
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influencing optical and charge transport properties by controlling the molecular interactions of merocyanine thin films†
Lukas
Böhner
a,
Philipp
Weitkamp
a,
Thorsten
Limböck
a,
Nora
Gildemeister
a,
Daniele
Fazzi
 ab,
Manuela
Schiek
c,
Ruth
Bruker
a,
Dirk
Hertel
a,
Roland
Schäfer
a,
Klas
Lindfors
a and
Klaus
Meerholz
*a
ab,
Manuela
Schiek
c,
Ruth
Bruker
a,
Dirk
Hertel
a,
Roland
Schäfer
a,
Klas
Lindfors
a and
Klaus
Meerholz
*a
aDepartment of Chemistry, University of Cologne, Greinstraße 4-6, 50939 Cologne, Germany. E-mail: klaus.meerholz@uni-koeln.de
bUniversita di Bologna, Dipartimento di Chimica “Giacomo Ciamician”, Via P. Gobetti 85, 40129 Bologna, Italy
cCenter for Surface and Nano-Analytics (ZONA), Johannes Kepler University, Altenberger Str. 69, Linz 4040, Austria
First published on 14th January 2025
Abstract
In amorphous organic semiconductors, charge transport typically takes place via slow hopping processes, but it is known that molecular aggregation can lead to enhanced exciton and charge transport through coupling of the transition dipole moments. In this work, we investigate the optical, morphological, and electronic properties of thin films of a merocyanine dye, which aggregates easily due to its dipolar character. Firstly, in spin-coated thin films the degree of aggregation can be tuned by thermal annealing, leading to strong spectral shifts alongside with Davydov splitting of >800 meV. At the same time, the mobility increases by approximately three orders of magnitude. We combine variable angle spectroscopic ellipsometry and polarization-resolved transmission spectroscopy with density functional theory to demonstrate that the aggregated molecules are oriented in an upright, standing configuration relative to the substrate surface. This arrangement involves a co-facial orientation of the molecular pi-systems, which is advantageous for lateral charge transport. Secondly, by utilizing highly oriented pyrolytic graphite as an ordered substrate and low-rate vacuum deposition, we are able to template the growth of the merocyanine layer and to substantially improve the in-plane morphological order. By combining atomic force microscopy and photoluminescence microspectroscopy we observe large oriented domains hundreds of μm2 in size, emitting linearly polarized light, whereby maintaining the edge-on molecular arrangement. This promises a further significant enhancement of lateral charge carrier mobility.
Introduction
Organic semiconducting materials as active thin films in electronic devices are usually present in a polycrystalline or amorphous state. They mostly exhibit lower charge carrier mobilities than single crystals, due to comparably low long-range order. The highest charge carrier mobility values for organic small molecules are usually obtained for single crystals of rod-, or disk-like molecules such as copper-phthalocyanine (1.0 cm2 V−1 s−1),1 tetracene (2.4 cm2 V−1 s−1),2 pentacene, or rubrene (40 cm2 V−1 s−1).3,4 Although highly dipolar molecules were long expected to show low charge carrier mobilities associated with increased energetic disorder, a mobility of 2.3 cm2 V−1 s−1 was measured for a merocyanine single crystal,5 demonstrating the concept of molecular dipole elimination upon centrosymmetric packing.6–8 However, the single-crystal growth process is lengthy and less feasible for utilization in organic electronic devices compared to amorphous or polycrystalline organic thin films, fabricated by, e.g., spin coating or thermal evaporation, especially for large area devices. Tremendous efforts have been undertaken into introducing a higher degree of order in such organic thin films to improve the charge transport performance while maintaining a homogeneous and dense film quality. Increasing order in organic thin films can be achieved by, e.g., templating substrates,9–12 external stimuli such as mechanical rubbing,13–15 or different liquid-phase deposition techniques which influence molecular orientation.16–18Additionally, it was shown that molecular aggregation of polar dyes like dipolar merocyanines or symmetric quadrupolar squaraines without ground state dipole moment can lead to improved charge carrier mobility.19–23 In these aggregates excitons are delocalized by coupling of the transition dipole moments (TDM) of the associated monomers.24,25 This leads to drastic changes of their optical properties compared to the monomeric form, which allows for monitoring the aggregation state by transmission or photoluminescence spectroscopy. The two best known forms of molecular aggregates are J- and H-aggregates.26 J-aggregates are known to show intense, red-shifted and narrow absorption peaks relative to the monomer. Further, photoluminescence (PL) with negligible Stokes shift can be observed. By contrast, H-aggregates usually show a broad, blue-shifted, and vibronically rich absorption spectrum, and the emission is strongly quenched. The differences in the spectra of J- and H-aggregates arise from the orientations of the TDMs of the aggregated molecules relative to each other. In J-aggregates the TDMs are oriented in a head-to-tail fashion, whereas in H-aggregates TDMs are oriented in parallel dimers.24–28
Merocyanine molecules possess large absorption coefficients which makes them interesting for applications such as solar cells or photodiodes.19,29 Additionally, the large dipole moment of merocyanines makes them prone to aggregation, provided that the molecular structure is sufficiently planar and does not prevent it. We exploit this to investigate a merocyanine molecule, namely HB238,6 with a ground-state dipole moment of 13.1 D (measured in 1,4-dioxane),6 which aggregates in a controlled way by tuning the experimental conditions. We use these aggregates as a model system to investigate the interdependence of morphological, optical, and charge transport properties. We fabricate and characterize spin-coated thin films on oxide surfaces, and we are able to show that the charge carrier mobility is significantly improved upon aggregation. By preparing thermally evaporated thin films on highly oriented pyrolytic graphite (HOPG) we enhance the structural order even further. We observe large, directionally oriented domains of hundreds of μm2 in size.
Results & discussion
The investigated merocyanine molecule with the trivial name HB2386 (chemical structure shown in the inset in Fig. 1a) is present in the monomeric (i.e., non-aggregated) form in acetone solution (c = 10−5 M). Calculation of the single molecule vibronic absorption spectrum and Huang-Rhys parameters, as carried out at the DFT/TDDFT level of theory (see Experimental techniques and methods), well resembles the experimental UV-Vis data, supporting that HB238 is present as non-aggregated monomeric species in solution (see Fig. S1†). A typical mirror-image of absorption and emission in the monomeric form is observed. When water is added, dramatic changes in the absorbance and photoluminescence spectra are observed (see Fig. 1a and b). We attribute these observations to the formation of HB238 aggregates resulting from the insolubility of HB238 in water. Upon aggregation, two distinct peaks arise in the absorption spectrum. One of the peaks is blue- [H-band: 2.47 eV, FWHM = 126 meV (1014 cm−1)] and the other one is red-shifted [J-band: 1.67 eV, FWHM = 49 meV (398 cm−1)] compared to the monomer absorption feature. A very weak absorption band reminiscent of the monomer transition can still be seen around 1.9 eV in the aggregate spectrum. Additionally, we observe a narrow emission peak [1.65 eV, FWHM = 40 meV (322 cm−1)] which is almost resonant to the red-shifted absorption peak of the J-band.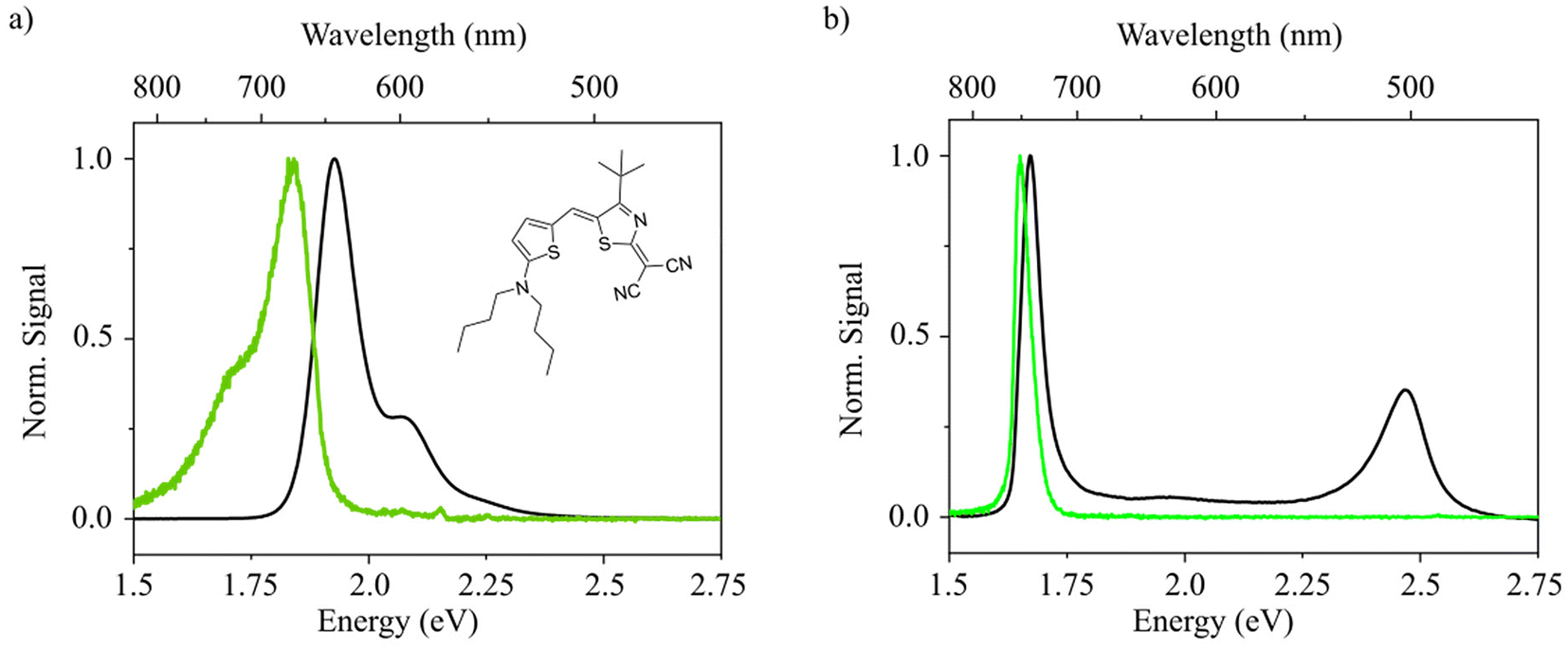 | ||
| Fig. 1 Optical properties of merocyanine HB238 in the monomeric and aggregated state. Normalized absorbance and emission spectra (black and green lines, respectively) of monomeric HB238 in acetone solution (a) and dispersed aggregates in a 10% acetone/90% water mixture (b). The inset in panel a shows the chemical structure of HB238. | ||
These findings correlate well with an aggregated state, in which the molecular TDMs are aligned to each other with an oblique angle, a model already proposed by Kasha et al. in 1965.24 The lower and higher lying states, resulting from the sum and difference of the oblique-angled TDMs, respectively, reveal the resulting exciton band bottom and top, giving rise to Davydov splitting (DS).30 Unlike pure J- or H-aggregates, both states gain oscillator strength. Emission, however, is only observed from the lowest state, i.e., the J-state, due to the fast internal non-radiative conversion. The observed DS is large (ca. 0.86 eV) in comparison with values reported for other organic compounds,31–35 ranging between approximately 0.14 eV for pentacene32 to 0.65 eV for squaraines.34 It demonstrates an unusually strong interaction between the molecules in our case with DS values similar to reports for oligo-phenylene-vinylenes and oligothiophenes.36 This interpretation is supported by the fact that the H-transition displays a spectrally narrow absorption peak without significant vibronic features, which is only observed for very strong coupling, when there is little spectral overlap between the monomer and H-band absorption (compare Fig. 1a and b, respectively).28
We now turn to spin-coated HB238 thin films necessary for device application. Spin coating on float glass substrates leads to thin films which show broad absorbance spectra. The spectra can be significantly altered by thermally annealing the samples.19 To investigate in detail the annealing-dependent optical properties, spin-coated thin films were annealed for ten minutes at different temperatures in the range of 50 °C to 160 °C. An additional unannealed [referred to as “as fabricated (a.f.)”] sample was prepared. For every annealing temperature a separate sample was fabricated. The spin-coated thin films further show pronounced uniaxial anisotropy which is discussed further below in the main text in more detail. All spectra were thus measured at an angle of incidence (AOI) of 45° and with p-polarized light, to address the ordinary and extra-ordinary optical components with equal electric field strengths. The results are shown in Fig. 2. The samples can be ordered into three regimes I, II, and III according to the annealing temperature. For clarity only a single absorbance spectrum for each regime is shown in Fig. 2a (all absorbance spectra are shown in Fig. S17†).
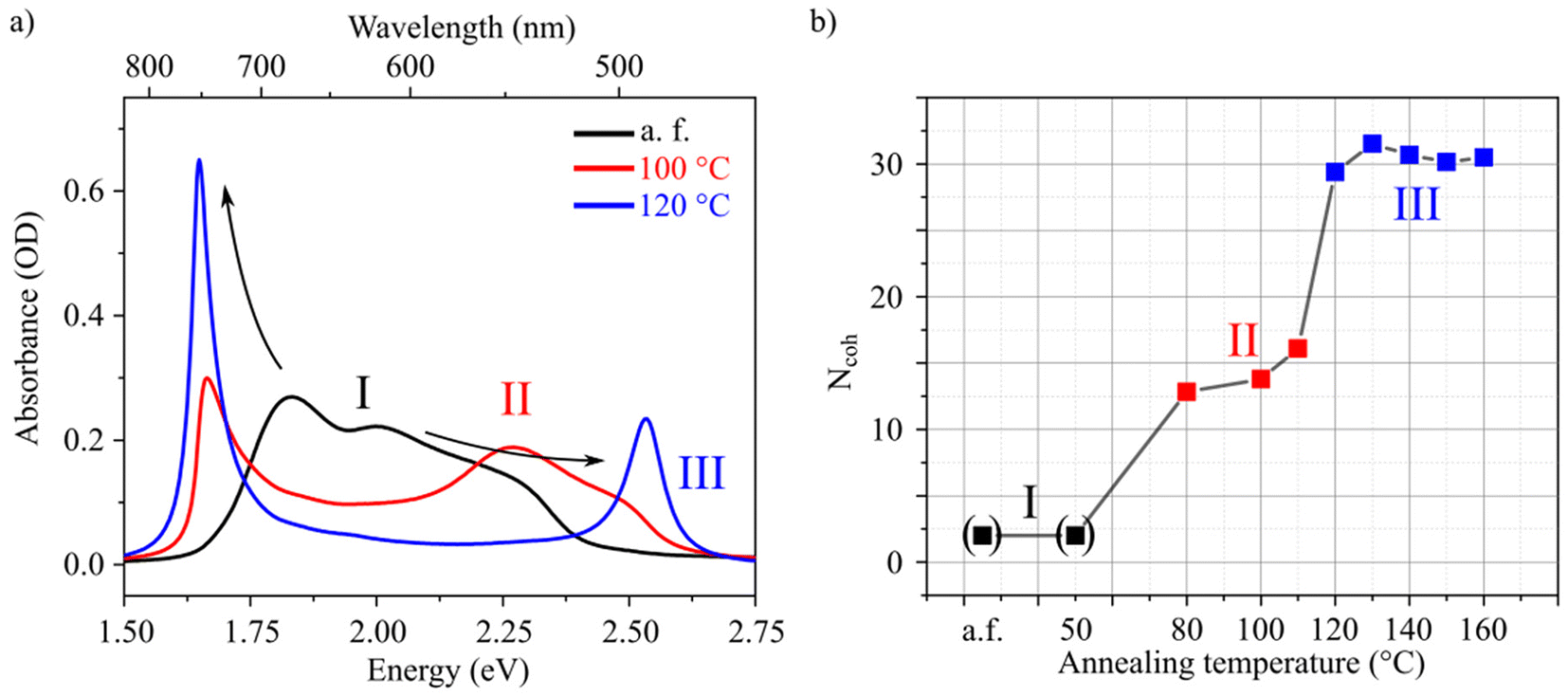 | ||
| Fig. 2 Annealing-induced aggregation of HB238. (a) Absorbance spectra illustrating the three different regimes (see main text) measured at 45° AOI using p-polarized light for the as fabricated sample (I, black), after annealing at 100 °C (II, red), and after annealing at 120 °C (III, blue). Black arrows are guides to the eye emphasizing spectral shifts. (b) Estimated number of coherently coupled molecules as a function of annealing temperature. The three different regimes can be distinguished and are highlighted with the same color as in (a). | ||
For regime I (low annealing temperatures; a.f. and 50 °C), a broad absorption band without clear peaks is observed. At intermediate temperatures (80 °C–110 °C), regime II, the spectra display the characteristic J-transition peak at ca. 1.65 eV. Additionally, the H-transition is observed as a spectral shoulder at around 2.50 eV. However, the spectra also show broad features, mainly at around 2.25 eV, which overlap with the H-transition absorption peak. Annealing at 120 °C or higher (here: up to 160 °C) leads to regime III. The absorbance spectra are dominated by distinct J- and H-transition peaks. The samples of regime III are thus in a similar aggregation state as the dispersed aggregates presented in Fig. 1b. This is further supported by 2θ XRD measurements of both samples (see Fig. S3†). From Fig. 2a it can be observed that, from regime I to III, the lowest energy vibronic peak at approximately 1.8 eV red shifts and finally results in the observed J-transition peak. Likewise, the higher energy vibronic transitions blue shift to finally evolve in the distinct H-transition peak. This evolution is indicated by the black arrows in Fig. 2a. A similar behavior has been described by Spano, and associated to increasing coupling strength.25 We thus attribute the observed changes in the absorbance spectra during the transitions between the regimes to increased intermolecular coupling. The spectral reorganization and thus coupling strength do not change gradually with annealing temperature, but stepwise between the three regimes. We attribute this finding to thermodynamic boundaries which are overcome, leading to molecular reorganization between 50 °C and 80 °C (I to II) and between 110 °C and 120 °C (II to III). This is further supported by 2θ XRD measurements of the thin films shown in Fig. S4.† In regime I there is no measurable diffraction peak, in regime II a diffraction peak of low intensity is observed, and from regime II to regime III the intensity of the peak suddenly increases by an order of magnitude. The peak intensity saturates in regime III in accordance with the unaltered spectral properties in regime III (see Fig. S17†).
To further quantify the degree of aggregation, we estimate the number of coherently coupled molecules (Ncoh) of each sample using the absorbance spectra. The parameter Ncoh describes the number of molecules over which, on average, the exciton is delocalized, and is thus not typically equal to aggregate, or crystal domain dimensions. To estimate this number from absorbance spectra, we utilize a method introduced by Knoester and Bakalis,37 which was developed for linear J-aggregates. In the method it is assumed that the spectral width of the J-peak [as half-width at half-maximum (HWHM)] corresponds to the energetic difference between the lowest (J-state) and the second lowest state of the exciton-band. With this assumption, the peak-width can be directly related to the number of coherently coupled molecules. For HB238-aggregates the mathematical description of a linear J-aggregate's exciton band is insufficient. As shown in, e.g., Fig. 1, HB238-aggregates display two distinct peaks in the absorbance spectrum, split asymmetrically around the monomer-absorption, with the H-transition absorption peak shifted further. These spectral features can be described using the exciton band description of two-dimensional, square herringbone aggregates developed by Spano.38
Using the model of ref. 38, the number of coherently coupled molecules Ncoh can be related to the spectral width of the J- and H-transition and expressed as
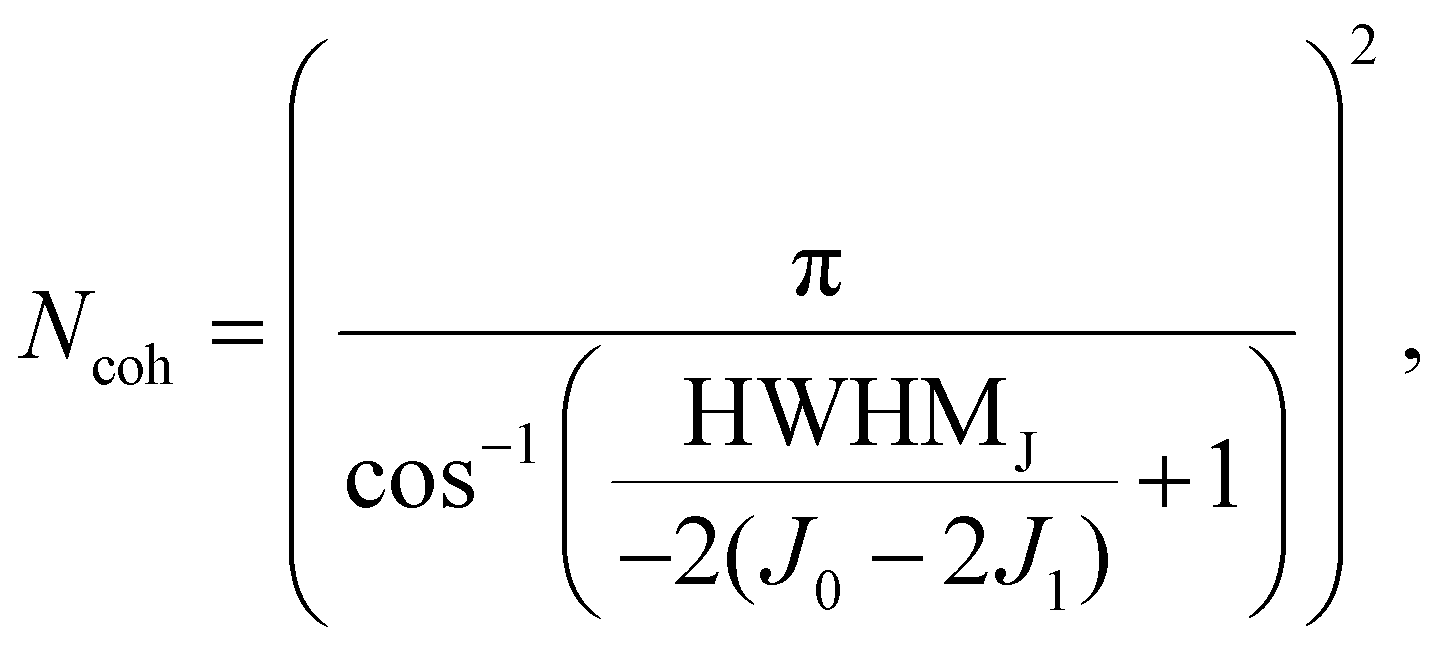 | (1) |
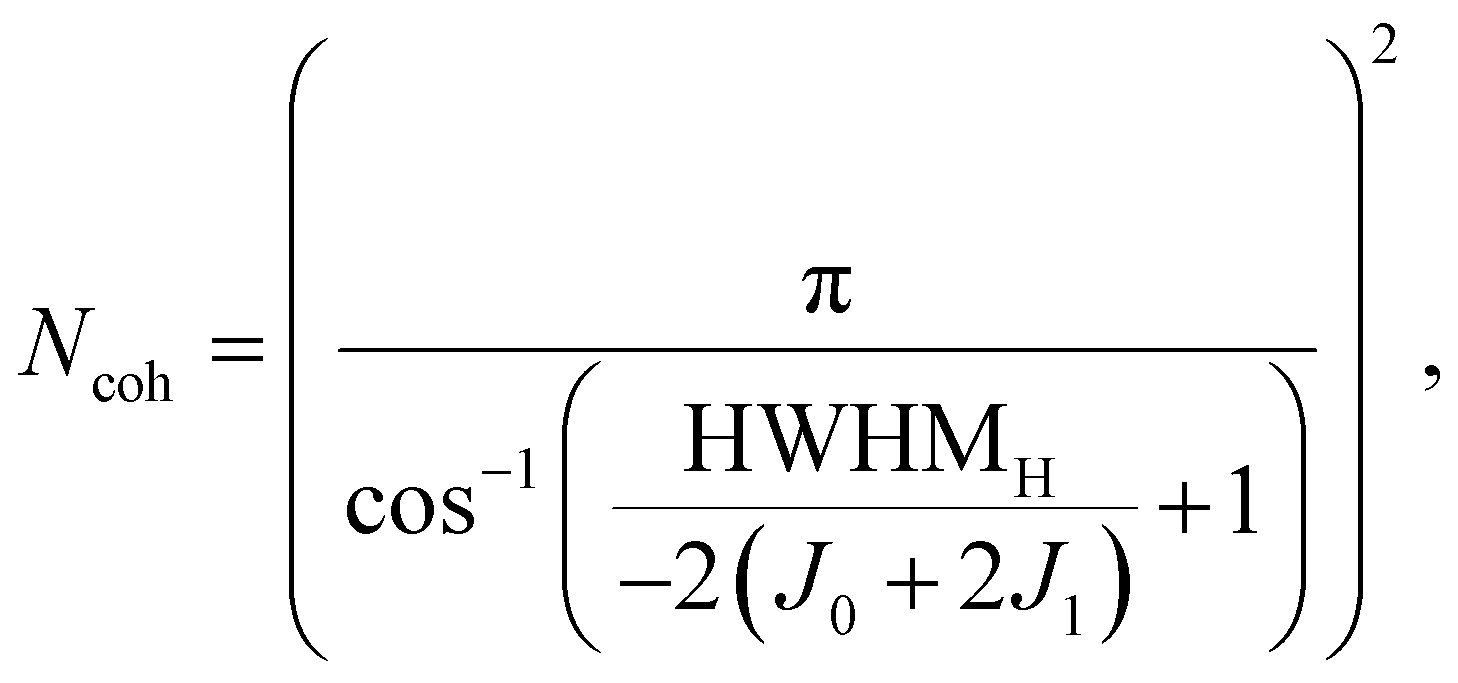 | (2) |
The advantage of this procedure is that all parameters are obtained from the same spectrum. It is not necessary to compare aggregate to monomer spectra which always includes uncertainties, due to the immeasurable, at least directly, liquid-to-crystal shift. The procedure yields an average number of coherently coupled molecules in a two-dimensional square aggregate. It has to be noted, however, that the described procedure includes some uncertainties. The HB238 aggregates might not be perfectly square. The AOI has a slight influence on the absorption peak-widths. Here all measurements were performed at an AOI of 45° with p-polarized light. The fitting of J- and H-transition absorption peaks of the samples annealed at 80 °C, 100 °C, and 110 °C is complicated by spectrally overlapping transitions. The obtained fits are still reasonable, evidenced by the fitted spectra shown in Fig. S21.† The estimated Ncoh of the spin-coated films are shown in Fig. 2b as a function of annealing temperature. Due to the above discussed reasons these values should be taken as an effective, relative measure for the exciton delocalization, instead of absolute values.
With the described procedure, it is not possible to properly estimate Ncoh for the samples of regime I due to the absence of pronounced J- and H-aggregate peaks (see Fig. 2a). We do however observe a slight anisotropic optical absorption behavior of these samples, which suggests that the thin films are not (entirely) composed of uncoupled monomers. Due to the high dipole moment of HB238 we assume the presence of a mixture of monomers and small molecular aggregates for the samples of regime I. For visualization, we set Ncoh = 2 for the samples of regime I. In regime II the samples exhibit Ncoh values of around 15 slightly increasing with increasing temperature. Suddenly, in regime III at an annealing temperature of 120 °C, Ncoh drastically increases to a value of ca. 30 where it stays approximately constant for higher annealing temperatures. We additionally measured the XRD diffraction patterns of all samples (see Fig. S4†). We observe only one significant diffraction peak at 2θ ≈ 5.0°, demonstrating that in all films in regime II and III a similar crystal structure is present and the dependence of its integrated intensity on the annealing temperature is in good agreement with the obtained Ncoh. In regime I no significant diffraction peaks were measured, suggesting amorphous films. The samples in regime II mark the start of crystallization, slowly progressing with increasing annealing temperature within regime II, evidenced by the appearance of slowly growing diffraction peaks. Finally, the integrated diffraction peak intensity increases drastically during the transition from regime II to III, as a threshold annealing temperature for molecular reorganization is reached somewhere between 110 °C and 120 °C.
We conclude, that an Ncoh of ca. 30 represents a saturation value for spin-coated thin films of HB238, determined by an average crystal defect density and disorder. In comparison, Ncoh was calculated with the above-described procedure to be 22 for the dispersed aggregates shown in Fig. 1. The lower Ncoh for dispersed aggregates might be explained with the instantaneous (kinetic) formation upon addition of water and an accompanied probability for packing defects.
We also tried annealing at higher temperatures. Starting from an annealing temperature of 170 °C, we observe pronounced depletion zones in the thin film morphology (dewetting), which become larger and more frequent at higher annealing temperatures (see Fig. S8c–f and S9a, b†). Concomitantly, the corresponding absorbance spectra show a noticeable high-energy spectral shoulder of the J-transition absorption peak (see Fig. S15 and S16a†), which complicates the analysis of Ncoh Finally, at 200 °C the material melted, leaving only mono- and few-layer islands on the surface (see Fig. S9c and d†). Because of these reasons the comparability with the results shown in Fig. 2 is not given and annealing temperatures higher than 160 °C are not further discussed here.
We further investigated the anisotropic optical properties of the fully annealed films (regime III). Fig. 3a shows the absorbance spectra of a spin-coated and annealed thin film recorded at different angles of incidence (θ) for p- and s-polarized light. For increasing angle of incidence, the spectra for p- and s-polarized illumination differ significantly from each other. The H-band absorption at 2.54 eV increases for increasing angle of incidence only for p-polarized excitation, whereas it is virtually absent at normal incidence for p- and generally for s-polarized excitation. At normal incidence the spectra for p- and s-polarized illumination are identical, as expected. The H-band absorption is thus only excited by an electric field that has an out-of-plane component with respect to the substrate (see sketch in Fig. 3c). These results imply the presence of uniaxial anisotropy and reveal a molecular orientation in out-of-plane direction (z-direction) extended over the entire thin film. We confirmed this finding by variable angle spectroscopic ellipsometry (VASE). Since standard spectroscopic ellipsometry in reflection is inherently limited in its sensitivity to the extraordinary components, we included transmission data (intensity as well as ellipsometry) and performed a multi-sample batch analysis.39,40,42 From this we obtained a uniaxial anisotropic complex refractive index consisting of ordinary (in-plane) and extra-ordinary (out-of-plane) components. These complex refractive indices [n (refractive index) and k (exctinction coefficient)] are shown in Fig. 3b.
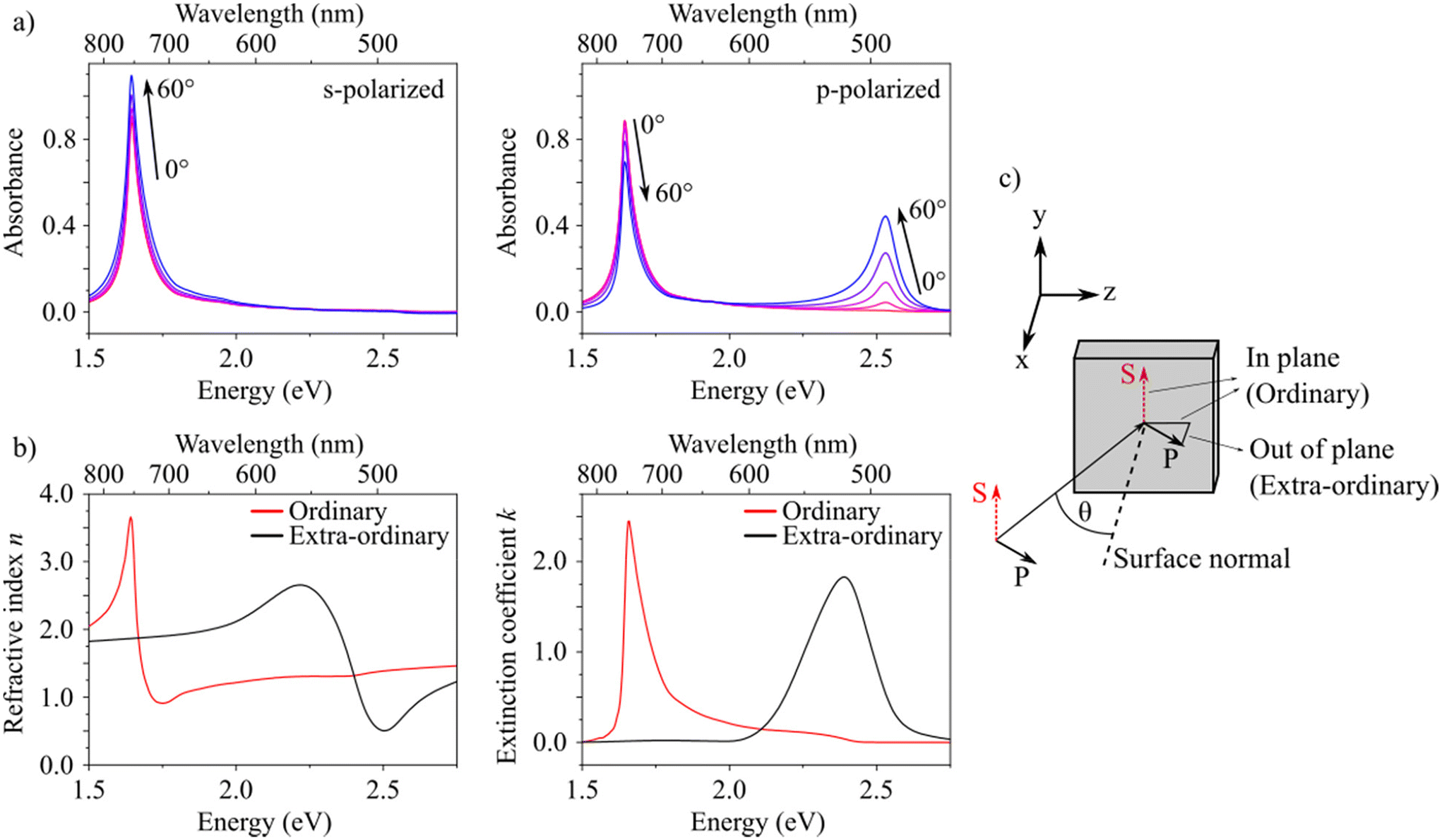 | ||
| Fig. 3 Anisotropic optical behavior of spin-coated and annealed (120 °C, 10 min) HB238 thin films on glass substrates. (a) Absorbance spectra at different angles of incidence (0°–60° in 15° steps) for s- (left) and p-(right) polarized light. (b) Uniaxial anisotropic complex refractive index n (real part, refractive index, left) and k (imaginary part, extinction coefficient, right) determined by variable angle spectroscopic ellipsometry (VASE). (c) Scheme describing the electric field vectors of incident s- and p-polarized light relative to the substrate (xy-plane). | ||
The data in Fig. 3 shows that the J-TDM is oriented in the substrate plane (xy-plane). These findings match our expectation for aggregates with oblique angled monomer's TDMs in which the J- and H-band are polarized perpendicular to each other.24,30,41 The lack of anisotropy in the substrate plane results from the random in-plane orientation of the crystallites (aggregates), since the macroscopic beam size in our experiments averages over many of them. The described morphology can also be observed in AFM images (see Fig. S6f†).
Note that the measured H-transition in the absorbance spectra is blue-shifted by ca. 0.15 eV compared to the peak of the extraordinary extinction coefficient of the H-transition as determined by VASE. The absorbance is calculated from the measured transmission T as the negative decadic logarithm, −log(T). Transmission T contains contributions from absorption A and reflection R, T = 1 − A − R, assuming that there is no scattering.42 The wavelength dependence of the transmission displayed as absorbance therefore does not resemble the extinction coefficient in a simple manner. A similar phenomenon was found in a study of self-organized oligothiophene films where absorbance peaks were calculated for transitions with different TDM orientations.43 There, it was found that absorbance peaks of transitions with out-of-plane TDMs are blue shifted compared to the peak in the complex refractive index used for the calculation. Absorbance peaks of transitions with in-plane TDMs did not show this shift. We assume that these considerations apply to our findings as well.
A series of merocyanine molecules with identical π-systems, but different aliphatic side-chains, including HB238, was investigated recently by means of optical spectroscopy regarding their different aggregation behaviors.19 There, only normal incidence measurements were performed, thus lacking the observation of HB238 H-transition absorption peaks. Instead, photoluminescence excitation measurements were performed, yielding information about the coupled nature of H- and J-transition for some of the investigated merocyanine aggregates.
Here, we prove the coupled nature of J- and H-bands by performing photoluminescence excitation measurements on the spin-coated and annealed films. A 0.8 NA objective was used for the measurement giving rise to AOIs of up to approximately 53°, making it possible to excite the H-transition. We found that the emission from the J-band was observed by exciting the H-band (see Fig. S2†), suggesting that our films consist of oblique-angled aggregates.
To gain further insights into the interdependence of structural and optical properties of HB238 spin-coated films, we computed via first-principles plane-wave based methods (see details in Experimental techniques and methods) the absorption spectrum of a HB238 single crystal. The crystals grown in water/acetone dispersion (Fig. 1) were too small for the determination of the single crystal structure to be used in the first principle calculations. HB238 shows pronounced polymorphism, depending on the crystal growth conditions.19,44,45 Single crystals grown from mesitylene solution44 represent the properties of the aggregates investigated in this study. Therefore, we assume a similar crystal structure amongst the thin films and this single crystal (see Fig. S3 and ESI† part I for details).
Fig. 4a displays the comparison between the experimental polarization-resolved absorbance spectra of spin-coated and annealed (120 °C) thin films at 45° angle of incidence, and the calculated polarization-resolved absorption spectra of the single crystal obtained from mesitylene solution. The good agreement between the experimental and calculated spectra supports our assumption that the crystal structure of the single crystal grown from mesitylene solution is identical to the one in the thin films. By analyzing the induced charge-density,46,47 namely the variation of the electron density upon an applied electric field, we can derive the spatial distribution of the exciton bands. The J-band at approximately 1.65 eV energy is related to a delocalized exciton whose induced charge density is polarized along the Cartesian y-axis (i.e., the b crystallographic axis). The identified H-band at approximately 2.5 eV energy, on the other hand, corresponds to a delocalized exciton along the Cartesian z-axis (i.e., the a crystallographic axis). The calculation shows an additional band around 1.9 eV for light polarized along the Cartesian z-axis. The induced charge density of this band corresponds to a localized exciton. This band is not observed in the thin film experiments. A reason for this could be related to the level of theory used here, which can lead to an overestimation of the intensity of some bands. Interestingly, this band can be observed in the absorption spectrum of dispersed aggregates (see Fig. 1b). Due to its energetic position (compare with Fig. 1a) and localized nature, we attribute this band to non-aggregated monomers.
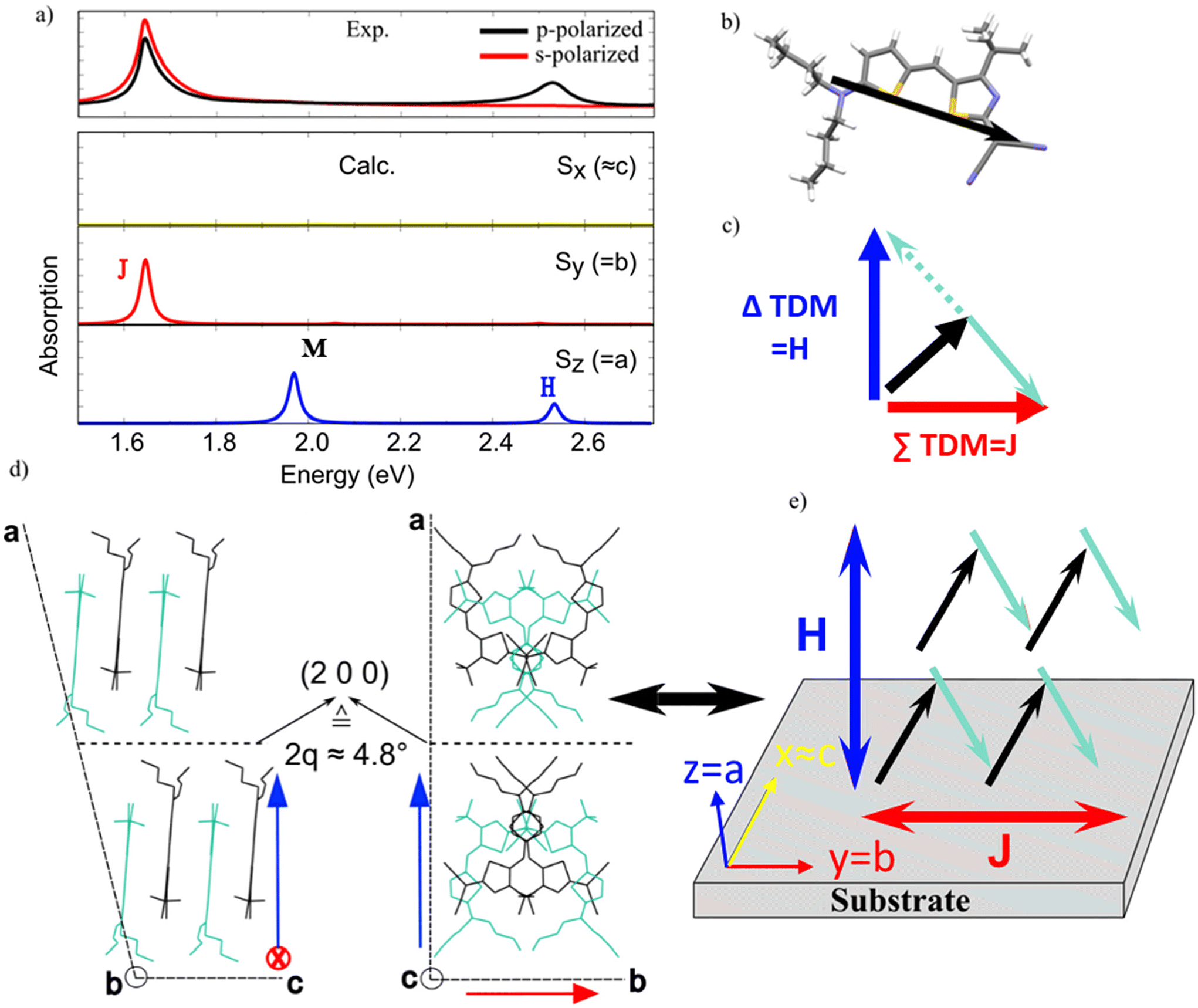 | ||
| Fig. 4 Calculated HB238 single crystal absorption spectrum (computed energies shifted by 0.4 eV) in comparison with experiment. (a) Experimental absorbance of a spin-coated and annealed (120 °C) thin film for p- (black) and s-polarized (red) light at an angle of incidence of 45° (top) and calculated polarized absorption spectra for HB238 single crystal (bottom). Yellow line – spectrum for light polarized along the Cartesian x-axis (i.e., similar to c crystallographic axis), red line – spectrum for light polarized along the Cartesian y-axis (i.e., b crystallographic axis), blue line – spectrum for light polarized along the Cartesian z-axis (i.e., a crystallographic axis). (b) HB238 molecular structure along with the calculated TDM (ωB97X-D/6-311G**, represented by black arrows). (c) Schematic representation of the perpendicular orientation of the TDM sum (red arrow) and difference (blue arrow) of two oblique angled TDMs (black and light blue). The concept is adopted from ref. 24. (d) Two representations of the single crystal unit cell showing the molecular packing. (e) Orientation of molecular TDMs (black and light blue arrows), and resulting J- and H-response (red and blue double arrows) relative to the substrate. In first approximation the substrate plane (see text and Fig. 3) closely coincides with the bc plane of the unit cell, leading to an overall out-of-plane (perpendicular) orientation for the H-band TDM and an in-plane orientation for the J-band TDM, i.e., edge-on orientation of the molecules. | ||
Given the good agreement between experimental and calculated absorption spectra, we can discuss the molecular organization of HB238 molecules in the spin-coated and annealed thin films relative to the substrate surface. Our calculated results can be linked to the molecular orientation inside the single crystal structure by regarding the intuitive molecular exciton model of Kasha et al.24 The monoclinic HB238 single crystal structure shows a shifted, anti-parallel packing between merocyanines whose backbones, and thus TDMs (see Fig. 4b) are oriented with an oblique angle relative to each other (black and light-green arrows in Fig. 4e). For oblique angled TDMs, the TDMs for J- and H-transitions can be constructed by the vector sum and difference of the TDMs of the individual molecules, respectively, and are inevitably oriented perpendicular to each other, which is schematically depicted in Fig. 4c for only two molecular TDMs for simplicity. Indeed, in our calculation, the J-band is polarized along the y Cartesian axis, coinciding with the b crystallographic axis, and the H-band is polarized along the z Cartesian axis, coinciding with the a crystallographic axis. We conclude the bc crystallographic plane is almost parallel to the substrate plane (xy). In this way the resulting orientation of the molecules relative to the substrate leads to a J- and H-transition oriented parallel, and perpendicular to the substrate plane, respectively, which agrees well with the measured ordinary and extraordinary complex refractive indices of the spin-coated and fully annealed films (see Fig. 3b).
The supramolecular orientation presented in Fig. 4d and e suggests high charge carrier mobilities in the plane of the substrate direction, due to the nearly perfect edge-on orientation of the molecules with respect to the substrate.48 In order to quantify the influence of aggregate formation on lateral charge carrier mobility, organic field effect transistors (OFETs) were fabricated by spin coating and annealing HB238 thin films (see details in Experimental techniques and methods). The gate dielectric is silicon-dioxide, i.e., chemically identical to the float glass and fused silica substrates used for the spectroscopic experiments shown in Fig. 2 and 3. The aggregation behavior of HB238 on the OFET and float glass substrates was confirmed independently by 2θ XRD (Fig. S3a and S4†) and AFM measurements (Fig. S5–S9†) and found to be essentially identical. Further, reflectance spectra of the OFET-samples were also measured for every annealing temperature at 50° AOI with p-polarized light, revealing a similar optically anisotropic behavior as for the studies on glass substrates (see Fig. S18†).
The samples used for the charge transport studies were annealed identically with the spectroscopy experiments on glass substrates. For every annealing temperature a separate sample was fabricated. The hole mobility was extracted from measurements of the drain-current in the linear regime for all samples and in the saturation regime for samples annealed at 120 °C or higher. The transistors did not show saturated drain currents for lower annealing temperatures (see Fig. S19 and S20†). The hole mobilities are shown in Fig. 5a as a function of the annealing temperature. As in the spectroscopic investigations, the samples can be ordered in the same three regimes I–III. To visualize the correlation of aggregate formation and charge carrier transport, the hole mobility (Fig. 5a) is shown as a function of Ncoh (Fig. 2b) in Fig. 5b.
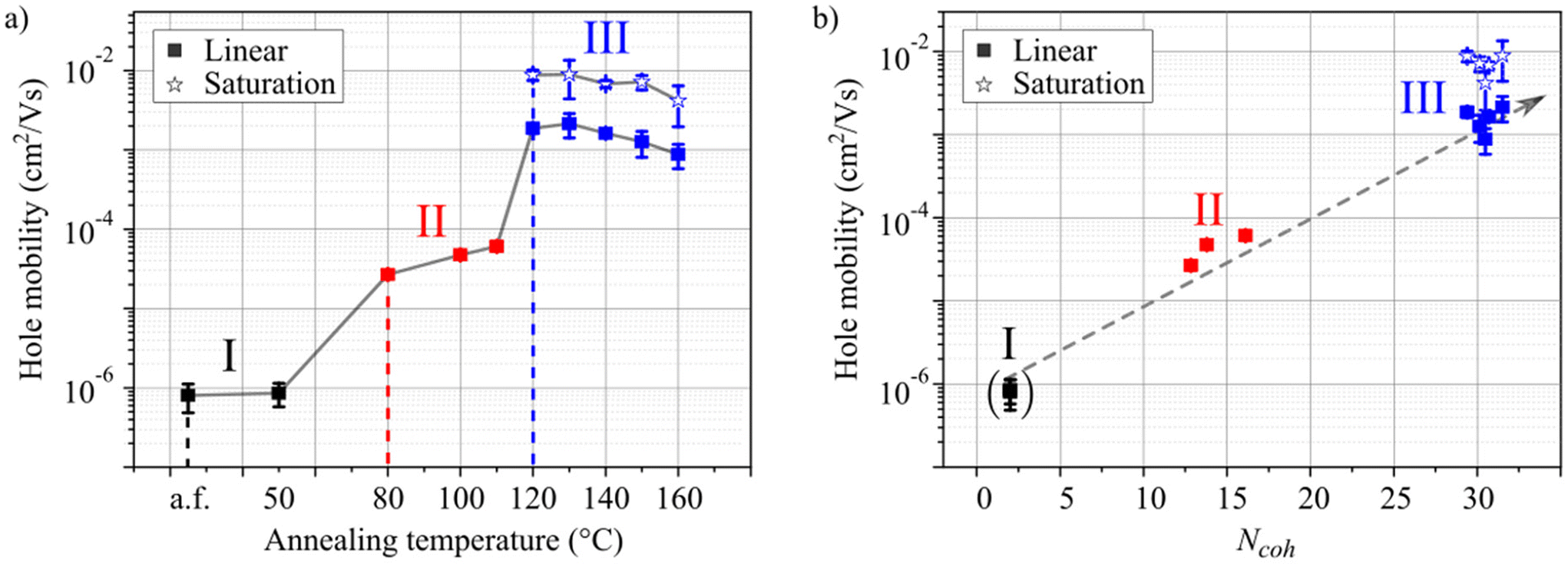 | ||
| Fig. 5 Hole mobility as a function of molecular aggregation. (a) Experimental hole mobilities of HB238 thin films spin-coated on OFET substrates for a non-annealed sample [as fabricated (a.f.)] and for samples after annealing at different temperatures between 50 °C and 160 °C for 10 minutes each. (b) Hole-mobilities as a function of the estimated number of coherently coupled molecules extracted from the absorbance spectra in Fig. 2a. The dashed grey arrow is a guide to the eye. | ||
As shown in Fig. 5a, the charge carrier mobility increases by more than three orders of magnitude as a consequence of the annealing-induced aggregation.44,45 The unannealed sample and the sample annealed at 50 °C exhibit low mobilities of approximately 10−6 cm2 V−1 s−1 (regime I). Upon annealing at 80 °C, 100 °C, or 110 °C the mobility increases by ca. one order of magnitude (regime II). Finally, annealing the samples at 120 °C or higher leads to a drastic increase of charge carrier mobility by more than three orders of magnitude in total, up to approximately 10−3 cm2 V−1 s−1 in the linear and even 10−2 cm2 V−1 s−1 in the saturation regime and remains approximately constant for higher annealing temperatures (regime III).
As observed in Fig. 5b the measured hole mobility and Ncoh appear to be strongly correlated. Both increase significantly during the transitions between the regimes, only gradually within regime II, and remain practically constant in regime III. The increasing hole mobility with increasing Ncoh can be attributed to a higher degree of crystallization with higher annealing temperature, as also evident from the XRD data (Fig. S4†). In crystalline domains charge transport proceeds via band or polaron transport,49 as opposed to the much slower hopping transport in amorphous domains. We attribute the saturation of both, Ncoh and hole mobility in regime III to a constant defect density for spin-coated thin films of HB238 after the threshold annealing temperature is reached (transition from regime II to III). This explanation is rationalized, by the fact that defects represent a localization site for excitons. At the same time, defects lead to either trap states or potential wells at which charge transport is hindered and might proceed by hopping.
The combined spectroscopic, XRD, and charge transport data proves that in regime III the spin-coated HB238 thin films entirely consist of nearly perfect edge-on oriented molecules (out-of-plane anisotropy), which significantly increases charge carrier mobility in a FET geometry. However, the crystallite orientation within the substrate plane is random (in-plane isotropy; see Fig. S6–S8†). In a computational study on the exactly same crystal structure, it was found, that the most efficient transport pathway for holes is along the b crystallographic axis,44,45 which interestingly coincides with the orientation of the TDM of the J-transition, schematically shown in Fig. 4. This coincidence allows the determination of the direction of most efficient hole transport by polarization-resolved microspectroscopic methods.
By growing larger and in-plane oriented crystalline domains the hole mobility can potentially be enhanced. The morphology of thin films can be improved by utilization of ordered substrates, acting as a “template” for the organic thin films. In order to obtain larger ordered domains, HOPG is used as a highly ordered substrate. We fabricated HB238 thin films on HOPG by thermal vapor deposition. The morphology and optical properties of these films were analyzed by means of spatially correlated AFM and PL microspectroscopy. AFM- and PL-micrographs were recorded at the same area of the sample. Furthermore, PL-spectra were measured at different positions within these areas. Fig. 6a and c show AFM topography images of a mono- and a multilayer HB238 thin film on HOPG, respectively (see Experimental techniques and methods for details). Corresponding linear height profiles, indicated by a white bar, are plotted in panel e. The positions where linear height profiles were extracted are highlighted by a white rectangle in panel a and c, displayed enlarged in panel e. The height profiles display approximately 2 nm steps for both the mono- and multilayer. Comparing this with the dimensions of the HB238 crystal structure discussed above (see panel e), we conclude that also here the molecules are oriented edge-on as in the spin-coated thin films in regime III. The step height is about half of the height of the unit cell if the crystallographic bc plane is parallel to the substrate. This corresponds to one layer of edge-on oriented molecules. The multilayer has a “wedding cake” like structure with stacked layers of equal height and a from layer-to-layer decreasing area as elucidated in Fig. 6e. Additionally, thicker crystallites are found on the surface of mono- and multilayer samples. The surface step heights as well as the spectroscopic properties of those differ from the dominating edge-on structure. Here the height profiles show approximately 0.5 nm steps. We therefore conclude that the molecules in these crystallites must be arranged in a “face-on” stacking (lying flat on the surface, see Fig. S23†).
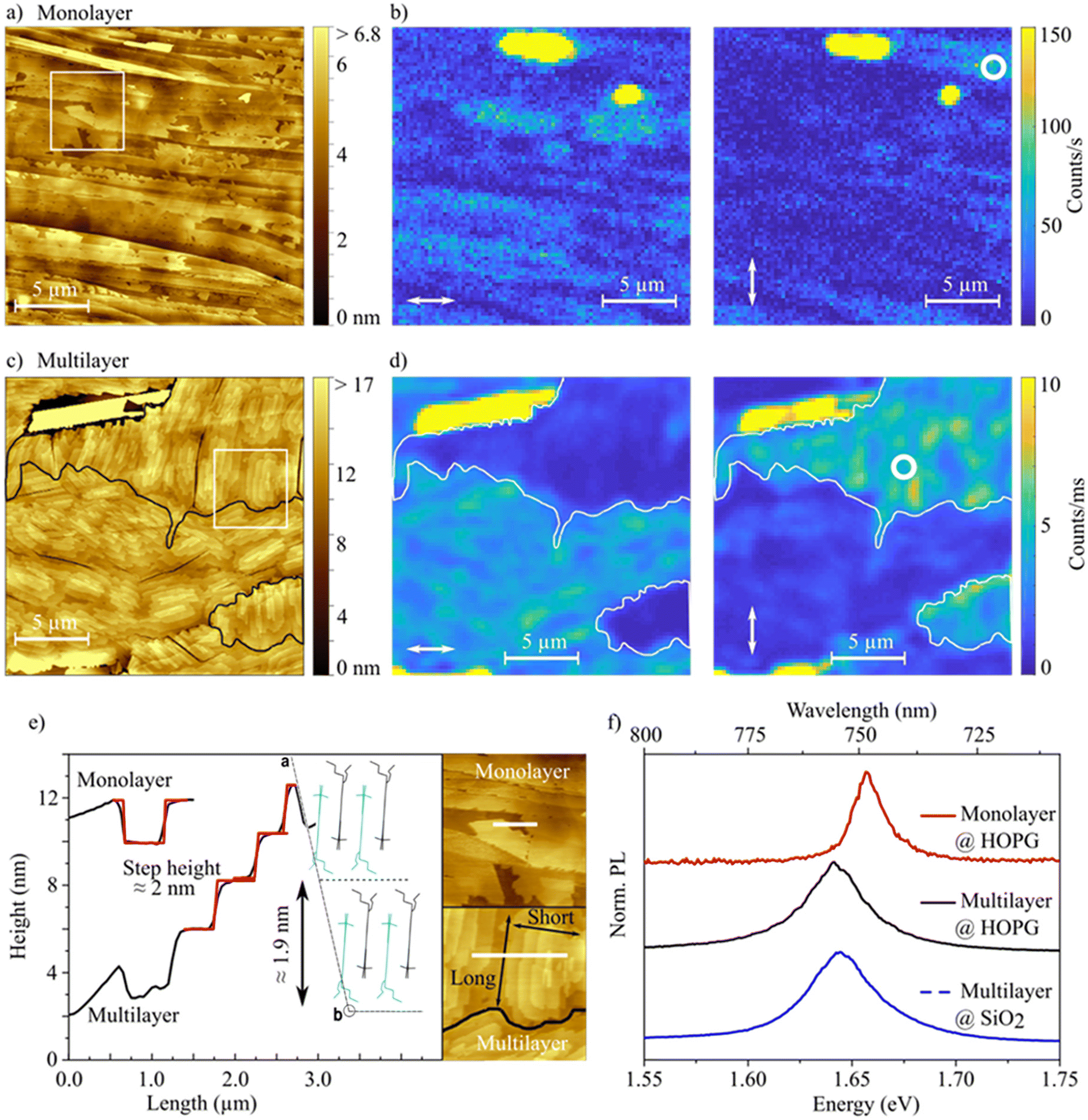 | ||
| Fig. 6 Correlated AFM and polarization-resolved PL study. (a) and (c) AFM images of a thermally evaporated HB238 mono- and multilayer on HOPG. The black solid lines in panel (c) indicate the domain boundaries and are guides to the eye. (b) and (d) Corresponding PL micrographs at the same position for different linear polarizations of the excitation light (indicated by white arrows). The white solid lines in panels (d) indicate the same domain boundaries as the black solid lines in panel (c). (e) Linear height profiles measured at the positions of the white bars indicated in the close-ups of the AFM height images on the right of panel (e). The areas of the close-ups are indicated with white squares in panels (a) and (c). The molecular orientation relative to the HOPG substrate surface is shown in the inset. (f) PL spectra of HB238 mono- (red) and multilayer (black) on HOPG, taken at positions indicated with white circles in panel b and d, compared to PL spectra of a spin-coated film on an OFET substrate annealed at 120 °C (blue). | ||
The evaporated films on HOPG and the spin-coated and annealed thin films on glass both with edge-on domains show similar optical properties. Polarization-resolved reflection spectra for different AOI (see Fig. S22†) display similar uniaxial anisotropy. Furthermore, 2θ XRD measurements confirm the same crystal phase in dispersed aggregates, spin-coated and annealed films, and on HOPG (see Fig. S3a†). Reflection and XRD measurements were performed over a macroscopic spot size and therefore include the face-on crystallites, too. However, since the HOPG surface is covered dominantly by the edge-on structures (as shown in Fig. 6a and c), we conclude that the reflection spectra and XRD-scans display mainly the properties of the edge-on structure. Finally, the PL spectra of the evaporated thin films on HOPG and of the spin-coated and annealed thin films on SiO2 are very similar and show only the characteristic J-transition emission at approximately 1.65 eV as shown in Fig. 6f. These results prove that the same aggregation type is present in all samples.
Fig. 6b and d show PL-micrographs for different linear polarizations of the excitation light indicated with white arrows, for the mono- and multilayer sample, respectively. White circles indicate positions at which spectra were recorded. The spectra are shown as solid lines in panel f. The observed micrographs are remarkably different for different excitation polarizations, best seen for the multilayer in panel d. Since the a crystallographic axis is oriented almost perpendicular to the substrate surface throughout the entire film (apart from face-on crystallites) and the J-transition polarization axis coincides with the b crystallographic axis (see Fig. 4c) we can determine the crystal unit cell orientation on the substrate surface from the PL brightness relative to the excitation polarization. For example, the comparably large edge-on domain on the top right in Fig. 6d is oriented with the b crystallographic axis roughly vertical with respect to the image, indicated by a much brighter PL for vertical excitation polarization. Additionally, by comparing with the AFM image, a similarly vertical crystallite in-plane orientation is identified, determined by the relative orientation of their long-edges, shown in panel c and in the inset in panel e. The black solid lines in the multilayer AFM image (panel c) separate the observed domains and are guides to the eye. With this information it is further possible to determine the directions or paths of most efficient hole mobility, since these coincide as well with the b crystallographic axis, just by observing the AFM or PL images.
Our combined investigations reveal that the aggregation state and molecular orientation determined for the spin-coated and annealed films, which improved charge transport properties by three orders of magnitude, is maintained in the ordered films on HOPG. The grain size of the oriented domains in the thin films on HOPG is orders of magnitude larger (10 s–100 s of μm2, see Fig. 6a and c) than the “crystallites” in spin-coated and annealed films on OFET substrates (<1 μm2, suggested by AFM topography in Fig. S6e–S8a†), which most likely results in significantly increased charge carrier mobility in thin films on HOPG. Since the lateral grain size is increased from less than 1 μm in the spin-coated films on amorphous substrates to more than 10 μm in the evaporated films on the templating substrate HOPG itself, we assume a likewise enhancement of lateral charge carrier mobility by more than an order of magnitude. However, due to the electric conductivity of HOPG it was not possible to fabricate OFETs from these films. The low emission intensity of the monolayer makes it more difficult to observe this effect in the polarized PL-scans (Fig. 6b). In addition, the physical orientation (i.e. the orientation of the long edge) cannot be determined for the monolayer due to the missing additional layers. However, we find large contiguous structures of at least tens of μm2 in size, limited by the substrate's step edges (Fig. 6a).
As a comparison, an HB238 thin film multilayer was evaporated on float glass using the exact same conditions as for the multilayer on HOPG. We observe the same aggregation state as on HOPG. However, the domains are much smaller (around 5 μm2), randomly oriented and separated by pronounced grain boundaries (see Fig. S24† for details). This proves that the large, directionally oriented domains found on HOPG are indeed a result of the ordered HOPG surface.
Fig. 6f shows PL-spectra from the evaporated mono- and multilayer thin film on HOPG, compared with PL spectra from a spin-coated and annealed (120 °C, 10 min) film on SiO2 (OFET substrate). The energetic positions of the J-band emission are similar between all samples and the slight energetic shifts are most likely attributed to dielectric phenomena, driven by the different energetic surroundings of the associated HB238 molecules. The molecules in the multilayer films on HOPG and SiO2 are mainly surrounded by other HB238 molecules and thus possess a similar transition energy. The molecules in the monolayer on HOPG are comparatively stronger interacting with the HOPG surface and we observe slightly blue-shifted emission. It can be recognized that the peak-width of the HOPG-monolayer PL-spectrum is significantly smaller (FWHM = 18 meV (145 cm−1)) than for the other spectra (FWHM = 35–40 meV (282–323 cm−1)). We attribute this finding to less disorder in the monolayer grown on HOPG. It is known from theoretical considerations, that in molecular aggregates disorder induces mixing of the bright lowest state with higher lying states, as well as localization of excitons, both broadening the electronic transition.50–53 It can be assumed, that the monolayer on HOPG exhibits higher order and thus a higher degree of exciton delocalization and potentially charge carrier mobility than the other samples.
Conclusion & outlook
We have studied the aggregation of the merocyanine HB238 and its impact on charge carrier mobility. We find that the mobility in an OFET with a spin-coated thin film of the organic semiconductor can be significantly increased by thermal annealing and accompanied aggregation. Beneficial for charge carrier transport, these films adopt a preferential out-of-plane molecular orientation and thus co-facial orientation of the molecular π-systems. To further enhance lateral charge transport properties, the in-plane morphological order of HB238 thin films was significantly improved by the utilization of HOPG as substrate. Evaporation of HB238 on HOPG led to ordered growth and we observe out-of-plane, as well as in-plane orientation in aggregated domains of up to 100 s of μm2 in size. Especially the monolayer edge-on structure on HOPG displays a high degree of order as evident from very sharp PL-spectra. We expect that the lateral charge carrier mobility of HB238 thin films on ordered HOPG substrates is enhanced by more than an order of magnitude compared to the films on amorphous substrates, according to the likewise enlargement of lateral grain size. This is a promising result on the way to high charge carrier mobility in ordered organic aggregate thin films.Our results show that the molecular arrangement in organic semiconductor thin films significantly influences device performance. The control over the molecular arrangement and grain size is one of the most important goals in the development of efficient organic electronic devices, which, as our results show, can be achieved by choosing a suitable templating substrate. The significantly increased domain size and order, as evidenced by the spectrally sharp PL-spectra, suggest that suitable templates may be used to realize organic electronics devices with superior performance. The here studied thin films further have interesting optical properties. For example, the highly anisotropic nature of HB238 thin films was recently applied in polarization-dependent strong light–matter coupling.53 The anisotropy may find further applications in polarization-sensitive devices and structures.
Data availability
The authors confirm that the data supporting the findings of this study are available within the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the RTG-2591 “TIDE – Template-designed Organic Electronics” (Deutsche Forschungsgemeinschaft, DFG) and by the University of Cologne through the Institutional Strategy of the University of Cologne within the German Excellence Initiative (QM2). K. L. and R. S. acknowledge funding from the DFG project LI 2633/5-1. M. S. is grateful to the PRO RETINA Stiftung for personal funding and the Linz Institute of Technology (LIT-2019-7-INC-313 SEAMBIOF). We thank F. C. Spano (Temple University) for very fruitful discussions about the estimation of the coherence length and S. Olthof (University of Cologne) for support discussing the XRD data. We acknowledge useful discussions with M. Reimer (University of Cologne).References
- R. Zeis, T. Siegrist and C. Kloc, Single-Crystal Field-Effect Transistors Based on Copper Phthalocyanine, Appl. Phys. Lett., 2005, 86, 2004–2006 CrossRef.
- C. Reese, W.-J. Chung, M. Ling, M. Roberts and Z. Bao, High-Performance Microscale Single-Crystal Transistors by Lithography on an Elastomer Dielectric, Appl. Phys. Lett., 2006, 89, 202108 CrossRef.
- O. D. Jurchescu, M. Popinciuc, B. J. Van Wees and T. T. M. Palstra, Interface-Controlled, High-Mobility Organic Transistors, Adv. Mater., 2007, 19, 688–692 CrossRef CAS.
- J. Takeya, M. Yamagishi, Y. Tominari, R. Hirahara, Y. Nakazawa, T. Nishikawa, T. Kawase, T. Shimoda and S. Ogawa, Very High-Mobility Organic Single-Crystal Transistors with in-Crystal Conduction Channels, Appl. Phys. Lett., 2007, 90, 102120 CrossRef.
- A. Liess, M. Stolte, T. He and F. Würthner, Single-Crystal Field-Effect Transistors of a Highly Dipolar Merocyanine Dye, Mater. Horiz., 2016, 3, 72–77 RSC.
- H. Bürckstümmer, E. V. Tulyakova, M. Deppisch, M. R. Lenze, N. M. Kronenberg, M. Gsänger, M. Stolte, K. Meerholz and F. Würthner, Efficient Solution-Processed Bulk Heterojunction Solar Cells by Antiparallel Supramolecular Arrangement of Dipolar Donor–Acceptor Dyes, Angew. Chem., Int. Ed., 2011, 50, 11628–11632 CrossRef PubMed.
- H. Bürckstümmer, N. M. Kronenberg, M. Gsänger, M. Stolte, K. Meerholz and F. Würthner, Tailored Merocyanine Dyes for Solution-Processed BHJ Solar Cells, J. Mater. Chem., 2010, 20, 240–243 RSC.
- F. Würthner and K. Meerholz, Systems Chemistry Approach in Organic Photovoltaics, Chem. – Eur. J., 2010, 16, 9366–9373 CrossRef PubMed.
- H. Wang, F. Zhu, J. Yang, Y. Geng and D. Yan, Weak Epitaxy Growth Affording High-Mobility Thin Films of Disk-like Organic Semiconductors, Adv. Mater., 2007, 19, 2168–2171 CrossRef CAS.
- S. Singha Roy, D. J. Bindl and M. S. Arnold, Templating Highly Crystalline Organic Semiconductors Using Atomic Membranes of Graphene at the Anode/Organic Interface, J. Phys. Chem. Lett., 2012, 3, 873–878 CrossRef CAS PubMed.
- K. Tanabe, H. Saijo and M. Shiojiri, Epitaxial Growth of Merocyanine Dye Molecules on AgBr Surfaces, J. Imaging Sci. Technol., 1998, 42, 434–439 CrossRef CAS.
- J. Yang and D. Yan, Weak Epitaxy Growth of Organic Semiconductor Thin Films, Chem. Soc. Rev., 2009, 38, 2634–2645 RSC.
- J. C. Ribierre, T. Tanaka, L. Zhao, Y. Yokota, S. Matsumoto, D. Hashizume, K. Takaishi, T. Muto, B. Heinrich, S. Méry, F. Mathevet, T. Matsushima, M. Uchiyama, C. Adachi and T. Aoyama, Simultaneous Edge-on to Face-on Reorientation and 1D Alignment of Small π-Conjugated Molecules Using Room-Temperature Mechanical Rubbing, Adv. Funct. Mater., 2018, 1707038, 1–12 Search PubMed.
- S.-J. Kang, Y.-Y. Noh, K.-J. Baeg, J. Ghim, J.-H. Park, D.-Y. Kim, J. S. Kim, J. H. Park and K. Cho, Effect of Rubbed Polyimide Layer on the Field-Effect Mobility in Pentacene Thin-Film Transistors, Appl. Phys. Lett., 2008, 92, 052107 CrossRef.
- T. Schembri, L. Kolb, M. Stolte and F. Würthner, Polarized, color-selective and semi-transparent organic photodiode of aligned merocyanine H-aggregates, J. Mater. Chem. C, 2024, 12, 4948–4953 RSC.
- Y. Yuan, G. Giri, A. L. Ayzner, A. P. Zoombelt, S. C. B. Mannsfeld, J. Chen, D. Nordlund, M. F. Toney, J. Huang and Z. Bao, Ultra-High Mobility Transparent Organic Thin Film Transistors Grown by an off-Centre Spin-Coating Method, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- C. W. Sele, B. K. C. Kjellander, B. Niesen, M. J. Thornton, J. B. P. H. Van Der Putten, K. Myny, H. J. Wondergem, A. Moser, R. Resel, A. J. J. M. Van Breemen, N. Van Aerle, P. Heremans, J. E. Anthony and G. H. Gelinck, Controlled Deposition of Highly Ordered Soluble Acene Thin Films: Effect of Morphology and Crystal Orientation on Transistor Performance, Adv. Mater., 2009, 21, 4926–4931 CrossRef CAS PubMed.
- W. Pisula, A. Menon, M. Stepputat, I. Lieberwirth, U. Kolb, A. Tracz, H. Sirringhaus, T. Pakula and K. Müllen, A Zone-Casting Technique for Device Fabrication of Field-Effect Transistors Based on Discotic Hexa-Perihexabenzocoronene, Adv. Mater., 2005, 17, 684–688 CrossRef CAS.
- A. Liess, A. Arjona-Esteban, A. Kudzus, J. Albert, A. M. Krause, A. Lv, M. Stolte, K. Meerholz and F. Würthner, Ultranarrow Bandwidth Organic Photodiodes by Exchange Narrowing in Merocyanine H- and J-Aggregate Excitonic Systems, Adv. Funct. Mater., 2019, 29(1–9), 1805058 CrossRef.
- A. R. Tameev, A. A. Kozlov, E. I. Mal'tsev, D. A. Lypenko, V. V. Bobinkin and A. V. Vannikov, Charge Carrier Transport in Aromatic Polyimides and Polyimide/J-Aggregate Composites, Proc. SPIE, 2001, 4105, 443–449 CrossRef CAS.
- G. Chen, H. Sasabe, W. Lu, X. F. Wang, J. Kido, Z. Hong and Y. Yang, J-Aggregation, of a Squaraine Dye and Its Application in Organic Photovoltaic Cells, J. Mater. Chem. C, 2013, 1, 6547–6552 RSC.
- M. Más-Montoya and R. A. J. Janssen, The Effect of H- and J-Aggregation on the Photophysical and Photovoltaic Properties of Small Thiophene–Pyridine–DPP Molecules for Bulk-Heterojunction Solar Cells, Adv. Funct. Mater., 2017, 27, 1605779 CrossRef.
- A. Arjona-Esteban, J. Krumrain, A. Liess, M. Stolte, L. Huang, D. Schmidt, V. Stepanenko, M. Gsänger, D. Hertel, K. Meerholz and F. Würthner, Influence of Solid-State Packing of Dipolar Merocyanine Dyes on Transistor and Solar Cell Performances, J. Am. Chem. Soc., 2015, 137, 13524–13534 CrossRef CAS PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, The Exciton Model in Molecular Spectroscopy, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- F. C. Spano, The Spectral Signatures of Frenkel Polarons in H- and J-Aggregates, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed.
- J-Aggregates, ed. T. Kobayashi, World Scientific, Singapore, 2012, vol. 2 Search PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, J-Aggregates: From Serendipitous Discovery to Supramolecular Engineering of Functional Dye Materials, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- A. Eisfeld and J. S. Briggs, The J-H-Bands, of Organic Dye Aggregates, Chem. Phys., 2006, 324, 376–384 CrossRef CAS.
- V. Steinmann, N. M. Kronenberg, M. R. Lenze, S. M. Graf, D. Hertel, K. Meerholz, H. Bürckstümmer, E. V. Tulyakova and F. Würthner, Simple, Highly Efficient Vacuum-Processed Bulk Heterojunction Solar Cells Based on Merocyanine Dyes, Adv. Energy Mater., 2011, 1, 888–893 CrossRef CAS.
- A. S. Davydov, The Theory of Molecular Excitons, Sov. Phys. Usp., 1964, 7, 145–178 CrossRef.
- B. L. Cannon, L. K. Patten, D. L. Kellis, P. H. Davis, J. Lee, E. Graugnard, B. Yurke and W. B. Knowlton, Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates, J. Phys. Chem. A, 2018, 122, 2086–2095 CrossRef CAS PubMed.
- C. Cocchi, T. Breuer, G. Witte and C. Draxl, Polarized Absorbance and Davydov Splitting in Bulk and Thin-Film Pentacene Polymorphs, Phys. Chem. Chem. Phys., 2018, 20, 29724–29736 RSC.
- A. Austin, N. J. Hestand, I. G. McKendry, C. Zhong, X. Zhu, M. J. Zdilla, F. C. Spano and J. M. Szarko, Enhanced Davydov Splitting in Crystals of a Perylene Diimide Derivative, J. Phys. Chem. Lett., 2017, 8, 1118–1123 CrossRef PubMed.
- M. Schulz, J. Zablocki, O. S. Abdullaeva, S. Brück, F. Balzer, A. Lützen, O. Arteaga and M. Schiek, Giant Intrinsic Circular Dichroism of Prolinol-Derived Squaraine Thin Films, Nat. Commun., 2018, 9, 2413 CrossRef PubMed.
- M. Muccini, M. Schneider, C. Taliani, M. Sokolowski, E. Umbach, D. Beljonne, J. Cornil and J. L. Brédas, Effect of Wave-Function Delocalization on the Exciton Splitting in Organic Conjugated Materials, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 6296–6300 CrossRef CAS.
- N. J. Hestand and F. C. Spano, Expanded Theory of H- and J-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- L. D. Bakalis and J. Knoester, ., Linear Absorption as a Tool to Measure the Exciton Delocalization Length in Molecular Assemblies, J. Lumin., 2000, 87, 66–70 CrossRef.
- F. C. Spano, L. Silvestri, P. Spearman, L. Raimondo and S. Tavazzi, Reclassifying Exciton-Phonon Coupling in Molecular Aggregates: Evidence of Strong Nonadiabatic Coupling in Oligothiophene Crystals, J. Chem. Phys., 2007, 127, 184703 CrossRef CAS PubMed.
- M. Campoy-Quiles, M. I. Alonso, D. D. C. Bradley and L. J. Richter, Advanced Ellipsometric Characterization of Conjugated Polymer Films, Adv. Funct. Mater., 2014, 24, 2116–2134 CrossRef CAS.
- J. Zablocki, M. Schulz, G. Schnakenburg, L. Beverina, P. Warzanowski, A. Revelli, M. Grüninger, F. Balzer, K. Meerholz, A. Lützen and M. Schiek, Structure and Dielectric Properties of Anisotropic N-Alkyl Anilino Squaraine Thin Films, J. Phys. Chem. C, 2020, 124, 22721–22732 CrossRef CAS.
- F. C. Spano, The Fundamental Photophysics of Conjugated Oligomer Herringbone Aggregates, J. Chem. Phys., 2003, 118, 981–994 CrossRef CAS.
- A. Kamptner, M. C. Scharber and M. Schiek, Accurate Determination of the Uniaxial Complex Refractive Index and the Optical Band Gap of Polymer Thin Films to Correlate Their Absorption Strength and Onset of Absorption, ChemPhysChem, 2024, 25, e202400233 CrossRef CAS PubMed.
- D. Oelkrug, H. J. Egelhaaf and J. Haiber, Electronic Spectra of Self-Organized Oligothiophene Films with “Standing” and “Lying” Molecular Units, Thin Solid Films, 1996, 284–285, 267–270 CrossRef CAS.
- N. Gildemeister, G. Ricci, L. Böher, J. Neudoerfl, D. Hertel, F. Wuerthner, F. Negri, K. Meerholz and D. Fazzi, Understanding the Structural and Charge Transport Property Relationships for a Variety of Merocyanine Single-Crystals: A Bottom up Computational Investigation, J. Mater. Chem. C, 2021, 9, 10851–10864 RSC.
- N. Gildemeister, S. Geller, R. Herzhoff, F. Negri, K. Meerholz and D. Fazzi, Impact of static and dynamic disorder effects on the charge transport properties of merocyanine single crystals, Mater. Adv., 2024, 5, 8475–8489 RSC.
- M. Guerrini, A. Calzolari and S. Corni, Solid-State Effects on the Optical Excitation of Push–Pull Molecular J-Aggregates by First-Principles Simulations, ACS Omega, 2018, 3, 10481–10486 CrossRef CAS PubMed.
- L. Bursi, A. Calzolari, S. Corni and E. Molinari, Light-Induced Field Enhancement in Nanoscale Systems from First-Principles: The Case of Polyacenes, ACS Photonics, 2014, 1, 1049–1058 CrossRef CAS.
- T. Schembri, J. H. Kim, A. Liess, V. Stepanenko, M. Stolte and F. Würthner, Semitransparent Layers of Social Self-Sorting Merocyanine Dyes for Ultranarrow Bandwidth Organic Photodiodes, Adv. Opt. Mater., 2021, 9, 2100213 CrossRef CAS.
- H. Oberhofer, K. Reuter and J. Blumberger, Charge Transport in Molecular Materials: An Assessment of Computational Methods, Chem. Rev., 2017, 117, 10319–103577 CrossRef CAS PubMed.
- F. C. Spano and S. Mukamel, Superradiance in Molecular Aggregates, J. Chem. Phys., 1989, 91, 683–700 CrossRef CAS.
- H. Fidder, J. Knoester and D. A. Wiersma, Optical Properties of Disordered Molecular Aggregates: A Numerical Study, J. Chem. Phys., 1991, 95, 7880–7890 CrossRef CAS.
- H. Fidder, J. Terpstra and D. A. Wiersma, Dynamics of Frenkel Excitons in Disordered Molecular Aggregates, J. Chem. Phys., 1991, 94, 6895–6907 CrossRef CAS.
- R. Schäfer, L. Böhner, M. Schiek, D. Hertel, K. Meerholz and K. Lindfors, Strong Light–Matter Interaction of Molecular Aggregates with Two Excitonic Transitions, ACS Photonics, 2024, 11, 111–120 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo02088j |
| This journal is © the Partner Organisations 2025 |