
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectrometric monitoring of CO2 electrolysis on a molecularly modified copper surface
Akiyoshi
Kuzume
*a and
Shoko
Kume
 *b
*b
aClean Energy Research Center, Yamanashi University, Kofu, 400-8510, Japan. E-mail: akuzume@yamanashi.ac.jp
bGraduate School of Advanced Science and Engineering, Hiroshima University, Higashi-Hiroshima, 739-8526, Japan. E-mail: skume@hiroshima-u.ac.jp
First published on 11th September 2024
Abstract
Since copper has been extensively studied due to its unique ability to reduce carbon dioxide to hydrocarbons and alcohols, it tends to yield a mixture of products. Among various efforts to improve the selectivity and efficiency of this catalysis, the introduction of organic molecules and polymers on the copper/electrolyte interface has proven to be an effective and promising way to improve surface activity, considering the variation and precise designability of organic structures. The role of surface molecular modifiers, however, is not as simple as that in homogeneous catalysts, and an understanding of a wide scale of interactions from the atomic scale to the whole electrode structure is required. This feature article classifies those different scale interactions caused by organic modifiers on copper catalysts, together with the experimental support by in situ vibrational spectroscopy which directly observes surface species and events. Based on these recent understandings, novel fabrication methods of organic structures on copper catalysts are also discussed.
 Akiyoshi Kuzume | Akiyoshi Kuzume is an Associate Professor at the Clean Energy Research Center in the University of Yamanashi, Japan. He received his BSc and MSC degrees from the University of Tokyo in 1999 and 2001, respectively, and PhD from Liverpool University, UK in 2004. He continued his research career in Alicante Univ., Keio Univ., Tohoku Univ., Bern Univ., and Tokyo Institute of Technology as a Post-Dr and Assistant and Associate Professors, before he was assigned the current position in 2020. His research focuses on the understanding of electrochemical reactions at the solid–liquid interfaces using spectroscopic and microscopic techniques. |
 Shoko Kume | Shoko Kume is an Associate Professor in the Graduate School of Advanced Science and Engineering, Hiroshima University. She received her PhD degree from the University of Tokyo in 2004 under the supervision of Prof. Hiroshi Nishihara. Her current research focuses on the interplay of metal catalysts with organic modifiers, molecular catalytic activity on the interfacial environment, and intelligent molecular catalysts responding to stimuli. |
1. Introduction
Carbon neutralization is an effort to achieve net-zero carbon dioxide (CO2) production by balancing the emission and absorption of carbon to and from the atmosphere. Because 90% of the total emitted greenhouse gases have been generated by the combustion of fossil fuels, considerable research has been conducted to decrease, fix, and recycle CO2 to realize carbon neutralization. Since it is unrealistic to lower CO2 emissions to zero, so much attention has been paid to developing technology to increase CO2 removal from the atmosphere through capture and storage. Electrochemical and chemical conversion technologies that can effectively convert CO2 to (bio)fuels, syngas, synthetic materials, and plastics are attracting interest from the perspective of sustainable development goals and carbon neutralization, not only for CO2 fixation but also to generate alternative fuels to petroleum. Electrochemical reduction of CO2, which has been studied since the 19th century,1 progresses via a multi-step reaction involving multiple electrons and protons (H+) and generates various chemicals such as carbon monoxide (CO), formate, hydrocarbons, and alcohols depending on the electrode material and solvent used.2 The CO2 reduction reaction (CO2RR) is accompanied by the hydrogen evolution reaction (HER) as a parasitic side reaction, which complicates the elucidation of the mechanism of the CO2RR. In the 1990s, Hori et al.3 roughly classified metal electrodes into four types according to the reaction selectivity and main products: (i) metals that mainly produce CO due to weak CO adsorption (Au, Ag, and Zr), (ii) metals that mainly produce formate (Pb, Hg, Cd, Tl, In, and Sn), (iii) metals that suppress the CO2RR due to CO poisoning (Pt, Ni, Fe, and Ti), which results in the dominant formation of hydrogen gas, and (iv) Cu, which can produce hydrocarbons such as methane, ethylene, and alcohol. The unique activity of Cu is caused by moderate stabilization of adsorbed CO, allowing it to be further reduced to various hydrocarbons. As a result, optimizing the Cu catalyst for the CO2RR to capture and effectively convert CO2 to useful materials has been extensively investigated for the last three decades.Since the first report of the Cu CO2RR producing hydrocarbons by Hori et al.,4,5 new aspects of this reaction have been discovered with development of new catalysts. In 2011, Kanan et al.6 prepared a Cu catalyst by reducing copper oxide which exhibited CO2 reduction with a much lower overpotential. Subsequent studies showed that Cu catalysts derived from oxides and hydroxides were effective for C2+ production.7,8 The favorable performance of Cu in the CO2RR has been ascribed to many factors such as (i) the adsorbed CO activity being affected by the remaining oxides and Cu+ species during the CO2RR,9–11 (ii) formation of active Cu atoms at grain boundaries,12,13 and (iii) faceting of the Cu surface.14 Bell et al.15 observed that the CO2RR was strongly dependent on the alkali metal cations present in the electrolyte, which revealed the importance of water molecules at the electrode surface acting as an H+ source in the CO2RR. Roldan Cuenya and co-workers showed that their Cu cube catalyst exhibited dynamic structural changes during the CO2RR even when Cu was zero-valent.16 The generation of C2+ products requires C–C bond formation on the catalyst surface, so it is also affected by the partial pressure of CO2.17
These discoveries on the CO2RR activity of nanostructured Cu surfaces have revealed the unique features of this electroreduction process. First, CO2 is hardly soluble in water and its supply to the electrode surface is as important as the catalyst surface activity in determining CO2RR activity. Second, CO2 reduction consumes H+ and produces hydroxide anions (OH−). Therefore, the behavior of water, the most common H+ supplier, can determine the catalytic efficiency. Third, the way H+ is supplied to the electrode surface and local pH affect CO2 reduction and determine the favored product. Fourth, the Cu surface is not robust during electroreduction; its structure often evolves to a more stable configuration under the applied cathodic potential and is affected by other chemical conditions. These interfacial insights notified researchers to carefully evaluate CO2RR performance.18
Along with the catalyst development, molecular processes of the CO2RR on Cu have been extensively discussed. They involve branching of the intermediates because Cu normally forms multiple carbonaceous products, even from a single crystal facet. These complex hydrocarbon-generation mechanisms and reaction pathways have been reviewed by Koper and colleagues (Fig. 1).19 Numerous experimental and theoretical studies have been conducted to elucidate the reaction mechanism and pathways of the electrochemical CO2RR and mechanistic insights for the design of efficient and selective Cu-based catalysts for the CO2RR have been outlined in multiple reviews.20–25
 | ||
| Fig. 1 Schemes of the possible reaction pathways of the CO2RR on transition metals: (a) pathway from CO2 to CO, CH4, CH3OH, and formate, (b) pathways from CO2 to ethylene and ethanol, and (c) pathway of CO2 insertion into a metal–H bond yielding formate. Adapted with permission from ref. 19. Copyright 2015 American Chemical Society. | ||
With these advances in Cu electrocatalysts, organic modification of Cu targeting CO2RR promotion has also been focused. In 2016, Wang's group reported the earliest attempt to modify the Cu catalyst surface with organic moieties to improve CO2RR activity by using various amino acids as modifiers.26 This and other early studies27–29 mainly focused on the local molecular interactions of CO2 and/or its reduction intermediates on Cu surfaces with introduced chemical moieties. In 2019, Toste et al.30 demonstrated the universal effect of various polymers and molecules on the Cu CO2RR, revealing that organic species affect catalysis by regulating the behavior of water on the Cu catalyst surface. In 2021, Goddard and co-workers showed that the components of a polymer modifier included in the catalytic system cooperatively improved the C2+ production selectivity.31 They suggested that the modifier is likely to induce a combination of nonspecific effects such as increased local CO2 concentration, increased porosity for gas diffusion, and a local electric field effect rather than the specific molecular interactions of the components affecting CO2RR activity.
Considering these findings, it came to be recognized that the effect of organic modifiers on CO2RR activity needs to be studied from a wider perspective, spanning the specific interactions between Cu atoms and reaction intermediates; the CO2 and H+ supply path to the active electrode surface; and larger-scale catalyst assemblies that involve three-phase interfaces (CO2 gas/liquid electrolyte/solid Cu).32,33 To evaluate this wide range of factors, direct observation of the catalyst surface during operation is essential. In situ observation with Raman and infrared (IR) spectroscopy is a powerful tool to elucidate the species present and events occurring on working catalyst surfaces. Vibrational spectroscopy techniques enable us to evaluate the reaction intermediate structure, the ratio of species involved in H+ transfer, and the state of the Cu surface. This direct information can help us to elucidate the role of organic modifiers in improving CO2RR activity.
In this feature article, we first introduce the use of vibrational spectroscopy to investigate the CO2RR process on catalyst surfaces. Next, recent research on the organic modification of Cu catalysts to increase CO2RR efficiency is presented. The studies are classified according to the role of the organic modifier, which is determined based on the in situ observation of the active catalyst surface. Finally, we introduce recent research on the development of organic modifiers to maximize their potential functions. Here we only cover studies that use vibrational spectroscopy; other reviews of Cu electrocatalysis and the role of organic modifiers in the CO2RR can be found elsewhere.34,35
2. Application of vibrational spectroscopy to the CO2RR on copper electrocatalysts
2.1 Vibrational spectroscopy for in situ interfacial analysis of electrocatalysis
Electrochemical reduction reactions of CO2 that occur on electrode surfaces involve multi-electron and multi-proton transfers, making in situ interfacial analysis invaluable for evaluating and understanding the reaction activity and mechanisms to characterize innovative high-performance catalysts. In particular, vibrational spectroscopy techniques such as Raman spectroscopy and IR absorption spectroscopy can provide chemical information about adsorbates, water molecules, and reaction intermediates during electrochemical reactions under applied potential, thus allowing elucidation of electron transfer and chemical reaction processes at solid–liquid interfaces. Vibrational spectroscopy is also sensitive to the local interfacial pH and electrode oxidation state, making it a powerful analytical method to evaluate the complex surface processes of the CO2RR.Raman scattering light36 contains information about the molecular vibrations of the material, but its scattering cross section (intensity) is much weaker than that of elastically scattered light (Rayleigh scattering). Surface-enhanced Raman spectroscopy (SERS) is a powerful tool to detect surface species with high sensitivity.37–39 Surface plasmon resonance (SPR), which typically occurs on rough and nanostructured surfaces of coinage metals (Au, Ag, and Cu), is the surface-localized resonance between incident light and the collective oscillations of the conduction electrons on a metal surface, inducing local electromagnetic fields that enhance Raman signals. The chemical enhancement effect, on the other hand, originates from electronic interactions, including charge transfer between the metal surface and adsorbed molecules; this effect is selective for the first layer of adsorbed species on a metal surface. These two enhancement effects allow SERS to be used as a surface-selective, non-destructive, and highly sensitive analytical method for solid surfaces.
Surface-enhanced infrared absorption spectroscopy (SEIRAS) is another type of vibrational spectroscopy that can detect adsorbed species with high selectivity and sensitivity.40,41 Adequate surface enhancement requires a nanostructured thin film of coinage metal adhered to the surface of an IR-transparent prism. The plasmonic fields generated on the coinage metal surface strongly contribute to the enhancement mechanism, selectively enhancing molecular signals in the immediate vicinity of the surface. In addition, SEIRAS in an attenuated total reflection (ATR) configuration allows for unrestricted mass transport to/from the electrode surface, making it suitable for investigating gas-evolving reactions such as the CO2RR.
SEIRAS detects the vibrational modes of molecules with large dipole moments, whereas Raman spectroscopy detects vibrational modes with high symmetry that have a large change in polarizability. Therefore, the combined use of SEIRAS and Raman spectroscopy in a complementary manner can provide comprehensive chemical information about a metal catalyst surface, such as the molecular structure, crystal structure, adsorption geometry, and adsorbate configuration, extracting mechanistic information and establishing structure–activity relationships. Both IR and Raman spectroscopies can be used for in situ observation of solid–liquid interfaces and therefore are widely utilized in the study of electrochemical CO2 reduction. It is important to point out that SEIRAS and SERS are frequently employed in the assumption that they provide largely overlapping information regarding reaction intermediates. To confirm this, Chang and co-workers investigated CO adsorption on Pt, Pd, Au, and Cu using both surface-enhanced spectroscopic techniques.42 SEIRAS and SERS provided similar spectral information in terms of peak position and the Stark tuning rate on strongly adsorbing surfaces (Pd and Pt), but probed different subpopulations of adsorbates on weakly adsorbing surfaces (Au and Cu) because of competitive adsorption of water molecules. Complementary density functional theory (DFT) calculations confirmed the lack of scaling between the derivatives of the dipole moment and polarizability, indicating that the peak intensities in SEIRAS and SERS spectra do not necessarily correlate with each other.
2.2 In situ observation of the electrochemical CO2RR on copper
Soon after the discovery of the SERS effect, Fleischmann's group performed in situ Raman spectroscopy measurements of the CO2RR, detecting surface-adsorbed CO2− and carboxyl groups on an electrochemically roughened Ag electrode.43 Since then, the detection of electrochemically generated species, especially adsorbed CO on metal surfaces, by SERS has provided valuable information for elucidating the mechanism of electrochemical CO2 reduction.As mentioned above, Cu is the only metal that mainly produces hydrocarbon and alcohol species from the electrochemical reduction of CO2. Using a Cu catalyst produces a wider range of intermediates than those obtained using other metals, which only produce CO or formate. It is known from the slope of the Tafel plot of the CO2RR that the rate-determining step is the first one-electron reduction process (CO2 + e− → CO2−). Considering the findings of in situ Raman spectroscopy and DFT calculations, Chernyshova et al.44 reported that the intermediate formed in the first step of the CO2RR is the carboxyl group η2(C,O)–CO2− adsorbed on the Cu surface with C and O (Fig. 2). The carboxyl group was further reduced to HCOO− and CO adsorbates depending on the presence of surface structures, such as defects and steps, and partial oxidation states of Cu. These intermediate adsorbates were finally converted to hydrocarbons, alcohols, and/or formate as the final products of the CO2RR.
 | ||
| Fig. 2 Operando SERS of the rough Cu surface in CO2-saturated 0.1 mol dm−3 NaHCO3 (pH 6.8). Spectra were measured in the cathodic direction from −0.1 V. Adapted with permission from ref. 44. Copyright 2018 National Academy of Sciences. | ||
Using a complementary combination of in situ Raman spectroscopy and IR spectroscopy, Ito and colleagues studied the spectral features of adsorbed species (C–O) and metal-adsorbed species (Cu–CO) on the Cu electrode surface during the CO2RR.45 At the onset of the CO2RR, the CO stretching vibration was observed around 2000 cm−1 on the Cu surface and the Cu–CO stretching vibration was observed around 360 cm−1. The intensities of both signals gradually decreased during a cathodic potential sweep accompanied by the decrease of adsorbed carbonate (CO32−). It is interesting to note that two signals assigned to CO adsorbed on terrace (around 2000 cm−1) and adatom defect Cu atoms (around 2100 cm−1)46 were observed when CO gas was introduced onto the Cu surface, while only the former appeared under in situ CO2RR conditions, demonstrating the ability of vibrational spectroscopy to monitor the effect of the surface structure of the Cu electrode on the CO2RR.
In situ Raman and IR spectroscopies are capable of not only detecting adsorbate species, but also monitoring the oxidation state of Cu, the role of interfacial water, and the effect of surface reconstruction, providing information that can be used to improve CO2RR efficiency and selectivity. Mandal et al. performed in situ Raman spectroscopy focusing on adsorbed CO and the oxidation state of the Cu surface during the CO2RR on various Cu2O nanostructures to clarify the effect of the CO adsorption affinity on the reaction efficiency and selectivity.47 The Cu2O surface was immediately reduced at the potential onset of the CO2RR and CO was adsorbed in an on-top configuration on the exposed metallic Cu sites. The selective formation of C2+ products proceeded only after the reduction of Cu2O to metallic Cu. In addition, the Raman signal intensity of adsorbed CO varied with the surface structures of Cu catalysts, and was related to the proportion of under-coordinated sites such as defects and boundaries on the metal surface. The increased population of adsorbed CO is likely to result in multi-carbon products.
The correlation between the potential-dependent CO coverage and selective production of C2+ was also demonstrated in a study of the CO2RR with Cu2O nanocubes using operando Raman spectroscopy.48 Zhan and co-workers suggested that the potential-dependent intensity ratio of the Cu–CO stretching band (around 350 cm−1) to the CO rotation band (around 280 cm−1) followed a volcano trend, similar to the Faraday efficiency of multi-carbon products (Fig. 3). This finding illustrated that there is a clear correlation between the CO coverage of catalyst surfaces and C–C coupling modes observed in vibrational spectra.
 | ||
| Fig. 3 Left half: Correlation between the potential-dependent intensity ratio of (P1: 350 cm−1/P2: 280 cm−1) and the Faraday efficiency of multi-carbon production showing a volcano trend in the function of potentials. Right half: (a) Raman spectra of adsorbed CO on Cu with different CO concentrations in the 0.1 mol dm−3 KHCO3 electrolyte. The experiments with different CO concentrations were repeated three times. (b) The intensity ratio of two peaks as a function of the CO concentration, where the red line shows the fitting result based on a Langmuir equation. (c) Theoretical benchmark of the peak ratio intensity vs. CO coverage. Adapted with permission from ref. 45. Copyright 2021 American Chemical Society. | ||
It is important to understand the roles of the oxidation state of the Cu surface and adsorbed intermediates in the electrochemical CO2RR. Chou et al.49 investigated the CO2RR mechanism with Cu catalysts in various oxidation states using in situ SEIRAS and X-ray absorption spectroscopy (Fig. 4). They showed that CO formed at the on-top position on the electrodeposited Cu surface, which primarily has the oxidation state of Cu(I), and a C1 hydrocarbon product was obtained during further reduction. The as-prepared Cu electrode exhibited an oxidation state of Cu(0), which promoted the generation of bridged CO, inhibiting the formation of a hydrocarbon product. In contrast, the Cu electrode pretreated by cyclic voltammetry contained both Cu(I) and Cu(0), where on-top CO and bridged CO, respectively, were observed during the CO2RR, enhancing selective C2 production. Therefore, the oxidation state of the Cu catalyst affected the electrocatalytic properties of the substrate–catalyst interface and modulated the CO2 reduction mechanism.
 | ||
| Fig. 4 Upper row: Schematic diagram of the detection of the reaction selectivity and final products on Cu surfaces with different oxidation states using IR and X-ray spectroscopy. Middle row: In situ SEIRAS of the (a) electrodeposited Cu, (b) as-prepared Cu, and (c) CV-treated Cu electrodes during cathodic and anodic scans in CO2-saturated 0.1 mol dm−3 KHCO3 electrolyte solution. Lower row: A table showing the vibrational frequencies and assignments for the species in the IR spectra (a)–(c) under an electrochemical CO2RR. Adapted with permission from ref. 46. Copyright 2020 American Chemical Society. The table shows the vibrational frequencies and assignments for the species in the electrochemical CO2RR. Adapted with permission from ref. 46. Copyright 2020 American Chemical Society. | ||
In the CO2RR, the presence of interfacial water directly affects the kinetics of hydrogenation steps along with the competing HER. However, the structural composition and dynamic evolution of interfacial water molecules are difficult to investigate because of the interference from bulk water and the bias dependence of interfacial water layers. Wang and co-workers used in situ ATR SEIRAS and in situ Raman spectroscopy combined with molecular dynamics simulations to probe the dynamic changes of interfacial water structure on an electrified surface and explore the mechanism of HER activity and selectivity with the goal of enhancing multi-carbon product formation.50 Their results demonstrated that the hydrogen-bonding structure of interfacial water played an important role in controlling reaction selectivity in the CO2RR.
In situ vibration spectroscopy can also be used to monitor the effect of surface morphology at the nanoscale on CO2RR efficiency and selectivity. Gunathunge et al.51 reported the first observation of the reversible formation of nanoscale Cu clusters on an electrode surface during the CO2RR induced by the adsorbed CO intermediate. A series of in situ SEIRA and SERS spectra of the on-top CO molecules were deconvoluted into two peaks: the low-frequency band (LFB; 2050 cm−1) and high-frequency band (HFB; 2080 cm−1) were assigned to the CO molecules adsorbed at terrace and defect sites, respectively. The potential-dependent spectra showed the reversible change of the band intensities of the HFB and LFB, suggesting the reversible formation of CO-stabilized Cu clusters containing under-coordinated Cu atoms. It is worth noting that while the sufficient detection of the SERS effect usually requires roughening of the copper surface, the use of silica encapsulated Au nanoparticles (Au@SiO2), as inert surface signal amplifiers, provides an effective strategy to conduct SERS investigations without the need to modify target surface morphologies.52,53 Zhao et al.54 applied this technique to monitor the single crystal low index Cu(hkl) surfaces on the CO2RR in KHCO3 solution using in situ Raman spectroscopy. The reaction intermediates such as*COOH, *CO, *OCCO and *CH2CHO were observed depending on the surface-crystal structure that determined the reaction selectivity. Combining with theoretical calculations, Cu(111) facilitates the generation of C1 products through the formation of intermediates *COOH and *CO, while Cu(110) further generates C2 through the pathway of *OCCO and *CH2CHO reaction intermediates.
3. Direct in situ observation of the electrochemical CO2RR on organically modified Cu surfaces
3.1 Inhibitory effects of molecular adsorption on active Cu sites
When introducing modifiers, the poisoning of active Cu sites owing to the adsorption of small molecules is a critical consideration. Although this phenomenon might be prevalent, it is often unreported because of activity losses. Waegele et al.55 elucidated this phenomenon by comparing the influence of the adsorption of 1-(4-tolyl)pyridinium (T-Pyr)56 and 1-(4-pyridyl)pyridinium (P-Pyr) on CO2RR activity (Fig. 5a). Surface modification with T-Pyr increased the production of C2+ compounds by increasing the interfacial pH. In contrast, surface modification with P-Pyr completely suppressed hydrocarbon production owing to the presence of the pyridine moiety (Fig. 5b). Notably, the P-Pyr-modified surface produced a comparable amount of hydrogen to the unmodified surface. SEIRAS analysis revealed the disappearance of the *COatop band (2070 cm−1, * denotes the adsorbed species on the surface) attributed to the uncoordinated Cu sites on the P-Pyr-modified surface (Fig. 5c). Conversely, the *COatop band at 2040 cm−1, attributed to highly coordinated Cu sites, was observed for the T-Pyr- and P-Pyr-modified and unmodified surfaces. These results indicate that the competitive adsorption between the nitrogen in pyridine and CO inhibits the activity at low coordination sites. | ||
Fig. 5 (a) Modifiers and reduced dimeric structure.41,42 (b) CO2RR selectivity on Cu electrodes with 10 mM modifiers in CO2-saturated 0.1 M KHCO3 at an applied potential of −1.1 V vs. an RHE.41 (c) Comparison of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching bands of COatop on Cu with different modifiers observed with SEIRAS at an applied potential of −1.5 V at the three different interfaces as indicated. The green dashed and solid lines represent a fit of the model. Reproduced with permission from ref. 55. Copyright 2019 American Chemical Society. O stretching bands of COatop on Cu with different modifiers observed with SEIRAS at an applied potential of −1.5 V at the three different interfaces as indicated. The green dashed and solid lines represent a fit of the model. Reproduced with permission from ref. 55. Copyright 2019 American Chemical Society. | ||
Lv et al.57 demonstrated that the modification using stearic acid was ineffective in any reduction process owing to its closely packed linear alkyl chain structure, whereas the sparser modification with an aromatic group was effective for producing large amounts of C2+ products with good selectivity. Alkanethiols with long alkyl chains generally form closely packed, self-assembled monolayers on metal surfaces, causing significant poisoning. However, adsorbed alkanethiol can desorb when a cathodic potential is applied (reductive desorption). Fontecave et al.32 used the reductive desorption of alkanethiol to expose surface Cu atoms while maintaining high surface hydrophobicity, which was achieved by modifying both the morphology (Cu dendrites) and 1-octadecanethiol. Only the top tip of the dendrite interacted with the electrolyte, which electrochemically activated it at the site where the reductive desorption of 1-octadecanethiol occurred. This site, located at the triple-phase boundary (gas/electrolyte/copper), is advantageous for the formation of C2+ products, as described in Sections 3.3 and 3.4. Dai et al.58 exploited alkanethiol in a similar phase-boundary strategy.
Buonsanti et al.59 introduced various surfactants on Cu nanocrystals and monitored their adsorption behaviour under cathodic potential. Dodecanethiol remained adsorbed on the nanocrystal surface and inhibited hydrocarbon production during the CO2RR, while the other ligands desorbed rapidly before the CO2RR, exerting negligible influence on product distribution.
3.2 Specific surface interactions between the modifier and CO2RR intermediates
Specific interactions between the introduced organic moieties and surface carbonaceous intermediates have been a primary focus since early studies, such as those by Verma et al.27 using solid polymer electrolytes and Wang et al.26 using amino acids for modification. The theoretical calculations further supported these findings.28,29 Etzold et al.60 introduced the ionic liquid 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([BMIm][NTf2]) as a chemical trapping agent. By analysing the product spectra under different conditions, the researchers elucidated the surface processes that led to the formation of multiple products on Cu. Although these studies demonstrated the effectiveness of organic modification and inspired subsequent research, direct observations of these reactions were not provided.Recently, in situ vibrational spectroscopy has become common for probing molecular interactions during CO2 reduction. Wang et al.61 introduced a 2,5-dimethoxy-1,4-benzoquinone (DMBQ) polymer onto a Cu surface (Fig. 6a), which enhanced the current density and C2+ product selectivity (Fig. 6c). The researchers attributed these improvements to the quinone groups, which facilitated CO2 reduction via the reduction of the quinone moiety. In situ ATR–SEIRAS measurements detected a DMBQ–CO2− adduct peak at 1529 cm−1 attributed to the vibration of the quinone ring skeleton in DMBQ. However, the 1582 cm−1 band for unmodified Cu, attributed to the asymmetric stretching vibration of *CO2−, was absent. They concluded that CO2 was reduced to *CO via the formation of an adduct with the reduced quinone moiety without directly forming *CO2− on the Cu surface. The increased intensity of the *CO peak (Fig. 6b) indicates a more efficient quinone-mediated reduction than direct CO2 reduction, thereby increasing the probability of *CO forming C–C bonds and producing C2+ products (Fig. 6a).
 | ||
| Fig. 6 (a) Schematic illustration of CO2 reduction over bare Cu (left) and Cu/pDMBQ (right). (b) Integral areas of the *CO band at different potentials observed by ATR-SEIRAS for CO2 reduction. (c) C2H4 partial current densities versus cell voltages for CO2/pure water coelectrolysis using an MEA electrolyzer with the Cu/pDMBQ GDE (purple dot) and Cu GDE (gray open square). Reproduced with permission from ref. 61. Copyright 2022 American Chemical Society. | ||
Lim et al.62 incorporated histidine into Cu2O and deposited it on a carbon electrode. Among various imidazolium-based modifiers, histidine is particularly effective for the formation of C2+ products, especially ethanol. In situ Raman spectroscopy revealed that histidine was adsorbed physically onto the Cu surface and persisted throughout the CO2RR, potentially owing to the formation of the carbamate cation via a CO2 adduct, enabling it to remain stable on the electrode in a cathodic environment. Interestingly, the absence of the characteristic Cu–CO and CO frustrated rotation bands (typically observed at 279 and 364![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−1, respectively) indicated the absence of *CO, a common intermediate for C2+ products. Although the CO stretching vibration might have been obscured by a histidine-related band at 2081cm−1, the spectroscopic evidence and theoretical calculations prompted the researchers to propose a reaction mechanism that proceeded via a histidine–CO2 adduct and not via *CO.
cm−1, respectively) indicated the absence of *CO, a common intermediate for C2+ products. Although the CO stretching vibration might have been obscured by a histidine-related band at 2081cm−1, the spectroscopic evidence and theoretical calculations prompted the researchers to propose a reaction mechanism that proceeded via a histidine–CO2 adduct and not via *CO.
Fontecave et al.63 reported that modification with 4-mercaptopyridine strongly favoured formate formation. Based on theoretical calculations, they proposed a mechanism in which the modifier suppresses one of the initial CO2 reduction pathways involving C–Cu bond formation, which typically leads to the formation of the *CO intermediate. Conversely, another pathway proceeding via O–Cu bond formation is unaffected and ends in formate formation. Interestingly, Liu et al.64 reported that 4-mercaptopyridine significantly affected the reduction step of externally supplied CO, and the product was dominated by acetate. In situ ATR–SEIRAS analysis reveals an increased amount of *CObridge upon modification. This *CObridge is more susceptible to being reduced to *CHO, which can then asymmetrically couple with *COatop to form *OCCHO, a plausible intermediate for acetate formation. The detection of the *CHO and *OCCHO peaks further supported the proposed reaction pathway. The theoretical calculations indicated that 4-mercaptopyridine modified the local reaction environment of Cu and stabilised intermediates by forming hydrogen bonds through the nitrogen atom.
Liu et al.65 introduced a common surfactant, dodecyl sulfonate, on the Cu surface by reducing dodecyl sulfonate-modified Cu(OH)2. The catalyst exhibited high selectivity for C2+ product formation (86% Faraday efficiency (FE)), and particularly a high proportion of ethanol (64% FE) was formed. In situ Raman spectroscopy revealed a higher wavenumber for the Cu–CO stretching (362 cm−1) for modified Cu than for unmodified Cu (354 cm−1), which may indicate differences in reactivity. They also analysed the reaction intermediate *HCCOH, which can be converted into ethylene and ethanol. The peak at 1197 cm−1 was attributed to the stretching vibration of C–OH in the *HCCOH intermediate, whereas it appeared at 1182 cm−1 for the unmodified catalyst, indicating that the interaction with dodecyl sulfonate strengthened the C–O bond. This phenomenon can be attributed to hydrogen bonding between the dodecyl sulfonate anion and the hydroxyl H of this intermediate. The stabilised C–O bond is difficult to cleave, leading to the formation of ethanol.
3.3 Effect of modifiers on the CO2RR involving surface water molecules
Modification via the electrodimerization of arylpyridinium on Cu (Fig. 5a) is a well-established example of promoting the CO2RR via organic modification. When first reported by Agapie et al. in 2017,56 the role of organic modification was unclear. Waegele et al.55 elucidated the promotion mechanism using SEIRAS and observed an increase in the local pH of the modified electrode by monitoring the carbonate peaks in solution. Sargent et al.66 exploited molecular design to expand this family of molecules by introducing substituents. By evaluating their performance, the researchers provided an explanation for CO2RR promotion, which was supported by theoretical calculations and in situ spectroscopy. They determined the *COatop/*CObridge ratio by in situ Raman spectroscopy and identified a volcano-type relationship between this ratio and the ethylene selectivity in the CO2RR. This finding indicates that the *CObridge–*CObridge or *COatop–*COatop pathway is responsible for efficient ethylene formation via the formation of C–C bonds between *COatop and *CObridge. A linear relationship between the Bader charge of N in tetrahydro-bipyridine and the *COatop/*CObridge ratio was demonstrated, and an interaction model mediated by H2O molecules was proposed.Wang et al.67 reported that modification with glutathione (Fig. 7a) selectively formed methane from CO2, with minimal formation of C2+ products (Fig. 7b). In situ Raman spectra (Fig. 7c) indicated the absence of *COatop, which is essential for C–C coupling and the formation of C2+ products. They also used the bicarbonate/carbonate peak ratio to indicate surface pH. A larger bicarbonate/carbonate peak ratio for glutathione-modified Cu indicated a lower local pH, possibly owing to the affinity of the deprotonated carboxylate anion (COO−) for H2O, which promoted proton release. This proton supply effect can also facilitate further reduction of *CO to methane before C–C bond formation and C2+ product generation.
 | ||
| Fig. 7 (a) Structure of modified Cu with glutathione (GSH). (b) Effect of GSH modification on the CO2RR product. (c) Operando Raman spectra for Cu/C and GSH-Cu/C in the potential range from the OCP to −0.8 V vs. RHE. Reproduced with permission from ref. 67. Copyright 2022 American Chemical Society. | ||
Toma et al.30 also modified a library of organic molecules and polymers and classified the modifiers based on their protic/aprotic substituents and hydrophobicity. A comparison of the products of the CO2RR under mild potential indicated that the products were limited to H2, CO, and formate. Protic functional groups promoted the HER. The contact angle of the surface exhibited a good relationship with the formic acid ratio and was insensitive to the chemical nature of the modifier. This finding indicates that the modifiers indirectly influenced the CO2RR by altering the behaviour of water on the surface. Adsorbed hydrogen is more stable on a hydrophobic surface than on a hydrophilic surface. The less stable adsorbed hydrogen can form H2 or react with CO2 to form formate; this process is more difficult on hydrophobic surfaces that produce CO as the major product. Although this study was not supported by in situ spectroscopic analysis, it indicated that the behaviour of surrounding water molecules significantly affects the CO2RR, which can be modulated by surface molecular modifiers.
Cui et al.68 observed surface water molecules on a hydrophobic octadecanethiol-modified Cu electrode using in situ Raman spectroscopy. The OH stretching band was deconvoluted into three peaks (Fig. 8a) corresponding to tetrahedrally coordinated H-bonded water at approximately 3200 cm−1, trigonally coordinated H-bonded water at approximately 3400 cm−1, and H-bonding-free water with dangling O–H bonds at approximately 3600 cm−1. The unmodified Cu electrode showed a strong dependence of the ratio of these components on the applied potential, with the amount of free water increasing as the potential became more negative. This finding indicates that the applied potential caused the water molecules to adopt two H-down configurations, cleaving the hydrogen-bond network among the surface water molecules and causing the water to dissociate into *H. In contrast, the hydrophobic octadecanethiol-modified Cu surface exhibited a relatively smaller OH stretching band, and the OH band was almost insensitive to the applied potential. This phenomenon leads to a lower tendency for surface *H formation. They also observed a *CO stretching band, indicating that the modification promoted *CO formation and enhanced its adsorption. This observation explains the performance of modified surfaces in the CO2RR, suppression of the HER, and the increase in methane formation (Fig. 8b).
 | ||
| Fig. 8 (a) In situ Raman spectra of the O–H stretching mode of interfacial water molecules with (hydrophobic) and without (hydrophilic) modification with 1-octadecanethiol. Peaks are deconvoluted. (b) Effect of modification on CO2RR selectivity. Reproduced with permission from ref. 68. Copyright 2021 American Chemical Society. | ||
Zheng et al.57 modified CuO nanosheets with toluene (T–Cu) via catalytic aerobic oxidation. The distance between immobilised toluene moieties on the Cu surface (5.1 Å) indicated a more dispersed structure of the hydrophobic surface than the stearic acid-modified Cu surface (S–Cu). Analysis of surface water molecules using in situ Raman spectroscopy indicated that the O–H stretching band of water could be deconvoluted into three peaks: 4-coordinated hydrogen-bonded water at approximately 3200 cm−1 (4-HB·H2O), 2-coordinated hydrogen-bonded water at approximately 3400 cm−1 (2-HB·H2O), and K+ ion-hydrated water at approximately 3600 cm−1 (K·H2O) (Fig. 9d). The increasing peak ratio for the K·H2O component with a more negative potential indicated the cleavage of hydrogen bonds and coverage of the surface by *H. It increased steeply for unmodified Cu; however, the increase was moderate for the toluene-modified catalyst (Fig. 9e). For closely packed S–Cu, the increase was the smallest. The difference in the behaviour of surface water was reflected in the CO2RR performance, with unmodified Cu producing a large amount of hydrogen (Fig. 9a), S–Cu consuming minimal total current for CO production (Fig. 9b), and T–Cu exhibiting the most efficient formation of C2+ products while suppressing hydrogen formation (Fig. 9c). These results indicate that maintaining the space between modifiers for effective proton transport and CO–CO dimerisation is essential for modifiers (Fig. 9f).
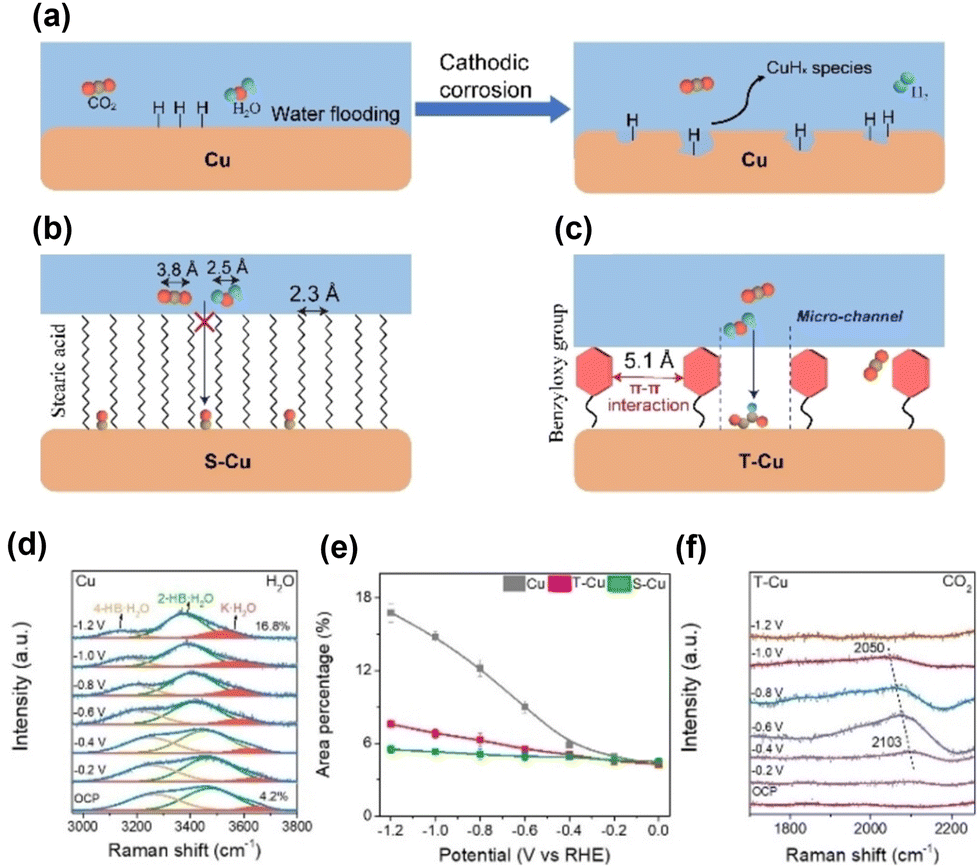 | ||
| Fig. 9 Schematic representation of surface processes for (a) unmodified Cu, (b) Cu modified with stearic acid (S–Cu) and (c) Cu modified with the benzyloxy group (T–Cu). (d) In situ Raman spectra of the interfacial water structure on Cu from 0 to −1.2 V. Peaks are deconvoluted. (e) Area percentages of K·H2O peaks at different applied potentials on Cu, T–Cu and S–Cu. (f) In situ Raman spectra of the CO adsorption peak on T–Cu. Reproduced with permission from ref. 57. Copyright 2023 Wiley. | ||
The strategy of introducing a molecular layer is a general method for suppressing the HER. Lu et al.69 studied the CO2RR under high-pressure CO2 conditions. Under these conditions, the carbonaceous product was converted to formate; however, some H2 was still produced. They introduced polypyrrole as a proton-resistant layer and succeeded in reducing H2 production.
Thoi et al.70 reported that the addition of a small amount of an ammonium surfactant (cetyltrimethylammonium bromide, CTAB) significantly suppressed the HER. The CO2RR performance is affected by the type of alkali cation in the electrolyte71,72 owing to the presence of surrounding hydrated water molecules. The addition of CTAB mitigated the difference between these alkali cations, decreasing the HER and increasing CO and formate formation. ATR–SEIRAS measurements revealed a decrease in the intensity of the O–H stretching band of surface water, which was also insensitive to the applied negative potential. This phenomenon can be attributed to the substitution of hydrated alkali cations with CTA+ cations, which led to the suppression of the HER. However, the disruption of the interaction between the hydrated cation and reaction intermediate also resulted in the inhibition of the pathway for C2+ product formation, with CO and formate being the major products.
The influence of CTAB on the CO2RR on Cu was also studied by Wang et al.,73 who revealed that formate formation was strongly increased in the low overpotential region (−0.5 V vs. RHE; Fig. 10a). In situ Raman spectroscopy revealed a band centred at approximately 2900 cm−1 attributed to C–H stretching in HCOO*, which is an intermediate for formate formation (Fig. 10b). The reductive desorption of HCOO* as formate can be considered a rate-determining step. They observed a change in the shape of this band (Fig. 10c), which was attributed to the substitution of HCOO* with CTA+ at the surface. Because cationic CTA+ is attracted as the potential shifts to negative values, the reductive desorption of HCOO* is promoted, resulting in a 56-fold increase in formate formation with an 86% selectivity in this low-overpotential region.
 | ||
| Fig. 10 (a) Dependence of the electrocatalytic CO2 reduction performance of Cu on the straight-chain alkyl group R of the NR(CH3)3Br additive. (b) and (c) In situ Raman spectra of Cu and Cu–CTAB in CO2-saturated 0.5 M KHCO3 under applied potentials, with the schematic illustration of formate formation. Reproduced with permission from ref. 73. Copyright 2020 American Chemical Society. | ||
Surendnarath et al.74 analysed the electrochemical kinetics of the CO2RR in an aprotic solvent, dimethylsulfoxide (DMSO), with phenol as a proton donor, indicating that different proton transfer processes significantly directed the reaction towards C2+ production. Jiao et al.75 studied the synergistic effect of DMSO, a hydrated alkaline cation. They primarily used constant-potential ab initio molecular dynamics simulations to explain the synergistic effect between the cation (Na+) and aprotic solvent (DMSO) in the electrolyte, resulting in an improvement in C2+ selectivity. The synergistic effect significantly reduced the connectivity of the water network, reducing the HER and formation of the C1 precursors *COH or *CHO, without affecting the hydrogenation of *OCCO*. These predictions were experimentally validated using in situ ATR-IR spectroscopy, which revealed more prominent *OCHO and *OCCOH with DMSO addition, even under a moderate potential of −0.3 V vs. RHE. This finding corroborates their prediction that C–C bond formation is favoured by the environment provided by the cation and aprotic solvent.
3.4 Effect of modification on the transportation of CO2, H2O, and protons
The poor solubility of CO2 molecules in water limits the intrinsic activity of Cu catalysts. In addition to the amount of CO2 consumed in the CO2RR process, the decomposition of CO2 into bicarbonate and carbonate anions under high local pH conditions is easily achieved by cathodic electrolysis. Excess water in CO2 also leads to a loss of electrical energy in the HER. In addition, the sparse adsorption of *CO tends to reduce the probability of C–C bond formation. The application of the gas diffusion electrode (GDE) in a flow cell, which directly introduces gaseous CO2 onto the electrode surface, significantly increases the geometric partial current density for C2+ products by one order of magnitude, from several 10 to 100 mA cm−1. Even when the catalyst surface is completely immersed in an aqueous electrolyte, the selective transport of CO2 except H2O at the microscopic level is expected to improve the production of C2+ products.Alkanethiols are frequently used modifiers due to their fairly stable adsorption even under the CO2RR operation. We already discussed their effect in Sections 3.1 and 3.3, and they also significantly affect the CO2 and H2O transportation onto the surface. Gong et al.76 systematically changed the length of the alkyl chain in alkanethiol modifiers and showed that stronger hydrophobicity with octadecanethiol enhanced CO2 mass transport, while shorter dodecanethiol can make the most efficient gas–liquid–solid contact affording large current density. They used in situ fluorescence electrochemical microscopy to evaluate the local CO2/H2O ratio under the CO2RR operation, which corresponds to the ratio of *CO and *H. The ratio of these adsorbed species influences not only the HER process but also the reaction branching between ethanol and ethylene.
Luo et al.77 compared different polymer binders for a needle-like CuO catalyst ink with different hydrophobicities (polyacrylic acid (PAA), Nafion, and fluorinated ethylene propylene (FEP)). Among the three binders, FEP exhibited the highest selectivity and current density of C2+ products. An evaluation of CO2-philicity and hydrophilicity by contact angle measurements indicated that the CO2/H2O ratio was the highest after modification with FEP. In situ Raman spectroscopy revealed that a high CO2/H2O ratio with the FEP binder corresponded to a wider potential range in which CO stretching was observed.
Zhuang et al.78 introduced polyaniline (PANI) onto the Cu surface (Fig. 11a), anticipating that the NH group in PANI has an affinity for CO2. This modification increased the current density of the C2+ products with high selectivity, indicating that a substantial amount of CO2 was increased on the Cu surface (Fig. 11b). They did not detect any differences in the Cu morphology or electronic state upon modification. The in situ ATR–SEIRAS measurements showed that the peak attributable to *COatop became more prominent relative to the OH stretching in H2O at the surface upon modification (Fig. 11c, top). They provided a more precise analysis of the *COatop stretching band (Fig. 11c, bottom). They revealed a greater *CO stretching wavenumber and weak Stark effect, indicating weaker Cu–C interactions, possibly because of close *CO packing. The narrow bandwidth of *CO also led the researchers to conclude that the modification caused an increased production of *CO, which must be packed in an ordered geometry.
 | ||
| Fig. 11 (a) Structure of PANI. (b) Modification effect on the CO2RR. (c) (top) In situ ATR-SEIRAS spectra of the CO2RR on the Cu and Cu–PANI electrodes, respectively, in CO2 saturated 0.1 M KHCO3. (bottom) Stark effects of the linearly adsorbed CO (COL) and full width at half-maximum (FWHM) of COL signals. Reproduced with permission from ref. 78. Copyright 2020 American Chemical Society. | ||
Daasbjerg et al.79 studied the electrochemically deposited 1,1′-di-p-tolyl-1,1′-,4,4′-bipyridine (T-bipyridine) on polycrystalline Cu. The thickness of the deposited film was varied by varying the applied potential during electrodeposition. As the thickness of the organic film increased, the selectivity of ethylene increased. They used in situ Raman spectroscopy and observed a notable difference in the wavenumber of *CO stretching between the modified and unmodified surfaces, with the former exhibiting a higher intensity. They concluded that the modification did not induce local reactivity on Cu, and greater *CO coverage was responsible for the increased production of C2+ products, which was significantly affected by the film thickness and porosity, and proposed that the higher CO partial pressure facilitated by the modification was a more significant factor than the surface hydrophobicity.
Xie et al.80 proposed that an anti-swelling anion exchange ionomer (AEI) could optimise the local environment for the electroreduction of CO2 to C2+ products at industrially relevant current densities on oxide-derived Cu. Unlike proton exchange ionomers, which typically have SO3− groups, the AEI can accumulate OH−via the –N(CH3)3+ group while preventing excessive water from intruding onto the Cu surface (Fig. 12a). The surface pH was estimated from the relative peak intensity of HCO3−/CO32− (Fig. 12b and c), revealing that the modification with AEI increased the pH of the Cu surface at a high current density. In situ Raman spectroscopy revealed the presence of *OH, Cu–CO, and *CO, whereas in situ FTIR spectroscopy revealed the presence of *CO, HCO3−, and COCO*. Based on these observations and theoretical calculations, they concluded that the increased surface pH facilitated the hydrogenation of COCO*.
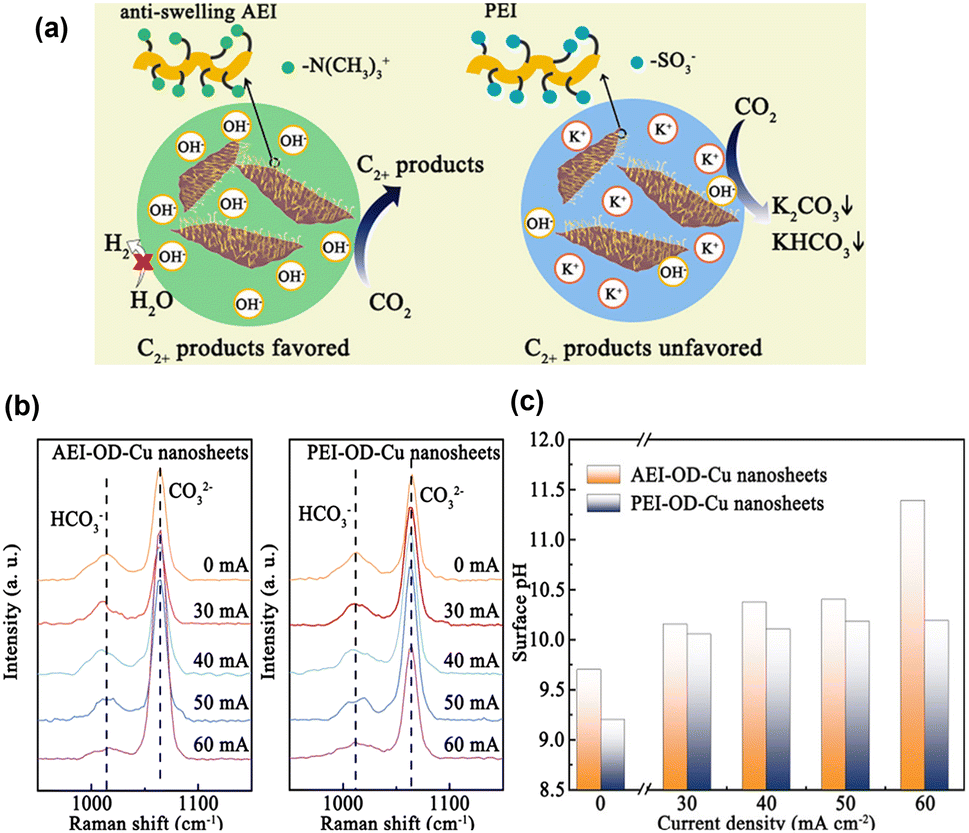 | ||
| Fig. 12 (a) Schematic illustration of CO2-to-C2+ activity and selectivity through local environment construction. (b) In situ Raman spectra obtained from the AEI-OD-Cu and PEI-OD-Cu nanosheets. (c) pH values calculated from the in situ Raman spectra. Reproduced with permission from ref. 80. Copyright 2022 American Chemical Society. | ||
Daasbjerg et al.81 analysed several polymer coatings of CuO electrodes and observed that the surface hydrophobicity of these polymers suppressed the HER. They also suggested that an increase in surface pH with restricted water diffusion could promote ethylene formation.
These effects on the transport of organic modifiers work in assembling high-current-density electrolysis cells. Introducing hydrophobic polymers to create a triple-phase catalyst surface, where the CO2 gas, electrolyte, and Cu surface meet at the active site, is extremely effective for increasing the current density for the formation of C2+ products.33
3.5 Effect of modification on the charged state and structure of surface Cu
Zhao et al.82 introduced fullerene (C60) onto CuO nanospindles. Because C60 is known to be a good electron acceptor, it reserves electrons under cathodic potential and stabilises the Cu+ state of nearby surface Cu atoms (Fig. 13a). The persistent Cu+ state under negative potential was monitored by in situ Raman spectroscopy (Fig. 13b). Modification with C60 significantly increased the formation of C2+ products (Fig. 13d) while suppressing the HER. The active surface was also monitored by in situ Raman spectroscopy (Fig. 13c). A persistent, higher-wavenumber peak corresponding to *COatop appeared, indicating a smaller electron donation from Cu+ to *CO. They also observed the reduction intermediates *CHO (1700 cm−1) and coupled *CO–CHO (1580 cm−1), which were only successfully detected on the C60-modified surface by in situ Raman spectroscopy and ATR–SEIRAS. Together with the theoretical calculations, they concluded that the preferential formation of *CHO and its reaction with *CO to form *CO–CHO provides an effective route for producing C2+ products. | ||
| Fig. 13 (a) Schematic illustration of the electroreduction of CuO–C60 precatalysts. (b) In situ Raman spectra of CuO and CuO–C60 precatalysts at a reduction potential of −0.4 V versus RHE. (c) In situ ATR-FTIR spectra under continuous CO2 flow over CuO and CuO–C60. (d) The Faraday efficiency ratios of C2+ to C1 products from the CO2RR. Reproduced with permission from ref. 82. Copyright 2023 Wiley. | ||
Recently, metal surface modification with N-heterocyclic carbene ligands has been investigated as a powerful tool for enhancing catalytic activity through strong interactions with surface metal atoms. Rosas-Hernández et al.83 electrodeposited Cu nanoparticles onto Cu foil in the presence of N-heterocyclic carbene–carbodiimide (NHC–CDI). This modification significantly increased the selectivity and current density of the carbonaceous products, and the performance showed a volcano-type dependence on the degree of modification. In situ Raman spectra showed that the *CO formed on the surface of unmodified Cu was mostly *CObridge; however, *COatop was formed exclusively on the modified surface. Close inspection of the *COatop peak revealed that Cu modified with a moderate amount of NHC–CDI contained two components, each assigned to *CO coordinated on terrace sites (LFB) or step sites (HFB). HFBs disappeared with increasing amounts of the modifier, possibly because of the occupation of the step site with the NHC–CDI ligand. These results indicate that the HFB was more readily dimerised and explain the superior CO2RR efficiency with moderate modification.
Chen et al.84 highlighted the facet-stabilising effect of 1-dodecanethiol. As described previously,32,68 modification with alkanethiol facilitated CO2 transportation and *CO accumulation on the Cu surface. They prepared nanostructured CuO treated with 1-dodecanethiol and observed the intense peak of *CO for the modified surface in the in situ Raman spectrum, which agreed with previous reports. In addition, the slightly lower wavenumber at negative potentials indicated stronger CO binding on the modified surface. They conducted other in situ measurements to elucidate that the modified surface was more prone to be reduced to expose the (100) plane of Cu. In situ ATR-FTIR spectroscopy exhibited a resemblance of the *CO state on the modified surface to Cu(100) and the unmodified surface to Cu(111). The intermediates *CHO and *CO–CHO were only observed on the modified surface, and theoretical calculations predicted that the C–C bond formation pathway from *CHO to *CO–CHO was favoured on the Cu(100) surface.
4. New fabrication methods for organic modifiers/Cu contact surfaces
In the previous sections, we reviewed the multifaceted effects of organic structures on the CO2RR activity on Cu surfaces and the role of in situ vibrational spectroscopy in monitoring surface events. Based on this understanding, it is evident that the fabrication method used for creating an organic/Cu contact surface significantly affects the CO2RR performance of the modified surface. Historically, modification was achieved using simple procedures, such as (i) casting a modifier solution onto the Cu catalyst surface and (ii) mixing the modifiers into the catalyst ink solution and depositing them onto the substrate surface. However, recent studies have shown that the CO2RR performance of the modified surface can be significantly improved by employing methods to control the structure built on the surface.Electrodeposition can modify the catalyst surface while maintaining its electroactivity because it does not function when the surface becomes electrochemically insulating owing to excessive deposition. It can also be used to introduce modifiers with poor solubility or a high tendency to aggregate, which make them difficult to be processed directly. Agapie et al.56 used electrodeposition to deposit neutral dimers by reducing water-soluble N-arylpyridinium salts across various pyridinium families. They also employed electrografting to covalently bond monomeric organic species, which enhanced the CO2RR activity towards C2+ products.85 Daasbjerg et al.79 reported that although the reduced N-arylpyridinium dimer is soluble in organic solvents, dissolving the electrodeposited film in acetone followed by complete drying diminished its effectiveness for the formation of C2+ products, owing to the transformation of the porous film into a closely packed structure. This result demonstrates the importance of the porous structure formed during electrodeposition for improving the catalytic activity of molecular films.
Restructuring the Cu surface by modification significantly influences the surface activity. During the cathodic dimerisation of N,N-ethylene-phenanthrolinium dibromide, Agapie et al.86 observed a cubic structure on the Cu surface. The incorporation of halogenide anions into Cu promotes Cu restructuring, resulting in the increased production of C2+ products during the CO2RR.87 This process was accompanied by the deposition of organic films via the reductive dimerisation of N,N-ethylene-phenanthrolinium (Fig. 14). The modified organic film played a dual role in enhancing the production of C2+ products: providing a preferential molecular reaction site66 and protecting cubic nanostructures with exposed {100} facets, which promoted the production of C2+ products but were unstable without protection.
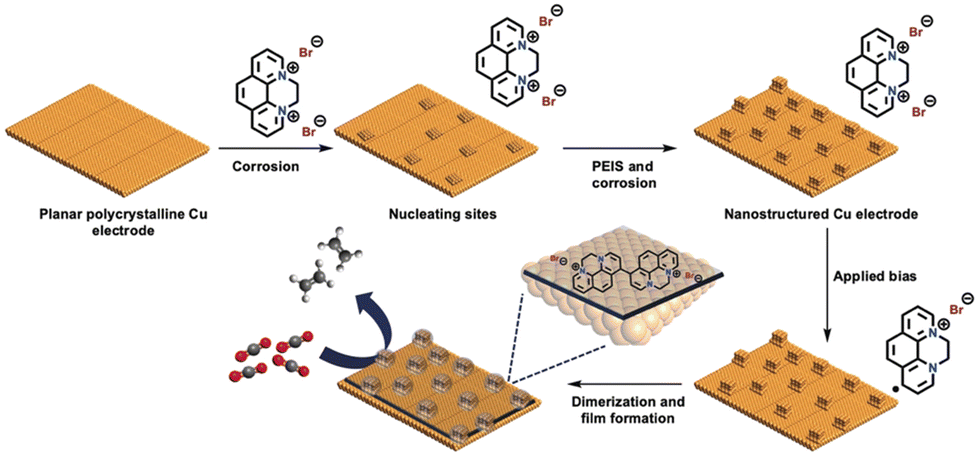 | ||
| Fig. 14 Pictorial representation of the model for nanostructuring of a polycrystalline copper electrode and film electrodeposition by dimerization of N,N′-ethylene-phenanthrolinium dibromide. The combination of film and nanostructuring leads to high C≥2 selectivity for CO2RR electrocatalysis. Reproduced with permission from ref. 86. Copyright 2019 Wiley. | ||
The protective effect of the organic layers on nanostructured Cu was also investigated in a study on the effect of the size of the Cu nanoparticles. Similar to other metal nanoparticle catalysts, the size of Cu nanoparticles affects the electrochemical CO2RR.88–90 In addition to the dispersed electronic state and number of low coordination sites in the nanoparticle state, C2+ formation in the CO2RR involved C–C bond formation, which was disadvantageous for subnanometer-sized particles with few cooperating surface atomic sites. Duan et al.91 investigated organic structures to elucidate the effect of Cu size. They simultaneously generated Cu0 and a graphdiyne scaffold by reacting an unstable copper(I)-σ-alkynyl complex at elevated temperatures (Fig. 15). The Cu species were kinetically trapped in the graphdiyne scaffold, and the size of the particles was controlled by the amount of cuprous precursor, ranging from single-atomic Cu sites to nanoclusters. They observed a very clear dependence of the C1/C2+ product ratio on particle size, predominantly forming C1 products at single-atom sites and C2+ products at 1–1.5-nm-sized particles.
 | ||
| Fig. 15 Schematic illustration of the synthesis of the Cu/graphdiyne scaffold and the size effect of Cu(0) catalysts on CO2RR selectivity. Reproduced with permission from ref. 91. Copyright 2022 Wiley. | ||
Recently, we developed a novel method for modifying catalytically active Cu surfaces.92–94 Cu-catalysed azide–alkyne cycloaddition (CuAAC) is a representative click chemistry technique that involves the formation of rigid covalent bonds between ethynyl and azide moieties. A distinctive aspect of this reaction is that Cu(I) is particularly active, with various Cu(I) species, salts, coordination compounds, and even insoluble inorganic solids known to catalyse this reaction. Initially, the metallic Cu electrode was electrochemically activated by anodisation to catalyse CuAAC between monomeric azides and ethynyl species (Fig. 16a). Although the reaction of these two monomers in solution afforded insoluble polymers that were difficult to process, anodisation of the Cu electrode effectively deposited the polymer. This modification resulted in the increased production of C2+ products with the effective inhibition of the HER.92 The modification with a small triazole molecule resulted in the inhibition of the CO2RR, demonstrating the importance of structural design beyond the kind of molecular groups. The feasibility of CuAAC also enabled the evaluation of different molecular moieties incorporated in the modified layer to demonstrate the special effect of tertiary amine moieties, which are effective proton carriers in the modified layer.93 We subsequently investigated layer growth from the same component on nanocrystalline Cu2O without electrochemical stimulation (Fig. 16b).94 The resulting layer on Cu2O was extremely flat and thin. An important feature of this method is that the layer growth point is fixed on the surface Cu(I) of Cu2O, thus rendering the growth self-regulating by preventing the further approach of the monomer on the Cu2O surface. The layer structure was independent of the surface Cu(I) activity but reliant on the degree of modification of the layer growth, leading to the formation of a uniform layer. Although the layer thickness was only a few nanometres, it effectively inhibited the HER. Another important feature of this modification is the protecting effect. Cu2O was mostly reduced to a Cu(0) species without losing its initial cubic structure. Transmission electron microscopy revealed that Cu2O was reduced within the formed layer, forming a hollow at the centre to compensate for the volume decrease (Fig. 16b). These results further motivated us to investigate the control of the structure of organic/Cu contact surfaces, where Cu exhibits a dynamic structure during catalysis.
 | ||
| Fig. 16 Schematic illustration of two CuAAC modifications: on anodized Cu (a) and on Cu2O nanocubes, with TEM images of structural transformation upon the CO2RR (b). Reproduced with permission from ref. 93 and 94. Copyright 2022 Wiley. | ||
5. Conclusions and outlook
Many studies have demonstrated the efficacy of organic modifiers on the activity of the CO2RR on Cu electrocatalysts, particularly in the production of C2+ products. With the progress in understanding general Cu electrocatalysts, the roles of organic modifiers should be examined from various perspectives. First, the diverse product range of the CO2RR on copper complicates reaction pathway elucidation compared to other cathodic electrocatalysts, as reaction conditions exert a greater influence on product formation. Second, the governing factor of the reaction is significantly different from that of homogeneous molecular catalysts, in which the activity is primarily understood by considering the nearest molecular interactions. Additionally, CO2 electrolysis relies on the transport of poorly soluble CO2 to water and other proton suppliers. Therefore, making the surface hydrophobic or creating a triple-phase boundary can improve the CO2RR performance. These aspects underscore the importance of direct in situ observation of the Cu surface during catalysis to better understand the role of the modifiers.This article collects the studies of the analysis of a Cu catalyst modified with organic compounds using in situ vibrational spectroscopy to classify the role of the organic modifiers. This technique enables the visualisation of the reduction intermediates, such as *CO, and reveals the states of water and Cu, which are important for understanding the effects of the modifiers. In addition, these bands can serve as indicators of local pH and electric field, providing extensive information about the key factors influencing the CO2RR. A deeper understanding of these methods will significantly facilitate research in this field.
The method of fabricating organic/Cu contact surfaces has significant potential for improving the CO2RR performance. We have highlighted successful examples in which the fabrication procedure was crucial for demonstrating the potential of the modifier. In addition, Cu exhibits a dynamic structure during electrocatalysis, which is significantly affected by the presence of modifiers that act as surface stabilisers. We discussed some of these studies, including our recent efforts to develop a new fabrication method in which Cu is not merely modified but actively participates in the modification process. The active participation of Cu leads to precisely controlled contact surface modification, the accumulation of more active Cu atoms on the surface, and the fabrication of effective and selective mass transport routes.
The designability of molecular modifiers has been expected to be a powerful tool for efficient and selective electrochemical CO2 conversion into various chemicals on copper. The development of an electrochemical CO2RR on copper revealed the importance of the total design of an electrochemical system, from the atomic scale to the interface structure and the whole cell assembly, which are entangled with one another. The enhancement of local activity can cause local CO2 depletion more easily, and it inevitably requires further improvement in CO2 transport. Also the works summarized in this review showed that a single molecular modifier can have multiple wide-ranging effects. The application of in situ vibrational spectroscopy is indispensable in dissecting these tangled factors. It can also provide important clues in designing molecular modifiers integrated with copper atom assembly, toward selective and effective production of a specific product among various optional chemicals from CO2.
Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by KAKENHI (grant no. 22K05129 and 21K05125) from Japan Society for the Promotion of Science (JSPS). AK acknowledges the financial support from the Cooperative Research Program of “Network Joint Research Center for Material and Devices” and the Ogasawara Memorial Foundation.References
- M. E. Royer, Comptes Rendus Acad. Sci., 1870, 70, 731–732 Search PubMed.
- L. V. Haynes and D. T. Sawyer, Anal. Chem., 1967, 39, 332–338 CrossRef.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39(11–12), 1833–1839 CrossRef.
- Y. Hori, K. Kikuchi and S. Suzuki, Chem. Lett., 1985, 1695–1698 CrossRef.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 897–898 CrossRef CAS.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y.-W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef.
- C. Liu, M. P. Lourenco, S. Hedstroem, F. Cavalca, O. Diaz-Morales, H. A. Duarte, A. Nilsson and L. G. M. Pettersson, J. Phys. Chem. C, 2017, 121, 25010–25017 CrossRef CAS.
- W. Zhang, C. Huang, Q. Xiao, L. Yu, L. Shuai, P. An, J. Zhang, M. Qiu, Z. Ren and Y. Yu, J. Am. Chem. Soc., 2020, 142, 11417–11427 CrossRef CAS PubMed.
- P. Qi, L. Zhao, Z. Deng, H. Sun, H. Li, Q. Liu, X. Li, Y. Lian, J. Cheng, J. Guo, Y. Cui and Y. Peng, J. Phys. Chem. Lett., 2021, 12, 3941–3950 CrossRef CAS PubMed.
- R. G. Mariano, K. McKelvey, H. S. White and M. W. Kanan, Science, 2017, 358, 1187–1192 CrossRef CAS PubMed.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijoo, P.-C. Chen, H. Wang, C. J. Pollock, X. Huang, Y.-T. Shao, C. Wang, D. A. Muller, H. D. Abruna and P. Yang, Nature, 2023, 614, 262–269 CrossRef CAS.
- Q. Lei, H. Zhu, K. Song, N. Wei, L. Liu, D. Zhang, J. Yin, X. Dong, K. Yao, N. Wang, X. Li, B. Davaasuren, J. Wang and Y. Han, J. Am. Chem. Soc., 2020, 142, 4213–4222 CrossRef CAS PubMed.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2018, 57, 6192–6197 CrossRef CAS.
- X. Wang, A. Xu, F. Li, S.-F. Hung, D.-H. Nam, C. M. Gabardo, Z. Wang, Y. Xu, A. Ozden, A. S. Rasouli, A. H. Ip, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 3525–3531 CrossRef CAS.
- E. L. Clark, J. Resasco, A. Landers, J. Lin, L.-T. Chung, A. Walton, C. Hahn, T. F. Jaramillo and A. T. Bell, ACS Catal., 2018, 8, 6560–6570 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Noerskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- F. Dattila, R. R. Seemakurthi, Y. Zhou and N. Lopez, Chem. Rev., 2022, 122, 11085–11130 CrossRef CAS.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Coord. Chem. Rev., 2021, 454, 214340 CrossRef.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef.
- K. C. Poon, W. Y. Wan, H. Su and H. Sato, RSC Adv., 2022, 12, 22703–22721 RSC.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- L. M. Aeshala, R. Uppaluri and A. Verma, Phys. Chem. Chem. Phys., 2014, 16, 17588–17594 RSC.
- S. Ahn, K. Klyukin, R. J. Wakeham, J. A. Rudd, A. R. Lewis, S. Alexander, F. Carla, V. Alexandrov and E. Andreoli, ACS Catal., 2018, 8, 4132–4142 CrossRef.
- S. Zhong, X. Yang, Z. Cao, X. Dong, S. M. Kozlov, L. Falivene, J.-K. Huang, X. Zhou, M. N. Hedhili, Z. Lai, K.-W. Huang, Y. Han, L. Cavallo and L.-J. Li, Chem. Commun., 2018, 54, 11324–11327 RSC.
- A. K. Buckley, M. Lee, T. Cheng, R. V. Kazantsev, D. M. Larson, W. A. I. I. I. Goddard, F. D. Toste and F. M. Toma, J. Am. Chem. Soc., 2019, 141, 7355–7364 CrossRef PubMed.
- J. Wang, T. Cheng, A. Q. Fenwick, T. N. Baroud, A. Rosas-Hernandez, J. H. Ko, Q. Gan, W. A. I. I. I. Goddard and R. H. Grubbs, J. Am. Chem. Soc., 2021, 143, 2857–2865 CrossRef.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef.
- F. P. Garcia de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- C. Lu, Y. Su, J. Zhu, J. Sun and X. Zhuang, Chem. Commun., 2023, 59, 6827–6836 RSC.
- P. Chen, Y. Wu, T. E. Rufford, L. Wang, G. Wang and Z. Wang, Mater. Today Chem., 2023, 27, 101328 CrossRef.
- C. V. Raman and K. S. Krishnan, Nature, 1928, 121, 501–502 CrossRef.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef.
- A. J. McQuillan, P. J. Hendra and M. Fleischmann, J. Electroanal. Chem., 1975, 65, 933–944 CrossRef.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215–5217 CrossRef.
- Y. X. Chen, A. Miki, S. Ye, H. Sakai and M. Osawa, J. Am. Chem. Soc., 2004, 125, 3680–3681 CrossRef.
- A. Miki, S. Ye, T. Senzaki and M. Osawa, J. Electroanal. Chem., 2004, 563, 23–31 CrossRef.
- X. Chang, S. Vijay, Y. Zhao, N. J. Oliveira, K. Chan and B. Xu, Nat. Commun., 2022, 13, 2656 CrossRef.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem., 1977, 84, 1–20 CrossRef.
- I. V. Chernyshova, P. Somasundaran and S. Ponnurangam, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9261–E9270 CrossRef.
- I. Oda, H. Ogasawara and M. Ito, Langmuir, 1996, 12, 1094–1097 CrossRef.
- H. Ogasawara, J. Inukai and M. Ito, Chem. Phys. Lett., 1992, 198, 389–394 CrossRef.
- L. Mandal, K. R. Yang, M. R. Motapothula, D. Ren, P. Lobaccaro, A. Patra, M. Sherburne, V. S. Batista, B. S. Yeo, J. W. Ager, J. Martin and T. Venkatesan, ACS Appl. Mater. Interfaces, 2018, 10, 8574–8584 CrossRef.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kuehl, R. Garcia-Muelas, N. Lopez and B. R. Cuenya, ACS Catal., 2021, 11, 7694–7701 CrossRef.
- T.-C. Chou, C.-C. Chang, H.-L. Yu, W.-Y. Yu, C.-L. Dong, J.-J. Velasco-Velez, C.-H. Chuang, L.-C. Chen, J.-F. Lee, J.-M. Chen and H.-L. Wu, J. Am. Chem. Soc., 2020, 142, 2857–2867 CrossRef PubMed.
- Y. Wang, J. Zhang, J. Zhao, Y. Wei, S. Chen, H. Zhao, Y. Su, S. Ding and C. Xiao, ACS Catal., 2024, 14, 3457–3465 CrossRef.
- C. M. Gunathunge, X. Li, J. Li, R. P. Hicks, V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2017, 121, 12337–12344 CrossRef.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef PubMed.
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 Search PubMed.
- Y. Zhao, X.-G. Zhang, N. Bodappa, W.-M. Yang, Q. Liang, P. M. Radjenovica, Y.-H. Wang, Y.-J. Zhang, J.-C. Dong, Z.-Q. Tian and J.-F. Li, Energy Environ. Sci., 2022, 15, 3968–3977 RSC.
- V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2019, 123, 24453–24460 CrossRef CAS.
- Z. Han, R. Kortlever, H.-Y. Chen, J. C. Peters and T. Agapie, ACS Cent. Sci., 2017, 3, 853–859 CrossRef CAS.
- Z. Liu, X. Lv, S. Kong, M. Liu, K. Liu, J. Zhang, B. Wu, Q. Zhang, Y. Tang, L. Qian, L. Zhang and G. Zheng, Angew. Chem., Int. Ed., 2023, 62, e202309319 CrossRef CAS.
- L. Xue, X. Wu, Y. Liu, B. Xu, X. Wang, S. Dai, P. Liu and H. Yang, Nano Res., 2022, 15, 1393–1398 CrossRef CAS.
- J. R. Pankhurst, P. Iyengar, A. Loiudice, M. Mensi and R. Buonsanti, Chem. Sci., 2020, 11, 9296–9302 RSC.
- G.-R. Zhang, S.-D. Straub, L.-L. Shen, Y. Hermans, P. Schmatz, A. M. Reichert, J. P. Hofmann, I. Katsounaros and B. J. M. Etzold, Angew. Chem., Int. Ed., 2020, 59, 18095–18102 CrossRef CAS PubMed.
- J. Li, F. Li, C. Liu, F. Wei, J. Gong, W. Li, L. Xue, J. Yin, L. Xiao, G. Wang, J. Lu and L. Zhuang, ACS Energy Lett., 2022, 7, 4045–4051 CrossRef CAS.
- C. Y. J. Lim, M. Yilmaz, J. M. Arce-Ramos, A. D. Handoko, W. J. Teh, Y. Zheng, Z. H. J. Khoo, M. Lin, M. Isaacs, T. L. D. Tam, Y. Bai, C. K. Ng, B. S. Yeo, G. Sankar, I. P. Parkin, K. Hippalgaonkar, M. B. Sullivan, J. Zhang and Y.-F. Lim, Nat. Commun., 2023, 14, 335 CrossRef CAS PubMed.
- C. E. Creissen, J. G. Rivera de la Cruz, D. Karapinar, D. Taverna, M. W. Schreiber and M. Fontecave, Angew. Chem., Int. Ed., 2022, 61, e202206279 CrossRef CAS PubMed.
- J. Ding, F. Li, X. Ren, Y. Liu, Y. Li, Z. Shen, T. Wang, W. Wang, Y.-G. Wang, Y. Cui, H. Yang, T. Zhang and B. Liu, Nat. Commun., 2024, 15, 3641 CrossRef CAS PubMed.
- Y. Wang, R. Zhao, Y. Liu, F. Zhang, Y. Wang, Z. Wu, B. Han and Z. Liu, Chem. Sci., 2024, 15, 4140–4145 RSC.
- F. Li, A. Thevenon, A. Rosas-Hernandez, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- Y. Shi, K. Sun, J. Shan, H. Li, J. Gao, Z. Chen, C. Sun, Y. Shuai and Z. Wang, ACS Catal., 2022, 12, 8252–8258 CrossRef CAS.
- S. Mu, L. Li, R. Zhao, H. Lu, H. Dong and C. Cui, ACS Appl. Mater. Interfaces, 2021, 13, 47619–47628 CrossRef CAS.
- L. Huang, G. Gao, C. Yang, X.-Y. Li, R. K. Miao, Y. Xue, K. Xie, P. Ou, C. T. Yavuz, Y. Han, G. Magnotti, D. Sinton, E. H. Sargent and X. Lu, Nat. Commun., 2023, 14, 2958 CrossRef CAS.
- S. Banerjee, Z.-Q. Zhang, A. S. Hall and V. S. Thoi, ACS Catal., 2020, 10, 9907–9914 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- X. Yang, H. Ding, S. Li, S. Zheng, J.-F. Li and F. Pan, J. Am. Chem. Soc., 2024, 146, 5532–5542 CrossRef CAS PubMed.
- Z. Tao, Z. Wu, Y. Wu and H. Wang, ACS Catal., 2020, 10, 9271–9275 CrossRef.
- A. T. Chu and Y. Surendranath, J. Am. Chem. Soc., 2022, 144, 5359–5365 CrossRef PubMed.
- X. Bai, C. Chen, X. Zhao, Y. Zhang, Y. Zheng and Y. Jiao, Angew. Chem., Int. Ed., 2024, 63, e202317512 CrossRef.
- Y. Lin, T. Wang, L. Zhang, G. Zhang, L. Li, Q. Chang, Z. Pang, H. Gao, K. Huang, P. Zhang, Z.-J. Zhao, C. Pei and J. Gong, Nat. Commun., 2023, 14, 3575 CrossRef.
- T. H. M. Pham, J. Zhang, M. Li, T.-H. Shen, Y. Ko, V. Tileli, W. Luo and A. Zuettel, Adv. Energy Mater., 2022, 12, 2103663 CrossRef.
- X. Wei, Z. Yin, K. Lyu, Z. Li, J. Gong, G. Wang, L. Xiao, J. Lu and L. Zhuang, ACS Catal., 2020, 10, 4103–4111 CrossRef.
- S. Zhao, O. Christensen, Z. Sun, H. Liang, A. Bagger, K. Torbensen, P. Nazari, J. V. Lauritsen, S. U. Pedersen, J. Rossmeisl and K. Daasbjerg, Nat. Commun., 2023, 14, 844 CrossRef PubMed.
- Y. Zhao, X. Zu, R. Chen, X. Li, Y. Jiang, Z. Wang, S. Wang, Y. Wu, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2022, 144, 10446–10454 CrossRef PubMed.
- H.-Q. Liang, S. Zhao, X.-M. Hu, M. Ceccato, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2021, 11, 958–966 CrossRef.
- B. Zhao, F. Chen, C. Cheng, L. Li, C. Liu and B. Zhang, Adv. Energy Mater., 2023, 13, 2204346 CrossRef.
- K. N. Kolding, M. Bretlau, S. Zhao, M. Ceccato, K. Torbensen, K. Daasbjerg and A. Rosas-Hernandez, J. Am. Chem. Soc., 2024, 146, 13034–13045 CrossRef PubMed.
- Y. Yao, T. Shi, W. Chen, J. Wu, Y. Fan, Y. Liu, L. Cao and Z. Chen, Nat. Commun., 2024, 15, 1257 CrossRef CAS.
- N. B. Watkins, Y. Wu, W. Nie, J. C. Peters and T. Agapie, ACS Energy Lett., 2023, 8, 189–195 CrossRef CAS.
- A. Thevenon, A. Rosas-Hernandez, J. C. Peters and T. Agapie, Angew. Chem., Int. Ed., 2019, 58, 16952–16958 CrossRef CAS.
- T. Kim and G. T. R. Palmore, Nat. Commun., 2020, 11, 3622 CrossRef CAS PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS PubMed.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS.
- W. Rong, H. Zou, W. Zang, S. Xi, S. Wei, B. Long, J. Hu, Y. Ji and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 466–472 CrossRef CAS.
- R. Igarashi, R. Takeuchi, K. Kubo, T. Mizuta and S. Kume, Front. Chem., 2019, 7, 860 CrossRef CAS PubMed.
- R. Takeuchi, R. Igarashi, K. Kubo, T. Mizuta and S. Kume, ChemElectroChem, 2020, 7, 2575–2581 CrossRef CAS.
- T. Umeda, T. Kurome, A. Sakamoto, K. Kubo, T. Mizuta, S. U. Son and S. Kume, Chem. Commun., 2022, 58, 8053–8056 RSC.
| This journal is © The Royal Society of Chemistry 2024 |