
Nickel-catalyzed reductive 1,2-alkylarylation of alkenes via a 1,5-hydrogen atom transfer (HAT) cascade†
Xi
Chen
a,
Qiang
Wang
b,
Xiao-Ping
Gong
a,
Rui-Qiang
Jiao
a,
Xue-Yuan
Liu
 *a and
Yong-Min
Liang
*a
*a and
Yong-Min
Liang
*a
aState Key Laboratory of Applied Organic Chemistry, School of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: liuxuey@lzu.edu.cn; liangym@lzu.edu.cn
bGuangdong Provincial Key Laboratory of Distributed Energy Systems, College of Chemical Engineering and Energy Technology, Dongguan University of Technology, Dongguan 523808, P.R. China
First published on 5th November 2024
Abstract
N-centered radical-mediated remote C(sp3)–H functionalization via HAT processes has been successfully applied in the difunctionalization of alkenes, serving as an elegant and robust method to convert readily available alkenes into various functionalized molecules. However, HAT strategy-enabled difunctionalization of alkenes using electrophiles as functionalizing reagents remains underexplored. In this study, we report a nickel-catalyzed regioselective reductive three-component 1,2-alkylarylation of alkenes with O-oxalate hydroxamic acid esters and aryl iodides. This radical addition/cross-coupling cascade reaction involves amidyl radical-triggered intramolecular 1,5-HAT and nickel-catalyzed reductive coupling processes under mild reaction conditions with good coupling efficiency. Additionally, this approach can be extended to the reductive 1,2-alkylarylation of alkynes, providing an efficient method for the synthesis of multi-substituted alkenes from easily accessible starting materials.
Introduction
Alkenes are among the most valuable and readily available building blocks and they have been extensively applied in organic synthesis, medicinal chemistry, and materials science.1 Along with the development of transition metal-catalyzed cross-coupling reactions, the difunctionalization of alkenes has proved to be an appealing and efficient strategy for the rapid construction of complex skeletons via the simultaneous formation of two new bonds across the π system.2 Among these methods, nickel-catalyzed reductive difunctionalization stands out, as it directly employs readily available electrophiles as functionalizing agents.3 This approach bypasses the need for unstable organometallics, which require additional synthesis steps and are sensitive to many functional groups, offering advantages in step economy, atom efficiency, and operational simplicity.4 For example, the Nevado group achieved nickel-catalyzed three-component reductive alkylarylation of alkenes using alkyl iodides and aryl iodides as radical precursors and coupling partners, respectively.5 Similarly, the Chu group reported intermolecular reductive carboacylation of alkenes with fluoroalkyl iodides and acyl chlorides as coupling agents.6 Despite significant progress made by these groups and others,7 further developing novel and efficient alkene difunctionalization protocols through nickel-catalyzed radical relay or concerted migratory insertion remains highly desirable.Aliphatic C(sp3)–H bonds are commonly found in nearly all organic substances, including pharmaceuticals, petroleum, and functional materials.8 Consequently, the direct functionalization of these widespread C(sp3)–H bonds for the rapid synthesis of valuable organic compounds has gained significant importance in organic synthesis.9 However, the high similarity in bond dissociation energies and electronic properties among multiple aliphatic C(sp3)–H bonds presents a major challenge for achieving site-specific C(sp3)–H bond functionalization.10 Over the past few years, inspired by the Hofmann–Löffler–Freytag (HLF) reaction,11 nitrogen-centered radical (e.g., amidyl or sulfonamidyl radicals) mediated intramolecular hydrogen atom transfer (HAT) processes emerged as a powerful strategy for the direct transformation of C(sp3)–H bonds at specific positions to various C(sp3)–C and C(sp3)–Y (Y = N, S, P, F, etc.) bonds.12 Mechanistically, a hydrogen atom is transferred from a carbon center to a nitrogen center, forming a highly reactive carbon-centered radical species, which is then trapped by a radical partner to achieve regioselective incorporation of a wide range of external functional groups (Scheme 1a).13 Given the growing interest of chemists in alkene difunctionalization,14 the Li group pioneered a copper-catalyzed fluoroamide-directed remote benzylic C(sp3)–H functionalization enabling the difunctionalization of alkenes with N-fluoro-2-methylbenzeneamides and nucleophiles, including alcohols, indoles, and pyrroles (Scheme 1b).15 Our group has also developed a copper-catalyzed alkylarylation of alkenes via N-centered radical-mediated remote benzylic C(sp3)–H activation using nucleophilic arylboronic acids as coupling partners.16 Nevertheless, HAT strategy-enabled alkene difunctionalization using electrophiles as functionalizing reagents remains underexplored. Building on our ongoing interest in remote C(sp3)–H functionalization and nickel-catalyzed cross-electrophile coupling reactions,17 we report herein a nickel-catalyzed intermolecular reductive 1,2-alkylarylation of alkenes using O-oxalate hydroxamic acid esters and aryl iodides in the presence of Mn (Scheme 1c), demonstrating good coupling efficiency and high regioselectivity.
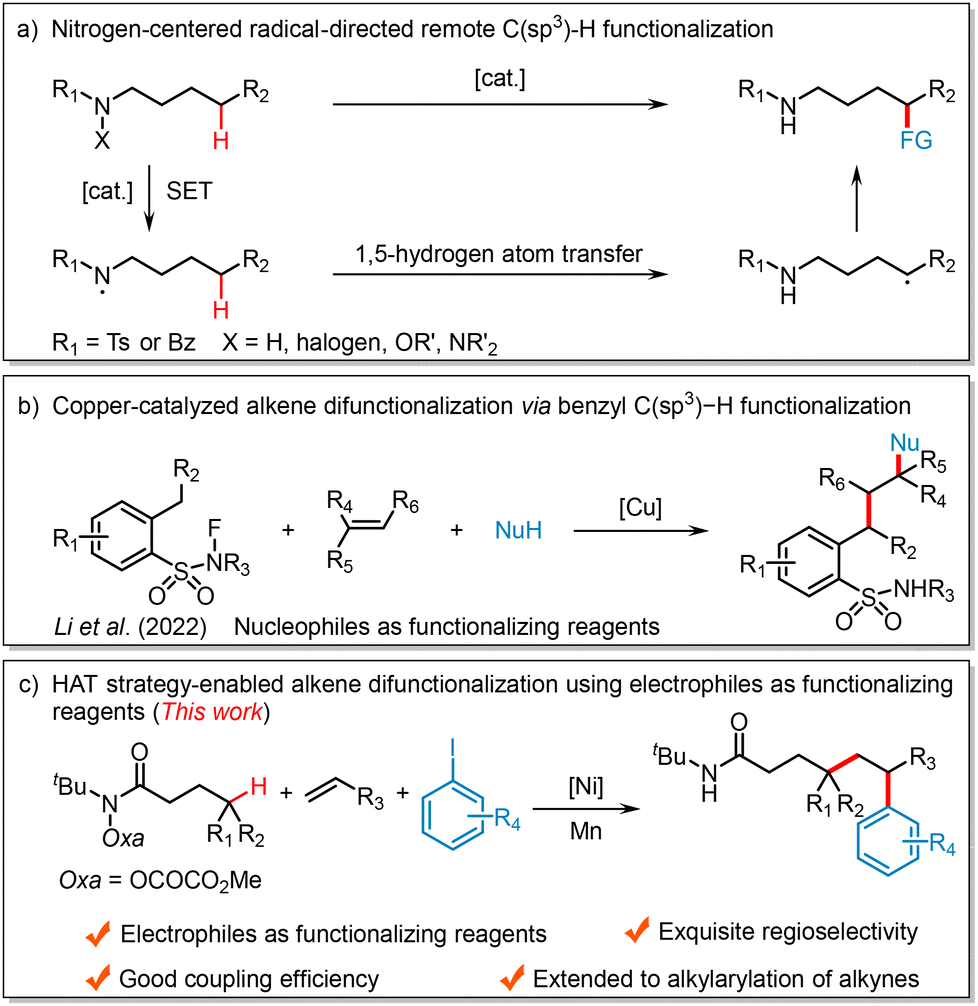 | ||
| Scheme 1 Background and reaction design. | ||
Results and discussion
The investigation began with the selection of O-oxalate hydroxamic acid ester 1a, benzyl acrylate 2a, and 4-iodobenzoic acid methyl ester 3a as model substrates (details provided in the ESI†). Using NiCl2·DME as the catalyst, bipyridine ligand L1, and Mn powder as the reductant, we were pleased to obtain the desired product 4a with a 13% yield (Table 1, entry 1). Given the critical role of solvents in cross-electrophile coupling, we initially tested several solvents (entries 2–5), discovering that a mixture of DMA/THF in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (v/v) was optimal. Screening of Ni salts revealed that the reactions could be catalyzed by both Ni(II) and Ni(0), with NiCl2·DME proving to be the most effective (entries 6 and 7). Further optimization of the reactant ratios increased the yield to 36% (entry 8). We also explored various bidentate N-containing ligands (L1–L7), finding that L2 provided the best efficiency (entries 9 and 10). The yield of 4a could be increased to 78% when using MgBr2 as an additive, which could accelerate the reduction of NiII intermediates or assist in activating the surface of Mn18 (entries 11–13). Control experiments confirmed that the Ni salt, ligand, and reductant are all essential components for the successful transformation (entries 14–16).
:1 ratio (v/v) was optimal. Screening of Ni salts revealed that the reactions could be catalyzed by both Ni(II) and Ni(0), with NiCl2·DME proving to be the most effective (entries 6 and 7). Further optimization of the reactant ratios increased the yield to 36% (entry 8). We also explored various bidentate N-containing ligands (L1–L7), finding that L2 provided the best efficiency (entries 9 and 10). The yield of 4a could be increased to 78% when using MgBr2 as an additive, which could accelerate the reduction of NiII intermediates or assist in activating the surface of Mn18 (entries 11–13). Control experiments confirmed that the Ni salt, ligand, and reductant are all essential components for the successful transformation (entries 14–16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ni cat. | Ligand | Additive | Solvent | Yieldc (%) |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (0.2 mmol), 3a (0.3 mmol, 1.5 equiv.), Ni cat. (10 mol%), ligand (12 mol%), Mn (3.0 equiv.), additive (1.0 equiv.), solvent (1.5 mL), Ar, 30 °C, and 18 h. b Oxa = OCOCO2Me. c Isolated yield. d 1a (0.4 mmol, 2.0 equiv.) was used. e Without Mn. | |||||
| 1 | NiCl2·DME | L1 | THF | 13 | |
| 2 | NiCl2·DME | L1 | DMA | 20 | |
| 3 | NiCl2·DME | L1 | Et2O | 0 | |
| 4 | NiCl2·DME | L1 | 1,4-Dioxane | 0 | |
| 5 | NiCl2·DME | L1 | DMA/THF = 2:1 |
27 | |
| 6 | NiI2 | L1 | DMA/THF = 2:1 |
11 | |
| 7 | Ni(COD)2 | L1 | DMA/THF = 2:1 |
19 | |
| 8d | NiCl2·DME | L1 | DMA/THF = 2:1 |
36 | |
| 9d | NiCl2·DME | L2 | DMA/THF = 2:1 |
45 | |
| 10d | NiCl2·DME | L3–L7 | DMA/THF = 2:1 |
25–44 | |
| 11d | NiCl2·DME | L2 | MgCl2 | DMA/THF = 2:1 |
65 |
| 12d | NiCl2·DME | L2 | LiCl | DMA/THF = 2:1 |
Trace |
| 13 | NiCl 2 ·DME | L2 | MgBr 2 |
DMA/THF = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
78 |
| 14d | L2 | MgBr2 | DMA/THF = 2:1 |
0 | |
| 15d | NiCl2·DME | MgBr2 | DMA/THF = 2:1 |
0 | |
| 16d,e | NiCl2·DME | L2 | MgBr2 | DMA/THF = 2:1 |
0 |
|
|||||
Having established the optimum reaction conditions (Table 1, entry 13), we explored the diversity and practicality of the reaction substrates (Scheme 2). We first examined the scope of electron-deficient alkenes. As expected, acrylates bearing alkyl, alkoxy, and succinimidyl groups were efficiently converted to the desired products (4a–4h) with yields ranging from 31% to 78%. Additionally, the gram-scale reaction of 2a produced 4a in a 62% yield, demonstrating robust scalability. Besides various acrylates, other alkenes, such as acrylamide, acrylonitrile, and vinyl phosphonate, also served as suitable coupling partners, yielding products 4i–4k in moderate yields. Notably, vinylboronic acid pinacol ester was readily incorporated into the reaction protocol, affording the corresponding product 4l in 67% yield, which offers potential for further derivatization through palladium-catalyzed Suzuki–Miyaura cross-coupling reactions. However, the reaction currently faces limitations with styrenes and unactivated alkenes. We subsequently evaluated different structural motifs surrounding the targeted C(sp3)–H bond. Both sterically hindered and oxygen-adjacent C(sp3)–H bonds were successfully functionalized, producing products 5a–5i with isolated yields of 38–66% and excellent regioselectivity. Introducing methyl or ethyl groups into the aliphatic backbone to create sterically hindered tertiary C(sp3)–H bonds did not significantly reduce product yields (5b and 5c). The amino acid-derived hydroxamic ester reacted smoothly, generating the corresponding product 5h in a relatively low yield. The secondary C(sp3)–H bond in the rigid adamantyl substrate can also be functionalized, affording target product 5j with a yield of 47%. The hydroxamic ester 1k containing acyclic secondary carbon reactive sites is also capable of this transformation under standard conditions (5k), but the yield is low and accompanied by the generation of a two-component by-product 5k′, which is probably due to the instability of the secondary alkyl radicals and their high propensity to undergo a direct coupling with aryl iodides. Finally, a series of aryl iodides were examined. Notably, when the substituent was located at the meta position of the phenyl group, the yield of product 6a was similar to that of 4a. However, when the substituent was in the ortho position, the yield of product 6b drastically decreased to 31%, indicating a significant steric hindrance caused by the ortho substituent. A range of electron-deficient substituents were tolerated, yielding the corresponding products 6c–6k in moderate to good yields. The reaction also showed compatibility with multi-substituted aryl iodides, producing products 6l and 6m in 36% and 65% yields, respectively. Compared to electron-deficient aryl iodides, electron-rich aryl iodides, such as OAc-substituted iodobenzene, were more challenging to use as coupling agents, yielding product 6n in lower yields. Additionally, heteroaryl iodides or aryl iodides derived from natural products or drugs were efficiently incorporated into the three-component reaction, as demonstrated by the successful isolation of products 6o–6q with satisfactory yields. Unfortunately, aryl bromides and aryl chlorides turned out to be unsuitable precursors for this reaction (see Table S3 in ESI† page S7 for the investigated substrates).
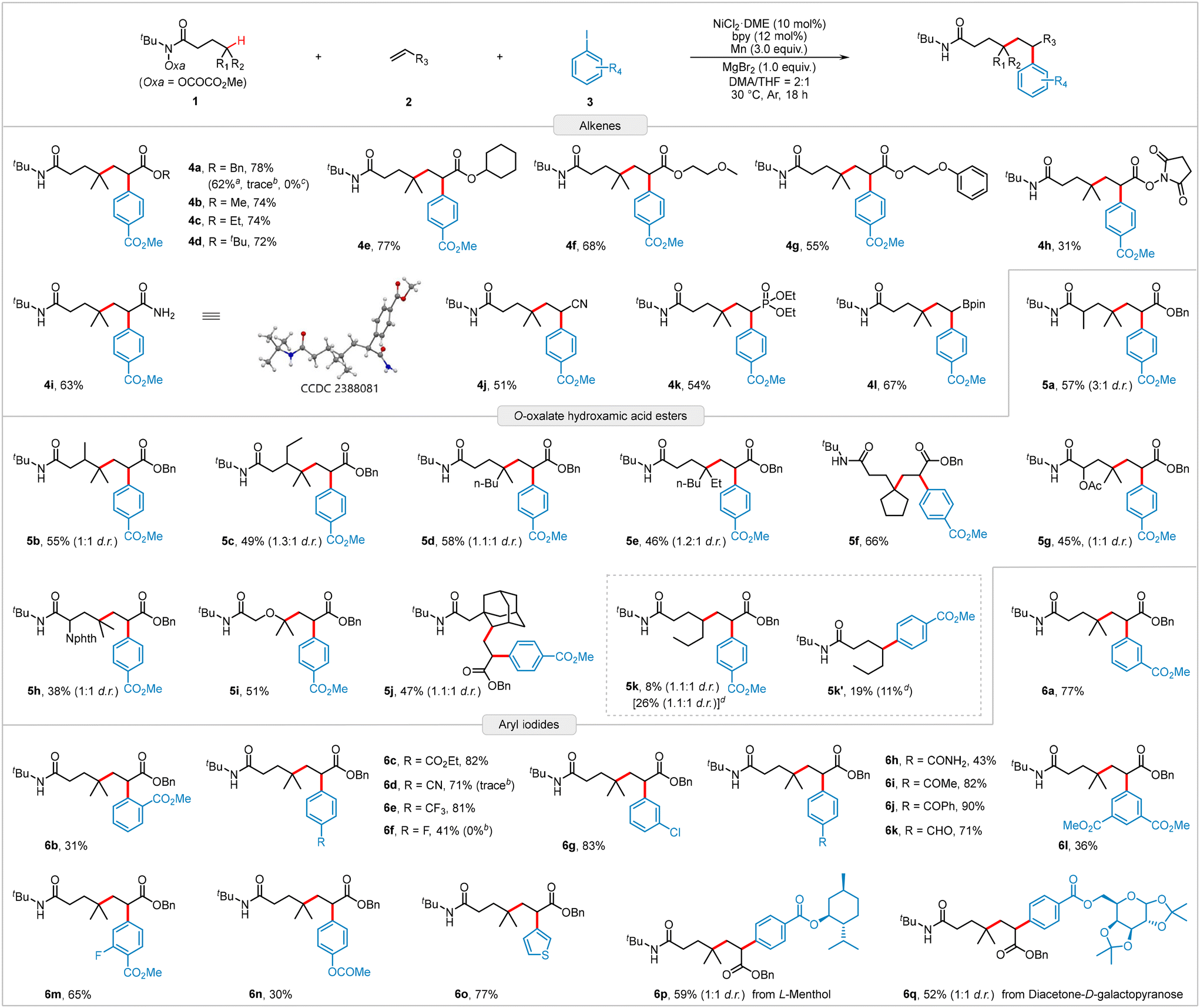 | ||
| Scheme 2 Substrate scope for the alkylarylation of alkenes. Reaction conditions: 1 (0.4 mmol, 2.0 equiv.), 2 (0.2 mmol), 3 (0.3 mmol, 1.5 equiv.), NiCl2·DME (10 mol%), bpy (12 mol%), Mn (3.0 equiv.), MgBr2 (1.0 equiv.), DMA/THF (2/1, v/v, 1.5 mL), Ar, 30 °C, and 18 h. Isolated yields. aThe reaction was carried out on a 3.0 mmol scale and reacted for 36 h. bAryl bromide was used. cAryl chloride was used. dUsing NiCl2 instead of NiCl2·DME to react at 50 °C for 24 h in a ratio of 1k:2a:3a = 1:2:1.5. | ||
Transition metal-catalyzed dicarbofunctionalization of alkynes is a powerful approach for constructing multi-substituted alkenes.19 However, there are only a few examples of nickel-catalyzed intermolecular reductive dicarbofunctionalization of alkynes using two different electrophiles.20 Building on the results described above, we investigated the extension of our methodology to the reductive 1,2-alkylarylation of alkynes. As illustrated in Scheme 3, using hydroxamic ester 1a and 4-iodobenzoic acid methyl ester 3a as model coupling partners, various aryl alkynes with both electron-withdrawing and electron-donating groups were successfully transformed under slightly modified reaction conditions (see Table S2 in ESI† pages S4 and S5 for optimized conditions), yielding the corresponding products 8a–8i in moderate yields. Heteroaryl-substituted alkynes, such as those containing thiophenes, also served as suitable substrates, producing heteroaromatic-containing products (8j and 8k) in 48% and 63% yields, respectively.
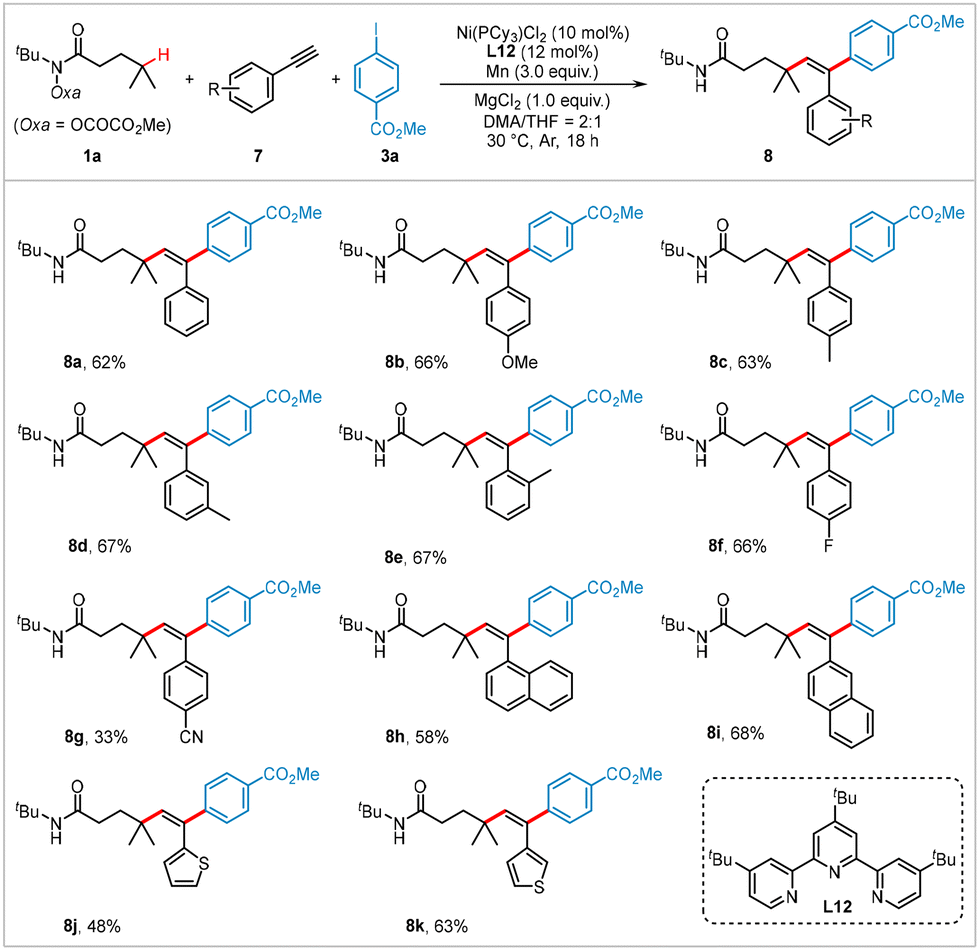 | ||
| Scheme 3 Substrate scope for the alkylarylation of alkynes. Reaction conditions: 1a (0.4 mmol, 2.0 equiv.), 7 (0.2 mmol), 3a (0.3 mmol, 1.5 equiv.), Ni(PCy3)Cl2 (10 mol%), L12 (12 mol%), Mn (3.0 equiv.), MgCl2 (1.0 equiv.), DMA/THF (2/1, v/v, 1.5 mL), Ar, 30 °C, and 18 h. Isolated yields. | ||
We conducted preliminary studies to gain further insights into the mechanism of this reaction. The addition of the radical trapping agent 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) completely suppressed the desired alkylarylation reaction, and the TEMPO adduct was detected via high-resolution mass spectrometry (HR-MS) (Scheme 4a). This result suggests that the alkylarylation reaction proceeds through a radical pathway. The TEMPO adduct was also detected when the reaction of 1a with TEMPO was conducted in the presence of Mn or stoichiometric Ni(COD)2 (Scheme 4b), indicating that both Mn and Ni(0) can reduce 1a. Additionally, we synthesized the Ar–NiII–I complex Ni-121 and evaluated its catalytic efficiency. In the stoichiometric reaction of Ni-1 with hydroxamic ester 1a and acrylate 2a, no detectable amount of 6c was observed. However, when 10 mol% of Ni-1 was used as the catalyst, the desired product 6c was obtained in a 52% yield (Scheme 4c). These results suggest that Ni-1 is unlikely to be a reactive intermediate in this cascade transformation, indicating that oxidative addition of the Ni(0) species to aryl iodide is improbable.22
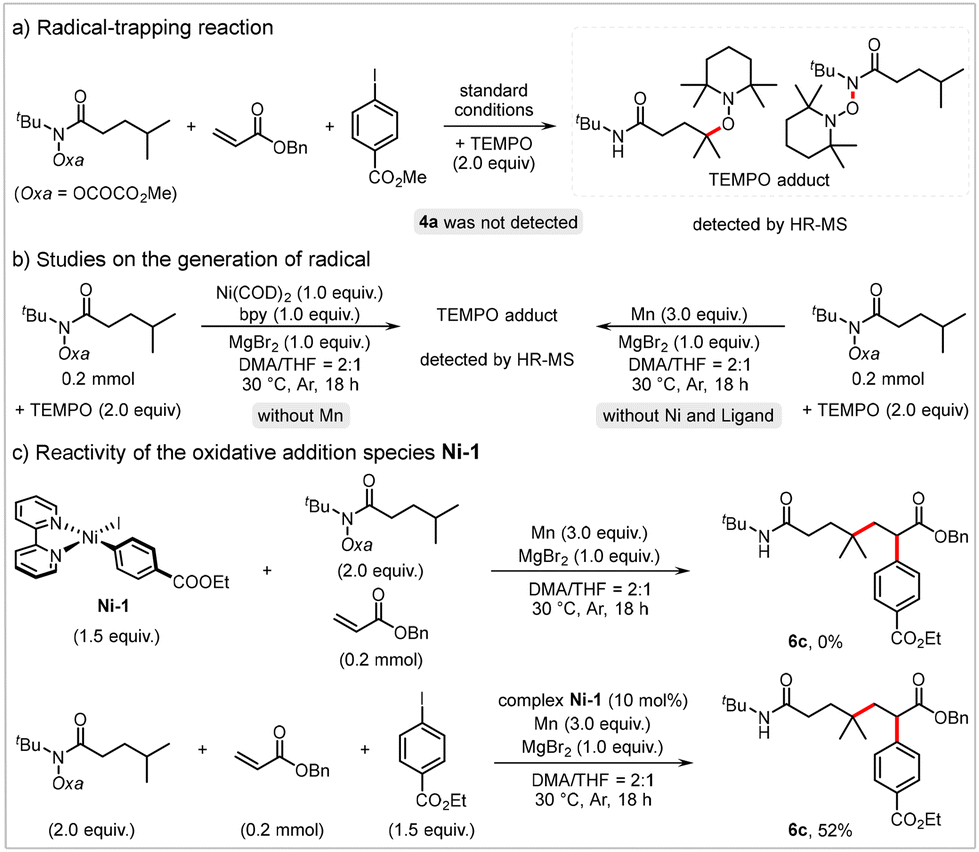 | ||
| Scheme 4 Mechanistic studies. | ||
Based on mechanistic studies and the relevant literature,23 a plausible mechanism is depicted in Scheme 5. The reduction of compound 1 by both Ni(0) and Mn via single-electron transfer (SET) processes generates Ni(I) and amide radical A. This radical undergoes an intramolecular 1,5-HAT to form carbon-centered radical B. The radical addition of radical B to alkene 2 produces radical C. Radical C is then captured by a Ni(I) species to yield Ni(II) intermediate D. Following reduction by Mn, the oxidative addition of Ni(I) intermediate E with aryl iodide 3 forms Ni(III) complex F. Reductive elimination of Ni(III) complex F generates the desired product along with Ni(I) species. Finally, the reduction of Ni(I) by Mn regenerates Ni(0) to finish the catalytic cycle.
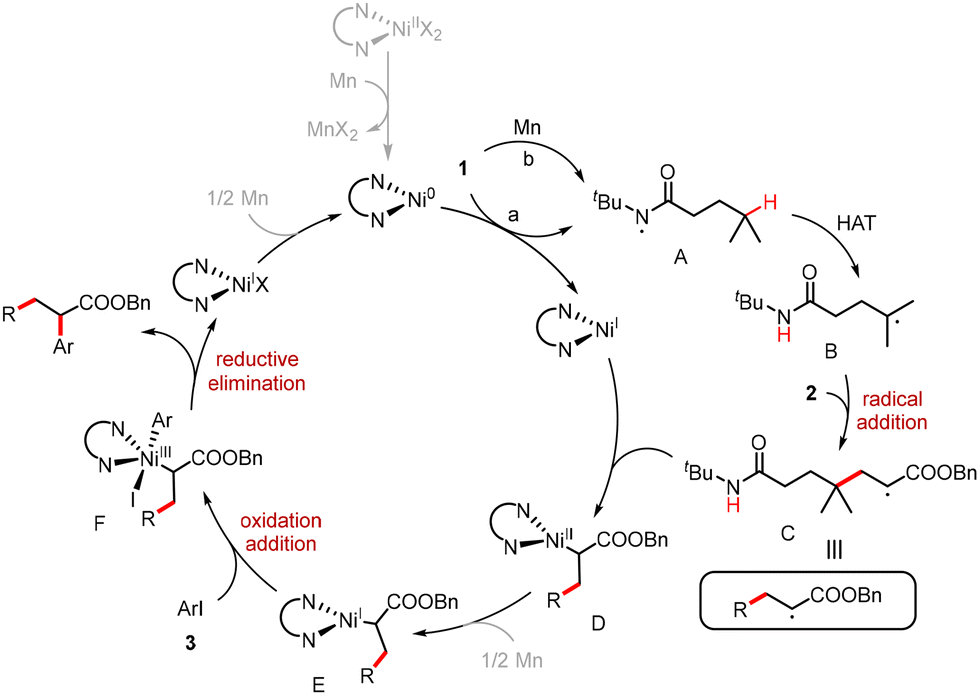 | ||
| Scheme 5 Proposed mechanism. | ||
Conclusions
In conclusion, a nickel-catalyzed 1,2-alkylarylation of alkenes with O-oxalate hydroxamic acid esters and aryl iodides has been successfully developed under reductive conditions. The reaction relies on amidyl radical-triggered 1,5-HAT and uses electrophiles as functionalizing reagents with good coupling efficiency and high regioselectivity. This mild protocol facilitates the sequential construction of two vicinal C–C bonds through the addition of two different electrophiles across the π-system, effectively avoiding the use of sensitive organometallic reagents and significantly expanding the scope of HAT strategy-enabled alkene difunctionalization reactions. Moreover, this protocol can be extended to the reductive 1,2-alkylarylation of alkynes, providing an alternative method to access important multi-substituted alkenes from readily available starting materials.Author contributions
Xi Chen performed the experiments and wrote the manuscript and ESI.† Qiang Wang coordinated the experiments and analyses. Xiao-Ping Gong and Rui-Qiang Jiao checked the manuscript and prepared some of the substrates and the ESI.† Xue-Yuan Liu and Yong-Min Liang conceived this project and provided guidance. All authors discussed the results and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSF 22171114 and 22371097).References
- (a) J. Lin, R.-J. Song, M. Hu and J.-H. Li, Recent Advances in the Intermolecular Oxidative Difunctionalization of Alkenes, Chem. Rec., 2019, 19, 440–451 CrossRef CAS PubMed; (b) S. Xu, H. Chen, Z. Zhou and W. Kong, Three-Component Alkene Difunctionalization by Direct and Selective Activation of Aliphatic C−H Bonds, Angew. Chem., Int. Ed., 2021, 60, 7405–7411 CrossRef CAS PubMed.
- (a) J.-S. Zhang, L. Liu, T. Chen and L.-B. Han, Transition-Metal-Catalyzed Three-Component Difunctionalizations of Alkenes, Chem. – Asian J., 2018, 13, 2277–2291 CrossRef CAS PubMed; (b) Z. Liu, Y. Gao, T. Zeng and K. M. Engle, Transition-Metal-Catalyzed 1,2-Carboboration of Alkenes: Strategies, Mechanisms, and Stereocontrol, Isr. J. Chem., 2020, 60, 219–229 CrossRef CAS PubMed; (c) L. M. Wickham and R. Giri, Transition Metal (Ni, Cu, Pd)-Catalyzed Alkene Dicarbofunctionalization Reactions, Acc. Chem. Res., 2021, 54, 3415–3437 CrossRef CAS PubMed; (d) Á. Velasco-Rubio and R. Martin, Recent Advances in Ni-Catalyzed 1,1-Difunctionalization of Unactivated Olefins, Adv. Synth. Catal., 2024, 366, 593–602 CrossRef.
- (a) R. Shrestha, S. C. M. Dorn and D. J. Weix, Nickel-Catalyzed Reductive Conjugate Addition to Enones via Allylnickel Intermediates, J. Am. Chem. Soc., 2013, 135, 751–762 CrossRef CAS PubMed; (b) Y. Ping and W. Kong, Ni-Catalyzed Reductive Difunctionalization of Alkenes, Synthesis, 2020, 979–992 Search PubMed; (c) X. Qi and T. Diao, Nickel-Catalyzed Dicarbofunctionalization of Alkenes, ACS Catal., 2020, 10, 8542–8556 CrossRef CAS; (d) S. Zhu, X. Zhao, H. Li and L. Chu, Catalytic three-component dicarbofunctionalization reactions involving radical capture by nickel, Chem. Soc. Rev., 2021, 50, 10836–10856 RSC; (e) H. Pellissier, Recent developments in enantioselective nickel(II)-catalyzed conjugate additions, Org. Chem. Front., 2022, 9, 6717–6748 RSC; (f) Y. Wei, L. Q. H. Lin, B. C. Lee and M. J. Koh, Recent Advances in First-Row Transition Metal-Catalyzed Reductive Coupling Reactions for π-Bond Functionalization and C-Glycosylation, Acc. Chem. Res., 2023, 56, 3292–3312 CrossRef CAS PubMed; (g) S. Zhu, H. Li, Y. Li, Z. Huang and L. Chu, Exploring visible light for carbon–nitrogen and carbon–oxygen bond formation via nickel catalysis, Org. Chem. Front., 2023, 10, 548–569 RSC.
- Y. Jin and C. Wang, Ni-catalysed reductive arylalkylation of unactivated alkenes, Chem. Sci., 2019, 10, 1780–1785 RSC.
- A. García-Domínguez, Z. Li and C. Nevado, Nickel-Catalyzed Reductive Dicarbofunctionalization of Alkenes, J. Am. Chem. Soc., 2017, 139, 6835–6838 CrossRef.
- X. Zhao, H.-Y. Tu, L. Guo, S. Zhu, F.-L. Qing and L. Chu, Intermolecular selective carboacylation of alkenes via nickel-catalyzed reductive radical relay, Nat. Commun., 2018, 9, 3488 CrossRef.
- (a) Y. Peng, X.-B. Xu, J. Xiao and Y.-W. Wang, Nickel-mediated stereocontrolled synthesis of spiroketals via tandem cyclization–coupling of β-bromo ketals and aryl iodides, Chem. Commun., 2014, 50, 472–474 RSC; (b) X.-X. Wang, X. Lu, S.-J. He and Y. Fu, Nickel-catalyzed three-component olefin reductive dicarbofunctionalization to access alkylborates, Chem. Sci., 2020, 11, 7950–7956 RSC; (c) T. Yang, X. Chen, W. Rao and M. J. Koh, Broadly Applicable Directed Catalytic Reductive Difunctionalization of Alkenyl Carbonyl Compounds, Chem, 2020, 6, 738–751 CrossRef CAS; (d) M. Belal, Z. Li, L. Zhu and G. Yin, Catalyst-controlled regiodivergent 1,2-difunctionalization of alkenes with two carbon-based electrophiles, Sci. China: Chem., 2022, 65, 514–520 CrossRef CAS.
- Q.-Z. Li, R. Zeng, Y. Fan, Y.-Q. Liu, T. Qi, X. Zhang and J.-L. Li, Remote C(sp3)−H Acylation of Amides and Cascade Cyclization via N-Heterocyclic Carbene Organocatalysis, Angew. Chem., Int. Ed., 2022, 61, e202116629 CrossRef CAS PubMed.
- K. Wu, N. Lam, D. A. Strassfeld, Z. Fan, J. X. Qiao, T. Liu, D. Stamos and J.-Q. Yu, Palladium(II)-Catalyzed C−H Activation with Bifunctional Ligands: From Curiosity to Industrialization, Angew. Chem., Int. Ed., 2024, 63, e202400509 CrossRef CAS.
- X. Wu and C. Zhu, Radical Functionalization of Remote C(sp3)–H Bonds Mediated by Unprotected Alcohols and Amides, CCS Chem., 2020, 2, 813–828 CrossRef CAS.
- (a) A. W. Hofmann, Ueber die Einwirkung des Broms in alkalischer Lösung auf die Amine, Ber. Dtsch. Chem. Ges., 1883, 16, 558–560 CrossRef; (b) K. Löffler and C. Freytag, Über eine neue Bildungsweise von N-alkylierten Pyrrolidinen, Ber. Dtsch. Chem. Ges., 1909, 42, 3427–3431 CrossRef; (c) M. E. Wolff, Cyclization of N-Halogenated Amines (The Hofmann-Löffler Reaction), Chem. Rev., 1963, 63, 55–64 CrossRef CAS.
- (a) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Catalytic alkylation of remote C–H bonds enabled by proton-coupled electron transfer, Nature, 2016, 539, 268–271 CrossRef CAS PubMed; (b) J. C. K. Chu and T. Rovis, Amide-directed photoredox-catalysed C–C bond formation at unactivated sp3 C–H bonds, Nature, 2016, 539, 272–275 CrossRef PubMed; (c) L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Remote C–H Functionalization via Selective Hydrogen Atom Transfer, Synthesis, 2018, 1569–1586 CAS; (d) N. Goswami and D. Maiti, An Update on Distal C(sp3)−H Functionalization Involving 1,5-HAT Emerging from Nitrogen Radicals, Isr. J. Chem., 2020, 60, 303–312 CrossRef CAS; (e) G. Kumar, S. Pradhan and I. Chatterjee, N-Centered Radical Directed Remote C−H Bond Functionalization via Hydrogen Atom Transfer, Chem. – Asian J., 2020, 15, 651–672 CrossRef CAS; (f) W.-J. Yue, C. S. Day and R. Martin, Site-Selective Defluorinative sp3 C–H Alkylation of Secondary Amides, J. Am. Chem. Soc., 2021, 143, 6395–6400 CrossRef CAS; (g) J. Wang, Y. Zhang, L. Zhu, X.-S. Xue and C. Li, Photoinduced Remote C(sp3)−H Phosphonylation of Amides, Angew. Chem., Int. Ed., 2024, 63, e202400856 CrossRef CAS.
- (a) H. Chen and S. Yu, Remote C–C bond formation via visible light photoredox-catalyzed intramolecular hydrogen atom transfer, Org. Biomol. Chem., 2020, 18, 4519–4532 RSC; (b) S. Sarkar, K. P. S. Cheung and V. Gevorgyan, C–H functionalization reactions enabled by hydrogen atom transfer to carbon-centered radicals, Chem. Sci., 2020, 11, 12974–12993 RSC; (c) W. Guo, Q. Wang and J. Zhu, Visible light photoredox-catalysed remote C–H functionalisation enabled by 1,5-hydrogen atom transfer (1,5-HAT), Chem. Soc. Rev., 2021, 50, 7359–7377 RSC; (d) Z. Zhang, X. Cao, X. Song, G. Wang, B. Shi, X. Li, N. Ma, L. Liu and G. Zhang, Metal-free nucleophilic 7,8-dearomatization of quinolines: Spiroannulation of aminoquinoline protected amino acids, Chin. Chem. Lett., 2023, 34, 107779 CrossRef CAS.
- (a) H. Mei, Z. Yin, J. Liu, H. Sun and J. Han, Recent Advances on the Electrochemical Difunctionalization of Alkenes/Alkynes, Chin. J. Chem., 2019, 37, 292–301 CrossRef CAS; (b) Z.-L. Li, G.-C. Fang, Q.-S. Gu and X.-Y. Liu, Recent advances in copper-catalysed radical-involved asymmetric 1,2-difunctionalization of alkenes, Chem. Soc. Rev., 2020, 49, 32–48 RSC; (c) Y. Zeng, Y. Li, D. Lv and H. Bao, Copper-catalyzed three-component oxycyanation of alkenes, Org. Chem. Front., 2021, 8, 908–914 RSC; (d) X. Lv, Y. Yang, L. Zhou and X. Zeng, Copper-Catalyzed Fluoroamide-Directed Remote C(sp3)-H Bond Functionalization Through Intramolecular Hydrogen Atom Transfer, Eur. J. Org. Chem., 2024, e202400027 CrossRef CAS; (e) T. Huang, J. Liu, Z. Wu, Z. Tian, L. Hai and Y. Wu, Photoredox-Catalyzed Alkylamination of Alkenes via Oxidative Radical-Polar Crossover and Site-Selective 1,5-Hydrogen Atom Transfer, Org. Lett., 2024, 26, 6847–6852 CrossRef CAS; (f) G.-Q. Tan, K.-D. Xu, H.-T. Qin and F. Liu, Nitrogen-directed unactivated γ-C(sp3)–H functionalization of amides toward 7-membered lactones via photoredox catalysis, Org. Chem. Front., 2024, 11, 3724–3728 RSC.
- L.-J. Zhong, Z.-Q. Xiong, X.-H. Ouyang, Y. Li, R.-J. Song, Q. Sun, X. Lu and J.-H. Li, Intermolecular 1,2-Difunctionalization of Alkenes Enabled by Fluoroamide-Directed Remote Benzyl C(sp3)–H Functionalization, J. Am. Chem. Soc., 2022, 144, 339–348 CrossRef CAS.
- R. Rui, B. Wang, X.-J. Xu, Z. Zhang, X. Chen and X.-Y. Liu, Cu-catalyzed alkylarylation of alkenes via N-directed remote C(sp3)–H functionalization, Org. Chem. Front., 2024, 11, 1484–1489 RSC.
- (a) Y.-T. Wen, X.-T. Kong, H.-C. Liu, C.-T. Wang, W.-X. Wei, B. Wang, X.-Y. Liu and Y.-M. Liang, Ni-Catalyzed Remote Radical/Cross-Electrophile Coupling Cascade for Selective C(sp3)–H Arylation, Org. Lett., 2022, 24, 2399–2403 CrossRef CAS; (b) X. Chen, Q. Wang, Z. Zhang, Z.-J. Niu, W.-Y. Shi, X.-P. Gong, R.-Q. Jiao, M.-H. Gao, X.-Y. Liu and Y.-M. Liang, Copper-Catalyzed Hydrogen Atom Transfer and Aryl Migration Strategy for the Arylalkylation of Activated Alkenes, Org. Lett., 2022, 24, 4338–4343 CrossRef CAS; (c) C.-T. Wang, P.-Y. Liang, M. Li, B. Wang, Y.-Z. Wang, X.-S. Li, W.-X. Wei, X.-Y. Gou, Y.-N. Ding, Z. Zhang, Y.-K. Li, X.-Y. Liu and Y.-M. Liang, Aryl-to-Vinyl 1,4-Nickel Migration/Reductive Cross-Coupling Reaction for the Stereoselective Synthesis of Multisubstituted Olefins, Angew. Chem., Int. Ed., 2023, 62, e202304447 CrossRef CAS; (d) X. Chen, Z. Zhang, W.-Y. Shi, Y.-N. Ding, Y.-Y. Luan, Y.-C. Huang, Q. Wang, X.-Y. Liu and Y.-M. Liang, Photoredox-Catalyzed N-Directed Regioselective Difluoroalkylation of Unactivated C(sp3)–H Bonds, Org. Lett., 2023, 25, 4456–4461 CrossRef CAS PubMed.
- Z. Zhu, Y. Gong, W. Tong, W. Xue and H. Gong, Ni-Catalyzed Cross-Electrophile Coupling of Aryl Triflates with Thiocarbonates via C–O/C–O Bond Cleavage, Org. Lett., 2021, 23, 2158–2163 CrossRef CAS PubMed.
- (a) J. Chen, J. Guo and Z. Lu, Recent Advances in Hydrometallation of Alkenes and Alkynes via the First Row Transition Metal Catalysis, Chin. J. Chem., 2018, 36, 1075–1109 CrossRef CAS; (b) W. Liu and W. Kong, Ni-Catalyzed stereoselective difunctionalization of alkynes, Org. Chem. Front., 2020, 7, 3941–3955 RSC; (c) S. Mondal, S. Ghosh and A. Hajra, Visible-light-induced redox-neutral difunctionalization of alkenes and alkynes, Chem. Commun., 2024, 60, 9659–9691 RSC.
- (a) Y. Jiang, J. Pan, T. Yang, Y. Zhao and M. J. Koh, Nickel-catalyzed site- and stereoselective reductive alkylalkynylation of alkynes, Chem, 2021, 7, 993–1005 CrossRef CAS; (b) S. Maiti and J. H. Rhlee, Reductive Ni-catalysis for stereoselective carboarylation of terminal aryl alkynes, Chem. Commun., 2021, 57, 11346–11349 RSC; (c) H. Li, F. Wang, S. Zhu and L. Chu, Selective Fluoromethyl Couplings of Alkynes via Nickel Catalysis, Angew. Chem., Int. Ed., 2022, 61, e202116725 CrossRef CAS; (d) Q.-Z. Li, Z.-H. Li, J.-C. Kang, T.-M. Ding and S.-Y. Zhang, Ni-catalyzed, enantioselective three-component radical relayed reductive coupling of alkynes: Synthesis of axially chiral styrenes, Chem. Catal., 2022, 2, 3185–3195 CrossRef CAS; (e) Y.-Z. Zhan, H. Meng and W. Shu, Rapid access to t-butylalkylated olefins enabled by Ni-catalyzed intermolecular regio- and trans-selective cross-electrophile t-butylalkylation of alkynes, Chem. Sci., 2022, 13, 4930–4935 RSC; (f) Y. Dai, F. Wang, S. Zhu and L. Chu, Selective Ni-catalyzed cross-electrophile coupling of alkynes, fluoroalkyl halides, and vinyl halides, Chin. Chem. Lett., 2022, 33, 4074–4078 CrossRef CAS.
- H. Chen, L. Hu, W. Ji, L. Yao and X. Liao, Nickel-Catalyzed Decarboxylative Alkylation of Aryl Iodides with Anhydrides, ACS Catal., 2018, 8, 10479–10485 CrossRef CAS.
- S. Zheng, Z. Chen, Y. Hu, X. Xi, Z. Liao, W. Li and W. Yuan, Selective 1,2-Aryl-Aminoalkylation of Alkenes Enabled by Metallaphotoredox Catalysis, Angew. Chem., Int. Ed., 2020, 59, 17910–17916 CrossRef CAS.
- (a) J. He, Y. Xue, B. Han, C. Zhang, Y. Wang and S. Zhu, Nickel-Catalyzed Asymmetric Reductive 1,2-Carboamination of Unactivated Alkenes, Angew. Chem., Int. Ed., 2020, 59, 2328–2332 CrossRef CAS; (b) H. Chen, H. Yue, C. Zhu and M. Rueping, Reactivity in Nickel-Catalyzed Multi-component Sequential Reductive Cross-Coupling Reactions, Angew. Chem., Int. Ed., 2022, 61, e202204144 CrossRef CAS PubMed; (c) W. Huang, S. Keess and G. A. Molander, One step synthesis of unsymmetrical 1,3-disubstituted BCP ketones via nickel/photoredox-catalyzed [1.1.1]propellane multicomponent dicarbofunctionalization, Chem. Sci., 2022, 13, 11936–11942 RSC; (d) Y. Li, Y. Li, H. Shi, H. Wei, H. Li, I. Funes-Ardoiz and G. Yin, Modular access to substituted cyclohexanes with kinetic stereocontrol, Science, 2022, 376, 749–753 CrossRef CAS PubMed; (e) Z.-Z. Zhao, X. Pang, X.-X. Wei, X.-Y. Liu and X.-Z. Shu, Nickel-Catalyzed Reductive C(sp2)−Si Coupling of Chlorohydrosilanes via Si−Cl Cleavage, Angew. Chem., Int. Ed., 2022, 61, e202200215 CrossRef CAS PubMed; (f) Z. Huang, M. E. Akana, K. M. Sanders and D. J. Weix, A decarbonylative approach to alkylnickel intermediates and C(sp3)-C(sp3) bond formation, Science, 2024, 385, 1331–1337 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2388081. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01875c |
| This journal is © the Partner Organisations 2025 |