
Implications for new particle formation in air of the use of monoethanolamine in carbon capture and storage†
Véronique
Perraud
 *,
Kanuri
Roundtree
,
Patricia M.
Morris
,
James N.
Smith
* and
Barbara J.
Finlayson-Pitts
*,
Kanuri
Roundtree
,
Patricia M.
Morris
,
James N.
Smith
* and
Barbara J.
Finlayson-Pitts
Department of Chemistry, University of California Irvine, Irvine, CA 92697, USA. E-mail: vperraud@uci.edu; jimsmith@uci.edu; Fax: +1 (949) 824-2420; Tel: +1(949) 824-2673 Tel: +1 (949) 824-9518
First published on 5th March 2024
Abstract
Alkanolamines are currently being deployed in carbon capture and storage (CCS) technology worldwide, and atmospheric emissions have been found to coincide with locations exhibiting elevated concentrations of methanesulfonic acid (MSA). It is thus critical to understand the fate and potential atmospheric reactions of these chemicals. This study reports the characterization of sub-10 nm nanoparticles produced through the acid–base reaction between gas phase monoethanolamine (MEA) and MSA, a product of organosulfur compound oxidation in air, using a flow reactor under dry and humid (up to ∼60% RH) conditions. Number size distribution measurements show that MEA is even more efficient than methylamine in forming nanoparticles on reaction with MSA. This is attributed to the fact that the MEA structure contains both an –NH2 and an –OH group that facilitate hydrogen bonding within the clusters, in addition to the electrostatic interactions. Due to this already strong H-bond network, water has a relatively small influence on new particle formation (NPF) and growth in this system, in contrast to MSA reactions with alkylamines. Acid/base molar ratios of unity for 4–12 nm particles were measured using thermal desorption chemical ionization mass spectrometry. The data indicate that reaction of MEA with MSA may dominate NPF under some atmospheric conditions. Thus, the unique characteristics of alkanolamines in NPF must be taken into account for accurate predictions of impacts of CCS on visibility, health and climate.
Introduction
Monoethanolamine (NH2CH2CH2OH, MEA) is a multifunctional amine currently deployed in carbon capture and storage (CCS) technology systems aimed at sequestering CO2 emissions before release into the atmosphere.1–5 The most widely used CCS media is a 30% aqueous solution of MEA.6 Briefly, the solvent medium chemically absorbs CO2 contained in the flue gas, which leads to a CO2-depleted gas stream exiting the stack. The solvent is subsequently regenerated and recycled back into the absorber column, while the CO2 is compressed and captured. A potential drawback from these technologies is the likely release of MEA into the air.4,5,7–14 For example, concentrations of MEA outside a CCS-equipped plant of the order of several ppb have been reported.7 MEA is also used as a solvent in various consumer products and industrial processes.15–19 In air, recognized fates of gas phase MEA to date include its reaction with O3 and OH,20–24 the formation of alkylaminium nitrate salts from its interaction with HNO321,24 and acid–base reactions with gas phase and particulate sulfuric acid.25,26Methanesulfonic acid (CH3SO3H, MSA) is a strong acid formed along with SO2 (a sulfuric acid precursor) in the oxidation of dimethyl sulfide (DMS) and dimethyl disulfide (DMDS)27–32 which have a variety of sources both natural and anthropogenic.33–59 Therefore, it is not surprising that the oxidation product MSA is detected in the gas phase and in ambient particles worldwide. Ambient gas phase atmospheric concentrations of MSA range from mid-104 to 107 molecules cm−3,60–69 and can reach levels similar to that of H2SO4, which is considered to be a major source of new particles. In some instances, the MSA concentration in air can actually surpass that of co-located H2SO4.68,70 MSA has also been detected in ambient particles worldwide, including in marine and coastal environments,71–77 in coastal areas affected by biomass burning plumes,78,79 and near agricultural regions as well as near urban centers.78,80,81 This acid has been detected in ultrafine and nucleation mode particles measured in the Arctic,76,82–85 at urban sites86,87 and in the Antarctic88 as well as in a boreal forest.67,89 Particulate MSA concentrations in the Arctic summertime have been observed to correlate well with new particle formation (NPF)85,90–94 suggesting a role for MSA in the earliest stages of NPF and growth. Chen and co-workers95 predicted that the total annual MSA budget would be 20 Gg S y−1 from DMS oxidation reactions alone. However, climate change is dramatically modifying the extent of ice sheet coverage, exposing more sea water, which increases phytoplankton productivity and DMS emissions and thus MSA in air.94,96–98 The significant contribution of MSA to atmospheric NPF is supported by both laboratory experiments99–107 and quantum chemical calculations.108–115
Amines and MSA are both found in ambient particles.67,80,116–118 This includes MEA, which has been detected as one of the most abundant amines in ambient particles in various locations around the globe,119–124 overlapping with sources of both DMS and MSA. MEA has also been detected in biomass burning aerosols collected in St John, Newfoundland, Canada125 and in both aerosol and precipitation samples over the North Atlantic Ocean.126
It is thought that NPF is responsible for a significant portion of the global cloud condensation nuclei budget.127 In addition to influencing cloud properties, airborne particles are well known to interact with solar radiation, thus playing a critical role in the Earth's climate.128,129 In a recent study, Hodshire et al.130 predicted, using a simplified DMS oxidation model, that inclusion of MSA formation and its role in aerosol processes (either acting as condensable non- or semi-volatile species, or participating in NPF) influenced the cloud-albedo aerosol indirect and the direct radiative effect.
While recent theoretical studies predicted that MEA may play an important role in NPF,108 to date there have been no direct experimental investigations of particle formation from MEA and MSA. We present the first measurements of 4–12 nm nanoparticles formed from this reaction, including their size distributions as a function of time and relative humidity, as well as their size-resolved chemical composition. For comparison, some data for the reaction of MSA with methylamine (MA), which is known to efficiently form particles,102,103,105,106 is also reported. It is shown that MEA is even more effective in forming new nanometer-sized particles than MA but surprisingly, is not very sensitive to the presence of water vapor. Such ultrafine particles are of particular concern as they can be deposited deep into the respiratory tract and even cross cellular membranes to reach other organs.131–136 Thus, this study has important implications for the potential impacts of CCS on climate,128,129 visibility137–140 and health.131,132,136,141
Experimental methods
Flow reactor description
Particles were produced from the reaction of gas phase MSA with gas phase MEA (or MA) in the presence or absence of water vapor in a 1-m long borosilicate glass flow reactor142 described in the ESI† (Fig. S1). Clean, dry air was provided by a purge air generator (Parker-Balston; model 75-62), and further purified by passing through carbon/alumina media (PermaPure, LLC) and a 0.01 μm inline filter (Parker Balston, BQ). Most of the air was supplied at the front end of the flow reactor through the perforated ring inlets as indicated in Fig. S1 (ESI†) (rings A, B and C). In experiments where water vapor was present, one or two bubblers filled with nanopure water (18.2 MΩ cm; Barnstead, Thermo Scientific) were used to humidify a fraction of the air introduced into the ring inlets. The bubblers were kept in a water bath to maintain a constant temperature of 22 °C (295 K). Experiments were carried out at relative humidities (RH) up to ∼60% as indicated by a humidity probe (Vaisala; model HMT 838) located at the end of the flow reactor. The reactants (MSA and MEA or MA) were introduced through the spoke inlets (spoke 2 and 3 respectively) located 60 cm downstream of the last ring inlet. The flow reactor was cleaned regularly with nanopure water and dried with clean hot air overnight (T = 343 K). After cleaning, the flow reactor was conditioned with gas-phase MSA for a least two days prior to an experiment. All experiments presented in this work were performed at 1 atm and at room temperature (T = 297 K).Reactants
Liquid monoethanolamine (NH2CH2CH2OH, Sigma Aldrich, >99.5%) was contained in a small 2-mL glass vial with a septum cap. Approximately ∼1 cm of PEEK tubing (1.59 mm O. D. × 0.18 mm I. D.) was inserted into the septum so that the MEA from the headspace diffused slowly into a stream of air. For comparison, parallel experiments were performed using MA (CH3NH2) with a commercial permeation tube (VICI Metronics). The amine vial (or permeation tube) was inserted into separate U-shaped glass tubes immersed into a water bath maintained at room temperature (T = 295 K). Glass beads were placed in the upstream arm of the U-shaped glass tubes to provide high surface area to keep the gas flow at a constant temperature. Air flowed through each tube at a rate of 215 cm3 min−1 for MEA and 93 or 211 cm3 min−1 for MA. For MSA, air (53 to 216 cm3 min−1) flowed directly over the pure liquid (Sigma Aldrich, >99.0%) contained in a glass trap which was maintained at room temperature using a water bath. Further details regarding the sampling, analysis and quantification of the gas phase reactants are given in the ESI† (Text S1 and Fig. S2). The initial concentrations of the reactants after dilution in the flow reactor were (1.7–6.8) × 1010 molecules cm−3 for MSA (0.7–2.8 ppb), (3.7–8.1) × 1010 molecules cm−3 for MEA (1.5–3.3 ppb) and (11.8–26.6) × 1010 molecules cm−3 for MA (4.8–10.8 ppb). Note that these concentrations represent upper limits as they do not account for potential wall losses.Particle size distribution measurements
Particle size distributions were continuously measured using a moveable stainless steel sampling line (0.64 cm O. D. × 0.46 cm I. D.) located inside the flow reactor along the centerline and placed at distances ranging from 3 to 43 cm away from spoke 2 (i.e., the MSA addition port). All particle size distributions reported in this study are number size distributions, unless stated otherwise. These distances correspond to reaction times in the reactor ranging from 0.3 to 4.5 s (total flow rate 23.4 L min−1) or 0.5 to 7.7 s (total flow rate 10.7 L min−1) based on a conversion factor determined in previous studies.99 Note that the amine addition port is introducing the reactant backward into the flow stream so that the reaction of MSA with MEA (MA) is occurring in between spoke 2 and 3, and we chose the MSA addition port as our t = 0 reaction time. It is expected that the residual reactants present in the stream exiting the flow reactor are lost to the walls of the small (I.D. 0.46 cm) sampling line. Therefore, the reaction times reported are those in the flow reactor, but these could be underestimated if the reaction continues in the sampling line (residence time ∼0.3–0.4 s) and the connection to the SMPS (residence time ∼0.8 s). Particle losses through the sampling lines to the SMPS were accounted for as described in the ESI† (Text S2 and Fig. S3). The shortest reaction time accessible (i.e. 0.3 s or 0.5 s depending on the total flow in the flow tube, that was 23.4 or 10.7 L min−1 respectively) is expected to be the most vulnerable to residence time artefacts. This was tested by sampling at different flow rates through the sampling line (2.4 to 4.8 L min−1), for a given experiment conducted at 0.5 s. Results presented in Fig. S4 (ESI†) show no change in the size distribution measured at all flow rates and suggest that reaction in the sampling line is not significant.Size distributions were measured using a scanning mobility particle sizer (SMPS) consisting of a 210Po radioactive source (10 mCi; NRD LLC; model P-2021), an electrostatic classifier (model 3080; TSI Inc.) equipped with a nano-differential mobility analyzer (nano-DMA; model 3085; TSI, Inc.), and a butanol-based ultrafine condensation particle counter (UCPC; model 3776; TSI, Inc.). To prevent buildup of the reactants in the SMPS during sampling, the sheath air inside the DMA was not recirculated, but instead air was provided by the purge air generator (15 L min−1) and a vacuum pump connected to the sheath air flow pulled the sheath air out of the DMA. The aerosol flow was set to 1.5 L min−1, which provided measurements of the size distributions over a mobility diameter range of 2.5 to 64 nm. The software AIM v9 (TSI, Inc.) was used to record and process the data. Particles were observed to be stable for long periods of time (Fig. S5, ESI†), allowing for size-resolved measurements that took up to 20 min per scan to yield enough mass for mass spectrometric analysis.
Size-resolved chemical composition measurements
Nanoparticles with diameters ranging from 4 to 12 nm were sampled using a thermal desorption chemical ionization mass spectrometer (TDCIMS)105,143–146 which was connected to the same sampling line as the SMPS. The particle stream was sampled through two inlets, each equipped with a 210Po unipolar charger (UPC)143,147 to generate negatively charged particles. At each inlet, particles were subsequently size-selected using a radial nano-DMA (rDMA) running in either high resolution mode with a sheath flow of 10 L min−1 and an aerosol flow of 1.0 L min−1 through each nano-rDMA, or low resolution mode with a sheath air flow of 5.0 L min−1 and an aerosol flow rate of 1.6 L min−1 through each nano rDMA.148 For both conditions, instead of recirculating the sheath gas within the nano-DMA, gaseous N2 produced from the headspace of a liquid N2 dewar was used as the sheath flow to prevent the accumulation of gas-phase MSA or MEA, and a vacuum pump was used at the DMA sheath flow outlet. The particles were collected on the tip of a Pt filament by electrostatic precipitation (applied high voltage of +3.5 kV). The filament was continuously flushed with an additional 1.25 L min−1 flow of N2 to minimize sampling artifacts from gas-phase species. To select particles with a defined mobility diameter, the voltage on each rDMA was varied from 30 to 325 V. Note that the use of two separate inlets, which merged at the collection wire region, increases the flux and mass of particles that are collected on the wire without sacrificing the rDMA resolution.The TDCIMS was run in positive ion mode to measure MEA with (H2O)nH+ as the reagent ions (n = 0–3), and in negative ion mode to measure MSA with (H2O)nO2− as the reagent ions from the presence of trace amounts of H2O and O2, respectively, in the carrier N2 gas. Monoethanolamine was detected as two major ions in the mass spectra, the parent [M + H]+ ion (m/z 62) and a fragment ion corresponding to [M + H–H2O]+ (m/z 44). The fragmentation of the parent [M + H]+ ion of MEA is consistent with early experimental and theoretical studies149,150 showing that although the amino group is the favored protonation site due to its higher proton affinity compared to the alcohol group,151 rearrangement and the loss of H2O dominates over the loss of NH3. The corresponding fragment ion associated with the loss of NH3 (m/z 45) was not observed in any of the mass spectra. MSA was detected in negative ion mode as the parent deprotonated [M − H]− ion (m/z 95) followed by a major fragment ion at m/z 80 (SO3−), with additional minor ions at m/z 64 (SO2−), m/z 96 (SO4−), m/z 97 (HSO4)− and m/z 112 (SO5−). Both positive and negative mass spectra are presented in Fig. S6 (ESI†). From the desorption profiles presented in Fig. S7 (ESI†), it is evident that MEA (and MA; data not shown) desorbs first from the filament followed by MSA, consistent with the differences in their respective saturation vapor pressures (Psat) at 298 K: Psat(MEA) = 3.4 × 10−4 atm152 and Psat(MSA) = 7.4 × 10−7 atm.153 Additional details on the TDCIMS analysis are described in the ESI† (Text S3–S5 and Fig. S6–S11).
Results and discussion
Fig. 1 represents the size distributions of particles from the MSA + MEA reaction under dry conditions, with each panel (A–F) representing a different reactant concentration condition. Varying reactant concentrations were achieved by either increasing or decreasing the flow of the reactant that was introduced into the flow tube, or by changing the total flow rate in the flow tube (23.4 L min−1 for panels (A–C); 10.7 L min−1 for panels (D–F)). Clearly, mixing gas phase MSA and MEA at low ppb levels results in rapid formation of particles. Corresponding plots of the evolution of the total number concentration as well as the geometric diameter as a function of the reaction time are presented in Fig. S12 (ESI†). Even at the smallest reactant concentrations (Fig. 1(A), 1.5 ppb MEA, 0.68 ppb MSA), particles measured at the shortest reaction time (t = 0.3 s; total flow rate 23.4 L min−1) are formed at a number concentration of 6.3 × 106 particles cm−3 with a geometric mean mobility diameter (GMD) of ∼ 4 nm. At 2.4 s, the number concentration increases by a factor of two with little change in size. At longer times, there is no further increase in the particle number concentration while the particles continue to grow to a GMD of ∼4.6 nm, suggesting that under these conditions there is a balance between nucleation, growth by addition of the reactants onto particles, and coagulation. Particle losses inside the flow tube were estimated using the particle loss calculator tool developed by von der Weinder et al.154 (using a density of 1 g cm−3), and was found to be small for all diameters (e.g. for a particle diameter of 2.5 nm, particle transmission is predicted to be 92 or 95% for a total flow rate inside the flow tube of 10.7 or 23.4 L min−1).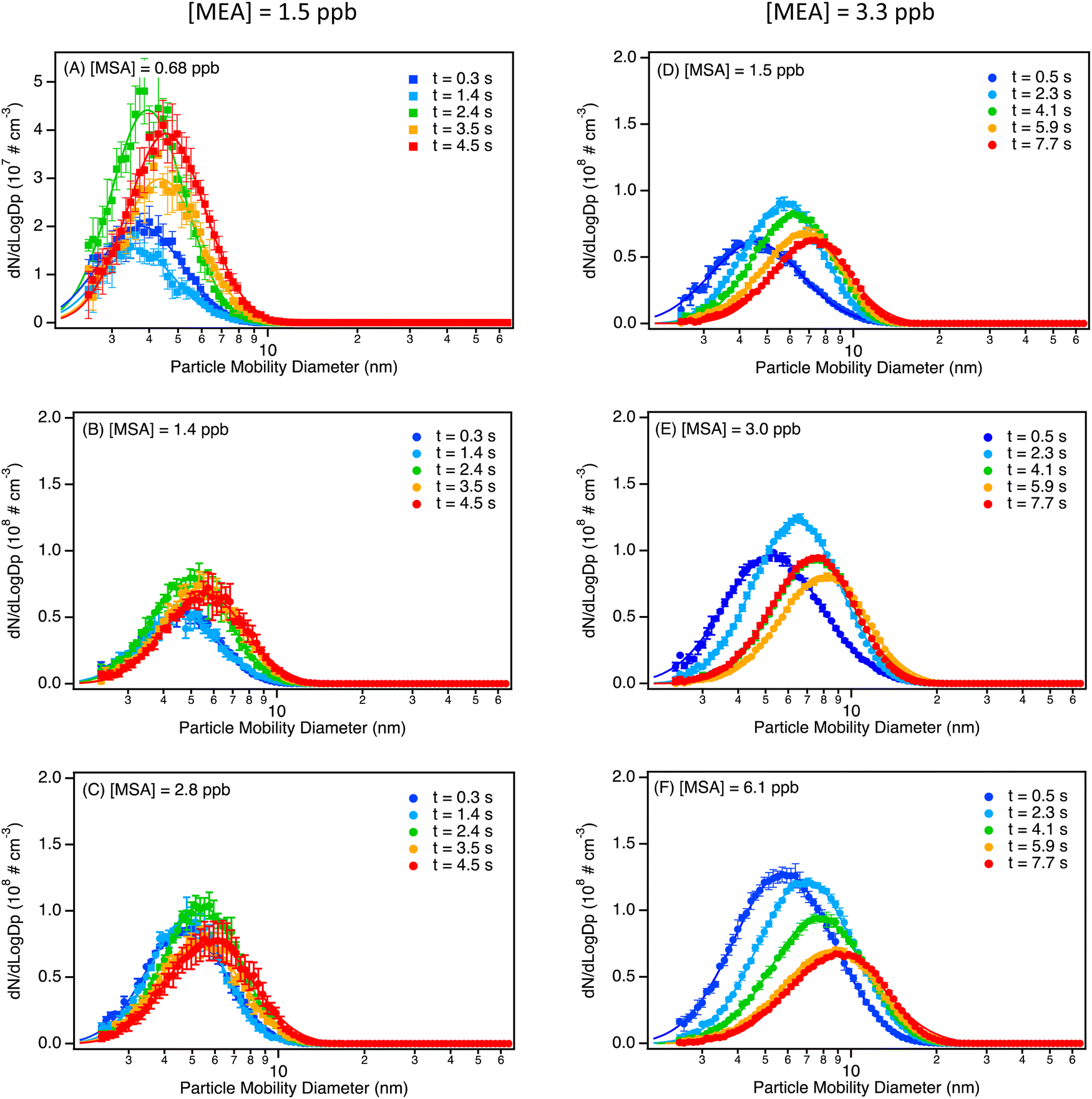 | ||
| Fig. 1 Size distributions of particles from MEA (1.5 ppb) reacting with (A) 0.68 ppb MSA, (B) 1.4 ppb MSA and (C) 2.8 ppb MSA, and size distribution from MEA (3.3 ppb) reacting with (D) 1.5 ppb MSA, (E) 3.0 ppb MSA and (F) 6.1 ppb MSA. Measurements were conducted at the same sampling ports distributed equally along the length of the flow tube, but experiments displayed in panel (A)–(C) were performed with a total flow rate of 23.4 L min−1 (resulting in reaction times between 0.3 and 4.5 s), while experiments displayed in panel (D)–(F) were performed with a total flow rate of 10.7 L min−1 (resulting in reaction times between 0.5 and 7.7s). All experiments were performed under dry conditions, and size distributions are the average of 3 to 8 replicates (error bars correspond to one standard deviation) for each reaction time. All size distributions were corrected for particle losses through the sampling lines. Total particle concentrations and geometric mean diameters as a function of reaction times are given in Fig. S12 (ESI†). | ||
Similar behavior is seen as the initial MSA concentration is increased, but with larger total particle number concentrations formed (Fig. 1(B), (C) and Fig. S12A, B, ESI†). In this case, at longer reaction times the particle number concentrations start to decrease and the GMD increases due to coagulation (Fig. 1(C)). Similar, but more pronounced, trends are seen at an initial MEA concentration of 3.3 ppb and increasing MSA concentrations (Fig. 1(D)–(F)). For approximately the same MSA concentration (Fig. 1(B), (D) and Fig. 1(C), (E)), doubling the concentration of MEA leads to an increase in total number concentration of a factor of ∼1.2–1.4 at 2.3–2.4 s reaction time, with an increase in diameter from 4.9 to 5.6 nm (MSA = 1.4–1.5 ppb) and from 5.3 to 6.3 nm (MSA = 2.8–3.0 ppb).
For the low concentration series, the formation of approximately half of the peak particle concentration at the first measurement time implies that the rate-determining step is fast. There is some uncertainty in the exact reaction time for this first data point since it does not take into account possible continued reaction in the sampling lines. However, a half-life of ∼0.5 s for the reaction of MSA with excess MEA at 1.5 ppb (Fig. 1(A)), is consistent with a gas phase bimolecular reaction rate constant for MEA with MSA of approximately 4 × 10−11 cm3 molecules−1 s−1.
The particle formation rate (J>2.5nm) was estimated using the total concentration of particles measured at ∼2.4 s (peak concentration) for all conditions, and dividing by the reaction time in seconds. Fig. 2 shows the resulting J>2.5nm values as a function of the product of the MEA and MSA initial concentrations. There is an initial rapid increase which is approximately linear out to [MEA] × [MSA] ∼ 2 ppb2, suggesting that the initial 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cluster formation is the rate-determining step. The drop-off at higher concentrations reflects coagulation. This is consistent with the TDCIMS measurements (Fig. 3), which show that the acid/base molar ratios in the particles from 4–12 nm remains within experimental error of one. All measurements were performed at 4.5 s reaction time and at an initial concentration of [MEA] of 1.5 ppb. There were no significant differences in the measured molar ratio across these experiments performed with MSA concentrations ranging from 0.68 to 2.8 ppb and the data obtained for all MSA concentrations was averaged together.
:1 cluster formation is the rate-determining step. The drop-off at higher concentrations reflects coagulation. This is consistent with the TDCIMS measurements (Fig. 3), which show that the acid/base molar ratios in the particles from 4–12 nm remains within experimental error of one. All measurements were performed at 4.5 s reaction time and at an initial concentration of [MEA] of 1.5 ppb. There were no significant differences in the measured molar ratio across these experiments performed with MSA concentrations ranging from 0.68 to 2.8 ppb and the data obtained for all MSA concentrations was averaged together.
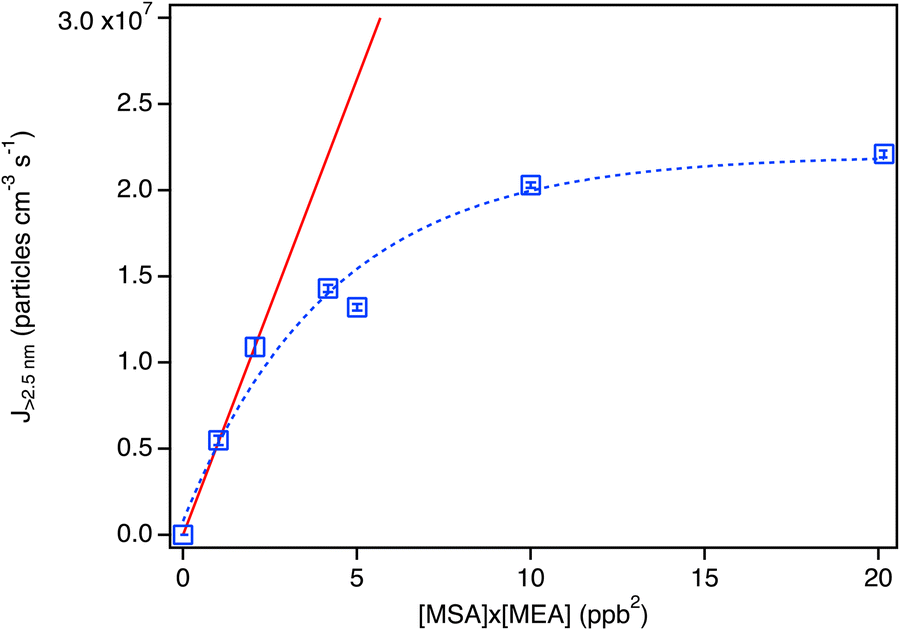 | ||
| Fig. 2 Particle formation rate (J>2.5nm) for the MSA + MEA system under dry conditions as a function of the product of the MSA and MEA mixing ratios in ppb. Each data point represents an average over 3 to 8 individual SMPS scans taken at 2.3–2.4 s reaction time, with error bars representing one standard deviation, and corrected for particle losses through the sampling lines. The red line is a linear fit to the data ([MSA] × [MEA] ≤ 2 ppb2) with a slope of (5.3 ± 0.03) × 106 particles cm−3 ppb2. | ||
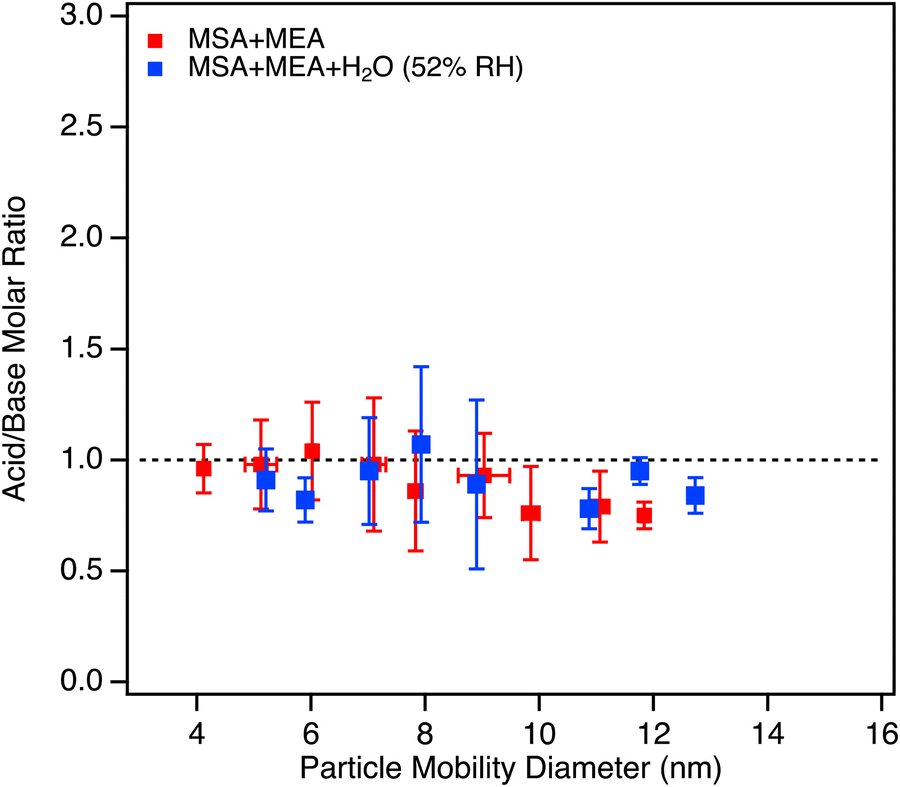 | ||
| Fig. 3 Acid/base molar ratios measured by the TDCIMS for the MSA + MEA system, under dry conditions (red data points) and at 52% RH (blue data points) collected at 4.5 s reaction time. MEA was measured in POS ion mode while MSA was measured in NEG ion mode, and the ion distributions in the MS spectra were similar at all reactant concentrations (Fig. S6, ESI†). Text S5 provides more detailed information on how the acid/base molar ratios were estimated. All measurements were performed with initial MEA concentration of 1.5 ppb. No significant difference was observed in the measured molar ratio across for experiments performed with [MSA] = 0.68 ppb, [MSA] = 1.4 ppb or [MSA] = 2.8 ppb, in either dry or humid conditions; thus the data points represent average values across the [MSA] concentrations range for each RH condition. For each data point, the error bars represent one standard deviation. The dashed line corresponds to an acid/base molar ratio of unity for reference. | ||
MSA concentrations in air can be as high as 107 molecules cm−3 (∼0.4 ppt)60,61,66,68 and MEA in the low ppb range has been recorded outside a CCS facility.7 The slope of the line in Fig. 2 at the lowest reactant concentrations is (5.3 ± 0.03) × 106 particles cm−3 ppb−2 so a NPF rate from the upper limit atmospheric concentrations of MEA (10 ppb) and MSA (0.4 ppt) of as much as ∼21200 particles cm−3 is predicted. This can be compared to a range of formation rates of particles >3 nm diameter (J3) from sulfuric acid of 0.001–105 cm−3 observed in different environments around the world.155 In short, even for conditions where MEA and MSA concentrations are less than the reported maxima, this single reaction system may contribute significantly to NPF and its importance may increase as MEA use in CCS increases, and MSA increases due to a warming climate.
Efficient particle formation from MEA and MSA is consistent with the excellent stability at room temperature and low vapor pressure of the MSA–MEA salt synthesized by Greaves and co-workers.156 Furthermore, MEA–MSA has been reported to have properties of a protic ionic liquid, even though it remains a solid at room temperature.156–159 Its properties include a glass transition of −44 °C, melting point of about 100 °C, and a thermal stability up to 286–323 °C for the fused salt.156
Tropospheric air contains significant amounts of water vapor, hence the impact of relative humidity (RH) on particle formation from MEA + MSA was also examined. Surprisingly, and in contrast to previous results obtained for small alkylamines, the addition of water vapor to the MSA + MEA system did not significantly increase the number concentration at RH below ∼20% as indicated in Fig. 4. Fig. 5(A) and (B) shows the evolution of the particle size distributions as a function of time at an RH of ∼50% for two different sets of precursor concentrations corresponding to the dry conditions presented in Fig. 1(A) and (C) respectively. The evolution of the size distributions as a function of time in the flow reactor is similar to that observed under dry conditions. To better compare the dry versus humid case, total particle concentrations and geometric mean diameters measured at 4.5 s over several repeated experiments were averaged and are shown in Fig. S13 (ESI†). The addition of water vapor increased the total number concentration by only a factor of 1.3–1.5 as indicated by the bars. Note that the enhancement factor (EF) measured at 4.5 s for the high MSA, high RH case is an underestimate as it already includes coagulation (Fig. 5(C)). At the peak particle concentration (t = 1.4 s reaction time), EF = 1.9. There is only a small increase in size (red squares) at the highest MSA concentrations.
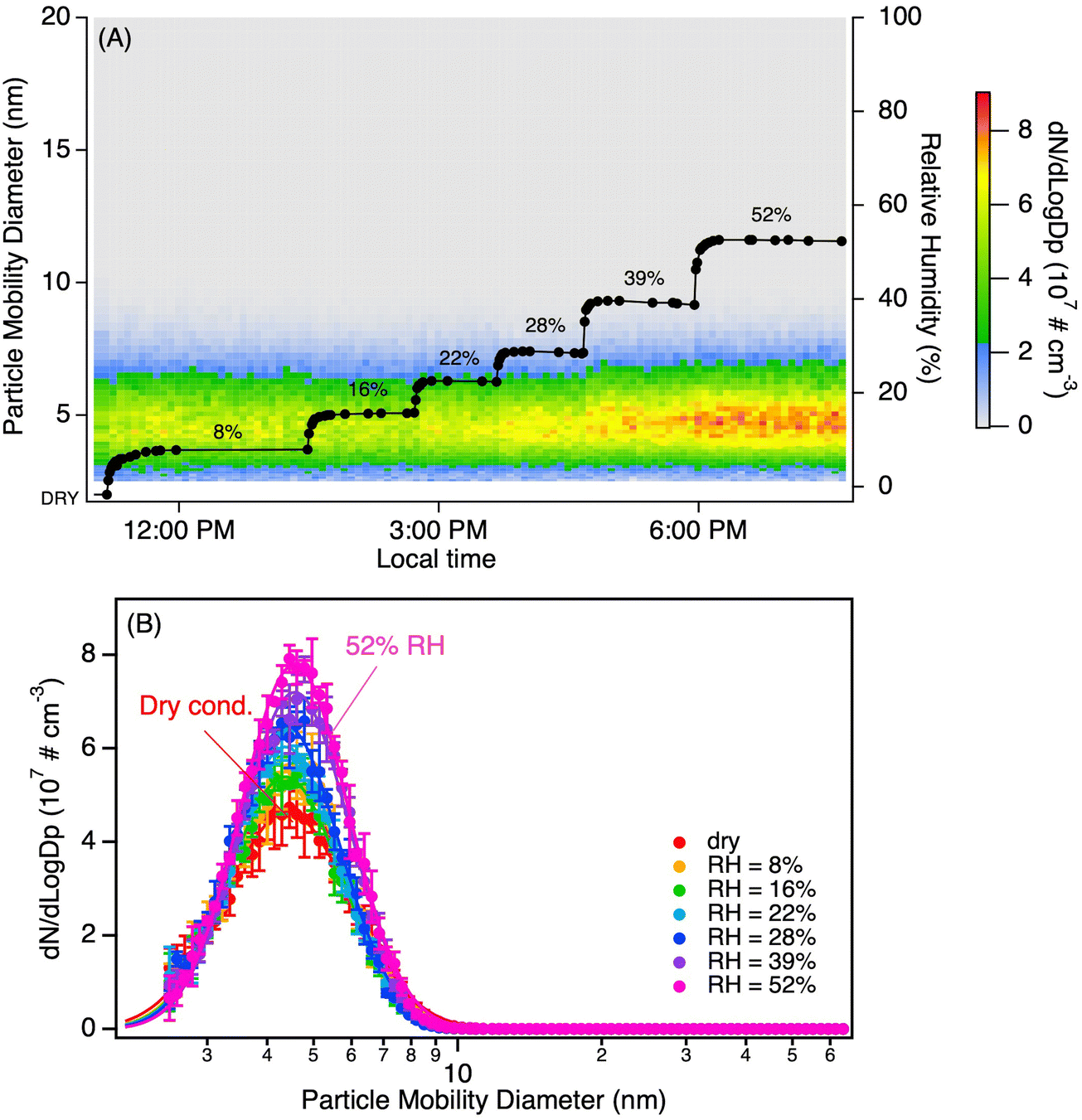 | ||
| Fig. 4 Evolution of the particle size distributions as a function of relative humidity (RH) from the reaction of MSA (0.7 ppb) with MEA (1.4 ppb). Panel (A) represents the evolution as a function of time while panel (B) represents a snapshot of the size distributions at a given RH (each distribution is an average over three SMPS scans with the error bars representing one standard variation). All measurements were performed at 4.5 s reaction time, and particle size distributions were corrected for particle loss through the sampling lines. | ||
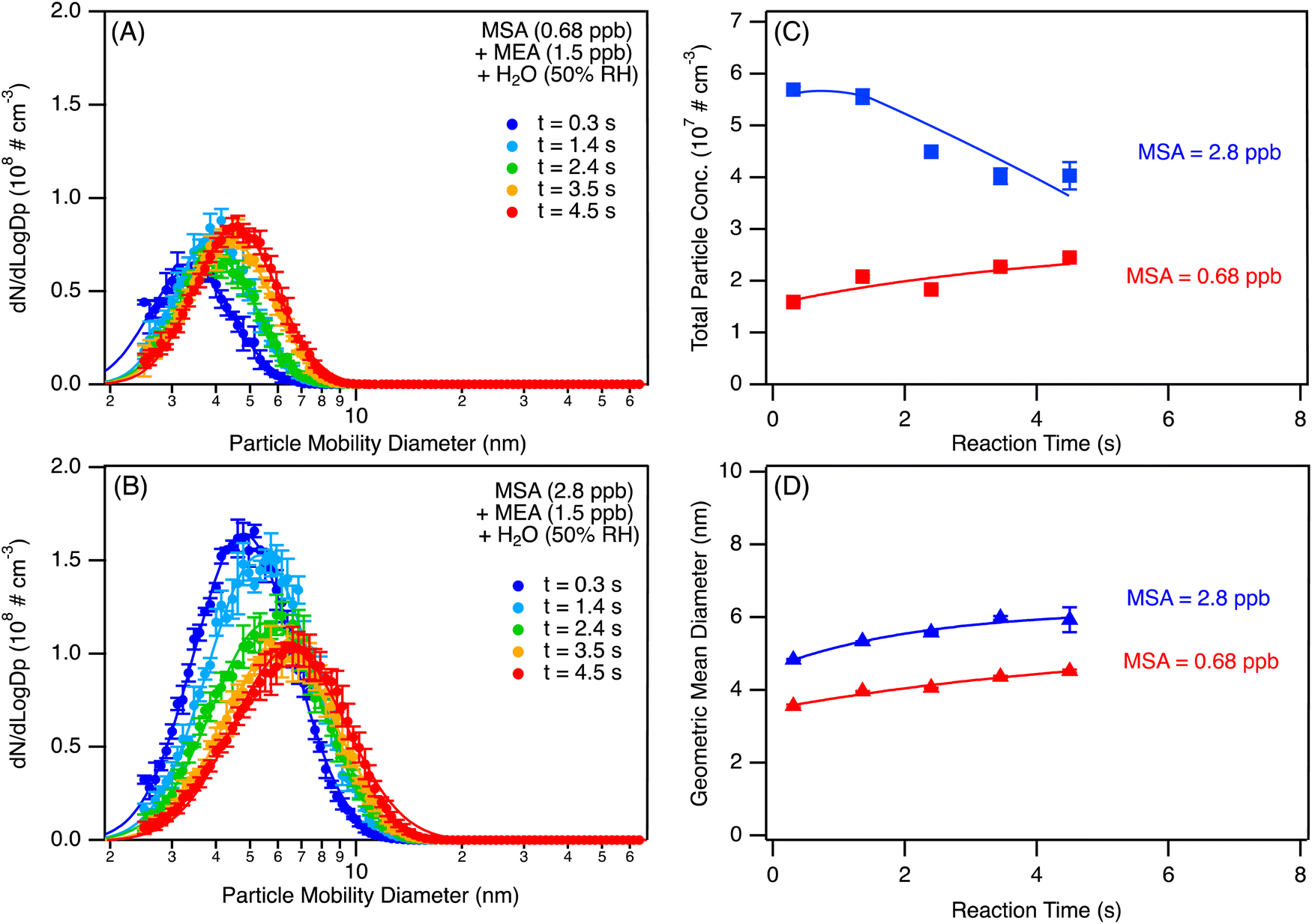 | ||
| Fig. 5 Size distributions (A) and (B) and evolution of the particle total concentrations and geometric mean diameters measured as a function of the reaction time (C) and (D) in the flow reactor for the MSA + MEA reaction system at 50% RH. All lines in panels C and D are guides to the eye. All data originate from replicate scans (n = 5) and are displayed with one standard deviation. All size distributions were corrected for particle losses through the sampling lines. | ||
Classical nucleation theory predicts that the number of water molecules in the critical cluster can, under some conditions, be obtained from the slope of a log–log plot of the formation rate of new particles versus the gas phase water concentration.160 However, this is highly dependent on a number of assumptions.161 As seen in Fig. 6, there is no significant correlation with H2O concentration. This could indicate that water is not a central ingredient in the critical cluster formed from MEA and MSA. Alternatively, it could be due to the absence of an energy barrier in the reaction so the slope simply reflects a lack of particle formation rate on the water concentration.161 Furthermore, no change in the acid/base molar ratio was observed in the TDCIMS measurements in the presence of water compared to the dry case (Fig. 3; blue data points) indicating that the particles remained neutral. This lack of dependence on water is in contrast to previous results obtained for the small alkylamines,102 where a slope of 1.3–2.3 in the log–log plot was observed.
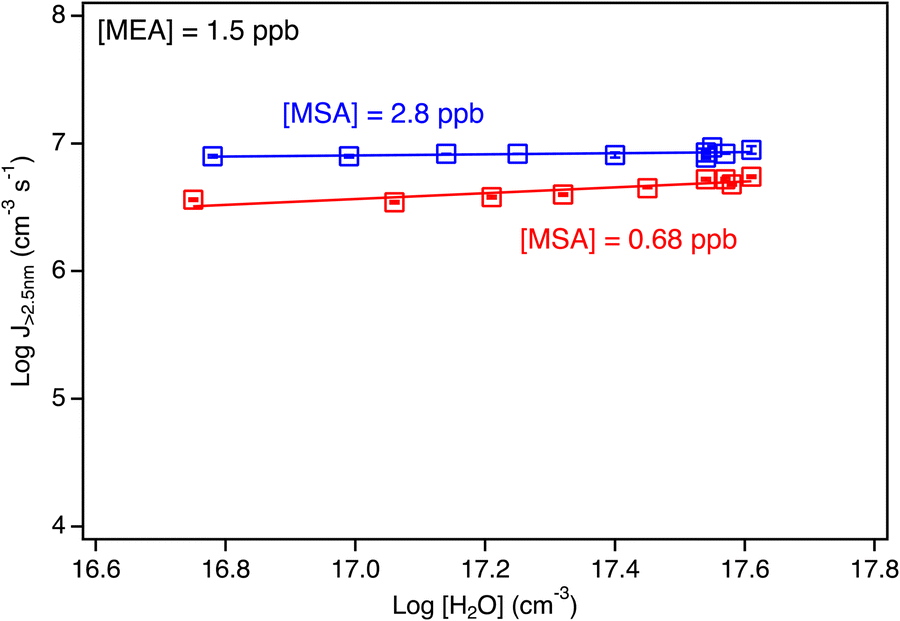 | ||
| Fig. 6 Logarithm of the particle formation rate (J>2.5 nm) for the MSA + MEA system under humid conditions (RH ranging from 8 to 56%) as a function of the log of the water concentration (in molecules cm−3). Each data point represents an average over 3 to 6 individual SMPS scans taken at 4.5 s reaction time, with error bars representing one standard deviation. All data points were corrected for particle loss through the sampling line. Red data are for [MSA] = 0.68 ppb and [MEA] = 1.5 ppb while the blue data are for [MSA] = 2.8 ppb and [MEA] = 1.5 ppb. The slopes of the lines are 0.04 for 2.8 ppb MSA and 0.2 for 0.68 ppb MSA. | ||
Shen et al.108 carried out computational studies of cluster formation from MEA and MSA, They showed that the 1:1 cluster was the least stable and hence formation of this cluster is the rate-determining step. This is consistent with the measured rates of particle formation depending on the product of the MEA and MSA concentrations and the 1:1 acid/base ratio of the particles. They demonstrated that the binding of MEA and MSA was determined by a combination of proton transfer from the acid MSA to the nitrogen of the MEA base, along with hydrogen bonding. MEA differs from simple amines in that it has both the –NH2 group as well as the –OH group, providing more than one hydrogen-bonding opportunity to MSA. Indeed, in all acid–base clusters, MSA acted as a H-bond donor and in many of the clusters, the –OH group of MEA acted as a H-bond donor to MSA. This results in strongly bound clusters held together by both electrostatic forces and a network of H-bonds, as illustrated in Fig. S14 (ESI†). It is interesting that the resulting structures have the –CH3 group of MSA on the edge of the cluster, making the cluster somewhat hydrophobic. The hydrate distribution reported by Shen et al.108 predicted that each cluster was predominantly hydrated by only one water molecule even at relatively high RH (80%). They also predicted that if water is present during cluster formation, it will enhance particle formation by about an order of magnitude at 50% RH due to a decrease in the evaporation rate of the initially formed 1:1 cluster. This predicted increase is significantly greater than the factor of 1.5–1.6 measured in these experiments.
In previous studies of NPF from MSA and amines, methylamine (MA) was shown to be the most efficient of the simple alkylamines in forming particles.102,103,105,106Fig. 7 compares the size distributions of particles formed from the reactions of 1.4 ppb MSA with 1.5 ppb MEA or 4.8 ppb MA under dry conditions. Even with three times the amine concentration, the total concentration of particles formed from MA is 17 times smaller than from MEA. This is consistent with previously reported theoretical calculations108,114,162 which predict a greater stability of the clusters with the increased H-bonding capability of MEA and, as a consequence, particle formation rates that are orders of magnitude higher for MSA + MEA compared to that for MSA + MA at similar concentrations. The gas phase basicity151 of MEA (896.8 kJ mol−1) compared to MA (864.5 kJ mol−1) also favors particle formation from MEA, along with the increased H-bonding opportunities.
 | ||
| Fig. 7 Representative averaged size distribution (red trace) from the reaction of MSA (1.4 ppb) with MEA (1.5 ppb). For comparison, a size distribution for MSA (1.4 ppb) reacting with MA (4.8 ppb) is also shown (green trace). Both size distributions were taken at ∼4 s reaction time. Each size distribution was averaged over five consecutive scans and the shaded area corresponds to one standard deviation uncertainty. The thick line corresponds to a log normal fit to the averaged data. Both size distributions have been corrected for particle losses through the sampling lines. | ||
In previous experimental studies, water had a dramatic effect on NPF from MSA reacting with small alkylamines,102,104–106,163 quite different from MEA. In the case of MA, the presence of water during particle formation led to a large increase in both number concentration and size starting at RH < 10% (Fig. S15, ESI†). Calculations indicated that the 4MSA-4MEA cluster with one water molecule, for example, resulted in a structure that had many potential hydrogen bonding sites available, allowing the cluster to grow via H-bonding with other species.163 For MEA, however, the clusters already have strongly hydrogen-bonded internal networks so that opportunities for further interactions with water molecules are reduced.
To compare the relative importance of the MEA and MA reactions with MSA for particle formation under atmospheric conditions, measurements under 10–50% RH were carried out with MA (Fig. S15, ESI†). Fig. 8 shows the NPF rate (J>2.5nm) for MEA compared to that of MA as a function of the product of the reactant concentrations. The slope of the linear fit through the data for the MEA reaction is more than four times that of the MA reaction. Thus, although NPF from MA + MSA is greatly enhanced in the presence of water, the MEA reaction is still more efficient under similar conditions. This highlights the significance of alkanolamines in NPF at low concentrations and points to H-bonding as a driver for NPF with MSA.
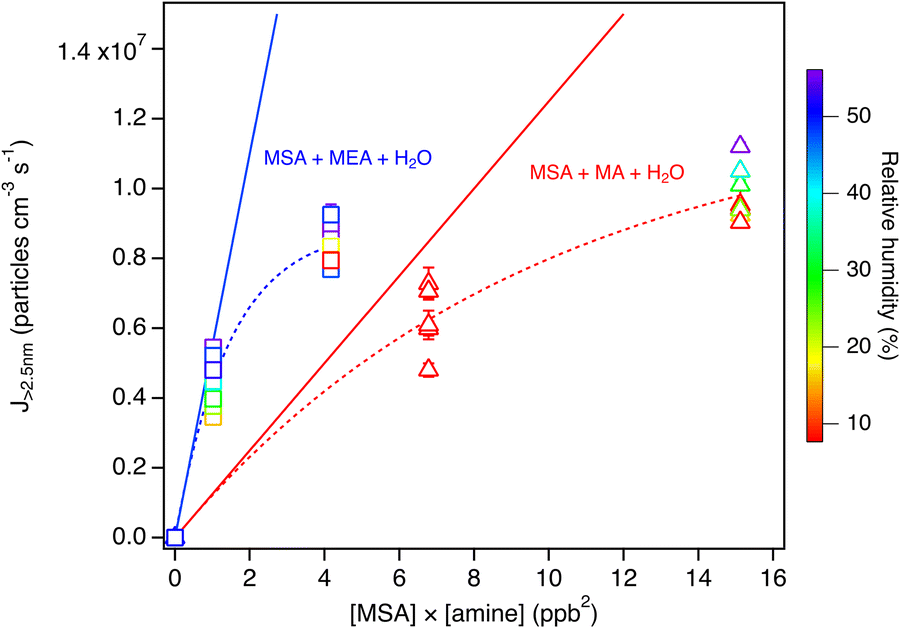 | ||
| Fig. 8 Comparison between particle formation rate (J>2.5 nm) for the MSA + MEA and the MSA + MA systems under humid conditions (RH ranging from 8 to 56%) as a function of the product of the MSA and the amine mixing ratios in ppb. Each data point represents an average over 3 to 6 individual SMPS scans taken at 4.5 s reaction time, with error bars representing one standard deviations. The dashed lines are exponential fits to the data and the solid lines are the tangent to the fits with slopes of 5.5 × 106 and 1.2 × 106 for the MSA + MEA and MSA + MA systems respectively. All data points were corrected for particle loss through the sampling lines. | ||
Conclusions
This study shows that the acid/base interaction of MSA with a short alkanolamine widely used in CCS is quite strong and produces sub-10 nm particles extremely well compared to a simple primary alkylamine, methylamine. Water vapor has a limited impact on NPF rates as MEA has OH− groups that already promote strong H-bonding network within the clusters. This is in contrast with previous work on alkylamines where water had a large impact on nucleation and growth of new particles. The particle composition from 4–12 nm showed an acid/base molar ratio close to unity, whereas those from the MA reaction contained more acid at the smaller diameters. These findings highlight that there is not a one-size-fits-all when it comes to treating amine interactions with MSA in atmospheric models.The overall contribution of MSA-initiated aerosol chemistry may become increasingly more important in the future.164 For example, there is a reduction of sea-ice coverage at the poles, leading to an increase in DMS emissions94,96–98 with an associated increase in MSA. At the same time, there has been a decline in anthropogenic SO2 emissions over few the past decades,165–170 with a related reduction in particulate sulfate in ambient particles in the Northern part of the globe.167,168,171,172 Thus, MSA acid–base mediated NPF will become increasingly more important in air in the near future.
Lastly, alkanolamines are being widely deployed in CCS technology which may lead to an increase in their abundance in the atmosphere. Thus, assessing and understanding the impacts of this acid–base driven chemistry on new particle formation in air is more important than ever.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Science Foundation (grants no. 1928252 and CHE-2004066) and the Army Research Office (grant no. W911NF2010064) for supporting this research. K. R. acknowledges support from the Lucille Foundation and the Chemistry Summer Undergraduate Research Fellowship (Chem-SURF). The authors thank Dr Paulus Bauer for helpful discussions during the preparation of this manuscript. The authors are also grateful to Dr Natalia Karimova and Dr R. Benny Gerber for their assistance in the DFT calculations and access to the Green-Planet Cluster at the University of California, Irvine.References
- G. T. Rochelle, Amine Scrubbing for CO2 Capture, Science, 2009, 325, 1652–1654 CrossRef CAS.
- C. H. Yu, C. H. Huang and C. S. Tan, A review of CO2 capture by absorption and adsorption, Aerosol Air Qual. Res., 2012, 12, 745–769 CrossRef CAS.
- S. A. Mazari, B. S. Ali, B. M. Jan, I. M. Saeed and S. Nizamuddin, An overview of solvent management and emissions of amine-based CO2 capture technology, Int. J. Greenhouse Gas Control, 2015, 34, 129–140 CrossRef CAS.
- L. Cuccia, J. Dugay, D. Bontemps, M. Louis-Louisy and J. Vial, Analytical methods for the monitoring of post-combustion CO2 capture process using amine solvents: A review, Int. J. Greenhouse Gas Control, 2018, 72, 138–151 CrossRef CAS.
- M. Corsten, A. Ramirez, L. Shen, J. Koornneef and A. Faaij, Environmental impact assessment of CCS chains – Lessons learned and limitations from LCA literature, Int. J. Greenhouse Gas Control, 2013, 13, 59–71 CrossRef CAS.
- M. Akram, K. Milkowski, J. Gibbins and M. Pourkashanian, Comparative energy and environmental performance of 40% and 30% monoethanolamine at PACT pilot plant, Int. J. Greenhouse Gas Control, 2020, 95, 102946, DOI:10.1016/j.ijggc.2019.102946.
- L. Zhu, G. W. Schade and C. J. Nielsen, Real-time monitoring of emissions from monoethanolamine-based industrial scale carbon capture facilities, Environ. Sci. Technol., 2013, 47, 14306–14314 CrossRef CAS.
- J. Mertens, J. Knudsen, M. L. Thielens and J. Andersen, On-line monitoring and controlling emissions in amine post combustion carbon capture: A field test, Int. J. Greenhouse Gas Control, 2012, 6, 2–11 CrossRef CAS.
- J. Mertens, H. Lepaumier, D. Desagher and M. L. Thielens, Understanding ethanolamine (MEA) and ammonia emissions from amine based post combustion carbon capture: Lessons learned from field tests, Int. J. Greenhouse Gas Control, 2013, 13, 72–77 CrossRef CAS.
- E. F. da Silva, H. Kolderup, E. Goetheer, K. W. Hjarbo, A. Huizinga, P. Khakharia, I. Tuinman, T. Mejdell, K. Zahlsen, K. Vernstad, A. Hyldbakk, T. Holten, H. M. Kvamsdal, P. van Os and A. Einbu, Emission studies from a CO2 capture pilot plant, Energy Procedia, 2013, 37, 778–783 CrossRef.
- J. Fagerlund, R. Zevenhoven, J. Thomassen, M. Tednes, F. Abdollahi, L. Thomas, C. J. Nielsen, T. Mikoviny, A. Wisthaler, L. Zhu, C. Biliyok and A. Zhurkin, Performance of an amine-based CO2 capture pilot plant at the Fortum Oslo Varme Waste to Energy plant in Oslo, Norway, Int. J. Greenhouse Gas Control, 2021, 106, 103242 CrossRef CAS.
- M. X. Fang, N. T. Yi, W. T. Di, T. Wang and Q. H. Wang, Emission and control of flue gas pollutants in CO2 chemical absorption system – A review, Int. J. Greenhouse Gas Control, 2020, 93, 102904 CrossRef CAS.
- A. K. Morken, B. Nenseter, S. Pedersen, M. Chhaganlal, J. K. Feste, R. B. Tyborgnes, O. Ullestad, H. Ulvatn, L. Zhu, T. Mikoviny, A. Wisthaler, T. Cents, O. M. Bade, J. Knudsen, G. de Koeijer, O. Falk-Pedersen and E. S. Hamborg, Emission results of amine plant operations from MEA testing at the CO2 Technology Centre Mongstad, Energy Procedia, 2014, 63, 6023–6038 CrossRef CAS.
- K. Veltman, B. Singh and E. G. Hertwich, Human and environmental impact assessment of postcombustion CO2 capture focusing on emissions from amine-based scrubbing solvents to air, Environ. Sci. Technol., 2010, 44, 1496–1502 CrossRef CAS.
- M. M. Fiume, B. A. Heldreth, W. F. Bergfeld, D. V. Belsito, R. A. Hill, C. D. Klaassen, D. C. Liebler, J. G. Marks, R. C. Shank, T. J. Slaga, P. W. Snyder and F. A. Andersen, Safety assessment of ethanolamine and ethanolamine salts as used in cosmetics, Int. J. Toxicol., 2015, 34, 84s–98s CrossRef CAS.
- B. C. McDonald, J. A. de Gouw, J. B. Gilman, S. H. Jathar, A. Akherati, C. D. Cappa, J. L. Jimenez, J. Lee-Taylor, P. L. Hayes, S. A. McKeen, Y. Y. Cui, S. W. Kim, D. R. Gentner, G. Isaacman-VanWertz, A. H. Goldstein, R. A. Harley, G. J. Frost, J. M. Roberts, T. B. Ryerson and M. Trainer, Volatile chemical products emerging as largest petrochemical source of urban organic emissions, Science, 2018, 359, 760–764 CrossRef CAS.
- J. Wooley, W. W. Nazaroff and A. T. Hodgson, Release of ethanol to the atmosphere during use of consumer cleaning products, J. Air Waste Manage. Assoc., 1990, 40, 1114–1120 CrossRef CAS.
- X. Ge, A. S. Wexler and S. L. Clegg, Atmospheric amines - Part I. A review, Atmos. Environ., 2011, 45, 524–546 CrossRef CAS.
- J.-A. Seo, I.-H. Bae, W.-H. Jang, J.-H. Kim, S.-Y. Bak, S.-H. Han, Y.-H. Park and K.-M. Lim, Hydrogen peroxide and monoethanolamine are the key causative ingredients for hair dye-related dermatitis and hair loss, J. Dermatol. Sci., 2012, 66, 12–19 CrossRef CAS.
- N. Borduas, J. P. D. Abbatt and J. G. Murphy, Gas phase oxidation of monoethanolamine (MEA) with OH radical and ozone: kinetics, products, and particles, Environ. Sci. Technol., 2013, 47, 6377–6383 CrossRef CAS PubMed.
- M. Karl, C. Dye, N. Schmidbauer, A. Wisthaler, T. Mikoviny, B. D'Anna, M. Muller, E. Borras, E. Clemente, A. Munoz, R. Porras, M. Rodenas, M. Vazquez and T. Brauers, Study of OH-initiated degradation of 2-aminoethanol, Atmos. Chem. Phys., 2012, 12, 1881–1901 CrossRef CAS.
- L. Onel, M. A. Blitz and P. W. Seakins, Direct determination of the rate coefficient for the reaction of OH radicals with monoethanol amine (MEA) from 296 to 510 K, J. Phys. Chem. Lett., 2012, 3, 853–856 CrossRef CAS.
- C. J. Nielsen, B. D’Anna, C. Dye, M. Graus, M. Karl, S. King, M. M. Maguto, M. Muller, N. Schmidbauer, Y. Stenstrom, A. Wisthaler and S. Pedersen, Atmospheric chemistry of 2-aminoethanol (MEA), Energy Procedia, 2011, 4, 2245–2252 CrossRef CAS.
- S. M. Murphy, A. Sorooshian, J. H. Kroll, N. L. Ng, P. Chhabra, C. Tong, J. D. Surratt, E. Knipping, R. C. Flagan and J. H. Seinfeld, Secondary aerosol formation from atmospheric reactions of aliphatic amines, Atmos. Chem. Phys., 2007, 7, 2313–2337 CrossRef CAS.
- X. M. Tian, Y. X. Chu and C. K. Chan, Reactive uptake of monoethanolamine by sulfuric acid particles and hygroscopicity of monoethanolaminium salts, Environ. Sci. Technol. Lett., 2022, 9, 16–21 CrossRef CAS.
- H. B. Xie, J. Elm, R. Halonen, N. Myllys, T. Kurten, M. Kulmala and H. Vehkamaki, Atmospheric fate of monoethanolamine: enhancing new particle formation of sulfuric acid as an important removal process, Environ. Sci. Technol., 2017, 51, 8422–8431 CrossRef CAS PubMed.
- B. Rosati, S. Christiansen, R. W. de Jonge, P. Roldin, M. M. Jensen, K. Wang, S. P. Moosakutty, D. Thomsen, C. Salomonsen, N. Hyttinen, J. Elm, A. Feilberg, M. Glasius and M. Bilde, New particle formation and growth from dimethyl sulfide oxidation by hydroxyl radicals, ACS Earth Space Chem., 2021, 5, 801–811 CrossRef CAS PubMed.
- P. Van Rooy, R. Drover, T. Cress, C. Michael, K. L. Purvis-Roberts, P. J. Silva, M. J. Nee and D. Cocker, Methanesulfonic acid and sulfuric acid aerosol formed through oxidation of reduced sulfur compounds in a humid environment, Atmos. Environ., 2021, 261, 118504 CrossRef CAS.
- E. H. Hoffmann, A. Tilgner, R. Schrodner, P. Brauera, R. Wolke and H. Herrmann, An advanced modeling study on the impacts and atmospheric implications of multiphase dimethyl sulfide chemistry, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11776–11781 CrossRef CAS.
- I. Barnes, J. Hjorth and N. Mihalopoulos, Dimethyl sulfide and dimethyl sulfoxide and their oxidation in the atmosphere, Chem. Rev., 2006, 106, 940–975 CrossRef CAS PubMed.
- R. Wollesen de Jonge, J. Elm, B. Rosati, S. Christiansen, N. Hyttinen, D. Ludemann, M. Bilde and P. Roldin, Secondary aerosol formation from dimethyl sulfide - improved mechanistic understanding based on smog chamber experiments and modelling, Atmos. Chem. Phys., 2021, 21, 9955–9976 CrossRef CAS.
- P. R. Veres, J. A. Neuman, T. H. Bertram, E. Assaf, G. M. Wolfe, C. J. Williamson, B. Weinzierl, S. Tilmes, C. R. Thompson, A. B. Thames, J. C. Schroder, A. Saiz-Lopez, A. W. Rollins, J. M. Roberts, D. Price, J. Peischl, B. A. Nault, K. H. Moller, D. O. Miller, S. Meinardi, Q. Y. Li, J. F. Lamarque, A. Kupc, H. G. Kjaergaard, D. Kinnison, J. L. Jimenez, C. M. Jernigan, R. S. Hornbrook, A. Hills, M. Dollner, D. A. Day, C. A. Cuevas, P. Campuzano-Jost, J. Burkholder, T. P. Bui, W. H. Brune, S. S. Brown, C. A. Brock, I. Bourgeois, D. R. Blake, E. C. Apel and T. B. Ryerson, Global airborne sampling reveals a previously unobserved dimethyl sulfide oxidation mechanism in the marine atmosphere, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4505–4510 CrossRef CAS PubMed.
- S. F. Watts, The mass budgets of carbonyl sulfide, dimethyl sulfide, carbon disulfide and hydrogen sulfide, Atmos. Environ., 2000, 34, 761–779 CrossRef CAS.
- R. J. Charlson, J. E. Lovelock, M. O. Andreae and S. G. Warren, Oceanic phytoplankton, atmospheric sulfur, cloud albedo and climate, Nature, 1987, 326, 655–661 CrossRef CAS.
- T. S. Bates, B. K. Lamb, A. Guenther, J. Dignon and R. E. Stoiber, Sulfur emissions to the atmosphere from natural sources, J. Atmos. Chem., 1992, 14, 315–337 CrossRef CAS.
- A. Lana, T. G. Bell, R. Simo, S. M. Vallina, J. Ballabrera-Poy, A. J. Kettle, J. Dachs, L. Bopp, E. S. Saltzman, J. Stefels, J. E. Johnson and P. S. Liss, An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean, Global Biogeochem. Cycles, 2011, 25 CrossRef CAS , GB1004.
- M. O. Andreae and P. J. Crutzen, Atmospheric aerosols: Biogeochemical sources and role in atmospheric chemistry, Science, 1997, 276, 1052–1058 CrossRef CAS.
- V. P. Aneja, Natural sulfur emissions into the atmosphere, J. Air Waste Manage. Assoc., 1990, 40, 469–476 CrossRef.
- K. Jardine, A. M. Yanez-Serrano, J. Williams, N. Kunert, A. Jardine, T. Taylor, L. Abrell, P. Artaxo, A. Guenther, C. N. Hewitt, E. House, A. P. Florentino, A. Manzi, N. Higuchi, J. Kesselmeier, T. Behrendt, P. R. Veres, B. Derstroff, J. D. Fuentes, S. T. Martin and M. O. Andreae, Dimethyl sulfide in the Amazon rain forest, Global Biogeochem. Cycles, 2015, 29, 19–32 CrossRef CAS.
- P. J. Crutzen, J. Williams, U. Poschl, P. Hoor, H. Fischer, C. Warneke, R. Holzinger, A. Hansel, W. Lindinger, B. Scheeren and J. Lelieveld, High spatial and temporal resolution measurements of primary organics and their oxidation products over the tropical forests of Surinam, Atmos. Environ., 2000, 34, 1161–1165 CrossRef CAS.
- J. Kesselmeier, F. X. Meixner, U. Hofmann, A. L. Ajavon, S. Leimbach and M. O. Andreae, Reduced sulfur compound exchange between the atmosphere and tropical tree species in Southern Cameroon, Biogeochemistry, 1993, 23, 23–45 CAS.
- S. Meinardi, I. J. Simpson, N. J. Blake, D. R. Blake and F. S. Rowland, Dimethyl disulfide (DMDS) and dimethyl sulfide (DMS) emissions from biomass burning in Australia, Geosphys. Res. Lett., 2003, 30, 1454 Search PubMed.
- C. E. Stockwell, P. R. Veres, J. Williams and R. J. Yokelson, Characterization of biomass burning emissions from cooking fires, peat, crop residue, and other fuels with high-resolution proton-transfer-reaction time-of-flight mass spectrometry, Atmos. Chem. Phys., 2015, 15, 845–865 CrossRef.
- V. Perraud, S. Meinardi, D. R. Blake and B. J. Finlayson-Pitts, Challenges associated with the sampling and analysis of organosulfur compounds in air using real-time PTR-ToF-MS and offline GC-FID, Atmos. Meas. Tech., 2016, 9, 1325–1340 CrossRef CAS.
- S. Trabue, K. Scoggin, F. Mitloehner, H. Li, R. Burns and H. Xin, Field sampling method for quantifying volatile sulfur compounds from animal feeding operations, Atmos. Environ., 2008, 42, 3332–3341 CrossRef CAS.
- P. Hobbs and T. Mottram, New Directions: Significant contributions of dimethyl sulphide from livestock to the atmosphere, Atmos. Environ., 2000, 34, 3649–3650 CrossRef CAS.
- J. S. Vandergheynst, D. J. Cogan, P. J. Defelice, J. M. Gossett and L. P. Walker, Effect of process management on the emission of organosulfur compounds and gaseous antecedents from composting processes, Environ. Sci. Technol., 1998, 32, 3713–3718 CrossRef CAS.
- Z. G. Yi, X. M. Wang, G. Y. Sheng and H. M. Fu, Exchange of carbonyl sulfide (OCS) and dimethyl sulfide (DMS) between rice paddy fields and the atmosphere in subtropical China, Agric., Ecosyst. Environ., 2008, 123, 116–124 CrossRef CAS.
- P. D. Goldan, W. C. Kuster, D. L. Albritton and F. C. Fehsenfeld, The measurement of natural sulfur emissions from soils and vegetation – 3 Sites in the Eastern-United-States revisited, J. Atmos. Chem., 1987, 5, 439–467 CrossRef CAS.
- J. Williams, N. Y. Wang, R. J. Cicerone, K. Yagi, M. Kurihara and F. Terada, Atmospheric methyl halides and dimethyl sulfide from cattle, Global Biogeochem. Cycles, 1999, 13, 485–491 CrossRef CAS.
- K. C. Li and D. Shooter, Analysis of sulfur-containing compounds in ambient air using solid-phase microextraction and gas chromatography with pulsed flame photometric detection, Int. J. Environ. Anal. Chem., 2004, 84, 749–760 CrossRef CAS.
- M. R. Ras, F. Borrull and R. M. Marce, Determination of volatile organic sulfur compounds in the air at sewage management areas by thermal desorption and gas chromatography-mass spectrometry, Talanta, 2008, 74, 562–569 CrossRef CAS PubMed.
- E. Smet and H. Van Langenhove, Abatement of volatile organic sulfur compounds in odorous emissions from the bio-industry, Biodegradation, 1998, 9, 273–284 CrossRef CAS PubMed.
- R. Raiswell and S. H. Bottrel, The disposal of flue gas desulphurisation waste: sulphur gas emissions and their control, Environ. Geochem. Health, 1991, 13, 119–126 CrossRef CAS.
- P. R. Mulay, P. Cavicchia, S. M. Watkins, A. Tovar-Aguilar, M. Wiese and G. M. Calvert, Acute illness associated with exposure to a new soil fumigant containing dimethyl disulfide – Hillsborough County, Florida, 2014, J. Agromedicine, 2016, 21, 373–379 CrossRef.
- D. D. Yan, A. C. Cao, Q. X. Wang, Y. Li, C. B. Ouyang, M. X. Guo and X. Q. Guo, Dimethyl disulfide (DMDS) as an effective soil fumigant against nematodes in China, PLoS One, 2019, 14, 1–9 Search PubMed.
- J. Fritsch, T. Fouillet, P. Charles, P. Fargier-Puech, C. Ramponi-Bur, S. Descamps, G. Du Fretay and A. Myrta, French experiences with dimethyl disulfide (DMDS) as a nematicide in vegetable crops, Acta Hortic., 2014, 1044, 427–433 CrossRef.
- F. L. Suarez, J. K. Furne, J. Springfield and M. D. Levitt, Morning breath odor: Influence of treatments on sulfur gases, J. Dent. Res., 2000, 79, 1773–1777 CrossRef CAS PubMed.
- S. Meinardi, K. B. Jin, B. Barletta, D. R. Blake and N. D. Vaziri, Exhaled breath and fecal volatile organic biomarkers of chronic kidney disease, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 2531–2537 CrossRef CAS PubMed.
- H. Berresheim, M. Adam, C. Monahan, C. O'Dowd, J. M. C. Plane, B. Bohn and F. Rohrer, Missing SO2 oxidant in the coastal atmosphere? - Observations from high-resolution measurements of OH and atmospheric sulfur compounds, Atmos. Chem. Phys., 2014, 14, 12209–12223 CrossRef.
- H. Berresheim, T. Elste, H. G. Tremmel, A. G. Allen, H. C. Hansson, K. Rosman, M. Dal Maso, J. M. Makela, M. Kulmala and C. D. O’Dowd, Gas-aerosol relationships of H2SO4, MSA, and OH: Observations in the coastal marine boundary layer at Mace Head, Ireland, J. Geophys. Res., 2002, 107, D19 CrossRef , 8100 (PAR 8105-8101 - PAR 8105-8112).
- F. L. Eisele and D. J. Tanner, Measurement of the gas-phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res., 1993, 98, 9001–9010 CrossRef CAS.
- A. Jefferson, D. J. Tanner, F. L. Eisele, D. D. Davis, G. Chen, J. Crawford, J. W. Huey, A. L. Torres and H. Berresheim, OH photochemistry and methane sulfonic acid formation in the coastal Antarctic boundary layer, J. Geophys. Res., 1998, 103, 1647–1656 CrossRef CAS.
- C. D. O'Dowd, K. Hameri, J. M. Makela, L. Pirjola, M. Kulmala, S. G. Jennings, H. Berresheim, H. C. Hansson, G. de Leeuw, G. J. Kunz, A. G. Allen, C. N. Hewitt, A. Jackson, Y. Viisanen and T. Hoffmann, A dedicated study of New Particle Formation and Fate in the Coastal Environment (PARFORCE): Overview of objectives and achievements, J. Geophys. Res., 2002, 107, 8108 Search PubMed.
- H. Bardouki, H. Berresheim, M. Vrekoussis, J. Sciare, G. Kouvarakis, K. Oikonomou, J. Schneider and N. Mihalopoulos, Gaseous (DMS, MSA, SO2, H2SO4 and DMSO) and particulate (sulfate and methanesulfonate) sulfur species over the northeastern coast of Crete, Atmos. Chem. Phys., 2003, 3, 1871–1886 CrossRef CAS.
- L. J. Beck, N. Sarnela, H. Junninen, C. J. M. Hoppe, O. Garmash, F. Bianchi, M. Riva, C. Rose, O. Perakyla, D. Wimmer, O. Kausiala, T. Jokinen, L. Ahonen, J. Mikkila, J. Hakala, X. C. He, J. Kontkanen, K. K. E. Wolf, D. Cappelletti, M. Mazzola, R. Traversi, C. Petroselli, A. P. Viola, V. Vitale, R. Lange, A. Massling, J. K. Nojgaard, R. Krejci, L. Karlsson, P. Zieger, S. Jang, K. Lee, V. Vakkari, J. Lampilahti, R. C. Thakur, K. Leino, J. Kangasluoma, E. M. Duplissy, E. Siivola, M. Marbouti, Y. J. Tham, A. Saiz-Lopez, T. Petaja, M. Ehn, D. R. Worsnop, H. Skov, M. Kulmala, V. M. Kerminen and M. Sipila, Differing mechanisms of new particle formation at two Arctic sites, Geosphys. Res. Lett., 2021, 48, e2020GL091334 CrossRef CAS.
- M. J. Lawler, M. P. Rissanen, M. Ehn, R. L. Mauldin, N. Sarnela, M. Sipilä and J. N. Smith, Evidence for diverse biogeochemical drivers of boreal forest new particle formation, Geosphys. Res. Lett., 2018, 45, 2038–2046 CrossRef.
- R. L. Mauldin, C. A. Cantrell, M. Zondlo, E. Kosciuch, F. L. Eisele, G. Chen, D. Davis, R. Weber, J. Crawford, D. Blake, A. Bandy and D. Thornton, Highlights of OH, H2SO4, and methane sulfonic acid measurements made aboard the NASA P-3B during Transport and Chemical Evolution over the Pacific, J. Geophys. Res., 2003, 108, 8796 Search PubMed.
- L. L. J. Quelever, L. Dada, E. Asmi, J. Lampilahti, T. Chan, J. E. Ferrara, G. E. Copes, G. Perez-Fogwill, L. Barreira, M. Aurela, D. R. Worsnop, T. Jokinen and M. Sipila, Investigation of new particle formation mechanisms and aerosol processes at Marambio Station, Antarctic Peninsula, Atmos. Chem. Phys., 2022, 22, 8417–8437 CrossRef CAS.
- D. Davis, G. Chen, P. Kasibhatla, A. Jefferson, D. Tanner, F. Eisele, D. Lenschow, W. Neff and H. Berresheim, DMS oxidation in the Antarctic marine boundary layer: Comparison of model simulations and field observations of DMS, DMSO, DMSO2, H2SO4(g), MSA(g), and MSA(p), J. Geophys. Res., 1998, 103, 1657–1678 CrossRef CAS.
- E. S. Saltzman, D. L. Savoie, R. G. Zika and J. M. Prospero, Methane sulfonic acid in the marine atmosphere, J. Geophys. Res.: Oceans, 1983, 88, 897–902 CrossRef.
- S. Huang, L. Poulain, D. van Pinxteren, M. van Pinxteren, Z. J. Wu, H. Herrmann and A. Wiedensohler, Latitudinal and seasonal distribution of particulate MSA over the Atlantic using a validated quantification method with HR-ToF-AMS, Environ. Sci. Technol., 2017, 51, 418–426 CrossRef CAS PubMed.
- L. Phinney, W. R. Leaitch, U. Lohmann, H. Boudries, D. R. Worsnop, J. T. Jayne, D. Toom-Sauntry, M. Wadleigh, S. Sharma and N. Shantz, Characterization of the aerosol over the sub-arctic north east Pacific Ocean, Deep Sea Res., Part II, 2006, 53, 2410–2433 CrossRef.
- A. Sorooshian, L. T. Padro, A. Nenes, G. Feingold, A. McComiskey, S. P. Hersey, H. Gates, H. H. Jonsson, S. D. Miller, G. L. Stephens, R. C. Flagan and J. H. Seinfeld, On the link between ocean biota emissions, aerosol, and maritime clouds: Airborne, ground, and satellite measurements off the coast of California, Global Biogeochem. Cycles, 2009, 23, 1–15 CrossRef.
- D. D. Huang, Y. J. Li, B. P. Lee and C. K. Chan, Analysis of organic sulfur compounds in atmospheric aerosols at the HKUST supersite in Hong Kong using HR-ToF-AMS, Environ. Sci. Technol., 2015, 49, 3672–3679 CrossRef CAS PubMed.
- M. D. Willis, J. Burkart, J. L. Thomas, F. Kollner, J. Schneider, H. Bozem, P. M. Hoor, A. A. Aliabadi, H. Schulz, A. B. Herber, W. R. Leaitch and J. P. D. Abbatt, Growth of nucleation mode particles in the summertime Arctic: a case study, Atmos. Chem. Phys., 2016, 16, 7663–7679 CrossRef CAS.
- L. C. Maudlin, Z. Wang, H. H. Jonsson and A. Sorooshian, Impact of wildfires on size-resolved aerosol composition at a coastal California site, Atmos. Environ., 2015, 119, 59–68 CrossRef CAS.
- A. Sorooshian, E. Crosbie, L. C. Maudlin, J. S. Youn, Z. Wang, T. Shingler, A. M. Ortega, S. Hersey and R. K. Woods, Surface and airborne measurements of organosulfur and methanesulfonate over the western United States and coastal areas, J. Geophys. Res., 2015, 120, 8535–8548 CrossRef CAS.
- C. Stahl, M. T. Cruz, P. A. Banaga, G. Betito, R. A. Braun, M. A. Aghdam, M. O. Cambaliza, G. R. Lorenzo, A. B. MacDonald, M. R. A. Hilario, P. C. Pabroa, J. R. Yee, J. B. Simpas and A. Sorooshian, Sources and characteristics of size-resolved particulate organic acids and methanesulfonate in a coastal megacity: Manila, Philippines, Atmos. Chem. Phys., 2020, 20, 15907–15935 CrossRef CAS.
- C. J. Gaston, K. A. Pratt, X. Y. Qin and K. A. Prather, Real-time detection and mixing state of methanesulfonate in single particles at an inland urban location during a phytoplankton bloom, Environ. Sci. Technol., 2010, 44, 1566–1572 CrossRef CAS PubMed.
- H. Yuan, Y. Wang and G. S. Zhuang, MSA in Beijing aerosol, Chin. Sci. Bull., 2004, 49, 1020–1025 CrossRef CAS.
- V. M. Kerminen, M. Aurela, R. E. Hillamo and A. Virkkula, Formation of particulate MSA: Deductions from size distribution measurements in the Finnish Arctic, Tellus B, 1997, 49, 159–171 CrossRef.
- M. J. Lawler, E. S. Saltzman, L. Karlsson, P. Zieger, M. Salter, A. Baccarini, J. Schmale and C. Leck, New insights into the composition and origins of ultrafine aerosol in the summertime high Arctic, Geosphys. Res. Lett., 2021, 48, e2021GL094395 CrossRef.
- J. Burkart, M. D. Willis, H. Bozem, J. L. Thomas, K. Law, P. Hoor, A. A. Aliabadi, F. Kollner, J. Schneider, A. B. Herber, J. D. Abbatt and W. R. Leaitch, Summertime observations of elevated levels of ultrafine particles in the high Arctic marine boundary layer, Atmos. Chem. Phys., 2017, 17, 5515–5535 CrossRef CAS.
- K. T. Park, S. Jang, K. Lee, Y. J. Yoon, M. S. Kim, K. Park, H. J. Cho, J. H. Kang, R. Udisti, B. Y. Lee and K. H. Shin, Observational evidence for the formation of DMS-derived aerosols during Arctic phytoplankton blooms, Atmos. Chem. Phys., 2017, 17, 9665–9675 CrossRef CAS.
- X. X. Li, Y. Y. Li, M. J. Lawler, J. M. Hao, J. N. Smith and J. K. Jiang, Composition of ultrafine particles in urban Beijing: Measurement using a thermal desorption chemical ionization mass spectrometer, Environ. Sci. Technol., 2021, 55, 2859–2868 CrossRef CAS PubMed.
- T. A. Pakkanen, V. M. Kerminen, C. H. Korhonen, R. E. Hillamo, P. Aarnio, T. Koskentalo and W. Maenhaut, Urban and rural ultrafine (PM(0.1)) particles in the Helsinki area, Atmos. Environ., 2001, 35, 4593–4607 CrossRef CAS.
- K. N. Fossum, J. Ovadnevaite, D. Ceburnis, M. Dall’Osto, S. Marullo, M. Bellacicco, R. Simo, D. T. Liu, M. Flynn, A. Zuend and C. O'Dowd, Summertime primary and secondary contributions to Southern ocean cloud condensation nuclei, Sci. Rep., 2018, 8, 13844 CrossRef PubMed.
- J. M. Makela, S. Yli-Koivisto, V. Hiltunen, W. Seidl, E. Swietlicki, K. Teinila, M. Sillanpaa, I. K. Koponen, J. Paatero, K. Rosman and K. Hameri, Chemical composition of aerosol during particle formation events in boreal forest, Tellus B, 2001, 53, 380–393 CrossRef.
- M. Dall’Osto, D. C. S. Beddows, P. Tunved, R. Krejci, J. Strom, H. C. Hansson, Y. J. Yoon, K. T. Park, S. Becagli, R. Udisti, T. Onasch, C. D. O'Dowd, R. Simo and R. M. Harrison, Arctic sea ice melt leads to atmospheric new particle formation, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- P. K. Quinn, T. L. Miller, T. S. Bates, J. A. Ogren, E. Andrews and G. E. Shaw, A 3-year record of simultaneously measured aerosol chemical and optical properties at Barrow, Alaska, J. Geophys. Res., 2002, 107, 4130 Search PubMed.
- W. R. Leaitch, S. Sharma, L. Huang, D. Toom-Sauntry, A. Chivulescu, A.-M. Macdonald, K. von Salzen, J. R. Pierce, A. K. Bertram, J. C. Schroder, N. C. Shantz, R. Y.-W. Chang and A.-L. Norman, Dimethyl sulfide control of the clean summertime Arctic aerosol and cloud, Elem. Sci. Anth., 2013, 1, 1–12 Search PubMed.
- M. Dall’Osto, R. Simo, R. M. Harrison, D. C. S. Beddows, A. Saiz-Lopez, R. Lange, H. Skov, J. K. Nojgaard, I. E. Nielsen and A. Massling, Abiotic and biotic sources influencing spring new particle formation in North East Greenland, Atmos. Environ., 2018, 190, 126–134 CrossRef.
- M. D. Willis, W. R. Leaitch and J. P. D. Abbatt, Processes controlling the composition and abundance of Arctic aerosol, Rev. Geophys., 2018, 56, 621–671 CrossRef.
- Q. Chen, T. Sherwen, M. Evans and B. Alexander, DMS oxidation and sulfur aerosol formation in the marine troposphere: a focus on reactive halogen and multiphase chemistry, Atmos. Chem. Phys., 2018, 18, 13617–13637 CrossRef CAS.
- M. Gali, E. Devred, M. Babin and M. Levasseur, Decadal increase in Arctic dimethylsulfide emission, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19311–19317 CrossRef CAS PubMed.
- S. Sharma, E. Chan, M. Ishizawa, D. Toom-Sauntry, S. L. Gong, S. M. Li, D. W. Tarasick, W. R. Leaitch, A. Norman, P. K. Quinn, T. S. Bates, M. Levasseur, L. A. Barrie and W. Maenhaut, Influence of transport and ocean ice extent on biogenic aerosol sulfur in the Arctic atmosphere, J. Geophys. Res., 2012, 117, D12209 Search PubMed.
- S. Becagli, L. Lazzara, C. Marchese, U. Dayan, S. E. Ascanius, M. Cacciani, L. Caiazzo, C. Di Biagio, T. Di Iorio, A. di Sarra, P. Eriksen, F. Fani, F. Giardi, D. Meloni, G. Muscari, G. Pace, M. Severi, R. Traversi and R. Udisti, Relationships linking primary production, sea ice melting, and biogenic aerosol in the Arctic, Atmos. Environ., 2016, 136, 1–15 CrossRef CAS.
- K. D. Arquero, R. B. Gerber and B. J. Finlayson-Pitts, The role of oxalic acid in new particle formation from methanesulfonic acid, methylamine, and water, Environ. Sci. Technol., 2017, 51, 2124–2130 CrossRef CAS PubMed.
- K. D. Arquero, J. Xu, R. B. Gerber and B. J. Finlayson-Pitts, Particle formation and growth from oxalic acid, methanesulfonic acid, trimethylamine and water: a combined experimental and theoretical study, Phys. Chem. Chem. Phys., 2017, 19, 28286–28301 RSC.
- H. Chen, M. J. Ezell, K. D. Arquero, M. E. Varner, M. L. Dawson, R. B. Gerber and B. J. Finlayson-Pitts, New particle formation and growth from methanesulfonic acid, trimethylamine and water, Phys. Chem. Chem. Phys., 2015, 17, 13699–13709 RSC.
- H. Chen, M. E. Varner, R. B. Gerber and B. J. Finlayson-Pitts, Reactions of methanesulfonic acid with amines and ammonia as a source of new particles in air, J. Phys. Chem. B, 2016, 120, 1526–1536 CrossRef CAS PubMed.
- H. H. Chen and B. J. Finlayson-Pitts, New particle formation from methanesulfonic acid and amines/ammonia as a function of temperature, Environ. Sci. Technol., 2017, 51, 243–252 CrossRef CAS PubMed.
- M. L. Dawson, M. E. Varner, V. Perraud, M. J. Ezell, R. B. Gerber and B. J. Finlayson-Pitts, Simplified mechanism for new particle formation from methanesulfonic acid, amines, and water via experiments and ab initio calculations, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18719–18724 CrossRef CAS PubMed.
- V. Perraud, X. X. Li, J. K. Jiang, B. J. Finlayson-Pitts and J. N. Smith, Size-resolved chemical composition of sub-20 nm particles from methanesulfonic acid reactions with methylamine and ammonia, ACS Earth Space Chem., 2020, 4, 1182–1194 CrossRef CAS.
- V. Perraud, J. Xu, R. B. Gerber and B. J. Finlayson-Pitts, Integrated experimental and theoretical approach to probe the synergistic effect of ammonia in methanesulfonic acid reactions with small alkylamines, Environ. Sci.: Processes Impacts, 2020, 22, 305–328 RSC.
- J. S. Johnson and C. N. Jen, Role of methanesulfonic acid in sulfuric acid-amine and ammonia new particle formation, ACS Earth Space Chem., 2023, 7, 653–660 CrossRef CAS PubMed.
- J. Shen, H.-B. Xie, J. Elm, F. Ma, J. Chen and H. Vehkamaki, Methanesulfonic acid-driven new particle formation enhanced by monoethanolamine: A computational study, Environ. Sci. Technol., 2019, 53, 14387–14397 CrossRef CAS PubMed.
- J. W. Shen, J. Elm, H. B. Xie, J. W. Chen, J. F. Niu and H. Vehkamaki, Structural effects of amines in enhancing methanesulfonic acid-driven new particle formation, Environ. Sci. Technol., 2020, 54, 13498–13508 CrossRef CAS PubMed.
- H. L. Zhao, X. T. Jiang and L. Du, Contribution of methane sulfonic acid to new particle formation in the atmosphere, Chemosphere, 2017, 174, 689–699 CrossRef CAS PubMed.
- F. R. Rasmussen, J. Kubecka and J. Elm, Contribution of methansulfonic acid to the formation of molecular clusters in the marine atmosphere, J. Phys. Chem. A, 2022, 126, 7127–7136 CrossRef CAS PubMed.
- J. Elm, Clusteromics II: Methansulfonic acid-base cluster formation, ACS Omega, 2021, 6, 17035–17044 CrossRef CAS PubMed.
- R. Zhang, J. Shen, H.-B. Xie, J. Chen and J. Elm, The role of organic acids in new particle formation from methanesulfonic acid and methylamine, Atmos. Chem. Phys., 2022, 22, 2639–2650 CrossRef CAS.
- S. Chee, K. Barsanti, J. N. Smith and N. Myllys, A predictive model for salt nanoparticle formation using heterodimer stability calculations, Atmos. Chem. Phys., 2021, 21, 11637–11654 CrossRef CAS.
- D. P. Chen, D. F. Li, C. W. Wang, F. Y. Liu and W. L. Wang, Formation mechanism of methanesulfonic acid and ammonia clusters: A kinetics simulation study, Atmos. Environ., 2020, 222, 117161 CrossRef CAS.
- M. van Pinxteren, B. Fiedler, D. van Pinxteren, Y. Iinuma, A. Kortzinger and H. Herrmann, Chemical characterization of sub-micrometer aerosol particles in the tropical Atlantic Ocean: marine and biomass burning influences, J. Atmos. Chem., 2015, 72, 105–125 CrossRef CAS.
- C. Muller, Y. Iinuma, J. Karstensen, D. van Pinxteren, S. Lehmann, T. Gnauk and H. Herrmann, Seasonal variation of aliphatic amines in marine sub-micrometer particles at the Cape Verde islands, Atmos. Chem. Phys., 2009, 9, 9587–9597 CrossRef.
- M. C. Facchini, S. Decesari, M. Rinaldi, C. Carbone, E. Finessi, M. Mircea, S. Fuzzi, F. Moretti, E. Tagliavini, D. Ceburnis and C. D. O'Dowd, Important source of marine secondary organic aerosol from biogenic amines, Environ. Sci. Technol., 2008, 42, 9116–9121 CrossRef CAS PubMed.
- X. F. Huang, C. R. Deng, G. S. Zhuang, J. Lin and M. X. Xiao, Quantitative analysis of aliphatic amines in urban aerosols based on online derivatization and high performance liquid chromatography, Environ. Sci.: Processes Impacts, 2016, 18, 796–801 RSC.
- H. Feng, X. N. Ye, Y. X. Liu, Z. K. Wang, T. X. Gao, A. Y. Cheng, X. F. Wang and J. M. Chen, Simultaneous determination of nine atmospheric amines and six inorganic ions by non-suppressed ion chromatography using acetonitrile and 18-crown-6 as eluent additive, J. Chromatogr. A, 2020, 1624, 461234 CrossRef CAS PubMed.
- M. Wang, Q. Y. Wang, S. S. H. Ho, H. Li, R. J. Zhang, W. K. Ran, L. L. Qu, S. C. Lee and J. J. Cao, Chemical characteristics and sources of nitrogen-containing organic compounds at a regional site in the North China Plain during the transition period of autumn and winter, Sci. Total Environ, 2022, 812, 151451 CrossRef CAS PubMed.
- A. P. Sullivan, K. B. Benedict, C. M. Carrico, M. K. Dubey, B. A. Schichtel and J. C. Collett, A quantitative method to measure and speciate amines in ambient aerosol samples, Atmosphere, 2020, 11, 808 CrossRef CAS.
- Q. Zhang and C. Anastasio, Free and combined amino compounds in atmospheric fine particles (PM2.5) and fog waters from Northern California, Atmos. Environ., 2003, 37, 2247–2258 CrossRef CAS.
- Z. Y. Liu, M. Li, X. F. Wang, Y. H. Liang, Y. R. Jiang, J. Chen, J. S. Mu, Y. J. Zhu, H. Meng, L. X. Yang, K. Y. Hou, Y. F. Wang and L. K. Xue, Large contributions of anthropogenic sources to amines in fine particles at a coastal area in northern China in winter, Sci. Total Environ, 2022, 839, 156281 CrossRef CAS PubMed.
- B. K. Place, A. T. Quilty, R. A. Di Lorenzo, S. E. Ziegler and T. C. VandenBoer, Quantitation of 11 alkylamines in atmospheric samples: separating structural isomers by ion chromatography, Atmos. Meas. Tech., 2017, 10, 1061–1078 CrossRef CAS.
- K. Gorzelska and J. N. Galloway, Amine nitrogen in the atmospheric environment over the north Atlantic ocean, Global Biogeochem. Cycles, 1990, 4, 309–333 CrossRef CAS.
- V. M. Kerminen, X. Chen, V. Vakkari, T. Petaja, M. Kulmala and F. Bianchi, Atmospheric new particle formation and growth: review of field observations, Environ. Res. Lett., 2018, 13, 103003 CrossRef.
- U. Poschl, Atmospheric aerosols: Composition, transformation, climate and health effects, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef PubMed.
- IPCC, in Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, ed. V. Masson-Delmotte, United Kindom and New York, NY, USA, 2021, p. 2391 Search PubMed.
- A. L. Hodshire, P. Campuzano-Jost, J. K. Kodros, B. Croft, B. A. Nault, J. C. Schroder, J. L. Jimenez and J. R. Pierce, The potential role of methanesulfonic acid (MSA) in aerosol formation and growth and the associated radiative forcings, Atmos. Chem. Phys., 2019, 19, 3137–3160 CrossRef CAS.
- C. A. Pope and D. W. Dockery, Health effects of fine particulate air pollution: Lines that connect, J. Air Waste Manage. Assoc., 2006, 56, 709–742 CrossRef CAS PubMed.
- M. R. Heal, P. Kumar and R. M. Harrison, Particles, air quality, policy and health, Chem. Soc. Rev., 2012, 41, 6606–6630 RSC.
- J. Lelieveld, Clean air in the Anthropocene, Faraday Discuss., 2017, 200, 693–703 RSC.
- J. Lelieveld, J. S. Evans, M. Fnais, D. Giannadaki and A. Pozzer, The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 2015, 525, 367–371 CrossRef CAS.
- A. L. Moreno-Ríos, L. P. Tejeda-Benítez and C. F. Bustillo-Lecompte, Sources, characteristics, toxicity, and control of ultrafine particles: An overview, Geosci Front., 2022, 13, 101147 CrossRef.
- S. W. Sang, C. Chu, T. C. Zhang, H. Chen and X. R. Yang, The global burden of disease attributable to ambient fine particulate matter in 204 countries and territories, 1990–2019: A systematic analysis of the Global Burden of Disease Study 2019, Ecotoxicol. Environ. Saf., 2022, 238, 113588 CrossRef CAS PubMed.
- X. J. Deng, X. X. Tie, D. Wu, X. J. Zhou, X. Y. Bi, H. B. Tan, F. Li and C. L. Hang, Long-term trend of visibility and its characterizations in the Pearl River Delta (PRD) region, China, Atmos. Environ., 2008, 42, 1424–1435 CrossRef CAS.
- D. Chang, Y. Song and B. Liu, Visibility trends in six megacities in China 1973–2007, Atmos. Res., 2009, 94, 161–167 CrossRef.
- A. Singh, W. J. Bloss and F. D. Pope, 60 years of UK visibility measurements: impact of meteorology and atmospheric pollutants on visibility, Atmos. Chem. Phys., 2017, 17, 2085–2101 CrossRef CAS.
- J. G. Watson, Visibility: science and regulation, J. Air Waste Manage. Assoc., 2002, 52, 628–713 CrossRef PubMed.
- A. L. Moreno-Rios, L. P. Tejeda-Benitez and C. F. Bustillo-Lecompte, Sources, characteristics, toxicity, and control of ultrafine particles: An overview, Geosci Front., 2022, 13, 101147 CrossRef CAS.
- M. J. Ezell, H. Chen, K. D. Arquero and B. J. Finlayson-Pitts, Aerosol fast flow reactor for laboratory studies of new particle formation, J. Aerosol Sci., 2014, 78, 30–40 CrossRef CAS.
- J. N. Smith, K. F. Moore, P. H. McMurry and F. L. Eisele, Atmospheric measurements of sub-20 nm diameter particle chemical composition by thermal desorption chemical ionization mass spectrometry, Aerosol Sci. Technol., 2004, 38, 100–110 CrossRef CAS.
- D. Voisin, J. N. Smith, H. Sakurai, P. H. McMurry and F. L. Eisele, Thermal desorption chemical ionization mass spectrometer for ultrafine particle chemical composition, Aerosol Sci. Technol., 2003, 37, 471–475 CrossRef CAS.
- M. J. Lawler, P. M. Winkler, J. Kim, L. Ahlm, J. Trostl, A. P. Praplan, S. Schobesberger, A. Kuerten, J. Kirkby, F. Bianchi, J. Duplissy, A. Hansel, T. Jokinen, H. Keskinen, K. Lehtipalo, M. Leiminger, T. Petaja, M. Rissanen, L. Rondo, M. Simon, M. Sipila, C. Williamson, D. Wimmer, I. Riipinen, A. Virtanen and J. N. Smith, Unexpectedly acidic nanoparticles formed in dimethylamine-ammonia-sulfuric-acid nucleation experiments at CLOUD, Atmos. Chem. Phys., 2016, 16, 13601–13618 CrossRef CAS.
- H. Chen, S. Chee, M. J. Lawler, K. C. Barsanti, B. M. Wong and J. N. Smith, Size resolved chemical composition of nanoparticles from reactions of sulfuric acid with ammonia and dimethylamine, Aerosol Sci. Technol., 2018, 52, 1120–1133 CrossRef CAS.
- D.-R. Chen and D. Y. Pui, A high efficiency, high throughput unipolar aerosol charger for nanoparticles, J. Nanopart. Res., 1999, 1, 115–126 CrossRef CAS.
- P. H. McMurry, A. Ghimire, H.-K. Ahn, H. Sakurai, K. Moore, M. Stolzenburg and J. N. Smith, Sampling nanoparticles for chemical analysis by low resolution electrical mobility classification, Environ. Sci. Technol., 2009, 43, 4653–4658 CrossRef CAS.
- D. V. Davis and R. G. Cooks, Site of protonation and bifunctional group-interactions in a,w-hydroxyalkylamines, Org. Mass Spectrom, 1981, 16, 176–179 CrossRef CAS.
- G. Bouchoux, N. Choret, F. Berruyer-Penaud and R. Flammang, Thermochemistry and unimolecular reactivity of protonated alpha,omega-aminoalcohols in the gas phase, Int. J. Mass Spectrom., 2002, 217, 195–230 CrossRef CAS.
- E. P. L. Hunter and S. G. Lias, Evaluated gas phase basicities and proton affinities of molecules: An update, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS.
- S. Kapteina, K. Slowik, S. P. Verevkin and A. Heintz, Vapor pressures and vaporization enthalpies of a series of ethanolamines, J. Chem. Eng. Data, 2005, 50, 398–402 CrossRef CAS.
- I. N. Tang and H. R. Munkelwitz, Determination of vapor-pressure from droplet evaporation kinetics, J. Colloid Interface Sci., 1991, 141, 109–118 CrossRef CAS.
- S.-L. von der Weiden, F. Drewnick and S. Borrmann, Particle loss calculator – A new software tool for the assessment of the performace of aerosol inlet systems, Atmos. Meas. Tech., 2009, 2, 469–494 Search PubMed.
- M. Kulmala, H. Vehkamäki, T. Petäjä, M. Dal Maso, A. Lauri, V. M. Kerminen, W. Birmili and P. H. McMurry, Formation and growth rates of ultrafine atmospheric particles: a review of observations, J. Aerosol Sci., 2004, 35, 143–176 CrossRef CAS.
- T. L. Greaves, A. Weerawardena, C. Fong, I. Krodkiewska and C. J. Drummond, Protic ionic liquids: Solvents with tunable phase behavior and physicoschemical properties, J. Phys. Chem. B, 2006, 110, 22479–22487 CrossRef CAS PubMed.
- G. Cai, S. Yang, Q. Zhou, L. Liu, J. Xu and S. Zhang, Physicochemical properties of various 2-hydroxyethylammonium sulfonate-based protic ionic liquids and their potential application in hydrodeoxygenation, Front. Chem., 2019, 7, 196 CrossRef CAS PubMed.
- X. Lu, J. M. Vincent-Luna, S. Calero, M. Roldan-Ruiz, R. Jimenez, M. L. Ferrer, M. C. Gutierrez and F. del Monte, Aqueous co-solvent in zwitterionic-based protic ionic liquids as electrolytes in 2.0 V supercapacitors, ChemSusChem, 2020, 13, 5983–5995 CrossRef CAS PubMed.
- S. J. Brown, D. Yalcin, S. Pnadiancherri, T. C. Le, I. O’rhan, K. Hearn, Q. Han, C. J. Drummond and T. L. Greaves, Characterising a protic ionic liquid library with applied machine learning algorithms, J. Mol. Liq., 2022, 367, 120453 CrossRef CAS.
- R. McGraw and A. Laaksonen, Scaling properties of the critical nucleus in classical and molecular-based theories of vapor-liquid nucleation, Phys. Rev. Lett., 1996, 76, 2754–2757 CrossRef CAS PubMed.
- O. Kupiainen-Maatta, T. Olenius, H. Korhonen, J. Malilal, M. Dal Maso, K. Lehtinen and H. Vehkamaki, Critical cluster size cannot in pracctice be determined by slope analysis in atmospherically relevant applications, J. Aerosol Sci., 2014, 77, 127–144 CrossRef CAS.
- Y. Liu, H. B. Xie, F. F. Ma, J. W. Chen and J. Elm, Amine-enhanced methanesulfonic acid driven nucleation: Predictive model and cluster formation mechanism, Environ. Sci. Technol., 2022, 56, 7751–7760 CrossRef CAS PubMed.
- J. Xu, V. Perraud, B. J. Finlayson-Pitts and R. B. Gerber, Uptake of water by an acid-base nanoparticle: theoretical and experimental studies of the methanesulfonic acid-methylamine system, Phys. Chem. Chem. Phys., 2018, 20, 22249–22259 RSC.
- V. Perraud, J. R. Horne, A. S. Martinez, J. Kalinowski, S. Meinardi, M. L. Dawson, L. M. Wingen, D. Dabdub, D. R. Blake, R. B. Gerber and B. J. Finlayson-Pitts, The future of airborne sulfur-containing particles in the absence of fossil fuel sulfur dioxide emissions, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13514–13519 CrossRef CAS PubMed.
- J. G. Murphy, P. K. Gregoire, A. G. Tevlin, G. R. Wentworth, R. A. Ellis, M. Z. Markovic and T. C. VandenBoer, Observational constraints on particle acidity using measurements and modelling of particles and gases, Faraday Discuss., 2017, 200, 379–395 RSC.
- Z. Klimont, S. J. Smith and J. Cofala, The last decade of global anthropogenic sulfur dioxide: 2000-2011 emissions, Environ. Res. Lett., 2013, 8, 1–6 Search PubMed.
- T. J. Breider, L. J. Mickley, D. J. Jacob, C. Ge, J. Wang, M. P. Sulprizio, B. Croft, D. A. Ridley, J. R. McConnell, S. Sharma, L. Husain, V. A. Dutkiewicz, K. Eleftheriadis, H. Skov and P. K. Hopke, Multidecadal trends in aerosol radiative forcing over the Arctic: Contribution of changes in anthropogenic aerosol to Arctic warming since 1980, J. Geophys. Res., 2017, 122, 3573–3594 CrossRef.
- G. M. Hidy and C. L. Blanchard, The changing face of lower tropospheric sulfur oxides in the United States, Elem. Sci. Anth., 2016, 4, 000138 CrossRef.
- D. I. Stern, Global sulfur emissions from 1850 to 2000, Chemosphere, 2005, 58, 163–175 CrossRef CAS PubMed.
- M. Amann, Z. Klimont and F. Wagner, Regional and global emissions of air pollutants: Recent trends and future scenarios, Ann. Rev. Environ. Resour., 2013, 38, 31–55 CrossRef.
- P. K. Quinn, G. Shaw, E. Andrews, E. G. Dutton, T. Ruoho-Airola and S. L. Gong, Arctic haze: current trends and knowledge gaps, Tellus B, 2007, 59, 99–114 CrossRef.
- D. Hirdman, J. F. Burkhart, H. Sodemann, S. Eckhardt, A. Jefferson, P. K. Quinn, S. Sharma, J. Strom and A. Stohl, Long-term trends of black carbon and sulphate aerosol in the Arctic: changes in atmospheric transport and source region emissions, Atmos. Chem. Phys., 2010, 10, 9351–9368 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods as well as 15 Fig. S1–S15. See DOI: https://doi.org/10.1039/d4cp00316k |
| This journal is © the Owner Societies 2024 |