
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpin state modulation and kinetic control of thermal contraction in a [Fe2Co2] discrete Prussian blue analogue†
Jyoti
Yadav
 and
Sanjit
Konar
*
and
Sanjit
Konar
*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Elements Building, Bhauri, Bhopal By-pass Road, Madhya Pradesh 462066, India. E-mail: jyoti17@iiserb.ac.in; skonar@iiserb.ac.in
First published on 13th November 2024
Abstract
Stimuli-responsive switchable molecules represent an important category of magnetic materials with significant potential for functional devices. However, engineering complexes with controlled switchability remains challenging due to their sensitivity to lattice interactions. Herein, we report a [Fe2Co2] square complex [FeTp(CN)3]2[Co{(F5-Ph)Py}2]2·2ClO4·4CH3OH·2H2O {1·4CH3OH·2H2O; (F5-Ph)Py = (Z)-N′-(perfluorophenyl) picolinimidamide and Tp = hydrotris(1-pyrazolyl)borate}, tailored with hydrogen bonding (HB) donor and acceptor moieties for effective lattice interactions. The alteration in HB interactions in crystal phases obtained via single-crystal-to-single-crystal (SC–SC) transformation (i.e., 1·4CH3OH·2H2O ↔ 1·2H2O ↔ 1) led to a remarkable change in magnetic properties. Complexes 1·4CH3OH·2H2O and 1 possess [FeIILS(μ-CN)CoIIILS] and [FeIIILS(μ-CN)CoIIHS] configurations, respectively. Meanwhile, 1·2H2O demonstrates multi-responsive (thermo-, photo- and pressure) reversible two-step electron transfer coupled spin transition (ETCST) accompanied by thermal contraction and expansion. The complete diamagnetic [FeIILS(μ-CN)CoIIILS] configuration in 1·2H2O was obtained at an intermediate temperature accompanied by thermal contraction, an unusual behaviour observed for the first time in cyanide-bridged systems. Additionally, 1·2H2O displayed temperature-induced excited spin state trapping (TIESST) of ∼70% of the paramagnetic [FeIIILS(μ-CN)CoIIHS] configuration at low temperatures. The isothermal relaxation of the thermally trapped paramagnetic state shows a much faster and complete conversion to a diamagnetic state at ∼140 K, compared to the relaxation observed at other temperatures (100–190 K), corroborating the observed unique magnetic behaviour. Hence, this result provides valuable insight into the strategic design of complexes with enhanced and controlled switchability for potential applications such as actuators and sensors.
Introduction
Prussian Blue Analogues (PBAs) have attracted great interest due to their potential applications in high-density memory devices, sensors and molecular switches.1–3 Among a range of magnetic properties observed in Fe/Co PBAs, the ETCST phenomenon is particularly intriguing due to the interconversion between diamagnetic [FeIILS(μ-CN)CoIIILS] and paramagnetic [FeIIILS(μ-CN)CoIIHS] configurations (LS = low spin and HS = high spin) in response to various external stimuli such as thermal, light, chemical, pressure, etc.4–15 The ETCST phenomenon shows sensitivity to internal factors, allowing the fine-tuning of electron transfer processes through strategic choices of ancillary ligands around Fe and Co centres, and regulation of secondary interactions involving anions, solvents or co-crystallised molecules.16–28Clerac and co-workers demonstrated the influence of solvent polarity on the ETCST behaviour of the [Fe2Co2] complex.23 In analogous [Fe2Co2] complexes, the disparity in electron transfer properties observed in both the solid and solution phases indicates the dependence of electron transfer on crystal packing, where intermolecular interactions play a crucial role.24,25 Later, Oshio and co-workers reported multi-step ETCST behaviour in a hydrogen bonding (HB) donor–acceptor system involving a [Fe2Co2] complex with 4-cyanophenol, where the diamagnetic [FeIILS(μ-CN)CoIIILS] (LS) and paramagnetic [FeIIILS(μ-CN)CoIIHS] (HS) states present strong and weak HB acceptor behaviour, respectively.26 The molecular level study in cyanide-bridged systems infers that any alteration in secondary interactions, particularly those involving the nitrogen atoms of terminal cyanide groups modulate the redox potential of the metal centres resulting in distinct magnetic behaviour, especially in their solvated and desolvated phases.27–33 However, in most cases, the loss of crystallinity upon desolvation limits their magneto-structural investigation.18,34,35 Therefore, preserving the crystalline nature of the complex with controlled magnetic bistability upon desolvation remains a formidable challenge. In this regard, the SC–SC transformations offer access to compounds that are challenging or impossible to obtain directly through solution reactions.36 But the reports of spin-state switching via SC–SC transformation in discrete molecular crystals are limited, and even scarce in cyanide bridged systems.29,37–40
The ETCST phenomenon generally displays thermodynamically stable LS and HS states at low and high temperatures, respectively,4,5 which are always accompanied by colossal positive thermal expansion and contraction.41,42 However, the observation of thermally induced ETCST accompanied by colossal negative thermal contraction at an intermediate temperature remains unprecedented to date. This thermo-mechanical effect of thermal expansion and contraction is promising for improving the performance of switchable materials as thermal actuators.43
In this study, we report a new [Fe2Co2] square complex 1·4CH3OH·2H2O synthesised using the rational design of ancillary ligands functionalised with –NH2 groups (HB donor) in addition to the nitrogen atom of the terminal cyanide (HB acceptor) of the [FeTp(CN)3] sub-unit (Fig. 1a). A profound effect of SC–SC transformation leads to variations in the HB interactions, thereby affecting the magnetic behaviour of the [Fe2Co2] complexes. In addition, an unusual and interesting negative thermal contraction is observed at an intermediate temperature for the first time.
 | ||
| Fig. 1 (a) Building units of the Fe2Co2 square complex showing H-bonding sites. Green circles (–CN) represent the HB acceptor and orange circles (–NH2) represent the HB donor site, Sol = solvent. (b) Molecular unit of complex showing the [Fe2Co2] square core and (c) structural framework with terminal cyanides in the antiparallel direction. Colour code: brown (Fe), pink (Co), grey (C), blue (N), light green (F) and yellow (B). Hydrogen atoms, solvents and anions are omitted for clarity. | ||
Results and discussion
Synthesis
A methanolic solution of the capping ligand ((F5-Bn)Py, L) and Co(ClO4)2·6H2O was added to a CH3OH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (2:1) solution of Na[FeTp(CN)3] and heated at 40 °C for 30 minutes; the mixture was filtered under hot conditions and kept for slow evaporation at 25 °C. In one week, the resulting filtrate yielded red plate-like crystals of complex 1·4CH3OH·2H2O. The crystals of 1·4CH3OH·2H2O eventually lost methanol molecules forming the partially desolvated complex 1·2H2O when kept at room temperature for ∼1 week. The complete desolvated complex 1 was obtained in a controlled vacuum from 1·4CH3OH·2H2O at 50 °C. The experimental and characterisation details are provided in the ESI.†
:H2O (2:1) solution of Na[FeTp(CN)3] and heated at 40 °C for 30 minutes; the mixture was filtered under hot conditions and kept for slow evaporation at 25 °C. In one week, the resulting filtrate yielded red plate-like crystals of complex 1·4CH3OH·2H2O. The crystals of 1·4CH3OH·2H2O eventually lost methanol molecules forming the partially desolvated complex 1·2H2O when kept at room temperature for ∼1 week. The complete desolvated complex 1 was obtained in a controlled vacuum from 1·4CH3OH·2H2O at 50 °C. The experimental and characterisation details are provided in the ESI.†
Structural analysis
The complex 1·4CH3OH·2H2O and the complexes obtained via SC–SC transformation upon successive loss of solvent molecules, 1·2H2O and 1, crystallise in the monoclinic P21/c space group. All the complexes consist of a dicationic [Fe2Co2] square core with Co and Fe centres positioned at alternate corners bridged by cyanide groups (Fig. 1b). Two ClO4− anions are present in the lattice forming a charge-neutral complex. Two of the cyanide groups of each [FeTp(CN)3]− sub-unit are involved in bridging with Co-centers, while the third cyanide group remains non-coordinated, oriented perpendicular to the [Fe2Co2] square core, in an antiparallel direction to the other (Fig. 1c). Each Fe and Co centre adopts a distorted octahedral geometry featuring a C3N3 and N6 coordination environment, respectively (Tables S4 and S10†).44 At low temperatures, the bond lengths around the Fe and Co-centres in 1·4CH3OH·2H2O correspond to [FeIILS(μ-CN)CoIIILS] linkages, while those in 1 confirm the [FeIIILS(μ-CN)CoIIHS] configuration (Table 1). However, the bond lengths around the metal centres in 1·2H2O are intermediate between the bond lengths observed for 1·4CH3OH·2H2O and 1, indicating the presence of an intermediate (IM) phase with contributions from both LS and HS phases (Table 1).26| Atoms | 1·4CH3OH·2H2O | 1·2H2O | 1 |
|---|---|---|---|
| 100 K (Å) | 110 K (Å) | 117 K (Å) | |
| Fe–C | 1.871 | 1.883 | 1.907 |
| Fe–N | 2.001 | 1.994 | 1.968 |
| Co–N | 1.918 | 1.997 | 2.115 |
The structural investigation of the three complexes shows similar crystal packing of the [Fe2Co2] square core which remained consistent during the SC–SC transformation upon the sequential loss of solvent molecules. In contrast, the HB interactions involving the lattice solvents, anions and the square core varied drastically. In complex 1·4CH3OH·2H2O, the HB interactions involve –NH2, –NCN, H2O, CH3OH and OClO4, forming an extended three-dimensional network (Fig. 2a, Table 2). During the SC–SC transformation from 1·4CH3OH·2H2O to 1·2H2O, although the three-dimensional extended HB network is retained, the loss of CH3OH molecules leads to the breakage of OH2O⋯OCH3OH, NCN⋯OCH3OH and OH2O⋯OCH3OH interactions and the formation of new HB interactions between NCN⋯OH2O (Fig. 2b, Table 2). However, in complex 1, the HB interactions are only limited to –NH2 and OClO4 groups (Fig. 2c, Table 2). The electron-withdrawing effect of the HB interaction with the terminal cyanide and the electron-donating effect of the HB interaction with –NH2 contribute to the positive and negative shifts in the redox potential of the Fe and Co-centres, respectively.26–28 Hence, the ambient potential difference between the two metal centres is expected to facilitate an efficient electron transfer process in 1·2H2O. The interplay between HB acceptor and HB donor interactions in 1·4CH3OH·2H2O results in a dominant positive shift of redox potential around the Fe-centre compared to the negative potential shift around the Co-centre. This stabilises the diamagnetic [FeIILS(μ-CN)CoIIILS] state. However, the weak HB interaction in 1 stabilises the paramagnetic [FeIIILS(μ-CN)CoIIHS] state (Table 2).26
 | ||
| Fig. 2 HB interactions in complexes (a) 1·4CH3OH·2H2O, (b) 1·2H2O and (c) 1. Colour code: brown (Fe), pink (Co), grey (C), blue (N), light green (F), dark green (Cl), red (O) and yellow (B). Hydrogen atoms not involved in HB interactions are omitted for clarity. | ||
| HB interactions*(Å) | 1·4CH3OH·2H2O | 1·2H2O | 1 |
|---|---|---|---|
| a All HB interactions were obtained with a maximum donor–acceptor distance of 2.9 Å and a minimum angle of 120°, except in 1. | |||
| NNH2⋯OClO4− | 2.857 Å | — | 2.866 Å |
| OH2O⋯OCH3OH | 2.698 Å | — | — |
| NNH2⋯OH2O | 2.875 Å | 2.807 Å | — |
| OH2O⋯OCH3OH | 2.762 Å | — | — |
| NCN⋯OCH3OH | 2.660 Å | — | — |
| NCN⋯OH2O | — | 2.800 Å | — |
The modulation of secondary interactions also results in variations in Co–N–C bend angles and octahedral distortions around the Co centre across the three complexes. The degree of distortion can be quantified using the parameters Σ (the sum of |90 − α| for the 12 cis-N–Co–N angles around the Co centre), Θ (the sum of deviations of 24 unique N–Co–N angles from 60°) and CShMCo (the continuous shape measure relative to the ideal octahedron at the Co-centre).44,45 At low temperatures, the values of Σ, Θ and CShMCo obtained for the Co ion in complex 1 are higher than those in 1·4CH3OH·2H2O and 1·2H2O, leading to a weaker ligand field around Co-centres in complex 1. This stabilises the Co-ion in the 2+ HS state resulting in the paramagnetic [FeIIILS(μ-CN)CoIIHS] configuration (Table 3).13,32 In contrast, the complex 1·4CH3OH·2H2O, with profound HB interactions shows the smallest values of Σ, Θ and CShMCo, indicating a stronger ligand field around the Co ion stabilising the 3+ LS state, resulting in the formation of the diamagnetic [FeIILS(μ-CN)CoIIILS] pairs. The complex 1·2H2O with weaker HB interactions compared to the complex 1·4CH3OH·2H2O, exhibits larger Co–N–C bend angles and distortion in the [Fe2(μ-CN)Co2] square core, although smaller than in complex 1. As a result, both 2+ and 3+ redox states of Co ions co-exist at low temperatures, with significant contributions of HS:3LS in complex 1·2H2O. Thus, the HB interactions and the distortions play a key role in the existence of ETCST in the complex 1·2H2O.32
| Complex | Σ (°) | Θ (°) | CShMCo |
|---|---|---|---|
| 1·4CH3OH·2H2O | 45.286 | 138.538 | 0.393 |
| 1·2H2O | 54.673 | 175.513 | 0.723 |
| 1 | 70.962 | 211.937 | 1.137 |
These studies show that the structural reorganizations at the molecular and supramolecular levels are important for inducing the thermo-responsive ETCST phenomenon in the cyanide-bridged systems.
Magnetic studies
The dc magnetic study was performed in the 2–300 K temperature range at a 1 T applied magnetic field. For complex 1·4CH3OH·2H2O, the χMT value at 300 K is found to be 0.37 cm3 K mol−1, which corresponds to the presence of diamagnetic [FeIILS(μ-CN)CoIIILS] linkages. This χMT value remains constant upon cooling to 2 K, indicating no significant change in the electronic and spin states of the diamagnetic linkages.17,23 Hence, the complex 1·4CH3OH·2H2O remains in a diamagnetic state in the whole temperature range (2–300 K).In complex 1·2H2O, the χMT value of 6.37 cm3 K mol−1 at 300 K aligns closely with the calculated spin-only value for a 2:2 magnetically isolated low spin FeIII (g = 2.7) and high spin CoII (g = 2.3) system. This suggests the presence of [FeIIILS(μ-CN)CoIIHS] linkages (HS state) at 300 K.17,23 Upon lowering the temperature below 200 K, the χMT value decreases abruptly to 3.5 cm3 K mol−1 which corresponds to 1:1 [FeIIILS(μ-CN)CoIIHS] and [FeIILS(μ-CN)CoIIILS] states (HS:LS, IM1 state). The χMT value decreases sharply to 1.92 cm3 K mol−1 upon further lowering the temperature below 168 K, indicating the presence of the HS:3LS fraction (IM2 state).26 A sharp decrease in the χMT value below 5 K is due to zero-field splitting (ZFS) of the residual [FeIIILS(μ-CN)CoIIHS] fractions present at low temperatures. During the heating mode, the magnetic profile follows a similar transition behaviour presenting a reversible two-step ETCST phenomenon possessing hysteresis widths of 8.6 K and 2.9 K for IM2-IM1 and IM1-HS transitions, respectively (Fig. 3). The contribution from the HS ([FeIIILS(μ-CN)CoIIHS]) and LS [FeIILS(μ-CN)CoIIILS] states in the IM phase was confirmed from the high spin fraction (γHS) calculations (Fig. S14b†). Furthermore, low-temperature variable-field magnetic measurements (2–100 K) verified the absence of ferromagnetic impurities in the sample (Fig. S15†).
 | ||
| Fig. 3 (a) χMT vs. temperature plot for complexes 1·4CH3OH·2H2O (black circles), 1·2H2O (red circles) and 1 (blue circles) @5 K min−1 and (b) the magnetic data of the complex 1·2H2O with the ageing of the sample. The magnetic data were measured on the amorphous sample obtained from direct mixing. | ||
The complex 1 displayed a χMT value of 6.1 cm3 K mol−1 at 300 K, corresponding to the presence of a paramagnetic [FeIIILS(μ-CN)CoIIHS] (HS) state. A slight increase in the χMT value up to 150 K was observed which decreased gradually below 80 K. The decrease in the χMT value becomes sharper below 5 K due to ZFS for the magnetically active FeIIILS and CoIIHS centres (Fig. 3). Hence, the complex 1 remains in a paramagnetic state. The reversible formation of complexes 1·4CH3OH·2H2O and 1·2H2O was investigated by keeping complex 1 in a CH3OH:H2O mixture for about a week. The structural characterisation was performed using infrared (IR) spectroscopy (Fig. S12†) and the solvent loss was assessed through thermogravimetric analysis (Fig. S10b†). Interestingly, upon resolvation, the complex 1·4CH3OH·2H2O was not isolated; instead, the obtained sample corresponded more closely to the formation of complex 1·2H2O. The magnetic study of the obtained complex shows the regained magnetic profile of the reversible two-step ETCST process like the one observed for complex 1·2H2O (Fig. S16†).
The magnetic properties of complex 1·2H2O were studied with the ageing of the sample, i.e., the sample was kept for a long time and magnetic measurement was performed at different intervals using the same set of samples. Over time, the initial two-step abrupt ETCST process became more gradual with an increased fraction of the paramagnetic state at low temperatures (Fig. 3b). The IR spectrum displayed similar νBH (∼2490 cm−1) and νCN (∼2154, 2144, 2135 cm−1) and mainly stretching frequencies in the fingerprint region (1500–400 cm−1), confirming the retained structural integrity of the complex with ageing (Fig. S13†). While the thermogravimetric analysis (TGA) followed by each of the magnetic measurements gave a quantitative analysis of solvent loss (Fig. S11†). Hence, the observed magnetic behaviour can be attributed to the complex 1·(2 − x)H2O (x = <2) showing sequential loss of the water molecules from the crystal lattice. This eventually resulted in the loss of HB interactions between the water molecules present in the lattice and –NH2 groups as well as the terminal cyanides of the [Fe2Co2] molecular framework. As a result, the paramagnetic [FeIIILS(μ-CN)CoIIHS] state in complex 1·2H2O becomes more stabilised with time. The inability to obtain crystal structures of the complex with a controlled and stepwise loss of water molecules hinders the precise determination of the quantity and positions of hydrogen bonding interactions in the system.
Temperature-dependent structural analysis
To gain insight into the structural changes in 1·2H2O during thermal-induced two-step ETCST, temperature-dependent structural studies were performed during both heating and cooling modes between 110 and 200 K. As discussed in the earlier section, the average bond lengths around Fe and Co-centres at 110 K were 1.940 and 1.997 Å, respectively, indicating the presence of an intermediate state (IM2) with LS:3HS states. With increasing temperature to 168 K, the average Co–N bond lengths increase to 2.004 Å, which is in agreement with the 1:1 LS:HS state.26 This is likely due to the presence of CoII in an intermixed spin state or corresponds to the average bond lengths for 1:1 [FeIIILS(μ-CN)CoIIHS] and [FeIILS(μ-CN)CoIIILS] units in the asymmetric structure, signifying the IM1 (LS:HS) phase. At 200 K, the complex exhibits the [FeIIILS(μ-CN)CoIIHS] configuration, evidenced by an average Co–N bond length of 2.113 Å.26 The Σ value for the Co-centre at 110 K is estimated to be 54.6°, which is slightly higher for CoIIILS, indicating the presence of an intermediate state. While the Σ value of 71.8° at 200 K is typical for CoIIHS ions. This indicates the complete conversion of the complex 1·2H2O into its complete HS state. With increasing temperature from 110 K to 200 K, the structural analysis shows an increase in NNH2⋯OH2O and NCN⋯OH2O distances by 0.05–0.06 Å along with 11° and 3° deviation in Co–N–C and Fe–C–N bond angles, respectively. The structural investigation during cooling shows a similar change in bond lengths around the Co and Fe-centres, justifying the reversible switching of the oxidation and spin states of Fe and Co-centres in complex 1·2H2O while maintaining the crystal symmetry. The crystallographic details are provided in the ESI.†
Thermal contraction and expansion
During the temperature-dependent structural analysis of complex 1·2H2O @5 K min−1, a gradual increase in the crystallographic cell axes b and c occurs with increasing temperature. However, the a-axis increases from 110 K to 140 K and decreases above 140 K. The combined effect leads to an increase in cell volume with increasing temperature (Fig. 4, @5 K min−1) showing a positive thermal expansion during heating. Similarly, the cell volume and b and c-axes show a contraction during the cooling mode (Fig. 4, @5 K min−1).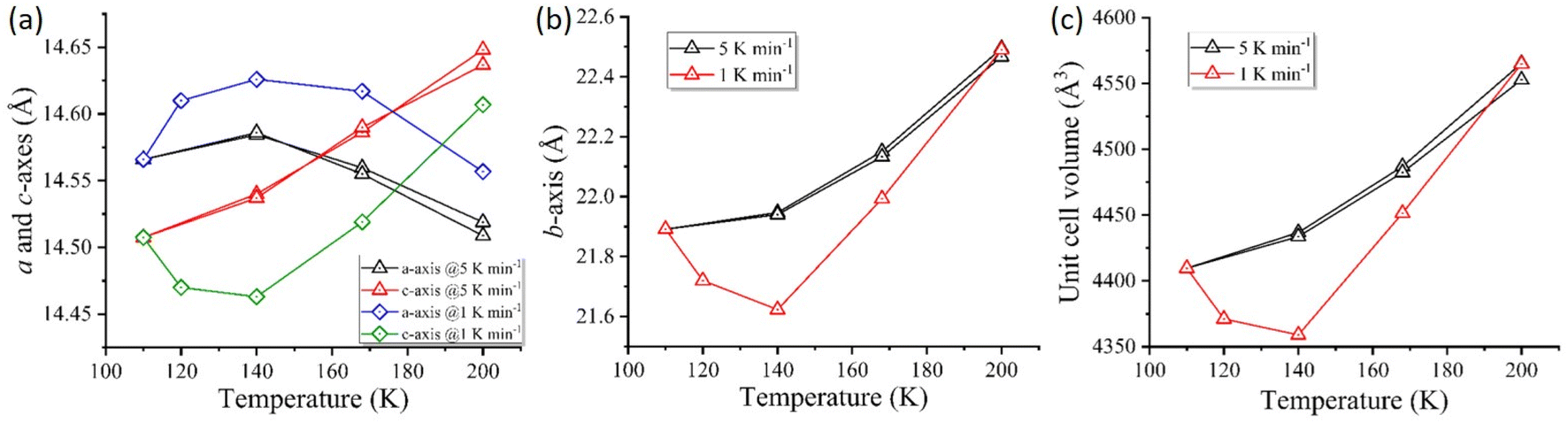 | ||
| Fig. 4 (a) Change in length of the b-axis with changes in temperatures at different scan rates. Black symbols represent the respective changes at 5 K min−1 (cooling and heating) and red symbols correspond to changes at 1 K min−1 (heating). (b) Change in a & c-axes with varying scan rates, and (c) change in unit cell volume showing thermal contraction and expansion during heating mode (red) and cooling and heating mode (black) in 1·2H2O. | ||
Interestingly, the structural investigation during heating at a slow scan rate of 1 K min−1, unveiled an anomalous negative thermal contraction at an intermediate temperature (∼140 K). At 140 K, the complex exhibited the shortest Co–N average bond length of 1.949 Å compared to the average bond length of 1.997 Å observed at 110 K (Table 4).
| Atoms | 110 K | 140 K | 168 K | 200 K |
|---|---|---|---|---|
| Fe–C | 1.883 | 1.875 | 1.893 | 1.915 |
| Fe–N | 1.994 | 1.993 | 1.984 | 1.965 |
| Co–N | 1.997 | 1.949 | 2.017 | 2.110 |
The crystallographic c and b axes shorten from 110 K to 140 K, followed by a subsequent elongation of axes with temperature reaching 200 K. In contrast, the crystallographic a-axis follows a reverse order showing an increase in axis length from 110 K to 140 K and a significant decrease while approaching 200 K (Fig. 4, @1 K min−1). As a result of the varying elongation and shortening in axis lengths (a, b and c), the cell volume (V) experiences an overall contraction from 110 K to 140 K and expansion up to 200 K (Table 5).
| Δcell parameter | Percentage change (±%) | ||
|---|---|---|---|
| 110 K | 140 K* | 200 K* | |
| a ‘+’ and ‘−’ symbols represent the increase and decrease in crystal cell parameters, respectively, during heating at different temperatures. *The values were calculated considering the 110 K structure as the standard. | |||
| Δa (Å) | 0 | +0.41 | −0.47 |
| Δb (Å) | 0 | −0.31 | +1 |
| Δc (Å) | 0 | −1.23 | +4.01 |
| ΔV (Å3) | 0 | −1.14 | +4.5 |
The quantitative analysis of HB distances during heating at 1 K min−1 shows shortening of NNH2⋯OH2O (2.784 Å) and NCN⋯OH2O (2.785 Å) distances observed at the same temperature with a heating rate of 5 K min−1 (NNH2⋯OH2O = 2.810 Å and NCN⋯OH2O = 2.811 Å). Similarly, the distortion parameter (Σ) and CShMCo for the Co-centre display smaller values at 140 K during the heating mode with different sweep rates: @1 K min−1; Σ = 49.318° and CShMCo = 0.499 and @5 K min−1; Σ = 55.763° and CShMCo = 0.706. This further justifies the stabilisation of the higher fraction of the diamagnetic [FeIILS(μ-CN)CoIIILS] state at an intermediate temperature (Table 6).
| Interactions/parameters | 110 K | 140 K | 168 K | 200 K |
|---|---|---|---|---|
| NNH2⋯OH2O (Å) | 2.807 | 2.784 | 2.812 | 2.855 |
| NCN⋯OH2O (Å) | 2.800 | 2.785 | 2.825 | 2.869 |
| Σ (°) | 54.673 | 49.318 | 59.067 | 70.438 |
| CShMCo | 0.723 | 0.499 | 0.786 | 1.107 |
This anomalous behaviour of the stability of the diamagnetic state at 140 K in complex 1·2H2O was also evident from the magnetic susceptibility data acquired during the heating mode @1 K min−1. The χMT value of 0.701 cm3 K mol−1 at 100 K, decreases to a minimum value of 0.05 cm3 K mol−1 as the temperature approaches 140 K. The observed χMT value at 140 K aligns well with the complete conversion of the [Fe2Co2] complex to its diamagnetic [FeIILS(μ-CN)CoIIILS] configuration (LS state).18,26 The χMT value further increases to 6.23 cm3 K mol−1 at 190 K and above temperatures, showing a complete conversion into the paramagnetic [FeIIILS(μ-CN)CoIIHS] configuration (HS state). The two-step ETCST behaviour of the complex 1·2H2O observed at 1 K min−1 shows a significant increase in the hysteresis widths of 16 K and 2.3 K from the LS-IM1 and IM1-HS phase, respectively, compared to the hysteresis widths obtained at the 5 K min−1 scan rate (Fig. 5).
 | ||
| Fig. 5 χ M T vs. temperature plot for complex 1·2H2O at different scan rates from 1 to 10 K min−1 at a 1 T applied magnetic field, showing complete conversion to the diamagnetic state achieved at 140 K. The measurement was performed on the freshly obtained crystals of the complex. | ||
TIESST and isothermal relaxation
The complex 1·2H2O shows temperature-induced excited spin state trapping (TIESST) of ∼50% and 70% of the paramagnetic [FeIIILS(μ-CN)CoIIHS] state upon rapid cooling to 100 K at 25 K min−1 and 30 K min−1, respectively (Fig. S17, ESI†). To gain insight into the stability of the diamagnetic state at intermediate temperature and justify that the observed behaviour is not due to the thermal regain of the diamagnetic configuration after the TIESST effect, but, an inherent property of the complex 1·2H2O, the sample was quickly cooled at 30 K min−1 to different temperatures and subjected to isothermal relaxation. This measurement technique maintains the uniformity in the percentage of high spin (paramagnetic) fraction (γHS) for isothermal relaxation measurements at different intervals of temperature ranging from 100 K to 190 K (Fig. 6a and b and S18†). A remarkable decay (within ∼2 hours) into its diamagnetic state (γHS = 0) was observed at 140 K, and the obtained relaxation curve (Fig. 6a and b) was fitted using the exponential law with the temperature-dependent rate constant k(T) as mentioned below:| γHS = Ae−k(T)t | (1) |
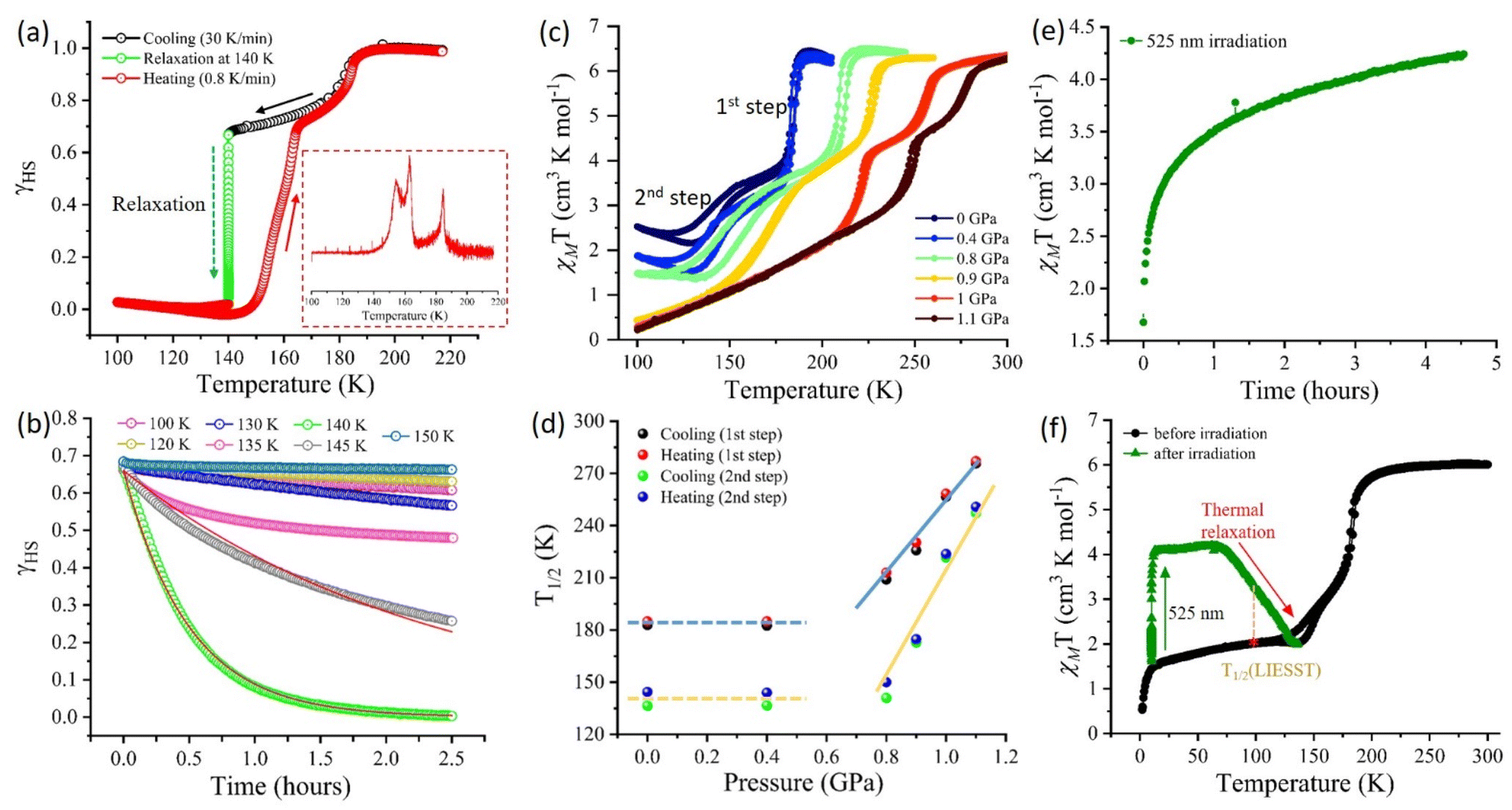 | ||
| Fig. 6 (a) γHSvs. time plot showing the relaxation study at 140 K (inset: 1st derivative plot during heating) and (b) γHS vs. time plot of relaxation at different temperatures for complex 1·2H2O. The solid red line represents the fitting of the respective relaxation data, (c) χMT vs. T plots with the application of pressure measured at a sweep rate of 3 K min−1. (d) T1/2vs. pressure plot following Clapeyron's equation above the threshold pressure of 0.8 GPa. (e) χMT vs. time plot upon irradiation with 525 nm of light, and (f) χMT vs. T plot upon irradiation with 525 nm light at 10 K and thermal relaxation @2 K min−1 for complex 1·2H2O at an applied dc field of 1 T. *The green symbols in (e) and (f) were collected using a fibre optic sample holder in a light setup, merged with the magnetic profile measured using a brass holder, represented in black symbols. *The pressure and light-responsive magnetic measurements were performed at a sweep rate of 5 K min−1 and an applied magnetic field of 1 T. | ||
Pressure-responsive ETCST in complex 1·2H2O
As discussed earlier, at ambient pressure, the complex retained its two-step transition behaviour with T1/2 values of 182.8 K and 136.5 K during cooling and 185 K and 144.5 K during the heating mode, for 1st and 2nd transitions, respectively (Fig. 6c). Interestingly, the application of 0.4 GPa hydrostatic pressure showed a significant increase in the diamagnetic [FeIILS(μ-CN)CoIILS] fraction at low temperatures from χMT = 2.55 cm3 K mol−1 @0 GPa to 1.9 cm3 K mol−1 @0.4 GPa at 100 K, while the transition temperatures remained unaffected. However, increasing the applied pressure of ∼0.8 GPa showed a sharp increase in T1/2, indicating a sensitive response to the applied pressure. The two-step ETCST behaviour was preserved with a linear increase in the transition temperatures up to the applied pressure of 1.1 GPa (Fig. 6c and d). As expected, the application of pressure shortens the metal–ligand distance and increases the ligand field strength around the metal centres, thereby favouring the diamagnetic [FeIILS(μ-CN)CoIILS] state, which is stabilised over a larger temperature range as the pressure increases.14,28,41,46,47 Remarkably, in complex 1·2H2O, the increase in applied pressure not only elevates the transition temperatures but also stabilises the diamagnetic [FeIILS(μ-CN)CoIIILS] state, as observed from the higher fraction of the LS state. The extracted T1/2 values plotted as a function of pressure shows two distinct regions, (i) below 0.8 GPa – with constant transition temperatures indicating a threshold pressure between 0 and 0.8 GPa, and (ii) above 0.8 GPa – bearing a linear relationship between the transition temperatures (heating T1/2↑ and cooling T1/2↓) and pressure (Fig. 6d). This behaviour follows the Clausius–Clapeyron equation (eqn (2)) as observed in any spin-crossover (SCO) complex.14,47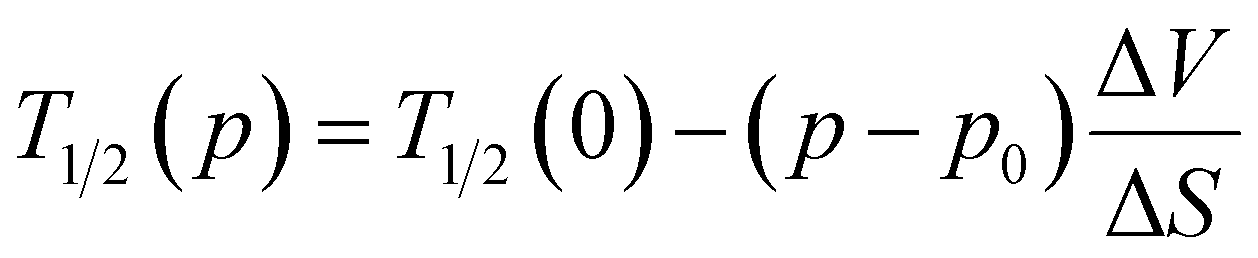 | (2) |
Photo-responsive ETCST in complex 1·2H2O
The light-induced excited spin state trapping (LIESST) of the complex 1·2H2O was studied by irradiating the complex with different wavelengths of light ranging from 436 to 850 nm at 10 K for ∼4–5 hours. The complex was initially present in the IM2 state with 1HS:3LS fractions and showed a significant photo-conversion (15–45%) to the metastable paramagnetic [FeIIILS(μ-CN)CoIIHS] state upon irradiation with light of 525, 640, and 740 nm wavelengths (Fig. 6e and S20†). Out of these, the photo conversion with 525 nm light was the highest, with ∼45% conversion to the metastable state (Fig. 6f). The photo-generated metastable state remained stable up to 75 K. With a further increase in temperature, thermal relaxation to its original IM2 phase was observed at ∼133 K with T1/2(LIESST) = 100 K (Fig. 6f). Above this temperature, the complex follows the initial two-step transition, reaching a complete paramagnetic [FeIIILS(μ-CN)CoIIHS] state at 200 K (Fig. 6f).
Conclusions
In summary, we have reported a [Fe2Co2] square complex functionalised with HB donor and acceptor moieties, showing SC–SC transformation with a sequential loss of solvent molecules from the crystal lattice. Interestingly, the solvatomorphic complexes 1·4CH3OH·2H2O, 1·2H2O and 1 exhibit varied HB interactions, leading to diverse magnetic behaviours. Complex 1·4CH3OH·2H2O adopts a diamagnetic [FeIILS(μ-CN)CoIIILS] phase, 1·2H2O exhibited a reversible multi-stimuli responsive (temperature, light and pressure) ETCST behaviour, while 1 displayed a stable paramagnetic [FeIIILS(μ-CN)CoIIHS] phase. An anomalous behaviour of the stability of the diamagnetic phase in 1·2H2O at an intermediate temperature during the heating mode is observed for the first time in the PBAs. The magnetic anomaly is accompanied by a two-step thermal contraction and expansion. Further studies on relaxation dynamics and structural analysis align well with the observed anomaly. Complex 1·2H2O also shows a thermal trapping (TIESST) of ∼50–70% of the paramagnetic [FeIIILS(μ-CN)CoIIHS] state at low temperatures. This work, with a comprehensive investigation, provides valuable insights, offering a foundation for the development of multifunctional molecular devices and thermal actuators, via modification and dynamic control of solid-state HB interactions, leading to step-wise thermo-mechanical effects.Data availability
Experimental and crystallographic details, additional structural views, FT-IR spectra, TGA data, and additional magnetic measurement information are provided in the ESI.†Author contributions
All authors approved the final version of the manuscript. JY and SK conceptualized the project. JY conducted the physical characterization and magnetic studies of the complexes, interpreted the data and working concept, and prepared the manuscript with SK.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Y. acknowledges the Council of Scientific and Industrial Research (CSIR), India, for providing the Senior Research Fellowship. S. K. thanks IISER Bhopal for the instrumentation facilities. This work was supported by SERB, India (Project no. CRG/2022/001676).Notes and references
- J. S. Miller and M. Drillon, Magnetism: molecules to materials I-V, Wiley, Hoboken, 2006 Search PubMed.
- M. A. Halcrow, Spin-crossover materials: properties and applications, Wiley, Hoboken, 2013 Search PubMed.
- Q. P. Xuan, J. Glatz, A. Benchohra, J.-R. Jimenez, R. Plamont, L.-M. Chamoreau, A. Flambard, Y. Li, L. Lisnard, D. Dambournet, O. J. Borkiewicz, M. L. Boillot, L. Catala, A. Tissot and R. Lescouezec, J. Mater. Chem. C, 2021, 9, 8882–8890 RSC.
- D. Aguila, Y. Prado, E. S. Koumousi, C. Mathoniere and R. Clerac, Chem. Soc. Rev., 2016, 45, 203–224 RSC.
- Y.-S. Meng, O. Sato and T. Liu, Angew. Chem., Int. Ed., 2018, 57, 12216–12226 CrossRef CAS.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 272, 704–705 CrossRef CAS.
- C. P. Berlinguette, A. Dragulescu Andrasi, A. Sieber, H. U. Gudel, C. Achim and K. R. Dunbar, J. Am. Chem. Soc., 2005, 127, 6766–6779 CrossRef CAS.
- T. Liu, D.-P. Dong, S. Kanegawa, S. Kang, O. Sato, Y. Shiota, K. Yoshizawa, S. Hayami, S. Wu, C. He and C.-Y. Duan, Angew. Chem., Int. Ed., 2012, 51, 4367–4370 CrossRef CAS PubMed.
- I.-R. Jeon, S. Calancea, A. Panja, D. M. P. Cruz, E. S. Koumousi, P. Dechambenoit, C. Coulon, A. Wattiaux, P. Rosa, C. Mathoniere and R. Clerac, Chem. Sci., 2013, 4, 2463–2470 RSC.
- J. Yadav, M. Nandi, R. Kharel, M. Mukherjee and S. Konar, Dalton Trans., 2024, 53, 8910–8914 RSC.
- J. Mercurol, Y. Li, E. Pardo, O. Risset, M. Seuleiman, H. Rousseliere, R. Lescouzec and M. Julve, Chem. Commun., 2010, 46, 8995–8997 RSC.
- E. S. Koumousi, I.-R. Jeon, Q. Gao, P. Dechambenoit, D. N. Woodruff, P. Merzeau, L. Buisson, X. Jia, D. Li, F. Volatron, C. Mathoniere and R. Clerac, J. Am. Chem. Soc., 2014, 136, 15461–15464 CrossRef CAS PubMed.
- C.-Q. Jiao, Y.-S. Meng, Y. Yu, W.-J. Jiang, W. Wen, H. Oshio, Y. Luo, C.-Y. Duan and T. Liu, Angew. Chem., Int. Ed., 2019, 58, 17009–17015 CrossRef CAS PubMed.
- Y. Li, A. Benchohra, B. Xu, B. Baptiste, K. Beneut, P. Parisiades, L. Delbes, A. Soyer, K. Boukheddaden and R. Lescouezec, Angew. Chem., Int. Ed., 2020, 59, 17272–17276 CrossRef CAS PubMed.
- Y. Zhang, D. Li, R. Clerac, M. Kalisz, C. Mathoniere and S. M. Holmes, Angew. Chem., Int. Ed., 2010, 49, 3752–3756 CrossRef CAS PubMed.
- M. Nihei, Chem. Lett., 2020, 49, 1206–1215 CrossRef CAS.
- Y.-Z. Zhang, P. Ferko, D. Siretanu, R. Ababei, N. P. Rath, M. J. Shaw, R. Clerac, C. Mathoniere and S. M. Holmes, J. Am. Chem. Soc., 2014, 136, 16854–16864 CrossRef CAS PubMed.
- J. Yadav, D. J. Mondal and S. Konar, Chem. Commun., 2021, 57, 5925–5928 RSC.
- R. Kharel, J. Yadav and S. Konar, Chem. Commun., 2024, 60, 839–842 RSC.
- N. Hoshino, F. Iijima, G. N. Newton, N. Yoshida, T. Shiga, H. Nojiri, A. Nakao, R. Kumai, Y. Murakami and H. Oshio, Nat. Chem., 2012, 4, 921–926 CrossRef CAS.
- L. Meng, Y.-F. Deng and Y.-Z. Zhang, Inorg. Chem., 2021, 60, 14330–14335 CrossRef CAS.
- J. Glatz, J.-R. Jimenez, L. Godeffroy, H. J. Bardeleben, L. Fillaud, E. Maisonhaute, Y. Li, L.-M. Chamoreau and R. Lescouezec, J. Am. Chem. Soc., 2022, 144, 10888–10901 CrossRef CAS.
- D. Siretanu, D. Li, L. Buisson, D. M. Bassani, S. M. Holmes, C. Mathoniere and R. Clerac, Chem.–Eur. J., 2011, 17, 11704–11708 CrossRef CAS.
- M. Nihei, Y. Sekine, N. Suganami, K. Nakazawa, A. Nakao, H. Nakao, Y. Murakami and H. Oshio, J. Am. Chem. Soc., 2011, 133, 3592–3600 CrossRef CAS PubMed.
- M. Nihei, K. Shiroyanagi, M. Kato, R. Takayama, H. Murakami, Y. Kera, Y. Sekine and H. Oshio, Inorg. Chem., 2019, 58, 11912–11919 CrossRef CAS.
- M. Nihei, Y. Yanai, I.-J. Hsu, Y. Sekine and H. Oshio, Angew. Chem., Int. Ed., 2017, 56, 591–594 CrossRef CAS.
- Y. Sekine, M. Nihei and H. Oshio, Chem.–Eur. J., 2017, 23, 5193–5197 CrossRef CAS.
- M. Nihei, Y. Yanai, D. Natke, R. Takayama, M. Kato, Y. Sekine, F. Renz and H. Oshio, Chem.–Eur. J., 2019, 25, 7449–7452 CrossRef CAS.
- C.-Q. Jiao, W.-J. Jiang, Y.-S. Meng, W. Wen, L. Zhao, J.-L. Wang, J.-X. Hu, G. G. Gurzadyan, C.-Y. Duan and T. Liu, Natl. Sci. Rev., 2018, 5, 507–515 CrossRef CAS.
- T. Liu, Y.-J. Zhang, S. Kanegawa and O. Sato, Angew. Chem., Int. Ed., 2010, 49, 8645–8648 CrossRef CAS PubMed.
- L. Cao, J. Tao, Q. Gao, T. Liu, Z. Xia and D. Li, Chem. Commun., 2014, 50, 1665–1667 RSC.
- C. Zheng, J. Xu, Z. Yang, J. Tao and D. Li, Inorg. Chem., 2015, 54, 9687–9689 CrossRef CAS.
- Y. Sekine, M. Nihei and H. Oshio, Chem. Lett., 2014, 43, 1029–1030 CrossRef CAS.
- C. P. Berlinguette, A. D. Andrasi, A. Sieber, H.-U. Gudel, C. Achim and K. R. Dunbar, J. Am. Chem. Soc., 2005, 127, 6766–6779 CrossRef CAS PubMed.
- R.-J. Wei, T. Shiga, G. N. Newton, D. Robinson, S. Takeda and H. Oshio, Inorg. Chem., 2016, 55, 12114–12117 CrossRef CAS.
- S. Xue, Y. Guo and Y. Garcia, CrystEngComm, 2021, 23, 7899–7915 RSC.
- C. Zheng, J. Xu, F. Wang, J. Tao and D. Li, Dalton Trans., 2016, 45, 17254–17262 RSC.
- R. Jankowski, M. Reczynski, S. Chorazy, M. Zychowicz, M. Arczynski, M. Koziel, K. Ogorzaly, W. Makowski, D. Pinkowicz and B. Sieklucka, Chem.–Eur. J., 2020, 26, 11187–11198 CrossRef CAS PubMed.
- M. You, D. Shao, Y.-F. Deng, J. Yang, N.-T. Yao, Y.-S. Meng, L. Ungur and Y.-Z. Zhang, Inorg. Chem., 2022, 61, 5855–5860 CrossRef CAS.
- S. Jia, L. Fan, C. Zheng, S. Jin and D. Li, Dalton Trans., 2022, 51, 12968–12974 RSC.
- H.-Y. Sun, Y.-S. Meng, L. Zhao, N.-T. Yao, P.-D. Mao, Q. Liu, F.-F. Yan, H. Oshio and T. Liu, Angew. Chem., Int. Ed., 2023, 62, e2023028 Search PubMed.
- J.-X. Hu, Y. Xu, Y.-S. Meng, L. Zhao, S. Hayami, O. Sato and T. Liu, Angew. Chem., Int. Ed., 2017, 56, 13052–13055 CrossRef CAS PubMed.
- S. Zhu, P. Wu, H. Yelemulati, J. Hu, G. Li, L. Li and Y. Tai, Matter, 2021, 4, 1832–1862 CrossRef CAS.
- S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Coord. Chem. Rev., 2005, 249, 1693–1708 CrossRef CAS.
- R. Ketkaew, et al., Octadist, 2019 Search PubMed.
- H. Zheng, Y.-S. Meng, G.-L. Zhou, C.-Y. Duan, O. Sato, S. Hayami, Y. Luo and T. Liu, Angew. Chem., Int. Ed., 2018, 57, 8468–8472 CrossRef CAS.
- P. Gutlich, V. Ksenofontov and A. B. Gaspar, Coord. Chem. Rev., 2005, 249, 1811–1829 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, experimental details, X-ray crystallography, shape analysis, octahedral distortion parameters, thermogravimetric analysis, and infrared spectroscopic data. CCDC 2321115–2321130. For ESI and crystallographic data in CIF or other electronic formats see DOI: https://doi.org/10.1039/d4sc05792a |
| This journal is © The Royal Society of Chemistry 2025 |