
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced α-phase stability of formamidinium lead iodide with addition of 5-ammonium valeric acid chloride†
Yanan
Li‡
a,
Abigale
Bahnick‡
a,
Patrick J.
Lohr
a,
Sean
Raglow
b and
Adam D.
Printz
 *ac
*ac
aDepartment of Chemical and Environmental Engineering, University of Arizona, Tucson, AZ 85721, USA
bDepartment of Physics, University of Arizona, Tucson, AZ 85721, USA
cDepartment of Materials Science and Engineering, University of Arizona, Tucson, AZ 85721, USA
First published on 3rd December 2024
Abstract
Formamidinium lead iodide (FAPbI3) is a metal halide perovskite composition that exhibits improved thermal stability and a more favorable band gap compared to the archetypical methylammonium lead iodide (MAPbI3). However, the photoactive α-phase is not thermodynamically stable at operating temperatures, which is a challenge that must be overcome for the viability of FAPbI3-based photovoltaics. This study explores the use of the ammonium acid additives 5-ammonium valeric acid iodide (5-AVAI) and 5-ammonium valeric acid chloride (5-AVACl), to stabilize the α-phase of FAPbI3. While both additives stabilize the photoactive α-phase and suppress the formation of the photoinactive δ-phase, increase grain size, reduce non-radiative recombination, and improve carrier lifetimes, the addition of 5-AVACl results in superior performance. The improvements with 5-AVACl added are possibly due to its unique ability to initiate formation of the α-phase of FAPbI3 prior to annealing. DFT calculations also show that the growth of moisture-stable (111) facets is more favorable with the addition of 5-AVACl. These property improvements result in a significant increase in the power conversion efficiency of solar cells, from 9.75 ± 0.61% for devices with pristine FAPbI3 to 13.50 ± 0.81% for devices incorporating 1 mol% 5-AVACl.
1. Introduction
There has been unprecedented progress made in the field of metal halide perovskite solar cells (PSCs) over the past decade, with power conversion efficiencies (PCEs) increasing to 26.7%, at parity with single crystalline silicon photovoltaics.1 The most investigated metal halide perovskite is methylammonium lead iodide (MAPbI3), but this composition is thermally unstable due to the volatile nature of the MA+ cation. Furthermore, MAPbI3 has a band gap between 1.55 and 1.6 eV, wider than the ideal band gap ∼1.34 eV necessary for the maximum theoretical power conversion (i.e., 33.7%) for a single-junction solar cell at AM 1.5G illumination according to the Shockley–Queisser limit.2 A promising alternative to MAPbI3 is formamidinium lead iodide (FAPbI3), which has improved thermal stability and a more favorable band gap compared to MAPbI3.3,4 The improved thermal stability of FAPbI3 has been attributed to enhanced hydrogen bonding between the FA+ and PbI6 octahedra.5 The more favorable band gap of FAPbI3 is due to the larger ionic radius of FA+ (253 pm) than of MA+ (217 pm), which yields a Goldschmidt tolerance factor closer to 1—indicative of better crystal symmetry—and results in the band gap narrowing to around 1.48 eV.4 More explicitly, the inclusion of the larger FA+ results in the perovskite forming a cubic or quasi cubic structure, that increases ionic character of the Pb–I bond and enhances spin–orbit coupling, consequently reducing the perovskite band gap compared to the octahedrally tilted tetragonal structure with MA+.5FAPbI3 exhibits two primary polymorphic phases near room temperature: the black, photoactive α-FAPbI3 phase and the yellow, photoinactive δ-FAPbI3 phase. The α-phase has a cubic crystal structure and is formed at temperatures around 150 °C, while the δ-phase is characterized by an octahedral plane-sharing hexagonal structure and is more thermodynamically stable at room temperature.6 While the δ-phase is more stable than the metastable α-phase at room temperature, the large band gap of the δ-phase (2.43 eV) makes it unsuitable for solar cell applications.7,8 Thus, to harness the potential of FAPbI3-based solar cells, the instability of the α-phase must be addressed.
The instability of the α-phase is caused by the relatively bulky FA+ inducing lattice distortion,9 but it has been shown that this distortion can be mitigated by the partial substitution of FA+ with smaller cations such as MA+ (ref. 10 and 11) and Cs+ (ref. 12 and 13) or by incorporating mixed halide anions such as Br−/I−.11,14 However, the strategy of partial substitution with smaller cations comes with a tradeoff of increased band gaps (i.e., blue-shifted light absorption), while the inclusion of mixed-halide perovskites has the deleterious consequence of phase segregation under light illumination that accelerates charge-carrier recombination and performance losses.15
Perhaps the most common strategy to improve the α-phase stability has been to incorporate methylammonium chloride (MACl) as an additive into precursor solutions.16–19 Kim et al. demonstrated that the Cl− in MACl induced p orbital localization of I− and enhanced the α-phase stability through stronger interactions between FA+ and I−.18 Devices comprising FAPbI3 with 40 mol% MACl achieved PCEs exceeding 24% and exhibited enhanced thermal and photostability, with over 90% PCE retention after 300 h of aging at 40 °C and 70% PCE retention after 300 h of 1-sun illumination without encapsulation. Although the MACl volatilized readily during the annealing process, the authors observed a small amount of residual MA+ in the FAPbI3 films. Later, Jeong et al. determined that when MACl was added at a concentration of 35 mol%, ∼5% of MA+ was incorporated into the FAPbI3 structure.19
It is thought that the larger band gap that is often coupled with improved α-phase stability can be avoided by using bulkier cations that do not substitute into the lattice for FA+. As one example, Park and coworkers studied the addition of propylammonium chloride (PACl) as an alternative to MACl to prepare a phase-pure and MA-free α-FAPbI3 and observed that the PACl improved both the PCE and stability of the α-phase compared to conventional MACl.20 It was argued that the large ionic radius of PA+ caused it to passivate the grain boundaries instead of substituting into the lattice structure; however, the band gap still slightly increased from 1.523 eV in pristine FAPbI3 to 1.531 eV with the addition of 20 mol% PACl, indicating some distortion of the lattice.20 Conversely, Grätzel and coworkers observed a red shifted absorption—i.e., decreased band gap—and improved crystallinity and α-phase stability with the addition of the bulky cation 5-ammonium valeric acid iodide (5-AVAI).21
We recently investigated the influence of ammonium acid additives—5-AVAI and 5-ammonium valeric acid chloride (5-AVACl)—on MAPbI3 perovskite and highlighted the critical role of halide composition on film properties and device performance.8 Both additives were found to slow down the crystallization process due to their strong interaction with perovskite, leading to increased grain size in MAPbI3 films. Critically, we observed significantly reduced non-radiative recombination with the inclusion of 5-AVACl that resulted in higher device efficiencies compared to pristine MAPbI3 or 5-AVAI-incorporating devices. While 5-AVAI has previously been reported to improve FAPbI3 device performance and stability,21 5-AVACl has not yet been studied. Given the beneficial effects of 5-AVACl in MAPbI3, we aimed to understand its influence in the more technologically relevant FAPbI3 perovskite. We found that 5-AVACl, like 5-AVAI, stabilized the α-phase and suppressed the formation of the δ-phase, resulting in improved film quality with larger grain size and longer carrier lifetimes than in pristine films. Furthermore, the 5-AVACl also encouraged more (111) growth, which has previously been shown to improve stability to moisture.22 At low concentrations (i.e., 1 mol%), neither additive shifted the band gap compared to pristine FAPbI3 films, while at higher concentrations (i.e., 5 mol%), the band gaps were slightly increased, which we attributed to the formation of low-dimensional perovskite. Notably, addition of 5-AVACl stabilized the intermediate phase and facilitated formation of the α-phase even without annealing, leading to films with fewer defects and increased photoluminescence and charge carrier lifetimes. As a result, device PCE increased from 9.90 ± 0.25% for pristine FAPbI3 to 13.16 ± 0.89% with 1 mol% 5-AVACl incorporated.
2. Experimental methods
2.1 Materials
Indium tin oxide (ITO)-coated glass slides (Rs ∼ 10 Ω □−1) were purchased from Xin Yan Technology. Formamidinium iodide (FAI, 99.99%) was purchased from GreatCell Solar Materials. Lead iodide (PbI2, 99.99% trace metals basis), 5-ammoniumvaleric acid chloride (5-AVACl, 98%), and 5-ammoniumvaleric acid iodide (5-AVAI, 97%) were purchased from TCI Chemicals. Tin(IV) oxide (SnO2, 15% in H2O colloidal dispersion), lithium bis(trifluoromethylsulfonyl)imide (LiTFSI, 98%), and anhydrous toluene (99.8%) were purchased from Alfa Aesar. 4-tert-butylpyridine (tBP, 96%) was purchased from Shanghai Shaoyuan Co., Ltd. Poly[bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA, Mw: 40 kDa) was purchased from Solaris Chem. Anhydrous N,N-dimethylformamide (DMF, 99.8%), and dimethylsulfoxide (DMSO, >99.5%) were purchased from Sigma-Aldrich. Anhydrous chlorobenzene (CB, 99.8%, extra dry) and anhydrous acetonitrile (99.8%, extra dry) were purchased from Acros Organics. 2-Propanol (certified ACS) and acetone (certified ACS) were purchased from Fischer Scientific. The DMF and toluene were dried with freshly activated 4 Å molecular sieves before use. All the other chemicals were used as received without further purification.2.2 Preparation of samples for XRD and PL
Microscope glass substrates (1′′ × 1′′) were cleaned by first brushing with a soft-bristle toothbrush and 1 wt% alconox detergent solution to remove any contaminations. The substrates were then sequentially cleaned by ultrasonication in 1 wt% alconox detergent solution, deionized (DI) water, acetone, and isopropanol for 10 min each. After each sonication treatment the substrates were rinsed with the solvent they were previously bathed in. Upon removal from the isopropanol, substrates were dried with nitrogen. Substrates were then exposed to oxygen plasma in an OptiGlow ACE plasma cleaner at 120 W for 5 min to remove residual organics and increase the wettability of the substrates immediately before film deposition.The 1.2 M FAPbI3 perovskite precursor solution was prepared in a N2-filled glovebox (O2 < 1 ppm and H2O < 0.2 ppm) by dissolving 553.2 mg mL−1 PbI2 and 206.4 mg mL−1 FAI in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 anhydrous DMF:DMSO and stirred (1000 rpm) on a hotplate at 70 °C for 1 h. For the FAPbI3 solutions with additives, 1–5 mol% (with respect to Pb2+) of 5-AVAI or 5-AVACl were directly mixed with the pristine precursor solution and then stirred for another 0.5 h on a hotplate at 70 °C. Next, the solutions were cooled for 1.5 h to room temperature and filtered with 0.2 μm polytetrafluoroethylene (PTFE) filters immediately before film deposition.
:1 anhydrous DMF:DMSO and stirred (1000 rpm) on a hotplate at 70 °C for 1 h. For the FAPbI3 solutions with additives, 1–5 mol% (with respect to Pb2+) of 5-AVAI or 5-AVACl were directly mixed with the pristine precursor solution and then stirred for another 0.5 h on a hotplate at 70 °C. Next, the solutions were cooled for 1.5 h to room temperature and filtered with 0.2 μm polytetrafluoroethylene (PTFE) filters immediately before film deposition.
In a N2-filled glovebox (O2 < 5 ppm and H2O < 0.2 ppm), 65 μL of FAPbI3 precursor solution was dispensed onto a plasma-treated substrate at room temperature. The films were deposited via a two-step spin-coating process, first at a slow spin speed of 1000 rpm (1000 rpm s−1 ramp rate) for 10 s, followed immediately by a fast spin speed of 6000 rpm (6000 rpm s−1 ramp rate) for 30 s. After 25 s of spinning during the second step, 150 μL of anhydrous chlorobenzene (i.e., the anti-solvent) was dripped onto the spinning film in a continuous stream within 1 s. During the anti-solvent drip, the pipette tip was positioned approximately 5 mm above the substrate, and at a ∼45° angle pointed to the center of the substrate. Subsequently, the as-prepared films were annealed at 150 °C for 30 min.
2.3 X-ray diffraction measurements
X-ray diffraction (XRD) was performed using a Phillips X’Pert Pro MPD X-ray diffractometer with Cu-Kα radiation (λ = 1.54 Å). XRD patterns were collected from 5–50° with a step size of 0.017° and a scan speed of 0.209° s−1.2.4 Scanning electron microscopy
The morphology of the perovskite films was characterized by a Hitachi S-4800 SEM, operating at an accelerating voltage of 10.0 kV and a current of 10 μA, with a working distance of 8–9 mm. Samples were coated with 5 nm of Pt prior to microscopy to improve conductivity and minimize image distortion by charging effects.2.5 Photoluminescence and UV-vis absorption spectroscopy
Steady-state PL spectra were measured at room temperature using a Horiba Fluorolog-3 Spectrofluorometer equipped with a 450-W Xenon lamp. The perovskite film was placed into the sample chamber at an angle of 30° to the incident light and excited with the 450 nm (3 nm bandpass). The emission spectra were collected from 470 to 890 nm with an increment of 1 nm.Time-resolved photoluminescence (TRPL) measurements were performed using the Horiba Fluorolog-3 and a DeltaHub TCSPC module with a NanoLED pulsed diode laser source at 639 nm (Horiba NanoLED DD-635L, 60 ps pulse duration) and a repetition rate of 20 MHz.
UV-vis absorption spectra of the sample films were collected using a Cary 5000 UV-vis-NIR Spectrophotometer in the range of 400–850 nm with 1 nm intervals. A blank glass substrate was used as the reference slide for baseline correction.
2.6 Preparation and characterization of solar cells
Patterned ITO-coated glass substrates (2 × 2 cm) were cleaned as described above, and then exposed to oxygen plasma at 120 W for 1 min right before deposition of SnO2 electron transport layer. A 15 wt% SnO2 colloidal dispersion was diluted with DI water to obtain a 4 wt% SnO2 solution. The diluted solution was stirred (1000 rpm) at room temperature for at least 0.5 h, then filtered using a 0.2 μm PTFE filter before deposition.In ambient conditions, 65 μL of the 4 wt% SnO2 solution was dispensed onto a plasma-treated substrate at room temperature and spin-coated at 4000 rpm (4000 rpm s−1 ramp rate) for 30 s, resulting in a layer thickness of ∼30 nm. A cotton swab was dipped into DI water and used to swab off SnO2 from the edges to expose the underlying cathode ITO for contacts. Once the SnO2 was swabbed, the layers were then annealed on a hotplate at 150 °C for 1 h. The substrates/films were subsequently cooled to room temperature before being transferred to a N2-filled glovebox, where the perovskite layers were deposited. The perovskite layer was deposited on top of the SnO2 films within 2 h to ensure good wetting using the spin-coating and annealing procedure discussed above (Section 2.2).
After deposition of the perovskite layer, the PTAA hole transport layer was also deposited in the glovebox. The PTAA solution was prepared by mixing 15 mg mL−1 of PTAA in anhydrous toluene, doped with 10 μL mL−1 of LiTFSI solution (170 mg mL−1 in anhydrous acetonitrile) and 6 μL mL−1 of tBP, stirred (1000 rpm) at room temperature for at least 0.5 h. The solution was filtered with a 0.2 μm PTFE filter and then left to settle for another 0.5 h to reduce formation of comets and pinholes in the subsequent PTAA layer. After the perovskite layer completely cooled to room temperature (i.e., after at least 2 h), 65 μL of the PTAA solution was spin-coated on top of the perovskite layer at 4000 rpm (2000 rpm s−1 ramp rate) for 45 s and then annealed at 75 °C for 5 min, resulting in a 30 nm thick doped PTAA film. To enhance hole transport, the substrates were then oxidized overnight in a desiccator with an environment comprising air at a humidity <10%.
The top contacts (150 nm of Ag) were deposited using a Temescal FC-2500 e-Beam Evaporator. The devices were placed into a vacuum deposition chamber with a pressure of < 5 × 106 torr, and the Ag vapor was deposited at a slow rate of 0.3 Å s−1 for the first 25 nm and then a faster rate of 1.0 Å s−1 for the remaining 125 nm to ensure high quality films. This process resulted in a perovskite solar cell with an ITO/SnO2/FAPbI3/PTAA/Ag architecture.
2.7 Solar cell characterization
Devices were masked to an active area of 0.12 cm2 and J–V measurements were collected using a Keithley 2400 source meter with a scan rate of 0.02 s per step (0.01 V) during simulated solar illumination (AM 1.5G, 100 mW cm−2, OAI TriSol TSS-100US AAA solar simulator), which was calibrated against a silicon reference cell.2.8 Modeling intermolecular interactions
Surface calculations were performed using the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA) with Grimme's DFT-D3 dispersion correction as implemented in the Vienna Ab initio Simulation Package (VASP).23–28 Valence–core interactions were treated with the projector augmented wave (PAW) method, and the energy cutoff for plane waves was 400 eV. All structures were fully relaxed using a conjugate-gradient algorithm until the force on each constituent atom was less than 0.01 eV Å−1. The convergence criterion for the self-consistent loop was set to 10−6 eV. A Γ-centered 3 × 3 × 1 and 8 × 8 × 8 Monkhorst–Pack k-point mesh was used for all slab calculations and unit cells, respectively.29 (100) and (111) PbI2-rich surfaces of FAPbI3 were prepared by cleaving pseudocubic FAPbI3 and introducing 20 Å of vacuum in the direction normal to the slab surface. The optimal cleaved surface area for adsorption was determined by increasing the supercell size until calculated energies were converged for all halide species. For all surface geometry optimizations, the bottom layer of unit cells was fixed, and the number of mobile layers was varied to ensure the convergence of relative adsorption energies. Dipole corrections were applied in the direction normal to the slab surface.When DFT calculations were performed on isolated molecules for subsequent adsorption calculations with VASP, geometrically optimized energies were produced by placing each adsorbate in the center of a cubic simulation cell of side length 30 Å. A Γ-only k-point mesh was used for Brillouin zone integration, and dipole corrections were applied in all Cartesian directions. All atomistic visualization was performed with the visualization for electronic and structural analysis (VESTA) software package.30
The unit cell of pseudocubic FAPbI3 perovskite was obtained from the materials project (mp-977014) and subsequently relaxed without constraints to prevent the introduction of stresses due to differences in functional and energy cutoff choice. Surfaces were prepared by cleaving a 2 × 2 × 3 supercell of pseudocubic FAPbI3 perovskite along the [100] and [111] lattice directions and introducing 25 Å of vacuum to prevent self-interactions along the direction normal to the surfaces.
The adsorption energy of each ammonium salt additive on FAPbI3 was calculated from the following:
| Eads = Eslab+additive − (Eslab + Eadditive) | (1) |
Similarly, we computed the change in surface formation energy induced by chloride substitution by the following:
| ΔEf = Eslab,Cl − Eslab,pristine + μI − μCl | (2) |
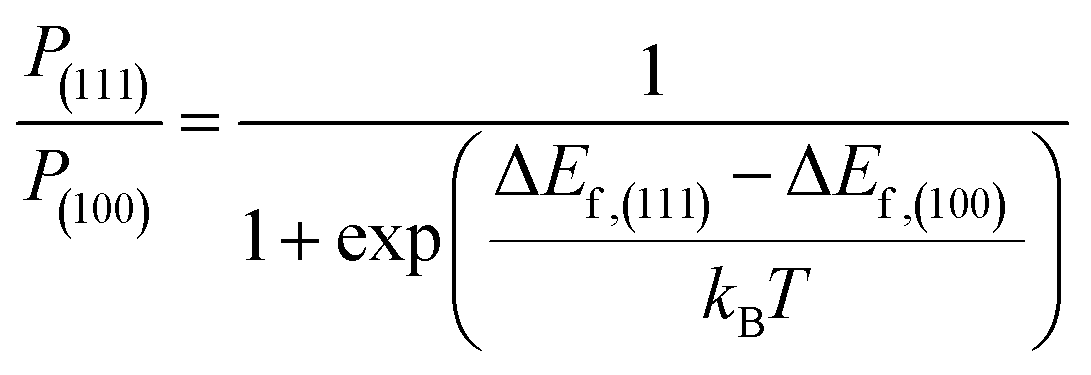 | (3) |
3. Results and discussion
In previous work, we demonstrated that inclusion of 5-AVAI and 5-AVACl enhanced the optoelectronic properties and stability of MAPbI3.31 In that study, we observed that 5-AVACl outperformed 5-AVAI as an additive when incorporated into MAPbI3, which we attributed to the reduced non-radiative recombination in the perovskite that frequently occurs with Cl-containing additives.20,31–33 We anticipated similar beneficial effects with 5-AVACl when added to FAPbI3 given its similar chemical composition and crystal structure to MAPbI3. In FAPbI3, the photoactive black phase (i.e., the α-phase) is thermodynamically unstable and is commonly stabilized by the addition of the ammonium-based additive MACl; however, the MA+ cation from this additive incorporates into the crystal lattice, causing distortion that results in an increased band gap that is deleterious to performance.16–20 We anticipated that the relatively larger size of the 5-AVA+ cation (745 pm) compared to MA+ (217 pm) and FA+ (253 pm) would inhibit its incorporation into the FAPbI3 lattice, thus minimizing the band gap change (Fig. 1). In this work, we added 5-AVACl or 5-AVAI at concentrations ranging from 1 to 5 mol% into FAPbI3 precursor solutions to study the effect of low concentrations of these bulky cations on the crystallization, phase stability, optoelectronic properties, and device performance of FAPbI3.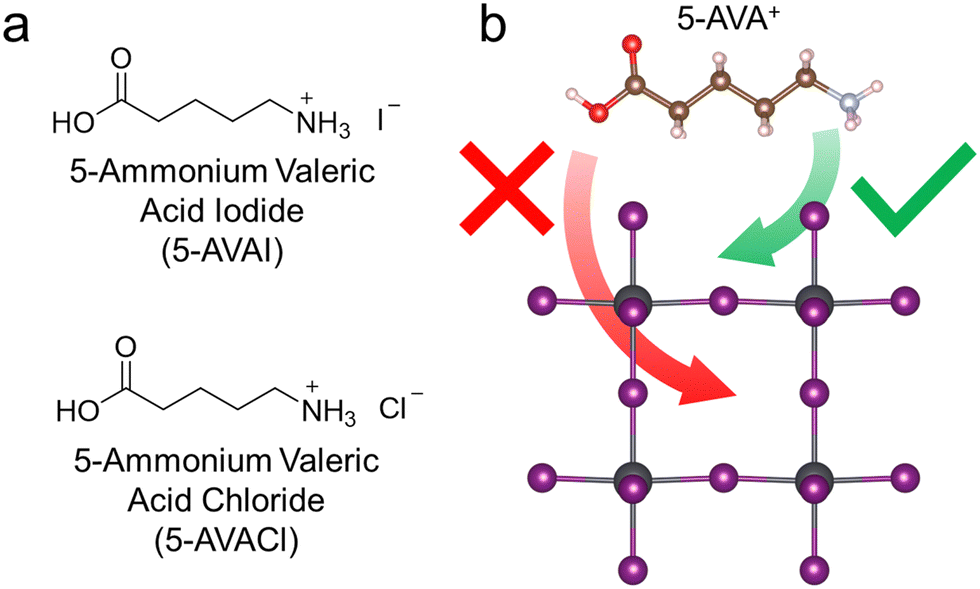 | ||
| Fig. 1 (a) Molecular structures of the additives used in this work, 5-AVAI and 5-AVACl. (b) Illustration of a FAPbI3 lattice (with cations removed) showing that the large 5-AVA+ cation can fit in the A-sites at the surface, but not within the crystal bulk. Because the 5-AVA+ cation remains at the surface rather than in the crystal bulk, it does not distort the lattice and cause a deleterious band gap increase. | ||
3.1 Additive influence on FAPbI3 crystallinity and growth
We performed X-ray diffraction (XRD) measurements to investigate the influence of 5-AVACl and 5-AVAI on the FAPbI3 crystal structure (Fig. 2a). In pristine FAPbI3, we observed three prominent 2θ peaks at 13.9, 28.0, and 31.4°, corresponding to the (100), (200), and (210) lattice planes typical of the photoactive α-phase.19 A diffraction peak was also observed at 11.75° which we assigned to the hexagonal non-perovskite δ-phase,34 and an additional small peak at 11.4° which we attributed to the monohydrate FAPbI3 phase.35 No peaks associated with PbI2 were observed. With the inclusion of the additives at a concentration of 1 mol%, the peak at 11.75° disappeared, indicating that both additives effectively suppressed δ-phase formation. Furthermore, we did not observe any peak shifts from lattice expansion or contraction, indicating no 5-AVA+ or Cl− substitution into the FAPbI3 unit cell. We also observed a decrease in the full width at half maximum (FWHM) of the (100) peak from 0.248 ± 0.007° for pristine FAPbI3 to 0.241 ± 0.002° with 5-AVAI added and 0.235 ± 0.005° with 5-AVACl added, indicating that the additives enhanced crystallinity.20 The improved crystallinity was possibly due to the structure-directing role of 5-AVA+, which has been shown to modulate the symmetry of α-FAPbI3 from cubooctahedral to more cubic by forming strong intermolecular hydrogen bonds with the perovskite lattice.21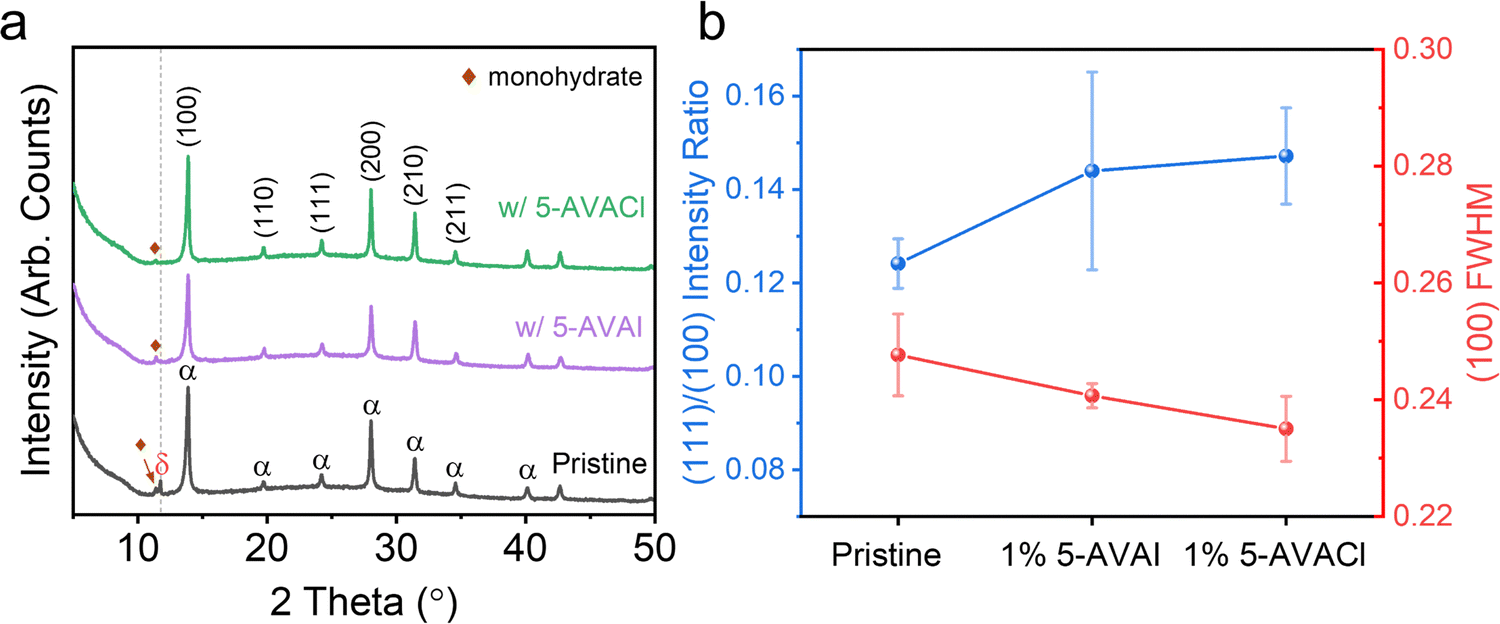 | ||
| Fig. 2 (a) XRD patterns of the pristine FAPbI3 film and the films prepared with 1 mol% 5-AVAI or 5-AVACl. (b) (111)/(100) intensity ratios and FWHM of the (100) peak in FAPbI3 films with and without additives. | ||
In FAPbI3, performance and stability have also been correlated with different crystal facets. Recently, Park and co-workers reported that the (100) facet contributed to enhanced optoelectronic properties, while the (111) facet was more stable against moisture.22 This improved stability of the (111) facets was attributed to the lower adhesion energy of H2O when compared to the (100) facets, indicating that water would preferentially interact and degrade the (100) facets.22 We calculated the intensity ratios of the (111) and (100) XRD peaks to evaluate how the additives affected facet orientation (Fig. 2b and Table S1, ESI†). The (111)/(100) ratio in pristine films was 0.124 ± 0.005, which increased to 0.144 ± 0.021 with 1 mol% 5-AVAI and to 0.147 ± 0.010 with 1 mol% 5-AVACl, indicating that films with additives might exhibit enhanced stability to moisture.
We tested the relative stability of FAPbI3 thin films to moisture by exposing them to damp air (25 °C, 85% RH) for 4 hours, performing XRD measurements afterwards to determine the how the crystalline structure evolved (Fig. S1, ESI†). After aging, degradation of the pristine FAPbI3 films was apparent with the emergence of strong δ-phase peaks, while in the films with 5-AVACl and 5-AVAI we observed greater retention of the α-phase with minimal transition to the δ-phase. We evaluated the degradation of the α-phase by integrating the area of the (100) peak before and after aging; we observed that 72.6% of the α-phase was retained for pristine FAPbI3, while 86.7% and 91.7% of the α-phase was retained with 5-AVAI and 5-AVACl, respectively (Table S2, ESI†).
The influence of additive concentrations on the crystallinity of FAPbI3 was also studied using XRD (Fig. 3). All films with 1 to 5 mol% additives exhibited identical peak locations of the α-phase as observed in the pristine films. As the additive concentration was increased, the (100) peak intensity increased and the FWHM narrowed, indicating improved crystallinity and potentially larger and more uniform grain size.20 Once the concentration was increased to 5 mol% additives, a new diffraction peak emerged at 7.9°, possibly due to the emergence of 2D perovskite.36,37 Interestingly, as the concentration of the additives increased, the 5-AVAI and 5-AVACl influenced the crystallographic orientation in different ways (Fig. 3 and Table S1, ESI†). Above concentrations of 1 mol%, both additives facilitated the growth of (100) planes, resulting in more intense (100) peaks than observed in pristine FAPbI3. However, the additives had drastically different influences on the growth of (111) planes reported to enhance moisture stability.22 With increasing concentration of 5-AVAI, the films exhibited a stronger enhancement in the (100) peak intensity than (111) peak intensity, leading to decreasing (111)/(100) ratios (Fig. 3a and c). Conversely, increasing the concentration of 5-AVACl resulted in films with a more pronounced increase in the (111) peak intensity and higher (111)/(100) ratios (Fig. 3b and d). The driving force for the different orientations when adding 5-AVAI and 5-AVACl was possibly due to the additives interacting preferentially with the (100) surface and consequently inhibiting FA+ insertion and growth of these planes.38,39 The relatively higher (111)/(100) ratio with 5-AVACl might be a further synergistic effect due to the Cl− counterion resulting in slow, controlled growth.38,40 Scanning electron micrographs also showed that the inclusion of both 5-AVAI and 5-AVACl yielded smoother-appearing surfaces with larger grain sizes, with increasing additive concentration resulting in enhancement of these effects (Fig. S2, ESI†).
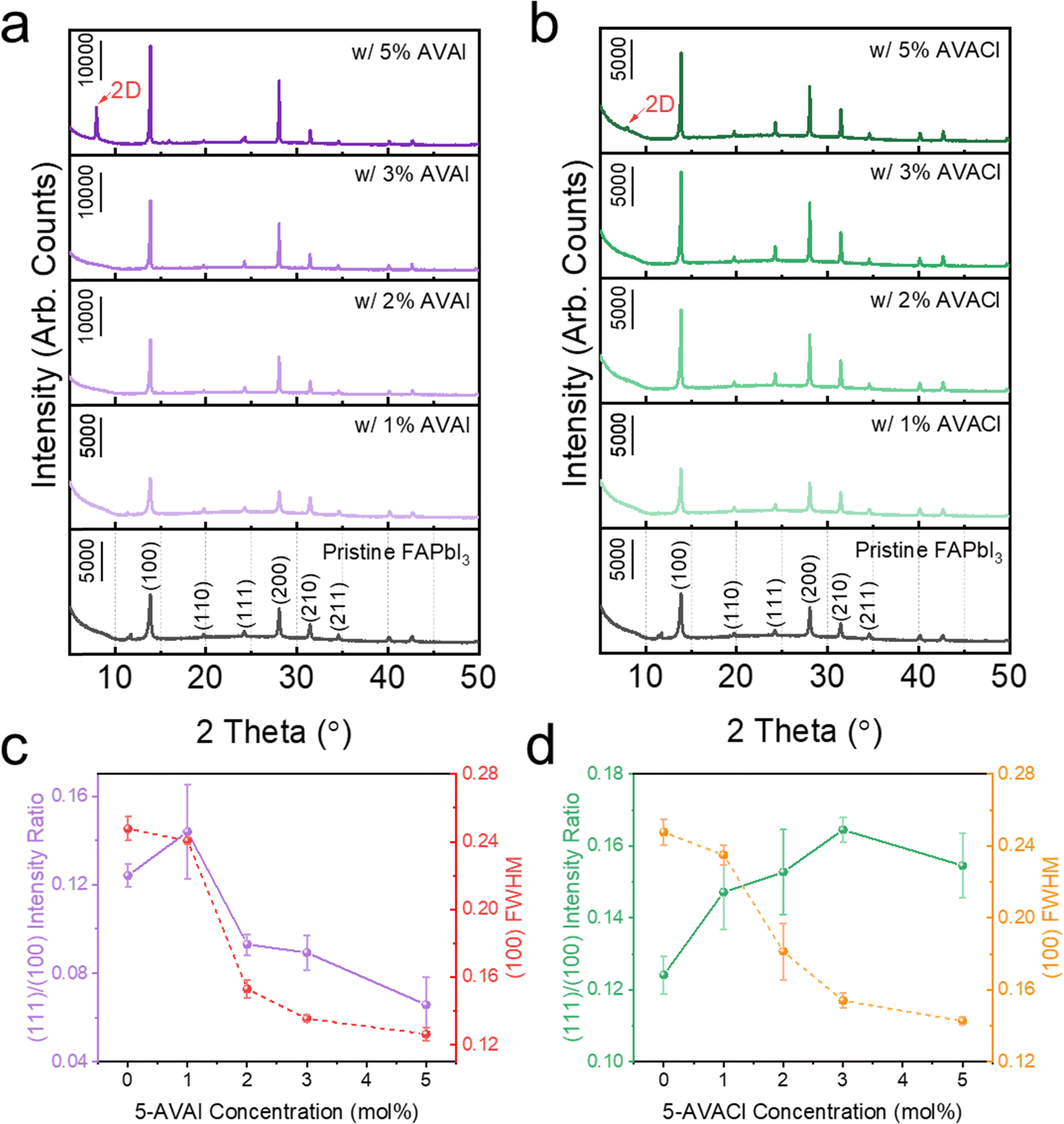 | ||
| Fig. 3 XRD patterns of the FAPbI3 films with 1–5 mol% of (a) 5-AVAI and (b) 5-AVACl, and their corresponding (c) and (d) intensity ratios and FWHM. | ||
To better understand how the additives influenced (111) and (100) growth, we created atomistic models of the (111) PbI3FA-rich and (100) PbI2-rich facets of FAPbI3 and explored the surface interactions with 5-AVAI and 5-AVACl using DFT (Fig. S3, ESI†). Our results indicated that 5-AVAI and 5-AVACl coordinated the (111) facet similarly through –NH3+⋯I− and X−⋯Pb2+ interactions with 5-AVAI being ∼88 meV more exergonic. However, the adsorption of 5-AVAI to the (100) facet was nearly 450 meV more exergonic than 5-AVACl, a result that we attributed to the large ionic radius of I−. Critically, the adsorption of both ammonium salts coordinated the (100) facet far more strongly than the (111) facet, potentially inhibiting the growth of this facet during crystallization and resulting in the observed increase of the (111)/(100) XRD peak intensity ratio relative to pristine films at low additive concentrations. This result agreed with recent work by Liu et al., who found that adding the ammonium salt butylammonium iodide (BAI) to FAPbI3 films resulted in the preferential growth of (111) facets by preventing FA+ cations from interacting with the (100) facet.38 The strong coordination of the (100) facet was also consistent with previous work by Jin and coworkers, who found that the thermal stability of α-FAPbI3 could be enhanced by reducing the surface energy through functionalization with long chain ammonium cations due to strong –NH3+⋯I− hydrogen bonding interactions.41 Later work by Yang et al. utilized this mechanism to stabilize α-FAPbI3 with a Ruddlesden–Popper (RP) 2D capping layer comprising phenylethylammonium spacer cations along grain boundaries of 3D FACsPbI3.42 As both 5-AVAI and 5-AVACl demonstrated preferential –NH3+⋯I− interactions on both the (100) and (111) facets, our observed suppression of δ-phase formation in annealed films could be partially attributed to a reduction in α-FAPbI3 surface energy caused by the adsorption of 5-AVAI and 5-AVACl. Despite the thermodynamic favorability of (111) facet growth with the addition of either 5-AVAI or 5-AVACl, we note that the (111)/(100) ratio decreased as the concentration of 5-AVAI increased—a surprising observation that warrants further study in the future.
We also investigated the possible incorporation of Cl− into FAPbI3 at the surfaces and found that chloride substitution into a I− lattice site was energetically favorable for both the (111) and (100) facet, a result that we attribute to the increased Cl−⋯Pb2+ bond strength compared to the I−⋯Pb2+ bond (Fig. S4, ESI†).18 Interestingly, the energy released by Cl− incorporation into the (111) facet was ∼116 meV higher than the energy released on the (100) facet. Using Boltzmann statistics, we found that this energetic difference corresponded to a ∼98.9% probability of the (111) Cl− substitution at room temperature, which indicated that the incorporation of Cl− could further increase the (111)/(100) ratio in FAPbI3 films incorporating 5-AVACl, which was in agreement with our XRD results in Fig. 3d.
Given the strong interactions of these additives with Pb2+, we anticipated that evidence of these interactions could be observed during the early stages of film growth. We used XRD to examine the as-cast—i.e., prior to annealing—films of FAPbI3 with and without 5-AVAI/5-AVACl to assess how these additives might influence film formation.43 The as-cast pristine film showed a strong peak associated with δ-phase at 11.8° and a tiny peak associated with α-phase at 13.91° (Fig. 4a), as well as some unknown peaks which we attributed to the intermediate phase according to literature.44 The XRD patterns of films with 1 mol% additives looked similar (Fig. S5, ESI†), so to amplify the signal and determine the effect of the additive interactions with the perovskite precursors, we increased the additive concentration to 5 mol%. A decrease in the intensity of the δ-phase peak was observed in films with 5-AVAI, while a large increase in the intensity of the α-phase peak was observed in films with 5-AVACl (Fig. 4a). Notably, the δ-phase peaks in all film types exhibited an identical position (11.8°), while the α-phase peak varied depending on composition (Fig. 4b). In the pristine films, the α-phase was located at 13.91°, while inclusion of 5-AVAI resulted in a slight downshift to 13.89° (Fig. 4b), possibly due to the lattice expansion induced by the large 5-AVA+ cation. Conversely, adding 5-AVACl resulted in a peak upshift to 13.94°, associated with lattice contraction (Fig. 4b), which suggested that the Cl− incorporated into the α-phase prior to annealing—possibly playing a role in the formation and stabilization of the α-phase in the early stages of crystallization.18 We note that we did not observe this α-phase peak shift in the annealed films, suggesting that the Cl− volatilized during annealing, aligning with the previous report that Cl− was only detected during the initial few minutes of annealing.20 The volatilization of Cl− from the perovskite is also consistent with our previous work with MAPbI3 which showed the decomposition of 5-AVACl into 5-AVA and HCl to be energetically favorable, hinting at a possible mechanism for the loss of Cl− during annealing.31 Critically, the use of a Cl− counterion suggested an alternative α-phase stabilization mechanism than the hydrogen bonding explanation put forth by Jin and coworkers.41 Given these observations, we believe that the stabilization of the α-phase in the early stages of film formation can be attributed to two consequences of the substitution of I− with the smaller Cl−: (1) released lattice strain within α-FAPbI3; and (2) induced p orbital localization of I− which enhanced the interaction of FA+ with I−.18,45
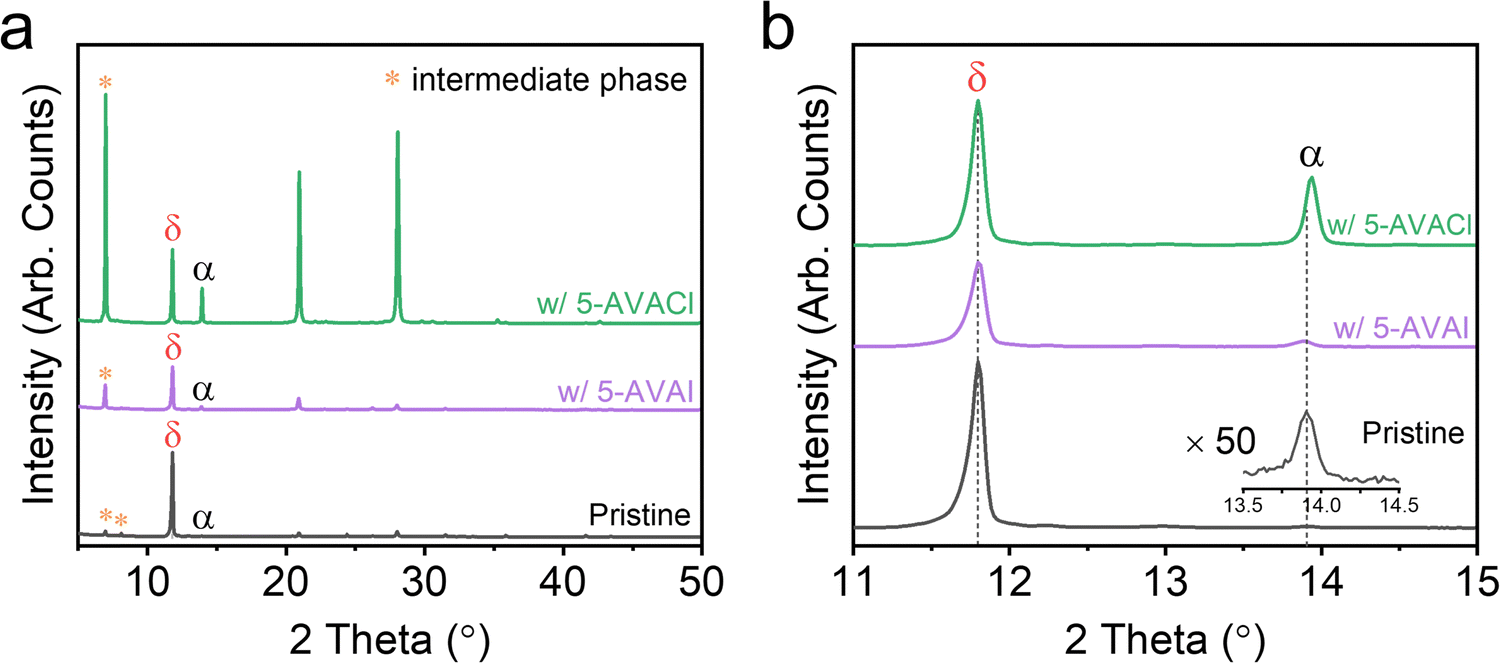 | ||
| Fig. 4 (a) Full XRD patterns and (b) zoomed-in view of the as-cast FAPbI3 films without additives and with 5 mol% 5-AVAI or 5-AVACl. | ||
3.2 Additive influence on FAPbI3 thin film optical properties
We also performed photoluminescence (PL) measurements to compare the extent of passivation of FAPbI3 surface defects by 5-AVAI and 5-AVACl. As shown in Fig. 5a, the PL intensity for the films with 1 mol% of 5-AVAI and 5-AVACl were ∼20 and 90-fold higher than pristine FAPbI3, implying that 5-AVAI and 5-AVACl both effectively passivated the defects in FAPbI3 perovskite. To verify that the enhanced PL intensity was not due to variations in film thickness, we used stylus profilometry to measure the film thickness; the pristine FAPbI3 film was 611.63 ± 24.91 nm thick while the films with 1 mol% 5-AVAI and 5-AVACl were 590.82 ± 12.22 nm and 603.20 ± 23.29 nm thick, respectively, similar in thickness. The small red shift observed in the films with 5-AVAI could be attributed to the increased crystallinity and larger grain size, consistent with a previous report by Grätzel and coworkers.21 Time-resolved photoluminescence (TRPL) was also measured to determine the photoluminescence lifetime of the films. The short lifetime, τ1, and long lifetime, τ2, parameters are related to trap-assisted recombination and radiative recombination, respectively. Based on the A1 and A2 values extracted from the TRPL measurements, films with 1 mol% 5-AVAI and 5-AVACl significantly decreased the non-radiative recombination from 45.11% for pristine FAPbI3 to 9.23% and 3.80%, respectively. Moreover, τ2 increased from 108.53 ns for pristine FAPbI3 films to 279.05 ns with 5-AVAI added and 347.54 ns with 5-AVACl added (Fig. 5b and Table S3, ESI†).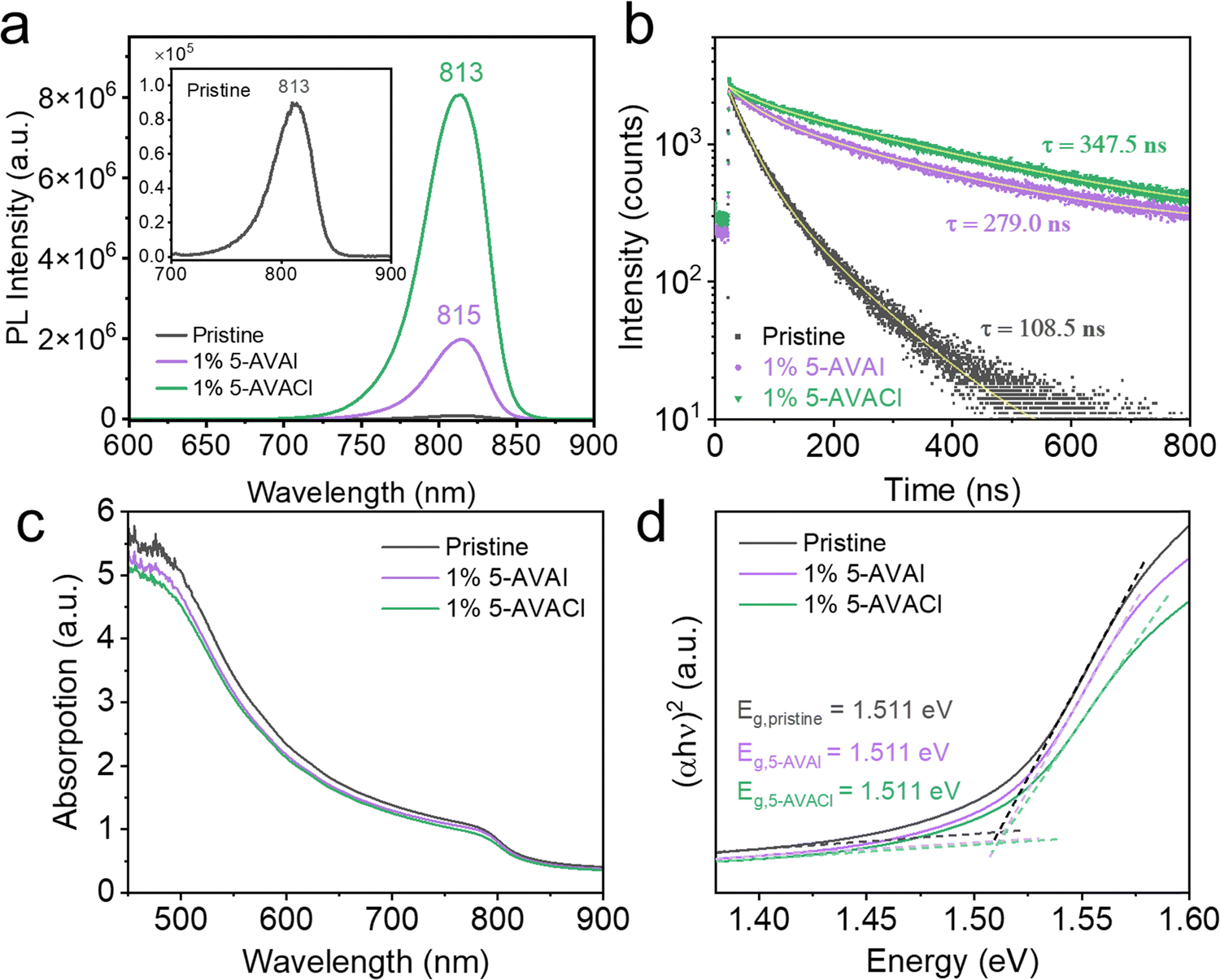 | ||
| Fig. 5 Optical properties of FAPbI3 films with and without additives. (a) Steady-state PL and (b) time-resolved PL of pristine FAPbI3 and FAPbI3 with 1 mol% 5-AVAI or 5-AVACl. Inset in (a) shows the zoomed-in view of pristine FAPbI3. (c) Absorption spectra and (d) Tauc plots of pristine FAPbI3 and FAPbI3 with 1 mol% 5-AVAI or 5-AVACl. | ||
We additionally performed UV-vis absorption spectroscopy to characterize any changes in the optical band gap caused by the introduction of 5-AVAI or 5-AVACl. A shift in optical band gap would suggest that the inclusion of these additives distorted the lattice and would likely limit device efficiency. The absorbance spectra showed similar band-edge absorption near ∼830 nm in all samples (Fig. 5c), indicating that there was no band gap shift; however, the absorbance from 450 to 850 nm was slightly decreased with the inclusion of 5-AVAI and 5-AVACl, which could limit current density. The optical band gaps were derived using Tauc plots, and all samples were found to have a band gap of 1.511 eV (Fig. 5d). The absence of a band gap shift was consistent with the XRD results, indicating that the additives did not negatively affect the FAPbI3 crystal structure.
3.3 Additive improvement of FAPbI3 photovoltaic device performance and stability
We fabricated FAPbI3 solar cells with an n–i–p architecture of ITO/SnO2/FAPbI3/PTAA/Ag with 5-AVAI and 5-AVACl to evaluate their effects on photovoltaic performance. The optimal concentration was 1 mol% for both additives, with efficiencies decreasing at higher concentrations, which we attributed to the insulating nature of the additives and possible formation of 2D/3D heterojunctions impeding charge transport (Fig. S6 and S7, ESI†). An interesting observation was that the addition of these additives resulted in an inverted hysteresis, suggesting that the additives introduced a chemical inductor effect, possibly by influencing surface recombination and ion migration within the perovskite layer.46–48 The forward scanned current density–voltage (J–V) curves of champion devices are shown in Fig. 6. The pristine FAPbI3 device had poor performance with an average PCE of 9.75 ± 0.61%, an open-circuit voltage (Voc) of 0.95 ± 0.03 V, a short-circuit current density (Jsc) of 18.93 ± 0.40 mA cm−2, and a fill factor (FF) of just 53.69 ± 2.77%. Both additives increased the Voc by ∼0.1 eV, leading to significantly improved PCEs; 1 mol% 5-AVAI increased the average PCE to 13.17 ± 0.39%, while 1 mol% 5-AVACl further enhanced the PCE to 13.50 ± 0.81%. The champion 5-AVACl device yielded a PCE of 14.45%—around 39% higher than the champion pristine device (10.41%)—which was attributed to significantly increased Voc (1.06 V), Jsc (21.29 mA cm−2), and FF (63.96%). The detailed parameters were summarized in Table 1.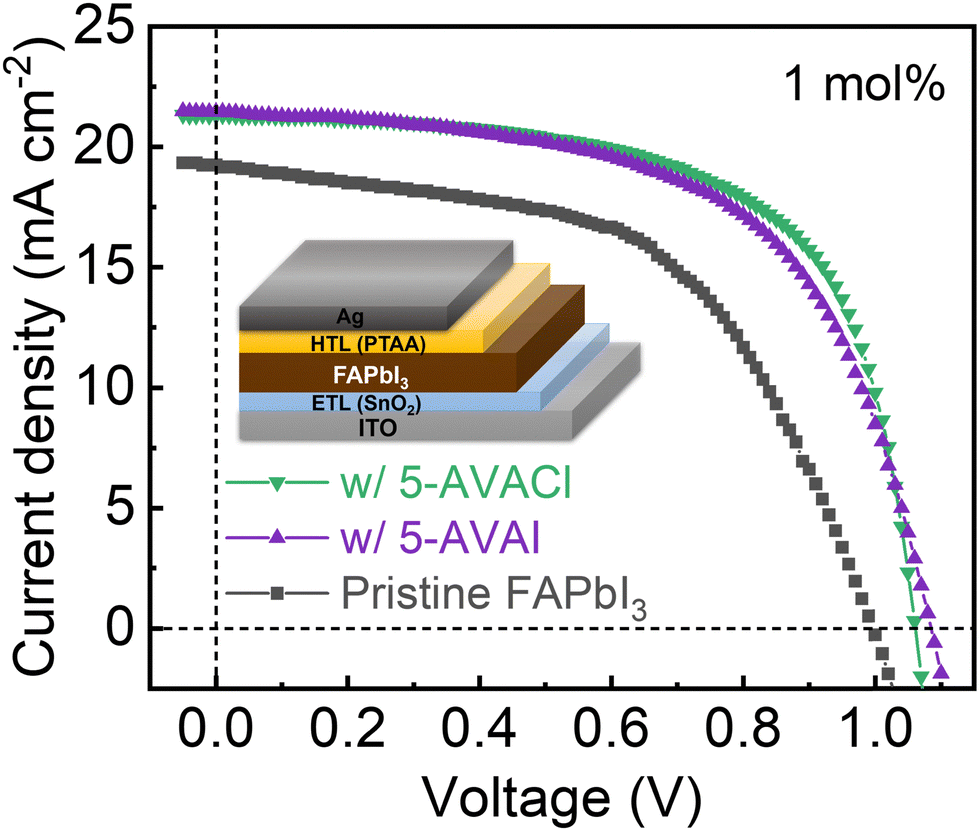 | ||
| Fig. 6 Forward scan J–V curves for the champion devices or pristine FAPbI3, and FAPbI3 with 1 mol% 5-AVAI or 5-AVACl added. Inset shows the device structure. | ||
| Additive | PCE (%) | V oc (V) | J sc (mA cm−2) | FF (%) |
|---|---|---|---|---|
| None | 9.75 ± 0.61 (10.41) | 0.95 ± 0.03 (0.99) | 18.93 ± 0.40 (19.22) | 53.69 ± 2.77 (52.83) |
| w/1% 5-AVAI | 13.17 ± 0.39 (13.74) | 1.06 ± 0.03 (1.08) | 21.12 ± 0.57 (21.44) | 59.92 ± 1.57 (59.08) |
| w/1% 5-AVACl | 13.50 ± 0.81 (14.45) | 1.05 ± 0.01 (1.06) | 21.00 ± 0.24 (21.29) | 61.11 ± 2.48 (63.96) |
As well as improving device efficiency, we also observed evidence of improved device stability. We compared the photovoltaic performance of representative devices directly after fabrication and after 7 days of aging in ambient conditions (15–20% relative humidity) at room temperature. As shown in Fig. S8 (ESI†), both additives significantly enhanced the stability of FAPbI3 devices. The pristine FAPbI3 device exhibited a noticeable 17.76% reduction in PCE after 7 days. In contrast, devices with 1 mol% 5-AVAI and 5-AVACl retained ∼98% of their initial PCEs.
4. Conclusion
Previous work has shown that the addition of 5-AVAI into FAPbI3 can stabilize the α-phase due to the bulkiness of the 5-AVA+ cation—which inhibits lattice substitution and distortion, maintaining the low band gap of FAPbI3—and its ability to hydrogen bond with the perovskite lattice.21 However, we observed that the benefits of adding a closely related additive, 5-AVACl, were even greater due to the synergistic effects of the 5-AVA+ cation and the Cl− counterion. We observed that including 5-AVACl resulted uniquely in the formation of the α-phase prior to annealing, which we speculated could be due to interactions between the Cl− counterion and the perovskite precursors. Furthermore, at a concentration of 1 mol%, both additives eliminated the formation of the δ-phase and increased the extent of the formation of moisture stable (111) facets; interestingly, as the concentration of the additives increased, the (111)/(100) peak intensity ratio increased for films with 5-AVACl, but decreased for films with 5-AVAI. This diverging phenomenon may be related to the thermodynamics of Cl− integration into FAPbI3, where our DFT calculations predicted a greater energy release for the incorporation of Cl− with the (111) facet than the (100) facet. At higher additive concentrations, 5-AVACl would continue to promote (111) growth. We also observed that the inclusion of 5-AVACl resulted in a 90-fold increase in photoluminescence and a nearly 3.5-fold increase in the charge carrier lifetime over pristine FAPbI3, which resulted in a 39% increase in photovoltaic champion device PCE from 10.41% for pristine FAPbI3 to 14.45% for FAPbI3 with 1 mol% 5-AVACl.Stabilizing and passivating α-FAPbI3 without the tradeoff of an increased band gap is a challenge of great importance for the viability of FAPbI3-based photovoltaic devices. Drawing inspiration from Alanazi et al. and our own previous work, we were the first to include 5-AVACl into FAPbI3, showing its promising potential.8,21 Although further optimization of devices comprising FAPbI3 with 5-AVACl is required, this work highlighted the importance of the species of the halide counterion in the crystallization dynamics, performance, and stability of FAPbI3-based solar cells.
Author contributions
A. P. conceived of the idea. A. P., Y. L., A. B., and P. L. designed the experiments. Y. L., A. B., P. L., and S. R. conducted the experiments. Y. L., A. B., P. L., and S. R. performed data analysis. Y. L., A. P., P. L., and S. R. wrote the manuscript draft. All authors approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under grant no. 2237211. The authors thank the following user facilities at the University of Arizona. The powder XRD measurements were performed at the XRD facility in the Department of Chemistry and Biochemistry of the University of Arizona, RRID:SCR_022886, on a Philips PANalytical X’Pert PRO MPD instrument. The authors acknowledge NASA grants #NNX12AL47G and #NNX15AJ22G and NSF grant #1531243 for funding of the instrumentation in the Kuiper Materials Imaging and Characterization Facility at the University of Arizona. UV-vis spectrometry was performed at the Micro/Nanofabrication Center at the University of Arizona. DFT calculations were performed upon High Performance Computing (HPC) resources supported by the University of Arizona TRIF, UITS, and Research, Innovation, and Impact (RII) and maintained by the University of Arizona Research Technologies Department. P. J. L. was supported by the financial and in-kind contributions of the Phoenix Chapter of the ARCS Foundation.References
- NREL, Best Research Cell Efficiencies, 2025.
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Formamidinium Lead Trihalide: A Broadly Tunable Perovskite for Efficient Planar Heterojunction Solar Cells, Energy Environ. Sci., 2014, 7(3), 982, 10.1039/c3ee43822h.
- A. Amat, E. Mosconi, E. Ronca, C. Quarti, P. Umari, Md. K. Nazeeruddin, M. Grätzel and F. De Angelis, Cation-Induced Band-Gap Tuning in Organohalide Perovskites: Interplay of Spin–Orbit Coupling and Octahedra Tilting, Nano Lett., 2014, 14(6), 3608–3616, DOI:10.1021/nl5012992.
- J. Y. Kim, J.-W. Lee, H. S. Jung, H. Shin and N.-G. Park, High-Efficiency Perovskite Solar Cells, Chem. Rev., 2020, 120(15), 7867–7918, DOI:10.1021/acs.chemrev.0c00107.
- A. Amat, E. Mosconi, E. Ronca, C. Quarti, P. Umari, M. K. Nazeeruddin, M. Grätzel and F. De Angelis, Cation-Induced Band-Gap Tuning in Organohalide Perovskites: Interplay of Spin-Orbit Coupling and Octahedra Tilting, Nano Lett., 2014, 14(6), 3608–3616, DOI:10.1021/nl5012992.
- S. Masi, A. F. Gualdrón-Reyes and I. Mora-Seró, Stabilization of Black Perovskite Phase in FAPbI 3 and CsPbI 3, ACS Energy Lett., 2020, 5(6), 1974–1985, DOI:10.1021/acsenergylett.0c00801.
- Y. An, J. Hidalgo, C. A. R. Perini, A.-F. Castro-Méndez, J. N. Vagott, K. Bairley, S. Wang, X. Li and J.-P. Correa-Baena, Structural Stability of Formamidinium- and Cesium-Based Halide Perovskites, ACS Energy Lett., 2021, 6(5), 1942–1969, DOI:10.1021/acsenergylett.1c00354.
- T. M. Koh, K. Fu, Y. Fang, S. Chen, T. C. Sum, N. Mathews, S. G. Mhaisalkar, P. P. Boix and T. Baikie, Formamidinium-Containing Metal-Halide: An Alternative Material for Near-IR Absorption Perovskite Solar Cells, J. Phys. Chem. C, 2014, 118(30), 16458–16462, DOI:10.1021/jp411112k.
- S.-H. Turren-Cruz, A. Hagfeldt and M. Saliba, Methylammonium-Free, High-Performance, and Stable Perovskite Solar Cells on a Planar Architecture, Science, 2018, 362(6413), 449–453, DOI:10.1126/science.aat3583.
- N. Pellet, P. Gao, G. Gregori, T. Yang, M. K. Nazeeruddin, J. Maier and M. Grätzel, Mixed-Organic-Cation Perovskite Photovoltaics for Enhanced Solar-Light Harvesting, Angew. Chem., Int. Ed., 2014, 53(12), 3151–3157, DOI:10.1002/anie.201309361.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Compositional Engineering of Perovskite Materials for High-Performance Solar Cells, Nature, 2015, 517(7535), 476–480, DOI:10.1038/nature14133.
- J.-W. Lee, D.-H. Kim, H.-S. Kim, S.-W. Seo, S. M. Cho and N.-G. Park, Formamidinium and Cesium Hybridization for Photo- and Moisture-Stable Perovskite Solar Cell, Adv. Energy Mater., 2015, 5(20), 1501310, DOI:10.1002/aenm.201501310.
- G. Kim, H. Min, K. S. Lee, D. Y. Lee, S. M. Yoon and S. I. Seok, Impact of Strain Relaxation on Performance of α-Formamidinium Lead Iodide Perovskite Solar Cells, Science, 2020, 370(6512), 108–112, DOI:10.1126/science.abc4417.
- W. Rehman, R. L. Milot, G. E. Eperon, C. Wehrenfennig, J. L. Boland, H. J. Snaith, M. B. Johnston and L. M. Herz, Charge-Carrier Dynamics and Mobilities in Formamidinium Lead Mixed-Halide Perovskites, Adv. Mater., 2015, 27(48), 7938–7944, DOI:10.1002/adma.201502969.
- D. J. Slotcavage, H. I. Karunadasa and M. D. McGehee, Light-Induced Phase Segregation in Halide-Perovskite Absorbers, ACS Energy Lett., 2016, 1(6), 1199–1205, DOI:10.1021/acsenergylett.6b00495.
- F. Xie, C.-C. Chen, Y. Wu, X. Li, M. Cai, X. Liu, X. Yang and L. Han, Vertical Recrystallization for Highly Efficient and Stable Formamidinium-Based Inverted-Structure Perovskite Solar Cells, Energy Environ. Sci., 2017, 10(9), 1942–1949, 10.1039/C7EE01675A.
- F. Ye, J. Ma, C. Chen, H. Wang, Y. Xu, S. Zhang, T. Wang, C. Tao and G. Fang, Roles of MACl in Sequentially Deposited Bromine-Free Perovskite Absorbers for Efficient Solar Cells, Adv. Mater., 2021, 33(3), 2007126, DOI:10.1002/adma.202007126.
- M. Kim, G.-H. Kim, T. K. Lee, I. W. Choi, H. W. Choi, Y. Jo, Y. J. Yoon, J. W. Kim, J. Lee, D. Huh, H. Lee, S. K. Kwak, J. Y. Kim and D. S. Kim, Methylammonium Chloride Induces Intermediate Phase Stabilization for Efficient Perovskite Solar Cells, Joule, 2019, 3(9), 2179–2192, DOI:10.1016/j.joule.2019.06.014.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Pseudo-Halide Anion Engineering for α-FAPbI3 Perovskite Solar Cells, Nature, 2021, 592(7854), 381–385, DOI:10.1038/s41586-021-03406-5.
- Y. Zhang, Y. Li, L. Zhang, H. Hu, Z. Tang, B. Xu and N. Park, Propylammonium Chloride Additive for Efficient and Stable FAPbI 3 Perovskite Solar Cells, Adv. Energy Mater., 2021, 11(47), 2102538, DOI:10.1002/aenm.202102538.
- A. Q. Alanazi, D. J. Kubicki, D. Prochowicz, E. A. Alharbi, M. E. F. Bouduban, F. Jahanbakhshi, M. Mladenović, J. V. Milić, F. Giordano, D. Ren, A. Y. Alyamani, H. Albrithen, A. Albadri, M. H. Alotaibi, J.-E. Moser, S. M. Zakeeruddin, U. Rothlisberger, L. Emsley and M. Grätzel, Atomic-Level Microstructure of Efficient Formamidinium-Based Perovskite Solar Cells Stabilized by 5-Ammonium Valeric Acid Iodide Revealed by Multinuclear and Two-Dimensional Solid-State NMR, J. Am. Chem. Soc., 2019, 141(44), 17659–17669, DOI:10.1021/jacs.9b07381.
- C. Ma, F. T. Eickemeyer, S.-H. Lee, D.-H. Kang, S. J. Kwon, M. Grätzel and N.-G. Park, Unveiling Facet-Dependent Degradation and Facet Engineering for Stable Perovskite Solar Cells, Science, 2023, 379(6628), 173–178, DOI:10.1126/science.adf3349.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775, DOI:10.1103/PhysRevB.59.1758.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- G. Kresse and J. Furthmüller, Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6(1), 15–50, DOI:10.1016/0927-0256(96)00008-0.
- G. Kresse and J. Hafner, Ab Initio Molecular Dynamics for Liquid Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47(1), 558–561, DOI:10.1103/PhysRevB.47.558.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104, DOI:10.1063/1.3382344.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin-Zone Integrations, Phys. Rev. B, 1976, 13(12), 5188–5192, DOI:10.1103/PhysRevB.13.5188.
- K. Momma and F. Izumi, VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data, J. Appl. Crystallogr., 2011, 44(6), 1272–1276, DOI:10.1107/S0021889811038970.
- Y. Li, P. J. Lohr, A. Segapeli, J. Baltram, D. Werner, A. Allred, K. Muralidharan and A. D. Printz, Influence of Halides on the Interactions of Ammonium Acids with Metal Halide Perovskites, ACS Appl. Mater. Interfaces, 2023, 15(20), 24387–24398, DOI:10.1021/acsami.3c01432.
- W. Wu, P. N. Rudd, Q. Wang, Z. Yang and J. Huang, Blading Phase-Pure Formamidinium-Alloyed Perovskites for High-Efficiency Solar Cells with Low Photovoltage Deficit and Improved Stability, Adv. Mater., 2020, 32(28), 2000995, DOI:10.1002/adma.202000995.
- H. Tan, A. Jain, O. Voznyy, X. Lan, F. P. García De Arquer, J. Z. Fan, R. Quintero-Bermudez, M. Yuan, B. Zhang, Y. Zhao, F. Fan, P. Li, L. N. Quan, Y. Zhao, Z.-H. Lu, Z. Yang, S. Hoogland and E. H. Sargent, Efficient and Stable Solution-Processed Planar Perovskite Solar Cells via Contact Passivation, Science, 2017, 355(6326), 722–726, DOI:10.1126/science.aai9081.
- S. Prathapani, D. Choudhary, S. Mallick, P. Bhargava and A. Yella, Experimental Evaluation of Room Temperature Crystallization and Phase Evolution of Hybrid Perovskite Materials, CrystEngComm, 2017, 19(27), 3834–3843, 10.1039/C7CE00402H.
- R. T. Wang, A. F. Xu, W. Li, Y. Li and G. Xu, Moisture-Stable FAPbI3 Perovskite Achieved by Atomic Structure Negotiation, J. Phys. Chem. Lett., 2021, 12(22), 5332–5338, DOI:10.1021/acs.jpclett.1c01482.
- Y. Li, H. Li, L. Tian, Q. Wang, F. Wu, F. Zhang, L. Du and Y. Huang, Vertical Phase Segregation Suppression for Efficient FA-Based Quasi-2D Perovskite Solar Cells via HCl Additive, J. Mater. Sci. Mater. Electron., 2020, 31(15), 12301–12308, DOI:10.1007/s10854-020-03775-z.
- J. Wang, L. Liu, S. Chen, G. Ran, W. Zhang, M. Zhao, C. Zhao, F. Lu, T. Jiu and Y. Li, Growth of 2D Passivation Layer in FAPbI3 Perovskite Solar Cells for High Open-Circuit Voltage, Nano Today, 2022, 42, 101357, DOI:10.1016/j.nantod.2021.101357.
- X. Liu, X. Jiang, Y. Yin, J. Zhang, H. Tian, J. Guo, X. Guo and C. Li, Dominating (111) Facets with Ordered Stacking in Perovskite Films, Energy Environ. Sci., 2024, 17, 6058–6067, 10.1039/D4EE01863J.
- S. Li, J. Xia, Z. Wen, H. Gu, J. Guo, C. Liang, H. Pan, X. Wang and S. Chen, The Formation Mechanism of (001) Facet Dominated α-FAPbI3 Film by Pseudohalide Ions for High-Performance Perovskite Solar Cells, Adv. Sci., 2023, 10(18), 2300056, DOI:10.1002/advs.202300056.
- J. Park, J. Kim, H.-S. Yun, M. J. Paik, E. Noh, H. J. Mun, M. G. Kim, T. J. Shin and S. I. Seok, Controlled Growth of Perovskite Layers with Volatile Alkylammonium Chlorides, Nature, 2023, 616(7958), 724–730, DOI:10.1038/s41586-023-05825-y.
- Y. Fu, T. Wu, J. Wang, J. Zhai, M. J. Shearer, Y. Zhao, R. J. Hamers, E. Kan, K. Deng, X.-Y. Zhu and S. Jin, Stabilization of the Metastable Lead Iodide Perovskite Phase via Surface Functionalization, Nano Lett., 2017, 17(7), 4405–4414, DOI:10.1021/acs.nanolett.7b01500.
- J.-W. Lee, Z. Dai, T.-H. Han, C. Choi, S.-Y. Chang, S.-J. Lee, N. De Marco, H. Zhao, P. Sun, Y. Huang and Y. Yang, 2D Perovskite Stabilized Phase-Pure Formamidinium Perovskite Solar Cells, Nat. Commun., 2018, 9(1), 3021, DOI:10.1038/s41467-018-05454-4.
- Y. Li, M. Dailey, P. J. Lohr and A. D. Printz, Performance and Stability Improvements in Metal Halide Perovskite with Intralayer Incorporation of Organic Additives, J. Mater. Chem. A, 2021, 9(30), 16281–16338, 10.1039/D1TA05252G.
- Z.-A. Nan, L. Chen, Q. Liu, S.-H. Wang, Z.-X. Chen, S.-Y. Kang, J.-B. Ji, Y.-Y. Tan, Y. Hui, J.-W. Yan, Z.-X. Xie, W.-Z. Liang, B.-W. Mao and Z.-Q. Tian, Revealing Phase Evolution Mechanism for Stabilizing Formamidinium-Based Lead Halide Perovskites by a Key Intermediate Phase, Chem, 2021, 7(9), 2513–2526, DOI:10.1016/j.chempr.2021.07.011.
- H. Min, M. Kim, S.-U. Lee, H. Kim, G. Kim, K. Choi, J. H. Lee and S. I. Seok, Efficient, Stable Solar Cells by Using Inherent Bandgap of α-Phase Formamidinium Lead Iodide, Science, 2019, 366(6466), 749–753, DOI:10.1126/science.aay7044.
- J. Bisquert, A. Guerrero and C. Gonzales, Theory of Hysteresis in Halide Perovskites by Integration of the Equivalent Circuit, ACS Phys. Chem. Au, 2021, 1(1), 25–44, DOI:10.1021/acsphyschemau.1c00009.
- J. Bisquert and A. Guerrero, Chemical Inductor, J. Am. Chem. Soc., 2022, 144(13), 5996–6009, DOI:10.1021/jacs.2c00777.
- J. Bisquert, Electrical Charge Coupling Dominates the Hysteresis Effect of Halide Perovskite Devices, J. Phys. Chem. Lett., 2023, 14(4), 1014–1021, DOI:10.1021/acs.jpclett.2c03812.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00527a |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2025 |