
Environmentally friendly Miyaura Borylations allowing for green, 1-pot borylation/Suzuki–Miyaura couplings†
Chandler B.
Nelson
 ,
Scott J.
L'Heureux
,
Madison J.
Wong
,
Simone L.
Kuhn
,
Erika
Ghiglietti
and
Bruce H.
Lipshutz
*
,
Scott J.
L'Heureux
,
Madison J.
Wong
,
Simone L.
Kuhn
,
Erika
Ghiglietti
and
Bruce H.
Lipshutz
*
Department of Chemistry and Biochemistry, University of California, Santa Barbara, CA 93106, USA. E-mail: lipshutz@chem.ucsb.edu; chandlernelson@chem.ucsb.edu
First published on 3rd September 2024
Abstract
Current routes to boronic acids and their corresponding esters to be used in subsequent Suzuki–Miyaura (SM) cross couplings impact the cost, waste, and safety concerns associated with generating these materials. A new method for installing the ethyl pinacol boronic ester, or B(Epin) derivative leads to stable borylated products under near-neat conditions using high concentrations of a green solvent and moderate reaction temperatures, catalyzed by relatively low palladium loadings. Alternatively, the newly fashioned Ar–B(Epin) can be generated in situ and used directly in the same pot for SM reactions leading to aromatic and heteroaromatic residues characteristic of the biaryl products being formed. An array of complex targets, including API-related products, can be generated via this green and environmentally responsible methodology.
1 Introduction
There are few chemical processes as omnipresent as the Suzuki–Miyaura (SM) coupling in modern organic synthesis. In fact, decades after the watershed paper by Suzuki and Miyaura1 the reaction was reported to be the second most utilized transformation in the pharmaceutical industry.2 Beyond this, the SM coupling has also been extensively used in the synthesis of agrochemicals,3–6 natural products,7–10 and in organic materials.6,11–13 Nonetheless, in cases where such a transformation has seen such widespread use due to its value to so many chemical processes, the cost, waste, hazards, and environmental impact14–18 associated with its use can also be considerable. While several recent developments serve to greatly improve the efficiency and environmental responsibility of the SM coupling reaction,19–23 a major hurdle that remains is the traditional manner in which the required boronic acid or ester coupling partner is obtained. While the Miyaura borylation24 is an attractive alternative to commonly used cryogenic lithium halogen exchange protocols,25,26 or C–H activation procedures involving non-sustainable levels of iridium-based catalysis,27–29 it often necessitates high loadings of palladium,24,30–32 high temperatures,30,32–35 and the use of relatively dilute and waste-generating organic solvents.30,32,33 Moreover, the prepared boronic esters or acids can be difficult to isolate, and have varying shelf lives and stabilities.36–38Each of these factors serve to increase the expense, energy demand, reliance on petroleum feedstocks, and organic waste associated with performing critical sp2–sp2 couplings, which are typically biaryl in nature. Hence, there is a definitive need for a more reliable and more environmentally responsible borylation protocol that not only allows for ease of preparation, but also is applicable to use in subsequent “green” 1-pot SM couplings that are environmentally friendly (Fig. 1).
 | ||
| Fig. 1 Borylation/Suzuki–Miyaura couplings. | ||
Among the recent advances towards more efficient Miyaura borylations, installation of the boronic acid residue via Pd-catalyzed couplings with bis-boronic acid (BBA) are perhaps the most favored.39,40 This is likely due to its reliance on moderate-to-low catalyst loadings, reasonable reaction temperatures (40–80 °C), and relatively green solvents (e.g., 2-MeTHF and EtOH). However, when these newly prepared boronic acids are then applied to 1-pot SM couplings, catalytic efficiency typically drops and loadings of 1 mol% or greater tend to be required,39,40 especially in cases involving highly functionalized substrates. Furthermore, though seemingly more atom economical, significant excesses of BBA are employed.39,40 Importantly, BBA is also known to decompose readily in the presence of heat, oxygen, Pd(II) species, and moisture.41,42 Beyond complicating handling, such decomposition can generate hydrogen gas posing significant safety concerns at scale.41,42 Additionally, the use of these generated boronic acids can be prone to protodeborylation and other unwanted side products,43,44 thereby lowering the efficiency of the overall process and requiring a potentially tedious separation.
The effect of base on Miyaura borylations is yet another notable area where recent improvements involving use of sterically hindered and electron-rich carboxylates allow for benign reaction conditions and more attractive palladium loadings.45,46 Work to date using this technology, however, lacks substrate complexity, and has shown limited options for use in 1-pot carbon–carbon bond formation.45,46 Hence, it is apparent that there exists a need for a green (and hence, safer), benign borylation protocol that provides access to stable, and if desirable, isolable borylated products that can also be formed in situ and used directly in 1-pot Suzuki–Miyaura couplings.
2 Results and discussion
2.1 Optimization
Initial optimization leading to a general and green borylation protocol focused on the use of micellar catalysis, where low loadings of Pd catalysis are available and lead to highly efficient couplings, even involving challenging educts.20,47 Utilization of bis(ethylpinacolato)diboron (B2Epin2) was selected, as a recent communication highlights the enhanced stability (when compared to boronic acid and Bpin derivatives) of these analogous boronic esters to water, as well as their reported ease of isolation and improved yields of SM couplings under traditional reaction conditions.44Initial ligand screening utilized potassium acetate, which is used broadly for Miyaura borylation procedures,24,30 along with 4-bromoanisole as a model substrate. Both AmPhos48 and t-Bu3P (as its [t-Bu3PH]BF4 complex)49 emerged as effective ligands on palladium for selectively producing the borylated Epin-containing product, along with only trace amounts of undesired homocoupled material (Table 1).‡
|
|
|||
|---|---|---|---|
| Entrya | Ligand | Yieldb (A%) | Yieldb (B%) |
| a Reactions run on a 0.25 mmol scale. b Yield determined via qNMR using 1,3,5-trimethoxybenzene as an internal standard. | |||
| 1 | QPhos | 46 | 9 |
| 2 | [t-Bu 3 PH]BF 4 | 80 | Trace |
| 3 | DPPF | 38 | Trace |
| 4 | XPhos | 25 | Trace |
| 5 | SPhos | 26 | Trace |
| 6 | DPPP | Trace | Trace |
| 7 | EvanPhos | Trace | Trace |
| 8 | AmPhos | 82 | Trace |
| 9 | DavePhos | 42 | Trace |
| 10 | RuPhos | 36 | Trace |
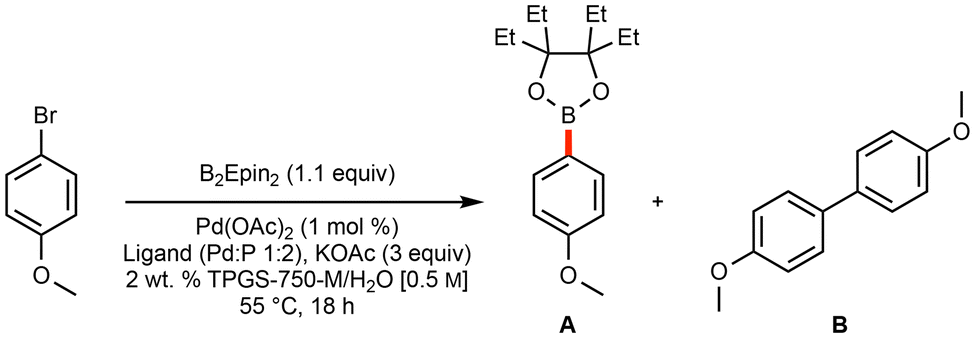
A comprehensive screening of bases (Table 2)‡ that included potassium triphosphate and potassium t-butoxide both led to significant levels of homocoupling. Use of potassium fluoride, although indicative of a good preference for the desired borylated product, led to a limited extent of conversion. Surprisingly, the electron-rich and sterically bulky potassium 2-ethylhexanoate (2-KEH), which has recently been shown to improve Miyaura borylations,41,46 differentiated the two ligands: use of [t-Bu3PH]BF4 performed markedly worse, while the yield using the AmPhos ligand-based catalytic system improved, leading to sole employment of pre-ligated Pd(AmPhos)2Cl2 going forward.50
|
|
||||
|---|---|---|---|---|
| Entrya | Ligand | Base | Yieldb (A%) | Yieldb (B%) |
| a Reactions run on a 0.25 mmol scale. b Yield determined via qNMR using 1,3,5-trimethoxybenzene as an internal standard. c Qualitative assessment based off TLC of crude reaction mixture (“sig.” = significant). d Potassium 2-ethylhexanoate. | ||||
| 1 | [t-Bu3PH]BF4 | KOAc | 80 | Trace |
| 2 | [t-Bu3PH]BF4 | K2CO3 | 40 | 10 |
| 3 | [t-Bu3PH]BF4 | KF | 34 | Trace |
| 4 | [t-Bu3PH]BF4 | K2HPO4 | 78 | Trace |
| 5 | [t-Bu3PH]BF4 | K3PO4 | Tracec | Sig.c |
| 6 | [t-Bu3PH]BF4 | 2-KEHd | 60 | Trace |
| 7 | [t-Bu3PH]BF4 | tBuOK | 19 | 33 |
| 8 | [t-Bu3PH]BF4 | TEA | 27 | 25 |
| 9 | AmPhos | NaOAc | Tracec | Tracec |
| 10 | AmPhos | K2HPO4 | 73 | 8 |
| 11 | AmPhos | 2-KEH | 87 | Trace |

Use of 2-KEH led to another remarkable observation: heating the reaction contents containing 2-KEH and B2Epin2 to 55 °C led to a readily stirred solution, suggesting that solvent-free, or highly concentrated reaction mixtures might be amenable. To test such conditions, solid 5-bromo-1-methyl-1H-indole was selected for this study. In the event, a high level of conversion and selectivity for the borylated product was realized by simply adding small amounts of EtOAc to facilitate stirring. Notably, a Pd. catalyst loading of only 0.25 mol% was needed.
2.2 Scope of Ar–BEpin products
The application of our optimized method using AmPhos allowed for the installation of the desired BEpin boronic ester to an array of functionalized (hetero)aryl halides. The moderate temperature of 55 °C, low palladium loading of 0.25 mol%, and concentrated EtOAc [2.5 M] led to moderate to excellent yields.Noteworthy is the application to several heteroaromatic compounds, and that each substrate was stable and isolable by column chromatography. There is no readily apparent trend between moderately electron-poor or -rich substrates; however, certain highly electron deficient substrates gave poor conversion (see compound 14 in Fig. 2). It is hypothesized that in substrates with heteroatoms that can coordinate to the catalyst (see compound 15 in Fig. 2), such coordination likely poisons the catalyst and is, thus, deleterious to conversion. Furthermore, ortho substitution provides significant steric hindrance towards installing the bulky ethyl pinacol boronic ester (see compound 16 in Fig. 2). Using 1-bromonaphthalene as a model for steric impedance, Fig. 3 shows that increasing reaction temperatures can overcome the activation barrier in such cases. At highly elevated temperatures (≥85 °C), however, competing side product (e.g. protodeborylation) formation was observed via proton NMR.
 | ||
Fig. 2 Scope of Ar–BEpin products. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Yield determined via qNMR using 1,3,5-trimethoxybenzene as an internal standard. bUtilized 0.5 mol% of Pd. cat. (Pd:P 1:4). EtOAc conc. of [1.25 M]. cDilution to [1.67 M] EtOAc d2 mmol scale. e0.3 mmol scale. f0.25 mmol scale. gUnsuccessful substrate. Yield determined via qNMR using 1,3,5-trimethoxybenzene as an internal standard. bUtilized 0.5 mol% of Pd. cat. (Pd:P 1:4). EtOAc conc. of [1.25 M]. cDilution to [1.67 M] EtOAc d2 mmol scale. e0.3 mmol scale. f0.25 mmol scale. gUnsuccessful substrate. | ||
 | ||
| Fig. 3 Temperature study Effect of temperature on conversion for a sterically hindered substrate (1H NMR conversions). | ||
2.3 Highly functionalized Ar–B(Epin) products
Borylations of highly functionalized substrates, such as X3 from the Merck Informer Library,51 indomethacin, and the precursors for abemaciclib52,53 and erdafitinib,54 required somewhat extended reaction times (24–48 h), a higher palladium loading (0.5 mol%), and/or higher reaction temperatures (70 °C). Moreover, these higher molecular weight substrates often required slight dilution to maintain stirring. Nonetheless, the BEpin moiety could be installed effectively for each of these complex substrates, further highlighting this method's applicability towards APIs and late-stage functionalized molecules. The scope of functionalized substrates (Fig. 4) includes selected aryl chlorides (see products 17 and 19 in Fig. 4), although, in harmony with recent literature,36,40 replacement of AmPhos with XPhos proved more amenable for aryl chlorides, loratadine and perphenazine (20 and 21). | ||
| Fig. 4 Scope of highly functionalized Ar–B(Epin) products. aXPhos utilized. bUtilized 0.5 mol% of Pd. cat. c(Pd:P 1:4). d70 °C. e0.25 mmol scale. f0.2 mmol scale. g0.125 mmol scale. | ||
2.4 Comparisons with existing literature
As illustrated in Table 3, several substrates were used for comparison purposes with existing literature procedures. Products 1, 22, and 18 show clear improvements in terms of yield, sustainability, and potentially, cost. In general, the need for dilute environmentally egregious solvents like dioxane55,56 is obviated, while metal economy is amply documented, e.g., with 18 being made using twenty times less palladium than the industrial process.57| This work | Literature | |
|---|---|---|
| a Selvita Pharmaceuticals patent (2016).55 b Astex Pharmaceuticals patent (2012).56 c Pfizer patent (2019).57 d Yield determined via qNMR using 1,3,5-trimethoxybenzene as an internal standard. | ||
| R = Et |

|
R = Me |
| 0.25 mol% | 1 mol% | |
| 55 °C | 90 °C | |
| EtOAc [2.5 M] | dioxane [0.68 M] | |
| overnight (18 h) | overnight (18 h) | |
| 84% (96%)d | 87%a | |
| R = Et |

|
R = Me |
| 0.5 mol% | 5 mol% | |
| 55 °C | 100 °C | |
| EtOAc [1.5 M] | dioxane [0.27 M] | |
| 24 h | 90 min | |
| 91% | 60%b | |
| R = Et |

|
R = Me |
| 0.5 mol% | 10 mol% | |
| 55 °C | 100 °C | |
| EtOAc [1 M] | DMSO [0.33 M] | |
| 44 h | 16 h | |
| 84% | 73%c | |
2.5 1-Pot cross electrophile couplings
By far, access to borylated aromatics and other sp2-containing C–B bonds is for use in SM couplings.2–13 Developing a 1-pot protocol towards this goal, however, proved to be more challenging than initially anticipated.The obvious choice regarding the medium for performing the SM coupling was the use of aqueous micellar catalysis, given procedures already in hand.20,47
Best results were obtained, therefore, when a different ligand (SPhos; 0.5 mol%) was added in the second, SM step. Unfortunately, with the exceptions noted in Fig. 5, the presence of excess B2Epin2 left over from the first reaction would borylate the bromide having been added in the second step, resulting in significant homocoupling of this halide. Increasing the equivalents of the second bromide added led to low levels of conversion. Attempts at utilizing different ligands, loadings of palladium, bases, surfactants, temperatures, and concentrations also proved to be ineffective. Notwithstanding these setbacks, where applicable, use of TPGS-750-M does allow for a 1-pot process that relies only on water, concentrated EtOAc, reaction temperatures of 55 °C, and an overall palladium loading of only 0.25 mol%. Nonetheless, a more generally applicable procedure was sought.
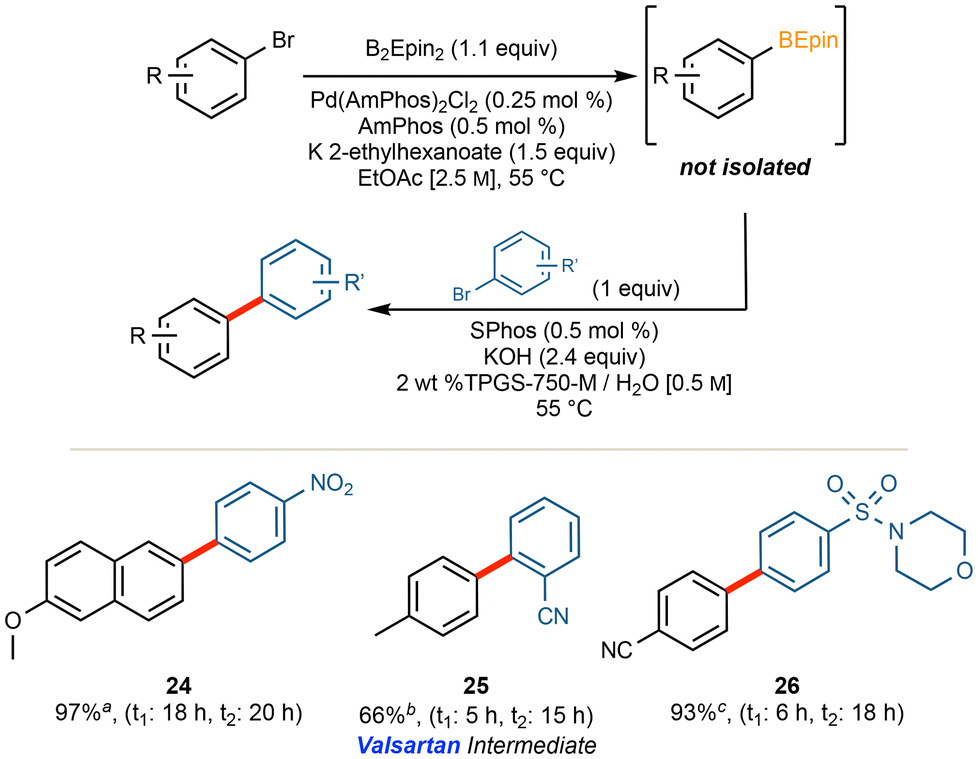 | ||
| Fig. 5 Initial studies of 1-pot cross-couplings with TPGS-750-M. aRun on a 0.5 mmol scale with 0.8 equiv. of bromide in the SM coupling. bRun on 0.3 mmol scale; conc. of [0.25 M] and 3 equiv. of K3PO4 in SM coupling. cRun on a 0.25 mmol scale. | ||
Another sustainable SM approach focused on an “on water”58–61 reaction utilizing tetrabutyl ammonium bromide (TBAB) as a phase transfer catalyst58 along with an inverted stoichiometry, leading to complete consumption of the BEpin ester. Although additional Pd. catalyst had to be added for the SM coupling step, the ligated Pd bearing the same AmPhos ligand could be used. Hence, a total loading of only 0.5 mol% of the same palladium-containing catalyst is involved for the 1-pot, 2-step process. Our developed two-step one-pot method showed applicability to a diverse set of substrates, as shown in Fig. 6. Even with the overall increased catalyst loading of 0.5 mol% and temperature of 70 °C for the SM reaction, heteroaromatic halides and complex substrates can be efficiently coupled in the same reaction vessel. This observation is supported by the quantitative late-stage formation of products 37 and 38 (Fig. 6). One limitation is brought about by the need for a relatively high base loading. This is illustrated by compound 39 where there is an acidic phenol and a base sensitive methyl ester. Nonetheless, and especially relevant to the generation of APIs in the final step of a synthesis, product 38 was analyzed via inductively coupled plasma mass spectrometry (ICP-MS), shown to have residual palladium to the extent of 7.9 ppm. This is within the FDA guidelines limit of 10 ppm palladium per dose per day62 and demonstrates that complex biaryl products can be made without anticipating further investments in time and materials for metal scavenging.
 | ||
| Fig. 6 General scope of 1-pot couplings. a1 equiv. of bromide used in the SM coupling. b1.5 equiv. of bromide used in the SM coupling. c0.5 mol% of Pd at 70 °C utilized in the borylation step. dXPhos (Pd:P 1:4) utilized in the borylation step. eUnsuccessful coupling (first step conversion confirmed via GC). fRun on a 0.5 mmol scale. gRun on a 0.125 mmol scale. | ||
2.6 Gram scale preparation of anidulafungin precursor
To show the potential scalability of this process, a gram scale preparation of compound 40, a significant precursor to the antifungal Anidulafungin, was sought. In the borylation step, only slight dilution with EtOAc was required for stirring purposes. Once completed, the SM coupling called for addition of 2-MeTHF (10% v/v) with addition of water to aid in stirring. The product was isolated in 99% yield via simple filtration (see ESI†), and the calculated environmental factor (E-Factor)63 of just 10.9 attests to the “greenness” of this new technology (Fig. 7).§ | ||
| Fig. 7 Gram scale 1-pot reaction and E-factor. aBorylation: EtOAc [2 M] utilized; 15.5 h. Suzuki: H2O [0.75 M] with 12.5% (v/v) 2-MeTHF; 16.5 h. | ||
2.7 Multistep sequence: boscalid
A sequence that includes a borylation/SM coupling that is representative of the options now available to the community is shown in Fig. 8. | ||
| Fig. 8 Multistep sequence: boscalid. | ||
In the first step, borylation of 1-bromo-4-chlorobenzene leads to an intermediate BEpin derivative that reacts exclusively at the bromide center. Without isolation, under SM coupling conditions using newly introduced 1-bromo-2-nitrobenzene, the targeted biaryl is formed. This coupling is done within four hours, notwithstanding a 1:1 ratio of the Epin boronic ester and second bromide reflecting steric hindrance of the ortho-nitro group preventing potentially competing borylation and homocoupling. Simple workup of the biaryl product led to crude material that underwent nitro reduction via carbonyl iron powder (CIP)64 in aqeuous micellar media. Upon completion, the penultimate material was then used crude (after filtration away from the CIP) in an amide coupling with 2-chloronicotinoyl chloride in EtOAc. This afforded boscalid in 65% overall yield.
2.8 Mechanistic commments
A final important observation was made with regard to the stability of these B(Epin) boronic esters. Generally, unreacted boronic esters remain stable in basic aqueous media at these reaction temperatures, which is consistent with the decomposition studies performed by Ikawa, Akai, and co-workers.44 This stability is attributed to the ethyl “arms” of a B(Epin), that provide steric hindrance to the empty p orbital on boron, translating into hydrolytic resistance. These observations have important mechanistic considerations for Suzuki–Miyaura couplings in which B(Epin) derivatives smoothly participate. Thus, the implication is that it is not the corresponding boronic acid that undergoes transmetallation, but rather the intact aryl–B(Epin). This notion is in agreement with the findings by Denmark, et al.65,66 in which their mechanistic studies found that glycol boronic esters can transmetallate without the need to first hydrolyze to the corresponding boronic acid. Nonetheless, more sterically hindered boronic esters haven't conclusively been determined to show similar behavior. Hence, studies to elucidate the mechanistic picture for sterically hindered Ar–B(Epins) are ongoing.3 Conclusion
A sustainable and environmentally responsible borylation protocol has been developed that allows for facile installation of the B(Epin) group into a variety of aromatic/heteroaromatic substrates. These species can be utilized in subsequent Suzuki–Miyaura cross couplings. There are several additional features associated with the new technology of note:• low loadings of Pd are involved
• green solvents/reaction media, such as recyclable EtOAc, or water, are used
• a borylation/Suzuki–Miyaura 1-pot sequence leads to both time and pot economies
• a scalable process
• low residual levels of Pd in the products, established via ICPMS, is to be expected
• multi-step sequences are amenable as illustrated by the synthesis of boscalid
Author contributions
All authors have given approval to the final version of the manuscript. C. B. N. conceived the project, optimized reactions conditions, carried out substrate scope, collected analytical data, and drafted the manuscript. S. J. L. assisted in optimization, substrate scope, and collecting analytical data. M. J. W. assisted in substrate scope and collecting analytical data. S. L. K. and E. G. perfomed early investigations into other sustainable borylation avenues. B. H. L. oversaw the work and assisted in drafting the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Financial support provided by the NSF (CHE-2152566) is warmly acknowledged. The authors express gratitude for assistance in collecting HRMS data from the UCSB Mass Spectrometry Facility staff, Dr Dezmond Bishop, and in collecting ICPMS data from the Nano and Pico Characterization Lab at the California NanoSystems Institute at UCLA, Dr Chong Hyun “Paul” Chang.References
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- J. Britton and T. F. Jamison, Eur. J. Org. Chem., 2017, 2017, 6566–6574 CrossRef CAS.
- L. Xiong, Y.-Q. Shen, L.-N. Jiang, X.-L. Zhu, W.-C. Yang, W. Huang and G.-F. Yang, Discovery and Synthesis of Crop Protection Products, American Chemical Society, 2015, vol. 1204, pp. 175–194 Search PubMed.
- J. B. Epp, P. R. Schmitzer and G. D. Crouse, Pest Manage. Sci., 2018, 74, 9–16 CrossRef CAS PubMed.
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- J. E. Zweig, D. E. Kim and T. R. Newhouse, Chem. Rev., 2017, 117, 11680–11752 CrossRef CAS PubMed.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed.
- A. Taheri Kal Koshvandi, M. M. Heravi and T. Momeni, Appl. Organomet. Chem., 2018, 32, e4210 CrossRef.
- M. A. Selepe and F. R. Van Heerden, Molecules, 2013, 18, 4739–4765 CrossRef CAS PubMed.
- G. Dhangar, J. L. Serrano, C. Schulzke, K. C. Gunturu and A. R. Kapdi, ACS Omega, 2017, 2, 3144–3156 CrossRef CAS PubMed.
- S.-H. Yang and C.-S. Hsu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2713–2733 CrossRef CAS.
- H. Iino and J.-i. Hanna, Polym. J., 2017, 49, 23–30 CrossRef CAS.
- B. H. Lipshutz, Is Organic Chemistry on Borrowed Time?, 2022, https://cen.acs.org/environment/green-chemistry/Opinion-organic-chemistry-borrowed-time/100/i34 Search PubMed.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- D. R. Joshi and N. Adhikari, J. Pharm. Res. Int., 2019, 1–18 Search PubMed.
- R. Ratti, SN Appl. Sci., 2020, 2, 263 CrossRef.
- S. Mukai and Y. Yamada, Knowledge, 2023, 3, 1–17 CrossRef.
- B. H. Lipshutz and A. R. Abela, Org. Lett., 2008, 10, 5329–5332 CrossRef CAS PubMed.
- J. R. A. Kincaid, M. J. Wong, N. Akporji, F. Gallou, D. M. Fialho and B. H. Lipshutz, J. Am. Chem. Soc., 2023, 145, 4266–4278 CrossRef CAS PubMed.
- Y. Hu, X. Li, G. Jin and B. H. Lipshutz, ACS Catal., 2023, 13, 3179–3186 CrossRef CAS PubMed.
- N. Akporji, R. R. Thakore, M. Cortes-Clerget, J. Andersen, E. Landstrom, D. H. Aue, F. Gallou and B. H. Lipshutz, Chem. Sci., 2020, 11, 5205–5212 RSC.
- V. Singhania, C. B. Nelson, M. Reamey, E. Morin, R. D. Kavthe and B. H. Lipshutz, Org. Lett., 2023, 25, 4308–4312 CrossRef CAS PubMed.
- T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508–7510 CrossRef CAS.
- J. A. Newby, L. Huck, D. W. Blaylock, P. M. Witt, S. V. Ley and D. L. Browne, Chem. – Eur. J., 2014, 20, 263–271 CrossRef CAS PubMed.
- D. S. Matteson, The Metal-Carbon Bond, John Wiley & Sons, Inc., Chichester, UK, 1987, vol. 4, pp. 307–409 Search PubMed.
- M. A. Larsen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4287–4299 CrossRef CAS PubMed.
- M. M. M. Hassan, S. Guria, S. Dey, J. Das and B. Chattopadhyay, Sci. Adv., 2023, 9, eadg3311 CrossRef CAS PubMed.
- M. Liang, C. Liu, W. Ju, S. Han, J. Zhang and Y. Zhao, Org. Lett., 2024, 26, 4224–4228 CrossRef CAS PubMed.
- W. K. Chow, O. Y. Yuen, P. Y. Choy, C. M. So, C. P. Lau, W. T. Wong and F. Y. Kwong, RSC Adv., 2013, 3, 12518–12539 RSC.
- F. D'Amico, C. Papucci, D. Franchi, G. Reginato, M. Taddei, A. Mordini, L. Zani, A. Dessì and M. Calamante, J. Org. Chem., 2024, 89, 6991–7003 CrossRef PubMed.
- K. L. Billingsley, T. E. Barder and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 5359–5363 CrossRef CAS PubMed.
- K. U. Rao and K. Venkateswarlu, Synlett, 2018, 1055–1060 CAS.
- M. Cai, C. Luo, C. Xu and B. Huang, Synthesis, 2022, 1339–1346 CrossRef CAS.
- L. Xu, Z. Dong, Q. Zhang, N. Deng, S.-Y. Li and H.-J. Xu, J. Org. Chem., 2022, 87, 14879–14888 CrossRef CAS PubMed.
- K. Billingsley and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3358–3366 CrossRef CAS PubMed.
- D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961–6963 CrossRef CAS PubMed.
- C. Achilli, A. Ciana, M. Fagnoni, C. Balduini and G. Minetti, Cent. Eur. J. Chem., 2013, 11, 137–139 CAS.
- G. A. Molander, S. L. J. Trice, S. M. Kennedy, S. D. Dreher and M. T. Tudge, J. Am. Chem. Soc., 2012, 134, 11667–11673 CrossRef CAS PubMed.
- C. Munteanu, T. E. Spiller, J. Qiu, A. J. DelMonte, S. R. Wisniewski, E. M. Simmons and D. E. Frantz, J. Org. Chem., 2020, 85, 10334–10349 CrossRef CAS PubMed.
- S. R. Gurung, C. Mitchell, J. Huang, M. Jonas, J. D. Strawser, E. Daia, A. Hardy, E. O'Brien, F. Hicks and C. D. Papageorgiou, Org. Process Res. Dev., 2017, 21, 65–74 CrossRef CAS.
- J. R. Coombs, R. A. Green, F. Roberts, E. M. Simmons, J. M. Stevens and S. R. Wisniewski, Organometallics, 2019, 38, 157–166 CrossRef CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2013, 43, 412–443 RSC.
- N. Oka, T. Yamada, H. Sajiki, S. Akai and T. Ikawa, Org. Lett., 2022, 24, 3510–3514 CrossRef CAS PubMed.
- S. Barroso, M. Joksch, P. Puylaert, S. Tin, S. J. Bell, L. Donnellan, S. Duguid, C. Muir, P. Zhao, V. Farina, D. N. Tran and J. G. de Vries, J. Org. Chem., 2021, 86, 103–109 CrossRef CAS PubMed.
- B. M. Klootwyk, J. C. Ruble, G. R. Cumming and E. M. Woerly, J. Org. Chem., 2024, 89, 5901–5904 CrossRef CAS PubMed.
- B. H. Lipshutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovskiy, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379–4391 CrossRef CAS PubMed.
- A. S. Guram, X. Wang, E. E. Bunel, M. M. Faul, R. D. Larsen and M. J. Martinelli, J. Org. Chem., 2007, 72, 5104–5112 CrossRef CAS PubMed.
- M. R. Netherton and G. C. Fu, Org. Lett., 2001, 3, 4295–4298 CrossRef CAS PubMed.
- It is noted that recent work from Woerly and colleagues showed that potassium 2-ethylhexanoate could be used catalytically with stoichiometric potassium carbonate in order to avoid work up and isolation issues associated with this base.46 However, in our work, we were able to effectively remove the base during work up, and using the base catalytically under our conditions, led to increased homocoupling.
- P. S. Kutchukian, J. F. Dropinski, K. D. Dykstra, B. Li, D. A. DiRocco, E. C. Streckfuss, L.-C. Campeau, T. Cernak, P. Vachal, I. W. Davies, S. W. Krska and S. D. Dreher, Chem. Sci., 2016, 7, 2604–2613 RSC.
- M. P. Goetz, M. Toi, M. Campone, J. Sohn, S. Paluch-Shimon, J. Huober, I. H. Park, O. Trédan, S.-C. Chen, L. Manso, O. C. Freedman, G. Garnica Jaliffe, T. Forrester, M. Frenzel, S. Barriga, I. C. Smith, N. Bourayou and A. Di Leo, J. Clin. Oncol., 2017, 35, 3638–3646 CrossRef CAS PubMed.
- S. Johnston, M. Martin, A. Di Leo, S.-A. Im, A. Awada, T. Forrester, M. Frenzel, M. C. Hardebeck, J. Cox, S. Barriga, M. Toi, H. Iwata and M. P. Goetz, npj Breast Cancer, 2019, 5, 1–8 CrossRef CAS PubMed.
- Y. Loriot, A. Necchi, S. H. Park, J. Garcia-Donas, R. Huddart, E. Burgess, M. Fleming, A. Rezazadeh, B. Mellado, S. Varlamov, M. Joshi, I. Duran, S. T. Tagawa, Y. Zakharia, B. Zhong, K. Stuyckens, A. Santiago-Walker, P. De Porre, A. O'Hagan, A. Avadhani and A. O. Siefker-Radtke, N. Engl. J. Med., 2019, 381, 338–348 CrossRef CAS PubMed.
- C.-H. R. Y. Fabritius, M. O. Nowak, K. A. Wiklik, A. B. Sabiniarz, M. D. Bień, A. M. Buda, P. S. Guzik, A. K. Białas, H. E. Pawlik and N. F. P. Boutard, EPO WO2016180537A1, 2016.
- G. Saxty, C. W. Murray, V. Berdini, G. E. Besong, C. C. F. Hamlett, S. J. Woodhead, Y. A. E. Ligny and P. R. Angibaud, EPO WO2012073017A1, 2012.
- P. Chen, S. Cho-Schultz, J. G. Deal, G. M. Gallego, M. Jalaie, R. S. Kania, S. K. Nair, S. Ninkovic, S. T. M. Orr and C. L. Palmer, EPO US2019330196A1, 2019.
- D. Badone, M. Baroni, R. Cardamone, A. Ielmini and U. Guzzi, J. Org. Chem., 1997, 62, 7170–7173 CrossRef CAS PubMed.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
- R. N. Butler and A. G. Coyne, Org. Biomol. Chem., 2016, 14, 9945–9960 RSC.
- D. G. Blackmond, A. Armstrong, V. Coombe and A. Wells, Angew. Chem., Int. Ed., 2007, 46, 3798–3800 CrossRef CAS PubMed.
- FDA, Q3D (R1) elemental impurities guidance for industry, 2022, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/elemental-impurities-drug-products-guidance-industry Search PubMed.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- N. R. Lee, A. A. Bikovtseva, M. Cortes-Clerget, F. Gallou and B. H. Lipshutz, Org. Lett., 2017, 19, 6518–6521 CrossRef CAS PubMed.
- A. A. Thomas, H. Wang, A. F. Zahrt and S. E. Denmark, J. Am. Chem. Soc., 2017, 139, 3805–3821 CrossRef CAS PubMed.
- C. P. Delaney, D. P. Marron, A. S. Shved, R. N. Zare, R. M. Waymouth and S. E. Denmark, J. Am. Chem. Soc., 2022, 144, 4345–4364 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03115f |
| ‡ For the complete list of screened ligands and bases, see the ESI.† |
| § See ESI† for E-Factor calculations. |
| This journal is © The Royal Society of Chemistry 2024 |