported by Fillaut et al. (Scheme 2).89 Mechanistically, the addition of aryldiazonium salt (1) to a solution of aryl sulfonyl hydrazones (2) generated a formazan intermediate (3). Deprotonation of the nitrogen attached to the arylsulfonyl group led to the formazanide anion (4). Further cyclization of (4) resulted in the formation of 2,5-disubstituted tetrazole derivatives (5) together with the elimination of the tosylsulfinate ion and isomerization. This developed method was compatible with sensitive functional groups such as coumarin, indole and carboxylic esters on both substrates and the desired tetrazoles were obtained in moderate yields.
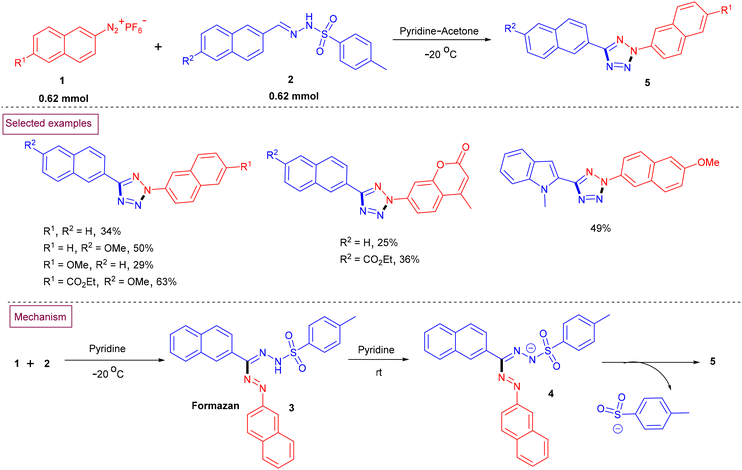 |
| | Scheme 2 Preparation of 2,5-disubstituted tetrazoles from arylsulfonyl hydrazones and aryldiazonium salts. | |
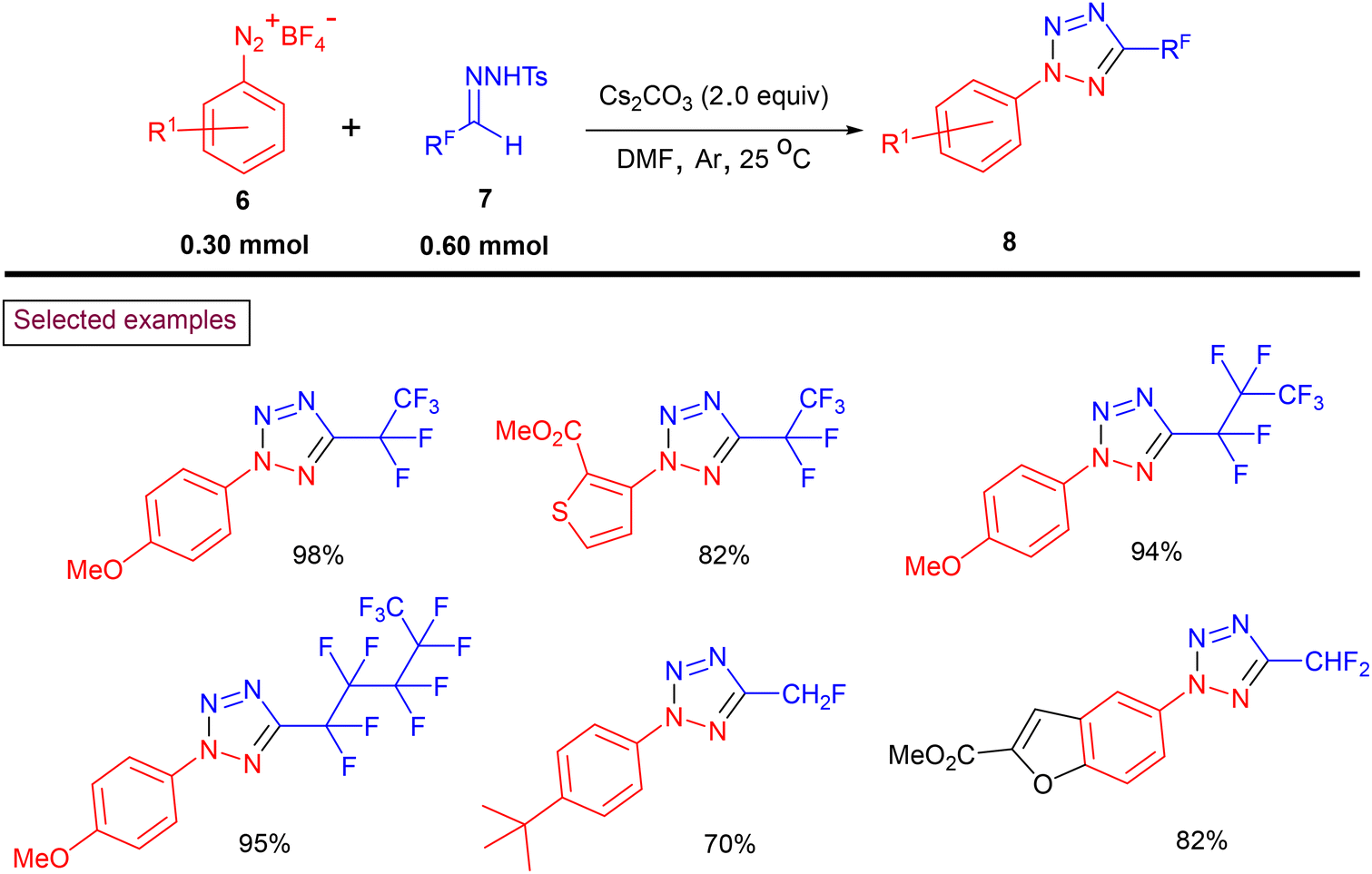
Recently, Ning and Liu et al. reported a general method to prepare fluoroalkylated tetrazoles (8) via the transition metal-free [3 + 2] cycloaddition of bench-stable fluoroalkyl N-sulfonylhydrazones (7) with aryldiazonium salts (6) (Scheme 3).90 A variety of mono, di, and perfluoroalkyl tetrazoles was obtained using an operationally safe procedure. The authors reported 77 unique fluoroalkylated tetrazoles in a step-economical fashion, which are difficult to obtain by any other known method. Furthermore, the notable features of this method include its scalability, mild conditions, functional group compatibility and one-pot operation.
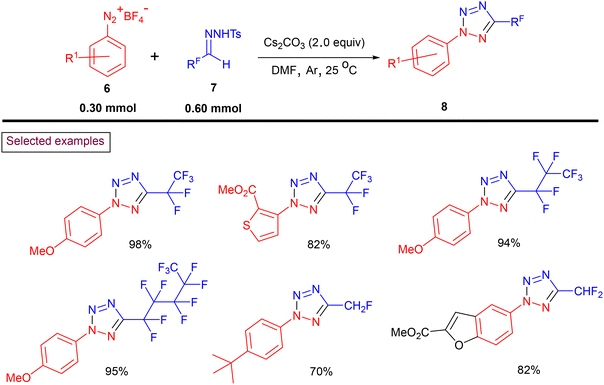 |
| | Scheme 3 Preparation of fluoroalkyl tetrazoles via annulation. | |
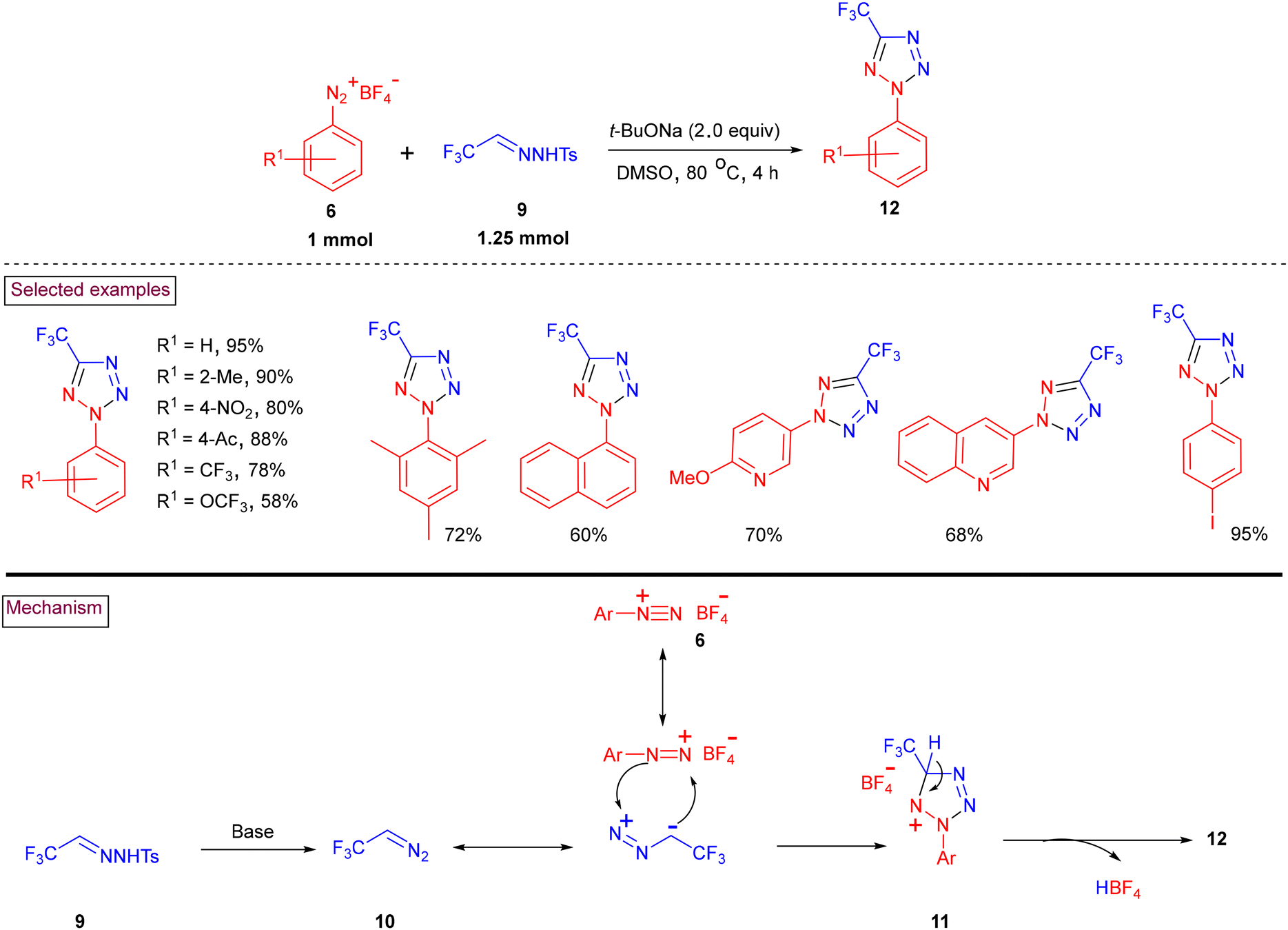
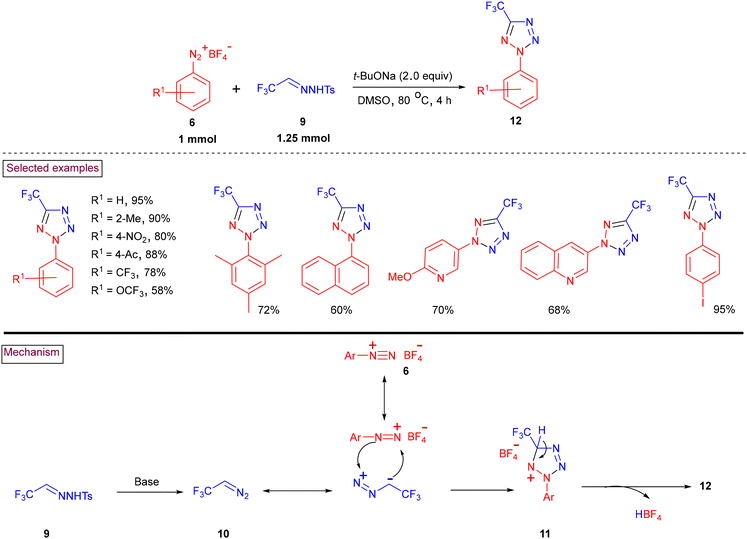
An aryldiazonium (6)-based transition-metal-free method to access 5-trifluoromethyltetrazole (12) using trifluoroacetaldehyde N-tosylhydrazone (9) as a convenient source of CF3CHN2 (10) was disclosed in 2022 (Scheme 4).91 Sterically challenging 2,4,6-trimethyl, 2,6-dimethyl and 2,4-dimethyl groups on the aryldiazonium salts participated in the cyclization process to yield the corresponding products (12) in moderate to good yields (72%–89%). It was observed that electron-donating groups were more reactive than electron-withdrawing groups such as –NO2, –CF3, –C(O)CH3, and –OCF3. Notably, heteroaryl-based pyridine and quinoline diazonium salts were also viable substrates in this cyclization process and the products were isolated in 60%–70% yield. This process involves the base-promoted decomposition of hydrazone derivative (9) to generate trifluorodiazoethane (10). Further 1,3-dipolarcycloaddition with aryldiazonium species (6) resulted in the formation of cyclized intermediate (11) and the desired product (12) was formed after the elimination of HBF4.
 |
| | Scheme 4 Preparation of 5-trifluoromethyltetrazole from aryldiazonium salts and CF3CHN2. | |
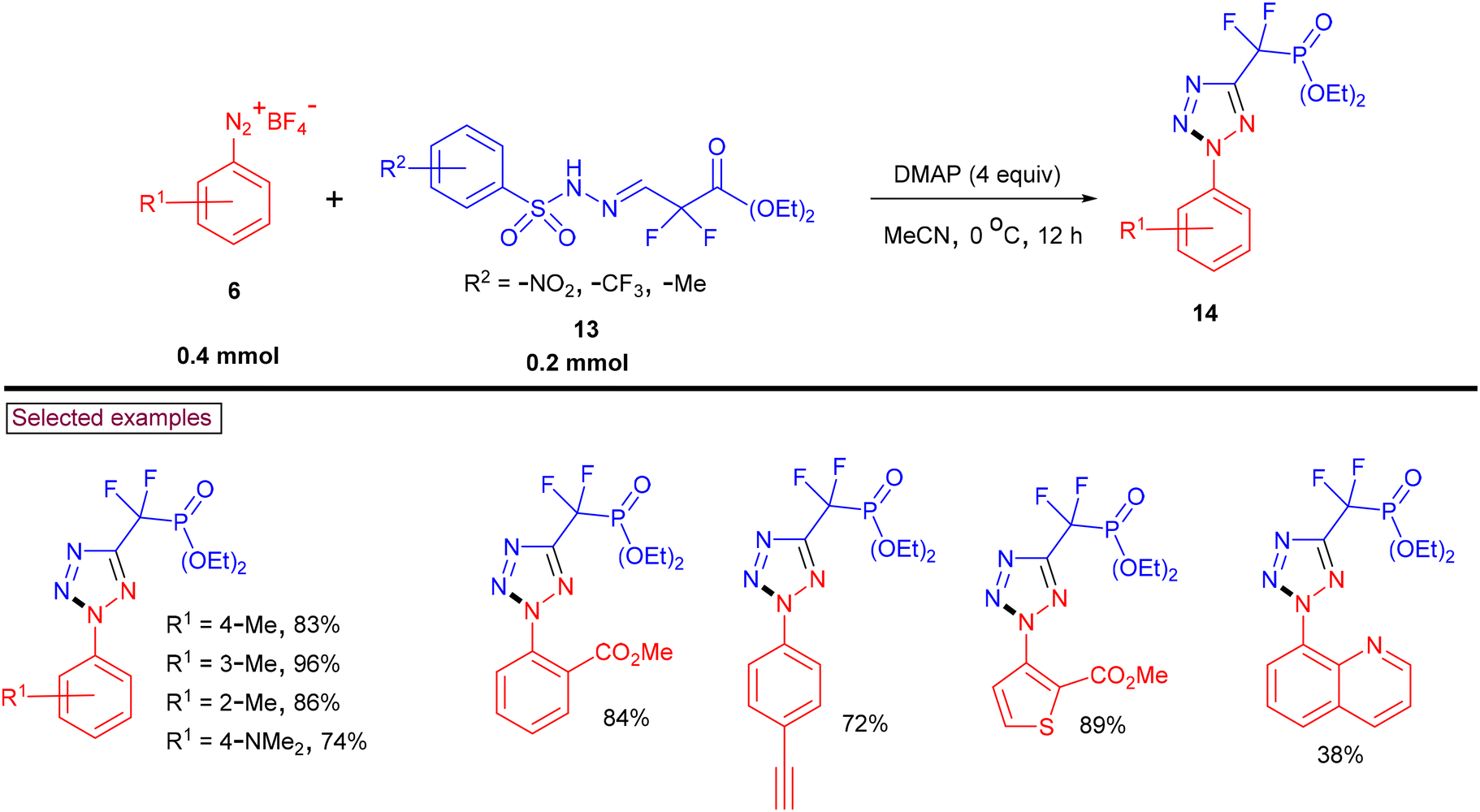
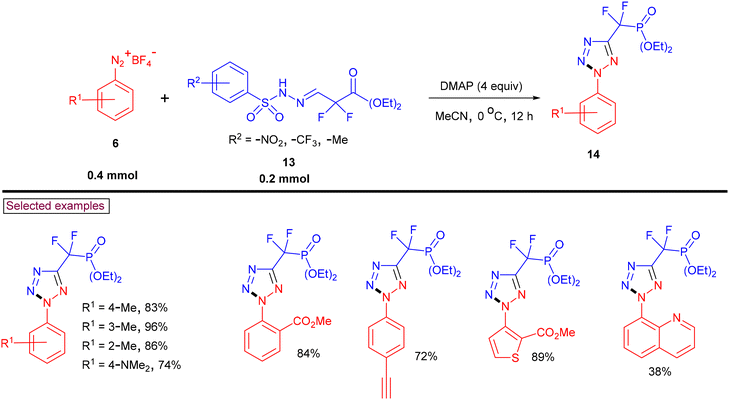
Stable difluoromethylene phosphonate hydrazones (13, DFMP) as azo precursors were reacted with aryldiazonium salts (6) under DMAP-promoted [3 + 2] cycloaddition conditions to afford a series of 2-aryl-2H-tetrazol-5-yl difluoromethylene phosphonates (14) (Scheme 5).92 Hydrazone derivatives bearing –CF3, –NO2 and –Me groups and aryldiazonium salts substituted with alkyl, alkoxyl, amino, halogens, alkenyl, and alkynyl, as well as strong electron-withdrawing groups such as –Ac, –CO2Me, –CN, and –CO2H reacted smoothly. Interestingly, aniline derivatives of Lapatinib and Pomalidomide were also successfully converted into the corresponding tetrazoles in good yields (66%–76%). The synthetic utility was further demonstrated by the hydrolyzing phosphonate group into phosphonic acid in the presence of TMSBr.
 |
| | Scheme 5 Preparation of phosphonate-tethered 2,5-disubstituted tetrazoles. | |
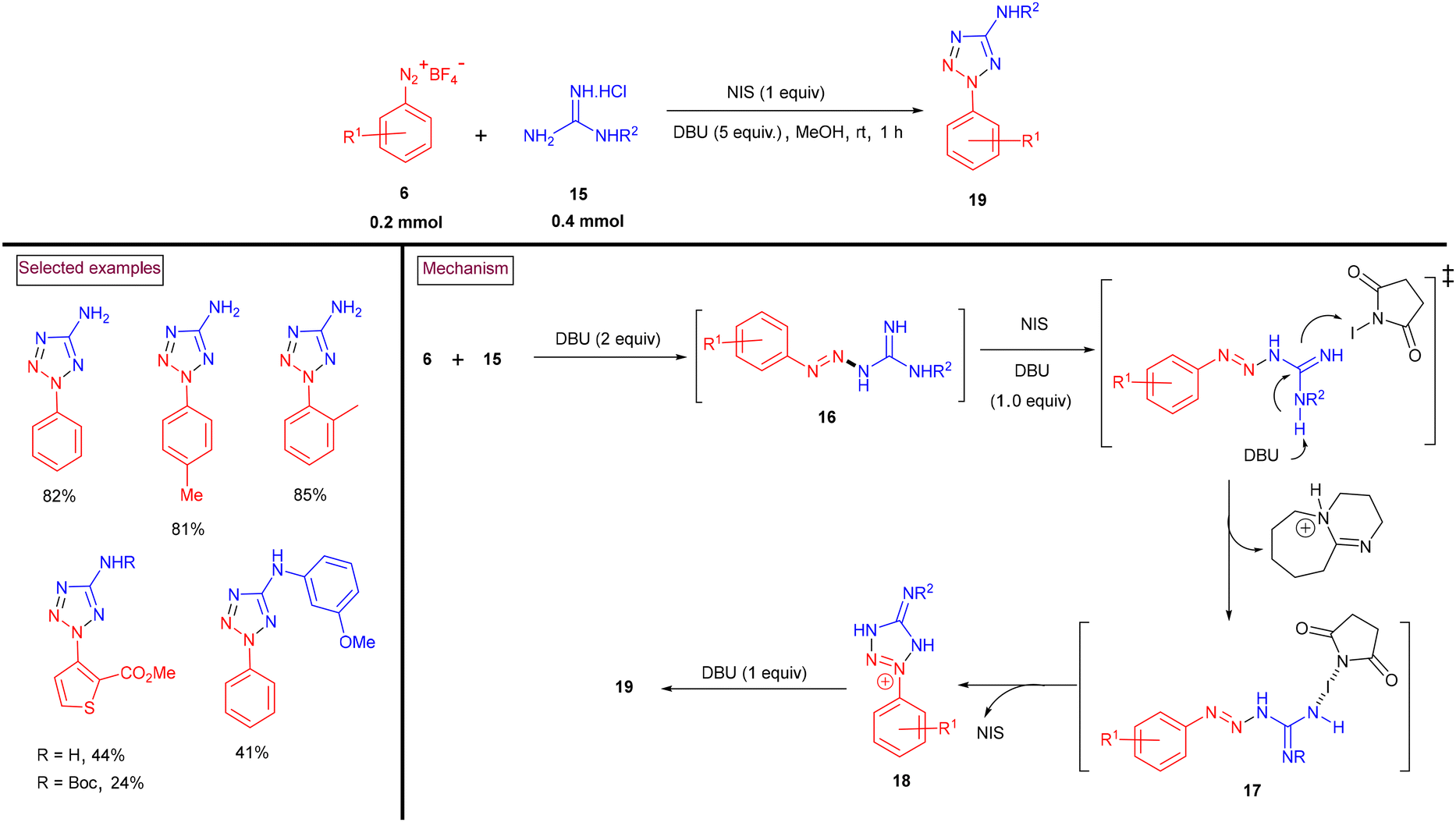
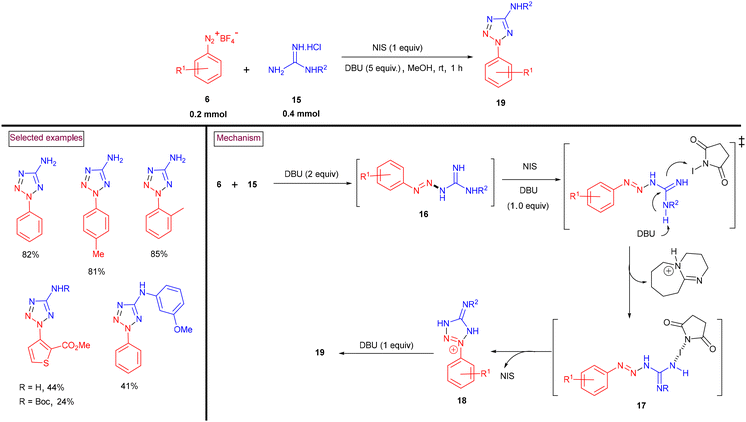
An N-iodosuccinimide-promoted green protocol for the elusive construction of 2-aryl-5-amino-2H-tetrazoles (19) via the [3 + 2] annulation sequence via the reaction of aryldiazonium salts (6) with guanidines (15) was developed (Scheme 6).93 A series of derivatizations of the target products was carried out to showcase the synthetic efficiency of this process. The proposed mechanism revealed that triazine intermediate (16) was generated via the nucleophilic addition of (15) to aryldiazonium salt (6). The in situ-generated HCl and HBF4 were neutralized by DBU (2 equiv.). Subsequent proton abstraction by DBU and the interaction of the resulting intermediate with NIS generated intermediate (17). Intermediate (18) was generated through intramolecular cyclization. Further deprotonation by DBU and aromatization yielded the tetrazole product (19).
 |
| | Scheme 6 Preparation of aminotetrazoles via NIS-promoted [3 + 2] annulation of aryldiazonium salts and guanidines. | |
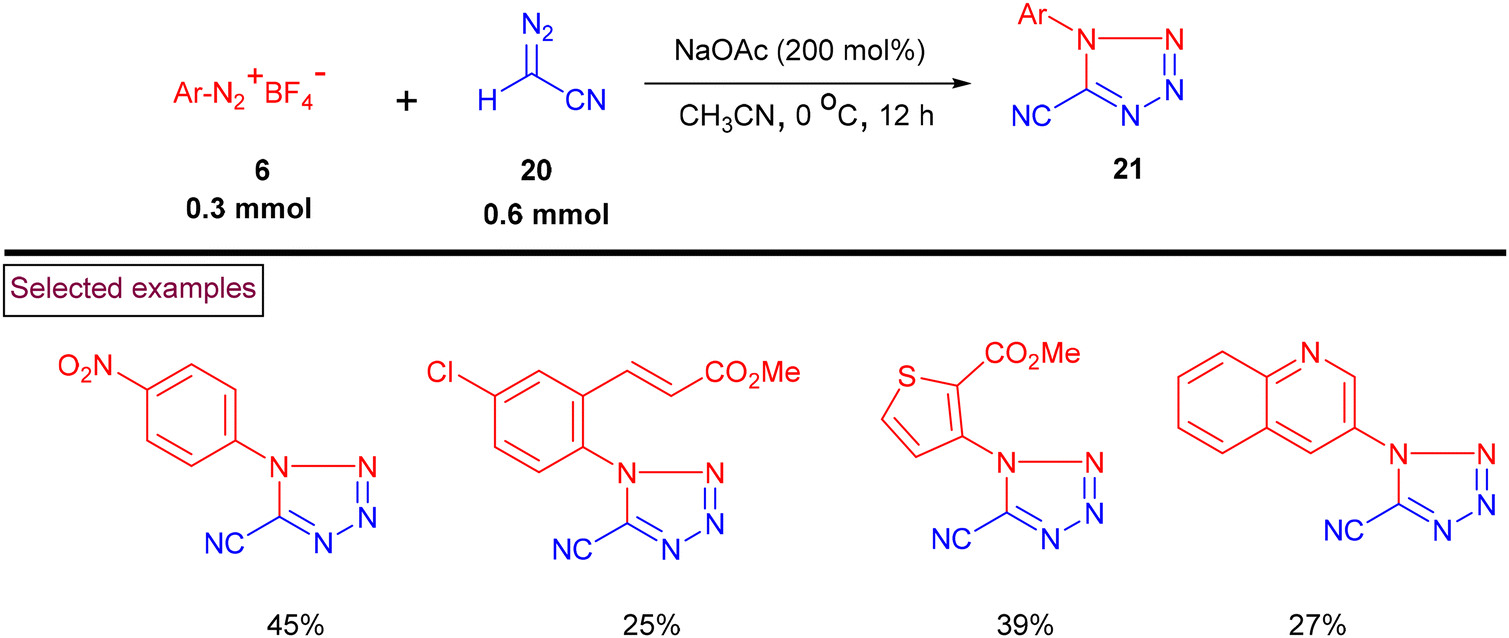
A regio-divergent cycloaddition approach for the construction of cyano-tetrazoles (21) from aryldiazonium salts (6) and diazoacetonitriles (20) was reported in 2020 (Scheme 7).94 This method enabled the synthesis of 1,5-disubstituted cyano-tetrazoles when the starting materials were treated with only NaOAc, whereas the corresponding 2,5-disubstituted cyano-tetrazoles were formed in the presence of catalytic AgOAc via the [3 + 2] cycloaddition process. Furthermore, a one-pot protocol for the synthesis of the corresponding tetrazole derivatives (21) from primary anilines has also been demonstrated.
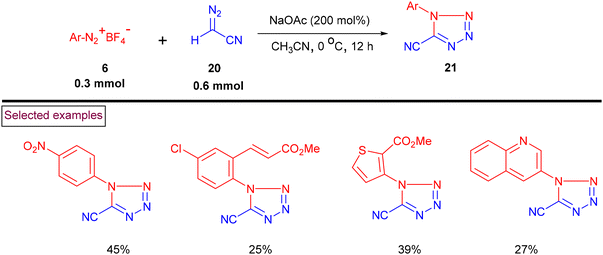 |
| | Scheme 7 Preparation of 1,5-disubstituted cyano-tetrazoles via cycloaddition. | |
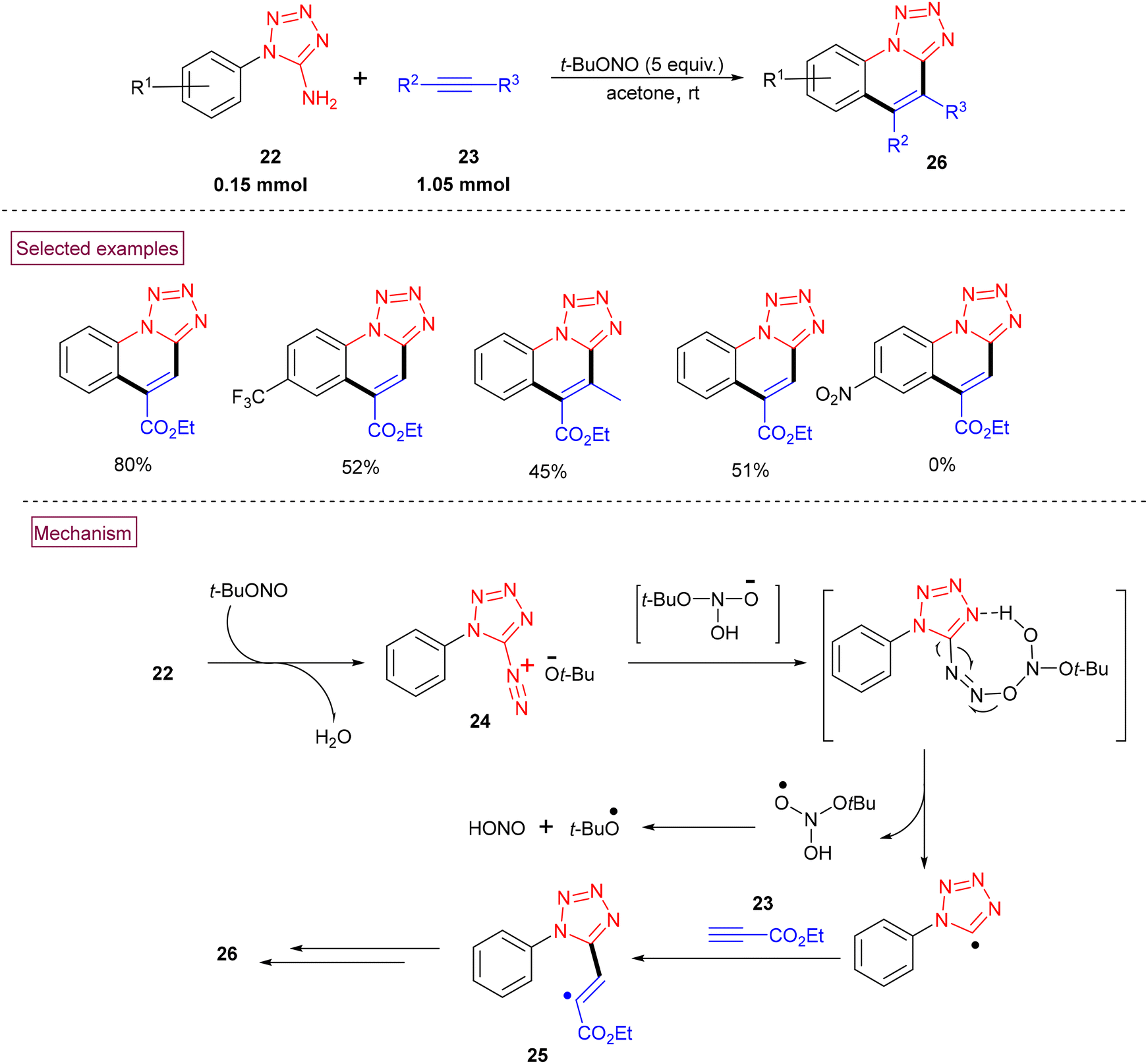
The efficient radical cyclization of 1H-tetrazole-5-amines (22) and alkynes (23) to afford tetrazolo[1,5-a]quinolines (26) via the in situ formation of tetrazolate-diazonium salt (24) mediated by tert-butyl nitrite was realized in 2019.95 This novel approach proceeded without using additives or metal catalysts under mild conditions (Scheme 8). According to the plausible mechanism, it was concluded that the in situ-formed diazonium salt (24) reacted with the deprotonated form of O-(tert-butyl)-N,N-dihydroxyl-amine (formed by the reversible reaction of excess tert-butyl nitrite with water) to afford a diazoether intermediate, which upon homolytic cleavage, followed by a reaction with alkyne (23) resulted in the formation of alkenyl radical (25). Further intramolecular radical cyclization, single-electron oxidation and deprotonation provided (26).
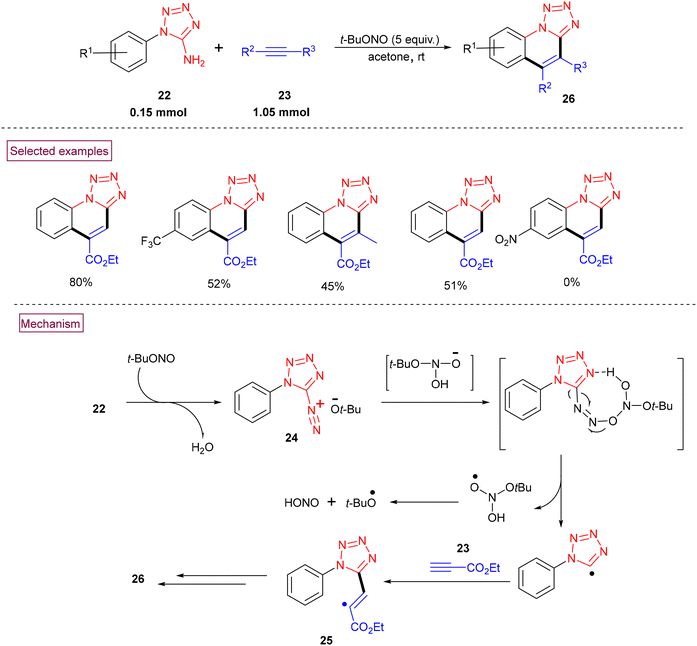 |
| | Scheme 8 Preparation of tetrazolo[1,5-a]quinolines via radical cyclization. | |
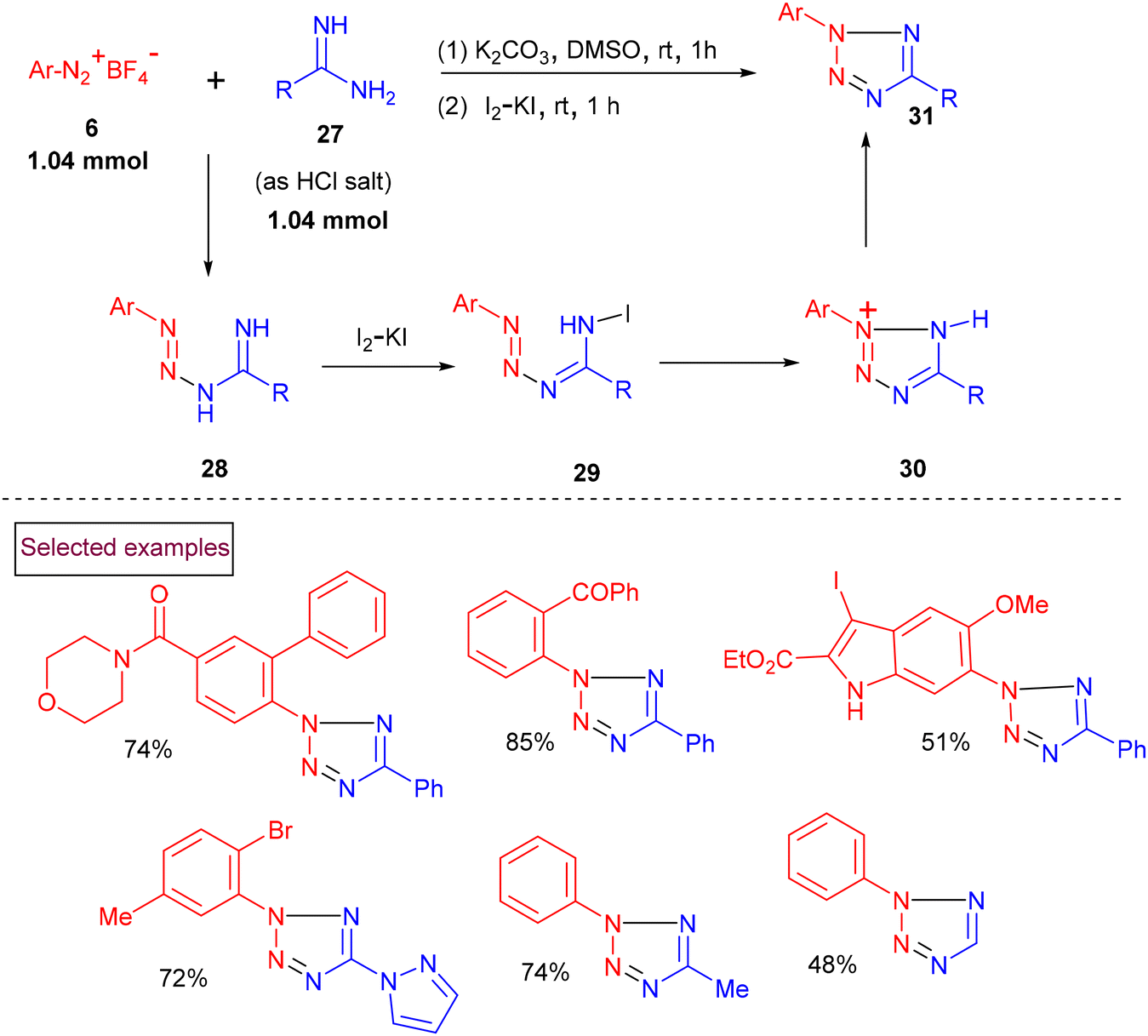
Kakehi et al. reported the synthesis of 2,5-diarylated tetrazole derivatives from phenylsulfonylhydrazone and aryldiazonium salts via a one-pot addition/cyclization sequence.96 Pioneered by this report, in 2015, we reported the atom-economic synthesis of highly substituted tetrazoles (31) from aryldiazonium salts (6) and amidines (27) (Scheme 9).97 The formed imino-triazines (28) were oxidatively cyclized to 2,5-disubstituted tetrazoles (31) using I2/KI under basic conditions. This one-pot approach delivered a wide range of tetrazole derivatives (31) in higher yields even on a multi-gram scale. The notable advantages of this strategy are mild reaction conditions, short reaction time and environmentally friendly conditions.
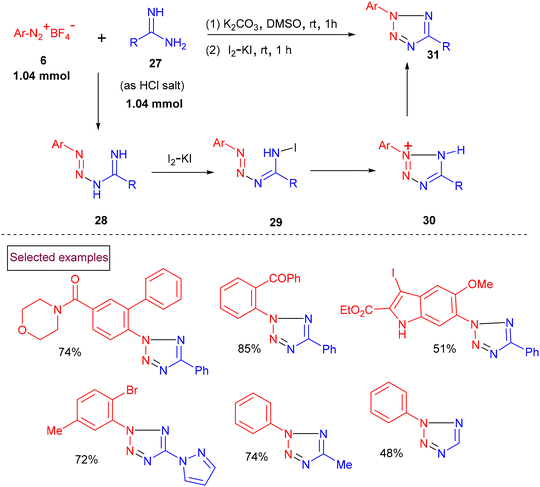 |
| | Scheme 9 One-pot synthesis of substituted tetrazoles from aryldiazonium salts and amidines. | |
3.2. 1,2,4-Triazole derivatives
The development of synthetic methods for the construction of 1,2,4-triazoles from aryldiazonium salts has increased in recent years due to their prevalent occurrence in numerous biologically active natural products.
Earlier this year, a convenient method to prepare multi-substituted 1,2,4-triazoles (39) and (39′) from the [3 + 2] cycloaddition of 2H-azirines (32) and aryldiazonium salts (6) in the presence of an organo-photo catalyst was reported by Rastogi et al.98 Regioisomeric mixtures of the triazole product (39 and 39′) were observed when unsymmetrically substituted azirines (32) were employed (Scheme 10). Interestingly, typical aryl radical generation from aryldiazonium salts (6) via visible light-mediated photocatalyzed conditions was avoided by choosing a reductive photocatalytic quenching cycle in the presence of an organic dye (9-mesityl-10-methylacridinium perchlorate) with strong oxidizing ability. This careful choice of reaction conditions furnished the desired multi-substituted 1,2,4-triazoles (39) via the intermediacy of azaallenyl radical cation (35). The presence of strong electron-withdrawing substituents (CN, NO2, COMe, CO2Et and CF3) in aryldiazonium salts (6) resulted in the formation of products in very low yields, whereas other substituents gave moderate to good yields. Mechanistically, the reaction was initiated via the single-electron reduction of the excited photocatalyst, resulting in the formation of azaallenyl radical cation (35) via azirinyl radical cation (33). The addition of this radical intermediate to the aryldiazonium salt (either pathway A or B) yielded azo radical cation intermediate (36) or (36′). Further SET oxidation, intramolecular cyclization, and base-mediated dehydration afforded products (39 or 39′).
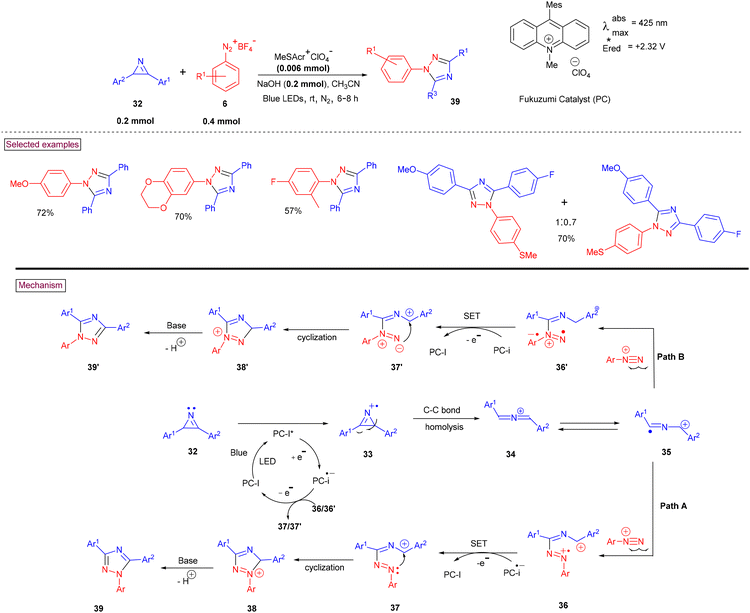 |
| | Scheme 10 Organo-photocatalyzed synthesis of 1,2,4-triazole derivatives. | |
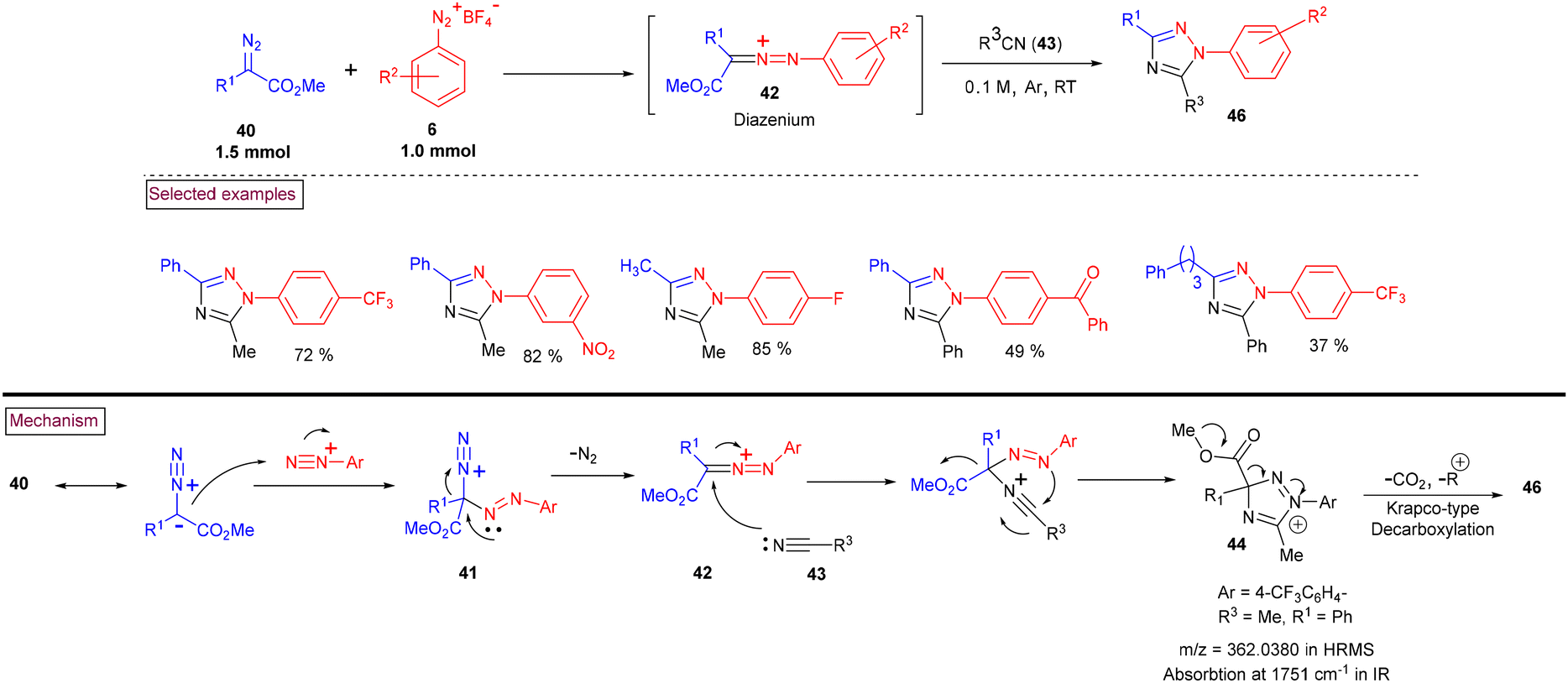
An attractive strategy to access highly substituted 1,2,4-triazole derivatives (46) via a donor/acceptor diazo activation strategy was reported in 2021 (Scheme 11).99 In this report, nucleophilic attack of α-substituted diazoester (40) on the terminal nitrogen of diazonium ion (6), followed by the extrusion of N2 afforded key diazenium intermediate (42). Further reaction with nucleophilic nitriles (43) and cyclization afforded intermediate (44). A sequential Krapcho-type decarboxylation and isomerization of intermediate (44) led to a diverse array of triazole derivatives (46). A similar Krapcho-type decarboxylation and isomerization was reported by Ma et al. (Scheme 12)100 Overall, this novel approach possesses notable functional group tolerance and proceeded with various substituted aryldiazonium salts (6), α-substituted diazoester (40) and nitriles (43).
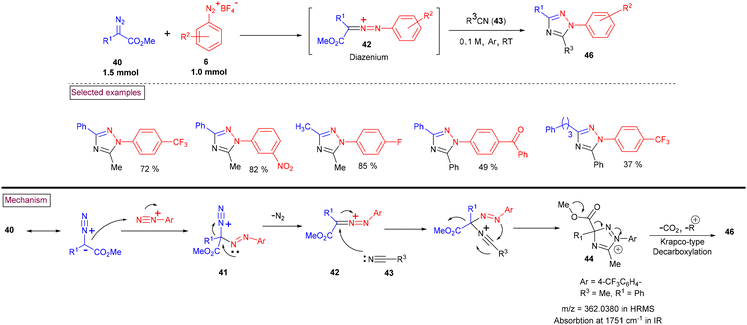 |
| | Scheme 11 Preparation of 1,2,4-triazole derivatives via donor/acceptor diazo activation. | |
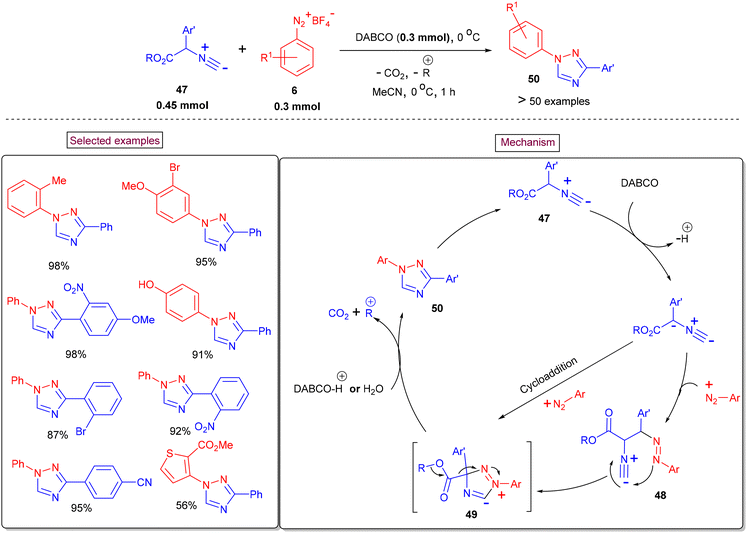 |
| | Scheme 12 Preparation of 1,2,4-triazole via decarboxylative annulation. | |
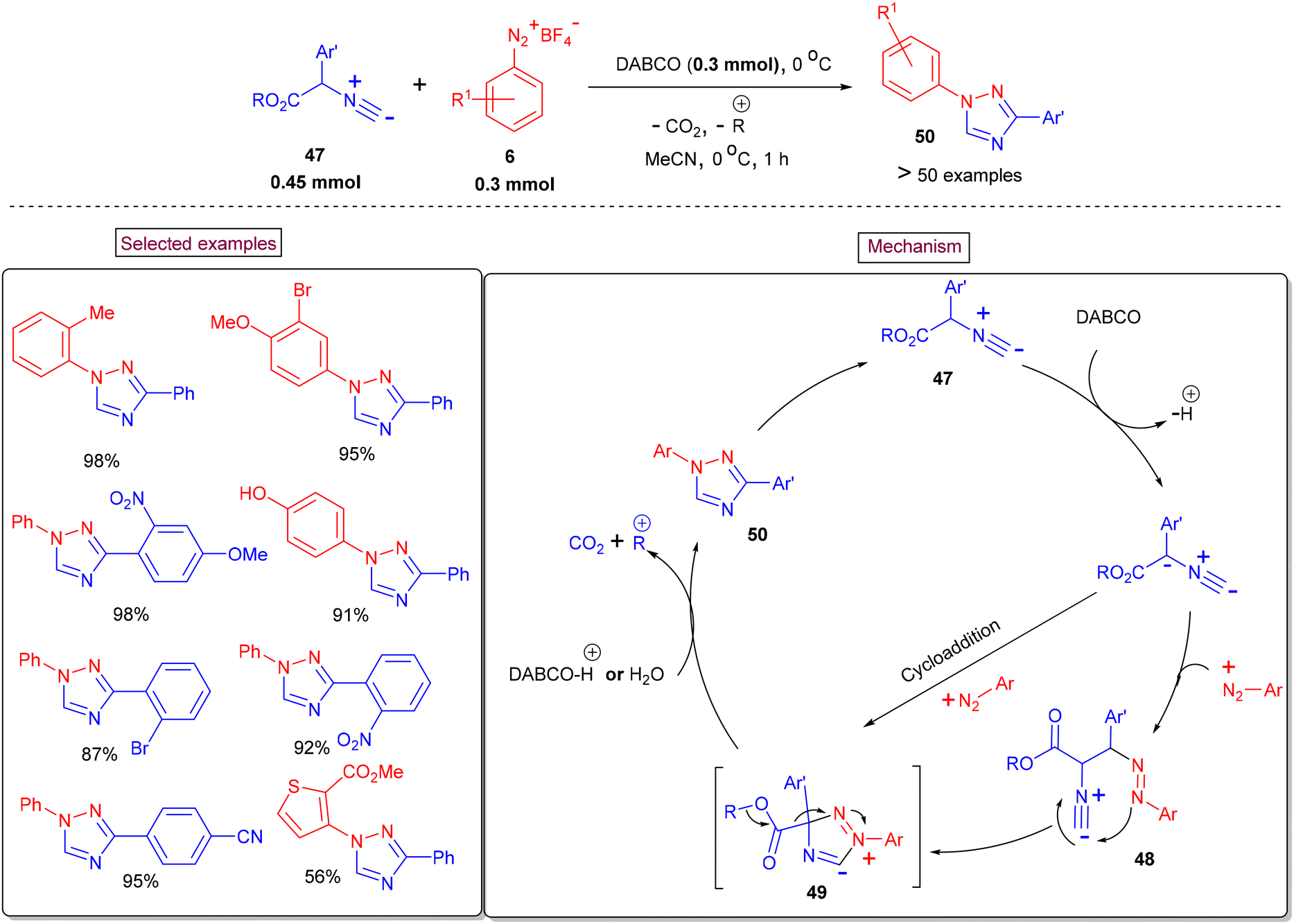
In continuing efforts to develop innovative methodologies for N-heterocycles, Ma et al. reported a decarboxylative annulation reaction of aryldiazonium salts (6) with 2-aryl-2-isocyanoacetates (47) to construct 1,3-diaryl-1,2,4-triazoles (50) under transition-metal-free conditions (Scheme 12).100 It was proposed that the anionic intermediate is formed due to the deprotonation of isocyanoacetate precursor (47), which then reacts with aryldiazonium salt (6) to give species (48). Further cyclization resulted in the formation of intermediate (49), which could also be obtained from the cycloaddition of aryldiazonium salt with an anionic intermediate. Subsequent Krapcho-type decarboxylation and isomerization furnished the desired product (50). Various synthetic utilities such as the synthesis of chiral triazolyl 1,1′-binaphthalene, an anti-tumor agent and potential inhibitor of vascular endothelial growth factor receptors 1 and 2, have also been demonstrated. In addition, this expeditious, regiospecific and base-promoted synthesis of 1,3-diaryl-1,2,4-triazoles (50) is scalable and enables one-pot operation from the corresponding aniline derivatives through in situ-generated diazonium salts.
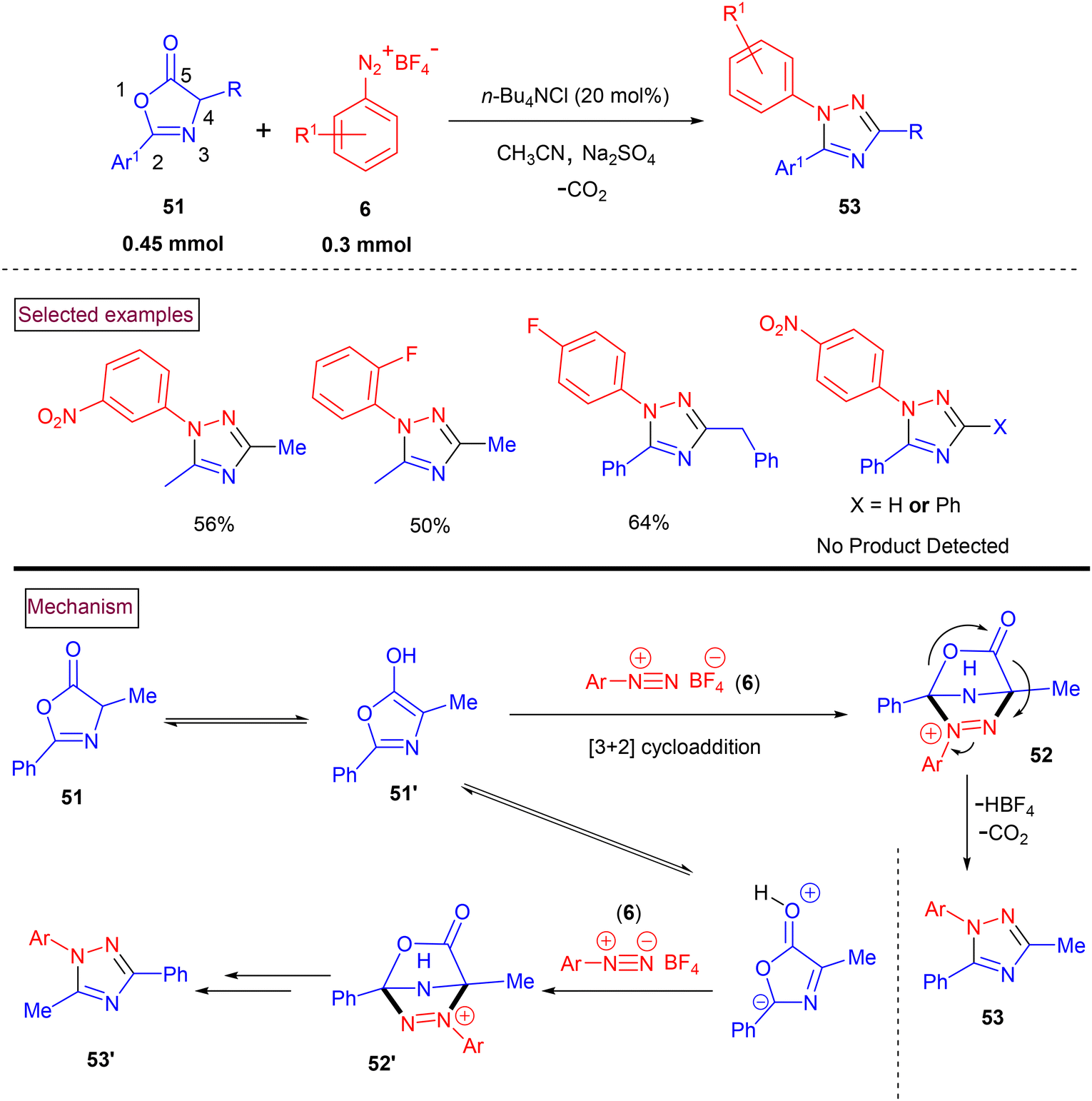
Chen's research group (2019) reported the facile synthesis of highly substituted 1,2,4-triazole derivatives (53) ranging from aryldiazonium salts (6) and azlactones (51) (Scheme 13).101 Various amino acid-derived azlactones including N-Boc-protected tryptophan resulted in the formation of the desired products in moderate yields. However, 4-Ph and 4-H-substituted azlactones gave no desired products. The scope of this method was also evaluated with a range of aryldiazonium salts (6) and the corresponding products (53) were obtained in acceptable yields. In a few cases, a mixture of regioisomeric products (53′) was found. The ratio of mixture was based on the addition site of azlactone (either C2 or C4). The plausible mechanistic study indicated that the reaction proceeded via a [3 + 2] cycloaddition and ring-opening decarboxylation cascade sequence. Accordingly, the enol tautomer of azlactone (51′) underwent cycloaddition with aryldiazonium salt (6) to yield cyclized intermediate (52) and/or (52′). Further decarboxylative ring opening resulted in the formation of the desired product (53) and/or (53′).
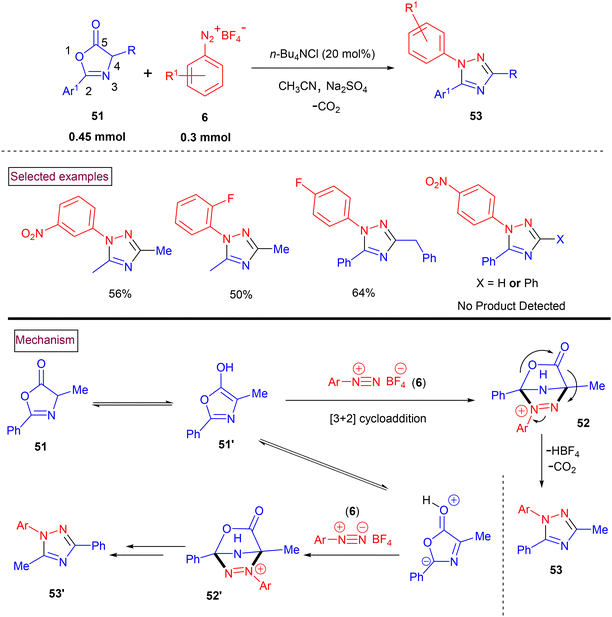 |
| | Scheme 13 Preparation of 1,2,4-triazoles via cycloaddition/decarboxylation cascade steps. | |
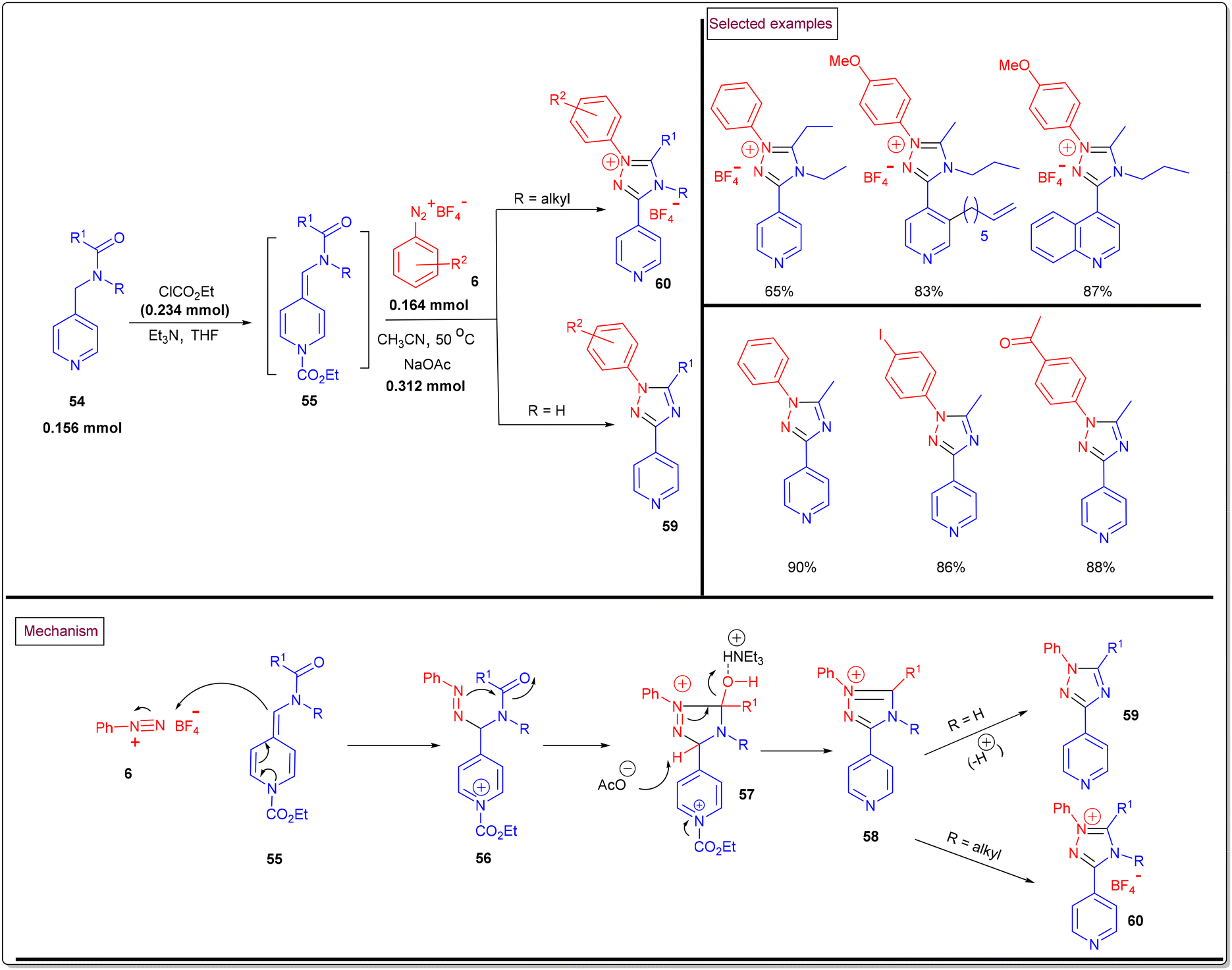
A novel route to access pyridine-tethered 1,2,4-triazole derivatives (59 and 60) through the [3 + 2] cyclo-condensation of alkylidene dihydropyridines 55 (generated via in situ dearomatization of pyridines/quinolines (54) upon reaction with chloroformate) and aryldiazonium salts (6) under mild conditions was reported in 2016 (Scheme 14).102 A wide range of dihydro analogues of pyridine and quinoline were involved in this transformation to afford the desired products in good yields (65%–98%). Notably, aryldiazonium salts bearing electron-releasing groups only participated in this process, whereas nitro-functionalized salts failed. Mechanistically, the nucleophilic addition of alkylidine–dihydropyridine (55) to diazonium salts (6) resulted in the formation of diazo intermediate (56). Further intramolecular cyclization of (56), benzylic deprotonation (57) and aromatization (58) processes led to 1,2,4-triazoles (59). When N-alkyl substituted dihydropyridines were employed, the corresponding triazolium salts (60) were obtained.
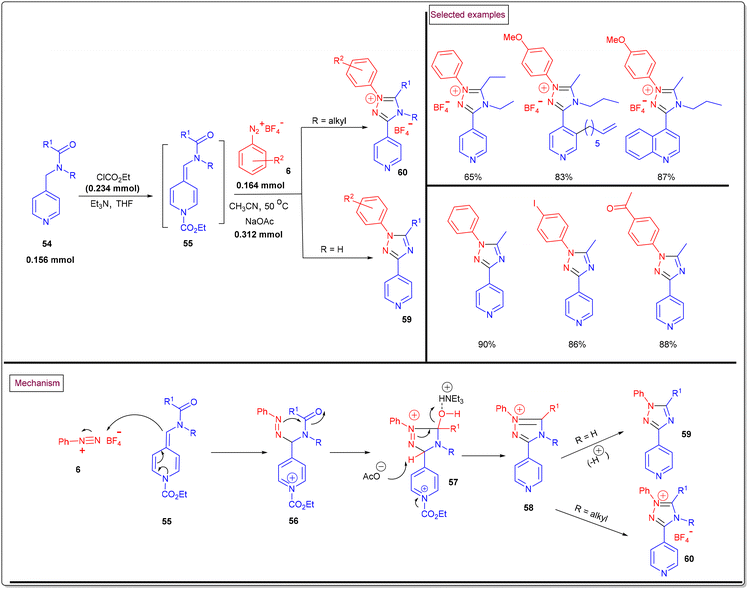 |
| | Scheme 14 Preparation of pyridine-tethered 1,2,4-triazole derivatives through [3 + 2] cyclo-condensation. | |
3.3. 1,2,3-Triazole derivatives
1,2,3-Triazoles represent a versatile framework that can be found in numerous bioactive molecules and materials.103 Important drugs such as rufinamide (anti-convulsant),104 cefatrizine, tazobactam (antibiotic),105,106 and carboxyamidotriazole (anti-cancer),107 possess this ubiquitous core in their frameworks. In addition, owing to their incredible applications in catalysis, carbene chemistry, and C–H activation, several novel approaches have been reported in recent years.108 The synthesis of 1,2,3-triazoles with N2–aryl substitution is highly challenging compared to the N1–aryl counterparts.
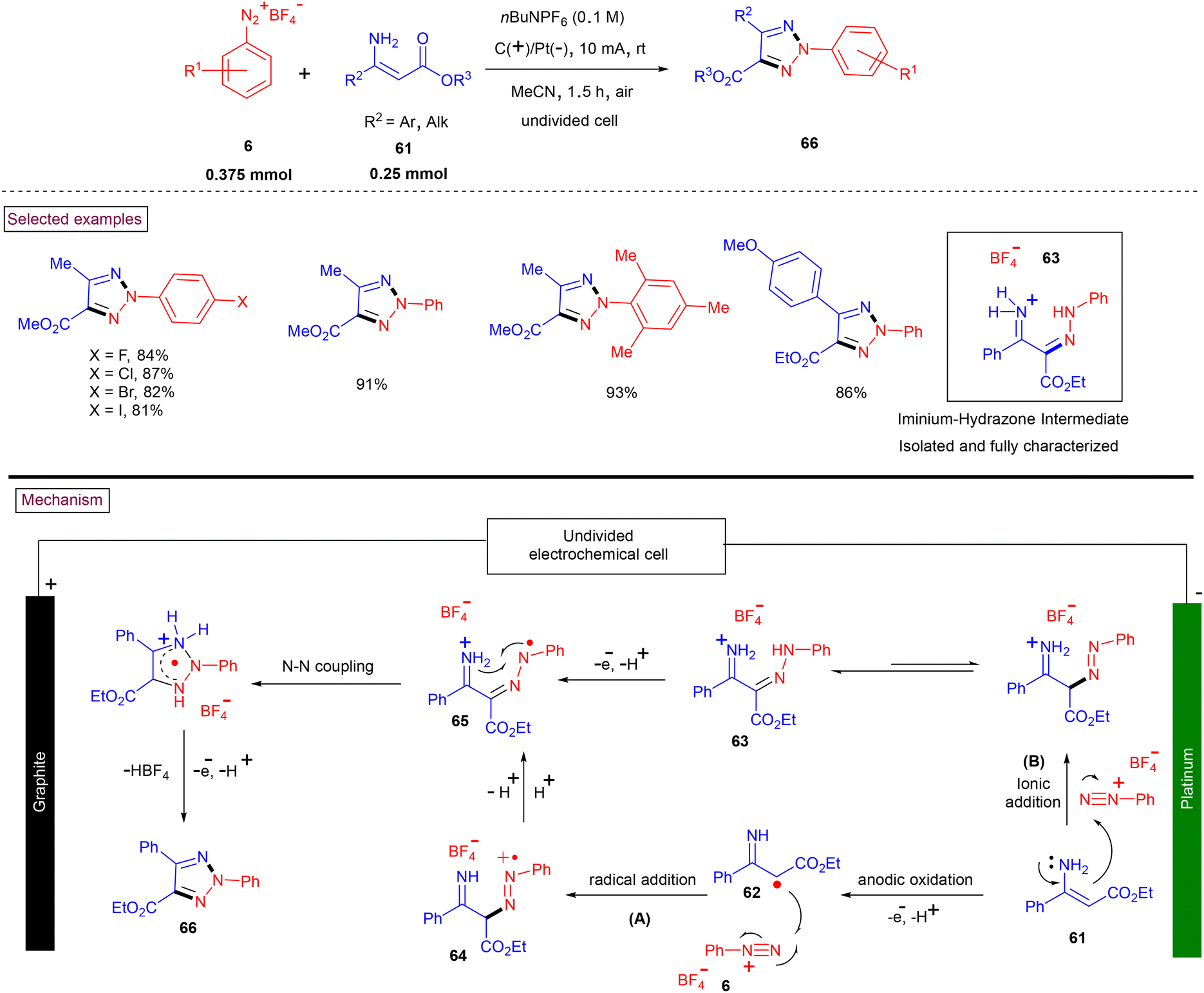
In recent years, aryldiazonium salts serve as one of the key precursors in several elegant reactions such as Cu-catalysed coupling of oxime acetates,109a Cu-catalysed intermolecular cyclization of arylamines,109b Cu(II)-catalyzed tandem C–H nitrogenation/annulation reaction of alkylthio enaminones,109c and Cu-mediated annulation with alkyl-3-aminoacrylates66 have been developed by several research groups. However, transition-metal-free aryldiazonium salts-based methods are scarce. To this end, highly regioselective N2-arylated 1,2,3-triazole derivates were synthesized by De Sarkar et al. via sequential C–N bond formation and electro-oxidative N–N coupling (Scheme 15).110 This chemical-oxidant and metal-free procedure for the preparation of N2-aryl 1,2,3-triazoles (66) proceeded at room temperature and afforded the functionalized triazoles in excellent yields from readily available enaminones (61) and aryldiazonium salts (6). The construction of the N–N bond was realized under electrochemical conditions and open-air atmosphere. It was proposed that radical cation (64) was formed (path A) via the anodic oxidation of (61) followed by the radical addition of (62) to (6), which upon H-shuffling yielded radical cation (65). Intermediate (65) was also proposed (path B) to be formed via the ionic addition of enamine (61) to diazonium salt (6) to generate imine-hydrazone intermediate (63), followed by a proton-coupled electron transfer process. The desired triazoles (66) were obtained via intramolecular N–N coupling and simultaneous liberation of HBF4.
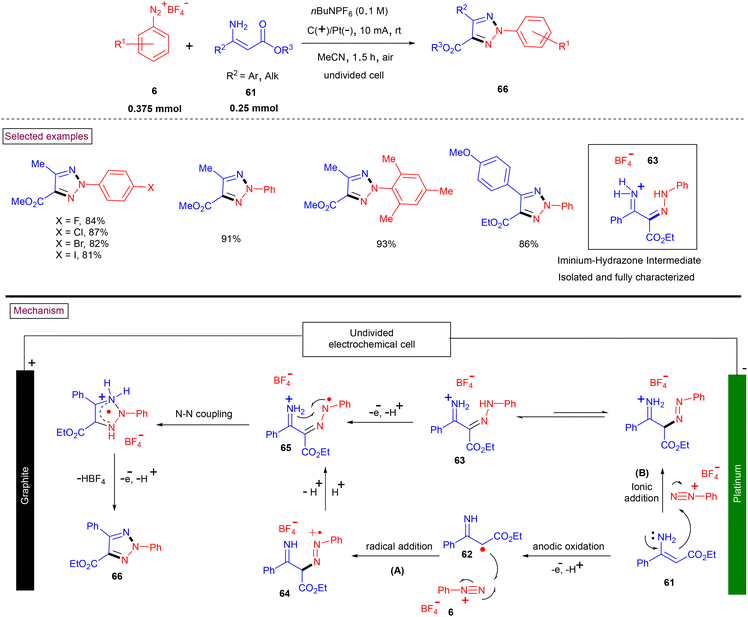 |
| | Scheme 15 Preparation of 1,2,3-triazole derivates via electro-oxidative N–N coupling. | |
3.4. Benzotriazole derivatives
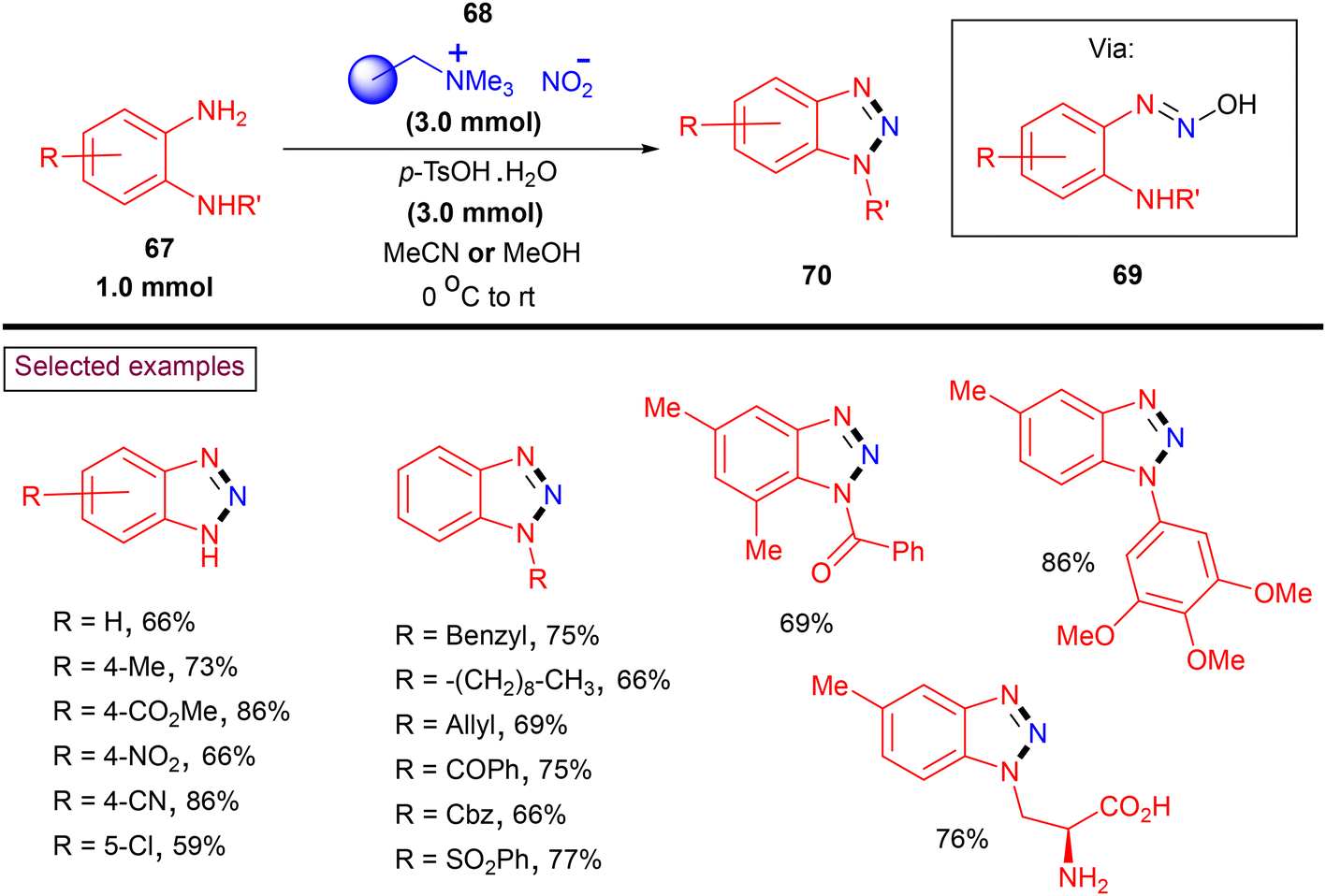
Benzotriazoles are widely applied, and especially in organic synthesis, these N-heterocyclic cores have been utilized as precursors to access pyridoacridines, carbazoles and indoles.111 Moreover, owing to their enzyme inhibition properties through π–π stacking or H-bonding of the triazole core, these scaffolds are widely used in drug chemistry.112 In 2019, Sutherland and co-workers reported a rapid diazotization and cyclization strategy to prepare both N-unsubstituted and N1-substitued benzotriazoles (70) from 1,2-aryldiamines (67) employing a polymer-supported nitrite (68) (Scheme 16).113 A wide range of N-unsubstituted and N-alkyl, aryl, allyl, benzyl, benzyloxy-carbonyl (Cbz), benzoyl, and sulfonated benzotriazoles (70) has been prepared via the intermediacy of (69) in moderate to good yields (up to 95%). Functional groups including –F, –CF3, and CO2Me survived under the reaction conditions. The application of this safe, operationally simple and one-pot method has been demonstrated by preparing various medicinally important benzotriazoles including an α-amino acid analogue.
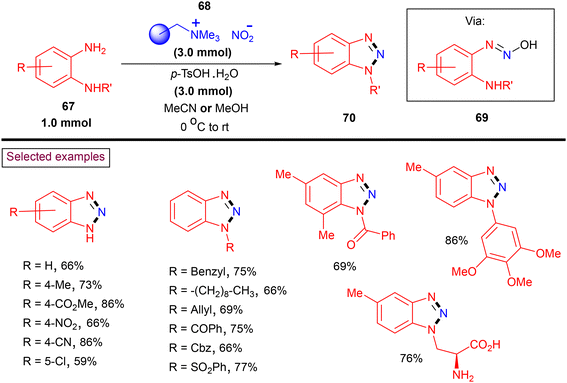 |
| | Scheme 16 Preparation of N1-substitued benzotriazoles from polymer-supported nitrite. | |
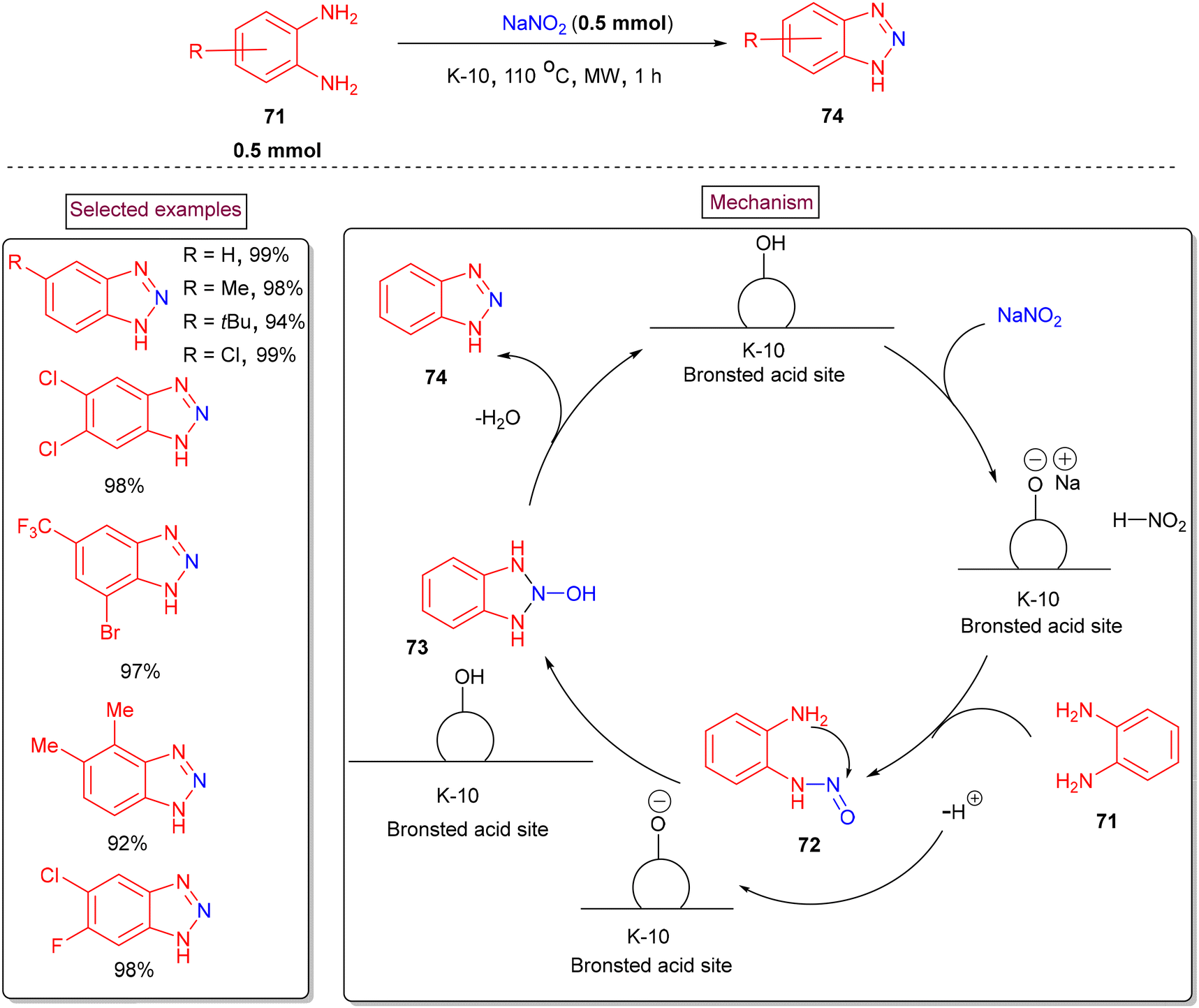
Török et al. demonstrated a cyclization of o-phenylenediamines to access functionalized benzotriazole derivatives via microwave-assisted environmentally friendly solid-phase diazotization process (2017).114 K-10 montmorillonite was employed as a catalyst and reaction medium owing to its high surface area and excellent microwave absorption capacity (Scheme 17). The reaction in the conventional heating-setup resulted in lower yields due to the formation of impurities. The scope of the reaction was studied with various multi-substituted o-phenylenediamines bearing groups such as –Me, –CF3, –tBu, –Cl, and –F and the corresponding products in excellent yields (92%–99%). Mechanistically, the in situ diazotization of o-phenylenediamine (71) resulted in the formation of nitroso compound (72), which furnished the desired benzotriazole derivatives (74) via an intramolecular cyclization (73) and dehydration.
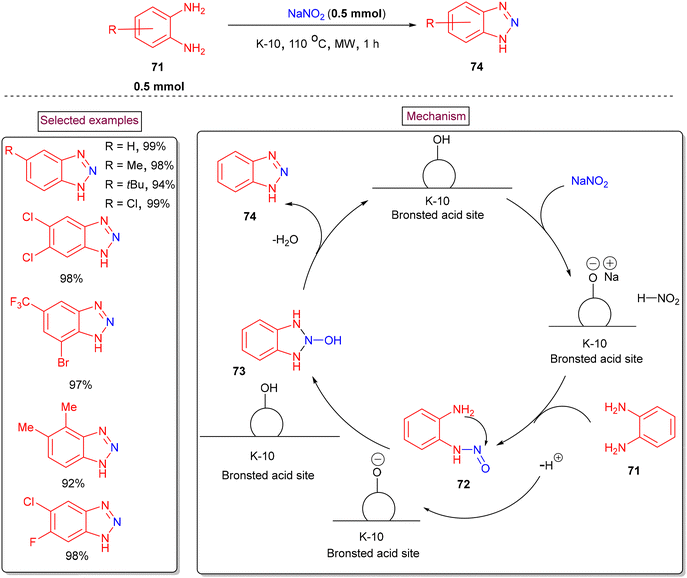 |
| | Scheme 17 Microwave synthesis of benzotriazoles via cyclization of o-phenylenediamines. | |
3.5. Benzimidazole derivatives
Benzimidazoles are intriguing N-heterocyclic compounds found in numerous biologically interesting compounds. Their vital structure in medicinal chemistry is recognized due to their valuable therapeutic activities such as antihypertensive,115a antiulcer,115b antiviral,115c anticancer,115d antifungal115e and antihistaminic.115f Moreover, substituted benzimidazole derivatives possess significant antiproliferative activities towards various cancer cell lines including MCF7, HepG2, A549, and HCT116. Access to densely functionalized benzimidazole derivatives is realized via metal-catalysed multicomponent reactions, oxidative cross-coupling of anilines or intramolecular C–H amidation strategies.116 The use of toxic azides or expensive catalysts often reduce the scope of these routes. Alternatively, the aryldiazonium-based one-pot and metal-free construction of highly substituted benzimidazole derivatives offers several advantages such as operationally simple, cost effective and greener processes.
The preparation of N-arylbenzimidazoles (78 and 78′) from aryldiazonium salts (6), nitriles and anilines (75) via the intermediacy of N,N′-diarylamidines, followed by oxidative cyclization using PhI(OAc)2 was realized by Youn and coworkers (Scheme 18).117 The limitations of this method were studied with various substituted aryldiazonium salts (6) and anilines (75), and the corresponding products (78) were obtained in moderate yields. The desired benzimidazoles were obtained by the substitution of aryldiazonium salts with nitriles to afford highly reactive N-arylnitrilium salts (76), which further underwent nucleophilic attack by anilines (75) and subsequent oxidative cyclization. Notably, regioselective preference was observed during oxidative cyclization on the electron-rich benzene rings compared to their electron-deficient counterparts.
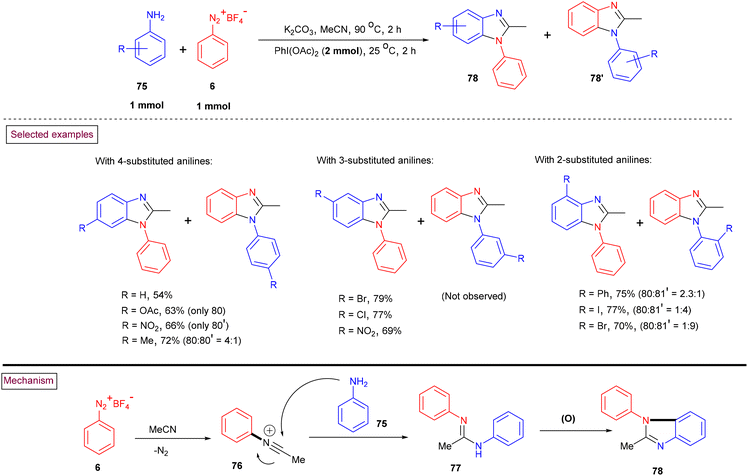 |
| | Scheme 18 Preparation N-arylbenzimidazoles via PhI(OAc)2-mediated oxidative cyclization. | |
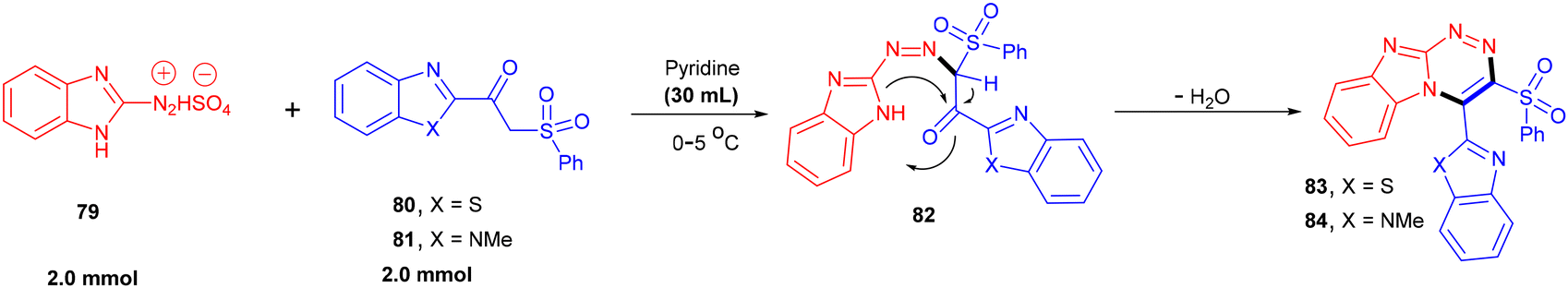
The diazonium salt of 2-aminobenzmidzaole (79) was reacted with benzimidazole (80) or benzothiazole based β-ketosulfones (81) to afford diazo derivatives (82) in good yields (Scheme 19).118 Further base-promoted cyclization and dehydration resulted in the formation of fused pyridazines (83 and 84). This dehydrative cyclization strategy was also applied to diazonium salts derived from 5-aminopyrazole and 5-amino[1,2,4]-triazole.
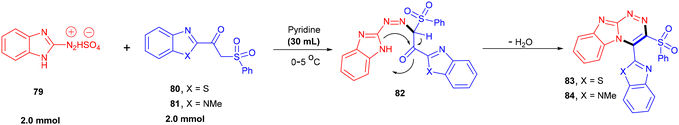 |
| | Scheme 19 Preparation of benzimidazole-fused pyridazines via the dehydrative approach. | |
3.6. Indazoles and pyrazoles
Multi-substituted pyrazoles and fused pyrazole derivatives are ubiquitous N-heterocycles that are found in a wide spectrum of pharmaceuticals, biologically active natural products and functional materials.119 The preparation of the pyrazole core is usually accomplished by the condensation of aryl aldehydes and hydrazines or cycloaddition of dialkyl azodicarboxylates with propargylamines. In recent years, several elegant reports have been disclosed, involving aryldiazonium salts to construct multi-substituted pyrazole scaffolds.
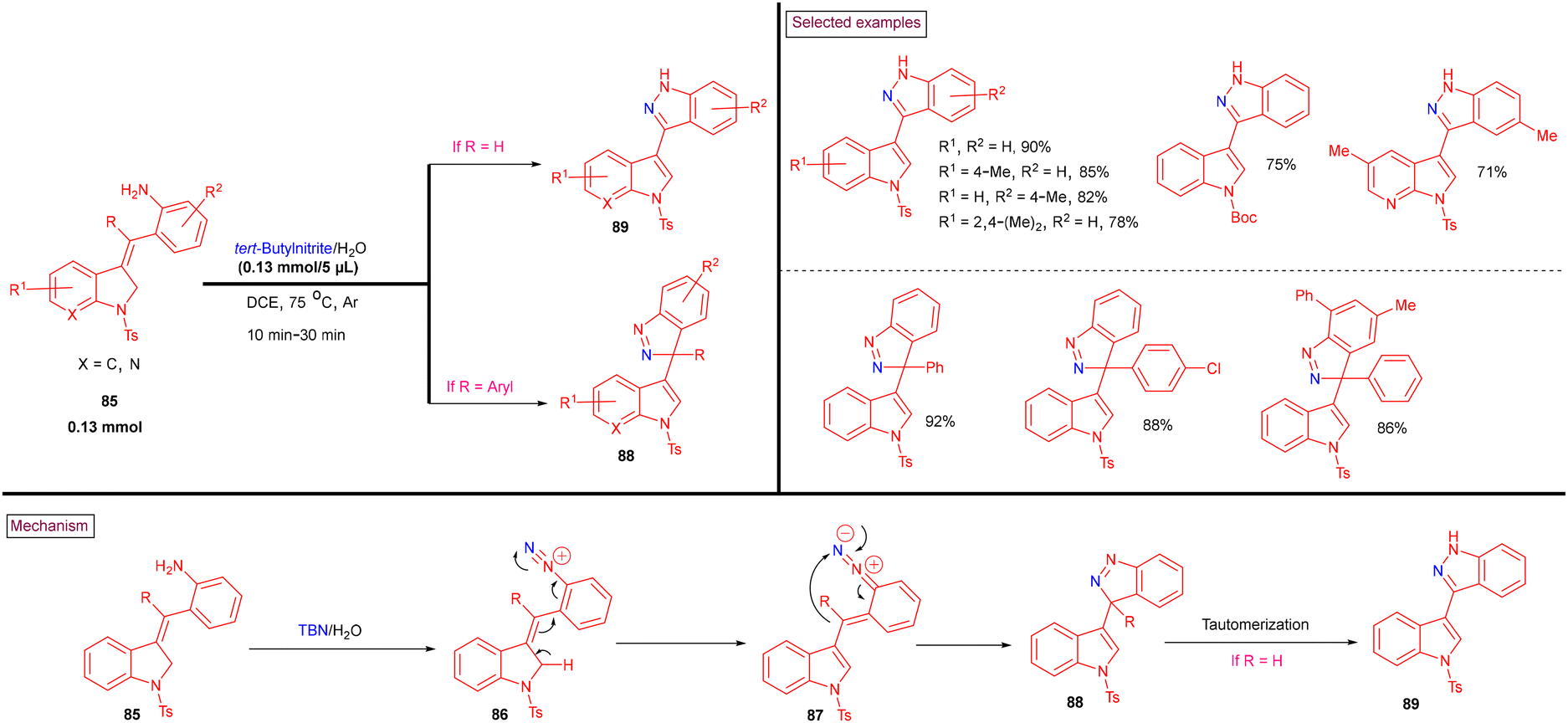
Recently, tert-butyl nitrite (TBN)-mediated C3-funtionalized indazole-indoles (89) were prepared from 2-(indolin-3-ylidenemethyl)anilines (88) via the in situ generation of diazonium salt (86) (Scheme 20).120 The reaction proceeded via the tandem isomerization of in situ-generated diazonium salt (86) and sequential C–N bond formation (88) via 5-endo-dig intramolecular cyclization. A broad range of 1H-indazole-indoles (89), 1H-indazole-azaindole (88), and 3H-indazole-indole hybrid structures were obtained with excellent functional group compatibility. The plausible mechanism was supported by control experiments. The notable advantages of this method are its gram-scale synthesis, mild conditions and good regioselectivity.
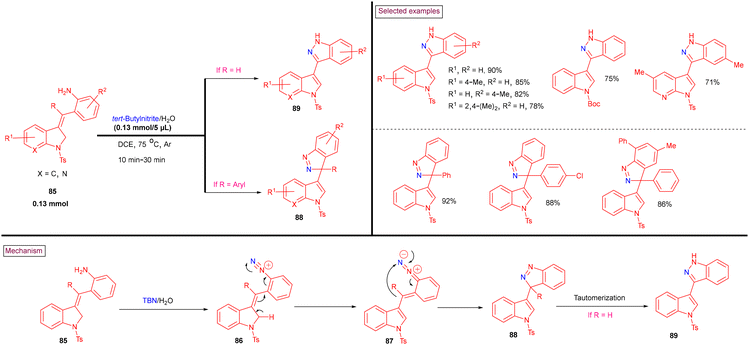 |
| | Scheme 20 TBN-mediated synthesis of C3-funtionalized indazole-indoles. | |
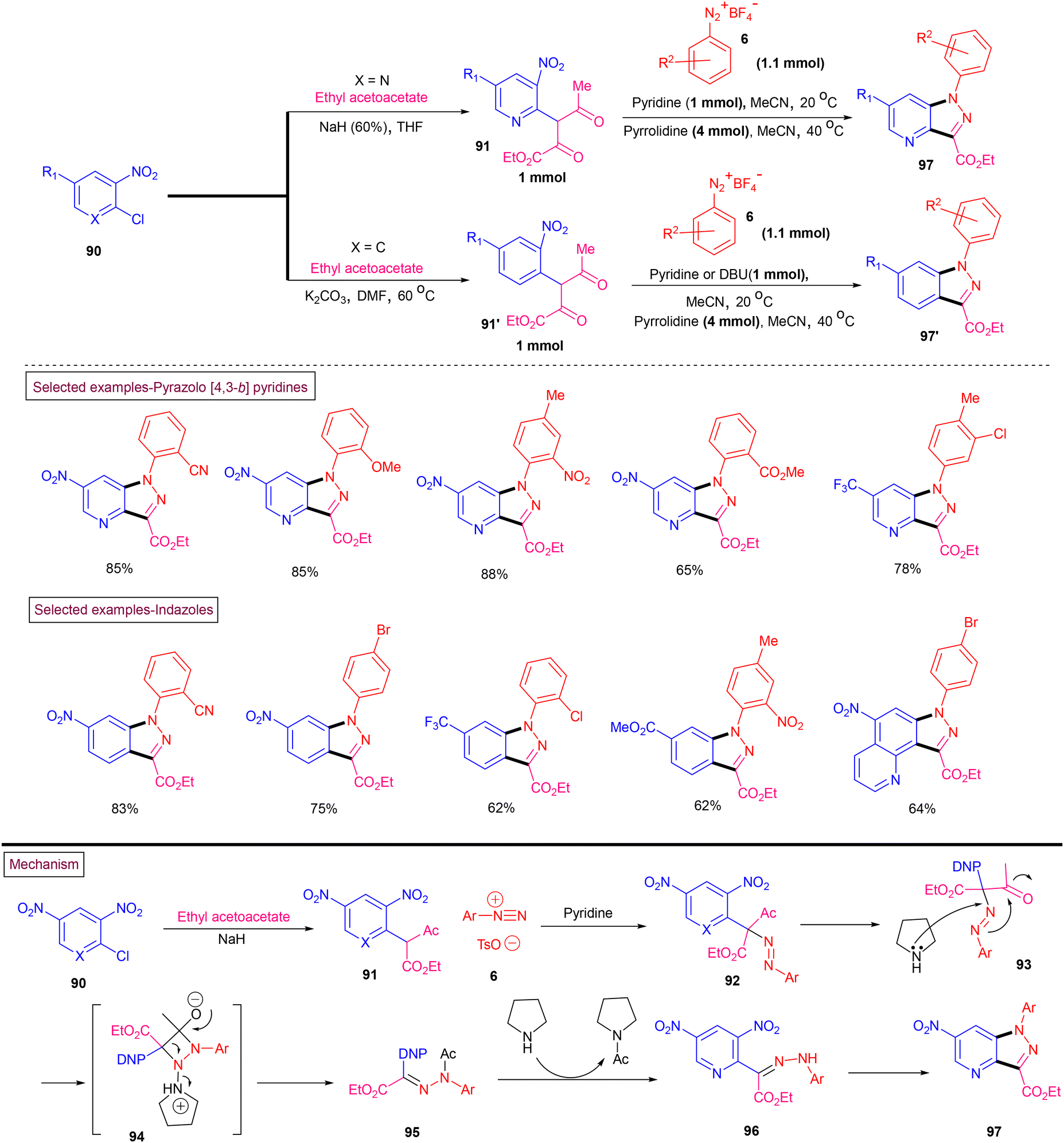
Starosotnikov and co-workers reported a one-pot method for the preparation of pyrazolo[4,3-b]pyridines (97) and indazoles (97′) from readily available substrates under transition metal-free conditions (Scheme 21).121 Commercially available 2-chloro-3-nitro-pyridines (90) were converted into pyridinyl keto esters (91) by reacting with an ethyl acetoacetate/NaH mixture via the SNAr process. Subjecting the keto esters to Japp–Klingemann conditions with aryldiazonium salt (6) resulted in the formation of hydrazone derivative (95). Further deacetylation and intramolecular nucleophilic substitution of the –NO2 group furnished cyclized pyrazolo[4,3-b]pyridines (97) in good yields. Interestingly, this method was also applied for the synthesis of indazoles (97′) by employing chloronitrobenzenes. Owing to their lower reactivity, phenyl keto esters (91′) were obtained by reacting the 1-chloro-2-nitrobenzenes with ethyl acetoacetate at elevated temperature (60 °C) in the presence of K2CO3. Final cyclization was executed using DBU as a base given that the cyclization was sluggish when pyrrolidine was utilized. A diverse range of electronically and sterically distinct aryldiazonium salts reacted smoothly under the reaction conditions to afford the corresponding products in moderate to good yields.
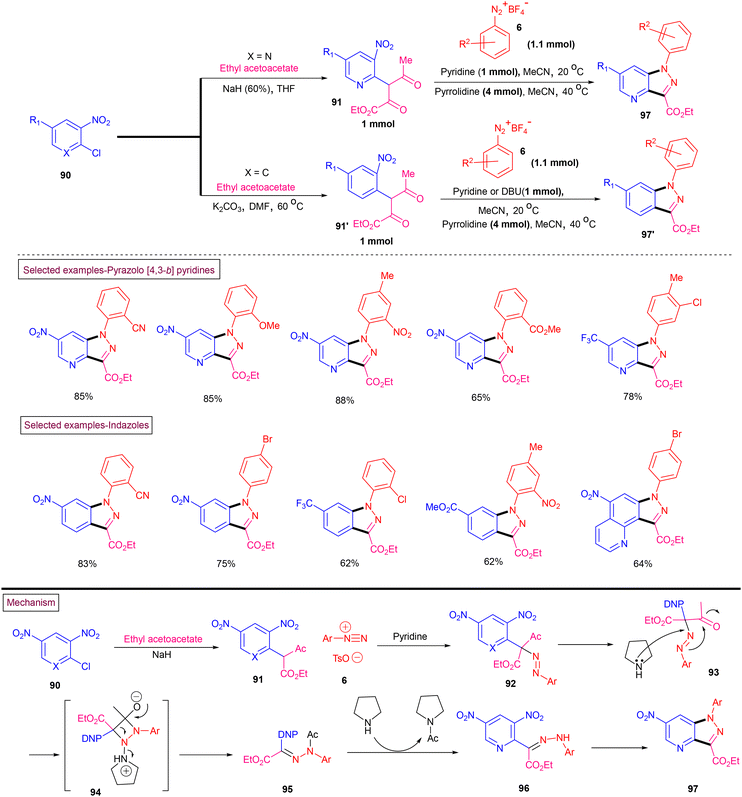 |
| | Scheme 21 Preparation of pyrazolo[4,3-b]pyridines and indazole derivatives. | |
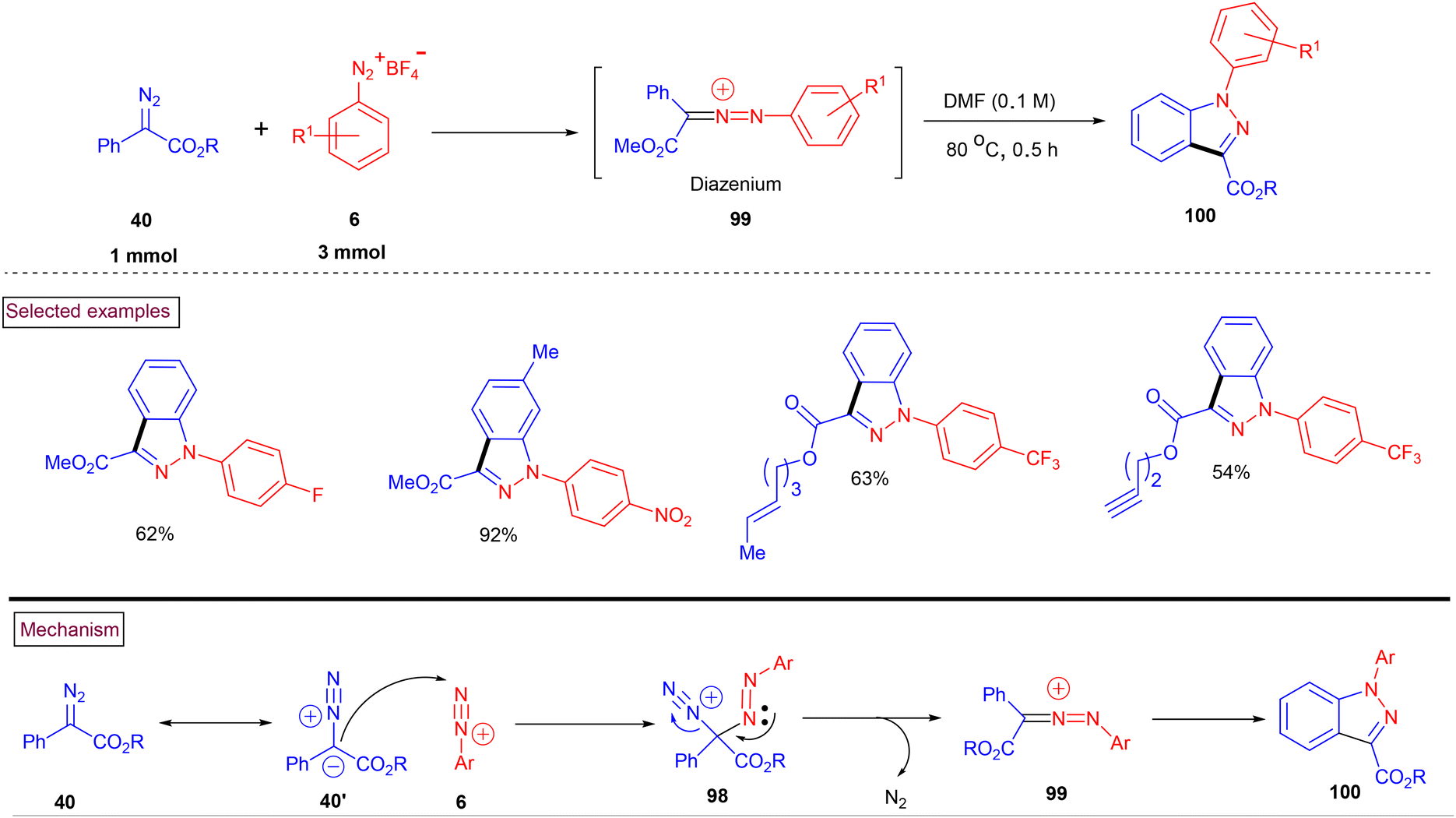
In 2020, Wang and Shi et al. reported a convenient metal-free strategy for the preparation of functionalized indazole derivatives (100) from aryldiazonium salts (6) and a diazo derivative (40) (Scheme 22).99 This transformation proceeded at 80 °C and exhibited excellent substrate scope. Aryldiazonium salts (6) with electron-withdrawing groups reacted more efficiently than electron-donating groups, which underwent rapid decomposition. Benzyl, alkyne, and menthol-derived esters (40) were compatible under the reaction conditions. The proposed reaction pathway was initiated by the nucleophilic addition of azo precursor (40′) to aryldiazonium salt (6), and subsequent denitrogenation to afford diazenium intermediate (99). Further intramolecular electrophilic cyclization resulted in the formation of indazole derivatives (100).
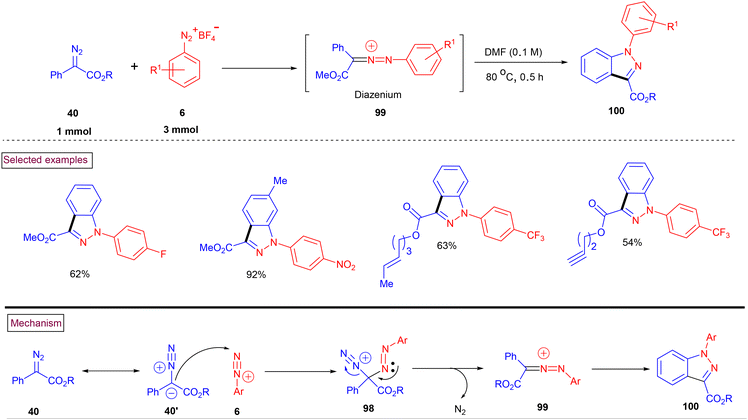 |
| | Scheme 22 Preparation of indazoles from aryldiazonium salts and diazo compounds. | |
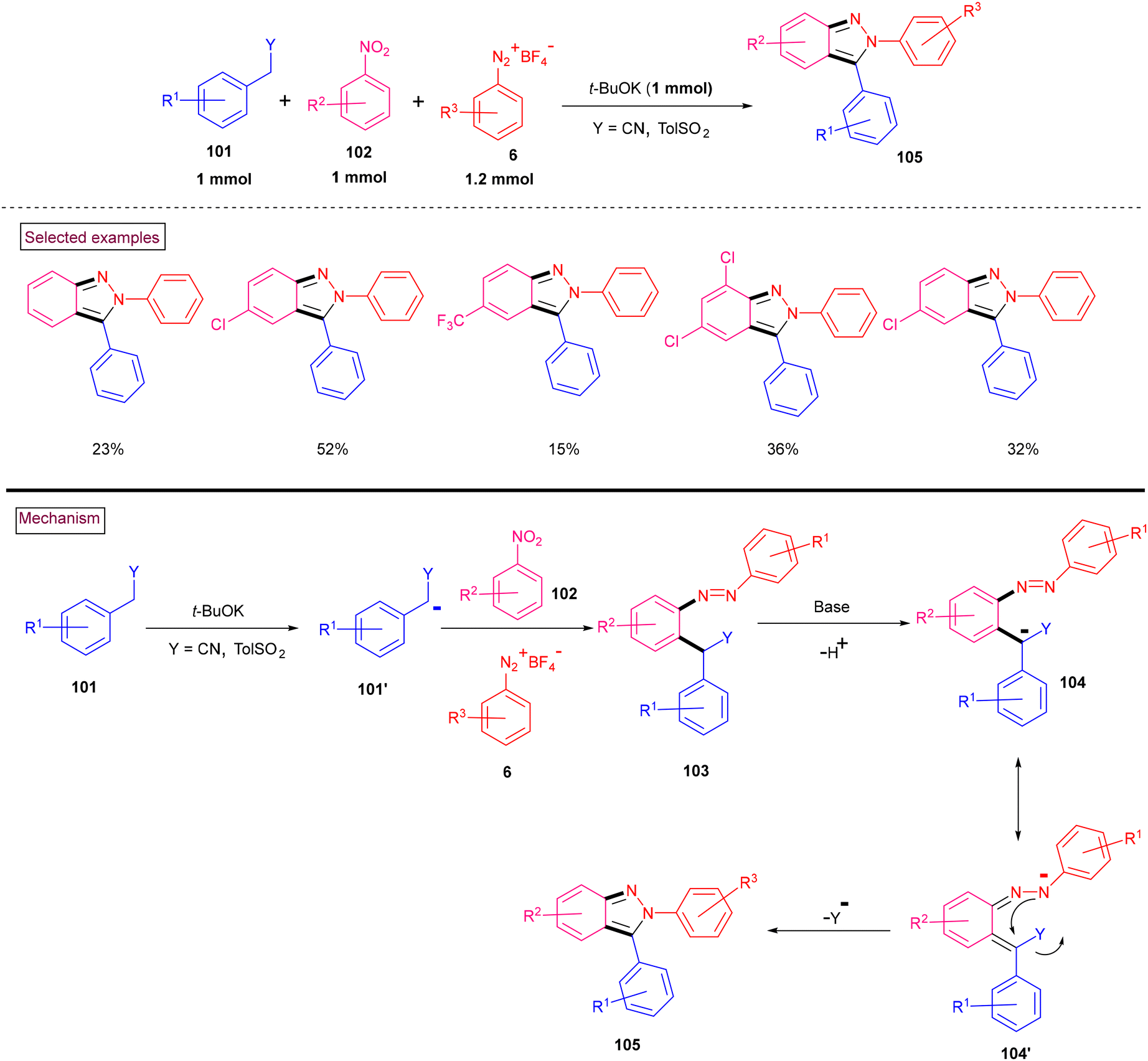
Stabilized carbanions of active methylene substrates bearing sulfonyl, alkoxycarbonyl and cyano groups (101′) reacted with nitrobenzenes (102) to generate a σH adduct (Wróbel et al. 2021) (Scheme 23).122 These unstable adducts were coupled with aryldiazonium substrates (6) preferentially at the NO2-substituted carbon atom (with the elimination of HNO2) or the meta-position (with the elimination of HCl), leading to azobenzenes. However, when less crowded secondary carbanions were employed, coupling took place at the ortho-center, and simultaneous base-induced in situ cyclization resulted in the formation of N-arylindazoles. Mechanistically, diazo compound (103) deprotonated under the basic conditions and the generated anion underwent electronic migration to provide (104′). Intramolecular cyclization with the elimination of cyanide or tosyl group yielded indazole derivatives (105). However, although the yields were moderate, the authors documented this as a novel procedure to access N-aryl indazoles.
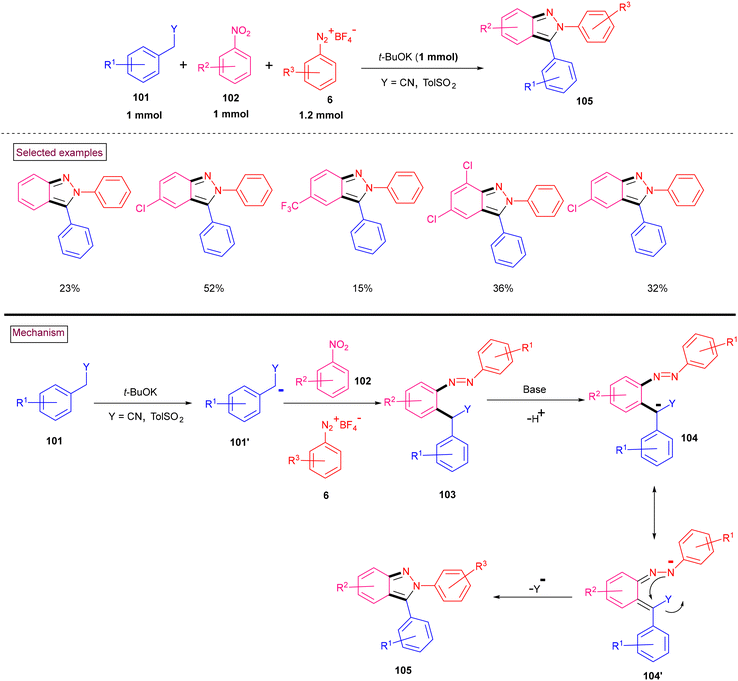 |
| | Scheme 23 Synthesis of indazoles from aryldiazonium salts and active methylenes via the σH adduct. | |
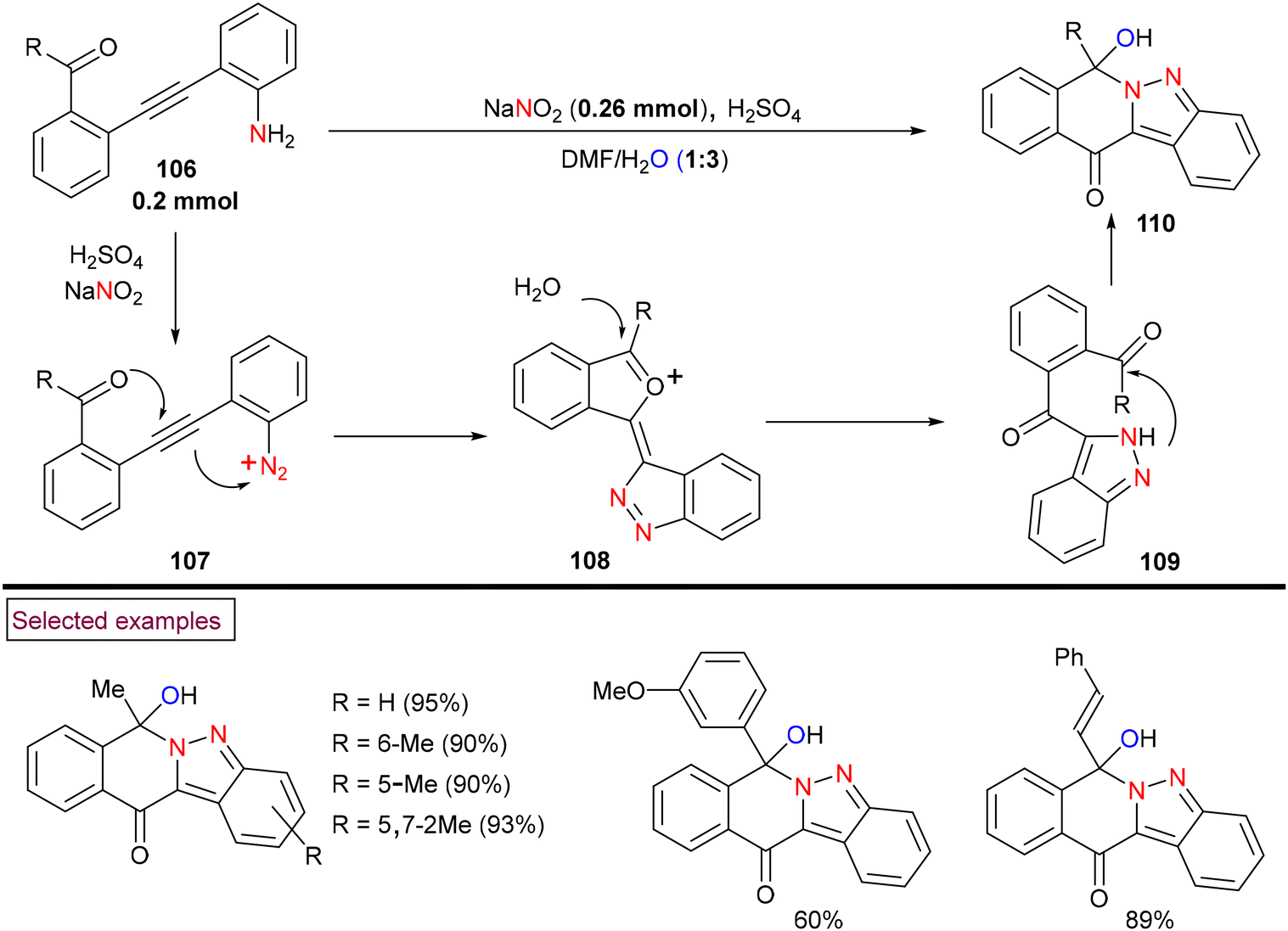
The preparation of fused tetracyclic 2H-indazoles (110) via acid-promoted tandem in situ diazotization/bicyclization of diaryl alkynes (106) was reported by Xu et al. (2018) (Scheme 24).123In situ-generated aryldiazonium salt (107) served as the nitrogen source in this unprecedented process and indazoles (110) were obtained in excellent yields with the consecutive formation of two C–N bonds, one C–O bond and one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. Diaryl alkynes bearing alkyl, aryl, benzyl and styryl ketones (106) reacted smoothly to yield the corresponding products (110) in good yields. The plausible mechanism was proposed by authors based on various control experiments. Accordingly, dual 5-exo-dig cyclization of in situ-generated aryldiazonium salt (107) followed by hydrolysis and intramolecular nucleophilic-addition of an amino group into the newly generated carbonyl group of (109) resulted in the formation of the final product. In addition, a series of post-synthetic modifications showcased the potential of this method.
O bond. Diaryl alkynes bearing alkyl, aryl, benzyl and styryl ketones (106) reacted smoothly to yield the corresponding products (110) in good yields. The plausible mechanism was proposed by authors based on various control experiments. Accordingly, dual 5-exo-dig cyclization of in situ-generated aryldiazonium salt (107) followed by hydrolysis and intramolecular nucleophilic-addition of an amino group into the newly generated carbonyl group of (109) resulted in the formation of the final product. In addition, a series of post-synthetic modifications showcased the potential of this method.
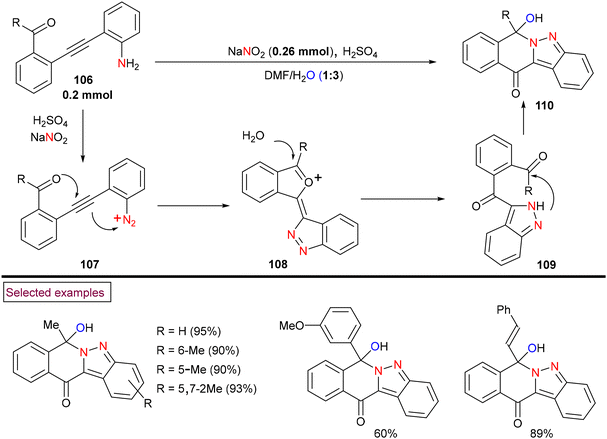 |
| | Scheme 24 Preparation of tetracyclic 2H-indazoles via acid-promoted tandem cyclization. | |
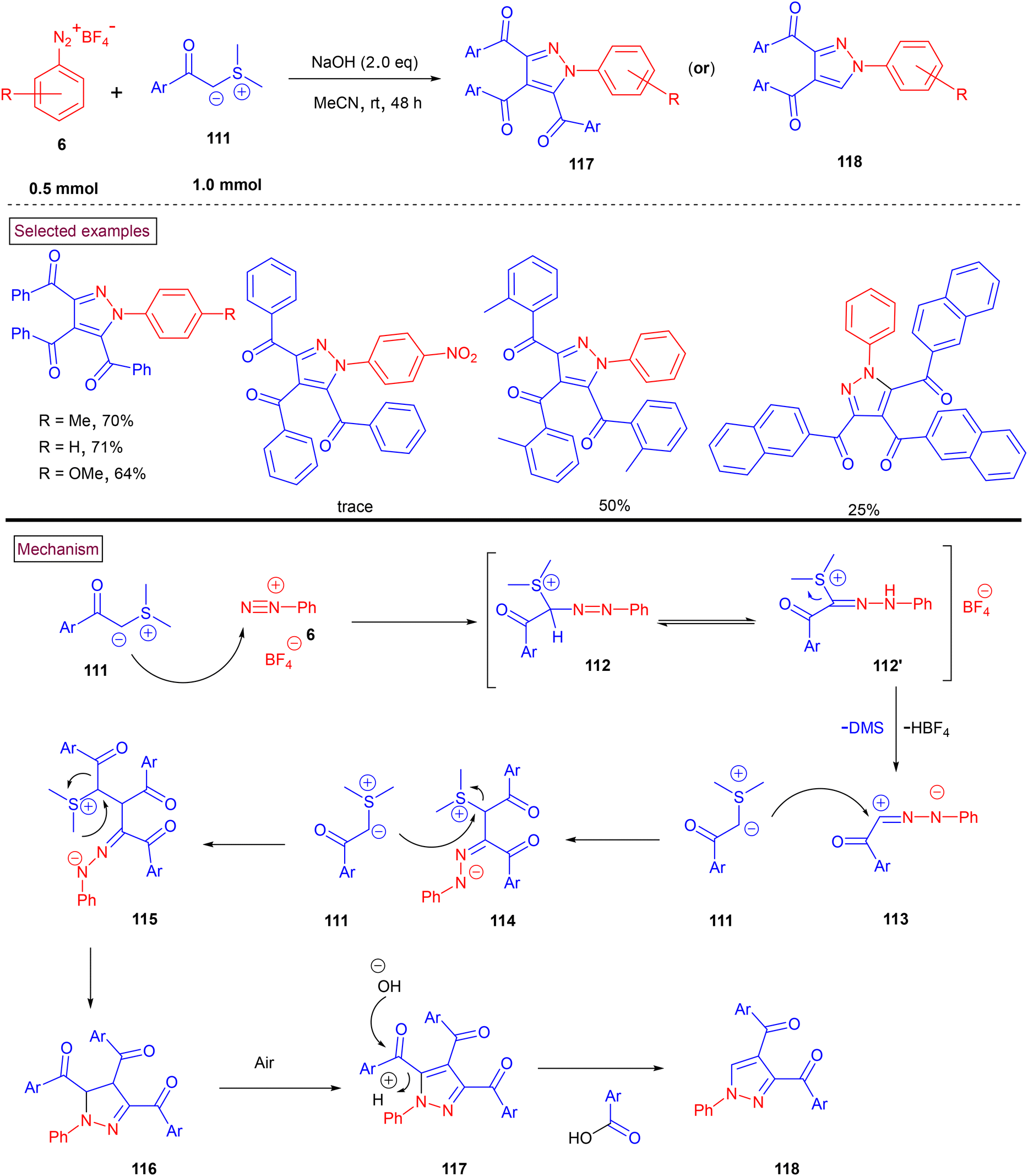
In 2021, a domino annulation strategy to access multi-substituted pyrazoles (117) or (118) via the (2 + 1 + 1 + 1) cyclization of sulfur ylides (111) and aryldiazonium salts (6) was reported by Zhang, Zhu and Shao et al. (Scheme 25).124 Five chemical bonds were formed in a one-pot manner using three equivalents of sulfur ylides (C1 synthon). The formation of tetrasubstituted indazoles (117) was accompanied with significant trisubstituted indazoles (118). A broad range of electron-withdrawing and releasing substituents on aryldiazonium salts (6) and sulfur ylides (111) were well-tolerated and provided the desired products in moderate to good yields. Comparatively better yields were obtained with aryldiazonium salts (6) and sulfur ylides (111) bearing electron-donating groups. Mechanistically, initial nucleophilic attack of ylide (111) on diazonium salt (6) resulted in the formation of intermediate 112/112′. The zwitterionic form of (113) was further reacted with other two equivalents of sulfur ylide through the SN2 pathway, yielding intermediate (115). Subsequent intramolecular nucleophilic attack and aromatization furnished pyrazole derivatives (117) or (118).
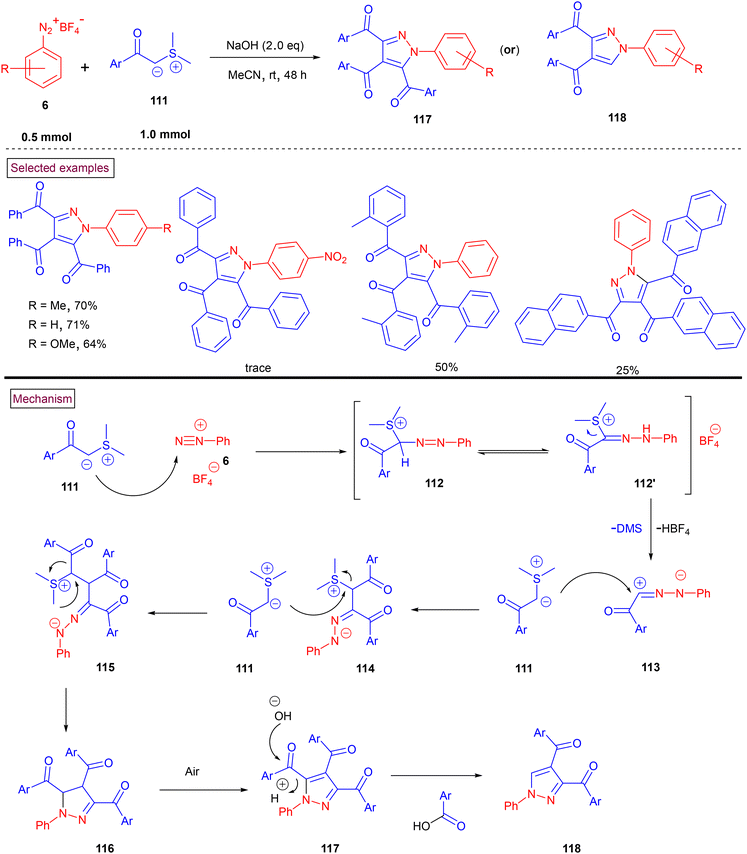 |
| | Scheme 25 Preparation of pyrazoles via (2 + 1 + 1 + 1) domino cyclization. | |
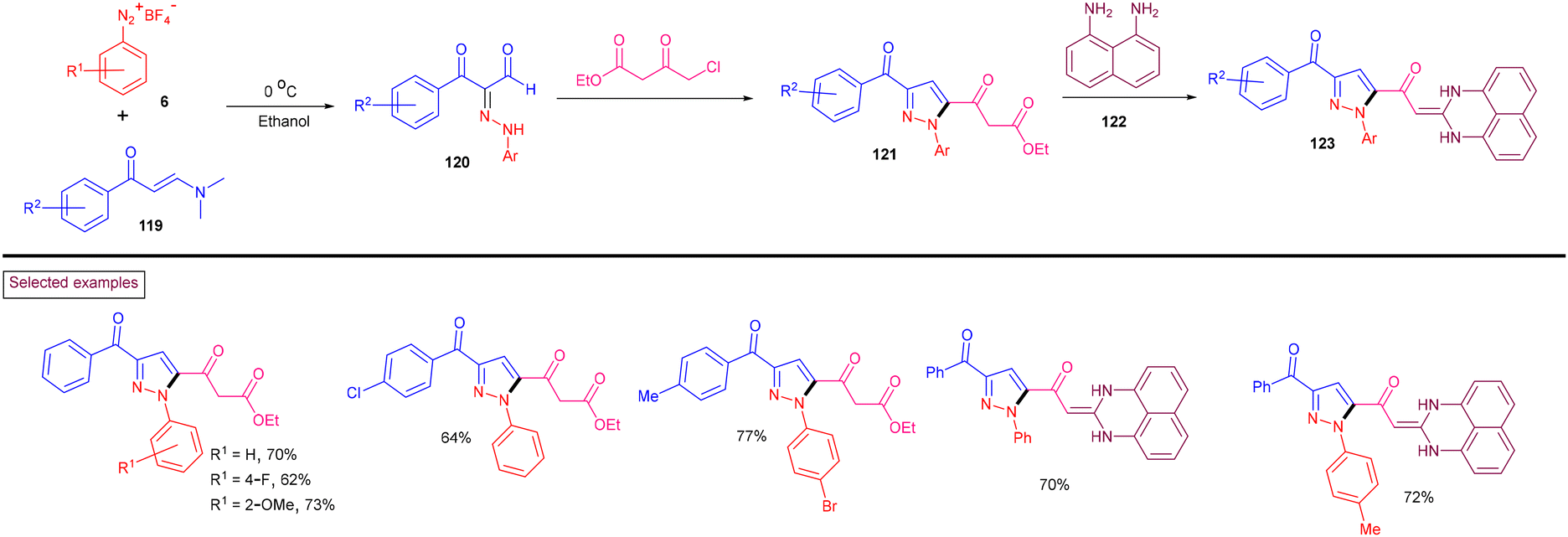
By exploiting the unique reactivity of aryldiazonium salts (6), enaminones (119) and β-ketoesters under transition-metal-free conditions, biologically important pyrazole derivatives (121) were obtained by Koca et al. (2020) (Scheme 26).125 These products were converted into hybrid perimidine-pyrazoles (123) and their mutagenic, genotoxic and carcinogenic effects were evaluated. Aryldiazonium salts (6) and enaminones (119) bearing various substituents were involved smoothly in this transformation and the pyrazoles were obtained in good yields. Mechanistically, aryldiazonium salt (6) reacted with enaminone (119) at 0 °C to afford hydrazone derivatives (120). Further condensation with β-ketoesters and intramolecular cyclization furnished pyrazoles (121). Condensation of (121) with diamine (122) delivered perimidine-pyrazoles (123).
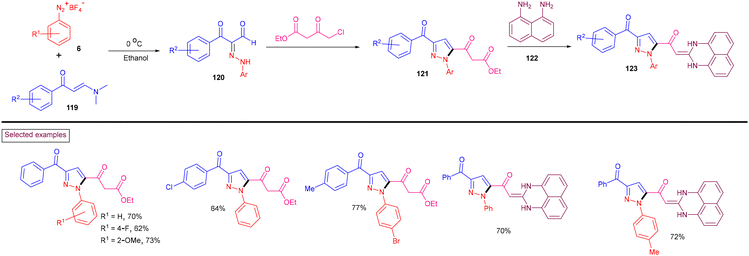 |
| | Scheme 26 Preparation of pyrazoles from aryldiazonium salts, enaminones and β-ketoesters. | |
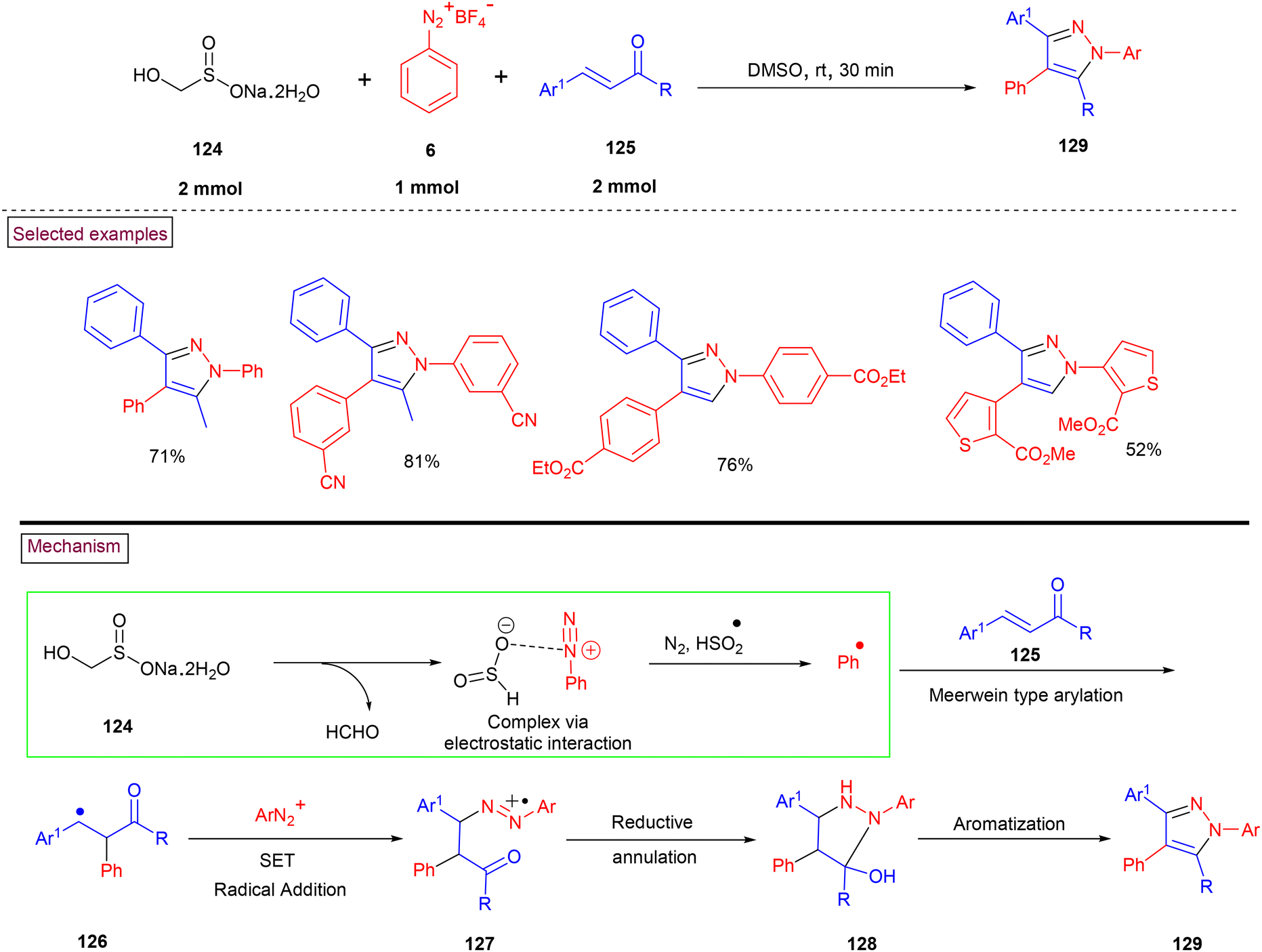
The efficient synthesis of multi-substituted pyrazole (129) was demonstrated by Wu and co-workers from aryldiazonium salts (6) and α,β-unsaturated aldehydes or ketones (125) via rongalite (124)-mediated three-component radical annulation process (Scheme 27).75 In this method, rongalite played a dual role as a simultaneous radical initiator and reducing agent in the reaction. In addition, aryldiazonium salt (6) served as precursors for both aryl radical and arylhydrazine unit in this reaction. Aryl and heteroaryl diazonium salts with substituents such as, –CN, CO2Me, and SO2Me furnished the desired multi-substituted pyrazoles in good yields (62%–81%). Moreover, this radical annulation was studied in detail with various α,β-unsaturated aldehydes or ketones. Based on the experimental investigations, a plausible radical pathway was proposed, in which rongalite decomposed to provide HSO2− with simultaneous liberation of formaldehyde and further complexation of HSO2− with (6) occurred via electrostatic interaction. The generation of aryl radical via the SET process and Meerwein-type addition to α,β-unsaturated ketone (125) led to radical intermediate (126). Further reaction with another molecule of aryldiazonium salt, reduction of azo intermediate (127) into a hydrazine derivative, intramolecular cyclization (128) and aromatization resulted in the formation of fully substituted pyrazolos (129).
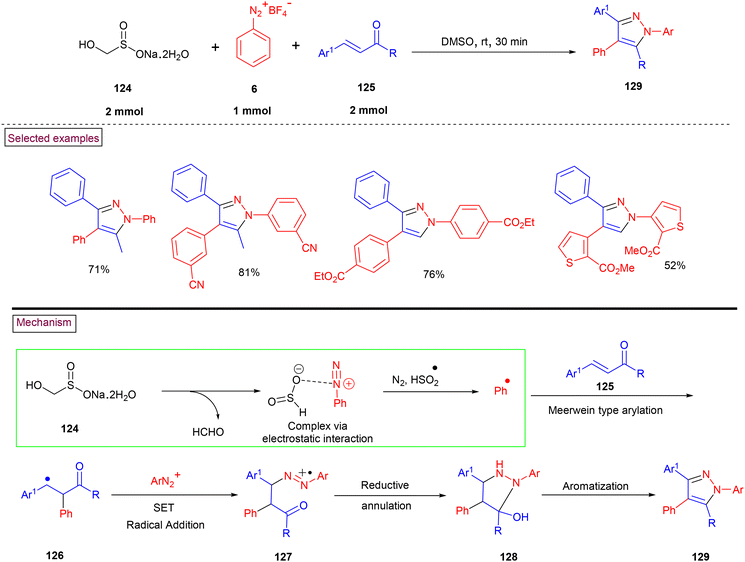 |
| | Scheme 27 Preparation of multi-substituted pyrazole via the rongalite-mediated radical annulation process. | |
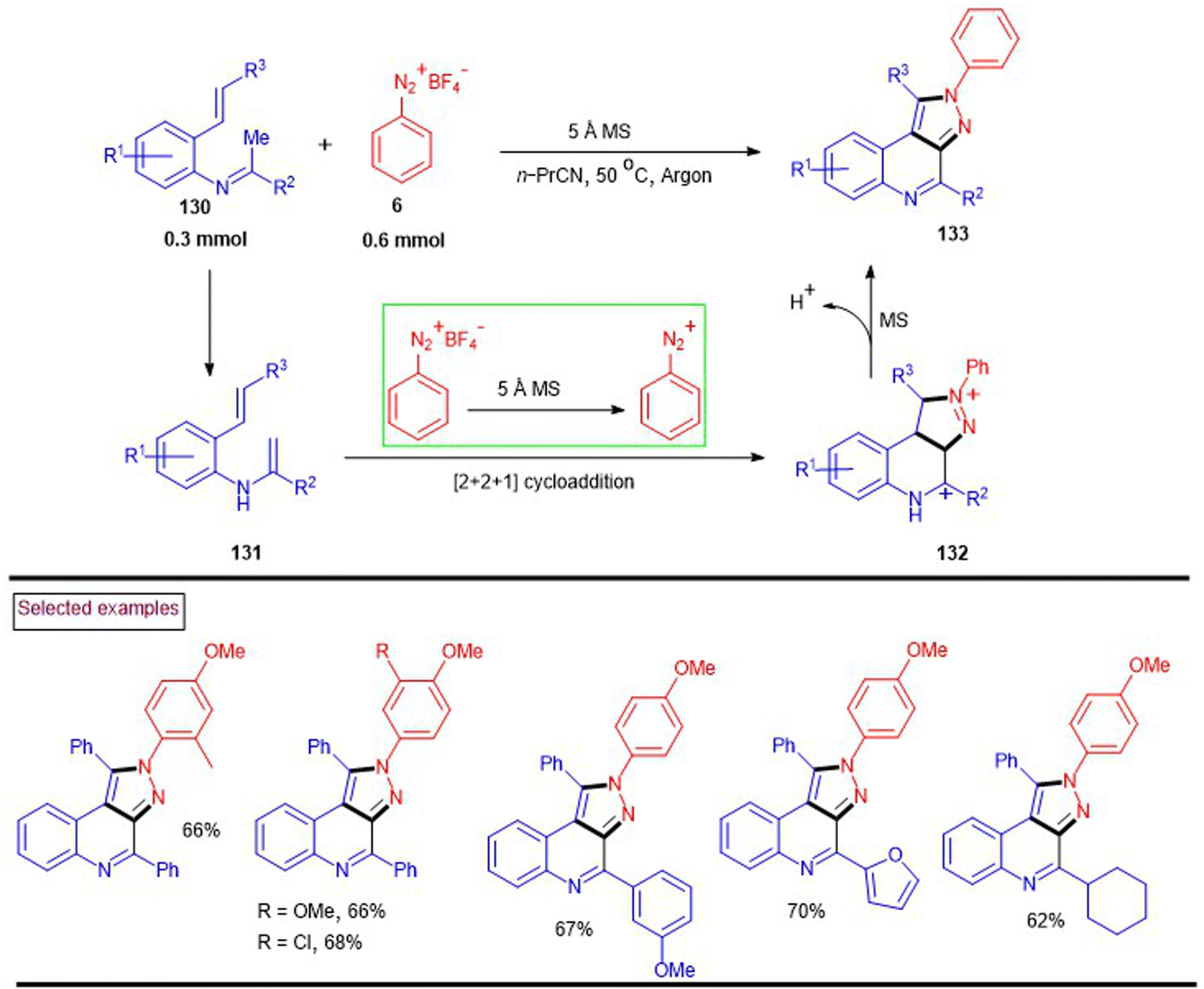
In 2016, Li et al. reported a straightforward method to access pyrazolo[3,4-c]quinolines (133) via the molecular sieve-mediated novel heteroannulation of N-(o-alkenylaryl)imines (130) with aryldiazonium tetrafluoroborates (6) (Scheme 28).126 In this dehydrogenative [2 + 2 + 1] annulation, molecular sieves served as a base to promote the consecutive formation of three new bonds (two C–N bond and one C–C bond). Electron-rich aryldiazonium salts afforded the desired products in greater yields than that with –Br and –NO2 groups. Alternatively, N-(o-alkenylaryl)imines (130)-bearing aryl and heteroaryl substituents at the terminal alkene were exhibited excellent reactivity. Similarly, aryl, heteroaryl and alkyl groups at the imine carbon were also tested and the desired products were isolated in appreciable yields (62%–70%). The mechanistic proposal stated that isomerization of the imine substrate to (131) was followed by a [2 + 2 + 1] annulation with diazonium species (132). Further isomerization and dehydrogenation provided the desired product (133).
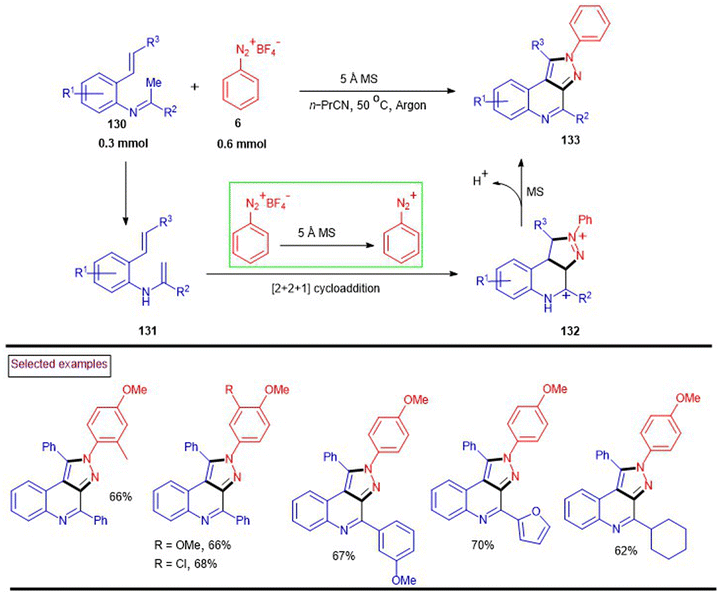 |
| | Scheme 28 Preparation of pyrazolo[3,4-c]quinolines via hetero annulation strategy. | |
3.7. Phenanthridine derivatives
Phenanthridine is an important fused N-heterocyclic motif prevalent in various alkaloids such as, nitidine, trispheridine, chelerythrine, sanguinarine, oxyvicine, calothrixin B and lycobetaine.127 Moreover, monofunctionalized platinum(II) compounds such as cisplatin and phenanthriplatin exhibited significant optoelectronic properties.128 Owing to these notable material and pharmaceutical applications, diverse synthetic methods have been invented in recent years. Most commonly, phenanthridine derivatives have been synthesized from ortho-aryl-functionalized isothiocyanates, isonitriles, azides, amides and amino derivatives under metal-catalysed conditions.129 Alternatively, transition metal-free methods are very scarce.
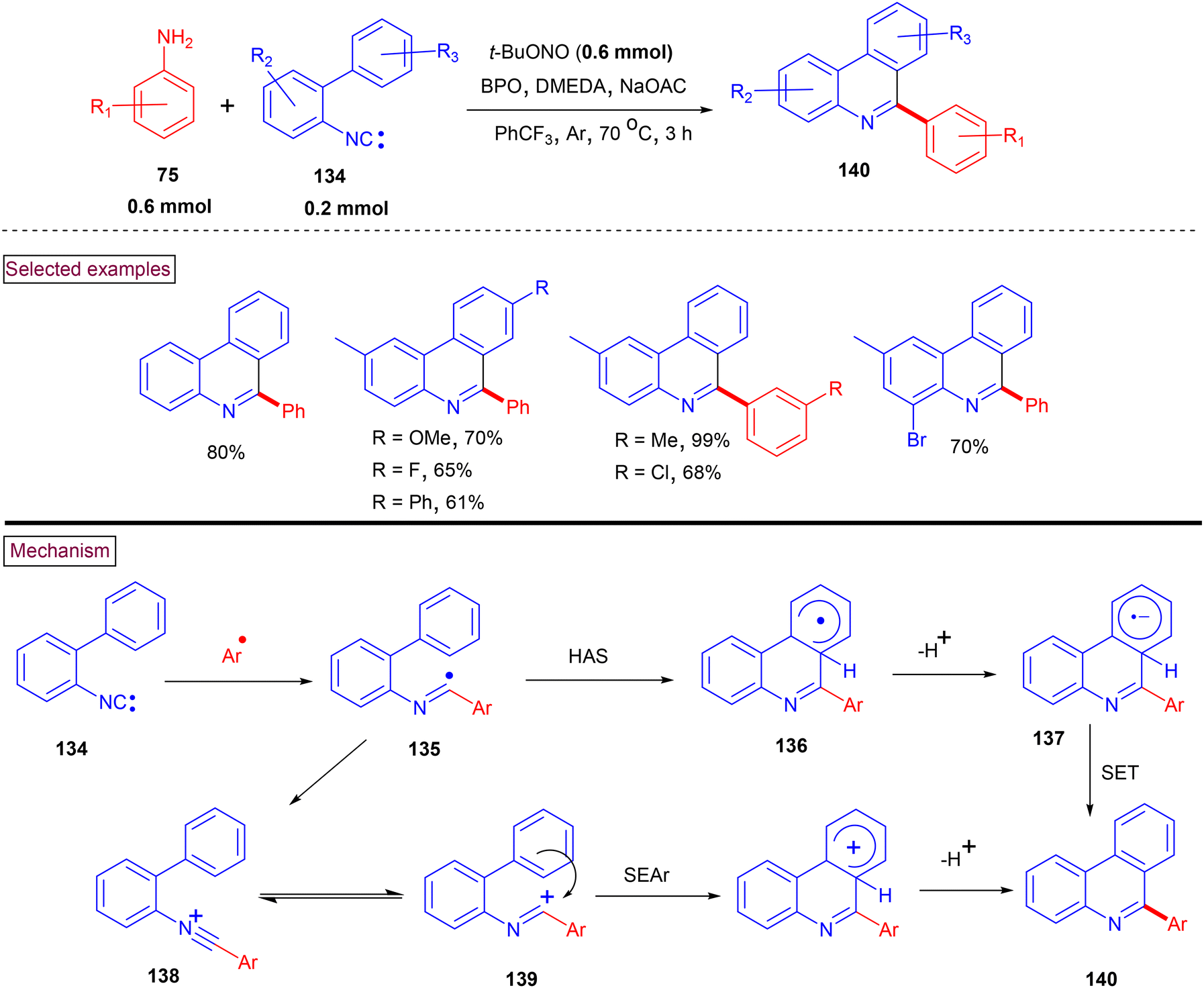
A one-pot arylative cyclization was reported to prepare 6-arylated phenanthridine derivatives (140) from 2-isocyanobiphenyls (134) and arylamines (75) (Zhu et al., Scheme 29).130 It is known that C6-functionalzied phenanthridines can be generated from imidoyl radical (135) via intramolecular homolytic aromatic substitution (HAS). Alternatively, the oxidation of the imidoyl radical to N-arylnitrilium ion (138) can also generate the corresponding phenanthridines via intramolecular arylation. This one-pot arylative cyclization smoothly proceeded with various anilines and hetero arylamines with –OMe, –tBu, –COMe, –CO2Et, –Br, and –NO2 groups and the corresponding products were isolated in good yields (61%–84%). However, the steric influence of ortho-substituents led to lower yields. In the case of meta-substituted biphenyls, regioisomeric mixture was formed in favour of the less hindered isomer. Mechanistic insights were obtained from a series of intramolecular competitive reactions and radical trapping experiments. The in situ-generated aryldiazonium salt decomposed to the aryl radical, which subsequently added to the terminal carbon of 2-isocyanobiphenyl (134). The resulting imidoyl radical (135) was converted to product (140) via intramolecular cyclization, deprotonation and SET process. Alternatively, N-arylnitrilium intermediate (138) could be generated via the SET oxidation of biphenyl imidoyl radical (135). Further arylative cyclization (SEAr) and dehydrative aromatization resulted in the formation of C6-phenanthridines (140). In 2015, our research group reported an efficient strategy for the preparation of substituted phenanthridine derivatives (140) from a reaction of varieties of nitriles with aryldiazonium salts under anhydrous and transition metal-free conditions.131 This transformation proceeded via the in situ formation of N-arylnitrilium intermediate (138), followed by intramolecular arylation to provide the corresponding phenanthridines (140). The scope and generality of the method was investigated with a variety of nitriles and a range 2-aryl benzenediazonium salts. This method enabled the synthesis of C6-aryl, alkyl (1° and 2°), benzyl and heteroaryl-substituted phenanthridines in moderate to good yields (61%–88%).
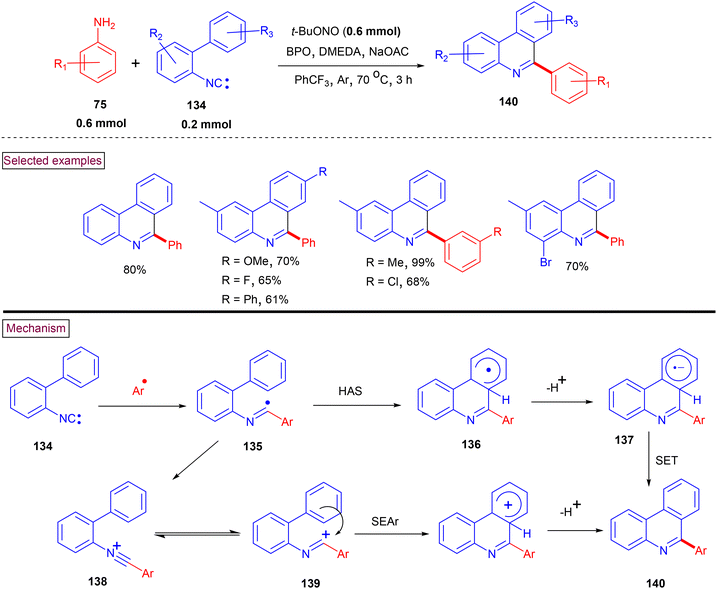 |
| | Scheme 29 Preparation of 6-arylated phenanthridines via one-pot arylative cyclization. | |
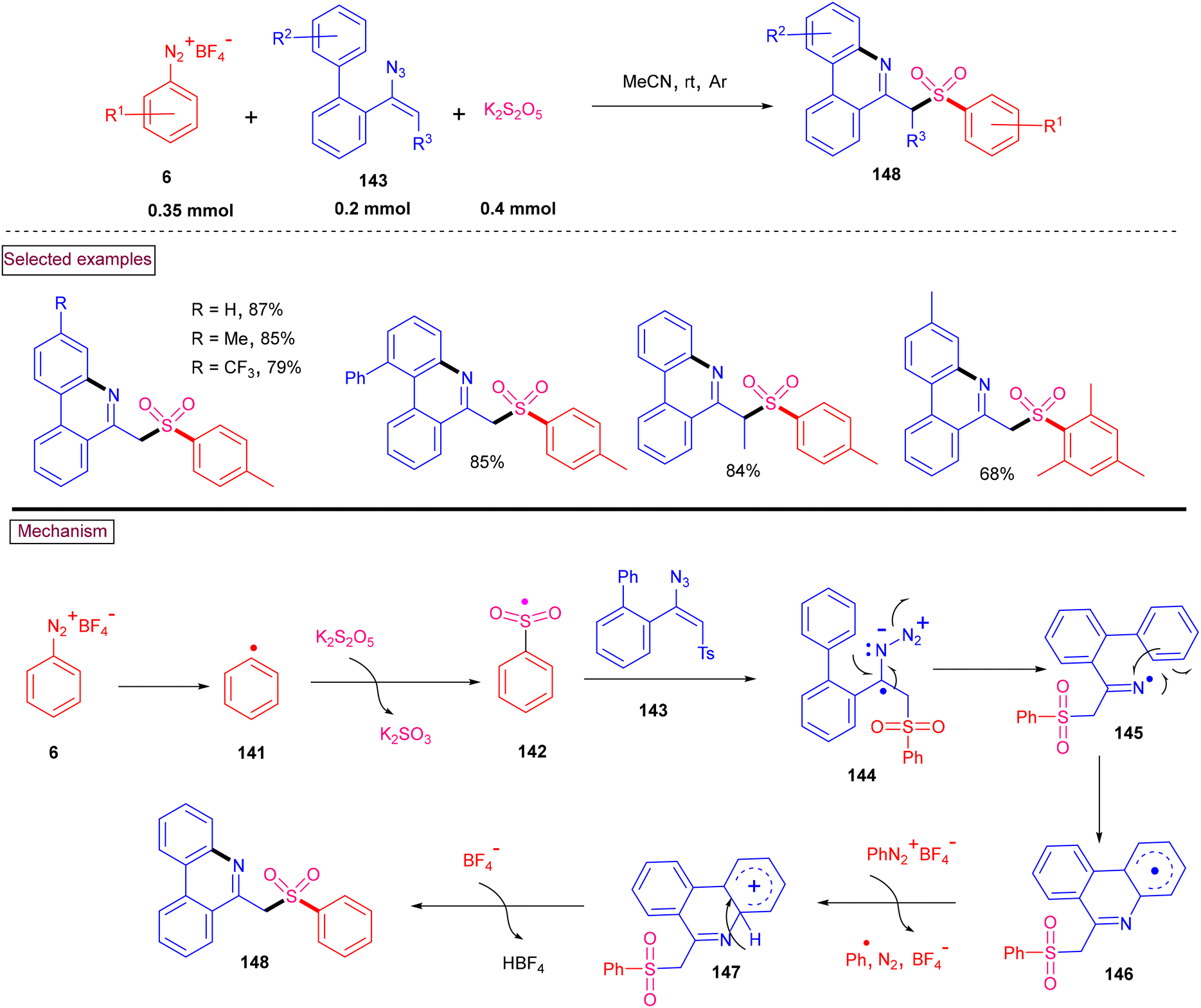
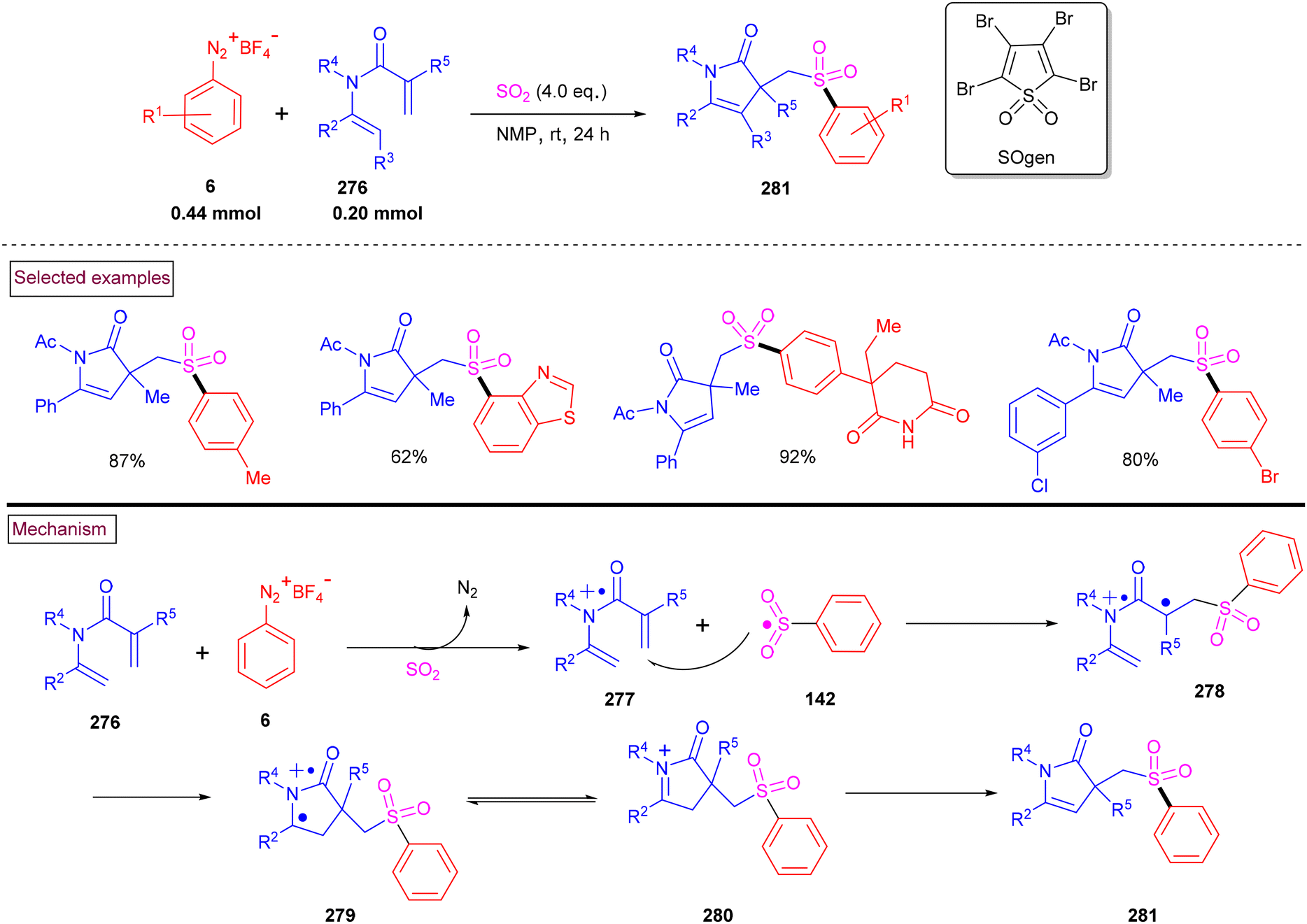
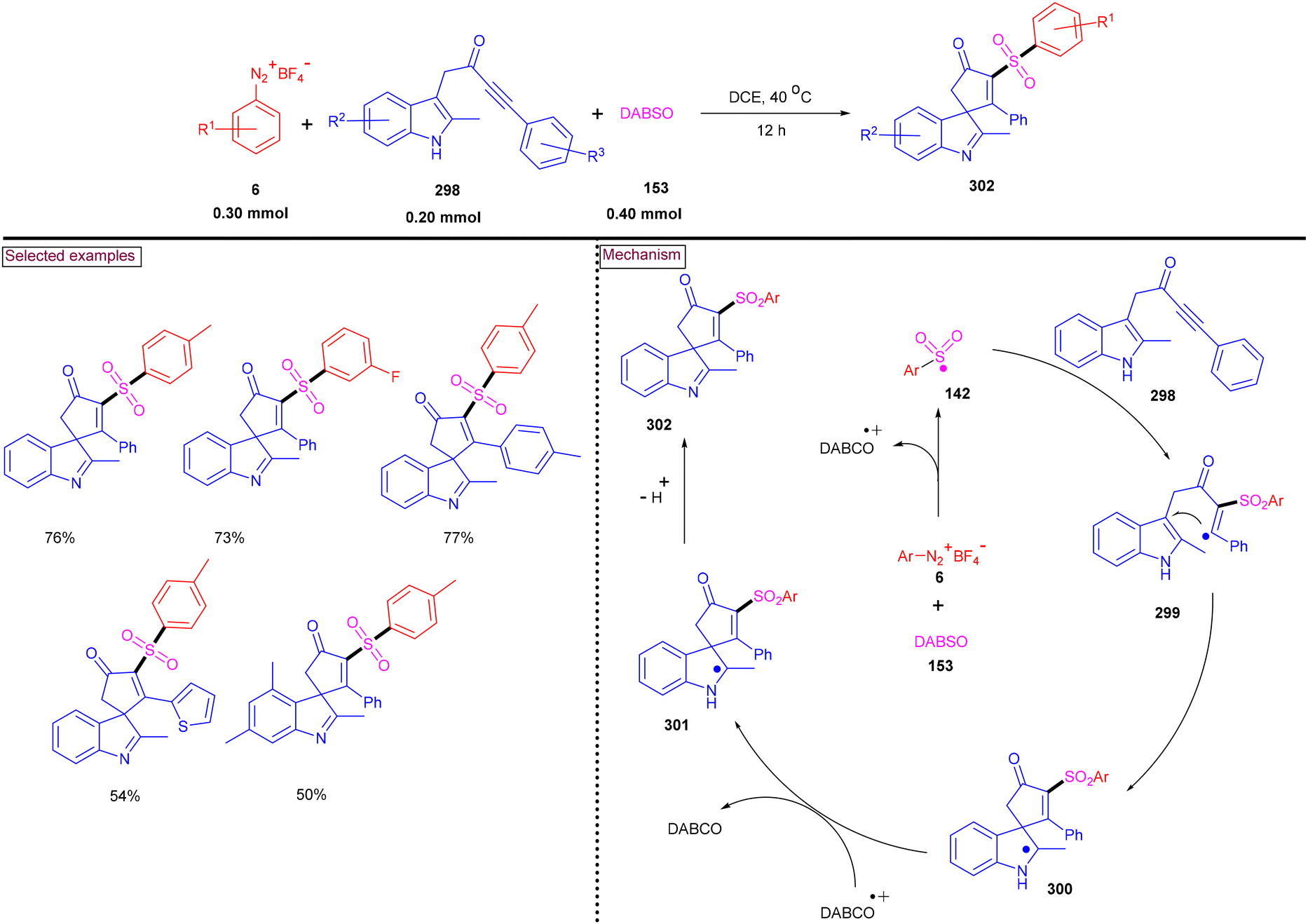
An alternative approach for the preparation of 6-(sulfonylmethyl)-phenanthridines (148) from the reaction of aryldiazonium salts (6) with vinyl azides (143) in the presence of K2S2O5 is discussed in Scheme 30.132 Compared to previous reports, this method utilized the economically affordable K2S2O5 as a sulfur surrogate. This tandem sulfonylation/cyclization strategy exhibited excellent functional group tolerance and the corresponding products were obtained in excellent yields. Vinyl azides (143) bearing different substituted aryl rings were tested smoothly to afford the corresponding phenanthridines (148) in good yields (79%–87%). Substitution at the alkenyl carbon of vinyl azide also worked well. Sterically and electronically distinct aryl/heteroaryl diazonium salts with functional groups such as, –F, –CN, and acetyl furnished phenanthridines (148) in excellent yields. According to the proposed pathway, initial decomposition of aryldiazonium salt (6) followed by SO2 insertion resulted in the formation of arylsulfonyl radical (142). The addition of (142) to vinyl azide (143) provided iminoyl radical (145). Intramolecular radical cyclization followed by SET oxidation in the presence of aryldiazonium salt gave the cationic intermediate, which upon deprotonation yielded the desired product (148). The gram-scale synthesis and Pd-catalysed direct arylative coupling were demonstrated.
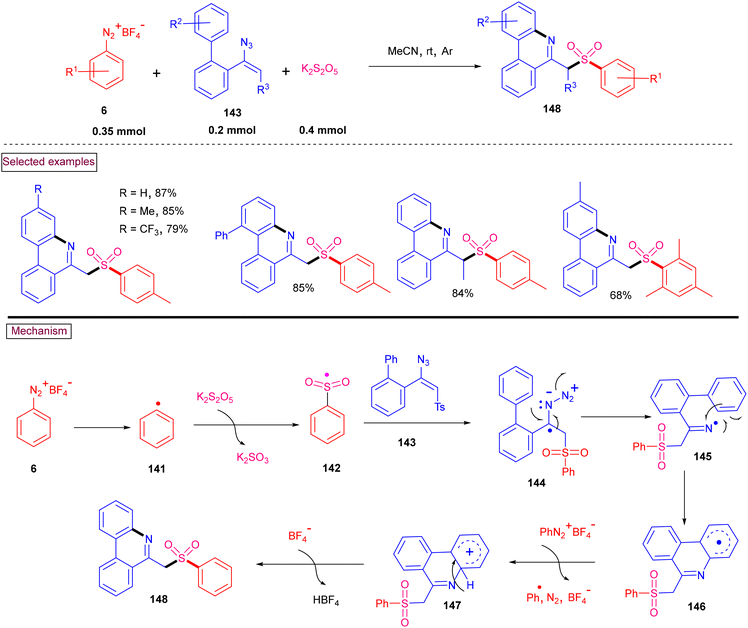 |
| | Scheme 30 Preparation of 6-(sulfonylmethyl)phenanthridines via radical sulfonylation cyclization using K2S2O5. | |
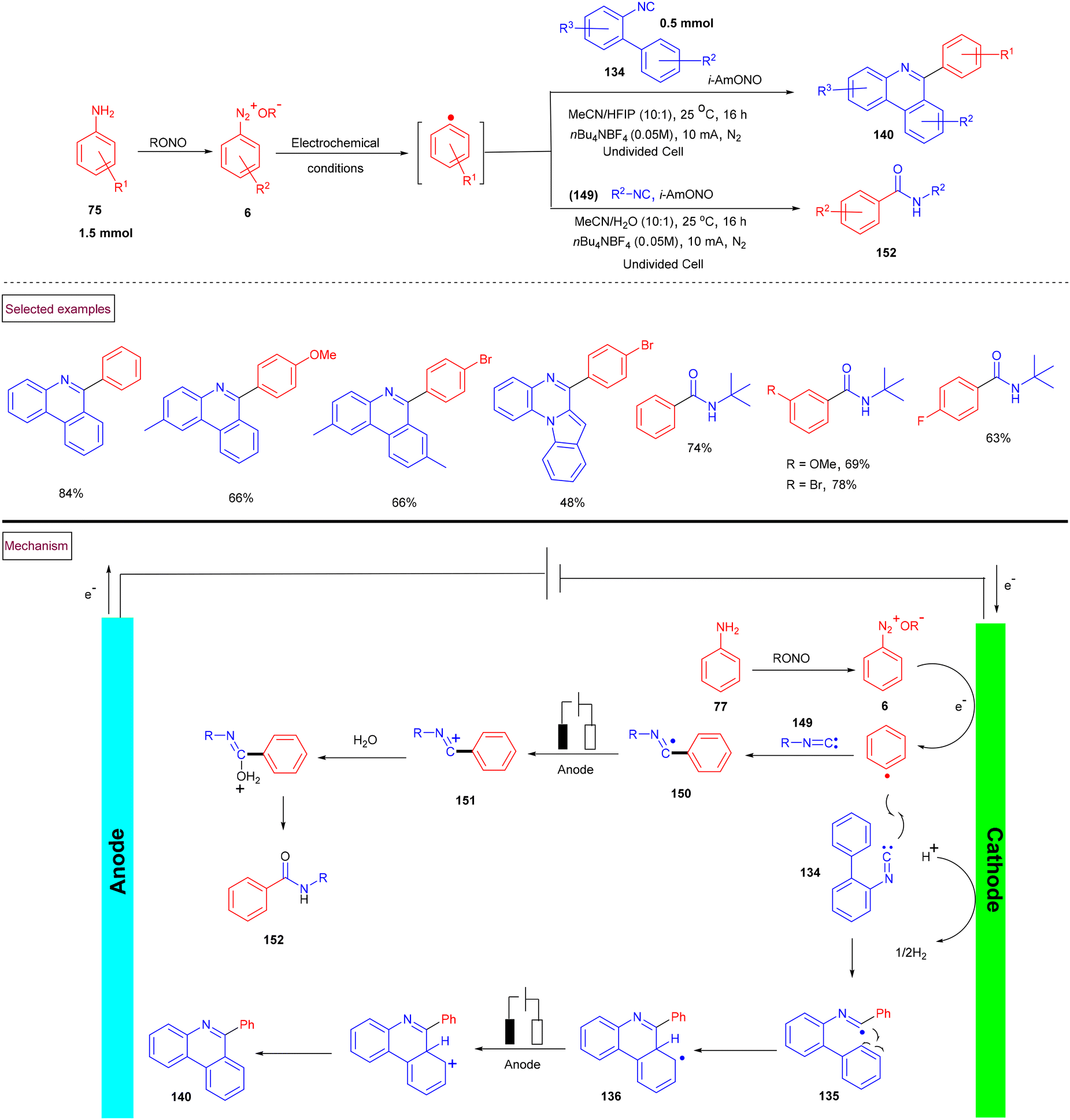
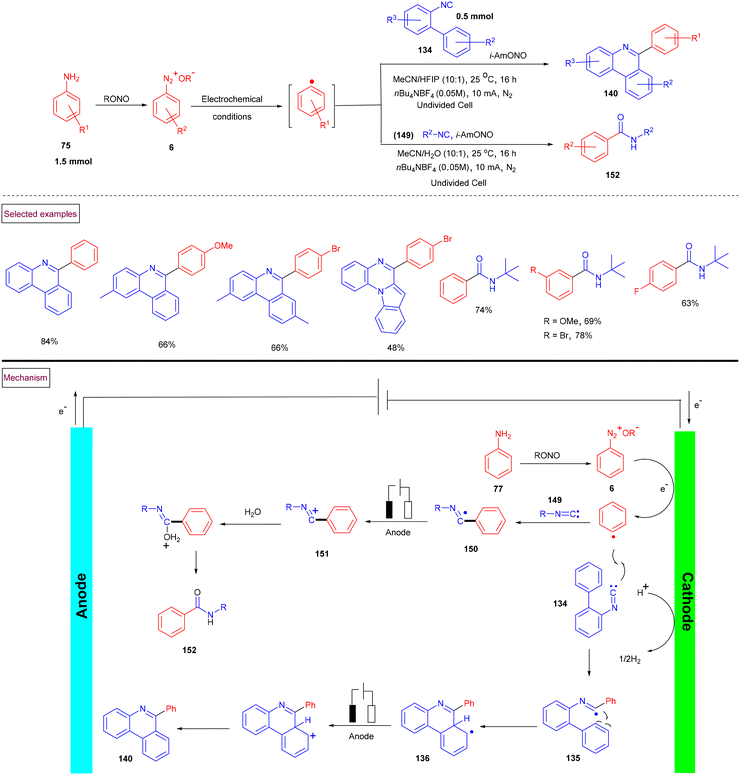
In 2021, Sharma et al. reported the first electrochemical synthesis of 6-aryl phenanthridines (140) from isocyanides (134) and aniline derivatives (75) (Scheme 31).133 Compared to Zhu's work (Scheme 30), this approach proceeded at ambient temperature. In situ generated aryldiazonium ion (6) and alkyl nitrite underwent cathodic reduction and resulted in the formation of aryl radical, which subsequently underwent oxidative cross-coupling with isocyanides (134) under metal and oxidant-free conditions. A series of differently functionalized phenanthridines (140) and amides (152) was achieved under undivided electrolytic conditions. This oxidative electrochemical cyclization strategy was extended further to synthesize various alkyl, aryl and benzyl amides in a solution of nBu4NBF4 (CH3CN/H2O = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Based on various control experiments and cyclic voltammetry studies, the authors proposed a plausible mechanism, wherein the aryl radical is initially generated by SET reduction of aryldiazonium salt (6). Further cross-coupling with isocyanide resulted in the formation of imidoyl radical (135), which upon intramolecular radical cyclization, oxidative aromatization and deprotonation produced phenanthridine derivatives (140). The addition of aryl radical to isocyanide (149) provided imidoyl intermediate (150), which on oxidation and hydration afforded amide derivatives (152).
:1). Based on various control experiments and cyclic voltammetry studies, the authors proposed a plausible mechanism, wherein the aryl radical is initially generated by SET reduction of aryldiazonium salt (6). Further cross-coupling with isocyanide resulted in the formation of imidoyl radical (135), which upon intramolecular radical cyclization, oxidative aromatization and deprotonation produced phenanthridine derivatives (140). The addition of aryl radical to isocyanide (149) provided imidoyl intermediate (150), which on oxidation and hydration afforded amide derivatives (152).
 |
| | Scheme 31 Preparation of 6-aryl phenanthridines via electrochemical synthetic strategy. | |
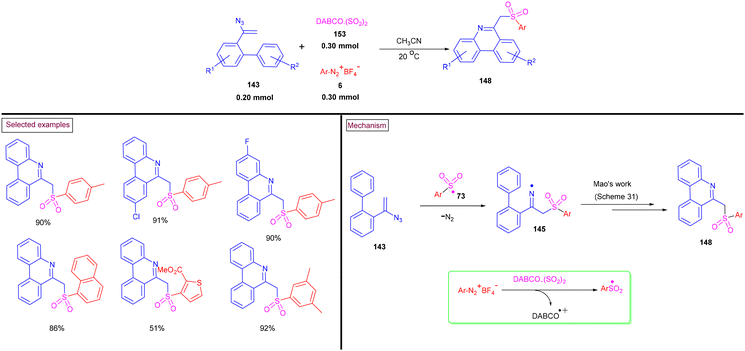
A unique approach for the construction of 6-(sulfonylmethyl)phenanthridines (148) was developed by Wu et al. (2018).134 This procedure was performed at room temperature with the use of aryldiazonium salts (6), DABCO.(SO2) (153) as a sulfur dioxide surrogate and vinyl azides (143) (Scheme 32). ortho-Substituted vinyl azides led to lower yields (65%) presumably due to the steric hindrance. Alternatively, multi-substituted aryl, heteroaryl and naphthyl diazonium salts (6) with functional groups such as halogens and ester afforded the desired phenanthridines in moderate yields (44%–96%). Based on the radical-trapping experiments and the related works, the authors proposed the mechanistic pathway via key imidoyl radical intermediate (145), which is similar to that of Mao's work discussed in Scheme 31.132
 |
| | Scheme 32 Preparation of 6-(sulfonylmethyl)phenanthridines via radical sulfonylation cyclization. | |
3.8. Pyridazine derivatives
Pyridazine is a six-membered heteroaromatic compound with two nitrogen atoms, commonly found in biologically important compounds and natural products. Herein, we discuss the synthetic methods to access pyridazine cores by employing aryldiazonium salt as a key precursor.
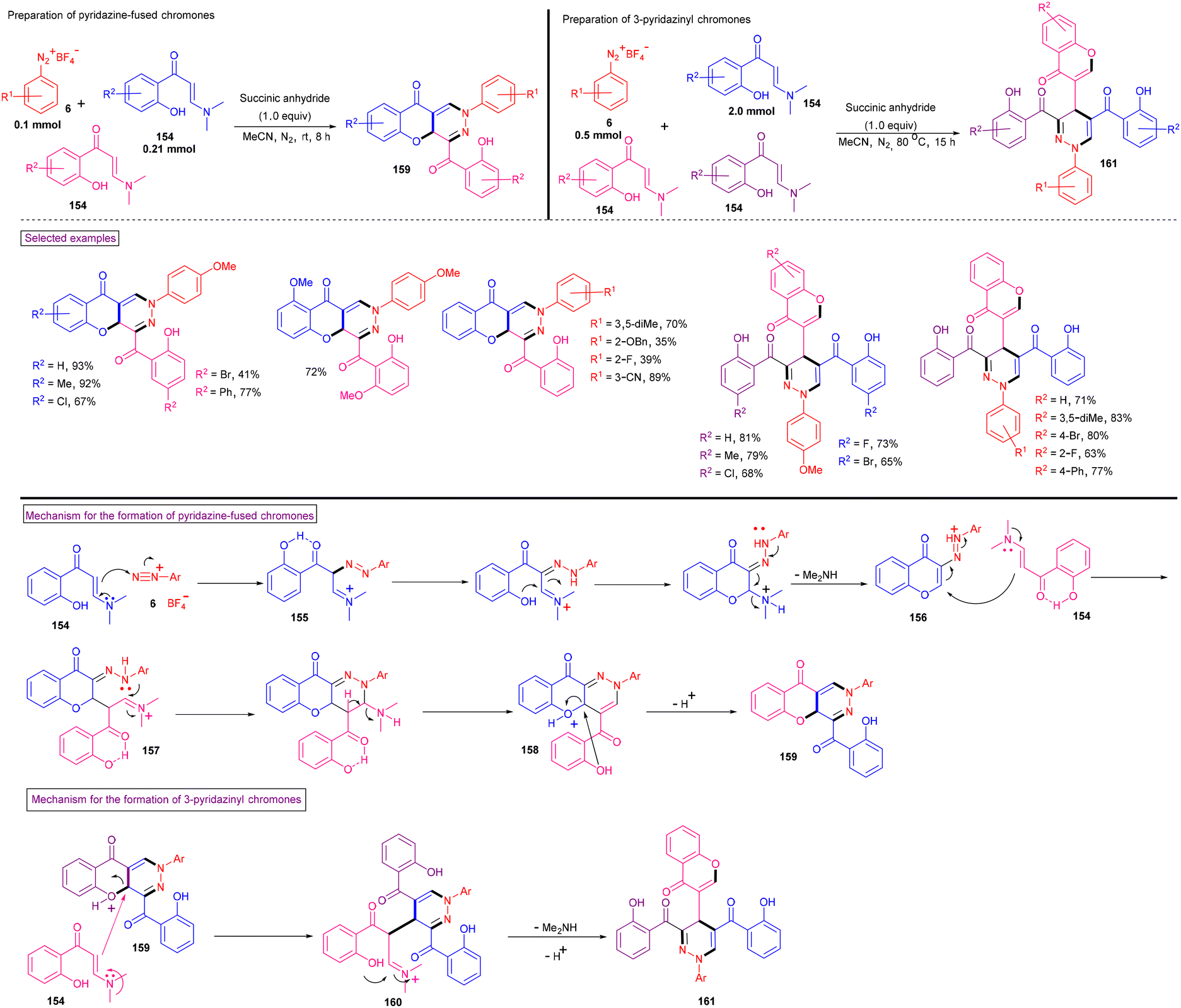
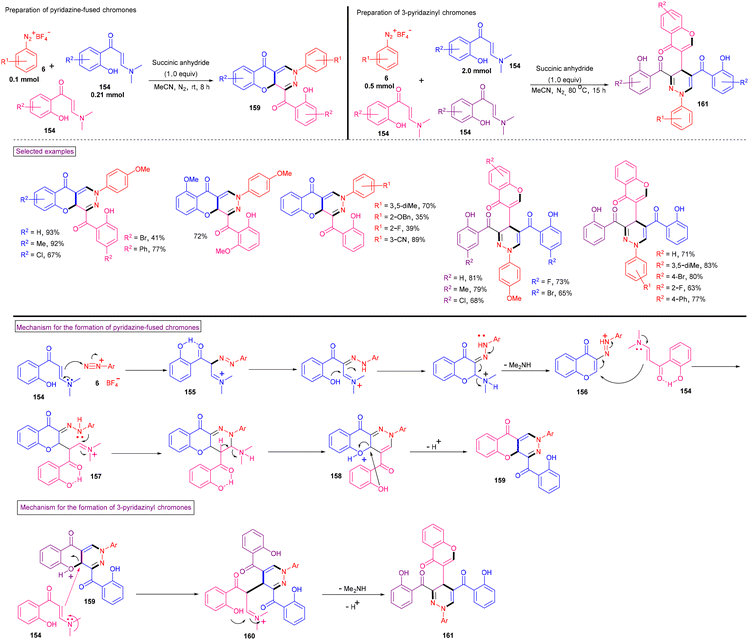
Recently, Yu et al. disclosed the skeleton-controllable annulation reaction of o-hydroxyphenylenaminones (154, o-HPEs) with aryldiazonium salts (6) to yield pyridazine fused chromones (159) and 3-pyridazinyl chromones (161) (Scheme 33).135o-HPEs bearing common substituents and halogens reacted smoothly with aryldiazonium salts (6) to afford pyridazine-fused chromones (159) in moderate to good yields (37%–93%). This switchable annulation is an applicable method for the synthesis of 3-pyridazinyl chromones (161). o-HPEs and aryldiazonium salts with different electronic properties and substitution patterns were found to be viable and the anticipated chromones were isolated in 65%–81% yield. The control experiments revealed that the process was initiated with the intermolecular addition of o-HPE to the diazonium precursor to give diazo intermediate (155). Further intramolecular cyclization and deamination afforded intermediate (156), which was attacked by another molecule of o-HPE followed by intramolecular cyclization, deamination and deprotonation to give the desired products (159). The plausible mechanism for the synthesis of 3-pyridazinyl-chromones (161) was also discussed. These pyridazine-fused products showed promising antiviral effects versus human coronavirus OC43 (HCoV-OC43).
 |
| | Scheme 33 Intermolecular chromone annulation strategy for pyridazine-fused chromones. | |
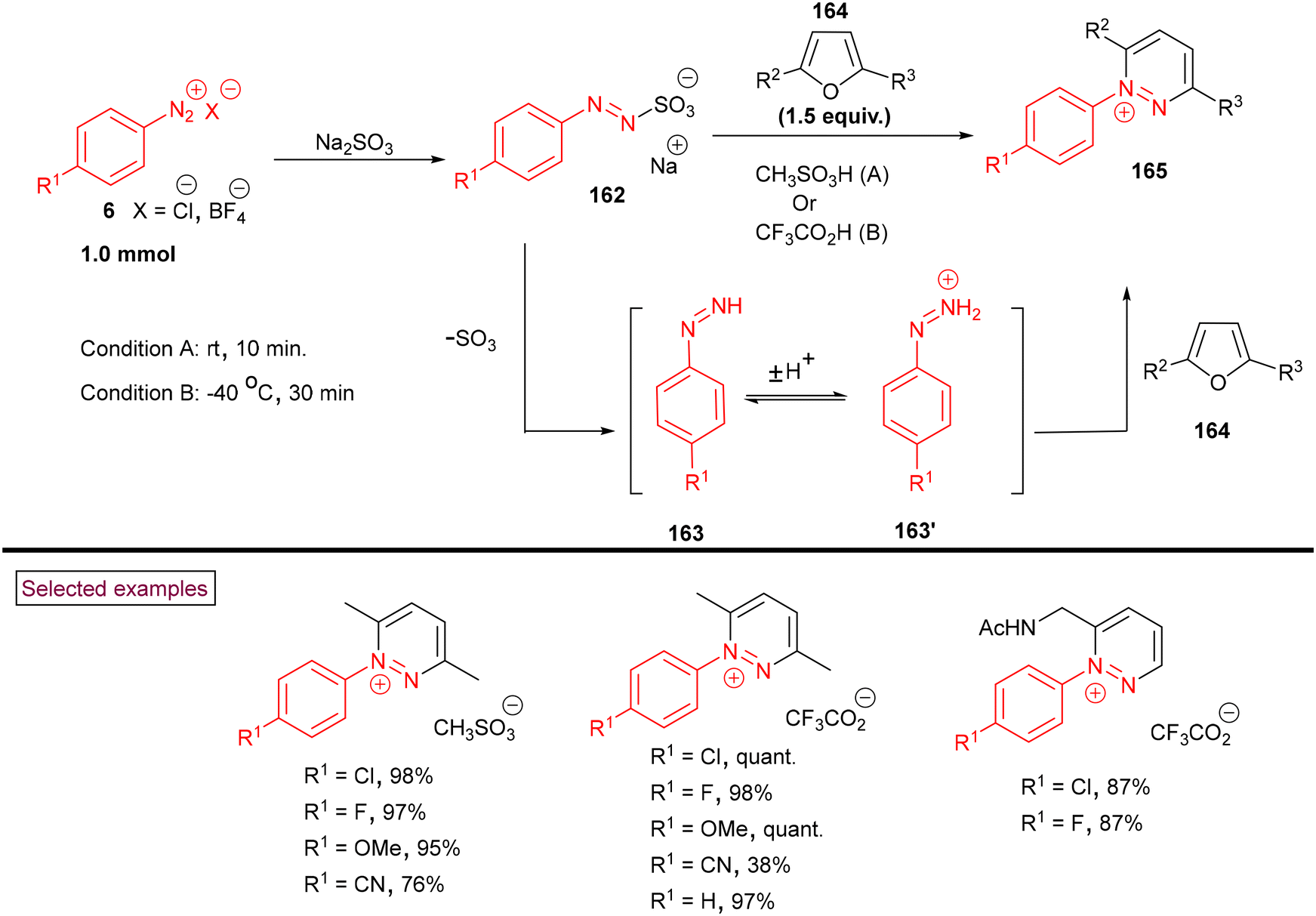
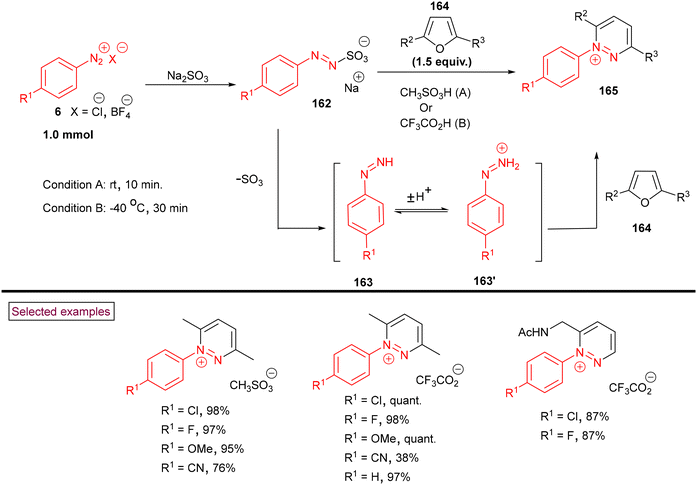
Very recently, Heinrich et al. reported a synthetic route to prepare pyridazinium salts (165) via the cycloaddition of in situ-generated phenyldiazenes (163 and 163′) with furans (164) (Scheme 34).136 In this strategy, aryldiazonium salts (6) served as the initial precursors and transformed into phenyldiazenes (163 and 163′) via a controlled reduction involving a formal two-electron transfer process with the intermediacy of phenylazosulfonates (162). Further, very fast cycloaddition with furan derivatives (164) and dehydration resulted in the formation of diversely substituted pyridazinium salts (165). This transformation was insensitive to oxygen and the presence of short-lived diazenium species promoted the cycloaddition, leading to higher yields. In addition, the authors demonstrated the post-synthetic hydrogenation of pyridazinium methanesulfonates to synthetically valuable tetrahydropyridazines in moderate yields.
 |
| | Scheme 34 Preparation of pyridazinium salts via in situ-generated phenyldiazenes. | |
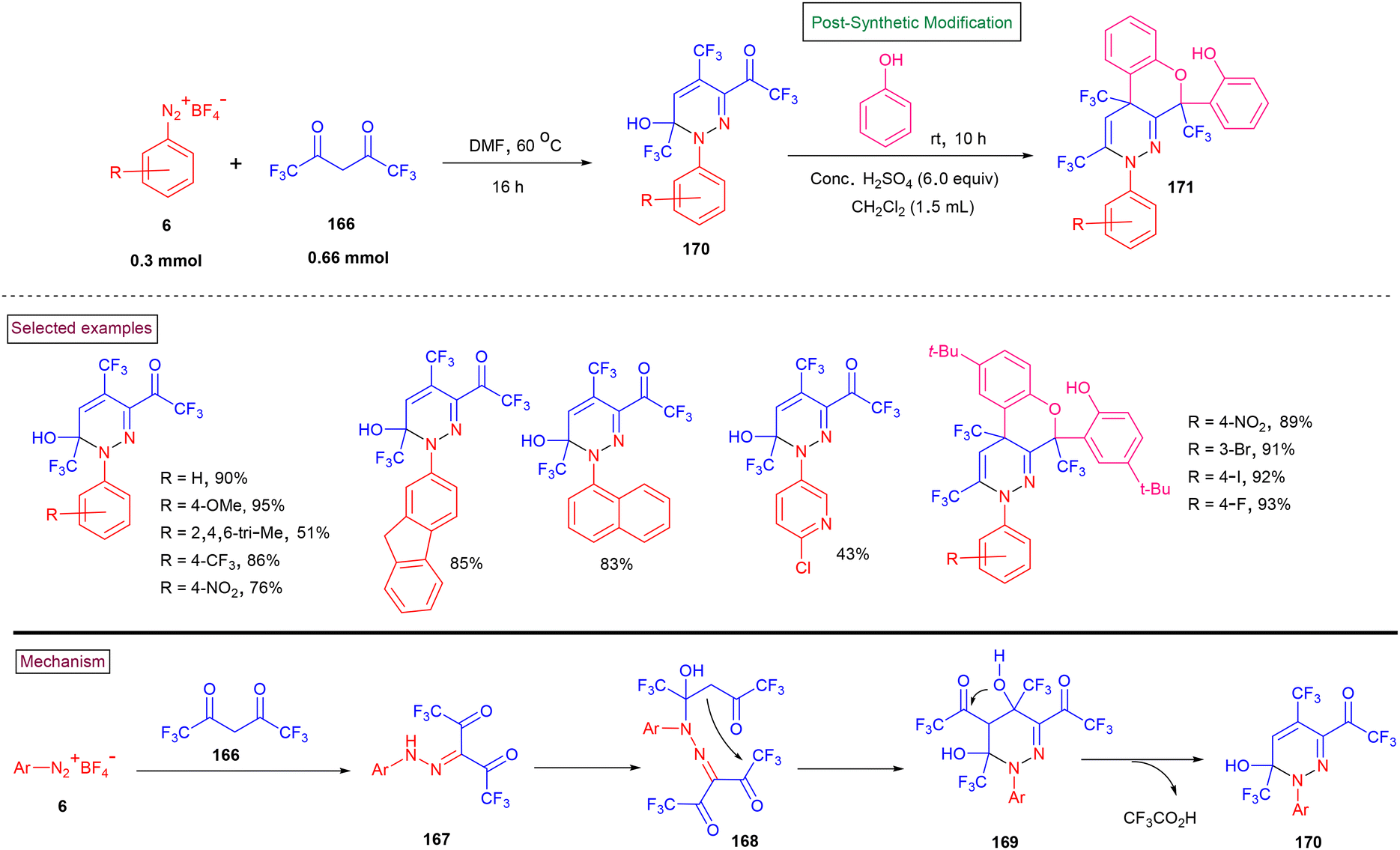
Per-trifluoromethyl pyridazines (170) were reported by Weng and Han et al. (2020) via the tandem cyclization of hexafluoroacetyacetone (166) and aryldiazonium salts (6) under mild conditions (Scheme 35).137 The reaction pathway was initiated by the condensation of aryldiazonium salt (6) with hexafluoroacetyacetone (166), affording hydrazone derivative (167). One of the ketone units underwent intramolecular nucleophilic addition to give the cyclized product (169). Another intramolecular nucleophilic addition, followed by a reductive elimination afforded the desired product (170). A broad range of aryl, heteroaryl, and polyaryl diazonium salts bearing diverse functionalities (–CF3, –NO2, –halo, and –acetyl) afforded pyridazines in moderate to good yields (45%–94%). These products were successfully converted into structurally diverse benzo[5,6]chromeno[3,4-c]pyridazines (171) in the presence of H2SO4.
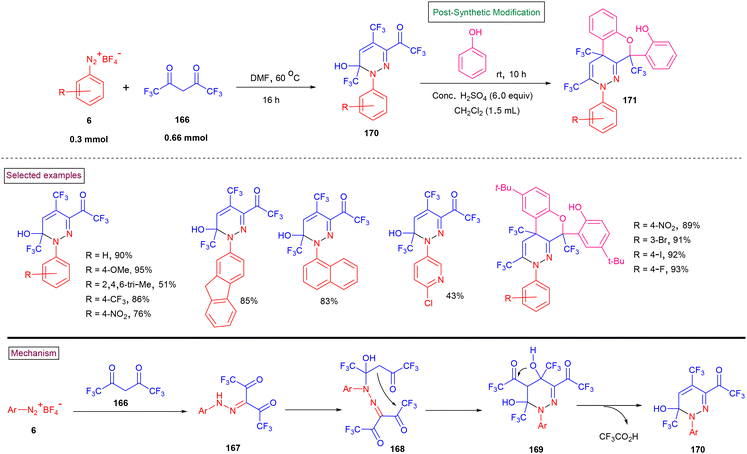 |
| | Scheme 35 Preparation of per-trifluoromethyl pyridazines from aryldiazonium salts. | |
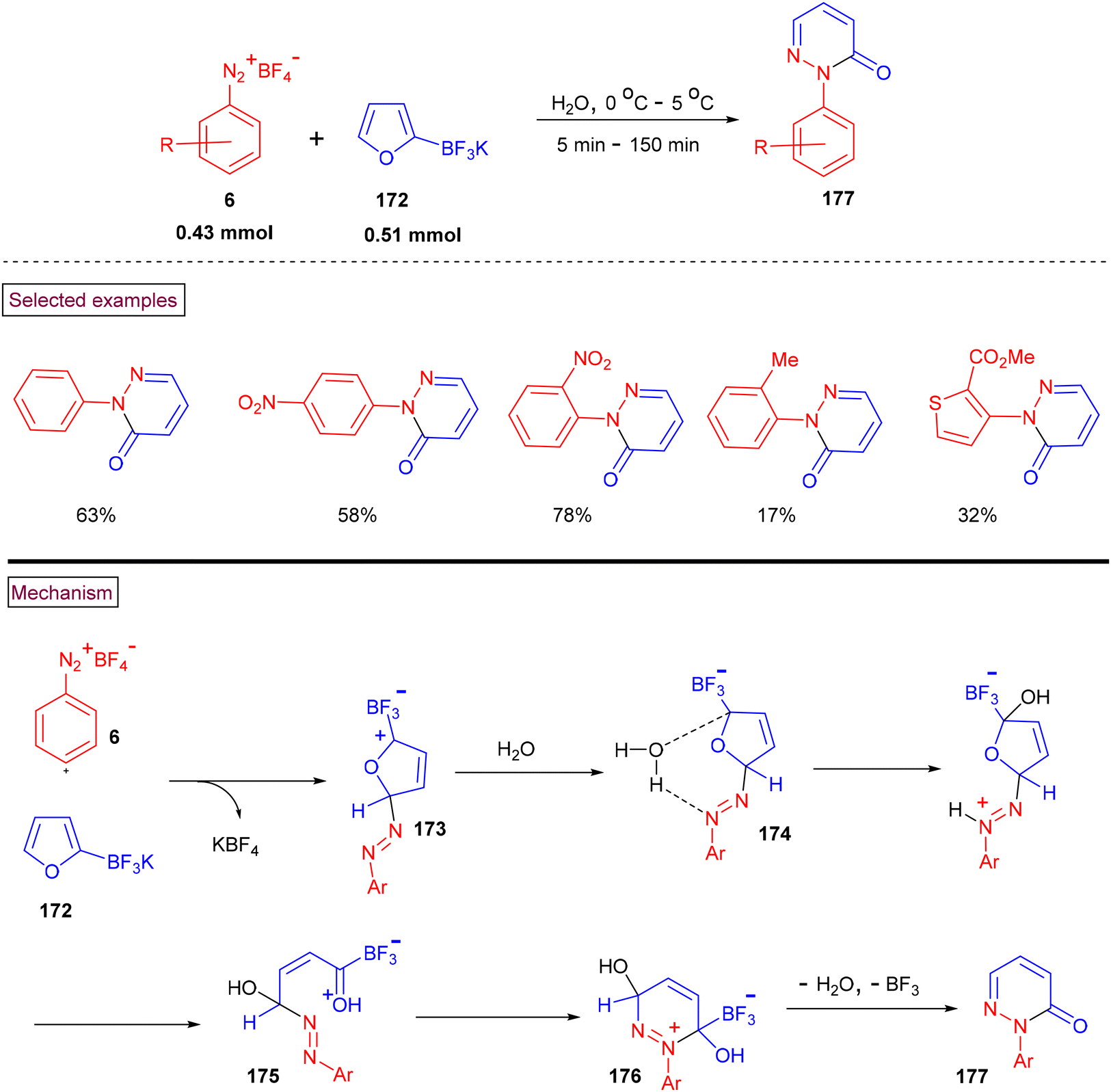
Roglans and co-workers (2014) reported an additive and catalyst-free method to prepare N-arylated pyridazinones (177) from aryldiazonium salts (6) and potassium 2-furantrifluoroborates (172) using water as the only solvent (Scheme 36).138 Steric factors were influential given that ortho-substituted aryldiazonium substrate (6) resulted in the formation of the desired products (177) in lower yields. Alternatively, an electron-withdrawing group (NO2) on the aryl ring favoured the reaction with higher yields (58%–78%) than electron-donating groups. The reaction mechanism involved the nucleophilic reaction of the furan substrate with diazonium species rather than Diels–Alder cycloaddition. The more electrophilic C5 position of (172) was attacked by the terminal nitrogen of (6), resulting in the formation of diazo intermediate (173). The participation of a water molecule in nucleophilic attack, protonation and ring opening yielded intermediate (175), which subsequently underwent intramolecular cyclization, and simultaneous elimination of BF3 and H2O furnished N-arylated pyridazinones (177).
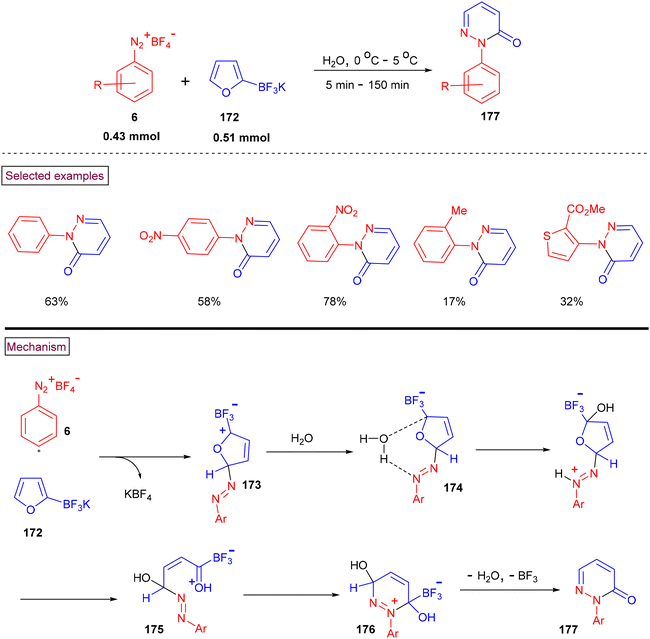 |
| | Scheme 36 Preparation of N-arylated pyridazinones from aryldiazonium salts. | |
3.9. Triazines derivatives
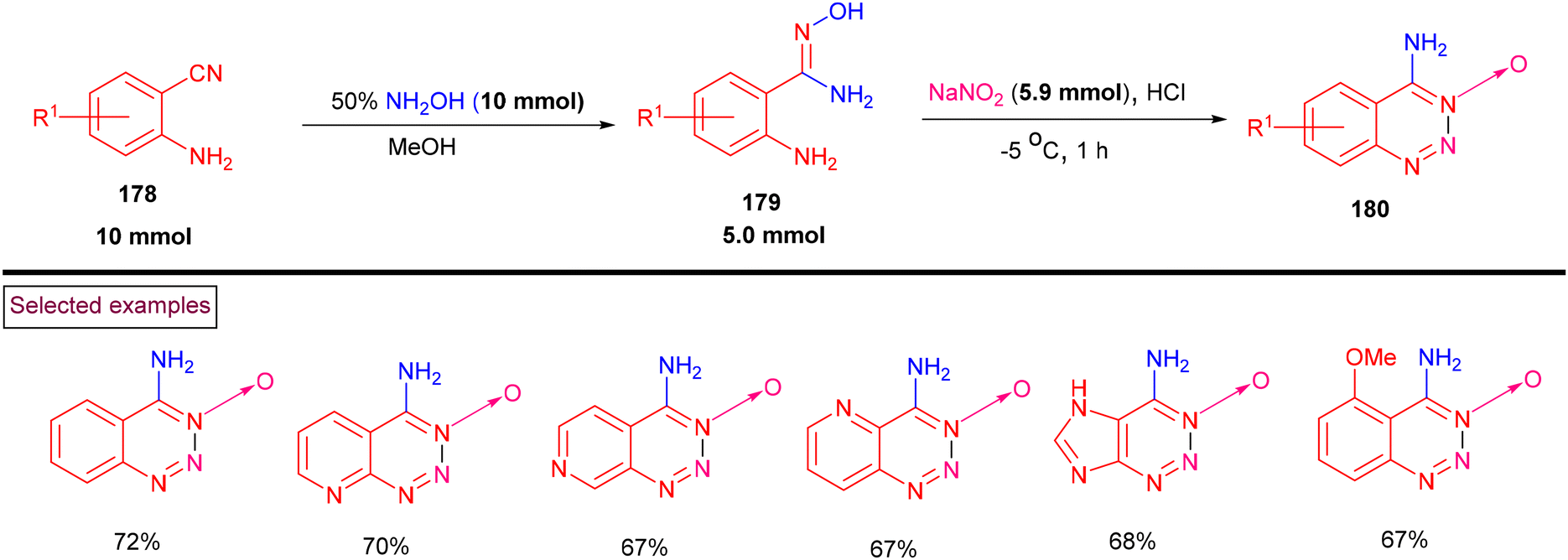
Zhang and Yin et al. (2021) achieved the synthesis of bicyclic 1,2,3-triazines 3-oxides (180) from a variety of o-aminoamidoximes (179) (Scheme 37).139 Mechanistically, o-aminocyanide substrates (178) were treated with hydroxylamine to afford the corresponding o-aminoamidoximes (179), which were further subjected to diazotization and cyclization to give 4-amino-1,2,3-triazine-3-oxides (180). A range of o-amino cyanides (178) including pyridyl, imidazole and pyrimidine derivatives was treated with NH2OH and the corresponding o-aminoamidoximes were isolated in yields of (12%–89%). Tandem diazotization/cyclization with a mixture of NaNO2/HCl afforded 1,2,3-triazines 3-oxides (180) in moderate yields. In the case of 1,2- and 1,4-diazine-based o-aminoamidoximes, chloroximes were obtained instead of 1,2,3-triazines 3-oxides owing to the lower reactivity of amines towards diazotization.
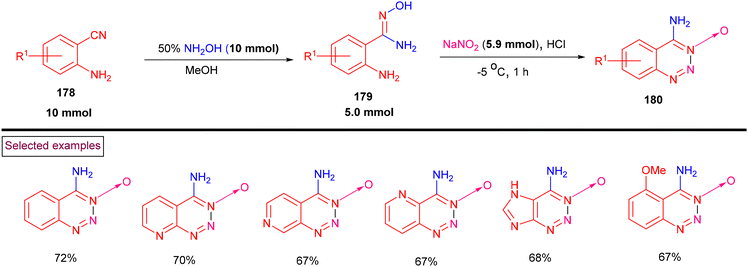 |
| | Scheme 37 Synthesis of 1,2,3-triazines 3-oxides from o-aminocyanide. | |
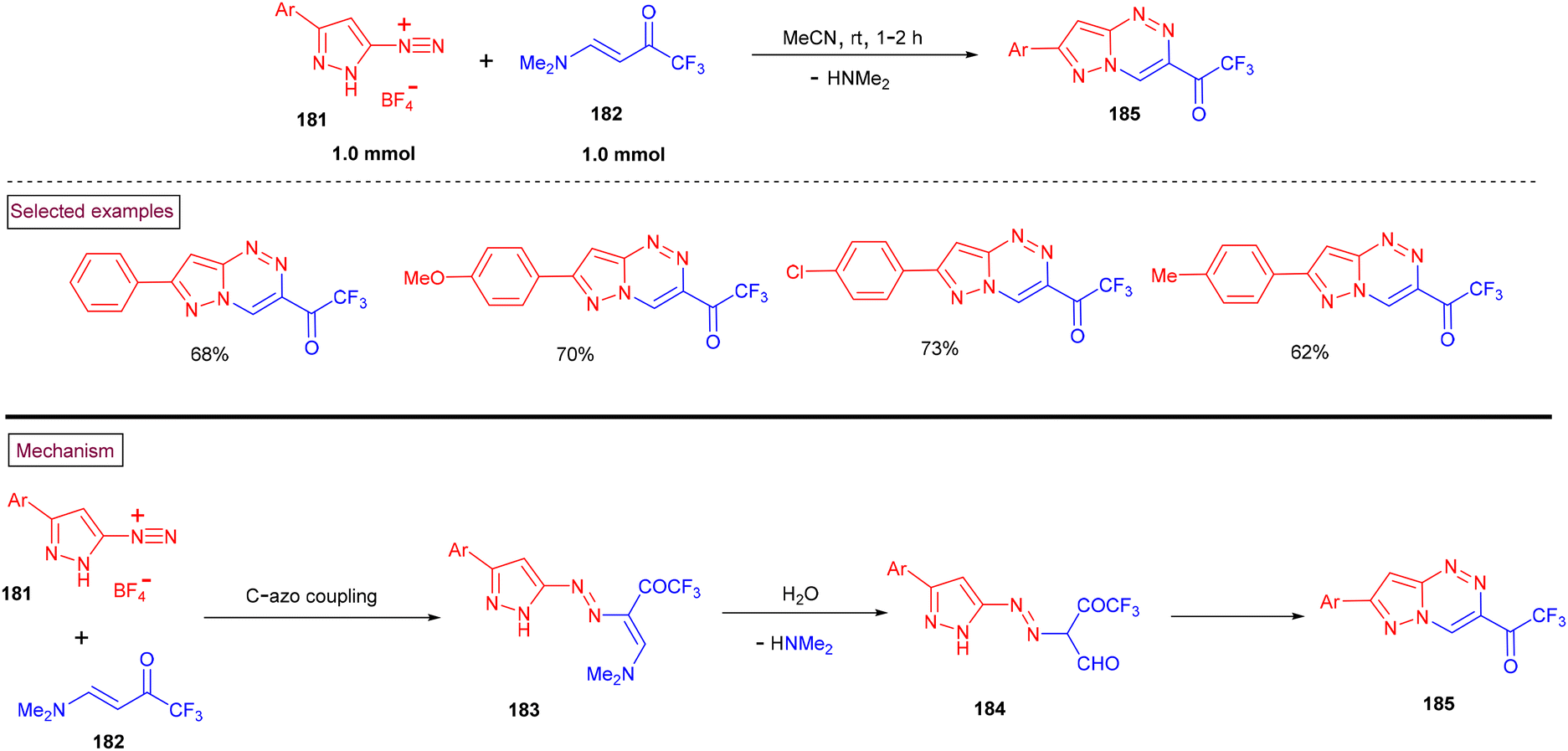
In 2020, Nenajdenko et al. reported a method to prepare pyrazolo[5,1-c][1,2,4]triazines (185) from 3-aryl-pyrazole-5-diazonium salts (181) and amino-enone substrates (182) (Scheme 38).140 A series of 7-aryl-3-(trifluoroacetyl)pyrazolo[5,1-c][1,2,4]triazines was formed at room temperature in moderate yields (62%–73%). According to the mechanistic rationale, the initial reaction between pyrazole-5-diazonium salt (181) and enaminone (182) afforded the direct C-azo-coupled intermediate (183). Rapid hydrolysis of the enamino group into a formyl unit resulted in the formation of aldehyde intermediate (184). Further intramolecular dehydrative cyclization furnished the desired pyrazolo[5,1-c][1,2,4]triazines (185).
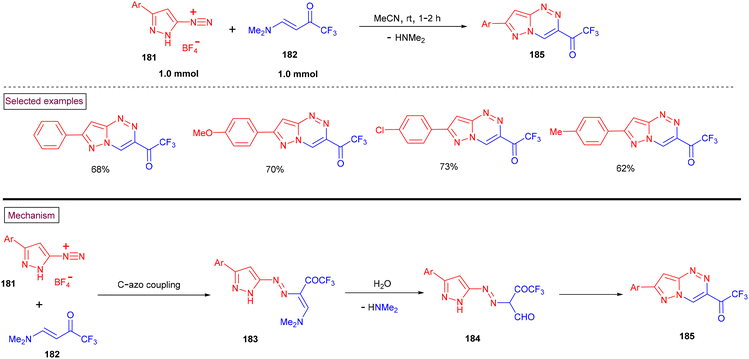 |
| | Scheme 38 Synthesis of pyrazolo[5,1-c][1,2,4]triazines. | |
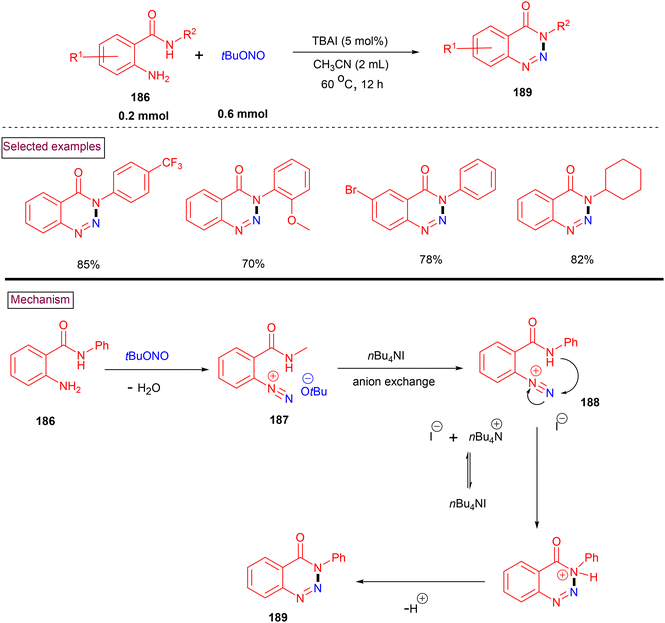
A TBAI-catalyzed method to prepare a diverse array of 1,2,3-benzotriazine-4-(3H)-ones (189) from 2-aminobenzamides (186) and tert-butyl nitrite was disclosed by Yan and Liu's team in 2016 (Scheme 39).141 Various substituted alkyl and aryl benzotriazin-4-(3H)-ones (189) were prepared in good to excellent yields (70%–99%). 2-Amino-N-arylbenzamides with common substituents such as –Me, –OMe, –tBu, –Br, –Cl, and –CF3 were suitable substrates under these conditions. Notably, ortho-functionalized substrates led to lower yields owing to steric factors. Mechanistically, nitrosation of primary amine (186) furnished diazonium salt (187). Further anion exchange in the presence of TBAI (188), intramolecular cyclization and dehydrogenation afforded 1,2,3-benzotriazine-4-(3H)-one (189) in a straightforward fashion.
 |
| | Scheme 39 TBAI-catalyzed synthesis of 1,2,3-benzotriazine-4-(3H)-ones. | |
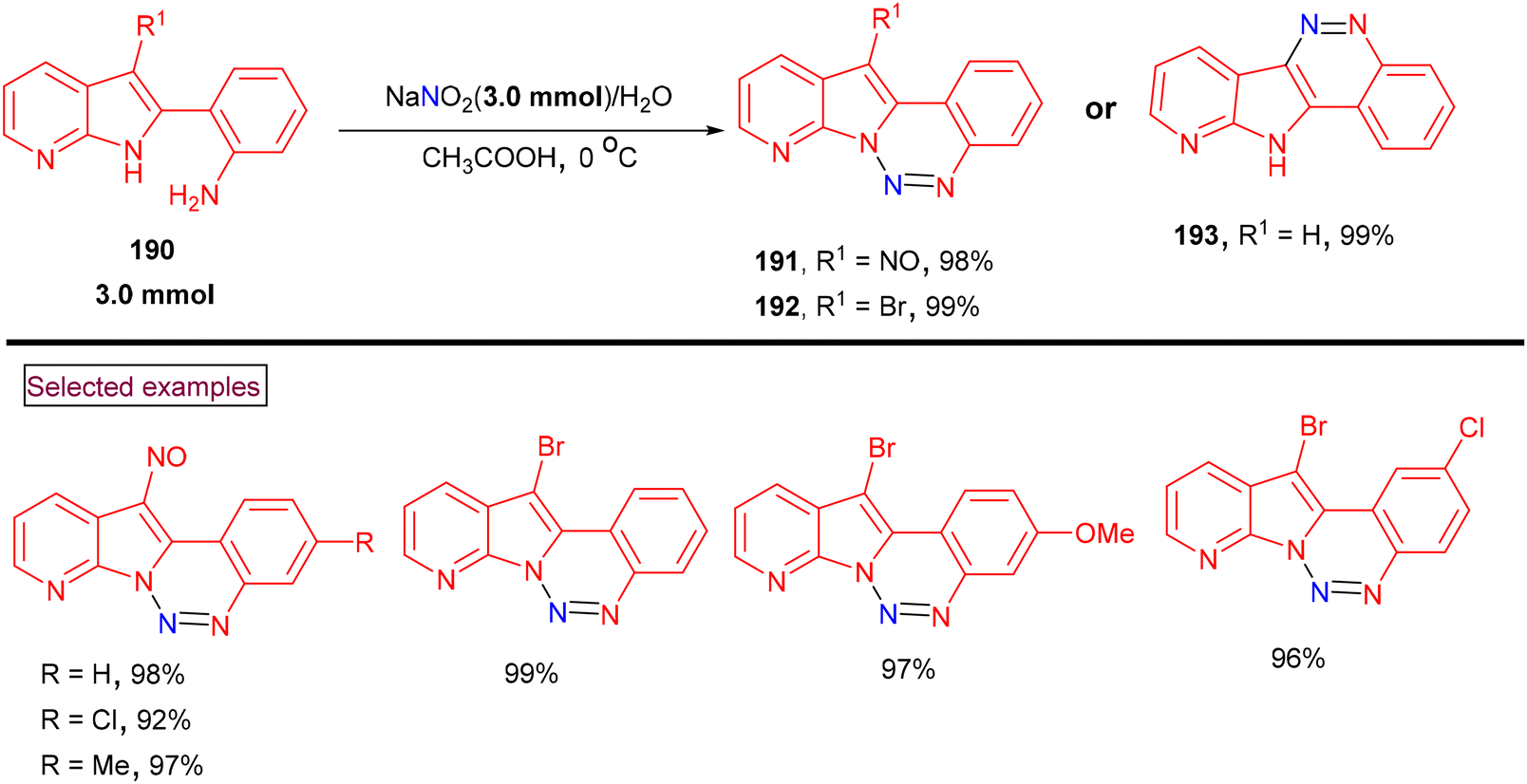
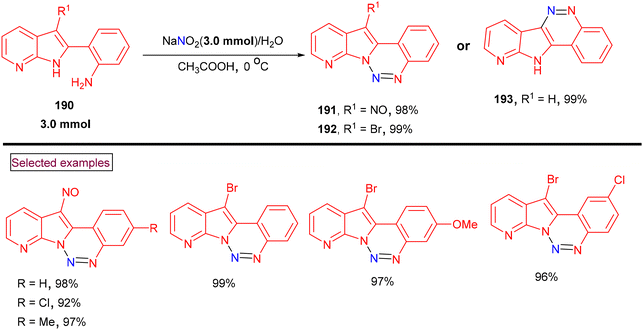
Diana and co-workers reported a one-pot method for the preparation of 12-nitrosopyrido[3′,2′:4,5]pyrrolo[1,2-c][1,2,3]benzotriazines (191) and 12-bromopyrido[3′,2′:4,5]pyrrolo[1,2-c][1,2,3]benzotriazines (192) using 2-(1H-pyrrolo[2,3-b]pyridin-2-yl)anilines (190) as a common precursor (Scheme 40).142 3-Unsubstituted, 3-nitroso and 3-bromo substrates (190) were prepared from 2-(1H-pyrrolo[2,3-b]pyridine-2-yl)anilines. A nitroso group was introduced at the indole C3-position selectively by employing acetyl-protected anilines, whereas NBS enabled direct bromination at the C3-position at room temperature. Key diazotization was performed at 0 °C using an equimolar quantity of sodium nitrite in acetic acid. Subsequent intramolecular amino-cyclization resulted in the formation of benzotriazine derivatives (191–193) in excellent yields. Some of these derivatives exhibited excellent anti-tumor activity against various human cancer cell lines (ovarian, melanoma, renal, breast and prostate tumor cell lines) with nanomolar GI50 values. Additionally, these compounds also showed activity in cells overexpressing P-gp.
 |
| | Scheme 40 Preparation of benzotriazines via intramolecular amino-cyclization. | |
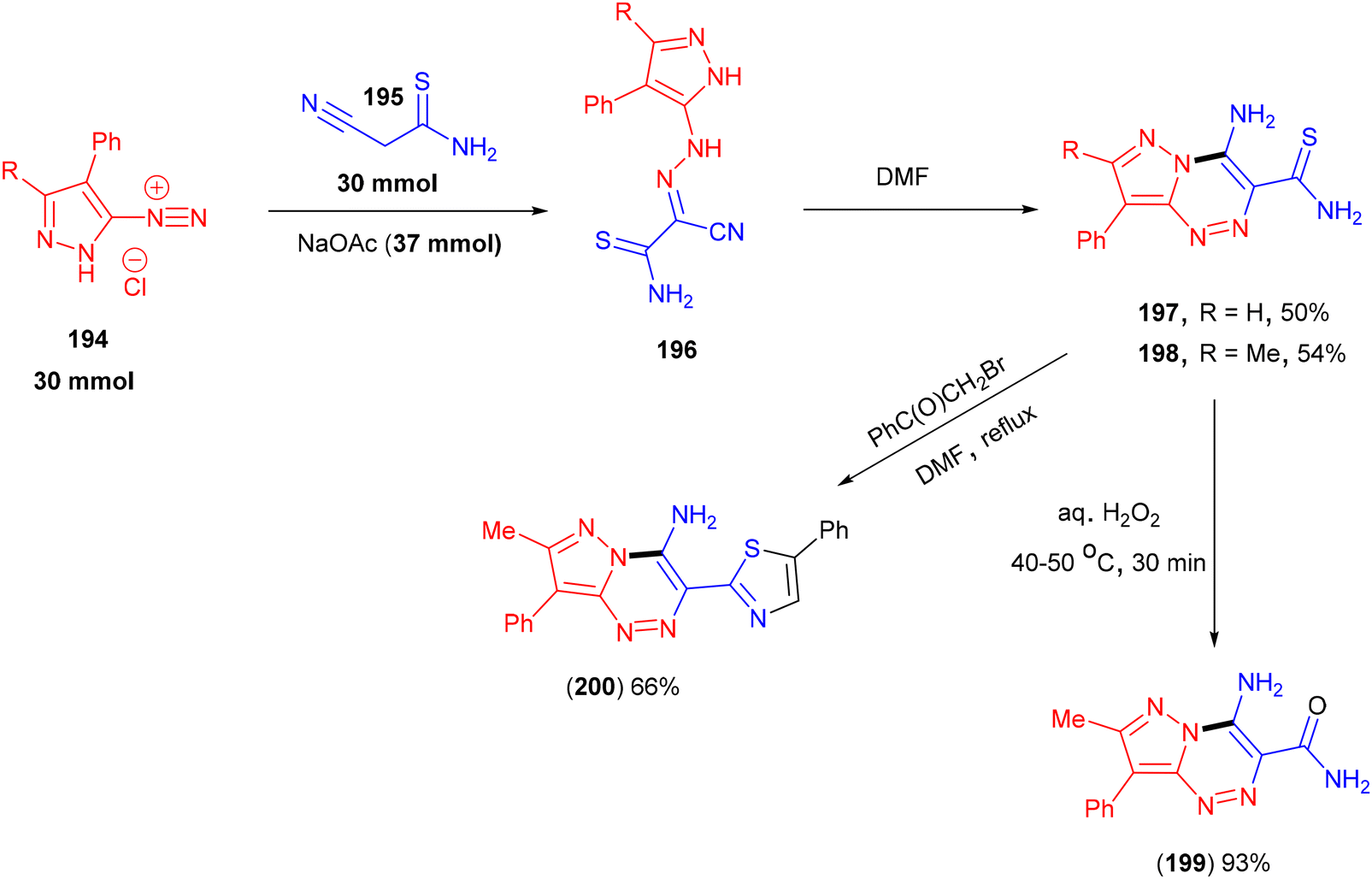
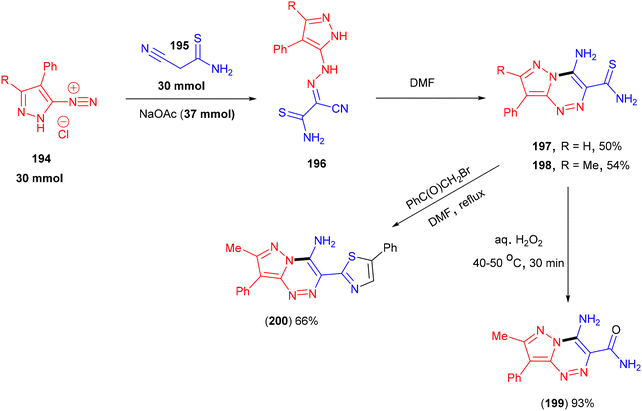
Pyrzolo[5,1-c][1,2,4]triazines (197 and 198) could be obtained from the reaction of pyrazole-3(5)-diazonium salts (194) with 2-cyanothioacetamide (195) in DMF (Scheme 41).143 Ledenyova and co-workers proposed that this process proceeded via the initial aza-coupling between (194) and (195) to afford unstable hydrazone intermediate (196), followed by intramolecular heterocyclization. Synthetically useful Hantzsch-type thiazoles (200) were obtained in good yields when these triazines were treated with α-halo ketones. Under oxidative conditions the in presence of H2O2, 4-aminopyrazolo[5,1-c][1,2,4]triazin-3-carboxamides (199) were isolated in excellent yield.
 |
| | Scheme 41 Synthesis of pyrazolo[5,1-c][1,2,4]triazines from active methylene compounds and aryldiazonium salts. | |
3.10. Quinazoline and quinazolinone derivatives
Functionalized quinazolines are one of the prestigious classes of N-heterocyclic core prevalent in many natural products and bioactive molecules including anticonvulsant, anti-inflammatory, anti-HIV, antimicrobial, antitumor and anti-pneumonia activities.144 In addition, quinazoline derivatives are also used in functional materials.145 In general, quinazolines are prepared by various methods, as follows: (i) catalytic sp3 C–H amination of 2-aminoarylaldehydes with benzylic amines,146a (ii) by o-iodoxybenzoic acid-mediated cascade reaction of o-aminobenzylamines with aldehydes,146b (iii) by Cu-catalysed sequential Ullmann coupling and aerobic oxidation of (2-bromophenyl) methylamines and amide derivatives146c and (iv) Cu-catalyzed (2 + 2 + 2) tandem annulation of nitriles with diaryliodonium salts.146d However, transition metal-free approaches using aryldiazonium salts for the synthesis of quinazolines are limited.
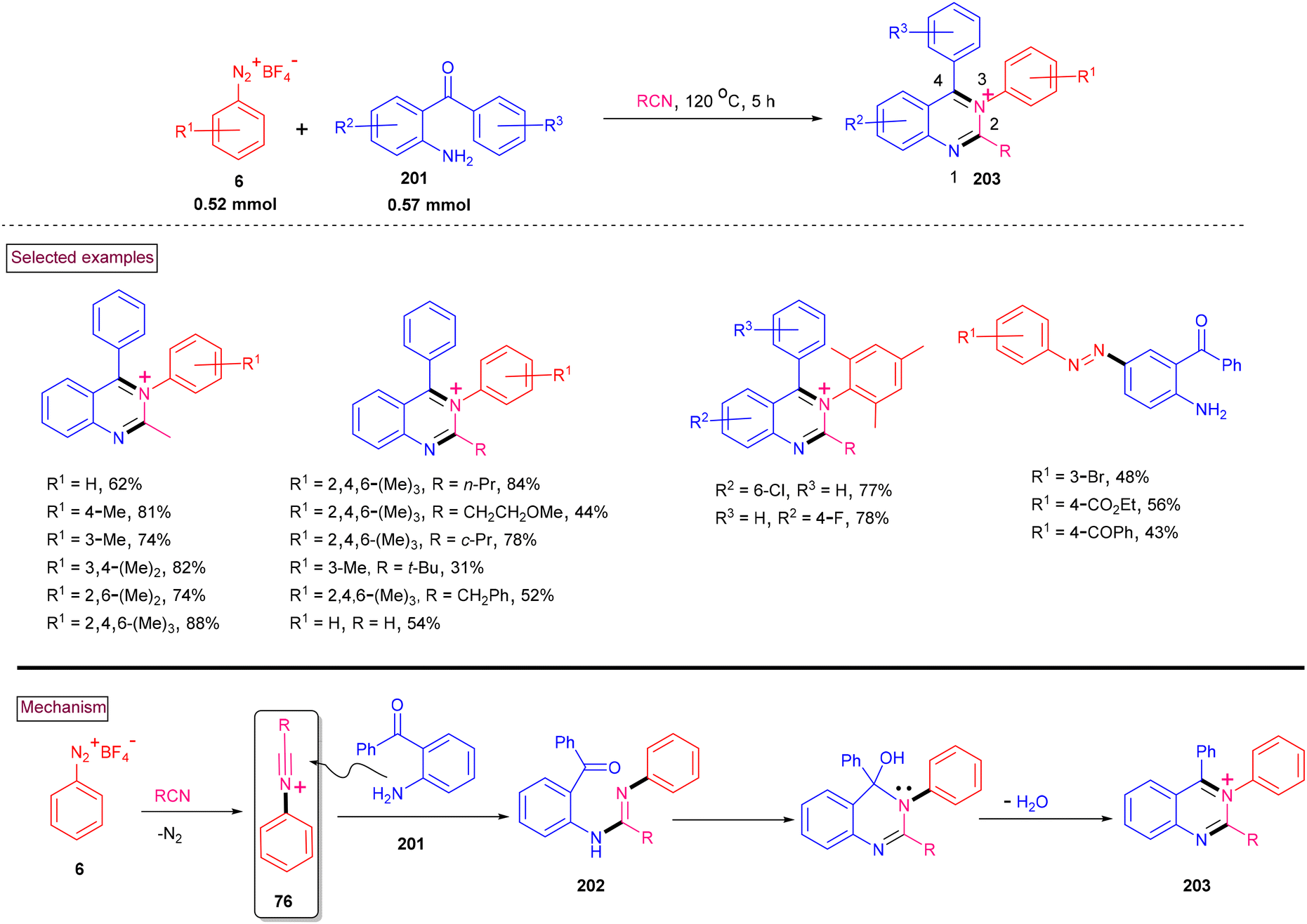
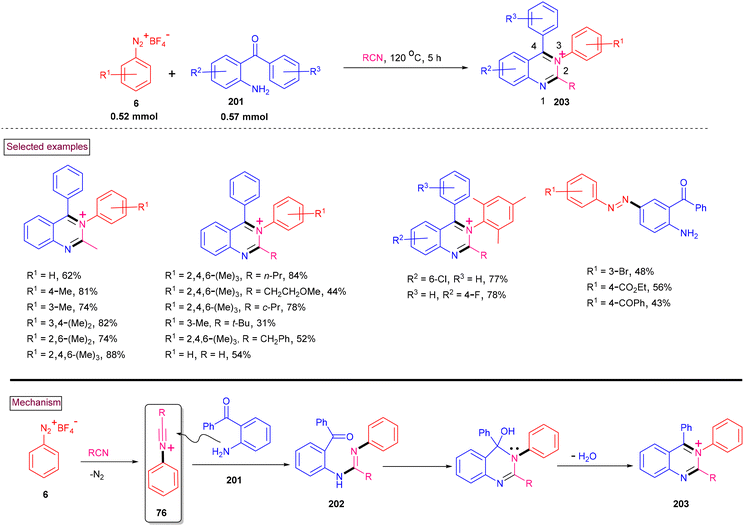
In continuation to our efforts to develop green synthetic methods for the construction of N-heterocycles, we reported the novel multi-component synthesis of functionalized N-arylquinazolinium salts (203) from aryldiazonium salts (6), 2-aminoarylketones (201) and nitriles (Scheme 42).147 Mechanistically, this process relied on the in situ formation of N-arylnitrilium intermediate (76), which subsequently underwent intermolecular amination (202), tandem cyclization and aromatization (203). Electron-donating groups (2,4,6-triMe, 3,4,5-triOMe) on the aryldiazonium salts favored the reaction and the corresponding products were isolated in excellent yields (76%–88%), whereas electron-withdrawing substituents such as, –F and –CO2Me resulted in the formation of diazo products. Various linear and branched alkyl nitriles and benzyl nitriles worked well under these conditions. Similarly, a range of multi-substituted 2-aminoarylketones (201) was found to be viable substrates to afford the N-arylquinazolinium salts in 73%–78% yields. The synthetic potential of this method was further demonstrated via nucleophilic addition of alkoxide (–OMe and –OEt) to the C4 position both in a stepwise and one-pot fashion. These types of cationic quinazolinium salts are usually obtained by N-substitution, whereas this method offers an alternative practical approach.
 |
| | Scheme 42 Multicomponent synthesis of N-arylquinazolinium salts. | |
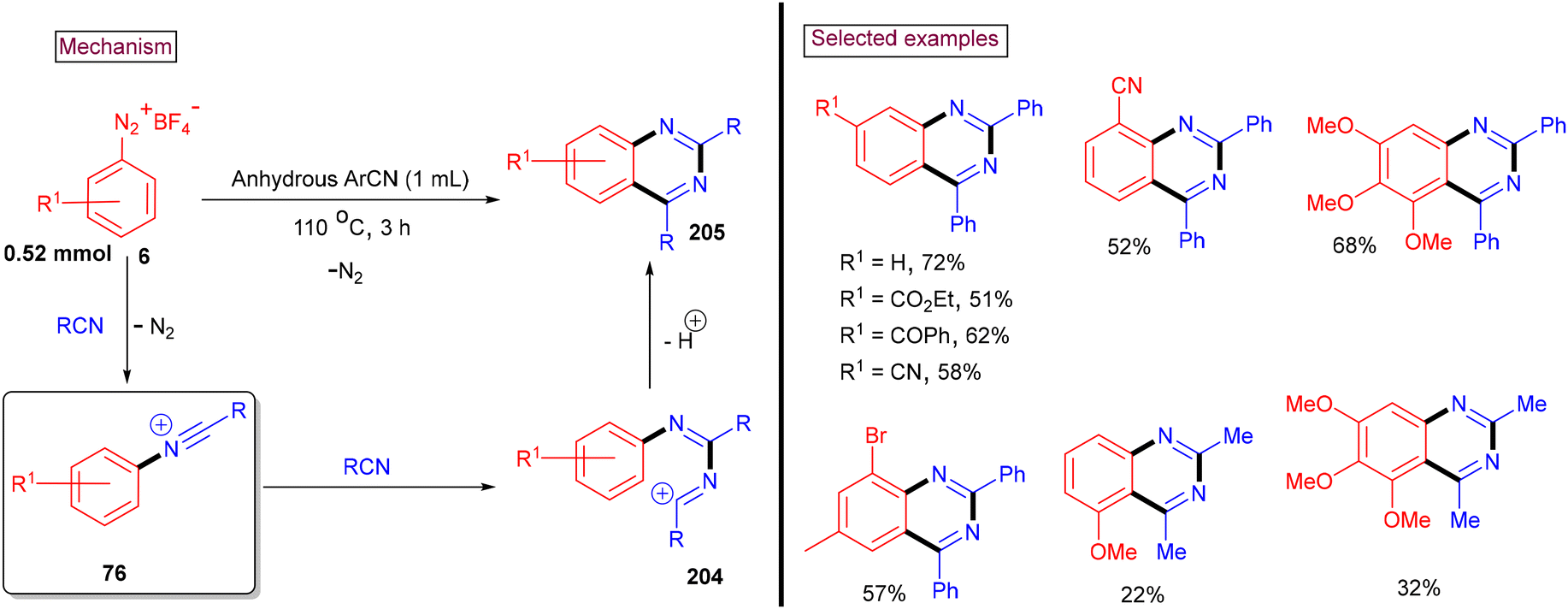
An intermolecular cascade annulation approach for the synthesis of 2,4-disubstituted quinazolines (205) from aryldiazonium salts (6) and nitriles was reported by our group in 2017 (Scheme 43).148 Alkyl nitriles (–iPr, –tBu, and CyH) led to the desired products in lower yields (38%–44%), whereas acetonitrile only reacted with activated aryldiazonium salts. This observation was due to the low stability of the cationic intermediates formed during this transformation. Alternatively, aryl and heteroaryl nitriles resulted in the formation of the corresponding quinazolines in good yields. Aryldiazonium salts with electron-donating and moderately withdrawing groups (–CO2Me, –COPh, –CF3, and –CN) worked well, whereas the strongly electron-withdrawing –NO2 group furnished the desired product in a lower yield (19%). In this method, in situ-generated N-arylnitrilium ion (76) (from the reaction of aryldiazonium salt and nitrile) reacted with another molecule of nitrile to afford (204), which was converted via a tandem addition/electrophilic cyclization to yield 2,4-disubstituted quinazolines (205) in moderate to good yields.
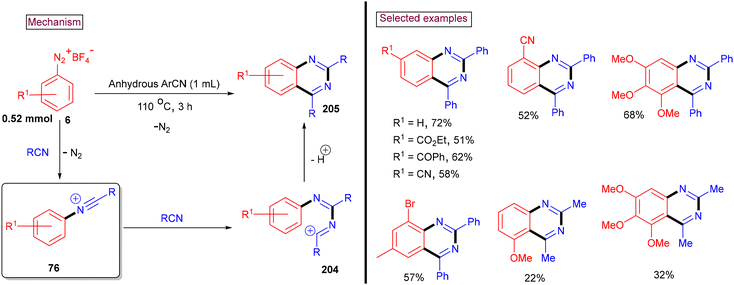 |
| | Scheme 43 Preparation of 2,4-disubstituted quinazolines via intermolecular cascade annulation. | |
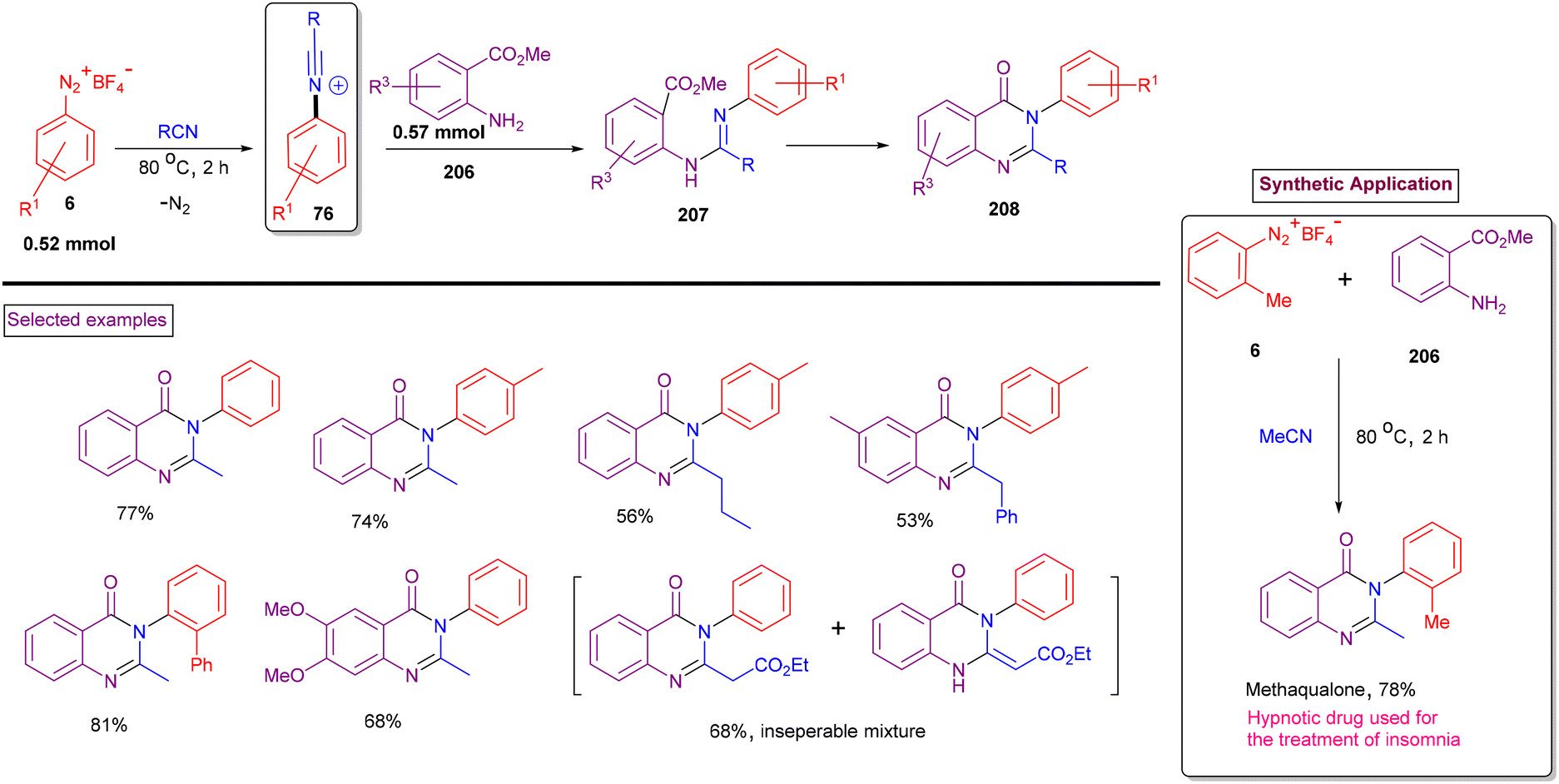
Quinazolinones are also highly prevalent units in a range of pharmaceuticals and natural products such as, methaqualone, afloqualone, terremide C, febrifugine and circumdatin K.149 In addition to their wide occurrence, quinazolinone derivatives exhibit diverse therapeutic activities including hypotensive, anticancer, and antiallergy. The synthesis of quinazolinone is commonly based either on the reaction of anthranilic acid, primary amine in the presence of formic acid or the transition metal-catalysed tandem reaction of 2-aminobenzamides with 1,3-diketones.150 Both methods often require the use of toxic reagents such as POCl3 and PCl3 and expensive transition metal catalysts. Based on our findings on the chemistry of aryldiazonium salts, we found a mild route to synthesise quinazolin-4(3H)-ones (208) via the reaction of aryldiazonium salts (6), 2-aminobenzoates (206) and nitriles (Scheme 44).151 In this case, in situ-formed N-arylnitrilium intermediate (76) underwent rapid amination with 2-aminobenzoate (206) and further cascade cyclization, yielding quinazolin-4(3H)-ones (208). In contrast to our previous report (Scheme 34), phenanthridines were not observed under these conditions with 2-phenyl aryldiazonium salts. It was observed that the intermolecular amination/cascade annulation of the N-arylnitrilium intermediate was superior to intramolecular arylation. An excellent substrate scope was observed with respect to aryldiazonium salt (6)-substituted anthranilates (206) and a variety of nitriles (alkyl, aryl, and benzyl) and the products were obtained in good isolated yields. The utility of this reaction was further demonstrated by preparing methaqualone, a well-known hypnotic drug.
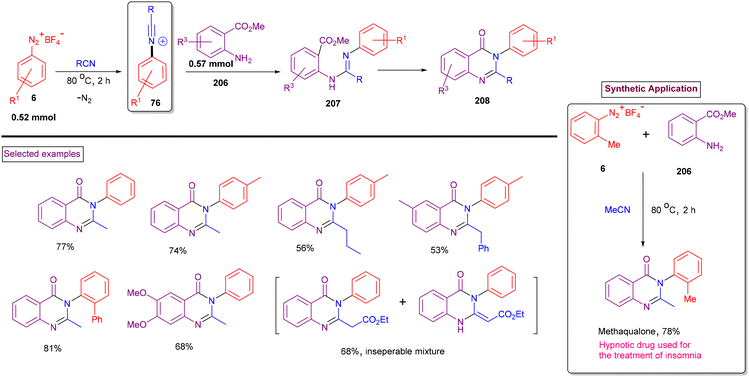 |
| | Scheme 44 Preparation of quinazolin-4(3H)-ones via in situ-generated N-arylnitrilium ion. | |
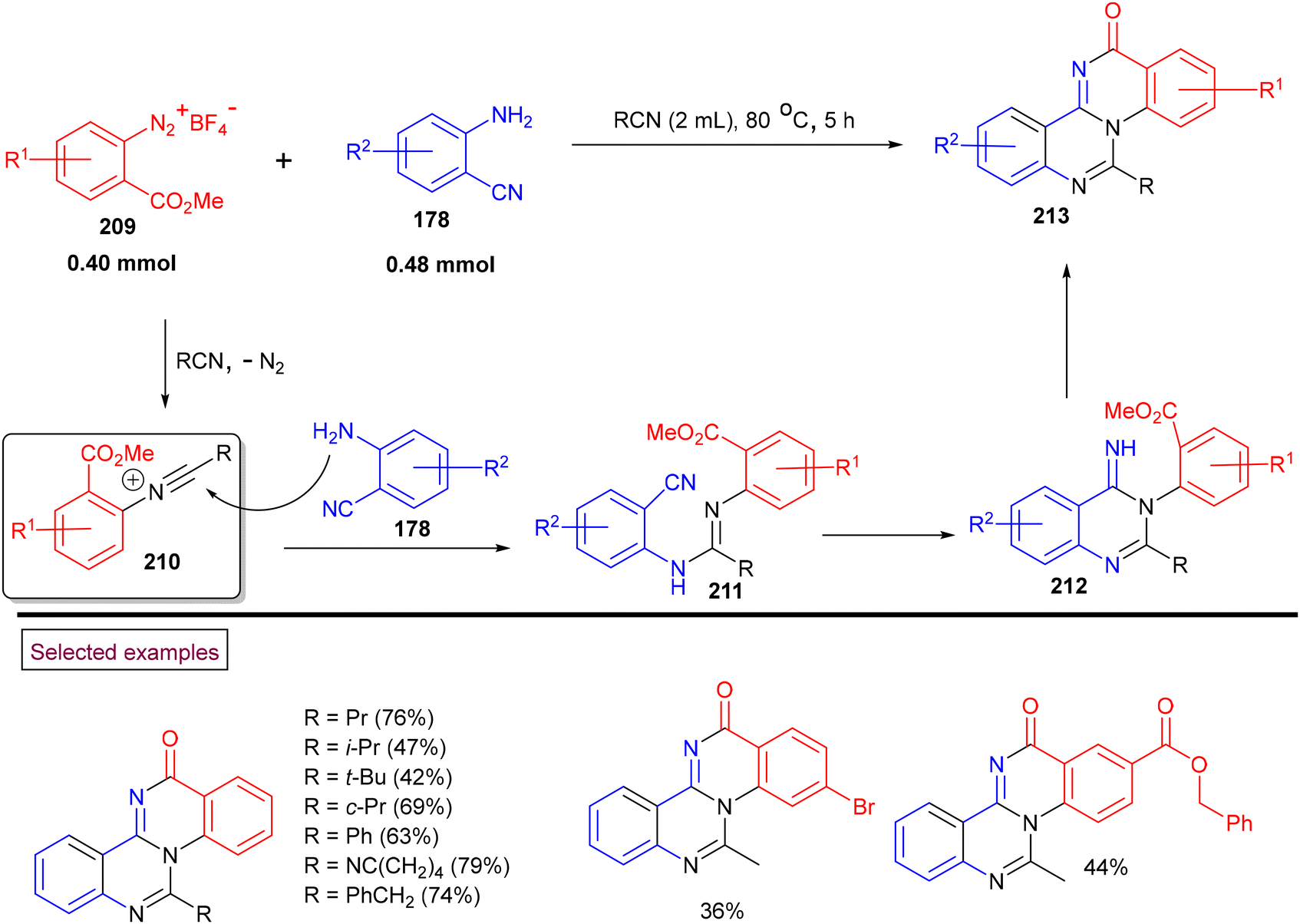
In 2018, we reported an unprecedented method to prepare quinazolino[3.4-a]-quinazolin-13-ones with excellent skeletal diversity (213) via the direct reaction of 2-cyanoanilines (178), nitriles and o-(methoxycarbonyl)benzenediazonium salts (209) (Scheme 45).152 Compared to other multistep literature methods, this process yielded the tetracyclic compounds in a one-pot operation with the sequential formation of four N–C bonds. This strategy proceeds through the initial formation of reactive N-arylnitrilium ion (210) and further amination/cascade cyclization/amidation to furnish the desired polynuclear N-heterocyclic compounds (213). In the case of ortho-substituted diazonium salts, quinazolin-4-(3H)-imine and N-arylquinazolin-3-(4H)-one were formed instead of the desired product due to steric effects. This protocol features several advantages including flexibility in the substitution patterns, one-pot operation, and mild, metal-free reaction conditions.
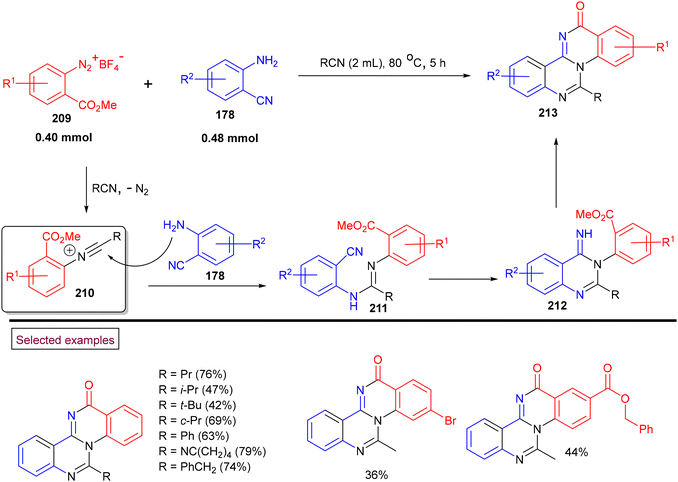 |
| | Scheme 45 Multicomponent synthesis of quinazolino[3.4-a]-quinazolin-13-ones. | |
3.11. Quinolines and quinazolin-4(3H)-imine derivatives
Quinazolin-4(3H)-imines are medicinally promising and biologically significant aza-heterocyclic frameworks with remarkable activities such as, antihypertensive, antimicrobial, antiproliferative, anticonvulsive, anti-viral, and α-glucosidase inhibitors.153 The Cu(OTf)2-catalysed tandem synthesis of 2-aminoaryl-functionalized quinazolin-4(3H)-imines from diaryliodonium salts and o-cyanoanilines was reported by Chen et al. in 2014.154 However, transition-metal-free procedures using various precursor were also reported in recent years. The Hünig's base-promoted preparation of 3-aryl-4-imino-3,4-dihydroquinazolin-2-carbonitriles was reported by Koutentis's group (2013) from 2-amino-N′-arylbenzamides and Appel salt (4,5-dichloro-1,2,3-dithazolium chloride).155a Rayat et al. (2011) disclosed the convenient preparation of 2-halo-3-aryl-4(3H)-quinazoliniminium salts via the intramolecular cyclization of functionalized carbodiimides in the presence of a Lewis acid and trace water.155b In addition, functionalized quinazolin-4(3H)-imines were also prepared from phenylchloroimines155c and 2-amino-N′-arylbenzamidines.155d However, despite these advancements, the requirement of pre-functionalization, harsh conditions, multistep procedures, and metal catalysts are limitations.
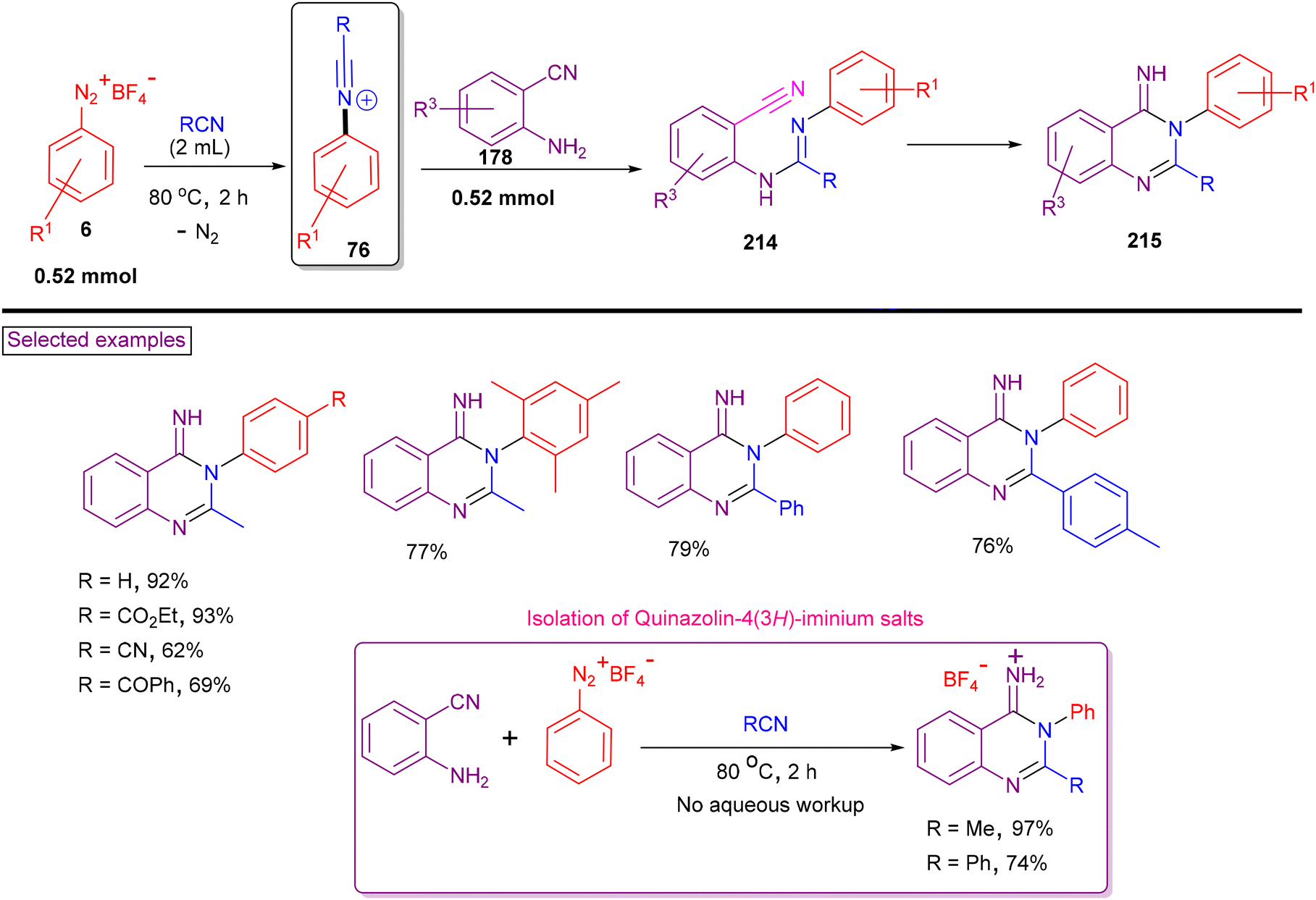
In 2017, our research group reported a facile method for the synthesis of quinazolin-4(3H)-imines (215) via intermolecular amination/tandem cyclization from aryldiazonium salts (6), 2-cyanoanilines (76) and nitriles in a one-pot manipulation (Scheme 46).156 Aryldiazonium salts with nitrile, ester, amide, ether, ketone and thioether were tested and the expected products (215) were isolated in good yields (up to 93%). Alternatively, alkyl, aryl, benzyl, heteroaryl nitriles and a few lab-prepared 2-cyanoanilines (178) were also tested to provide the products in good yields (68%–88%). In situ-generated N-arylnitrilium ion (76) was aminated with 2-cyanoaniline (178), resulting in the formation of amidine (214). Intramolecular aminative cyclization with an adjacent cyano group yielded the product (215). Without the need for aqueous workup, quinazolin-4(3H)-iminium salts were isolated in high yields.
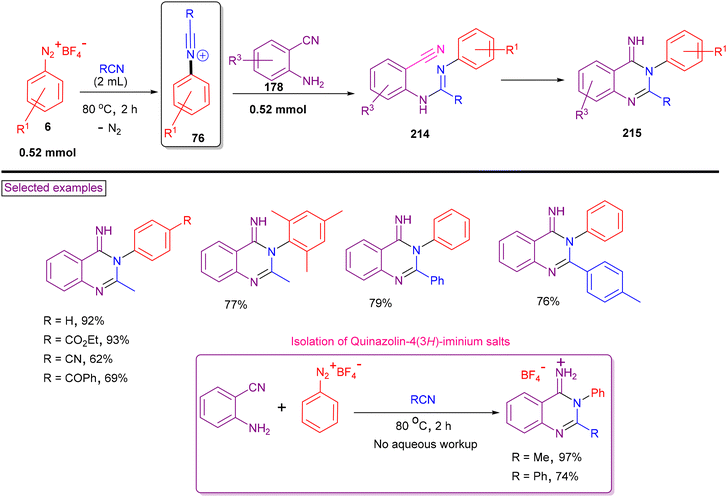 |
| | Scheme 46 Preparation of quinazolin-4(3H)-imines via intermolecular amination/tandem cyclization. | |
Quinoline is also a vital scaffold for drug discovery leads and plays a crucial role in medicinal chemistry. Quinoline derivatives have been tested for their diverse biological activities such as antifungal, antiviral, antimalarial, anticancer, CNS effects, cardiovascular, analgesic, anticonvulsant, and anti-inflammatory.157 Together with well-known classical syntheses, the construction of functionalized quinolines is achieved via transition metal-catalysed reactions,158 ultrasound irradiation methods,159 and ionic liquid-mediated reactions.160
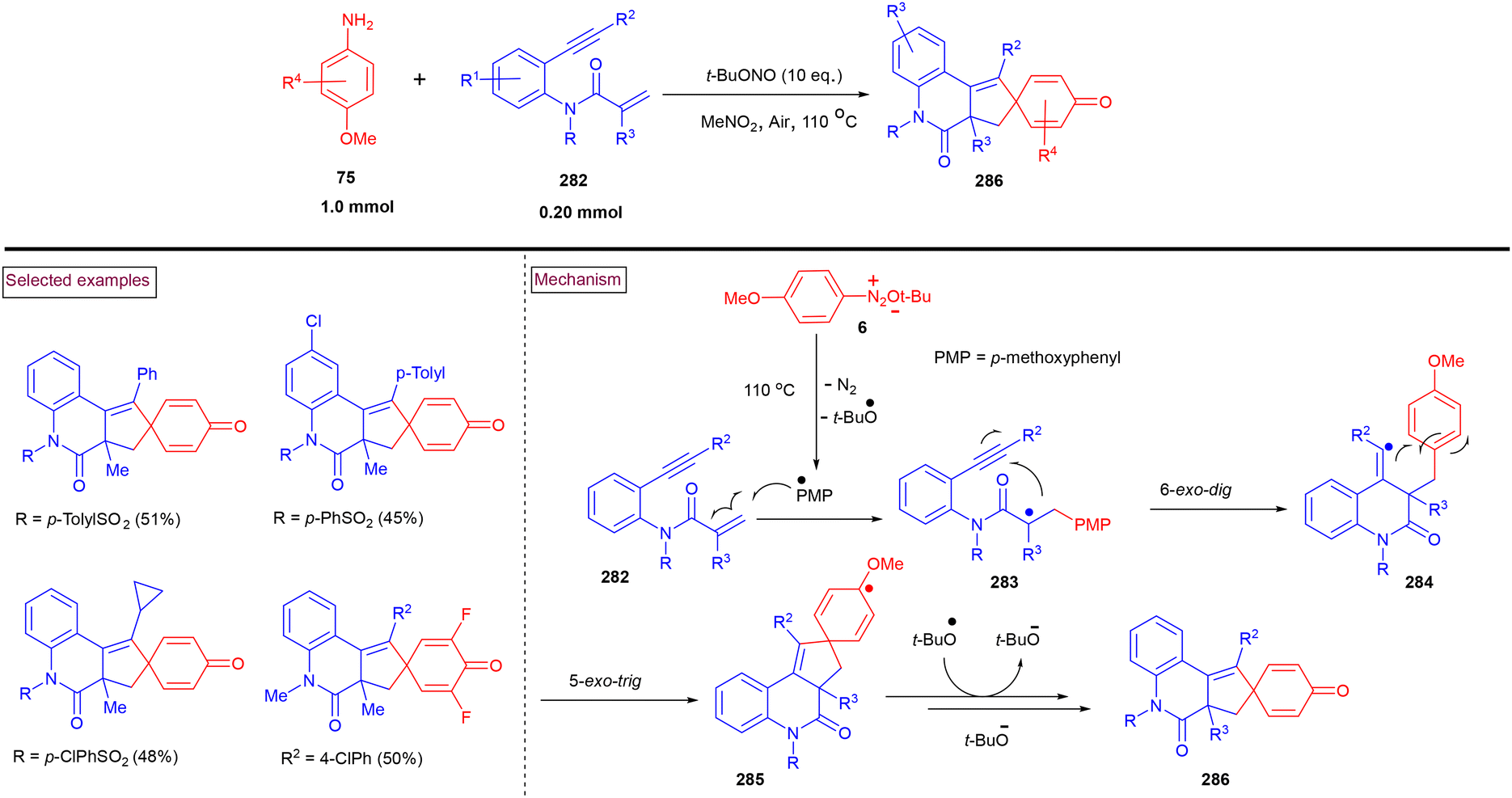
In 2022, Batra et al. realized a [4 + 2] benzannulation approach for the synthesis of azole-fused quinolines (223) from alkynes (221) (terminal and internal) and 5/2 amino-N-phenyl azoles (216) in the presence of t-BuONO (Scheme 47).161 A radical mechanism was proposed by the authors, in which the initial formation of aryldiazonium salt (217) from the amino precursor (216) and its reaction with deprotonated intermediate (218) resulted in the formation of stabilized intermediate (219). The further formation of radical intermediate (220) via homolytic cleavage and radical addition to alkyne, and radical cyclization followed by aromatization yielded the cyclized product (223). Interestingly, the authors also demonstrated the synthesis of pyrrolo[1,5-a]quinolines and 1,2,3-triazolo[1,5-a]quinolines. It was proven that a functional group to accept the hydrogen must be attached to the carbon next to the amino group to stabilize the transition state during this transformation.
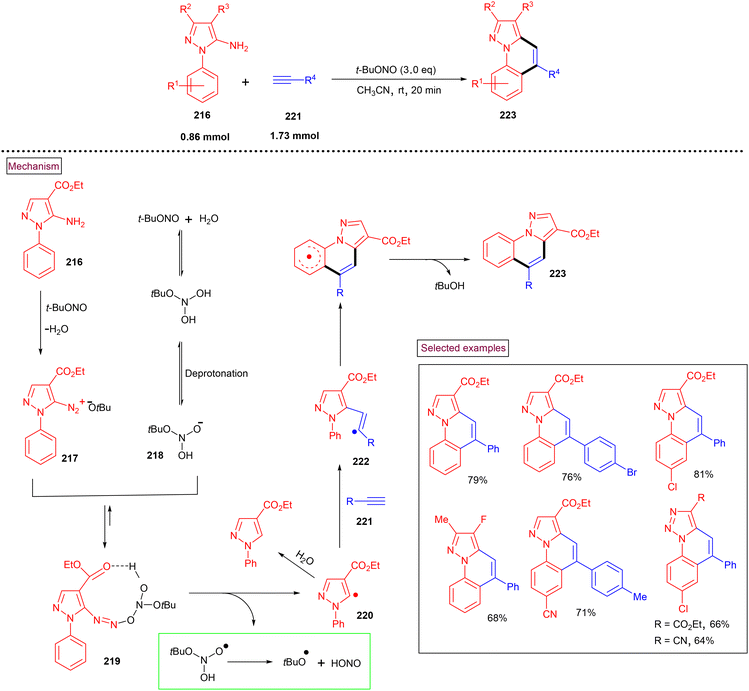 |
| | Scheme 47 Preparation of azole-fused quinolines via the [4 + 2] benzannulation approach. | |
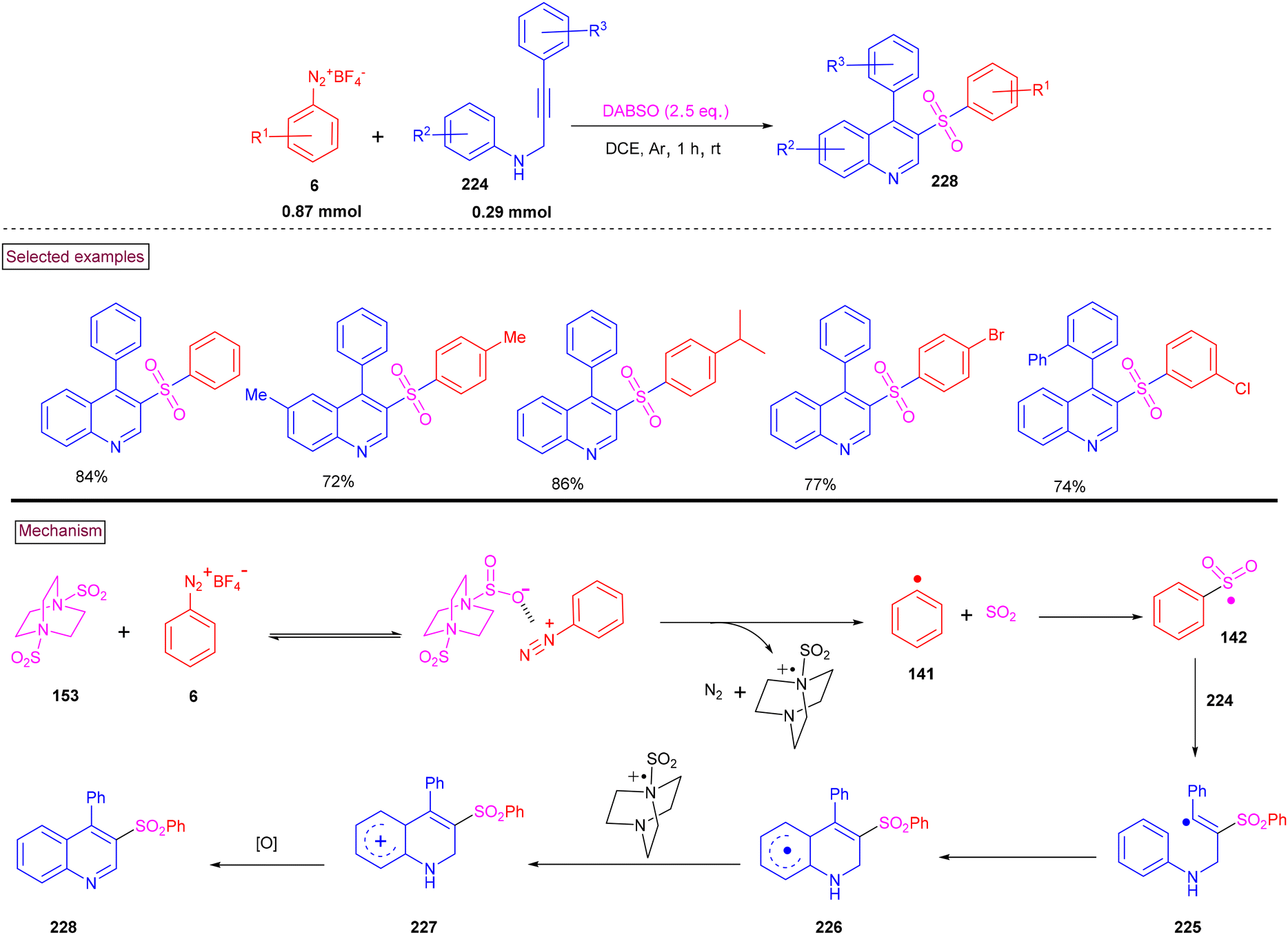
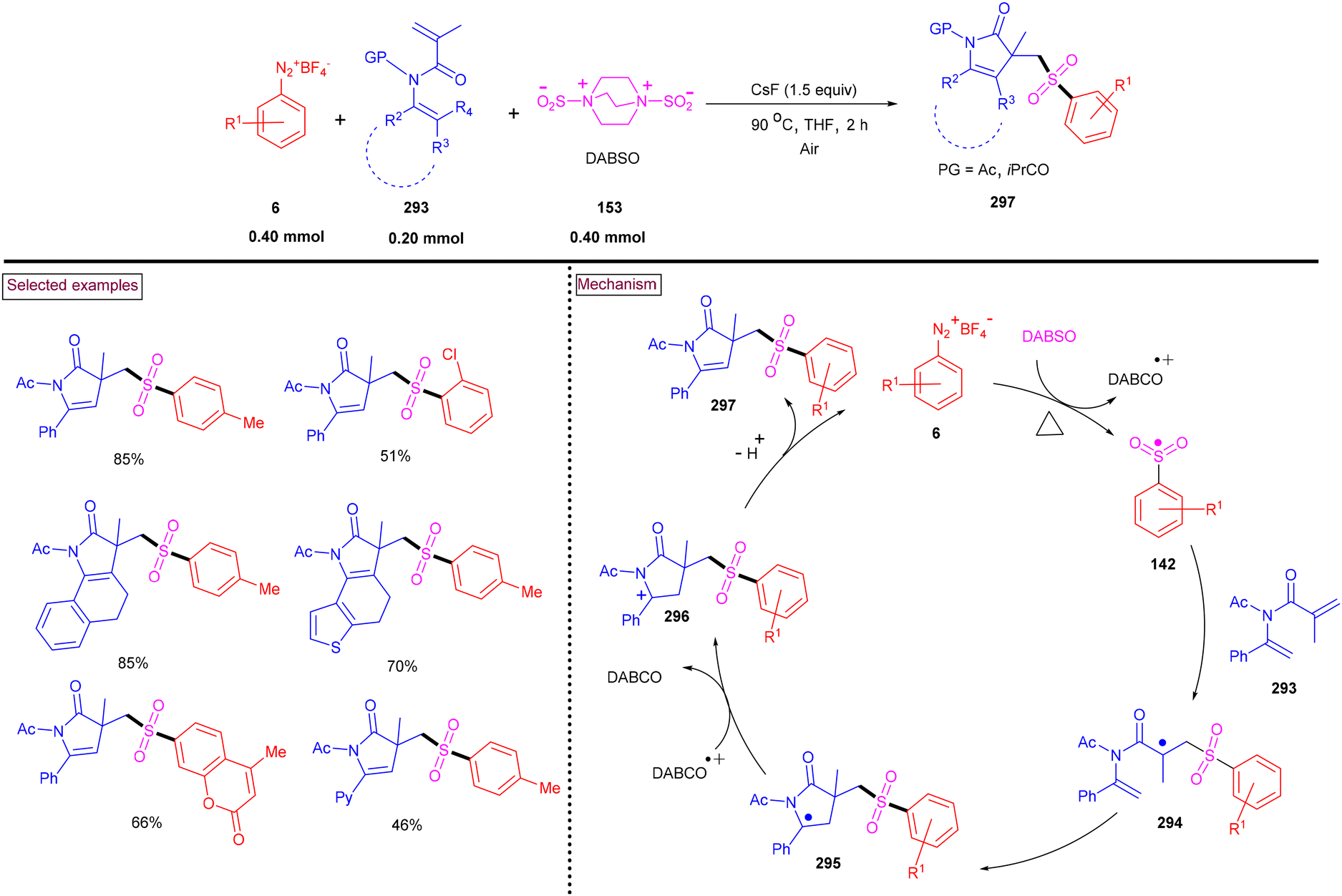
By performing a three-component reaction involving aryldiazonium salt (6), N-propargylamine (224) and DABSO under inert conditions, 3-arylsulfonylquinolines (228) were obtained via oxidative sulfonylation (Scheme 48).162 Mal et al. (2022) studied the substrate scope with variously substituted N-propargylamines (224) and aryldiazonium salts (6). Alkyl-, aryl-, and heteroaryl-bearing N-propargylamines reacted well and the desired products were obtained in good isolated yields. Based on the control experiments, the reaction pathway was proposed to proceed via a radical pathway. Mechanistically, the in situ generation of aryl sulfonyl radical (142), followed by its reaction with the triple bond of N-propargylamine (224) generated intermediate (225). Further intramolecular cyclization and aromatization resulted in the formation of 3-aryl sulfonyl quinoline (228). During this transformation, DABSO played a dual role as a sulfone source and oxidant. In addition, 3-arylsulfonylquinolines (228) bearing halogens were subjected to a series of post-synthetic transformations involving Pd-catalyzed Suzuki coupling, Sonogashira coupling, and Heck coupling, demonstrating its synthetic potential.
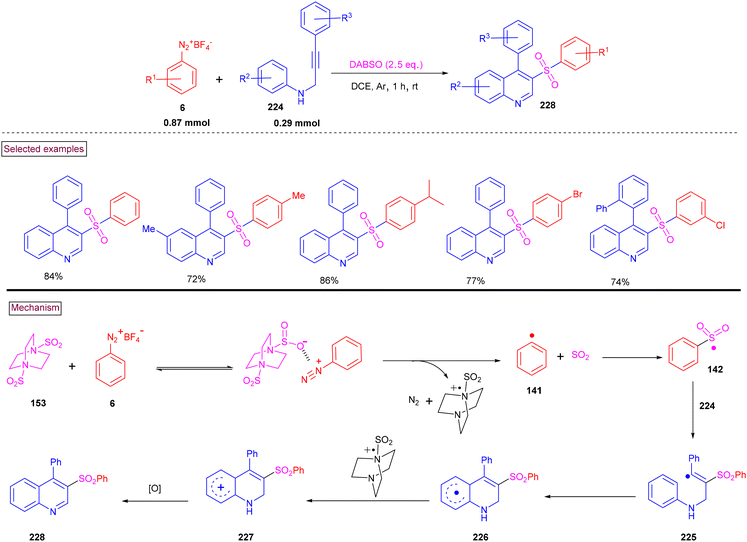 |
| | Scheme 48 Preparation of 3-arylsulfonylquinolines via oxidative sulfonylation. | |
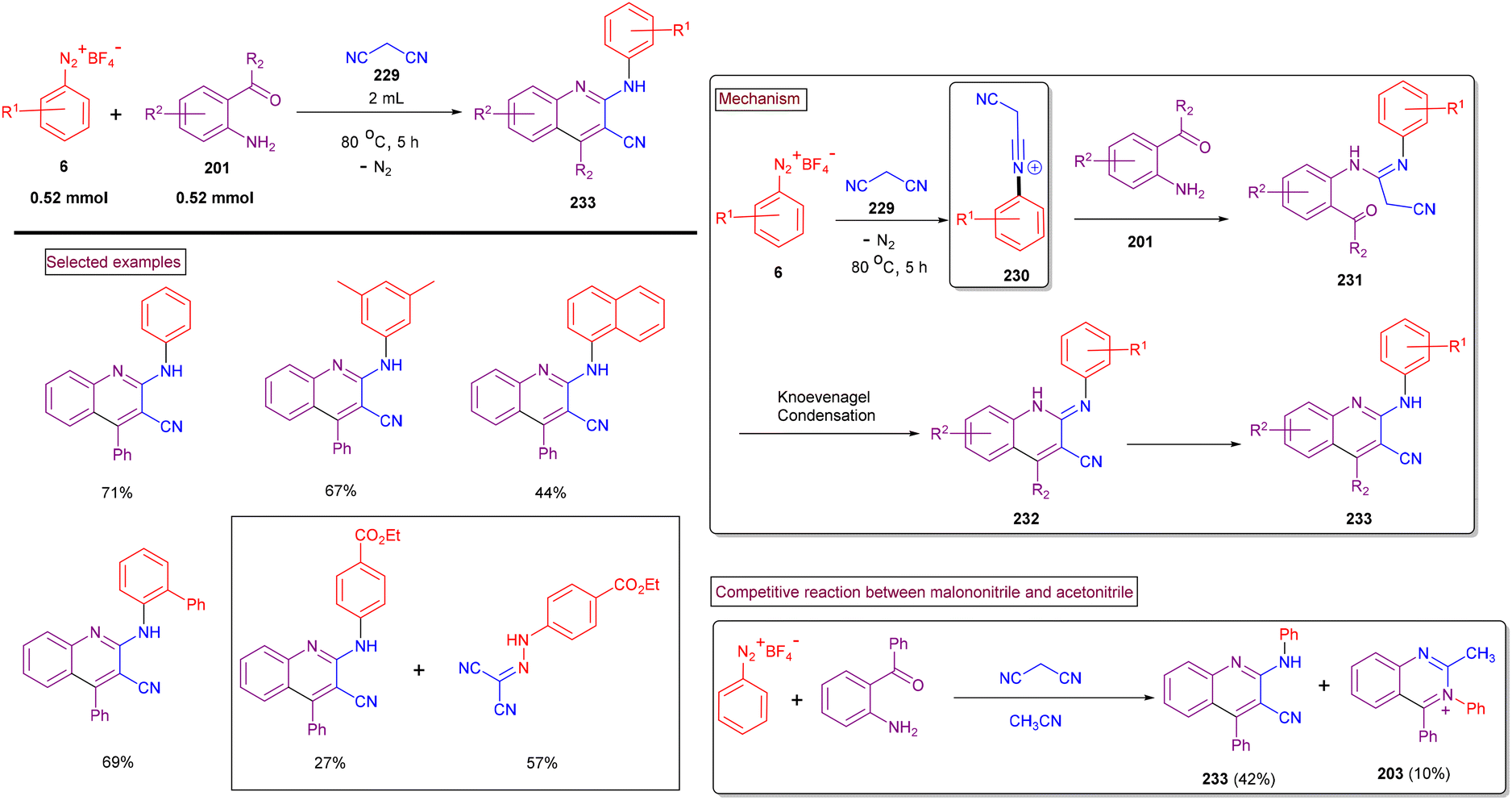
The preparation of multi-functionalized quinolines is often a challenge and requires a multi-step procedure or metal catalysts. Thus, to circumvent this issue, in 2020, our research group developed a direct method to access 2-arylamino-3-cyano-4-aryl/alkyl quinolines (233) through a cascade reaction involving aryldiazonium salts (6), 2-aminoarylketones (201), and malononitrile (229) under metal-free conditions (Scheme 49).163 The scope of this method was studied with multi-substituted aryldiazonium salts (6) including sterically demanding ortho-substitutions and quinolines (233) were obtained in good isolated yields (54%–72%). Electron-withdrawing groups (–F and –CO2Me) led to direct azo coupling with malononitrile in appreciable quantities. Mechanistically, we postulated that the initially formed N-arylnitrilium intermediate (230) from the direct reaction of malononitrile (229) and aryldiazonium salts (6) was trapped by amino precursor (205), followed by Knoevenagel condensation and aromatization to afford the desired products (233) in moderate to good yields. The competitive formation of (233) and (203) occurred in the presence of a mixture of nitriles.
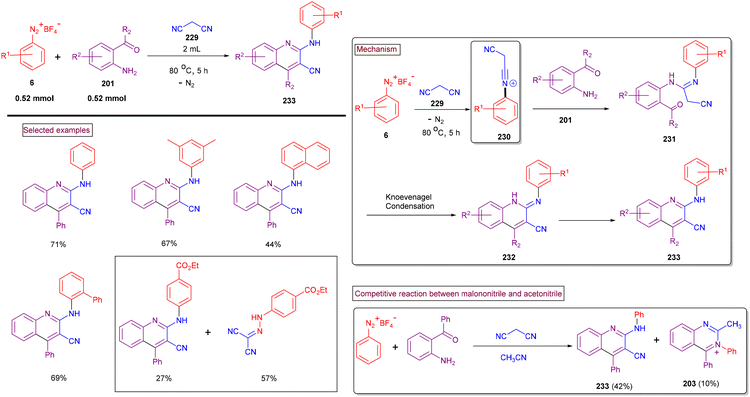 |
| | Scheme 49 Preparation of 2-arylamino-3-cyano-4-aryl/alkyl quinolines via cascade cyclization. | |
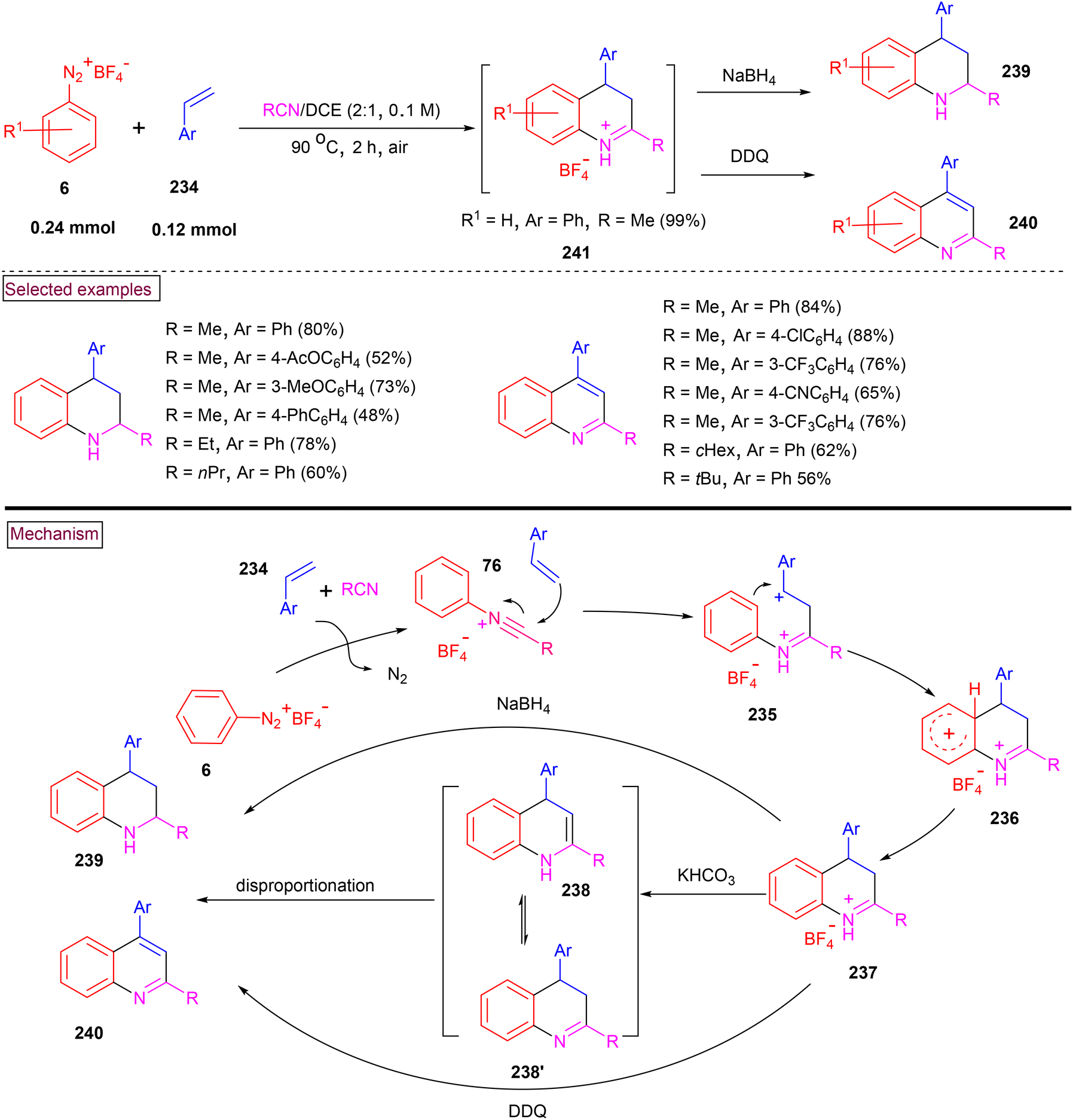
In 2018, Youn et al. reported a one-pot and convenient procedure for the assembly of quinolines (240) and tetrahydroquinolines (239).164 In this method (Scheme 50), the formation of N-arylnitrilium intermediate (76) was realized from the in situ reaction of nitriles and aryldiazonium salts (6) under anhydrous conditions. Further electrophilic addition of alkene (234), generation of benzylic carbocation (235), cyclization and deprotonation resulted in the formation of 3,4-dihyroquinolinum salts (237), which served as a common intermediate. This intermediate could be further transformed selectively to tetrahydroquinolines (239) in the presence of NaBH4 or quinolines (240) in the presence of DDQ. Alkyl, aryl, heteroaryl, disubstituted, and internal cyclic alkene precursors were evaluated under these conditions and the desired products (239 and 240) were obtained in excellent yields. 1,1-Disubstituted alkenes resulted in the formation of quinolines with 1,2-aryl migration. In the same year (2018), we developed a method for the preparation of various functionalized 3,4-dihydroquinolinium salts (239) from the direct reaction of aryldiazonium salts (6) and alkene (234) in a nitrile solvent.165 Diverse substituted styrenes and aryldiazonium salts smoothly reacted in the presence of a nitrile solvent to afford the 3,4-dihydroquinolinium salts in good yields (up to 81%). These salts are stable, isolable, and could be further converted into valuable 3-hydroxyquinoline. Additionally, it was observed that quinoline derivatives were formed by intramolecular trapping of the N-arylnitrilium intermediate with o-vinyl group.
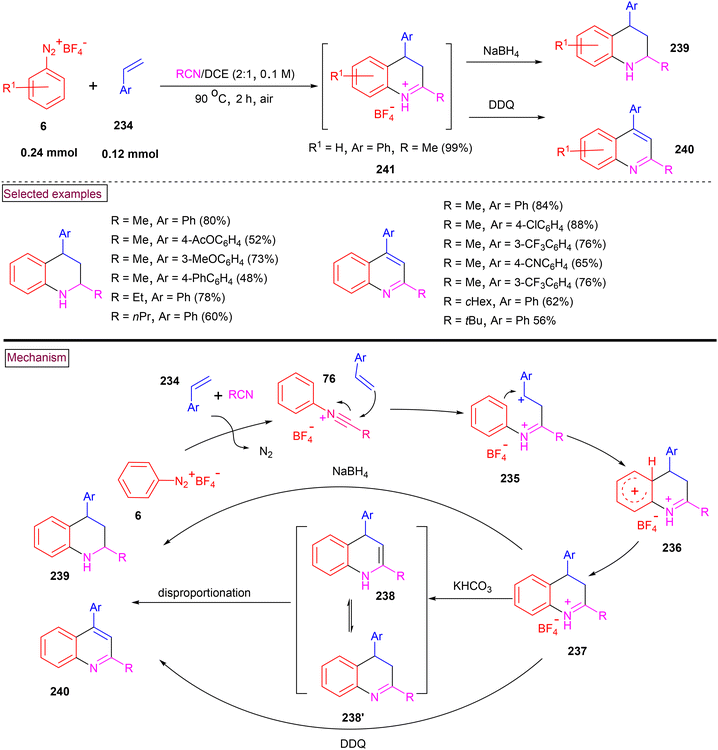 |
| | Scheme 50 One-pot preparation of quinolines and tetrahydroquinolines using N-arylnitrilium ions. | |
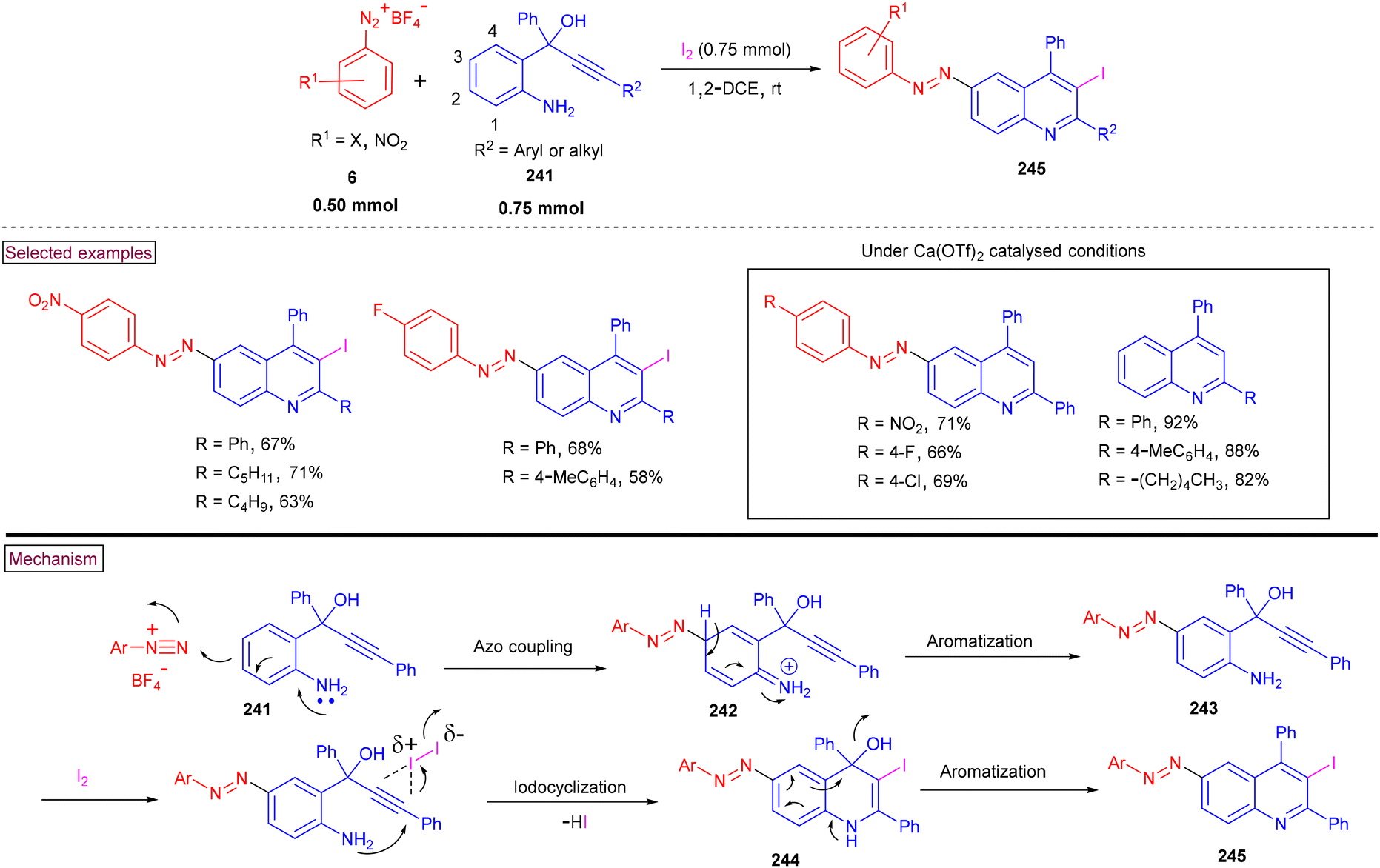
The one-pot synthesis of 6-(aryldiazenyl)-3-iodoquinolines (245) was reported from aryldiazonium salts (6), 2-aminoaryl propargyl alcohol (241) and iodine at ambient temperature (Scheme 51).166 This unique three-component reaction proceeded via azo-coupling to give intermediate (242), which underwent iodine-promoted cyclization on the alkyne moiety, followed by aromatization to yield the desired product (245). Propargyl alcohols (241) with aryl, alkyl and alkyne moieties was tested against various aryldiazonium salts and the corresponding iodocyclization process provided 6-aryldiazenyl-3-iodoquinolines (245) in good yields. When the C-3 position of substrate (241) was blocked, 3-iodoquinolines were obtained in moderate yields. Additionally, 6-aryldiazenylquinolines were obtained via Ca(OTf)2-promoted azo coupling/cyclization reactions in the absence of I2, whereas intramolecular cyclization of 2-aminoaryl propargyl alcohols gave the 2,4-difunctionalized quinolines under Ca(OTf)2-catalysed conditions. Detailed control experiments were carried out to explore the reaction mechanism and the 3-iodoquinolines were subjected to various cross-coupling sequences such as Heck reaction, Sonogashira coupling, and Suzuki coupling reactions.
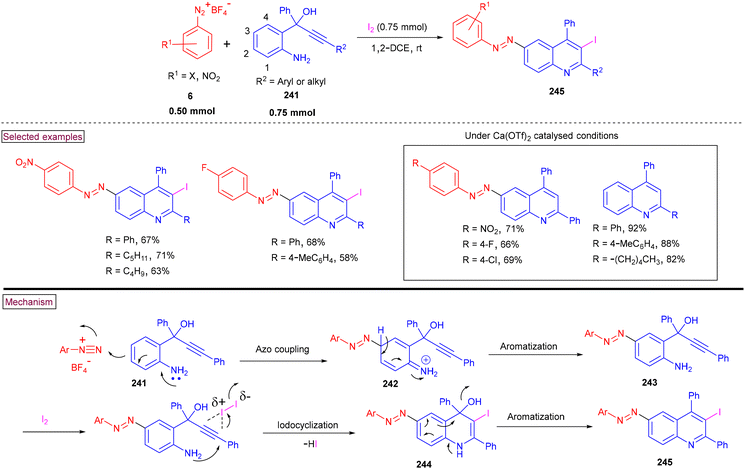 |
| | Scheme 51 Preparation of 6-(aryldiazenyl)-3-iodoquinolines from aryldiazonium salts. | |
In 2017, Yu et al. reported the convenient synthesis of multi-substituted quinolines (247) from aryldiazonium salts (6), alkynes (23) and nitriles (Scheme 52).167 In comparison to the traditional annulation of iminium salts with alkynes and oxidation, N-arylnitrilium ions annulated directly to the quinolines without the need for oxidation. Aryl, heteroaryl and aliphatic alkynes (23, both internal and terminal) smoothly reacted to afford quinolines (247) in good yields. In the case of aryldiazonium salts with electron-withdrawing groups, the formation of amide was observed owing to the hydrolysis of in situ-generated N-arylnitrilium intermediate (76). Based on the control experiments, the authors proposed the reaction pathway in which the in situ-generated N-arylnitrilium intermediate (76) (from the direct reaction of aryldiazonium salt (6) and anhydrous nitrile) reacted with alkyne (23) to give vinyl cation (246). Further Friedel–Crafts-type cyclization resulted in the formation of the desired quinoline derivative (247). In the same year, our research group developed a regiospecific method for accessing 3-haloquinolines via a three-component annulation of nitriles, haloalkynes (Br, Cl, and I) and aryldiazonium salts (6).168 Aryldiazonium salts with sensitive groups such as ester, benzoyl, alkyne, ether, cyano and halogens were involved in this transformation and provided the functionalized halo-quinolines in good yields. A similar mechanistic pathway was explained. Furthermore, the gram-scale formal synthesis of pitavastatin was also demonstrated.
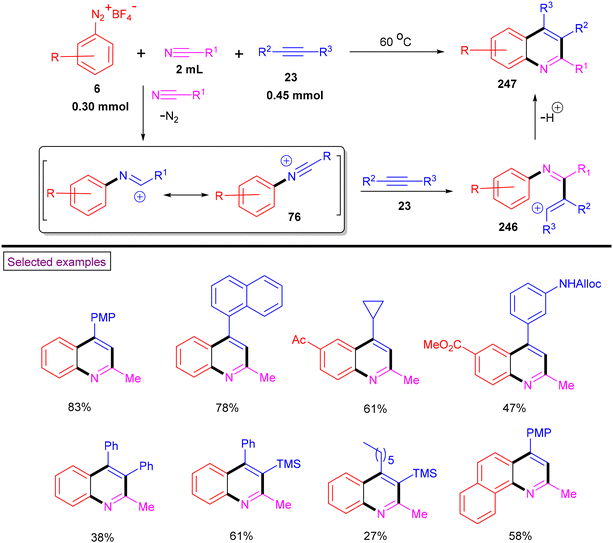 |
| | Scheme 52 Preparation of quinolines from aryldiazonium salts, alkynes and nitriles. | |
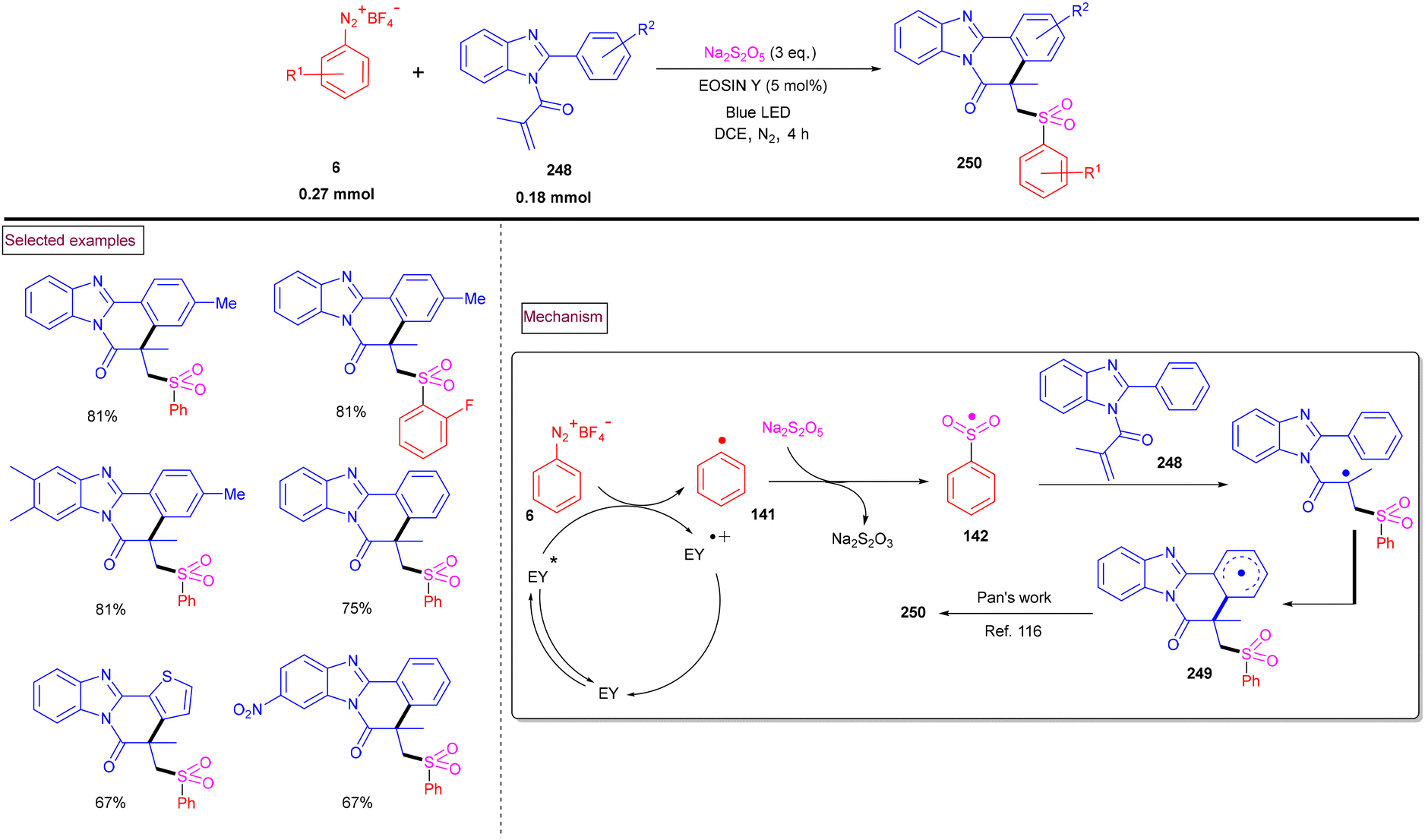
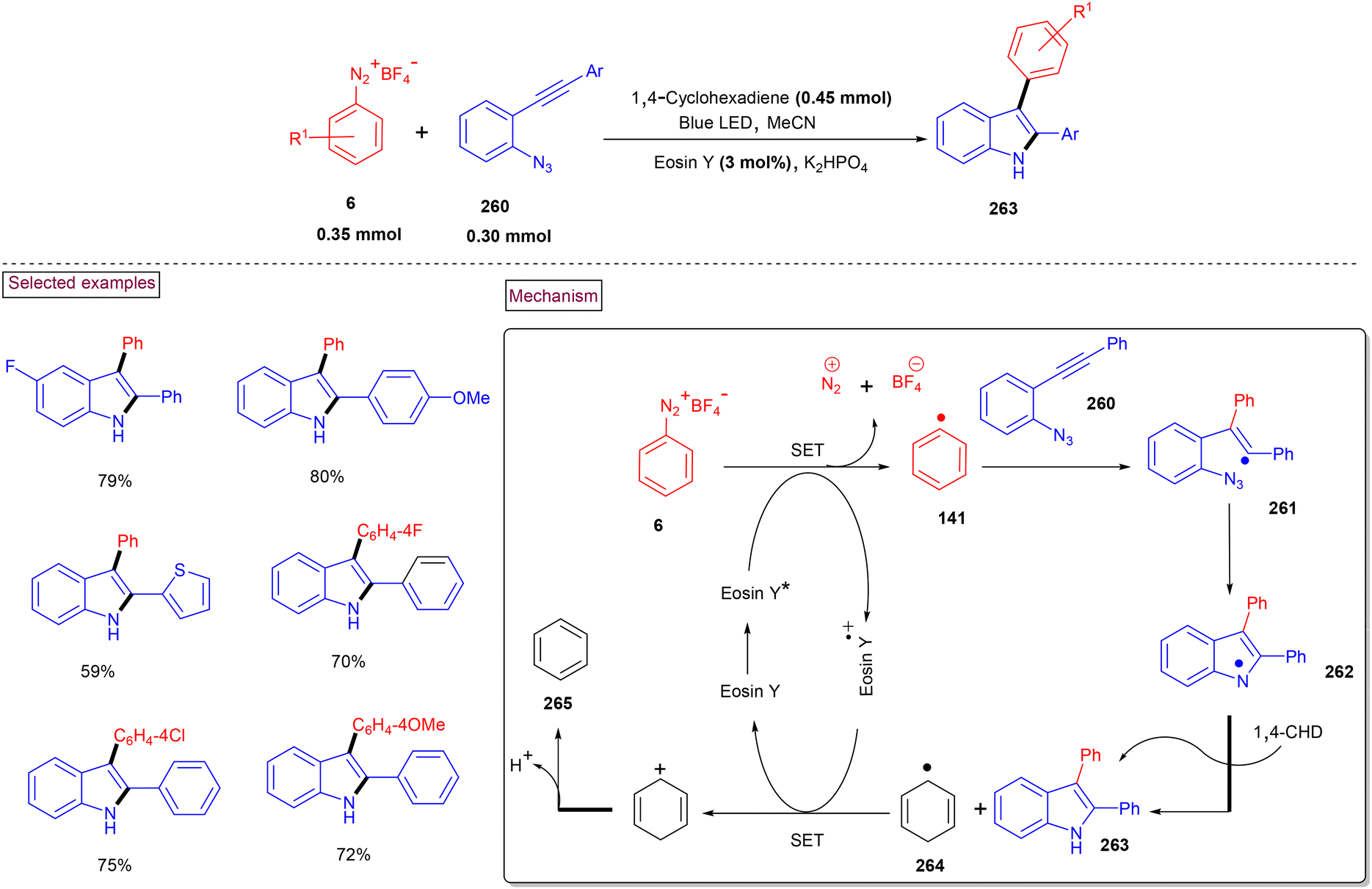
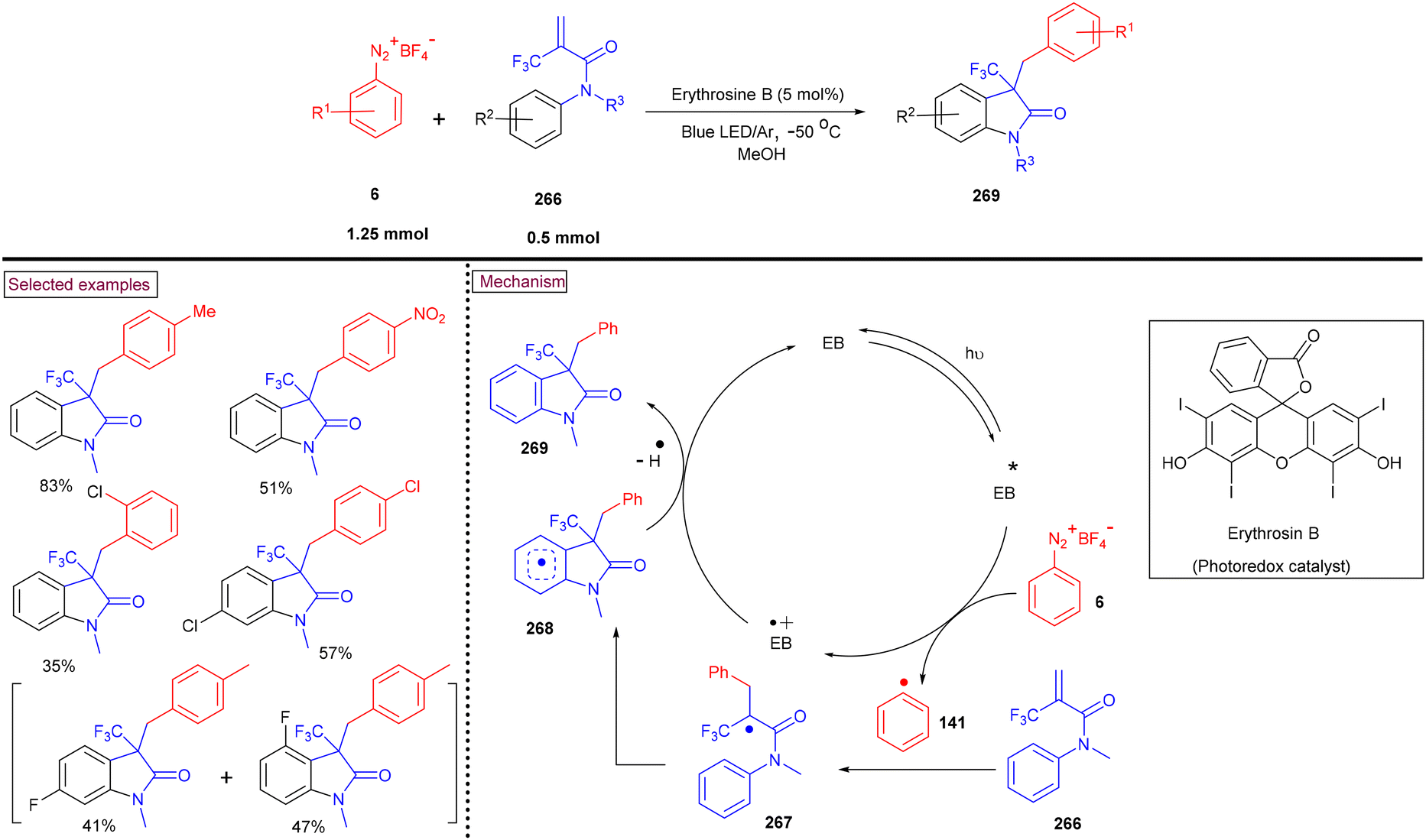
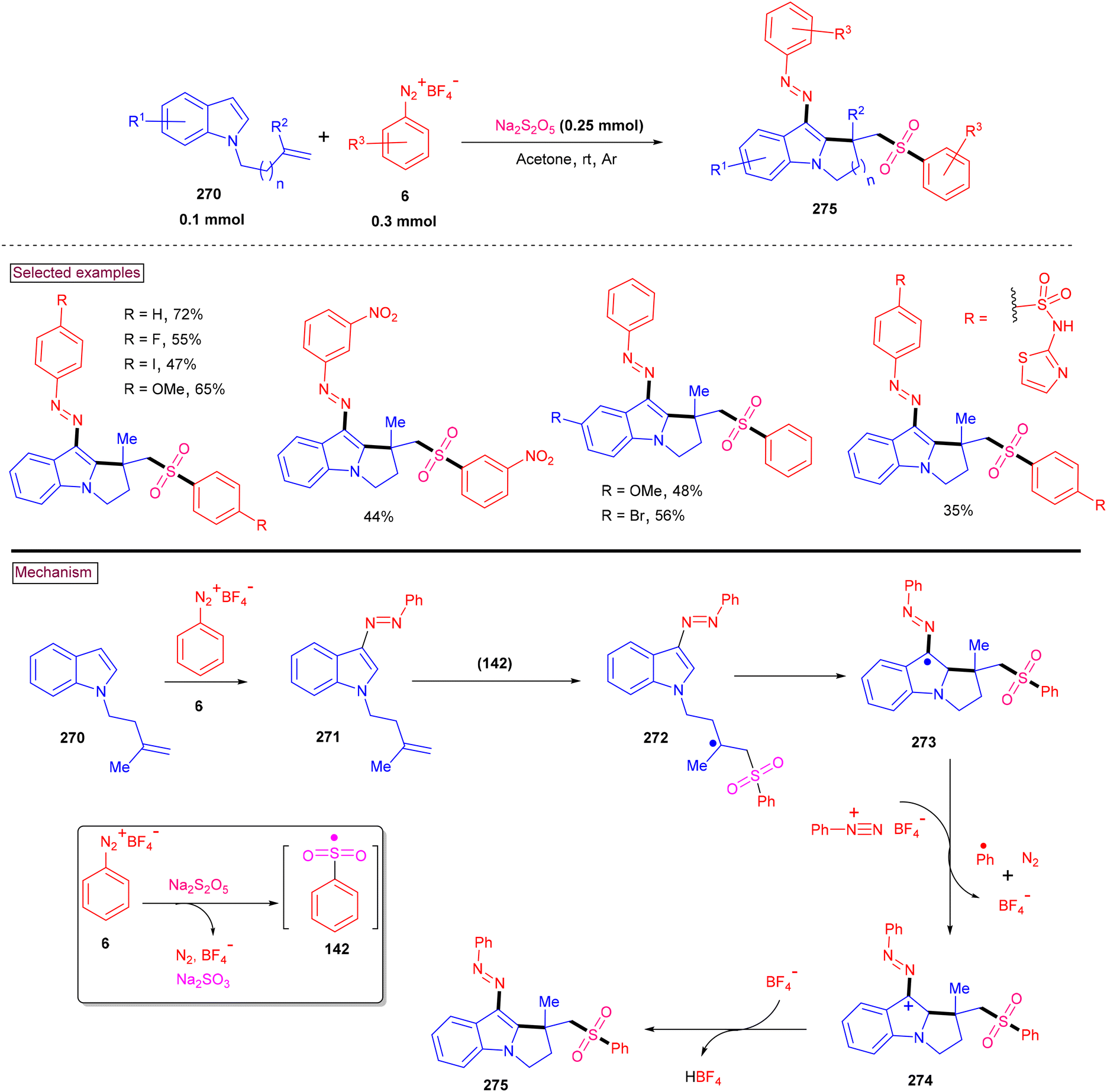
4. Transition metal-free visible light-mediated photo redox catalysis involving aryldiazonium salts
Comprehensive photocatalytic routes to assemble N-heterocyclic skeletons based on metal complexes are well-established. Alternatively, visible light-mediated photoreactions using metal-free catalysts to construct N-heterocyclic scaffolds are of special interest owing to their low-cost, low toxicity and higher stability.169 In this context, aryldiazonium salts are widely utilized as arylating precursors in photo redox catalysis due to their broad substrate scope, ease of preparation, atom economy and lower redox potential.47 Compared to aryl halides and organometallic reagents, these salts serve as good alternatives and used in numerous organic transformations.46 A wide range of organophotoredox catalysts such as eosin Y, rose Bengal, and erythrosine B has been developed in recent years with excellent reactivity, distinct activation modes and low cost compared to the metal-based photocatalysts, (–Ir, –Ru, and more). In this section, we discuss a few recent developments, focusing on the preparation of N-heterocycles with the combination of aryldiazonium salts (as arylating agents) and organophotocatalysts.
In 2022, Gupta and Singh et al. reported a visible light-promoted three-component annulation to prepare sulfonylated benzamido or indolo[2,1-a]isoquinolin-6(5H)-ones (250) from aryldiazonium salt (6), acrylamides (248) and Na2S2O5 in the presence of Eosin as an organophotocatalyst (Scheme 53).170 A wide spectrum of aryldiazonium salts and acrylamides with electronically distinct substituents participated in the cascade radical annulation and provided the desired products in excellent yields. It was observed that aryldiazonium salts (6) with electron-withdrawing groups and ortho-substitutions resulted in the formation of the desired products in diminished yields. Alternatively, electron-withdrawing groups on the benzimidazole ring as well as the C2-aryl ring produced the desired products (250) in excellent yields (up to 90%). By performing various radical trapping experiments, competition experiments and radical clock experiments, the authors proposed the detailed mechanism. Aryl radical (141) was produced from diazonium species (E1/2 = −0.2 V) through oxidative quenching of the excited Eosin catalyst (E1/2 = −1.11 V). Further trapping of (141) with SO2 resulted in the formation of phenylsulfonyl radical (142), which underwent reaction with acrylamide derivative (248) and generated an alkyl radical. A similar three-component synthesis of arylsulfonyl-substituted indolo[2,1-a]isoquinolinones and benzimidazo-[2,1-a]isoquinolin-6(5H)-ones (250) via a thermally generated radical cascade cyclization was reported by Pan's research group.171 In this method, 2-aryl-N-methacryloyl indoles (248) were subjected to arylsulfonylation/cyclization with aryldiazonium salts (6) and readily available potassium metabisulfite, which served as an SO2 surrogate. Aryldiazonium salts 2-aryl-N-acryloyl indoles/benzimidazoles exhibited excellent substrate scope and the corresponding products were formed in good yields (62%–80%). A similar mechanism through radical annulation was proposed. However, this organo-photocatalyzed transformation avoids the expensive DABSO and utilized the inexpensive Na2S2O5 as an SO2 surrogate.
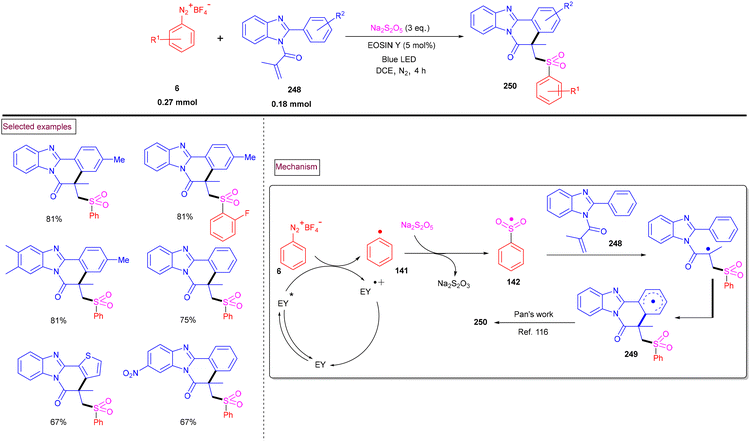 |
| | Scheme 53 Visible light-mediated preparation of sulfonylated indolo[2,1-a]isoquinolin-6(5H)-ones. | |
By using aryldiazonium salts (6) or N-hydroxyphthalimide esters (251) as an aryl or alkyl radical source and acrylamide (248) as a radical acceptor, a range of imidazo-isoquinolinone derivatives (259) was prepared by Xu and Hammond et al. in the presence of aromatic ketone photocatalyst (252) (Scheme 54).172 Notably, in the absence of DMAc solvent the reaction outcome was poor and this observation may be due to the formation of DMAc radical and its reaction with excited-state photocatalyst. According to the detailed mechanism, N-hydroxyphthalimide esters act as the alkyl radical source for this radical transformation. For convenience, we present the mechanistic cycle using both aryldiazonium salt (6) and N-hydroxyphthalimide ester (251). Under blue light irradiation, excited-catalyst (253) was generated, which reacted with DMAc and produced DMAc radical and (254) via hydrogen atom transfer. The interaction of DMAc radical with (6) or (251) produced aryl radical (141) or alkyl radical (255). Alkyl radical (256) was formed after the decarboxylation process. The addition of (256) or (141) to the terminal alkene of (248) afforded alkyl radical (257). Further intramolecular radical cyclization and oxidation delivered the desired product (259).
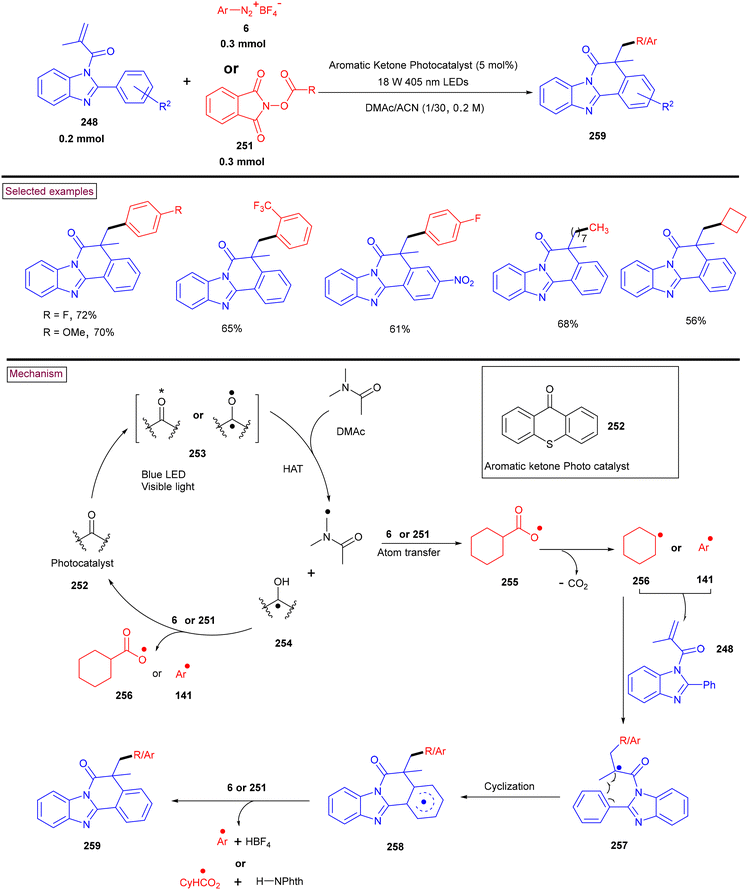 |
| | Scheme 54 Preparation of imidazo-isoquinolinone via photochemical radical cyclization. | |
A visible light-promoted method for the preparation of unsymmetrical 2,3-diarylated indole derivatives (263) from readily available o-azidoarylalkynes (260) and aryldiazonium salts (6) was demonstrated by Jin and Cheng's group (Scheme 55).173 This cascade reaction was initiated by the activation of photocatalyst eosin Y at room temperature. o-Azidoarylalkynes (260) bearing terminal aryl and heteroaryl groups with –Cl, –F, –OMe, and –Me substituents reacted smoothly to afford indole derivatives (263) in moderate to good yields (54%–79%). However, the alkyl analogue failed to react under these reaction conditions. Alternatively, different functionalized aryldiazonium salts (6) were also amenable substrates in this process, giving the products in good yields (up to 75%). Mechanistically, aryl radical (141) was generated from aryldiazonium salt (6) via SET reduction and added to the alkyne of (260) to produce alkenyl radical (261). Further cyclization with the extrusion of N2 gas and H-abstraction from 1,4-CHD resulted in the formation of title compound (263). Radical (264) was oxidized to benzene (265) via the SET process with Eosin Y˙+.
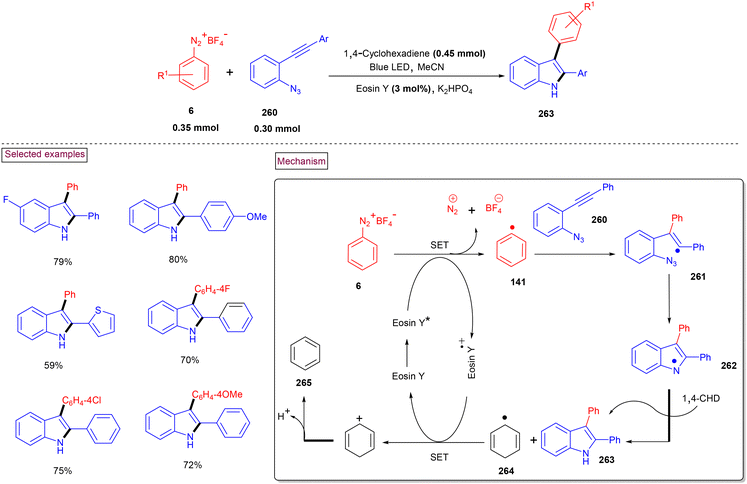 |
| | Scheme 55 Visible light-mediated preparation of 2,3-diarylated indole derivatives. | |
A visible light-mediated approach was developed for the preparation of 3-(trifluoromethyl)indolin-2-ones (269) from functionalized N-alkyl-N-aryl-2-(trifluoromethyl)acryl-amides (266) (Scheme 56).174 This arylation–cyclization sequence was initiated by aryl radical (141) generated by aryldiazonium salt (6) in the presence of the erythrosine B photo-redox catalyst. It is worth noting that a strongly electron withdrawing –NO2 substituent at the para-position of (6) provided the desired product in 51% yield, whereas moderately electron-withdrawing –Cl group at the ortho and meta-positions led to lower yields (34%–35%), stating that the steric factor was crucial in this process. The electronic effects of the substituents in acrylamides (266) were significantly affected the reaction outcome given that electron-deficient substrates poorly reacted compared to the electron-donating substitutions. N-(3-Fluorophenyl)acrylamide reacted with various aryldiazonium salts to produce the regioisomeric products. The proposed mechanism revealed that the reaction of excited-state erythrosine B with aryldiazonium salt (6) resulted in the formation of aryl radical (141), which further added to the alkene unit of acrylamide derivatives (266) and produced alkyl radical (267). Further intramolecular cyclization and oxidation yielded the desired product (269).
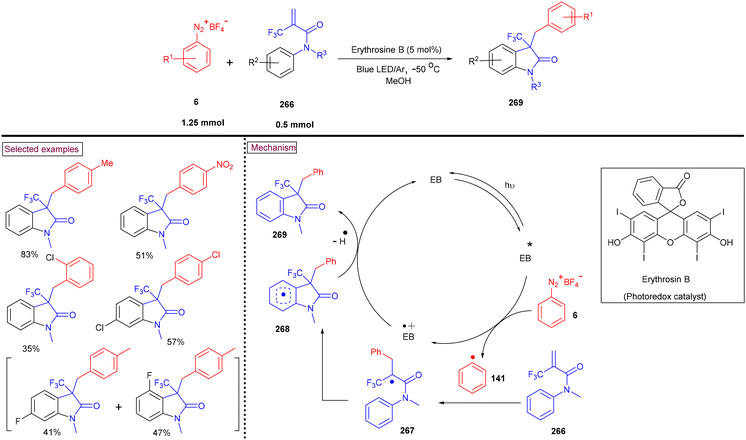 |
| | Scheme 56 Visible light-mediated preparation of 3-(trifluoromethyl)indolin-2-ones. | |
The photocatalytic transformations discussed in this section share a few common processes such as generation of aryl/alkyl radical in the presence of a photoexcited organocatalyst, addition of the radical to an unsaturated substrate followed by intramolecular cyclization and regeneration of the photocatalyst. On this common base, diverse substrates are reacted in the presence of various organo photocatalysts and structurally unique N-heterocycles are obtained.
5. Transition metal-free radical cyclization involving aryldiazonium salts
An arylsulfonyl radical-induced cascade cyclization approach was reported to prepare various 2,3-dihydro-1H-pyrrolo[1,2-a]indoles (275) from unactivated alkene substrates (270) and three equivalents of aryldiazonium salts (6) in the presence of Na2S2O5 (Das et al. 2023).175 This elegant method utilized readily available substrates and enabled the installation of azo and sulfonyl groups in the N-heterocyclic scaffold (Scheme 57). Various substituted aryldiazonium salts (6) including a sulfathiazole-based precursor reacted to afford the desired products in 26%–77% yields. Alternatively, indoles (270) bearing unactivated alkenes of various lengths reacted smoothly to give a series of azo and sulfonylated pyrrolo[1,2-a]indoles (275). As described in Schemes 30 and 32, in situ-generated aryl sulfonyl radical (142) was added to aza substrate (271) to provide alkyl radical (272). Intramolecular radical cyclization (273) and oxidation (274), followed by deprotonation resulted in the formation of indole derivative (275). Noteworthy, three equivalents of (6) were necessary to introduce aryl-aza and aryl sulfonyl units. In addition, (6) was also crucial to oxidize (273) into (274).
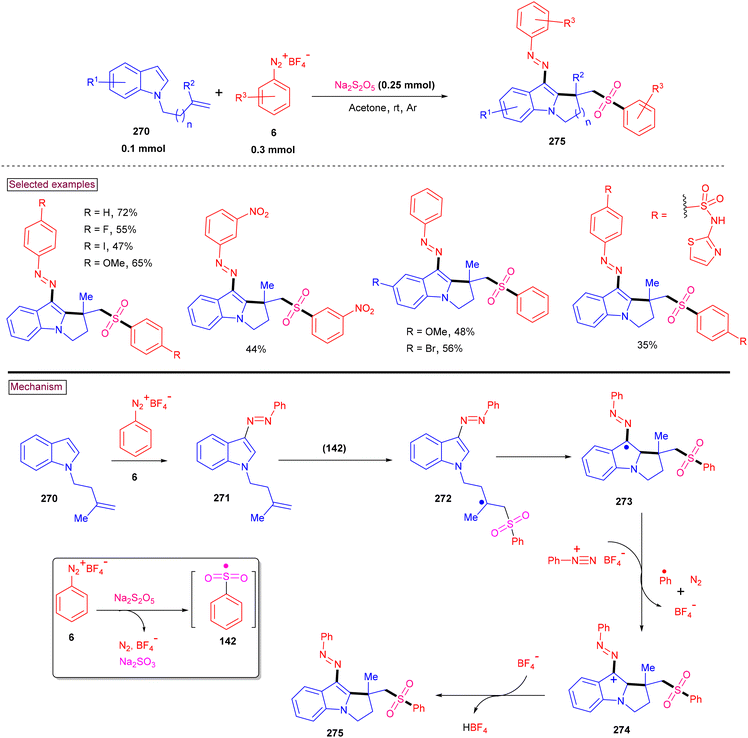 |
| | Scheme 57 Preparation of 2,3-dihydro-1H-pyrrolo[1,2-a]indoles via a radical cascade cyclization. | |
In 2022, Zhang and Lian et al. reported a one-pot, regioselective method for the synthesis of sulfonylated pyrrolin-2-ones (281) in good yields (up to 95%).176 This reaction proceeded via a cascade sulfonylation and cyclization between aryldiazonium salts (6) and 1,5-dienes (276) (Scheme 58). Equal reactivity was observed with aryldiazonium salts bearing electron-donating and withdrawing-substituents. However, sterically hindered ortho-substitutions slightly lowered the yields (67%). Diene component (276) with differently substituted aryl group exhibits excellent reactivity to afford the desired products (281) in high yields. Based on the control experiments, the authors proposed that the transformation via a 5-endo-trig sulfonylative cyclization is the key step. The initial interaction between diene precursor (276) and aryldiazonium salt (6) through electron transfer resulted in the formation of an aryl radical and radical cation species (277). Trapping of the aryl radical by sulfur dioxide yielded aryl sulfonyl radical (142), which added to the alkene unit of diene (276). Further intramolecular cyclization, oxidation and tautomerization furnished the desired product (281).
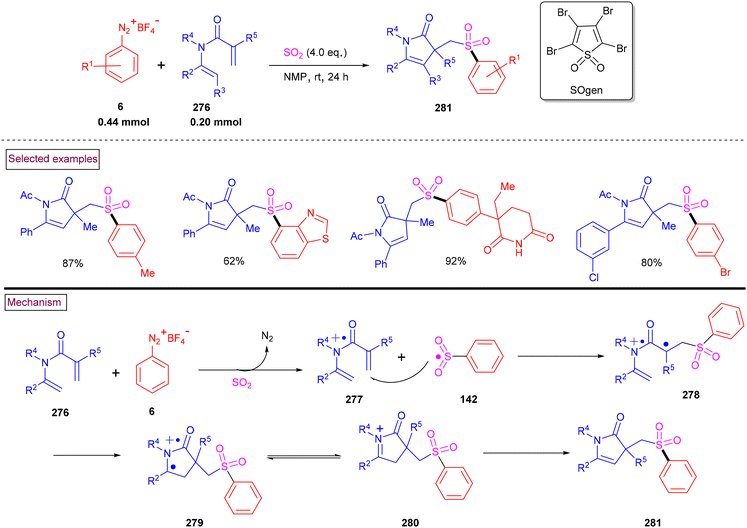 |
| | Scheme 58 Preparation of sulfonylated pyrrolin-2-ones via 5-endo-trig sulfonylative cyclization. | |
Wang and Jiang et al. combinedly prepared a wide range of spirocyclohexadienones bearing cyclopenta[c]quinoline-4-ones (286) via an oxidant-free, radical triggered deaminative ipso-cyclization of anilines (75) with N-tethered 1,7-enynes (282) (Scheme 59).177 Various substituted aryl alkyne-tethered 1,7-enyne substrates smoothly participated in this oxidative bicyclization to afford the corresponding products in moderate yields (up to 60%). A unique example of cyclopropyl alkyne was also reactive in this process (48%). Both alkyl and aryl substituted alkenes were found to be amenable substrates in this transformation. As proposed in the mechanism, the p-methoxyphenyldiazonium radical generated under thermal decomposition was added to the alkyne unit of 1,7-enynes (282), followed by 6-exo-dig/5-exo-trig cyclization (284 and 285), single-electron transfer (SET) oxidation and demethylation furnished the spiro-product (286) with two all-carbon quaternary stereo centres.
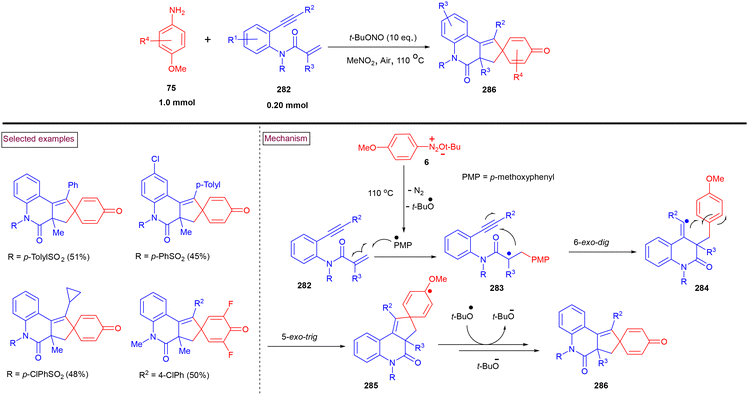 |
| | Scheme 59 Preparation of spirocyclohexadienones via de-aminative ipso cyclization. | |
Studer et al. (2016) realized a novel method for the preparation of polycyclic γ-lactams (292) via the radical cascade cyclization of 1,6-enynes (287) and anilines (75) (Scheme 60).178 This developed method showed excellent substrate scope and sequential formation of three C–C bonds. In addition, polycyclic pyrrole and γ-butyrolactones could also be obtained by employing structurally related substrates. Without substantial changes in the yields, both electron-donating and withdrawing-groups on substituted anilines (75) smoothly participated in this transformation (up to 64% yield). However, sterically demanding ortho and meta substituents lowered the yields (34%–35%). 1,6-Enynes embedded with aryl, heteroaryl and alkyl units were reacted with aryldiazonium salts (6) to afford a diverse range of γ-lactams in synthetically useful yields. The mechanistic illustration revealed that the LiI-mediated aryl radical was generated from in situ-prepared aryldiazonium salt (6), followed by its addition to the activated alkyne unit of 1,6-enynes (287) to afford tertiary alkyl radical (288). Further 5-exo cyclization and intramolecular radical cyclization provided aryl radical (290) Deprotonation and anion-oxidation processes resulted in the formation of polycyclic γ-lactams (292). N-Ts deprotection, ester hydrolysis and Ag-catalysed oxidative decarboxylation were performed on polycyclic γ-lactams (292) to showcase the synthetic potential of this method. Adopting a similar procedure, Studer et al. disclosed a convenient protocol for the preparation of a benzo-fused tetracyclic skeleton with an extended π-system from commercially available anilines and aryl-tethered 1,6-enynes.179a Anilines with common substituents (–OMe, –Cl, –Br, and –H) afforded the desired products in moderate yields (36%–58%). Alternatively, aryl, heteroaryl and alkyl moieties on the alkyne part of 1,6-enynes were investigated to furnish the desired products in moderate yields. This approach serves as a complementary strategy to previously known Ru and Ir-photo redox catalysed transformations.179b–d
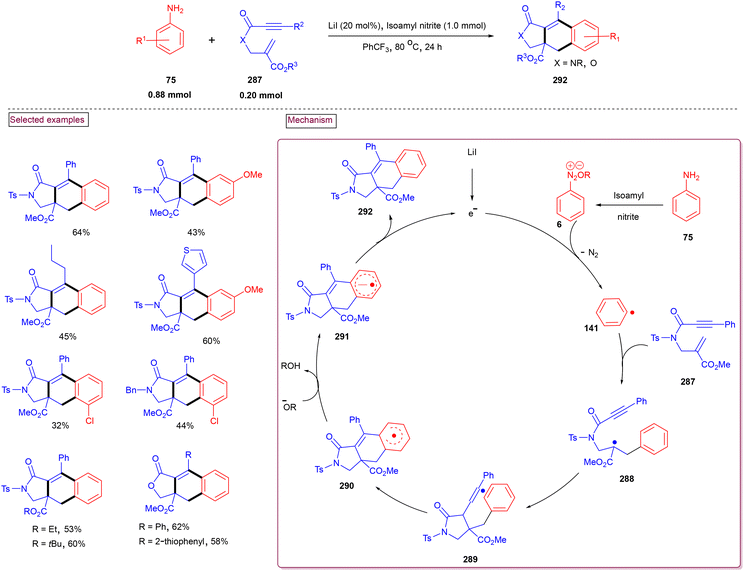 |
| | Scheme 60 Preparation of polycyclic γ-lactams via a radical cascade cyclization. | |
6. Transition metal-free radical cyclization involving aryldiazonium salts and DABSO reagent
Over the past several years, the utility of bench-stable DABSO [DABCO·[SO2]2] or 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) as the source of SO2 has stimulated numerous research on the insertion of sulfur dioxide into various molecular skeletons.180 To this end, a combination of DABSO and aryldiazonium salts offers a wide opportunity for the preparation of sulfonylated heterocycles under transition-metal-free conditions.
Zeng, Wu and Chen et al. (2022) together described a convenient method to prepare sulfonylated pyrrolidones (297) from 1,5-dienes (293), aryldiazonium salts (6) and DABSO (153) via a transition-metal free sulfonylation/cyclization (Scheme 61).181a Compared to the Cu-catalysed amino-sulfonylation of vinyl amides with sodium sulfinates181b or regioselective Cu(I)-catalysed sulfonylation/cyclization of pre-functionalized vinylamides using sulfonyl chloride,181c this transition metal-free method to access a pyrrolidinone skeleton possessing several advantages from a green synthetic perspective. Fused aryl and heteroaryl groups on 1,5-diene substrates bearing acetate, nitro, ketone, and halogens were studied and the corresponding pyrrolidones (297) were obtained in moderate to good yields (46%–84%). Variations in aryldiazonium salts (6) were also studied and the expected products were isolated in good yields (66%–88%). The authors proved that an arylsulfonyl radical-based chemo and regioselective sulfonylation/cyclization mechanism is involved in this transformation. Under thermal conditions, aryl sulfonyl radical (142) was generated from aryldiazonium salt and DABSO and added to electron-deficient alkene (293) regioselectively to provide alkyl radical (294). Further intramolecular cyclization, oxidation in the presence of DABCO radical cation and deprotonation yielded the desired product (297). Gram-scale synthesis and post-synthetic transformations (bromination and Stille coupling) were also demonstrated.
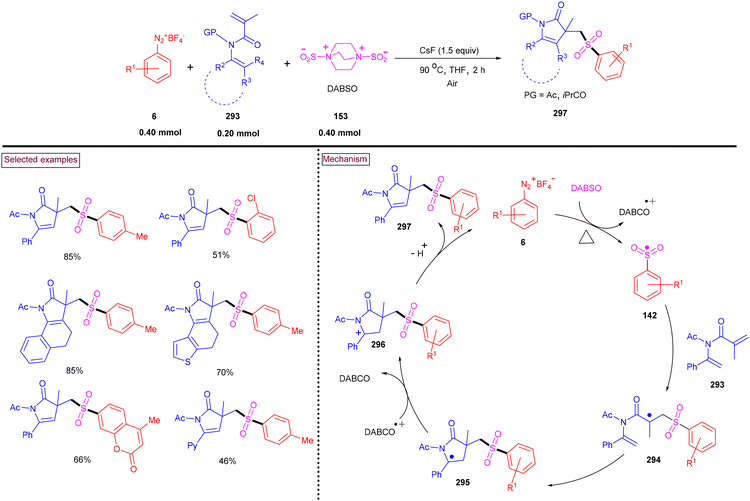 |
| | Scheme 61 Preparation of sulfonylated pyrrolin-2-ones via sulfonylation/cyclization using DABSO reagent. | |
A de-aromative cascade spiro-cyclization was demonstrated for the preparation of sulfonated spiro-indole derivatives (302) via a three-component reaction involving aryldiazonium salts (6), indolyl ynones (298) and DABSO (Scheme 62).182 This strategy was compatible with various substituted aryldiazonium salts (6) and indolyl ynones (298). Mono- and disubstituted aryldiazonium salts bearing –Me, –F, –Cl, –CF3, and –CO2Et delivered the corresponding indoles in good isolated yields (up to 82%). Alternatively, indolyl ynones with aryl and heteroaryl alkyne moieties reacted smoothly to afford the products in moderate yields, whereas their alkyl counterpart was unreactive. Notably, in the absence of a substituent at the C2 position of the indole unit, no desired product was obtained presumably due to the stability of the radical and cationic intermediates. Arylsulfonyl radical (142) and the DABCO radical cation were generated from aryldiazonium salt (6) in the presence of DABSO. The addition of (142) to an alkyne unit provided vinyl radical (299). Further radical spiro-cyclization at the C3 position of (300), followed by oxidation resulted in the formation of cationic intermediate (301). Base-assisted deprotonation yielded the desired product (302).
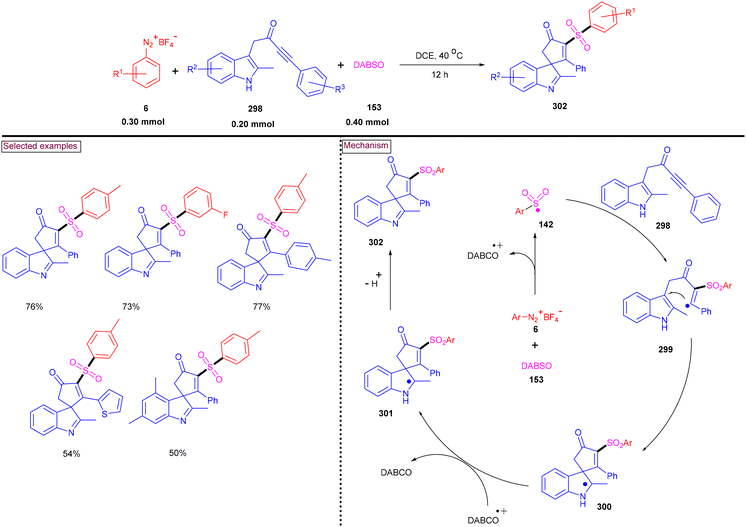 |
| | Scheme 62 Preparation of sulfonated spiro-indoles via de-aromative cascade spiro-cyclization. | |
In 2020, Volla et al. described an efficient transition metal-free method to synthesize azaspiro[4,5]-decanes (307) and azaspiro[4,5]-trienones (312) via the cascade radical spirocyclization of N-benzylacrylamides (303) and N-arylpropiolamides (308) (Scheme 63).183 A fair scope was exerted with a diverse range of aryl diazonium salts (6) with excellent isolated yields in most cases (up to 90%). Unsubstituted and substituted alkenes in acrylamide derivatives (303) were found equally reactive. Similarly, the effect of substituents was studied on N-arylpropiolamides (308) and a range of azaspiro[4,5]-trienones (312) were obtained via sulfonylative spirocyclization. Mechanistically, key intermediate arylsulfonyl radical (142) was generated from DABSO and aryldiazonium salt (6), which was added to the unsaturated substrate to generate (304) or (309). Further transformations were similar to the mechanism described in Scheme 62. Demethylation in the final stage occurred via coordination to DABCO.
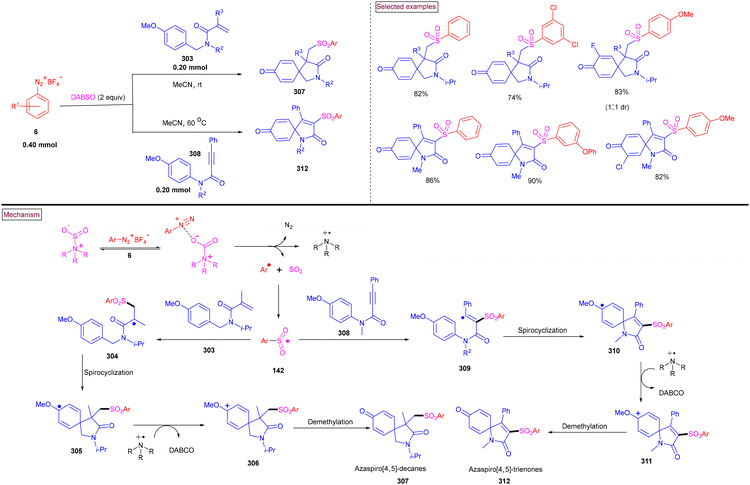 |
| | Scheme 63 Preparation of azaspiro compounds via cascade radical spirocyclization of N-benzylacrylamides. | |
Wu's research group (2019) developed a three-component procedure to access sulfonated 9H-pyrrolo[1,2-a]indoles (318) from aryldiazonium salts (6), 1-(prop-2-yn-1-yl)indoles (313) and DABSO (Scheme 64).184 The addition of in situ-generated radical intermediate (142) to an alkyne unit resulted in the formation of alkenyl radical intermediate (314). Following a similar mechanism to that discussed in Scheme 61, and the desired product (318) was formed. The authors demonstrated the efficiency of this procedure by preparing widely substituted sulfonated 9H-pyrrolo[1,2-a]indoles (318) under mild conditions.
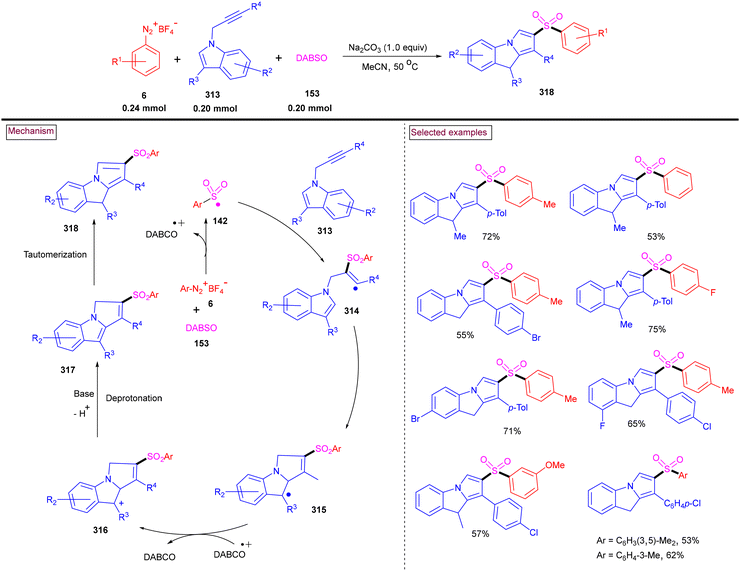 |
| | Scheme 64 Preparation of sulfonated 9H-pyrrolo[1,2-a]indoles via the tandem radical process. | |
7. Transition metal-free synthesis of valuable synthetic intermediates using aryldiazonium salts
Besides the remarkable contribution of aryldiazonium salts in preparing a diverse range of N-heterocycles, these salts serve as a precursor to achieve a range of synthetically useful intermediates through various processes such as, sulfonylation, selenolation, carboamination, and diazo-insertion.185 These intermediates are obtained from aryldiazonium salts in a highly efficient way. Herein, we summarize several transition-metal-free methods reported from 2015 onwards.
7.1. Functionalized aza-compounds from aryldiazonium salts
A unique method for the preparation of C2, C3, C5-multi-substituted indole derivatives (325) from aryldiazonium salts (6), azoles (323) and indolines (319) was reported by Liang's group (Scheme 65).186 This method offered a wide range of 5-azo-2-azoyl-3-iodinated indole derivatives (325) with the consecutive formation of C–I and C–N bonds in a one-pot operation. The mechanistic analysis revealed initial electrophilic substitution of aryldiazonium species installed the diazo unit at C5 (320), which was oxidized to indole (321). The formation of iodonium intermediate (322) and further reaction with azole (323) and iodination at C3 furnished the desired products (325) with excellent regioselectivity. Alkyl and benzyl-protected indolines reacted smoothly to give the products in 52%–75% yields, whereas N-acetyl and benzoyl-protected indolines exhibited diminished reactivity. A broad range of aryl, heteroaryl and polyaryl diazonium salts (6) afforded multi-substituted indoles (325) in appreciable yields (51%–71%). Alternatively, indazole, and benzotriazole were also viable in this process, furnishing the corresponding products in reasonable yields.
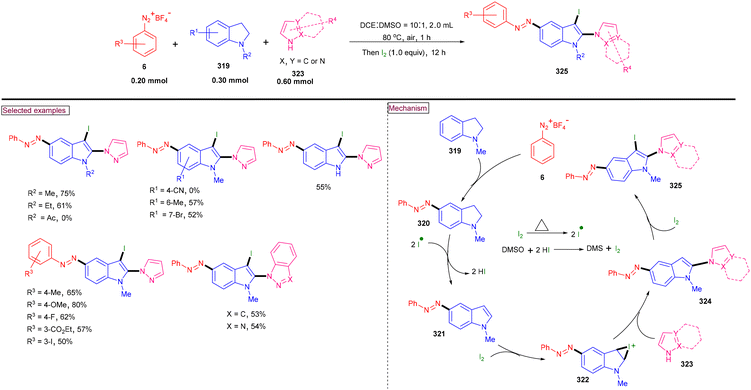 |
| | Scheme 65 Synthesis of multi-substituted aza-indoles from aryldiazonium salts and indolines and azoles. | |
In 2024, Ouyang et al. reported a multicomponent carbonylazotization approach from aryldiazonium salts (6), polyhalomethanes, alcohols (328) and indole (323) or pyrroles (330) via a halogen-atom-transfer (XAT) process (Scheme 66).187 The control experiments proved that installation of a diaza unit on pyrrole or indole via electrophilic aromatic substitution was the initial step. Aryl radical (141) generated from aryldiazonium salt (6) under Cu(I)-catalysis reacted with perhalomethane via the XAT process to generate a trihalomethyl radical, which then reacted with indole or pyrrole to yield intermediates (327 or 333), respectively. Further alcoholysis delivered multi-functionalized pyrrole (334) or indole (329). Aryldiazonium salts with electron-withdrawing groups (–NO2 and –CF3) lowered the product yields (41%–47%). Pyrroles and indoles with N-alkyl, aryl, allyl or benzyl substituents were compatible in this process. Conversely, N-unsubstituted pyrroles/indoles were moderately reactive, whereas those with electron-deficient substituents such as –Ts and –Ac groups were found to be inert. Only aliphatic alcohols (328) (–MeOH, –EtOH, and –iPrOH) were involved in this reaction, presumably due to the steric reason during the esterification.
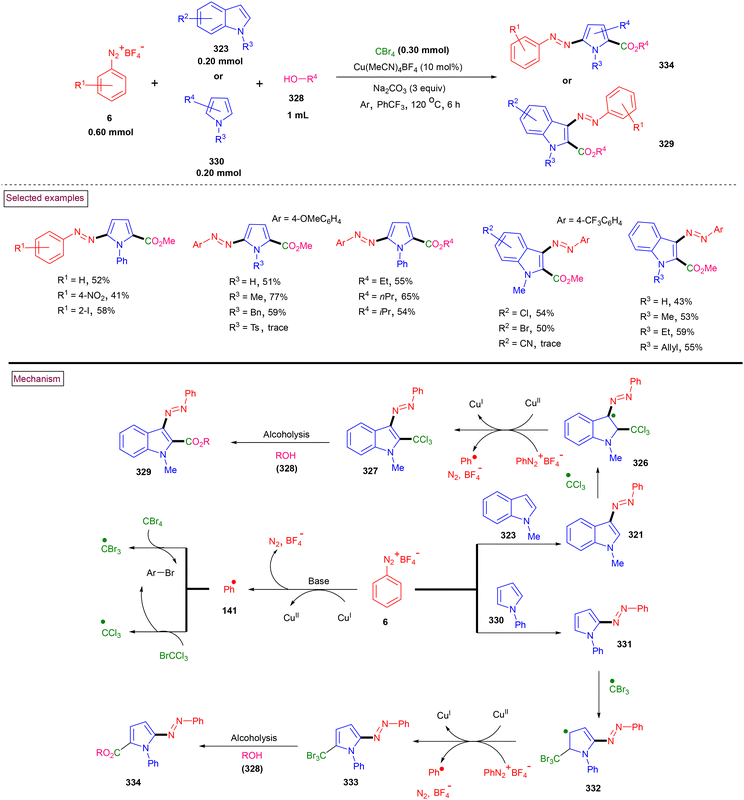 |
| | Scheme 66 Carbonylazotization of indoles or pyrroles from aryldiazonium salts via the halogen-atom-transfer (XAT) process. | |
A set of arylazobiindoles (339) with novel photo-switching properties was prepared via the transition metal-free reaction of aryldiazonium salts (6) with indoles (323) in a nonpolar aprotic solvent (Scheme 67).188 This three-component reaction occurred via an initial azo coupling (339), followed by the nucleophilic addition of another indole unit to indolium species (337). Aromatization led to hydrazine (338) and subsequent oxidation resulted in the formation of the desired products (339) in good yields. Diazonium salts (6) with alkyl, –CF3 and halogens reacted well, yielding the corresponding products in good yields. The steric influence exerted by the ortho-substituents lowered the yield. N-Unsubstituted indoles did not effectively participate in this reaction, leading to 25% yield, whereas the electron-withdrawing benzoyl group led to no product formation.
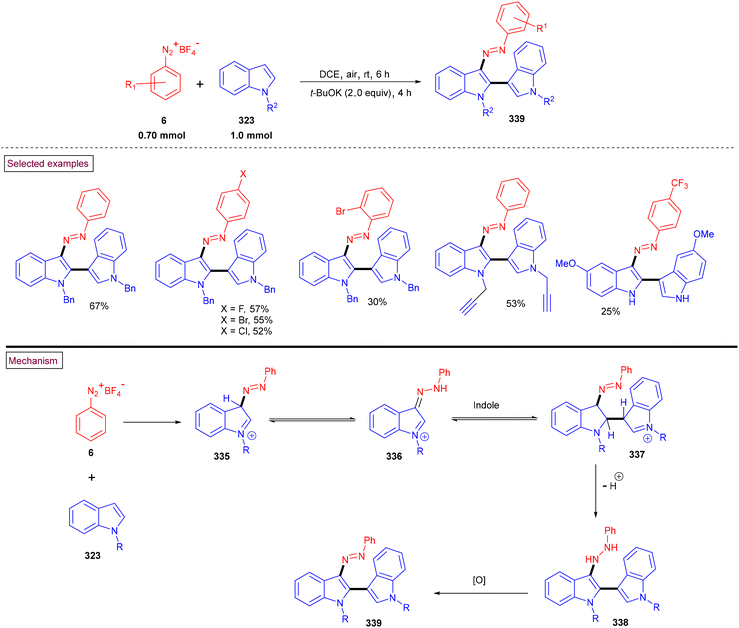 |
| | Scheme 67 Synthesis of novel arylazobiindoles from aryldiazonium salts and indoles. | |
Wang et al. (2023) reported a direct method to access a wide range of trifluoromethyl formazans (344 and 345) via the base-mediated azo coupling of trifluoromethyl N-aryl/N-acyl hydrazones (341 or 340) and a wide range of aryldiazonium salts (6) (Scheme 68).189 The aryl unit of N-acylhydrazones (340) with electron-donating groups (–Me and –OMe) favoured the reaction with higher yields than that with electron-withdrawing substituents. Notably, linear and cyclic alkyl, heteroaryl, styryl and conjugated alkenes were employed, successfully affording the corresponding formazans (344) in moderate to good yields (47%–96%). Mechanistically, the base-mediated deprotonation of N-acylhydrazones (340), followed by a reaction with aryldiazonium salt (6) resulted in the formation of aza-intermediate (343). Further 1,3-H migration afforded the desired products (344).
 |
| | Scheme 68 Synthesis of trifluoromethyl formazans via base-mediated azo coupling. | |
The Cs2CO3-promoted synthesis of arylalkyldiazenes (350) from diazonium salts (6) and alkyl iodides (347) via the halogen-atom transfer (XAT) process under transition-metal-free conditions was reported by Shao and Lu et al. (Scheme 69).190 Aryldiazonium salts (6) served as both the amination reagent and alkyl radical initiator, enabling the rapid formation of diverse C(sp3)–N bonds. Aryldiazonium salts with diverse functionalities such as, –CF3, –F, CO2Me, –NO2, –CN, and –COMe resulted in good yields. A wide range of primary, secondary, and tertiary alkyl iodides (347) bearing –CN, CHO, –NO2, COMe, amide and ester reacted smoothly to provide the anticipated arylalkyldiazenes (350) in good yields. Moreover, chemoselectivity towards the iodide group was observed in the presence of chloride. In addition, gram-scale synthesis in batch mode and continuous-flow mode was achieved in good yields. The synthetic applications were demonstrated via the three-step synthesis of melatonin (352) and deprotection of the diazo unit into free amine (353).
 |
| | Scheme 69 Synthesis of arylalkyldiazenes from diazonium salts and alkyl iodides. | |
A series of unsymmetrically substituted organo azide compounds (358) was prepared via the photoinduced carbo-azidation of unactivated alkenes (354) with aryldiazonium salts (6) and TMSN3 (355) under oxidant-free conditions (Zhang et al., 2021).191 This multi-component process occurred under transition-metal free conditions and the rapid construction of two C–N bonds was realized (Scheme 70). Linear and branched alkenes (354) with functional groups such as alcohol, ester, halogens, sulfonyl and ethers participated well in this transformation and the desired products (358) were obtained in excellent yields (75%–95%). Similarly, various functionalized aryldiazonium salts were involved smoothly under the reaction conditions. Mechanistically, single-electron transfer (SET) from the excited photocatalyst to TMSN3 (355) resulted in an azido radical and PC anion. The addition of the azido radical to alkene generated an alkyl radical, which was rapidly trapped by aryldiazonium species (6) to give radical cation (357). Another SET from radical cation (357) to PC anion led to product (358) and regenerated the PC. Importantly, alkenes derived from natural products such as, (+)-nootkatone and (−)-β-citronellol also furnished the corresponding products in good yields.
 |
| | Scheme 70 Photoinduced carboazidation of alkenes using aryldiazonium salts and TMSN3. | |
The eosin-Y-catalysed azo coupling of aryldiazonium salt (6) and imidazo-heteroarenes (359–360) and arenes (361) under photo-redox conditions was realized via a C(sp2)–H bond azo coupling strategy to afford biologically relevant azo-products (364–366) in good to excellent yields (76%–100%).192 Diverse functionalized imidazo[1,2-a]pyridines (359–360) reacted with aryldiazonium salts (6) of different electronic nature and the desired azo-coupled derivatives were obtained in excellent yields (Scheme 71). Imidazo[2,1-b]thiazole cores (360) with substitutions on thiazole and imidazole participated smoothly to afford the anticipated products in good yields (81%–100%). Although other N-heterocycles such as indole, 1,3,5-oxadiazole, and benzothiazole were not involved in this transformation, various aniline derivatives smoothly converted into the corresponding azo products in good yields. A radical reaction pathway was confirmed by various control experiments. The excited-state photocatalyst (EY*) converted aryldiazonium species (6) into a radical, which was trapped by (359) and resulted in radical intermediate (362). Another SET oxidation and deprotonation generated the desired product (364).
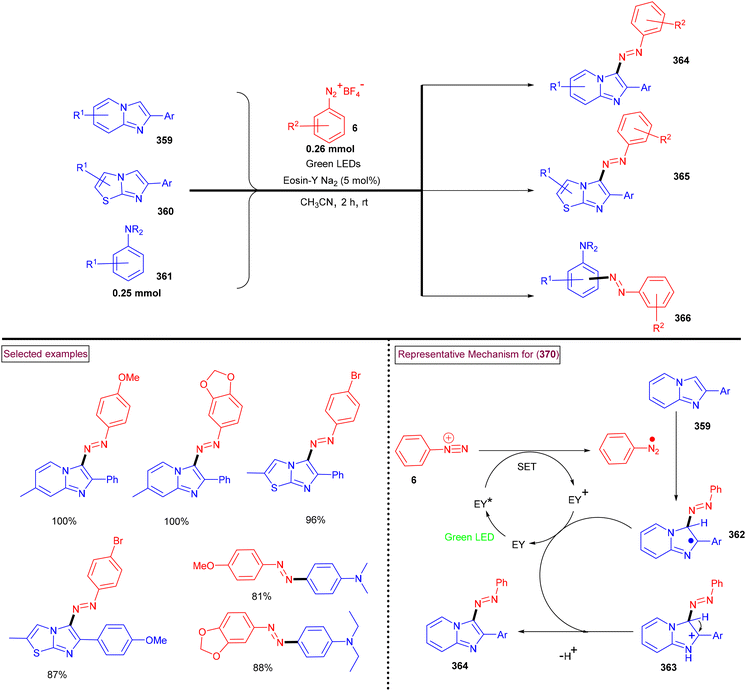 |
| | Scheme 71 Azo coupling of anilines and imidazo-heteroarenes using aryldiazonium salts. | |
Heinrich et al. (2015) demonstrated a Meerwein-type carboamination of alkene precursors (368) using aryldiazonium salts (6) under base-mediated conditions (Scheme 72).193 This carboamination strategy utilized aryldiazonium salt as the aryl radical source and radical scavenger (nitrogen centered). It was observed that alkenes bearing ketones, esters, phenyl ethers, azides, and cyclic acetals led to the desired carboaminated products (372) in good yields. When an alcohol-tethered alkene was employed, pyrazolidinone (373) was formed in many cases as a side product especially in the presence of weaker bases such as potassium acetate. Through a series of step-wise transformations, (6) was homolytically cleaved into aryl radical (141) via diazoanhydride (367). Further addition to alkene (368) and trapping with (6) resulted in the formation of radical cation (370). Diazo compound (372) was obtained via the reduction of (370), whereas sequential oxidation and intramolecular cyclization led to pyrazolidinone (373).
 |
| | Scheme 72 Base-mediated radical carboamination of alkenes using aryldiazonium salts. | |
7.2. Functionalized amidines from aryldiazonium salts
In 2018, our group reported a facile method for the synthesis of N-arylketimines (375) from aryldiazonium salts (6), nitriles and arenes (374) (Scheme 73).194 Mechanistically, this method proceeded via the initial formation of N-arylnitrilium intermediate (76), followed by an intermolecular arylation by various activated arene precursors. Desired ketimines (375) were obtained in good to excellent yields in one-pot with the formation of C–C and N–C bonds. Fused and multi-substituted aryldiazonium salts (6) bearing keto, ether, ester and halogens were smoothly involved in the reaction. Primary, secondary, tertiary, cyclic aliphatic and aryl nitriles worked well and afforded the corresponding products in acceptable yields. Ketimines (375) were obtained with excellent Z selectivity due to the steric effects. The synthesis of a few fungicidal compounds was also demonstrated using this method.
 |
| | Scheme 73 Three-component synthesis of N-arylketimines from diazonium salt nitriles and activated arenes. | |
Another method for the synthesis of N-sulfonyl amidines (377) via the intermolecular nucleophilic addition of sulfonamides (376) to in situ-generated N-arylnitrilium intermediate (76) was disclosed by Chen et al. (2018) (Scheme 74).195 Aryldiazonium salts (6) substituted with different functional groups and aryl, heteroaryl as well as aliphatic sulfonamides (376) were conveniently used in this process to access the N-sulfonyl amidines (377) in good yields. Notably, N-Me-protected sulfonamides furnished the products in 71%–74% yield, whereas N-Ts protection afforded the desired product in a lower yield (26%) owing to steric hinderance.
 |
| | Scheme 74 Synthesis of N-sulfonyl amidines from diazonium salts nitriles and sulfonamides. | |
7.3. Functionalized organo-selenium compounds from aryldiazonium salts
The biological and physiochemical behaviours of Se-containing compounds can be significantly enhanced, and thus the development of synthetic methods leading to these compounds has attracting interest from chemists.196 At lower dosage levels, organoselenium compounds exhibit excellent biological activities.197 In recent years, numerous synthetic approaches to prepare organoselenium products have been reported. Unfortunately, most of these methods require harsh conditions and metal-catalysed process. Herein, we compile a few synthetic processes that utilize aryldiazonium salts for the preparation of organoselenium compounds.
Zhao and Lu et al. (2020) described the synthesis of Se-(difluoromethyl)-4-methylbenzenesulfonoselenoate (378, TsSeCF2H) for the first time and demonstrated its utility in the difluoromethyl selenolation of aryl amines (75) through in situ-generated aryldiazonium salts (6) under photocatalytic conditions.198 A series of aniline derivatives (75) with diverse electronic nature and different degrees of steric hinderance was well-tolerated and the corresponding products (383) were obtained in moderate to good yields (Scheme 75). Structurally complex arylamines embedded with urea, acetal, cyclic ketone, lactone, and imide were also smoothly converted into the desired products in acceptable yields. Notably, the incorporation of the difluoromethylselenyl unit was successfully demonstrated on biologically important skeletons such as, pomalidomide, clofibrate, L-menthol and sulfamethoxazole derivatives. Under visible light irradiation, the photocatalyst (rose Bengal) played a crucial role in generating aryl radical (141) from the aryldiazonium salt and difluoromethylselenyl radical from (380).
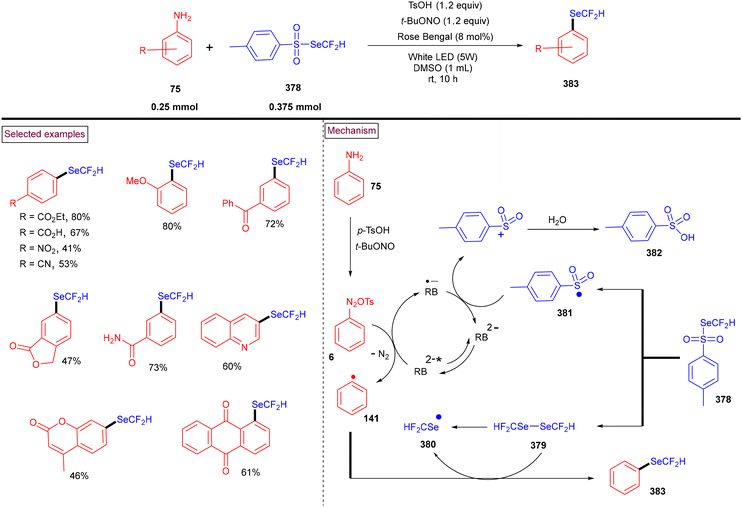 |
| | Scheme 75 Difluoromethyl selenolation of aryl amines through in situ-generated aryldiazonium salts. | |
A one-pot approach to prepare selenide ethers (385) was reported from in situ-generated aryldiazonium salts (6) and diselenides (384) by Lee et al. in 2019.199 Anilines (75) substituted with –Me, –OMe, –NO2, and halogens reacted with diphenyl diselenides to afford the corresponding products in 45%–99% yield (Scheme 76). Sterically hindered anilines led to moderate yields. Similarly, diaryl diselenides (384) with substituents such as, –Me and –CF3 smoothly led to C–Se bond formation in good yields. A few drug molecules such as sulfacetamide, sulfameter and sulfamethoxazole were obtained in the late-stage selenylation process. According to the plausible mechanism, in situ-generated aryldiazonium salt (6) reacted with diaryl diselenide (384) to afford aryl radical (141) and selenyl radical. The combination of these two radicals provided selenide ethers (385). The authors also demonstrated the formation of thioethers from anilines and thiols.
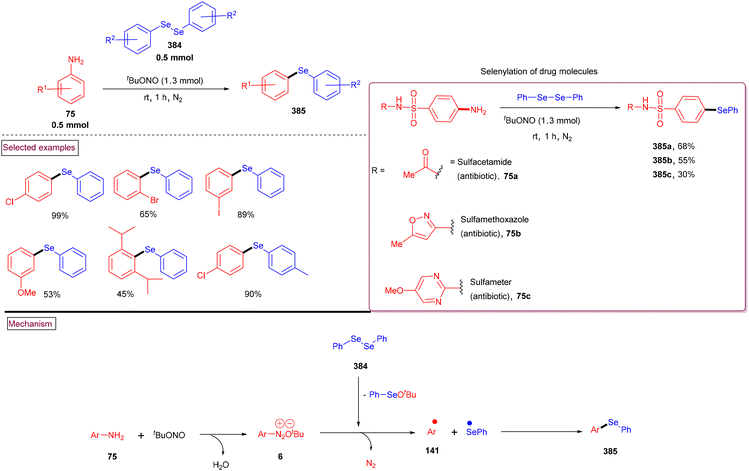 |
| | Scheme 76 Synthesis of selenide ethers from in situ-generated aryldiazonium salts and diselenides. | |
The preparation of trifluoromethylselenyl arenes (389) was demonstrated by the reaction of trifluoromethyl tolueneselenosulfonates (386) with aryldiazonium salts (6) in the presence of eosin Y under visible light irradiation (Scheme 77).51 Generation of the C(sp2)–SeCF3 bond was realized with a broad functional group tolerance. A series of aryldiazonium salts (6) with functionalities such as, –COOH, –CO2Et, –NO2, –CONH2, and halogens was tested and the desired trifluoromethylselenolated products (389) were obtained in good yields (45%–78%). Interestingly, this trifluoromethylselenolation was successful with higher homologues such as CF2CF3 and CF2CF2CF3. The reaction pathway initiated with the SET reduction of aryldiazonium salt (6) by excited eosin Y and its reaction with (CF3Se)2 (formed via homolysis of 386) to afford trivalent selenium radical species (386). Further SET oxidation in the presence of (6) and interaction with DMSO afforded the trifluoromethylselenolated products (389). A similar type of trifluoromethyl selenoethers was reported by Zhang and He et al. (2018) from aryldiazonium salts (6) and [Me4N][SeCF3].200 Noteworthily, this process proceeded through nucleophilic substitution at −20 °C and the corresponding products were obtained in moderate yields. Interestingly, this substitution pattern was applied for alkyl halides, α-diazocarbonyls, and diaryliodonium triflates to yield the corresponding seleno ethers.
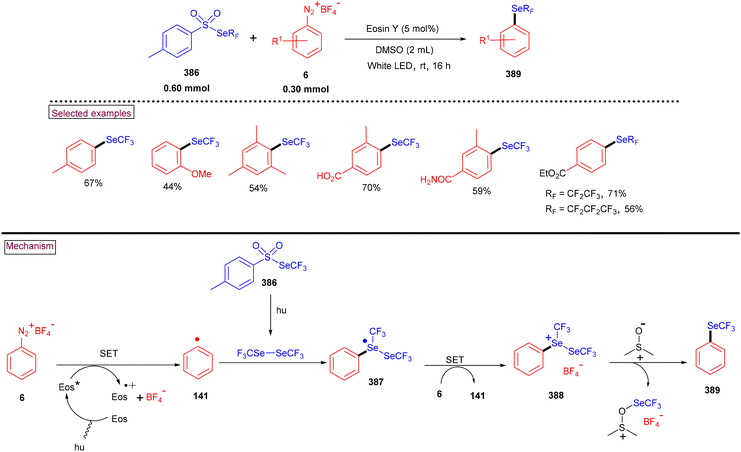 |
| | Scheme 77 Visible light-mediated synthesis of trifluoromethylselenyl arene. | |
7.4. Functionalized sulfonyl compounds from aryldiazonium salts
Lian and Zhang et al. (2021) described a convenient method to access geometrically well-defined (E)-β-amidovinyl sulfones (395) from enamides (390), aryldiazonium salts (6) and externally generated SO2 in good to excellent yields via direct C(sp2)–H sulfonylation.201 Excellent stereo and regioselectivity were realized from both secondary and tertiary enamides (390) (Scheme 78). A variety of benzamides, alkenyls, alkyls, pyridyls and those derived from pregnenolone, L-menthol and cholesterol were reacted with aryldiazonium salts (6) bearing electronically diverse substituents to give the desired products (395) in good yields. Similar to the mechanism discussed in Scheme 61, a plausible mechanism involving key arylsulfonyl radical (73) was reported. Complex (391) resulted from the coordination of aryldiazonium salt (6), SO2 and enamide (390). Extrusion of N2 from the complex led to arylsulfonyl radical (142) and radical cation (392). The addition of the arylsulfonyl radical to (392) generated radical intermediate (393), which was in equilibrium with (394). The desired product (395) was formed via tautomerization.
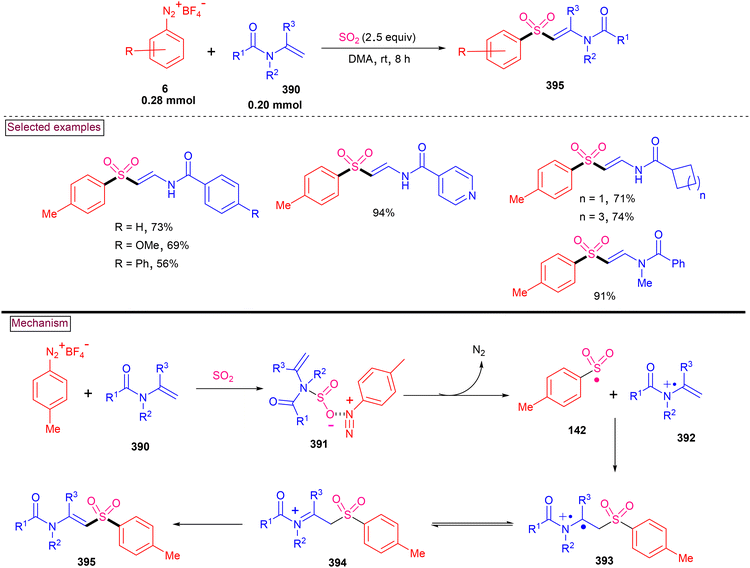 |
| | Scheme 78 Synthesis of (E)-β-amidovinyl sulfones via direct C(sp2)–H sulfonylation. | |
Hydroarylsulfonylation of styrenes could be achieved via the treatment of substrates with aryldiazonium salts (6), thiophenol and DABSO under mild conditions (Scheme 79).202 Thiophenol served as the HAT (hydrogen atom transfer) reagent. This methodology was further extended to obtain the alkoxyarylsulfonylated product (398) in the presence of MeOH and 1,4-dicyanobenzene as an oxidant. These methoxylated products underwent convenient elimination to afford vinyl sulfone (399) in the presence of a base. As illustrated in section 6 (Schemes 61–64), in situ-generated arylsulfonyl radical (142) was added to alkene (234) to generate the more stable benzyl radical (404) in an anti-Markovnikov fashion. H-transfer from thiol to (396) resulted in the formation of hyroarylsulfonylated product (400). Alternatively, SET oxidation of the benzylic radical by 1,4-DCB yielded quinine methide (397), which reacted with alcohol to afford alkoxyarylsulfonylated product (398). Base-mediated elimination furnished the unsaturated product (399).
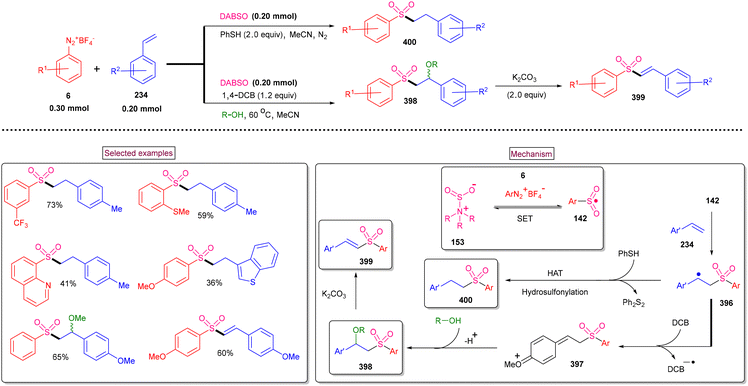 |
| | Scheme 79 Hydroarylsulfonylation of vinyl arenes using HAT reagent. | |
A cascade approach to synthesize β-keto sulfones (407) from the reaction of alkynes/alkenes and aryldiazonium salts (6) with DABSO was developed under transition-metal-free conditions (Scheme 80).20318O labeling studies revealed that the incoming oxygen on the keto functionality was sourced from the open air. Additionally, sodium metabisulfite was also identified as an inexpensive sulfone source in this process. In situ-generated aryl sulfonyl radical (142) reacted with alkyne (221) or alkene (234) to afford radical intermediates (401 and 402), which reacted with oxygen to give dimers (403 and 404). Dimerization and homolytic cleavage resulted in the formation of oxy radicals (405 and 406). Further hydroxylation and isomerization led to β-keto sulfones (407).
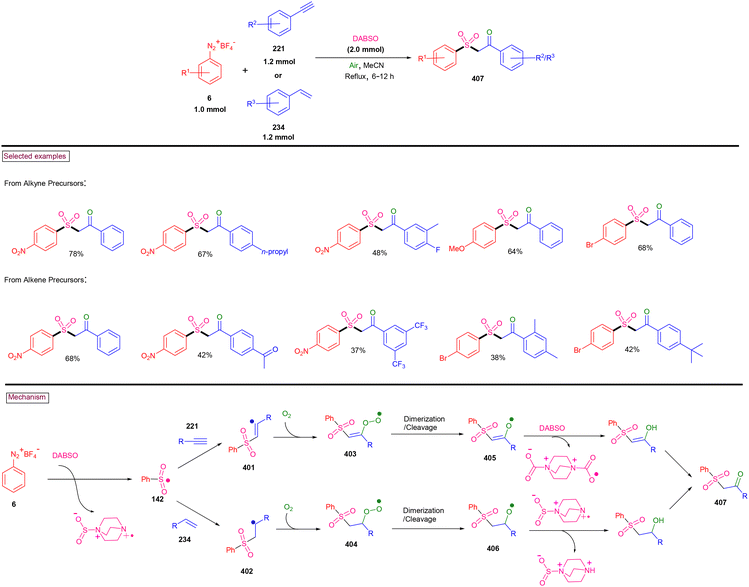 |
| | Scheme 80 Synthesis of β-keto sulfones from aryl diazonium salts, DABSO and alkynes/alkenes. | |
Weng et al. (2020) described a Sandmeyer-type fluoro-sulfonylation approach to access arylsulfonyl fluorides (409) under Cu-free conditions, in which the direct conversion of NH2 into SO2F was realized.204 Sodium metabisulfite and Selectfluor (408) were used as the SO2 and fluorine source, respectively (Scheme 81). A broad range of functionalized aryldiazonium salts (6) was used in this process including functional groups such as, –CO2H, –Br, –CN, ketones, and esters and the corresponding products (409) were obtained in excellent yields. Importantly, a simplified one-pot approach to get aryl sulfonyl fluorides (409) from arylamines was also developed without any significant drop in yield via in situ diazotization. This deaminative fluorosulfonylation was successfully demonstrated on the gram scale.
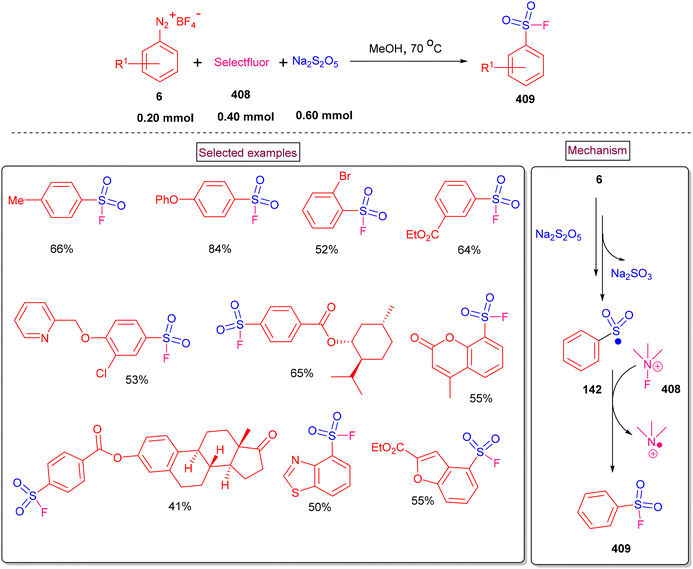 |
| | Scheme 81 Synthesis of arylsulfonyl fluorides via deaminative fluorosulfonylation. | |
Quinoline-N-oxides (410) were transformed into 2-sulfonylquinolines (413) in the presence of aryldiazonium salts (6) and Na2S2O5 (Xia et al., 2019).205 Aryldiazonium salts (6) bearing electron-donating groups reacted more efficiently than their electron-withdrawing analogues (Scheme 82). It was observed that pyridine-N-oxide and quinoxaline-N-oxides were not involved in this sulfonylation process. However, isoquinoline-N-oxide was smoothly converted into the corresponding product in good yields. The involvement of arylsulfonyl radical (142) was confirmed by a radical trapping experiment. Quinoline-N-oxide (410) was reacted with in situ-generated (73) through a Minisci-like radical process to give oxy radical (411). Aryl sulfonyl radical-induced aromatization produced the product (413). Another arylsulfonylation strategy to prepare a range of arylsulfones was reported by Wu and Fan et al. (2015) from aryliodonium salts, aryldiazonium salts (6) and SO2 under transition-metal-free conditions.206 The optimization studies revealed that an alcohol solvent increased the yield dramatically. The scope was investigated in detail with various substituted (–F, –Cl, –OMe, and esters) aryliodonium and aryldiazonium salts in the presence of a base and NaHSO3.
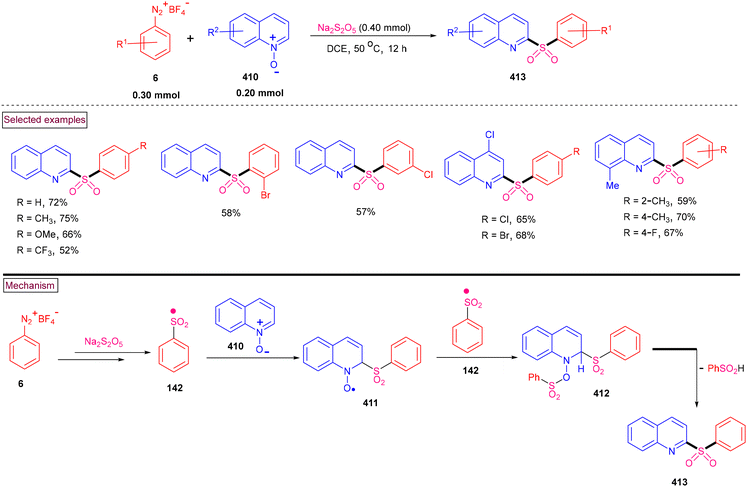 |
| | Scheme 82 C2-Arylsulfonylation of quinoline-N-oxides using aryldiazonium salts. | |
A three-component approach from aryldiazonium salts (6), hydroxylamines (414) and DABSO was developed to prepare O-aminosulfonates (415) in moderate to good yields (Wu and Li et al., 2017).207 Parallelly, sulfonamides (416) were obtained when amine was employed in the presence of N-hydroxybenzotriazole (Scheme 83). Aryldiazonium salts (6) bearing electronically distinct functional groups (–Me, –OMe, –CF3, –NO2, –Cl, –Br, and –CO2Et) were found to be compatible and the desired products were obtained in excellent yields. However, a minimal steric effect was observed in the case of ortho-substituted aryldiazonium salts (6). Alternatively, a variety of hydroxylamines (414) including 1H-benzo[d][1,2,3]triazol-1-ol, N-hydroxy-N,4-dimethylbenzenesulfonamide, and 3-hydroxybenzo[d][1,2,3]triazin-4(3H)-one were tested and the desired O-aminosulfonates (415) were obtained in moderate to good yields. In addition, primary, secondary and aryl amines participated in the production of sulfonamide derivatives (416) in the yield of 44%–83%. The oxy/amino radical of N-hydroxy precursors directly coupled with an in situ-generated arylsulfonyl radical to provide the desired products.
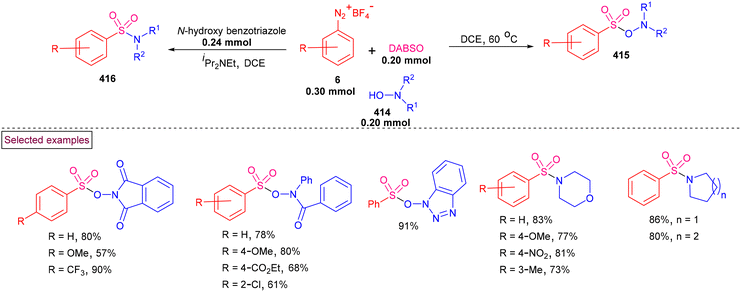 |
| | Scheme 83 Synthesis of O-aminosulfonates from aryldiazonium salts, hydroxylamines and DABSO. | |
8. Conclusions
Aryldiazonium salts as a readily accessible and reactive N2-bearing synthons, have been proven to be very promising substrates to prepare N-heterocyclic cores with a high degree of skeletal and structural diversity. In this review, the synthetic progress in the construction of a wide range of five- and six-membered N-heterocycles such as tetrazoles, triazoles, indazoles, pyrazoles, phenanthridines, pyridazines, triazines, quinolines, quinazolines, and quinazolin-4(3H)imines from aryldiazonium salts under transition metal-free conditions was discussed. Aryldiazonium precursor-based cascade annulations, cycloadditions, addition/oxidative cyclization, photocatalytic reactions, electrochemical reactions, and radical annulations were specifically highlighted. These aryldiazonium salt-derived synthetic strategies discussed herein showcased appreciable merits such as excellent functional group tolerance, high selectivity, ambient conditions, scalability, and more importantly one-pot operations. We believe that this review will enable chemists to develop novel synthetic methods by utilizing aryldiazonium salts as key precursors and contribute significantly to the synthetic community.
Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Ziad Moussa is grateful to the United Arab Emirates University (UAEU) and to the Research Office for supporting the research developed in his laboratory and reported herein (UPAR Grant code G00004605). Mani Ramanathan thank Arutperunjothi Thiruarutprakasa Vallalar for the grace and Prof. Shiuh-Tzung Liu for the constructive suggestions and discussion.
References
- D. Ye, H. Lu, Y. He, Z. Zheng, J. Wu and H. Wei, Rapid syntheses of N-fused heterocycles via acyl-transfer in heteroaryl ketones, Nat. Commun., 2022, 13, 3337 CrossRef PubMed.
- M. M. Heravi and V. Zadsirjan, Prescribed drugs containing nitrogen heterocycles: an overview, RSC Adv., 2020, 10, 44247–44311 RSC.
- R. D. Taylor, M. MacCoss and A. D. Lawson, Rings in drugs, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Modern advances in heterocyclic chemistry in drug discovery, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC.
-
J. J. Li, Heterocyclic Chemistry in Drug Discovery, Wiley-VCH, Weinheim, 2013, ISBN: 978-1-118-14890-7 Search PubMed.
- N. Schneider, D. M. Lowe, R. A. Sayle, M. A. Tarselli and G. A. Landrum, Big data from pharmaceutical patents: A computational analysis of medicinal chemist's bread and butter, J. Med. Chem., 2016, 59, 4385–4402 CrossRef CAS PubMed.
- D. G. Brown and J. Bostrom, Analysis of past and present synthetic methodologies on medicinal chemistry: Where have all the new reactions gone?, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, A review on recent advances in nitrogen-containing molecules and their biological applications, Molecules, 2020, 25, 1909 CrossRef CAS PubMed.
- P. Yin, Q. Zhang and J. M. Shreeve, Dancing with energetic nitrogen atoms: Versatile N-functionalization strategies for N-heterocyclic frameworks in high energy density materials, Acc. Chem. Res., 2016, 49, 4–16 CrossRef CAS PubMed.
- M. C. Rios, N. F. Bravo, C. C. Sanchez and J. Portilla, Chemosensors based on N-heterocyclic dyes: Advances in sensing highly toxic ions such as CN− and Hg2+, RSC Adv., 2021, 11, 34206–34234 RSC.
- O. O. Ajani, O. Y. Audu, D. V. Aderohunmu, F. E. Owolabi and O. A. Olomieja, Undeniable pharmacological potentials of quinazoline motifs in therapeutic medicine, Am. J. Drug Discovery Dev., 2017, 7, 1–24 CrossRef CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S.FDA approved pharmaceuticals, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- B. R. Smith, C. M. Eastman and J. T. Njardarson, Beyond C, H, O, and N! Analysis of the elemental composition of U. S. FDA approved drug architectures, J. Med. Chem., 2014, 57, 9764–9773 CrossRef CAS PubMed.
- P. Martins, J. Jesus, S. Santos, L. R. Raposo, C. Roma-Rodrigues, P. V. Baptista and A. R. Fernandes, Heterocyclic anticancer compounds: Recent advances and the paradigm shift towards the use of nanomedicine's tool box, Molecules, 2015, 20, 16852–16891 CrossRef CAS PubMed.
- M. Dinodia, N-heterocycles: Recent advances in biological applications, Mini-Rev. Org. Chem., 2023, 20, 735–747 CrossRef CAS.
- A. Kumar, A. K. Singh, H. Singh, V. Vijayan, D. Kumar, J. Naik, S. Thareja, J. P. Yadav, P. Pathak, M. Grishina, A. Verma, H. Khaliluallah, M. Jaremko, A. H. Emwas and P. Kumar, Nitrogen containing heterocycles as anticancer agents: A medicinal chemistry perspective, Pharmaceuticals, 2023, 16, 299 CrossRef CAS PubMed.
- T. Jayakumar, C.-M. Yang, T.-L. Yen, C.-Y. Hsu, J.-R. Sheu, C.-W. Hsia, M. Manubolu, W.-C. Huang, C.-Y. Hsieh and C.-H. Hsia, Anti-inflammatory mechanism of an alkaloid Rutaecarpine in LTA-stimulated RAW 264.7 cells: Pivotal role on NF-kB and ERK/p38 signalling molecules, Int. J. Mol. Sci., 2022, 23, 5889 CrossRef CAS PubMed.
- P. Yadav and K. Shah, Quinolines, a perpetual, multipurpose scaffold in medicinal chemistry, Bioorg. Chem., 2021, 109, 104639 CrossRef CAS PubMed.
- V. Kamat, R. Santhosh, B. Poojari, S. P. Nayak, B. K. Kumar, M. Sankaranarayanan, Faheem, S. Khanapure, D. A. Barretto and S. K. Vootla, Pyridine- and Thiazole- based hydrazides with promising anti-inflammatory and antimicrobial activities along with their in-silico studies, ACS Omega, 2020, 5, 25228–25239 CrossRef CAS PubMed.
- R. Reddyrajula and U. K. Dalimba, Structural modificationof zolpidem led to potent antimicrobial activity in imidazo[1,2-a]pyridine/pyrimidine-1,2,3-triazoles, New J. Chem., 2019, 43, 16281–16299 RSC.
- H. A. Flores Toque, F. B. M. Priviero, C. E. Teixeira, E. Perissutti, F. Fiorino, B. Severino, F. Frecentese, R. Lorenzetti, J. S. Baracat, V. Santagada, G. Caliendo, E. Antunes and G. De Nucci, Synthesis and pharmacological evaluations of sildenafil analogues for treatment of erectile dysfunction, J. Med. Chem., 2008, 51, 2807–2815 CrossRef CAS PubMed.
- L. E. Rodriguez, C. H. House, K. E. Smith, M. R. Roberts and M. P. Callahan, Nitrogen heterocycles form peptide nucleic acid precursors in complex prebiotic mixtures, Sci. Rep., 2019, 9, 9281 CrossRef PubMed.
- M. F. Jacobsen, M. M. Knudsen and K. V. Gothelf, Efficient N-arylation and N-alkenylation of the five DNA/RNA nucleobases, J. Org. Chem., 2006, 71, 9183–9190 CrossRef CAS PubMed.
- A. Kirschning, Coenzymes and their role in the evolution of life, Angew. Chem., Int. Ed., 2021, 60(12), 6242–6269 CrossRef CAS PubMed.
- W. Guo, M. Zhao, W. Tan, L. Zheng, K. Tao and X. Fan, Developments towards synthesis of N-heterocycles from amidines via C-N/C-C bond formation, Org. Chem. Front., 2019, 6, 2120–2141 RSC.
- S. Majee, Shilpa, M. Sarav, B. K. Banik and D. Ray, Recent advances in the green synthesis of active N-heterocycles and their biological activities, Pharmaceuticals, 2023, 16, 873 CrossRef CAS PubMed.
- C.-M. T. Vo and J. W. Bode, Synthesis of saturated N-heterocycles, J. Org. Chem., 2014, 79, 2809–2815 CrossRef CAS PubMed.
- H. Wang, Y. Zheng, H. Xu, J. Zou and C. Jin, Metal-free synthesis of N-heterocycles via intramolecular electrochemical C-H aminations, Front. Chem., 2022, 10, 950635 CrossRef CAS PubMed.
- N. Kaur, Review on the synthesis of six membered N, N-heterocycles by microwave irradiation, Synth. Commun., 2015, 45, 1145–1182 CrossRef CAS.
-
(a) B. Nie, W. Wu, H. Jiang and J. Zhang, Recent advances in the synthesis of bridgehead (or ring-junction) nitrogen heterocycles via transition metal-catalyzed C–H bond activation and functionalization, Org. Chem. Front., 2020, 7, 3067–3099 RSC;
(b) F.-F. Xu, J.-Q. Chen, D.-Y. Shao and P.-Q. Huang, Catalytic enantioselective reductive alkynylation of amides enables one-pot syntheses of pyrrolidine, piperidine and indolizidine alkaloids, Nat. Commun., 2023, 14, 6251, DOI:10.1038/s41467-023-41846-x;
(c) X. Peng, M.-M. Zheng, P. Qin, X.-S. Xue, F.-G. Zhang and J.-A. Ma, Silver-catalysed [3 + 2] annulation reaction of aryldiazonium salts with allenes enabled by boronate direction, Org. Chem. Front., 2023, 10, 74–82 RSC;
(d) Z. Chen, H. Zhang, S.-F. Zhou and X. Cui, Photoredox-catalyzed synthesis of sulfonated oxazolines from N-allylamides through the insertion of sulfur dioxide, Org. Chem. Front., 2022, 9, 364–369 RSC;
(e) A. K. Singh and J. Kandasamy, Palladium catalysed stereocontrolled synthesis of C-aryl glycosides using glycals and arenediazonium salts at room temperature, Org. Biomol. Chem., 2018, 16, 5107–5112 RSC;
(f) Q. Rao, Y. Zhang, Y.-P. Liu, B. Jiang, X. Wang, S.-J. Tu and W.-J. Hao, Azofuran activation for annulative rearrangement enabled by gold(I)/Brønsted acid relay catalysis, Chem. Commun., 2023, 59, 5725–5728 RSC;
(g) J. Y. Ouyang, F.-F. Shen, H.-Q. Zhao, J.-J. Chen, Z.-D. Wen, H.-M. Jiang, J.-H. Qin, Q. Sun, J.-H. Li and X.-H. Ouyang, Aryldiazonium Salt-Triggered [2 + 2 + 1] Heteroannulation of Indoles by an Arylhydrazone Radical-Relayed 1,5-Hydrogen Atom Transfer, Org. Lett., 2023, 25, 6549–6554 CrossRef CAS PubMed.
- F. Mo, D. Qiu, L. Zhang and J. Wang, Recent development of aryl diazonium chemistry for the derivatization of aromatic compounds, Chem. Rev., 2021, 121, 5741–5829 CrossRef CAS PubMed.
- J. Liu, J. Jiang, L. Zheng and Z.-Q. Liu, Recent advances in the synthesis of nitrogen heterocycles using arenediazonium salts as nitrogen sources, Adv. Synth. Catal., 2020, 362, 4876–4895 CrossRef CAS.
- F. G. Zhan, Z. Chen, C. W. Cheung and J. A. Ma, Aryl diazonium salt-triggered cyclization and cycloaddition reactions: past, present, and future, Chin. J. Chem., 2020, 38, 1132–1152 CrossRef.
- F. Mo, G. Dong, Y. Zhang and J. Wang, Recent applications of arene diazonium salts in organic synthesis, Org. Biomol. Chem., 2013, 11, 1582–1593 RSC.
- J. Chen, X. Xie, J. Liu, Z. Yu and W. Su, Revisiting aromatic diazotization and aryl diazonium salts in continuous flow: highlighted research during 2001–2021, React. Chem. Eng., 2022, 7, 1247–1275 RSC.
- N. Oger, E. L. Grognec and F.-X. Felpin, Handling diazonium salts in flow for organic and material chemistry, Org. Chem. Front., 2015, 2, 590–614 RSC.
- V. D. Filimonov, M. Trusova, P. Postnikov, E. A. Krasnokutskaya, Y. M. Lee, H. Y. Hwang, H. H. Kim and K.-W. Chi, Unusually stable, versatile, and pure arenediazonium tosylates: their preparation, structures, and synthetic applicability, Org. Lett., 2018, 10, 3961–3964 CrossRef PubMed.
- J. P. Griess, Vorlaufige Notiz uber die Einwirkung von salpetriger Saure auf Amidinitround aminitrophenylsaure, Justus Liebigs Ann. Chem., 1858, 106, 123–125 CrossRef.
- T. Sandmeyer, The substitution of the amine group with chlorine atom in aromatic systems, Ber. Dtsch. Chem. Ges., 1884, 17, 1633–1635 CrossRef.
- G. Balz and G. Schiemann, On aromatic fluoric compounds, I A new method for its representation, Ber. Dtsch. Chem. Ges., 1927, 60, 1186–1190 CrossRef.
- M. Gomberg and W. E. Bachmann, The synthesis of biaryl compounds by means of the diazo reaction, J. Am. Chem. Soc., 1924, 46, 2339–2343 CrossRef CAS.
- K. Kikukawa, Reaction of diazonium salts with transition metals-III: Palladium (0)-catalyzed arylation of unsaturated compounds with arenediazonium salts, Tetrahedron, 1981, 37, 31–36 CrossRef CAS.
- K. Kikukawa and T. Matsuda, Reaction of diazonium salts with transition metals. I. Arylation of olefins with arenediazonium salts catalyzed by zero valent palladium, Chem. Lett., 1977, 6, 159–162 CrossRef.
- R. Pschorr, Neue synthese des phenanthrens und seiner derivate, Ber. Dtsch. Chem. Ges., 1896, 29, 496–501 CrossRef CAS.
- H. Meerwein, E. Buchner and K. van Emster, Uber die Einwirkung aromatischer Diazoverbindungen auf α,β-ungesattigte Carbonylverbindungen, J. Prakt. Chem., 1939, 152, 237–266 CrossRef CAS.
- F. Mo, D. Qiu, Y. Zhang and J. Wang, Renaissance of Sandmeyer-type reactions: conversion of aromatic C-N bonds into C-X bonds (X = B, Sn, P, or CF3), Acc. Chem. Res., 2018, 51, 496–506 CrossRef CAS PubMed.
- S. S. Babu, P. Muthuraja, P. Yadav and P. Gopinath, Aryldiazonium salts in photoredox catalysis-Recent trends, Adv. Synth. Catal., 2021, 363, 1782–1809 CrossRef CAS.
- D. Qiu, H. Meng, L. Jin, S. Wang, S. Tang, X. Wang, F. Mo, Y. Zhang and J. Wang, Synthesis of aryl trimethylstannanes from aryl amines: A Sandmeyer-type stannylation reaction, Angew. Chem., Int. Ed., 2013, 52, 11581–11584 CrossRef CAS PubMed.
- C. Matheis, V. Wagner and L. J. Goosen, Sandmeyer-type trifluoromethylthiolation and trifluoromethylselenolation of (hetero)aromatic amines catalyzed by copper, Chem. – Eur. J., 2016, 22, 79–82 CrossRef CAS PubMed.
- D. Tanini, L. Ricci and A. Capperucci, Rongalite-promoted on water synthesis of functionalized tellurides and ditellurides, Adv. Synth. Catal., 2020, 362, 1323–1332 CrossRef CAS.
- C. Ghiazza, V. Debrauwer, C. Monnereau, L. Khrouz, M. Medebielle, T. Billard and A. Tlili, Visible-light-mediated metal-free synthesis of trifluoromethylselenolated arenes, Angew. Chem., Int. Ed., 2018, 57, 11781–11785 CrossRef CAS PubMed.
- Y. Gu, D. Chang, X. Leng, Y. Gu and Q. Shen, Organometallics, 2015, 34, 3065–3071 CrossRef CAS.
- E. S. Zhilin, L. L. Fershtat, D. M. Bystrov, A. S. Kulikov, A. O. Dmitrienko, I. V. Ananyev and N. N. Makhova, Renaissance of 1,2,5-oxadiazolyl diazonium salts: synthesis and reactivity, Eur. J. Org. Chem., 2019, 4248–4259, DOI:10.1002/ejoc.201900622.
- A. Chakraborty, S. Jana, G. Kibriya, A. Dey and A. Hajra, tert-butyl nitrite mediated azo coupling between anilines and imidazoheterocycles, RSC Adv., 2016, 6, 34146–34152 RSC.
- J. Rong, H. Jiang, S. Wang, Z. Su, H. Wang and C. Tao, Metal-free cascade reactions of aziridines with arylalkynes and aryldiazoniums: facile access to arylazopyrrolines, Org. Biomol. Chem., 2020, 18, 3149–3157 RSC.
- C. Zhu, H. Zeng, F. Chen, Z. Yang, Y. Cai and H. Jiang, Intermolecular C(sp3)-H amination promoted by internal oxidants: synthesis of trifluoroacetylated hydrazones, Angew. Chem., Int. Ed., 2018, 57, 17215–17219 CrossRef CAS PubMed.
- W.-C. Gao, Y.-F. Cheng, Y.-Z. Shang, H.-H. Chang, X. Li, R. Zhou, Y. Qiao and W.-L. Wei, Copper(II)-catalyzed four-component oxysulfonylation/diazenylation: synthesis of α-arylhydrazo-β-keto sulfones, J. Org. Chem., 2018, 83, 11956–11962 CrossRef CAS PubMed.
- Y. Shao, H. Zheng, J. Qian and X. Wan, In situ generation of nitrilimines from aryldiazonium salts and diazo esters: synthesis of fully substituted pyrazoles under room temperature, Org. Lett., 2018, 20, 2412–2415 CrossRef CAS PubMed.
- X. Peng, M.-Y. Xiao, J.-L. Zeng, F.-G. Zhang and J.-A. Ma, Construction of difluoromethylated tetrazoles via silver-catalyzed regioselective [3 + 2] cycloadditions of aryl diazonium salts, Org. Lett., 2019, 21, 4808–4811 CrossRef CAS PubMed.
- S.-J. Zhai, X. Peng, F.-G. Zhang and J.-A. Ma, Catalytic direct regioselective synthesis of phosphonylated tetrazoles from aryl diazonium salts and seyferth-gilbert regent, Org. Lett., 2019, 21, 9884–9888 CrossRef CAS PubMed.
- M.-Y. Xiao, Z. Chen, F.-G. Zhang and J.-A. Ma, Regioselective synthesis of carboxylic and fluoromethyl tetrazoles enabled by silver-catalyzed cycloaddition of diazoacetates and aryl diazonium salts, Tetrahedron, 2020, 76, 131063 CrossRef CAS.
- J.-Q. Liu, X. Shen, Y. Wang, X.-S. Wang and X. Bi, [3 + 2] cycloaddition of isocyanides with aryl diazonium salts: catalyst-dependent regioselective synthesis of 1,3-and 1,5-disubstituted 1,2,4-triazoles, Org. Lett., 2018, 20, 6930–6933 CrossRef CAS PubMed.
- H. Li, X. Wu, W. Hao, H. Li, Y. Zhao, Y. Wang, P. Lian, Y. Zheng, X. Bao and X. Wan, [3 + 2] cycloaddition of nitrile ylides with diazonium salts: copper-catalyzed one-pot synthesis of fully substituted 1,2,4-triazoles, Org. Lett., 2018, 20, 5224–5227 CrossRef CAS PubMed.
- X.-Y. Yu, W.-J. Xiao and J.-R. Chen, Synthesis of trisubstituted 1,2,4-triazoles from azlactones and aryldiazonium salts by a cycloaddition/decarboxylation cascade, Eur. J. Org. Chem., 2019, 6994–6998 CrossRef CAS.
- Z. Liu, H. Ji, W. Gao, G. Zhu, L. Tong, F. Lei and B. Tang, Copper(I)-mediated carboamination of vinyl azides by aryldiazonium salts: synthesis of N2-substituted 1,2,3-triazoles, Chem. Commun., 2017, 53, 6259–6262 RSC.
- H.-N. Liu, H.-Q. Cao, C. W. Cheung and J.-A. Ma, Cu-mediated expeditious annulation of alkyl 3-aminoacrylates with aryldiazonium salts: Access to alkyl N2-aryl 1,2,3-triazole-carboxylates for druglike molecular synthesis, Org. Lett., 2020, 22, 1396–1401 CrossRef CAS PubMed.
- C. Zhu, H. Zeng, F. Chen, C. Liu, R. Zhu, W. Wu and H. Jiang, Copper-catalyzed coupling of oxime acetates and aryldiazonium salts: an azide-free strategy toward N-2-aryl-1,2,3-triazoles, Org. Chem. Front., 2018, 5, 571–576 RSC.
- C. Zhu, H. Zeng, F. Chen, C. Liu and H. Jiang, Copper-catalyzed cyclization of aryl amines and aryldiazonium salts under air: access to N-2-aryl-naphthotriazoles, Adv. Synth. Catal., 2019, 361, 5149–5159 CrossRef CAS.
- P. Wu, Y. He, H. Wang, Y.-G. Zhou and Z. Yu, Copper(II)-catalyzed C-H nitrogenation/annulation cascade of ketene N, S-acetals with aryldiazonium salts: a direct access to N2-substituted triazole and triazine derivatives, Org. Lett., 2020, 22, 310–315 CrossRef CAS PubMed.
- Q. Ding, M. Li, Y. Sun, Y. Yu, J. B. Baell and F. Huang, Copper-catalyzed [4 + 2] annulation reaction of β-enaminones and aryl diazonium salts without external oxidant: synthesis of highly functionalized 3H-1,2,4-triazines via homogeneous or heterogeneous strategy, Org. Chem. Front., 2020, 7, 457–463 RSC.
- S. Kindt, K. Wicht and M. R. Heinrich, Base-induced radical carboamination of nonactivated alkenes with aryldiazonium salts, Org. Lett., 2015, 17, 6122–6125 CrossRef CAS PubMed.
- J. Liu, E. Xu, J. Jiang, Z. Huang, L. Zheng and Z.-Q. Liu, Copper-mediated tandem ring-opening/cyclization reactions of cyclopropanols with aryldiazonium salts: synthesis of N-arylpyrazoles, Chem. Commun., 2020, 56, 2202–2205 RSC.
- X.-L. Yu, J.-R. Chen, D.-Z. Chen and W.-J. Xiao, Visible-light-induced photocatalytic azotrifluoromethylation of alkenes with aryldiazonium salts and sodium triflinate, Chem. Commun., 2016, 52, 8275–8278 RSC.
- R. A. Angnes, C. Potnis, S. Liang, C. R. D. Correia and G. B. Hammond, Photoredox-catalyzed synthesis of alkylaryldiazenes: formal deformylative C-N bond formation with alkyl radicals, J. Org. Chem., 2020, 85, 4153–4164 CrossRef CAS PubMed.
- M. Wang, B.-C. Tang, J.-C. Xiang, X.-L. Chen, J.-T. Ma, Y.-D. Wu and A.-X. Wu, Aryldiazonium salts serve as a dual synthons: construction of fully substituted pyrazoles via Rongalite-mediated three-component radical annulation reaction, Org. Lett., 2019, 21, 8934–8937 CrossRef CAS PubMed.
-
(a) A. Roglans, A. Pla-Quintana and M. Moreno-Manas, Diazonium salts as substrates in palladium-catalyzed cross-coupling reactions, Chem. Rev., 2006, 106, 4622–4643 CrossRef CAS PubMed;
(b) L. Wang and G. Liu, One-pot Negishi cross-coupling reaction of aryldiazonium salts via Ni catalysis induced by visible-light, Catal. Commun., 2019, 131, 105785 CrossRef;
(c) L. S. Varnedoe, B. D. Angel, J. L. McClellan and J. M. Hanna Jr, Pd/C-catalyzed cross-coupling of arenediazonium salts with potassium aryltrifluoroborates, Lett. Org. Chem., 2010, 7, 1–6 CrossRef CAS PubMed.
- Z. Xia and Q. Zhu, A transition-metal-free synthesis of arylcarboxyamides from aryl diazonium salts and isocyanides, Org. Lett., 2013, 15, 4110–4113 CrossRef CAS PubMed.
- M. Hartmann, Y. Li and A. Studer, Transition-metal-free oxyarylation of alkenes with aryl diazonium salts and TEMPONa, J. Am. Chem. Soc., 2012, 134, 16516–16519 CrossRef CAS PubMed.
- C. Jin, L. Su, D. Ma and M. Cheng, Transition-metal-free, visible-light-mediated cyclization of o-azidoarylalkynes with aryl diazonium salts, New J. Chem., 2017, 41, 14053–14056 RSC.
- R. Abrams, Q. Lefebvre and J. Clayden, Transition metal free cycloamination of prenyl carbamates and ureas promoted by aryldiazonium salts, Angew. Chem., Int. Ed., 2018, 57, 13587–13591 CrossRef CAS PubMed.
- S. Leyva and J. Cardoso-Ortiz, Recent developments in the synthesis of tetrazoles and their pharmacological relevance, Curr. Org. Chem., 2020, 25, 388–403 CrossRef.
- P. Zhan, Z. Li, X. Liu and E. De Clercq, Sulfanyltriazole/tetrazole: a promising class of HIV-1 NNRTIs, Mini-Rev. Med. Chem., 2009, 9, 1014–1023 CrossRef PubMed.
- F. Gao, J. Xiao and G. Huang, Current scenario of tetrazole hybrids for antibacterial activity, Eur. J. Med. Chem., 2019, 184, 111744 CrossRef CAS PubMed.
- L. V. Myznikov, A. Hrabalek and G. I. Koldobskii, Drugs in the tetrazole series, Chem. Heterocycl. Compd., 2007, 43, 1–9 CrossRef CAS , and reference cited therein.
- C.-X. Wei, M. Bian and G.-H. Gong, Tetrazolium compounds: synthesis and applications in medicine, Molecules, 2015, 20, 5528–5553 CrossRef CAS PubMed.
- Y. Zou, L. Liu, J. Liu and G. Liu, Bioisosteres in drug discovery: focus on tetrazole, Future Med. Chem., 2019, 12, 91–93 CrossRef PubMed.
- S.-Q. Wang, Y.-F. Wang and Z. Xu, Tetrazole hybrids and their antifungal activities, Eur. J. Med. Chem., 2019, 170, 225–234 CrossRef CAS PubMed.
- V. A. Ostrovskii, R. E. Trifonov and E. A. Popova, Medicinal chemistry of tetrazoles, Russ. Chem. Bull., 2012, 61, 768–780 CrossRef CAS.
- N. Fournier-Le Ray, N. Joly and J.-L. Fillaut, Optimising the synthesis of 2,5-diaryltetrazoles: The decisive choice of the reaction solvent, Tetrahedron, 2023, 143, 133560 CrossRef CAS.
- Z. Fang, Y. Zhang, H. Wang, G. Zanoni, J. Li, X. Li, Z. Liu and Y. Ning, Straightforward access to fluoroalkyl tetrazoles from fluoroalkyl N-sulfonylhydrazones, Org. Chem. Front., 2022, 9, 5606–5610 RSC.
- X. Lin, B. Luo, W. He, Q. Lu, L. Li, H. Zheng, W. Wu and Z. Weng, Metal-free synthesis of 5-trifluoromethyltetrazoles, Tetrahedron Lett., 2022, 95, 153717 CrossRef CAS.
- S.-J. Zhai, D. Cahard, F.-G. Zhang and J.-A. Ma, Metal-free regioselective construction of 2-aryl-2H-tetrazol-5-yl difluoromethylene phosphonates, Chin. Chem. Lett., 2022, 33, 863–866 CrossRef CAS.
- Y.-L. Yang, S. Li, F.-G. Zhang and J.-A. Ma, N-iodosuccinimide-promoted [3 + 2] annulation reaction of aryldiazonium salts with guanidines to construct aminotetrazoles, Org. Lett., 2021, 23, 8894–8898 CrossRef CAS PubMed.
- M.-Y. Xiao, M.-M. Zheng, X. Peng, X.-S. Xue, F.-G. Zhang and J.-A. Ma, Catalytic direct construction of cyano-tetrazoles, Org. Lett., 2020, 22, 7762–7767 CrossRef CAS PubMed.
- T. Liu, Y.-G. Ji and L. Wu,
tert-butyl nitrite-mediated radical cyclization of tetrazole amines and alkynes toward tetrazolo[1,5-a]quinolines, Org. Biomol. Chem., 2019, 17, 2619–2623 RSC.
-
(a) S. Ito, Y. Tanaka and A. Kakehi, Synthesis of 2,5-diaryltetrazoles from N-phenylsulfonylbenzhydrazidoyl chlorides and arylhydrazines, Bull. Chem. Soc. Jpn., 1976, 49, 762 CrossRef CAS;
(b) S. Ito, Y. Tanaka, A. Kakehi and K.-I. Kondo, A Facile Synthesis of 2,5-Disubstituted tetrazoles by the reaction of phenylsulfonylhydrazones with Arenediazonium Salts, Bull. Chem. Soc.
Jpn., 1976, 49, 1920 CrossRef CAS.
- M. Ramanathan, Y.-H. Wang and S.-T. Liu, One-pot reactions for synthesis of 2,5-substituted tetrazoles from aryldiazonium salts and amidines, Org. Lett., 2015, 17, 5886–5889 CrossRef CAS PubMed.
- P. Mishra, P. Kumar, O. S. Srivastava and N. Rastogi, Organophotoredox-mediated Formal [3 + 2]-Cycloaddition of 2H-Azirines with Aryldiazonium Salts: Direct Access to Trisubstituted 1,2,4-Triazoles, Chem. – Asian J., 2023, 18, e202300007, DOI:10.1002/asia.202300007.
- X. Li, X. Ye, C. Wei, C. Shan, L. Wojtas, Q. Wang and X. Shi, Diazo activation with diazonium salts: synthesis of indazole and 1,2,4-triazole, Org. Lett., 2020, 22, 4151–4155 CrossRef CAS PubMed.
- Y.-T. Tian, F.-G. Zhang, J. Nie, C. W. Cheung and J.-A. Ma, Metal-free Decarboxylative Annulation of 2-Aryl-2-isocyano-acetates with Aryldiazonium Salts: General Access to 1,3-Diaryl-1,2,4-triazoles, Adv. Synth. Catal., 2021, 363, 227–233 CrossRef CAS.
- X.-Y. Yu, W.-J. Xiao and J.-R. Chen, Synthesis of trisubstituted 1,2,4-triazoles from azlactones and aryldiazonium salts by a cycloaddition/decarboxylation cascade, Eur. J. Org. Chem., 2019, 6994–6998 CrossRef CAS.
- M. S. Joshi and F. C. Pigge, Construction of 1,2,4-triazole derivatives via cyclocondensation of alkylidene dihyropyridines and aryldiazonium salts, Org. Lett., 2016, 18, 5916–5919 CrossRef CAS PubMed.
-
(a) D. P. Vala, R. M. Vala and H. M. Patel, Versatile synthetic platform for 1,2,3-triazole chemistry, ACS Omega, 2022, 7, 36945–36987 CrossRef CAS PubMed;
(b) M. J. Vaishnani, S. Bijani, M. Rahamathulla, L. Baldaniya, V. Jain, K. Y. Thajudeen, M. M. Ahmed, S. A. Farhana and I. Pasha, Biological importance and synthesis of 1,2,3-tirazole derivatives: a review, Green Chem. Lett. Rev., 2024, 17, 2307989 CrossRef.
- M.-X. Song and X.-Q. Deng, Recent developments on triazole nucleus in anticonvulsant compounds: a review, J. Enzyme Inhib. Med. Chem., 2018, 33, 453–478 CrossRef CAS PubMed.
- C. Deng, H. Yan, J. Wang, K. Liu, B.-S. Liu and Y.-M. Shi, 1,2,3-Triazole-containing hybrids with potential antibacterial activity against ESKAPE pathogens, Eur. J. Med. Chem., 2022, 244, 114888 CrossRef CAS PubMed.
- D. R. Ferreira, P. C. Alves, A. M. Kirillov, P. Rijo and V. Andre, Silver(I)-tazobactam frameworks with improved antimicrobial activity, Front. Chem., 2022, 9, 1–12 RSC.
- R. Ju, L. Guo, J. Li, L. Zhu, X. Yu, C. Chen, W. Chen, C. Ye and D. Zhang, Carboxyamidotriazole inhibits oxidative phosphorylation in cancer cells and exerts synergistic anti-cancer effect with glycolysis inhibition, Cancer Lett., 2016, 370, 232–241 CrossRef CAS PubMed.
-
(a) R. Maity and B. Sarkar, Chemistry of compounds based on 1,2,3-triazolylidine-type mesoionic carbenes, JACS Au, 2022, 2, 22–57 CrossRef CAS PubMed;
(b) M. Zurro and O. G. Mancheno, 1,2,3-triazole-based catalysts: from metal to supramolecular organic catalysis, Chem. Rec., 2017, 17, 485–498 CrossRef CAS PubMed;
(c) X. Ye, Z. He, T. Ahmed, K. Weise, N. G. Akhmedov, J. L. Petersen and X. Shi, 1,2,3-triazoles as versatile directing group for selective sp2 and sp3 C-H activation: cyclization vs substitution, Chem. Sci., 2013, 4, 3712–3716 RSC.
-
(a) C. Zhu, H. Zeng, F. Chen, C. Liu, R. Zhu, W. Wu and H. Jiang, Copper-catalyzed coupling of oxime acetates and aryldiazonium salts: an azide-free strategy toward N-2-aryl-1,2,3-triazoles, Org. Chem. Front., 2018, 5, 571–576 RSC;
(b) P. S. Gribanov, A. N. Philippova, M. A. Topchiy, L. I. Minaeva, A. F. Asachenko and S. N. Osipov, General method of synthesis of 5-(Het)arylamino-1,2,3-triazoles via Buchwald-Hartwig reaction of 5-amino-or 5-halo-1,2,3-triazoles, Molecules, 2022, 27, 1999 CrossRef CAS PubMed;
(c) P. Wu, Y. He, H. Wang, Y.-G. Zhou and Z. Yu, Copper(II)-catalyzed C-H nitrogenation/annulation cascade of ketene N, S-acetals with aryldiazonium salts: a direct access to N2-substituted triazole and triazine derivatives, Org. Lett., 2020, 22, 310–315 CrossRef CAS PubMed.
- M. Baidya, S. Mallick and S. De Sarkar, Regioselective synthesis of N2-aryl 1,2,3-triazoles via electro-oxidative coupling of enamines and aryldiazonium salts, Org. Lett., 2022, 24, 1274–1279 CrossRef CAS PubMed.
- J. Yua, A. S. Singh, G. Yan, J. Yua and V. K. Tiwari, Recent developments on denitrogenative functionalization of benzotriazoles, Synthesis, 2020, 3781–3800 Search PubMed.
- I. Briguglio, S. Piras, P. Corona, E. Gavini, M. Nieddu, G. Boatto and A. Carta, Benzotriazole: an overview on its versatile biological behaviour, Eur. J. Med. Chem., 2015, 97, 612–648 CrossRef CAS PubMed.
- R. J. Faggyas, N. L. Sloan, N. Buijs and A. Sutherland, Synthesis of structurally diverse benzotriazoles via rapid diazotization and intramolecular cyclization of 1,2-aryldiamines, Eur. J. Org. Chem., 2019, 5344–5353, DOI:10.1002/ejoc.201900463.
- A. Kokel and B. Török, Microwave-assisted solid phase diazotation: a method for the environmentally benign synthesis of benzotriazoles, Green Chem., 2017, 19, 2515–2519 RSC.
-
(a) Y. Zhang, J. Xu, Y. Li, H. Yao and X. Wu, Design, synthesis and pharmacological evaluation of novel NO-releasing benzimidazole hybrids as potential antihypertensive candidate, Chem. Biol. Drug Des., 2015, 85, 541–548 CrossRef CAS PubMed;
(b) A. M. Ganie, A. M. Dar, F. A. Khan and B. A. Dar, Benzimidazole derivatives as potential antimicrobial and antiulcer agents: a mini review, Mini-Rev. Med. Chem., 2019, 19, 1292–1297 CrossRef CAS PubMed;
(c) M. Tonelli, F. Novelli, B. Tasso, I. Vazzana, A. Sparatore, V. Boido, F. Sparatore, P. La Colla, G. Sanna, G. Giliberti, B. Busonera, P. Farci, C. Ibba and R. Loddo, Antiviral activity of benzimidazole derivatives. III. Novel anti-CVB-5, anti-RSV and anti-Sb-1 agents, Bioorg. Med. Chem. Lett., 2014, 22, 4893–4909 CrossRef CAS PubMed;
(d) S. Yadav, B. Narasimhan and H. Kaur, Perspectives of benzimidazole derivatives as anticancer agents in the new era, Anti-Cancer Agents Med. Chem., 2016, 16, 1403–1425 CrossRef CAS PubMed;
(e) E. Guzel, U. A. Cevik, A. E. Evren, H. E. Bostanci, U. D. Gul, U. Kayis, Y. Ozkay and Z. A. Kaplancikli, Synthesis of benzimidazole-1,2,4-triazole derivatives as potential antifungal agents targeting 14α-demethylase, ACS Omega, 2023, 8, 4369–4384 CrossRef CAS PubMed;
(f) O. Ebenezer, F. Oyetunde-Joshua, O. D. Omotoso and M. Shapi, Benzimidazole and its derivatives: recent advances (2020–2022), Results Chem., 2023, 5, 100925 CrossRef CAS.
-
(a) N. T. Chung, V. C. Dung and D. X. Duc, Recent achievements in the synthesis of benzimidazole derivatives, RSC Adv., 2023, 13, 32734–32771 RSC;
(b) S. Venugopal, B. Kaur, A. Verma, P. Wadhwa and S. K. Sahu, A review on modern approaches to benzimidazole synthesis, Curr. Org. Synth., 2023, 20, 595–605 CrossRef CAS PubMed.
- S. W. Youn and E. M. Lee, Metal-free one-pot synthesis of N, N′-diarylamidines and N-arylbenzimidazoles from arenediazonium salts, nitriles, and free anilines, Org. Lett., 2016, 18, 5728–5731 CrossRef CAS PubMed.
- A. F. Darweesh, A. E. M. Mekky, A. A. Salman and A. M. Farag, Synthesis of novel benzimidazole and benzothiazole derivatives, Heterocycles, 2014, 89, 113–125 CrossRef CAS.
- I. A. El Hassani, K. Rouzi, H. Assila, K. Karrouchi and M. Ansar, Recent advances in the synthesis of pyrazole derivatives: A review, Reactions, 2023, 4, 478–504 CrossRef.
- A. Kar, G. Rana, R. Sahoo, S. Ghosh and U. Jana, Design and synthesis of indazole-indole hybrid via tert-butyl nitrite mediated cascade diazotization/isomerization/cyclization, J. Org. Chem., 2024, 89, 7295–7302 CrossRef CAS PubMed.
- V. V. Nikol'skiy, M. E. Minyaev, M. A. Bastrakov and A. M. Starosotnikov, Straight forward and efficient protocol for the synthesis of pyrazolo[4,3-b]pyridines and indazoles, Int. J. Mol. Sci., 2023, 24, 1758 CrossRef PubMed.
- Z. Wróbel, B. Wilk and A. Kwast, Substitution of a nitro group by diazonium salts in σH adducts of carbanions to mono-nitrobenzenes. Formation of substituted azobenzenes and indazoles, Tetrahedron, 2021, 90, 132186 CrossRef.
- C. Zhang, S. Chang, S. Dong, L. Qiu and X. Xu, Acid-promoted bicyclization of diaryl alkynes: synthesis of 2H-indazoles with in situ generated diazonium salt as nitrogen source, J. Org. Chem., 2018, 83, 9125–9136 CrossRef CAS PubMed.
- F. Qiu, Y. Cao, L. Zeng, C. Zhang, L. Fu, J. Zhang, H. Zhu and J. Shao, (2 + 1 + 1 + 1)-annulation reactions of aryldiazonium tetrafluoroborates with sulfur ylides to polysubstituted pyrazoles, J. Org. Chem., 2021, 86, 8997–9006 CrossRef CAS PubMed.
- M. Gümüş, N. Gümüş, H. E. Eroğlu and I. Koca, Design, synthesis and cytotoxic activities of pyrazole-perimidine hybrids, ChemistrySelect, 2020, 5, 5916–5921 CrossRef.
- G.-B. Deng, H.-B. Li, X.-H. Yang, R.-J. Song, M. Hu and J.-H. Li, Dehydrogenative [2 + 2 + 1] heteroannulation using a methyl group as a one-carbon unit: Access to pyrazolo[3,4-c]quinolines, Org. Lett., 2016, 18, 2012–2015 CrossRef CAS PubMed.
- S.-Q. Qin, L.-C. Li, J.-R. Song, H.-Y. Li and D.-P. Li, Structurally simple phenanthridine analogues based on nitidine and their antitumor activity, Molecules, 2019, 24, 437 CrossRef PubMed.
- J. D. Monroe, S. A. Moolani, E. N. Irihamye, K. E. Lett, M. D. Hebert, Y. Gibert and M. E. Smith, Cisplatin and phenanthriplatin modulate long-noncoding RNA expression in A549 and IMR90 cells revealing regulation of microRNAs, Wnt/β-catenin and TGF-β signaling, Sci. Rep., 2021, 11, 10408, DOI:10.1038/s41598-021-89911-z.
-
(a) V. Talukdar, A. Vijayan, N. K. Katari, K. V. Radhakrishnan and P. Das, Recent trends in the synthesis and mechanistic implications of phenanthridines, Adv. Synth. Catal., 2021, 363, 1202–1245 CrossRef CAS;
(b) A. Dey, V. Kumar, R. Chatterjee, A. Behera, R. K. Maurya, A. G. Burra, S. Kumar, M. Katravath and R. Dandella, Recent advancements in synthesis of phenanthridine via 2-isocyanobiphenyls, Asian J. Org. Chem., 2023, 12, e202300369, DOI:10.1002/ajoc.202300369.
- Z. Xia, J. Huang, Y. He, J. Zhao, J. Lei and Q. Zhu, Arylative cyclization of 2-isocyanobiphenyls with anilines: one-pot synthesis of 6-arylphenanthridines via competitive reaction pathways, Org. Lett., 2014, 16, 2546–2549 CrossRef CAS PubMed.
- M. Ramanathan and S.-T. Liu, Preparation of substituted phenanthridines from the coupling of aryldiazonium salts with nitriles: A metal free approach, J. Org. Chem., 2015, 80, 5329–5336 CrossRef CAS PubMed.
- F. Chen, L.-X. Quan, A. Zhou, C. Ji, Y. Li, X. Zhu, L.-L. Mao and J.-P. Wan, Tandem sulfonylation/cyclization of vinyl azides with aryldiazonium tetrafluoroborates and SO2, Eur. J. Org. Chem., 2023, e202201269, DOI:10.1002/ejoc.202201269.
- B. K. Malviya, K. Singh, P. K. Jaiswal, M. Karnatak, V. P. Verma, S. S. Badara and S. Sharma, Catalyst-free synthesis of phenanthridines via electrochemical coupling of 2-isocyanobiphenyls and amines, New J. Chem., 2021, 45, 6367–6378 RSC.
- X. Wang, Y. Li, G. Qiu and J. Wu, Synthesis of 6-(sulfonylmethyl)phenanthridines through a reaction of aryldiazonium tetrafluoroborates, sulfur dioxide, and vinyl azides, Org. Chem. Front., 2018, 5, 2555–2559 RSC.
- S. Song, Z. Zhang, M. Peng, X. Xia, S. Dong, Y. Wang and F. Yu, Selective synthesis of pyridazine-fused chromones and 3-pyridazinyl chromones through intermolecular chromone annulation of o-hydroxyphenyl enaminones with aryldiazonium salts, Org. Chem. Front., 2024, 11, 3906–3912 RSC.
- S. Gradl, J. Köckenberger, J. Oppl, M. Schiller and M. R. Heinrich, Synthetic route to phenyl diazenes and pyridazinium salts from phenylazosulfonates, J. Org. Chem., 2021, 86, 6228–6238 CrossRef CAS PubMed.
- W. Wu, X. Han and Z. Weng, Synthesis of pertrifluoromethyl pyridazine derivatives via tandem reaction of aryldiazonium salts with hexafluoroacetylacetone, Org. Chem. Front., 2020, 7, 3499–3504 RSC.
- O. El Bakouri, D. Cassú, M. Solá, T. Parella, A. Pla-Quintana and A. Roglans, A new mild synthetic route to N-arylated pyridazinones from aryldiazonium salts, Chem. Commun., 2014, 50, 8073–8076 RSC.
- Y. Liu, X. Qi, W. Zhang, P. Yin, Z. Cai and Q. Zhang, Construction of bicyclic 1,2,3-triazine N-oxides from aminocyanides, Org. Lett., 2021, 23, 734–738 CrossRef CAS PubMed.
- E. V. Sadchikova, D. L. Alexeeva and V. G. Nenajdenko, Trifluoroacetyl substituted pyrazolotriazines: synthesis and pathways of formation, Mendeleev Commun., 2020, 30, 180–182 CrossRef CAS.
- Y. Yan, H. Li, B. Niu, C. Zhu, T. Chen and Y. Liu, Mild and efficient TBAI-catalyzed synthesis of 1,2,3-benzotriazine-4-(3H)-ones from tert-butyl nitrite and 2-aminobenzamides under acid-free conditions, Tetrahedron Lett., 2016, 57, 4170–4173 CrossRef CAS.
- B. Parrino, A. Carbone, M. Muscarella, V. Spano, A. Montalbano, P. Barraja, A. Salvador, D. Vedaldi, G. Cirrincione and P. Diana, 11H-Pyrido[3′,2′:4,5]pyrrolo[3,2-c]cinnoline and Pyrido[3′,2′:4,5]pyrrolo[1,2-c,][1,2,3]benzotriazine: Two New Ring Systems with Antitumor Activity, J. Med. Chem., 2014, 57, 9495–9511 CrossRef CAS PubMed.
- I. V. Ledenyova, V. V. Didenko, V. V. Dotsenko and K. S. Shikhaliev, Azo-coupling of pyrazole-3(5)-diazonium chlorides with cyanothioacetamide: a convenient synthesis of pyrazolo[5,1-c,][1,2,4]triazine-3-carbothioamides, Tetrahedron Lett., 2014, 55, 1239–1242 CrossRef CAS.
- V. Alagarsamy, K. Chitra, G. Saravanan, V. Raja Solomon, M. T. Sulthana and B. Narendhar, An overview of quinazolines: Pharmacological significance and recent developments, Eur. J. Med. Chem., 2018, 151, 628–685 CrossRef CAS PubMed.
- G. N. Lipunova, E. V. Nosova, V. N. Charushin and O. N. Chupakhin, Functionalized quinazolines and pyrimidines for optoelectronic materials, Curr. Org. Synth., 2018, 15, 793–814 CrossRef CAS.
-
(a) D. S. Deshmukh and B. M. Bhanage, Molecular Iodine Catalysed Benzylic sp3 C-H Bond Amination for the Synthesis of 2-Arylquinazolines from 2-Aminobenzaldehydes, 2-Aminobenzophenones and 2-Aminobenzyl Alcohols, Synlett, 2018, 979–985 CAS;
(b) S. Hati and S. Sen, Synthesis of Quinazolines and Dihydroquinazolines: o-Iodoxybenzoic Acid Mediated Tandem Reaction of o-Aminobenzylamine with Aldehydes, Synthesis, 2016, 1389–1398 CAS;
(c) C. Wang, S. Li, H. Liu, Y. Jiang and H. Fu, Copper-catalyzed synthesis of quinazoline derivatives via Ullmann-type coupling and aerobic oxidation, J. Org. Chem., 2010, 75, 7936–7938 CrossRef CAS PubMed;
(d) C. M. Cao, J. Sheng and C. Chen, Cu-Catalyzed Cascade Annulation of Diaryliodonium Salts and Nitriles: Synthesis of Nitrogen-Containing Heterocycles, Synthesis, 2017, 5081–5092 CAS.
- M. Ramanathan, J. Wan and S.-T. Liu, Preparation of N-arylquinazolinium salts via a cascade approach, J. Org. Chem., 2019, 84, 7459–7467 CrossRef CAS PubMed.
- M. Ramanathan and S.-T. Liu, Preparation of quinazolines via 2 + 2 + 2 annulation from aryldiazonium salts and nitriles, J. Org. Chem., 2017, 82, 8290–8295 CrossRef CAS PubMed.
-
(a) A. M. Alsibaee, H. M. Al-Yousef and H. S. Al-Salem, Quinazolinones, the winning horse in drug discovery, Molecules, 2023, 28, 978 CrossRef CAS PubMed;
(b) E. Jafari, M. R. Khajouei, F. Hassanzadeh, G. H. Hakimelahi and G. A. Khodarahmi, Quinazolinone and quinazoline derivatives: recent structures with potent antimicrobial and cytotoxic activities, Res. Pharm. Sci., 2016, 11, 1–14 Search PubMed.
- B. Borah, S. Swain, M. Patat and L. R. Chowhan, Recent advances and prospects in the organocatalytic synthesis of quinazolinones, Front. Chem., 2022, 10, 1–26, DOI:10.3389/fchem.2022.991026.
- M. Ramanathan, M.-T. Hsu and S.-T. Liu, Preparation of 4(3H)-quinazolinones from aryldiazonium salt, nitriles and 2-aminobenzoate via a cascade annulation, Tetrahedron, 2019, 75, 791–796 CrossRef CAS.
- M. Ramanathan and S.-T. Liu, Preparation of quinaolinoquinazolinones via a cascade approach, J. Org. Chem., 2018, 83, 14138–14145 CrossRef CAS PubMed.
-
(a) B. S. Kuarm, Y. T. Reddy, J. V. Madhav, P. A. Crooks and B. Rajitha, 3-[Benzimidazo- and 3-[benzothiadiazoleimidazo-(1,2-c)quinazolin-5-yl]-2H-chromene-2-ones as potent antimicrobial agents, Bioorg. Med. Chem. Lett., 2011, 21, 524–527 CrossRef CAS PubMed;
(b) V. Alagarsamy, Antibacterial and antifungal activities of some novel 2,6-disubstituted quinazolin-4(3H)-ones, Indian J. Pharm. Sci., 2002, 64, 600–603 CAS;
(c) P. B. Reddy, S. M. Reddy, K. L. Reddy and P. Lingaiah, Indian Phytopathol., 1985, 38, 361–364 CAS.
- X. Pang, C. Chen, X. Su, M. Li and L. Wen, Diverse Tandem Cyclization Reactions of o-Cyanoanilines and Diaryliodonium Salts with Copper Catalyst for the Construction of Quinazolinimine and Acridine Scaffolds, Org. Lett., 2014, 16, 6228–6231 CrossRef CAS PubMed.
-
(a) S. I. Mirallai, M. J. Manos and P. A. Koutentis, One-step conversion of 2-amino-N’-arylbenzamidines into 3-aryl-4-imino-3,4-dihydroquinazoline-2-carbonitriles using 4,5-dichloro-1,2,3-dithiazolium chloride, J. Org. Chem., 2013, 78, 9906–9913 CrossRef CAS PubMed;
(b) V. K. Naganaboina, K. L. Chandra, J. Desper and S. Rayat, An easy entry into 2-Halo-3-aryl-4(3H)quinazoliniminium Halides from Heteroenyne-Allenes, Org. Lett., 2011, 13, 3718–3721 CrossRef CAS PubMed;
(c) C. Quintero, M. Valderrama, A. Becerra, C. G. Daniliuc and R. S. Rojas, Synthesis of 4(3H)quinazolinimines by reaction of (E)-N-(aryl)-acetimidoyl or -benzimidoyl chloride with amines, Org. Biomol. Chem., 2015, 13, 6183–6193 RSC;
(d) A. Bodtke and P. Langer, Efficient synthesis of tricyclic quinazolines by one-pot cyclizations of 2-(dichloroisocyanido)benzonitrile, Tetrahedron Lett., 2004, 45, 8741–8743 CrossRef CAS.
- M. Ramanathan, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Synthesis of substituted quinazolin-4(3H)-imines from aryldiazonium salts, nitriles, and 2-cyanoanilines via a metal-free tandem approach, Org. Lett., 2017, 19, 5840–5843 CrossRef CAS PubMed.
- B. S. Matada, R. Pattanashettar and N. G. Yernale, A comprehensive review on the biological interest of quinoline and its derivatives, Bioorg. Med. Chem., 2021, 32, 115973 CrossRef CAS PubMed.
- R. Sharma, P. Kour and A. Kumar, A review on transition-metal mediated synthesis of quinolines, J. Chem. Sci., 2018, 130, 73, DOI:10.1007/s12039-018-1466-8.
- O. F. Elebiju, O. O. Ajani, G. O. Oduselu, T. A. Ogunnupebi and E. Adebiyi, Recent advances in functionalized quinoline scaffolds and hybrids—Exceptional pharmacophore in therapeutic medicine, Front. Chem., 2022, 10, 1074331 CrossRef CAS PubMed.
- Z.-G. Le, M. Liang, Z.-S. Chen, S.-H. Zhang and Z.-B. Xie, Ionic liquid as an efficient medium for the synthesis of quinoline derivatives via α-chymotrypsin-catalysed Friedlander condensation, Molecules, 2017, 22, 762 CrossRef PubMed.
- D. S. Barak, D. J. Dahatonde and S. Batra, Metal– and photoredox–catalyst free unified approach for the synthesis of azole–fused quinolines via tert–butyl nitrite–mediated regioselective annulation, Asian J. Org. Chem., 2022, 11, 331–338 CrossRef.
- A. Mathuri, M. Pramanik and P. Mal, 3-Arylsulfonylquinolines from N-Propargylamines via Cascaded Oxidative Sulfonylation Using DABSO, J. Org. Chem., 2022, 87, 6812–6823 CrossRef CAS PubMed.
- M. Ramanathan, J. Wan, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Synthesis of 2-arylamino-3-cyanoquinolines via a cascade reaction through nitrilium intermediate, Org. Biomol. Chem., 2020, 18, 975–982 RSC.
- S. W. Youn, H. J. Yoo, E. M. Lee and S. Y. Lee, Metal-Free One-Pot Synthesis of (Tetrahydro)Quinolines through Three-Component Assembly of Arenediazonium Salts, Nitriles, and Styrenes, Adv. Synth. Catal., 2018, 360, 278–283 CrossRef CAS.
- M. Ramanathan, J. Wan and S.-T. Liu, Preparation of 3-hydroxyquinolines from direct oxidation of dihydroquinolinium salts, RSC Adv., 2018, 8, 38166–38174 RSC.
- S. Yaragorla, A. Pareek, R. Dada and P. L. Saini, Electrophilic cyclization of 2-aminophenylprop-1-yn-3-ols into 3-iodo-6-(aryldiazenyl) quinolines by using a one-pot azo-coupling and iodocyclization sequence, Eur. J. Org. Chem., 2018, 1863–1871 CrossRef CAS.
- H. Wang, Q. Xu, S. Shen and S. Yu, Synthesis of Quinolines through Three-Component Cascade Annulation of Aryl Diazonium Salts, Nitriles, and Alkynes, J. Org. Chem., 2017, 82, 770–775 CrossRef CAS PubMed.
- M. Ramanathan and S.-T. Liu, Cascade annulations of aryldiazonium salts, nitriles and halo-alkynes leading to 3-haloquinolines, Tetrahedron, 2017, 73, 4317–4322 CrossRef CAS.
- R. Pawlowski, F. Stanek and M. Stodulski, Recent advances on metal-free, visible-light- induced catalysis for assembling nitrogen- and oxygen-based heterocyclic scaffolds, Molecules, 2019, 24, 1533 CrossRef PubMed.
- G. C. Upreti, T. Singh, S. Ranjan, R. K. Gupta and A. Singh, Visible light mediated three component cascade sulfonylative annulation, ACS Omega, 2022, 7, 29728–29733 CrossRef CAS PubMed.
- J. Zhang, Z. Yang, J.-T. Yu and C. Pan, Three-component synthesis of Arylsulfonyl-substituted indolo[2,1-a]isoquinolinones and benzimidazo-[2,1-a]isoquinolin-6(5H)-ones by SO2 insertion and radical cascade cyclization, Org. Biomol. Chem., 2022, 20, 3067–3071 RSC.
- B. Zhao, G. B. Hammond and B. Xu, Aromatic ketone-catalyzed photochemical synthesis of imidazo-isoquinolinone derivatives, J. Org. Chem., 2021, 86, 12851–12861 CrossRef CAS PubMed.
- C. Jin, L. Su, D. Ma and M. Cheng, Transition-metal-free, visible-light-mediated cyclization of o-azidoarylalkynes with aryl diazonium salts, New J. Chem., 2017, 41, 14053–14056 RSC.
- Z. Gonda, F. Beke, O. Tischler, M. Petro, Z. Novak and B. L. Toth, Erythrosine B Catalyzed Visible-Light Photoredox Arylation–Cyclization of N-Alkyl-N-aryl-2-(trifluoromethyl)acrylamides to 3-(Trifluoromethyl)indolin-2-one Derivatives, Eur. J. Org. Chem., 2017, 2112–2117, DOI:10.1002/ejoc.201601493.
- P. Kumar, A. Rai, M. Kumar, G. R. Krishna and U. Das, Radical cascade cyclization of unactivated alkene-tethered indoles with aryldiazonium salt and sodium metabisulfite to access azo-and sulfonylated-2,3-dihydro-1H-pyrrolo[1,2-a]indoles, J. Org. Chem., 2023, 88, 9123–9129 CrossRef CAS PubMed.
- X. Wang, F. You, B. Xiong, L. Chen, X. Zhang and Z. Lian, Metal- and base-free tandem sulfonylation/cyclization of 1,5-dienes with aryldiazonium salts via the insertion of sulfur dioxide, RSC Adv., 2022, 12, 16745–16750 RSC.
- J. Li, W.-W. Zhang, X.-J. Wei, F. Liu, W.-J. Hao, S.-L. Wang, G. Li, S.-J. Tu and B. Jiang, Radical deaminative ipso-cyclization of 4-methoxyanilines with 1,7-enynes for accessing spirocyclohexadienone-containing cyclopenta[c]quinolin-4-ones, J. Org. Chem., 2017, 82, 6621–6628 CrossRef CAS PubMed.
- J. Xuan, C. G. Daniliuc and A. Studer, Construction of polycyclic γ-lactams and related heterocycles via electron catalysis, Org. Lett., 2016, 18, 6372–6375 CrossRef CAS PubMed.
-
(a) J. Xuan, D. G. Abradelo, C. A. Strassert, C.-G. Daniliuc and A. Studer, Radical cascade cyclization: Reaction of 1,6-enynes with aryl radicals by electron catalysis, Eur. J. Org. Chem., 2016, 4961–4964 CrossRef CAS;
(b) J. W. Tucker, J. D. Nguyen, M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Tin-free radical cyclization reactions initiated by visible light photoredox catalysis, Chem. Commun., 2010, 46, 4985–4987 RSC;
(c) G.-B. Deng, Z.-Q. Wang, J.-D. Xia, P.-C. Qian, R.-J. Song, M. Hu, L.-B. Gong and J.-H. Li, Tandem cyclizations of 1,6-enynes with Arylsulfonyl chlorides by using visible-light photoredox catalysis, Angew. Chem., Int. Ed., 2013, 52, 1535–1538 CrossRef CAS PubMed;
(d) Y. Li, B. Liu, R.-J. Song, Q.-A. Wang and J.-H. Li, Visible light-initiated C(sp3)-Br/C(sp3)-H functionalization of α-carbonyl alkyl bromides through hydride radical shift, Adv. Synth. Catal., 2016, 358, 1219–1228 CrossRef CAS.
-
(a) M. S. Hashtroudi, V. F. Vavsari and S. Balalaie, DABSO as a SO2 gas surrogate in the synthesis of organic structures, Org. Biomol. Chem., 2022, 20, 2149–2163 RSC;
(b) J. A. Andrews and M. C. Willis, DABSO-A reagent to revolutionize organosulfur chemistry, Synthesis, 2022, 1695–1707 CAS.
-
(a) F. Liu, J. Huang, X. Wu, F. Du, L. Zeng, J. Wu and Z. Chen, Regioselective radical-relay sulfonylation/cyclization protocol to sulfonylated pyrrolidones under transition-metal-free conditions, J. Org. Chem., 2022, 87, 6137–6145 CrossRef CAS PubMed;
(b) W. Rao, L. Jiang, X. Liu, M. Chen, F. Chen, X. Jiang, J. Zhao, G. Zou, Y. Zhou and L. Tang, Copper(II)-catalysed alkene aminosulfonylation with sodium sulfinates for the synthesis of sulfonylated pyrrolidones, Org. Lett., 2019, 21, 2890–2893 CrossRef CAS PubMed;
(c) R. Ding, Y. Liu, H. Hao, C. Chen, L. Liu, N. Chen, Y. Guo and P. Wang, Regioselective, Copper(I)-catalysed tandem sulfonylation-cyclization of 1,5-dienes with sulfonyl chlorides, Org. Chem. Front., 2021, 8, 3123–3127 RSC.
- Z. Chen, H. Zhang, S. F. Zhou and X. Cui, Metal-free sulfonylative spirocyclization of indolyl-ynones via insertion of sulfur dioxide: Access to sulfonated spiro[cyclopentenone-1,3′-indoles], Org. Lett., 2021, 23, 7992–7995 CrossRef CAS PubMed.
- A. M. Nair, I. Halder, S. Khan and C. M. R. Volla, Metal free sulfonylative spirocyclization of alkenyl and alkynyl amides via insertion of sulfur dioxide, Adv. Synth. Catal., 2020, 362, 224–229 CrossRef CAS.
- H. Chen, M. Liu, G. Qiu and J. Wu, A three-component reaction of aryldiazonium tetrafluoroborates, sulfur dioxide, and 1- (prop-2-yn-1-yl)indoles under catalyst-free conditions, Adv. Synth. Catal., 2019, 361, 146–150 CrossRef CAS.
-
(a) E. L. S. De Souza and C. C. Oliveira, Selective radical transformations with aryldiazonium salts, Eur. J. Org. Chem., 2023, 26, e202300073 CrossRef CAS;
(b) P. Das, S. Das and R. Jana, Aryldiazonium salts and DABSO: A versatile combination for three-component sulfonylative cross-coupling reactions, Chem. – Asian J., 2022, 17, e202200085 CrossRef CAS PubMed;
(c) D. Kundu, Synthetic strategies for aryl/heterocyclic selenides and tellurides under transition-metal-catalyst free conditions, RSC Adv., 2021, 11, 6682–6698 RSC;
(d) X. Zhang, Y. Mei, Y. Li, J. Hu, D. Huang and Y. Bi, Visible-light-mediated functionalization of aryldiazonium salts, Asian J. Org. Chem., 2021, 10, 453–463 CrossRef CAS.
- Y. Liu, X. Gu, X. Zhang, M. Xu, Z. Zhang and T. Liang, Iodine-mediated oxidative triple functionalization of indolines with azoles and diazonium salts, Chem. Commun., 2024, 60, 4613–4616 RSC.
- T.-B. Zhang, F. Wang, J.-Y. Ouyang, Z.-W. Luo, J.-H. Qin, J.-H. Li and X.-H. Ouyang, Aryldiazonium-salt-triggered carboxylative azotization of pyrroles or indoles with polyalomethanes via halogen-atom transfer (XAT), Org. Lett., 2024, 26, 461–466 CrossRef CAS PubMed.
- V. L. Bondarev, A. A. Festa, O. A. Storozhenko, N. E. Golantsov, V. Pappula, A. G. Tskhoverebov, A. V. Varlamov and L. G. Voskressensky, Azo coupling of indoles revisited: Synthesis of biindolyl photoswitches via the azo-coupling/C-H functionalization domino approach, J. Org. Chem., 2023, 88, 12949–12957 CrossRef CAS PubMed.
- K.-H. Wang, C. Bian, X. Liang, W. Luo, J. Wang, D. Huang and Y. Hu, Synthesis of trifluoromethyl formazans by reaction of trifluoromethyl N-acylhydrazones/N-aryl hydrazones and aryldiazonium tetrafluoroborates, J. Fluor. Chem., 2023, 272, 110211 CrossRef CAS.
- J. Zhang, M. Jiang, C.-S. Wang, K. Guo, Q.-X. Li, C. Ma, S.-F. Ni, G.-Q. Chen, Y. Zong, H. Lu, L.-W. Xu and X. Shao, Transition-metal free C-N bond formation from alkyl iodides and diazonium salts via halogen-atom transfer, Nat. Commun., 2022, 13, 7961 CrossRef CAS PubMed.
- J. Shen, J. Xu, L. He, Y. Ouyang, L. Huang, W. Li, Q. Zhu and P. Zhang, Photoinduced rapid multicomponent cascade reaction of aryldiazonium salts with unactivated alkenes and TMSN3, Org. Lett., 2021, 23, 1204–1208 CrossRef CAS PubMed.
- S. Saba, C. R. Dos Santos, B. R. Zavarise, A. A. S. Naujorks, M. S. Franco, A. R. Schneider, M. R. Scheide, R. F. Affeldt, J. Rafique and A. L. Braga, Photo-induced direct C(sp2)-H bond azo coupling of imidazo-heteroarenes and anilines with aryldiazonium salts catalysed by Eosin Y, Chem. – Eur. J., 2020, 26, 4461–4466 CrossRef CAS PubMed.
- S. Kindt, K. Wicht and M. R. Heinrich, Base-induced radical carboamination of nonactivated alkenes with aryldiazonium salts, Org. Lett., 2015, 17, 6122–6125 CrossRef CAS PubMed.
- M. Ramanathan, Y.-H. Wang, Y.-H. Liu, S.-M. Peng, Y.-C. Cheng and S.-T. Liu, Preparation of ketimines from aryldiazonium salts, arenes, and nitriles via intermolecular arylation of N-arylnitrilium ions, J. Org. Chem., 2018, 83, 6133–6141 CrossRef CAS PubMed.
- Y. Zhang and Z. Chen, Three component reaction of aryl diazonium salt with sulfonamide and acetonitrile to synthesize N-sulfonyl amidine, Tetrahedron, 2018, 59, 4183–4186 CrossRef CAS.
-
(a) J. M. Sonego, S. I. De Diego, S. H. Szajnman, C. G. Rodriguez and J. B. Rodriguez, Organoselenium compounds: Chemistry and applications in organic synthesis, Chem. – Eur. J., 2023, 29, e202300030 CrossRef CAS PubMed;
(b) M. V. Musalov, V. A. Potapov, M. V. Musalova, S. V. Amosova and L. B. Krivdin, Recent Developments in the Synthesis of Organoselenium Compounds Based on the Reactions of Organic Diselenides with Acetylenes, Catalysts, 2023, 13, 1369, DOI:10.3390/catal13101369.
- R. Mamgain, M. Kostic and F. V. Singh, Synthesis and antioxidant properties of organoselenium compounds, Curr. Med. Chem., 2023, 30, 2421–2448 CrossRef CAS PubMed.
- K. Lu, Q. Li, X. Xi, T. Zhou and X. Zhao, Metal-free difluoromethylselenolation of arylamines under visible-light photocatalysis, J. Org. Chem., 2020, 85, 1224–1231 CrossRef CAS PubMed.
- Y.-C. Shieh, K. Du, R. S. Basha, Y.-J. Xue, B.-H. Shih, L. Li and C.-F. Lee, Synthesis of thioethers and selenide ethers from anilines, J. Org. Chem., 2019, 84, 6223–6231 CrossRef CAS PubMed.
- T. Dong, J. He, Z.-H. Li and C.-P. Zhang, Catalyst-and additive-free trifluoromethylselenolation with [Me4N][SeCF3], ACS Sustainable Chem. Eng., 2018, 6, 1327–1335 CrossRef CAS.
- L. Chen, M. Zhou, L. Shen, X. He, X. Li, X. Zhang and Z. Lian, Metal-and base-free C(sp2)-arylsulfonylation of enamides for synthesis of (E)-amidovinyl sulfones via the insertion of sulfur dioxide, Org. Lett., 2021, 23, 4991–4996 CrossRef CAS PubMed.
- P. Das, S. Das, K. Varalaxmi and R. Jana, Metal-free, multicomponent anti-Markovnikov hydroarylsulfonylation and alkoxyarylsulfonylation of vinyl arenes, Adv. Synth. Catal., 2021, 363, 575–584 CrossRef CAS.
- M. Kumar, R. Ahmed, M. Singh, S. Sharma, T. Thatikonda and P. P. Singh, Functionalization of alkynes and alkenes using a cascade reaction approach: Synthesis of β-keto sulfones under metal-free conditions, J. Org. Chem., 2020, 85, 716–725 CrossRef CAS PubMed.
- T. Zhong, M.-K. Pang, Z.-D. Chen, B. Zhang, J. Weng and G. Lu, Copper-free Sandmeyer-type reaction for the synthesis of sulfonyl fluorides, Org. Lett., 2020, 22, 3072–3078 CrossRef CAS PubMed.
- G. You, D. Xi, J. Sun, L. Hao and C. Xia, Transition-metal- and oxidant-free three-component reaction of quinoline-N-oxides, sodium metabisulfite and aryldiazonium tetrafluoroborates via a dual radical coupling process, Org. Biomol. Chem., 2019, 17, 9479–9488 RSC.
- X. Liu, W. Li, D. Zheng, X. Fan and J. Wu, Synthesis of sulfones via a reaction of aryldiazaonium tetrafluoroborates, sulfur dioxide, and aryliodoniums, Tetrahedron, 2015, 71, 3359–3362 CrossRef CAS.
- T. Liu, D. Zheng, Z. Li and J. Wu, Route to O-aminosulfonates and sulfonamides through insertion of sulfur dioxide and hydrogen atom transfer, Adv. Synth. Catal., 2017, 359, 2653–2659 CrossRef CAS.
|
| This journal is © the Partner Organisations 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab and
Ziad
Moussa
*ab and
Ziad
Moussa