
A frontier-orbital view of the initial steps of lytic polysaccharide monooxygenase reactions†
Erna Katharina
Wieduwilt
 a,
Leila
Lo Leggio
b and
Erik Donovan
Hedegård
*a
a,
Leila
Lo Leggio
b and
Erik Donovan
Hedegård
*a
aDepartment of Physics, Chemistry, and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense M, Denmark. E-mail: erdh@sdu.dk
bDepartment of Chemistry, University of Copenhagen, 2100 Copenhagen, Denmark
First published on 26th February 2024
Abstract
Lytic polysaccharide monooxygenases (LPMOs) are copper enzymes that oxidatively cleave the strong C–H bonds in recalcitrant polysaccharide substrates, thereby playing a crucial role in biomass degradation. Recently, LPMOs have also been shown to be important for several pathogens. It is well established that the Cu(II) resting state of LPMOs is inactive, and the electronic structure of the active site needs to be altered to transform the enzyme into an active form. Whether this transformation occurs due to substrate binding or due to a unique priming reduction has remained speculative. Starting from four different crystal structures of the LPMO LsAA9A with well-defined oxidation states, we use a frontier molecular orbital approach to elucidate the initial steps of the LPMO reaction. We give an explanation for the requirement of the unique priming reduction and analyse electronic structure changes upon substrate binding. We further investigate how the presence of the substrate could facilitate an electron transfer from the copper active site to an H2O2 co-substrate. Our findings could help to control experimental LPMO reactions.
Introduction
Lytic polysaccharide monooxygenases (LPMOs) are copper-dependent enzymes that catalyse the oxidative degradation of several polysaccharide substrates, such as cellulose, chitin, starch, and pectin.1–9 LPMOs work by activating (otherwise inactive) C–H bonds in the glycosidic bonds that link the monomers in the respective polysaccharides. This activation ultimately leads to oxidation and cleavage of the glycosidic bond.10–12 Several mechanistic pathways have been suggested for LPMOs, and we have summarized the most recent suggestions in Fig. 1.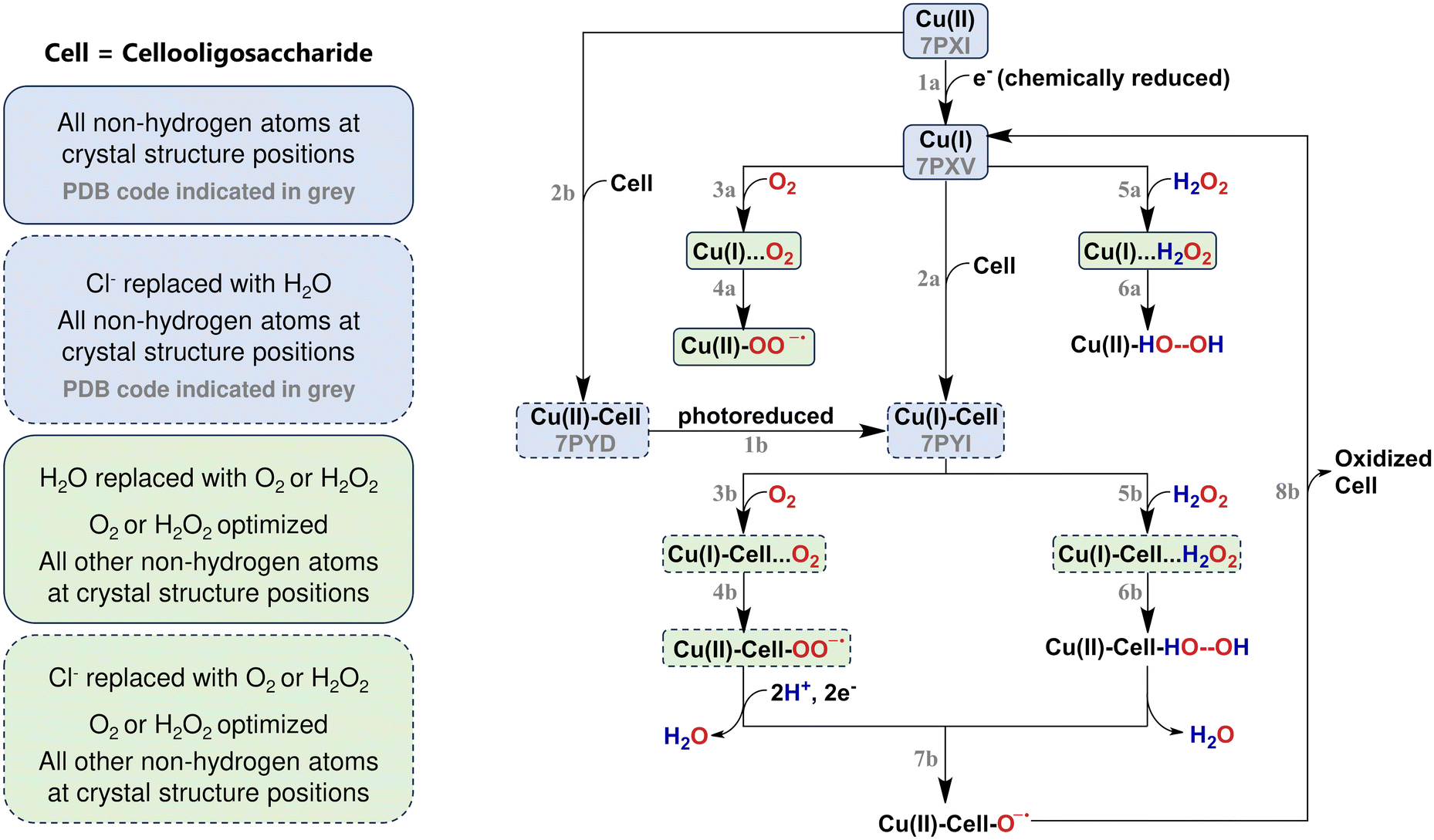 | ||
| Fig. 1 Simplified reaction mechanism of LPMOs with boxes highlighting the structures that were investigated in this study. Corresponding structures are shown in Fig. 2 and Fig. S1.† | ||
Originally, LPMOs were discovered in bacteria and fungi. Today, they have been found across all kingdoms of life (though not yet in mammals) and exhibit a growing number of functions.10,13 For instance, LPMOs and LPMO-like proteins were recently suggested to also play crucial roles in plant, insect and human pathogenesis such as blood diseases and meningitis.13,14,15 However, the function that has so far attracted the most attention is a significant boosting effect on biomass degradation. Nowadays, LPMOs are employed in industrial enzyme cocktails to produce biofuels and other value-added products.16,17
To date, eight different LPMO families have been discovered, namely AA9-AA17 (AA12 does not contain LPMOs).18 All members share a common active site: a copper centre coordinated by two histidine residues in a motif termed the histidine brace (see Fig. 2).1 Initially, LPMOs were thought to be strictly monooxygenases,1,19,20 catalysing the reaction
| R-H + O2 + 2H+ + 2e− → R-OH + H2O | (1) |
| R-H + H2O2 → R-OH + H2O | (2) |
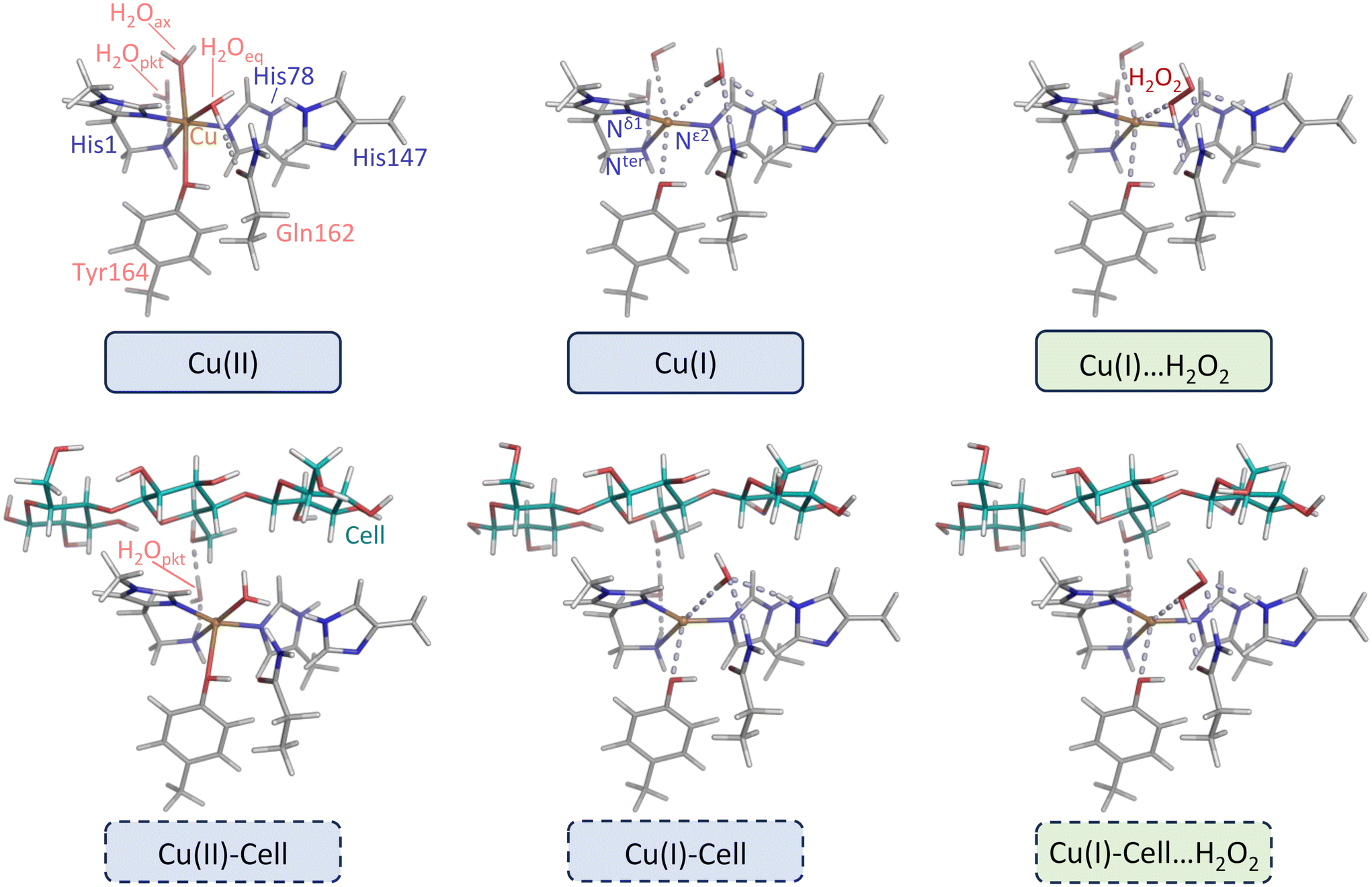 | ||
| Fig. 2 Left and middle: Substrate-free and substrate-bound structures directly derived from four crystal structures of LsAA9A. Right: Structures with pre-bound H2O2 (see products of reactions (5a) and (5b) in Fig. 1) derived from replacing water or chloride in the two crystal structures of LsAA9A with oxidation state I. Blue and green boxes correspond to the ones denoted correspondingly in Fig. 1. Description of the geometry: the active site of LPMOs consists of a copper center coordinated by two histidines (His1 and His78), forming the histidine brace.1,10,13–17 His1 is the N-terminal residue that binds bidentate through the amine of the N-terminus and the imidazole side chain. His78 binds only through the side chain. In LPMOs of fungal origin, His1 is often methylated (this is also the case for LsAA9A). Apart from the histidine brace, there are no residues that are strictly conserved across all LPMOs, however, many LPMOs have a generally conserved tyrosine (Tyr164 in LsAA9A) as axial ligand in the oxidation state II. Two water ligands (one in axial and one in equatorial position) complete the octahedral Jahn–Teller distorted active site with axially elongated ligands. In the second coordination sphere, we included Gln162 and His147, which are both important in stabilizing the pre-bound H2O2 structures.40–42 If cellotetraose is present, it binds from subsite −2 to +2, and forms a hydrogen bond to the so-called “pocket water”43 that in turn hydrogen bonds to the terminal NH2 group of His1. Geometrical changes upon reduction and substrate binding are described in the main text and in detail in ref. 44. | ||
The peroxygenase reaction circumvents the requirement for external protons and electrons12,21 (see reactions (5b)–(7b) in Fig. 1), and also the requirement by certain monooxygenases reaction mechanisms to reduce the Cu(II) to Cu(I) at every cycle, thus limiting the reducing agent requirement.22 Furthermore, for the LPMOs where mono- and peroxygenase reactions have been compared, the latter is orders of magnitude faster.21,22 For a few cases, the catalytic reaction has also been performed anaerobically, showing that these LPMOs indeed exclusively use H2O2 as co-substrate23,24 (according to eqn (2) or reactions (5b)–(7b) in Fig. 1). However, whether this is true for all LPMOs is still controversial. Moreover, it is also unknown if some LPMOs can use H2O2 as a “peroxide shunt” pathway as some iron-heme enzymes (see further below).25
A complicating factor for experimental investigations concerning the co-substrate is that LPMOs can catalyse the off-path oxidase reaction
| O2 + 2H+ + 2e− → H2O2 | (3) |
Regardless of whether LPMOs react according to eqn (1) as monooxygenases or according to eqn (2) as peroxygenases, or variations thereof, it has been firmly demonstrated that an initial reduction of the Cu(II) resting states is required for catalytic turnover (see reactions (1a) and (1b) in Fig. 1).32 This is also true when LPMOs react as oxidases without substrate and produce H2O2 according to eqn (3). This initial reduction has been denoted a “priming” reduction, for which a variety of reductants can be employed.2,11,33,34
It has been shown that both the Cu(I) state and the Cu(II) resting state can bind the substrate (according to reactions (2a) and (2b) in Fig. 1), but some experimental studies show that the reduced LPMO has a 2 to 10-fold higher affinity for cellulose.23,35 Therefore, it has been proposed that metal reduction precedes substrate binding.
The nature of the priming reduction separates LPMOs from other metalloenzymes that also activate inactive C–H bonds – but whose reaction cycle otherwise shows similarities to the LPMOs’ catalytic cycle:36 the iron-heme enzyme cytochrome P450 usually forms a highly oxidizing species (Cpd1) from O2. The resting state Fe(III)-heme unit is first reduced after substrate binding,37,38 and O2 binds to this Fe(II)-heme unit (similar to the O2 pathway with reactions (3b) and (4b) in Fig. 1). The Fe(III)–O2− species formed from initial O2 binding is further reduced and protonated into a Fe(III)-OOH intermediate (Cpd0). The HOO− unit in Cpd0 is heterolytically cleaved with help from nearby residues that can participate in acid/base chemistry under oxidation of Fe(III) to the oxidative species (Cpd1), responsible for C–H activation. Thus, cytochrome P450 needs a continuous flow of electrons in its catalytic cycle. This is different from LPMOs, which can perform up to 20 catalytic cycles after one priming electron has been added39 (when they work as peroxygenases). The iron-heme unit is also used by peroxidases, such as cytochrome c peroxidase and horseradish peroxidase (HRP). Unlike most cytochrome P450s, the peroxidases form the oxidative species (Cpd1) from H2O2 binding to the ferric Fe(III)-heme resting state. Thus, this is similar to the H2O2 pathway with reactions (5a)/(5b) and (6a)/(6b) in Fig. 1, with the important difference that it is the oxidized form of the heme unit that interacts with H2O2, and no reduction is required for H2O2 activation. Instead, the binding of H2O2 generates the Fe(III)-OOH intermediate (after deprotonation of H2O2).45 The P450 cytochromes can use a similar mechanism known as the “peroxide shunt” pathway, but this generally leads to a one-electron reduced form of Cpd1, denoted Cpd2, and a protein radical.38
In general, LPMO reactions with O2 and H2O2 have been intensively studied experimentally and computationally.10,12,13,43,46 Unlike the iron-heme proteins, computational studies focused mainly on events after binding of the co-substrate,18,19,47,48 whereas less attention has been paid to the priming reduction,49,50 substrate binding and the events before the co-substrate interacts with Cu(I). However, a recent combined experimental/theoretical study51 by Lim et al. interrogated the Cu(I) species of an AA9 LPMO through core spectroscopy and density functional theory (DFT) calculations: they found the Cu(I) species to have a dx2−y2 frontier orbital that was proposed to be optimally oriented to react with an incoming co-substrate (H2O2 in ref. 51). Lim et al. further argue that the dx2−y2-orbital is significantly higher in energy (1.1–1.4 eV) compared to the remaining occupied copper d-orbitals, bringing it closer to the LUMO (the H2O2 σ*-orbital). This facilitates the one-electron transfer from the copper to H2O2 in reaction (6b) in Fig. 1.51 Yet, this investigation did not explicitly include a saccharide substrate. Meanwhile, other studies have suggested that substrate binding has specific impacts on LPMO reactivity: the nature of the substrate has been proposed to take part in determining the mechanism for the generation of the oxidative species48,52–54 and substrate binding has been proposed to directly influence the orbitals of d-character.9 Further, several electron paramagnetic resonance (EPR) studies on AA9 and AA10 LPMOs report the appearance of super-hyperfine couplings and changes in the EPR spectra upon substrate binding.21,55–57 Different explanations for these observations were given. For example, conformational changes such as a distortion of the copper-coordinating amino acids and a reorganization of coordinating water molecules.56,57 It has also been proposed that substrate-induced electronic structure changes “pre-dispose” the active site to form a stable Cu(II)-Cell-O2 intermediate.56 Some of the discussed electronic structure changes include, for example, a stronger interaction between the copper and the coordinating nitrogen atoms21,55 and an increase in energy of the dx2−y2 frontier orbital.21,56
To reveal further details of the first steps of the LPMO reaction and the effects of reduction and substrate binding, we use a frontier orbital approach. Analysing frontier molecular orbitals (MOs) has previously been used to understand the reactivity of both iron-heme enzymes38,45 and copper enzymes involving a Cu(II)–O2 moiety.58–60 The orbitals around the HOMO and LUMO are here taken as acceptors for incoming co-substrates. We analyse changes in the frontier MOs in the presence and absence of substrate and further how reduction and incoming O2 and H2O2 influence the electronic structure. We use the structures from a recent crystallographic study by Tandrup et al.22 to directly link the frontier MOs to accurate experimental structures: In the structures obtained in ref. 22, a controlled photoreduction of the copper in the X-ray beam61 was exploited to obtain structures that can be assigned Cu(I) and Cu(II) oxidation states,22,62 both with and without bound cellooligosaccharide substrate (abbreviated as Cell). Thus, with outset in these crystal structures,22 we investigate how the observed changes in active-site geometry due to reduction and substrate binding influence the electronic structure through analysis of the frontier orbitals. Moreover, we consider also how the frontier orbitals change due to incoming oxidative species in the forms of O2 or H2O2.
Computational details
Our calculations are anchored in the experimental geometry of four different LsAA9A crystal structures measured by Tandrup et al. (pdb codes 7PXI, 7PXV, 7PYI and 7PYD, see structures in blue boxes in Fig. 1).22 In the structures 7PXI and 7PXV no substrate is bound, and they will be labelled as Cu(II) and Cu(I) respectively. Cellotetraose is bound in the structures 7PYD and 7PYI, and they will be labelled Cu(II)-Cell-Cl− and Cu(I)-Cell-Cl−, respectively. Note that Tandrup et al. obtained the Cu(I) crystal structure (pdb code 7PXV) through chemical reduction with ascorbic acid, while the Cu(I)-Cell-Cl− structure (pdb code 7PYI) was photoreduced by increasing the radiation dose.22Since all four crystal structures contain only non-hydrogen atoms, hydrogens were added with the program Maestro63 (which is part of the Schrödinger suite). From the resulting structures, we extracted the copper, the side chains of His1 and His78 (constituting the histidine brace), the side chains of His147, Gln162 and Tyr164, as well as any copper coordinating water molecules (labelled H2Oeq and H2Oax). Tyr164 is an axial ligand to the copper in AA9 and other LPMOs, while His147 and Gln162 are important residues in the second coordination sphere (see top left panel in Fig. 2). His1 is methylated1 and the methylation was always kept. His147 was always protonated in the Nε2 position.64 From the crystal structures with the substrate, three units of the cellotetraose, the chloride anion and the so-called pocket water21 (labelled H2Opkt) were extracted additionally. The latter binds to the terminal NH2 group as described in ref. 21. In the Cu(II) and Cu(I) structures without substrate, we further included a water molecule in a similar position to the pocket water (also labelled H2Opkt). In all extracted structures, His78, His147, Gln162 and Tyr164 were cut between Cα and Cβ, while the N-terminal coordinating His1 was cut between the backbone carbon atom (C) and Cα. Except for His1, only the side chain atoms of the residues were kept, and dangling bonds were saturated with hydrogen atoms. For the structures with cellotetraose, the three saccharide units closest to the copper centre were extracted, and the glycosidic bond was cut between the oxygen and the fourth saccharide unit. The resulting systems are shown in Fig. 2 and Fig. S8† (with optimized hydrogen positions, see below).
The hydrogen positions were optimized with the quantum chemistry software ORCA 5.0.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 65 using the DFT functional TPSS66 and the def2-SV(P) basis set67 with D3 dispersion correction,68 Becke–Johnson damping69 and the resolution of identity (RI) approximation.70 The auxiliary basis set corresponding to def2-SV(P) was used for the RI approximation (def2/J). The positions of all non-hydrogen atoms were fixed at their crystal structure positions. All energies, orbitals, orbital energies and spin densities were obtained from single-point calculations on these structures with the DFT functionals B3LYP71–73 and a def2-TZVPP67 basis set. We use the default version of B3LYP in ORCA, where the calculation of Coulomb (J) and exchange (X) terms is done with the split RI-J and chain-of-spheres for exchange (RIJCOSX) approximation.74 We additionally calculated the orbital energies with the TPSS66 functional. Since B3LYP and TPSS generally gave similar trends, we show only the B3LYP results in the main text and refer to section S3 in the ESI† for the TPSS results.
65 using the DFT functional TPSS66 and the def2-SV(P) basis set67 with D3 dispersion correction,68 Becke–Johnson damping69 and the resolution of identity (RI) approximation.70 The auxiliary basis set corresponding to def2-SV(P) was used for the RI approximation (def2/J). The positions of all non-hydrogen atoms were fixed at their crystal structure positions. All energies, orbitals, orbital energies and spin densities were obtained from single-point calculations on these structures with the DFT functionals B3LYP71–73 and a def2-TZVPP67 basis set. We use the default version of B3LYP in ORCA, where the calculation of Coulomb (J) and exchange (X) terms is done with the split RI-J and chain-of-spheres for exchange (RIJCOSX) approximation.74 We additionally calculated the orbital energies with the TPSS66 functional. Since B3LYP and TPSS generally gave similar trends, we show only the B3LYP results in the main text and refer to section S3 in the ESI† for the TPSS results.
In both substrate-bound structures (Cu(I)-Cell-Cl− and Cu(II)-Cell-Cl−) a chloride anion is present (it coordinates to copper in the Cu(II)-Cell-Cl− structure). The results for the structures with chloride are discussed in section S2 in the ESI.† We substituted chloride with water, keeping the oxygen atom fixed at the position of the chloride atom. The structures with water are labelled Cu(I)-Cell and Cu(II)-Cell (see Fig. 2) and their hydrogen positions were optimized as described above.
We further investigated the effect of H2O2 binding on the electronic structure. H2O2 was added to Cu(I) and Cu(I)-Cell-Cl−, replacing either the equatorial9,18,75 water or chloride (note that with 4.03 Å and 3.84 Å respective distances to copper, neither the water nor the chloride is in coordination distance). In all structures, coordinates of H2O2 were freely optimized together with the hydrogen atoms, employing the procedure described above. The Cu(I)-Cell⋯H2O2 structure is shown in Fig. 2. Instead of obtaining a pre-bound Cu(I)⋯H2O2 (i.e. the structure without substrate) with the procedure described above, we could only obtain a structure reminiscent of the “caged” state (indicated as Cu(II)-HO--OH in Fig. 1), where H2O2 coordinates to copper and the O–O distance is elongated to distances of approximately 2 or more ångström.18–20 The pre-bound form has previously been calculated with both TPSS and B3LYP using a QM/MM embedding scheme to mimic the protein environment18–20 and it was also obtained by Lim et al.51 who employed a QM-cluster approach. Therefore, we optimized the Cu(I)⋯H2O2 structure with the same functional (B3LYP) that Lim et al.51 used to obtain the pre-bound state, and obtained a similar orientation for H2O2 (see Fig. 2). Yet, the Cu–H2O2 distance is smaller in our Cu(I)⋯H2O2 structure (2.54 Å) compared to the structure by Lim et al.51 (2.93 Å), which could result from the fact that we kept the protein non-hydrogen atoms fixed at their crystal structure positions, while Lim et al.51 optimized the complete model.
Prompted by a previous investigation56 of the influence of the potential coupling of the substrate and the binding of O2, we further investigated the binding of O2 to substrate-free and substrate-bound structures (resulting in the structures in green boxes in the left part of Fig. 1, see Fig. S1†).
We mainly use a frontier MO approach to evaluate electronic structure changes. The characters of the MOs were estimated by Löwdin reduced orbital populations; they provide a percentage for the contribution of the atomic orbitals to each molecular orbital. From a computational perspective, Cu(I) is in a closed-shell singlet state, where all electrons are paired, i.e. α- and β-electrons within a given spatial orbital have the same energy. This is not the case for intermediates with Cu(II) oxidation state (or Cu(I) with O2), which are open-shell (doublet or triplet) states. In these states, α- and β-orbitals may differ in energy, and we refer separately to the α and β frontier orbitals for these calculations. Structures, individual orbitals and spin densities were plotted using PYMOL76 and the ORCA_PLOT tool. Atomic spin populations were calculated using Löwdin partitioning and quantum theory of atoms in molecules (QTAIM) atomic charges were calculated with AIMAll.77
Results & discussion
Effect of the priming electron on frontier orbitals
We start by investigating the first step of the LPMO mechanism, namely the reduction of the copper centre in the structures without substrate (reaction (1a) in Fig. 1). Upon reduction from Cu(II) to Cu(I), Tandrup et al. observe that the copper loses the equatorial and axial water molecules, thereby changing from elongated hexacoordinated to T-shaped geometry. In the T-shaped form, Cu(I) is only coordinated by the nitrogen atoms of the protein ligands. In the here studied structures without substrate (see Cu(II) and Cu(I) structures in Fig. 2 and Table 1), the Cu–O distances increase from 2.2 and 2.7 Å to 4.0 and 3.5 Å for the equatorial and axial water molecules, respectively, upon reduction.22| Structure | PDB entry | Nδ1 (Å) | Nter (Å) | Nε2 (Å) | OTyr (Å) | Equatorial ligand (Å) | Axial ligand (Å) | O–O (Å) |
|---|---|---|---|---|---|---|---|---|
| a The equatorial water (not in coordinating distance) is replaced with H2O2 and H2O2 is optimized. b Chloride is replaced with H2O and oxygen is constrained to the position of chloride. c Chloride is replaced with H2O2 and H2O2 is optimized. | ||||||||
| Cu(II) | 7pxi | 1.87 | 2.18 | 1.96 | 2.74 | 2.16 | 2.71 | n/a |
| Cu(I) | 7pxv | 1.84 | 2.29 | 1.99 | 2.81 | 4.03 | 3.49 | n/a |
| Cu(I)⋯H2O2 | 7pxv | 2.54 | 1.50 | |||||
| Cu(II)-Cell | 7pyd | 2.00 | 2.26 | 2.02 | 2.48 | 2.25 | n/a | n/a |
| Cu(I)-Cell | 7pyi | 1.89 | 2.50 | 2.00 | 2.69 | 3.84 | n/a | n/a |
| Cu(I)-Cell⋯H2O2 | 7pyi | 2.39 | 1.72 | |||||
The orbital energy diagrams of Cu(II) and Cu(I) are shown in Fig. 3 for B3LYP (and in Fig. S10† for TPSS). We focus first on the diagrams without substrate, i.e. the ones labelled Cu(II) and Cu(I). Concerning the frontier orbitals in the Cu(II) structure, both the α-and β-HOMOs have the character of tyrosine orbitals (see also Table S7†), and the (occupied) frontier orbitals are all of ligand character. The occupied orbitals of copper d-character are considerably (2.2–8.2 eV) lower in energy than the HOMO. The fact that the orbitals of d-character are removed from the frontier means that they are unlikely to take part in a reaction with an incoming co-substrate. On the other hand, the β-LUMO is mostly (to 62%) a copper d-orbital, namely the dx2−y2-orbital (see Fig. 4), making it the most likely orbital to accept an electron from a reductant.
 | ||
| Fig. 3 Normalized orbital energies and Löwdin reduced orbital populations per molecular orbital for Cu(II) and Cu(II)-Cell (left panels) as well as Cu(I) and Cu(I)-Cell structures (right panels), obtained from B3LYP/def2-TZVPP calculations (and directly derived from the crystal structures, see blue boxes Fig. 1). The colour indicates the residue with the largest Löwdin reduced orbital population (see legend). Löwdin reduced orbital populations are reported if the population of the copper d-orbitals is larger than 20%. Only these orbitals are shown in the top panels, whereas the bottom panels include all orbitals. The orbital energies are normalized to the HOMO energy. | ||
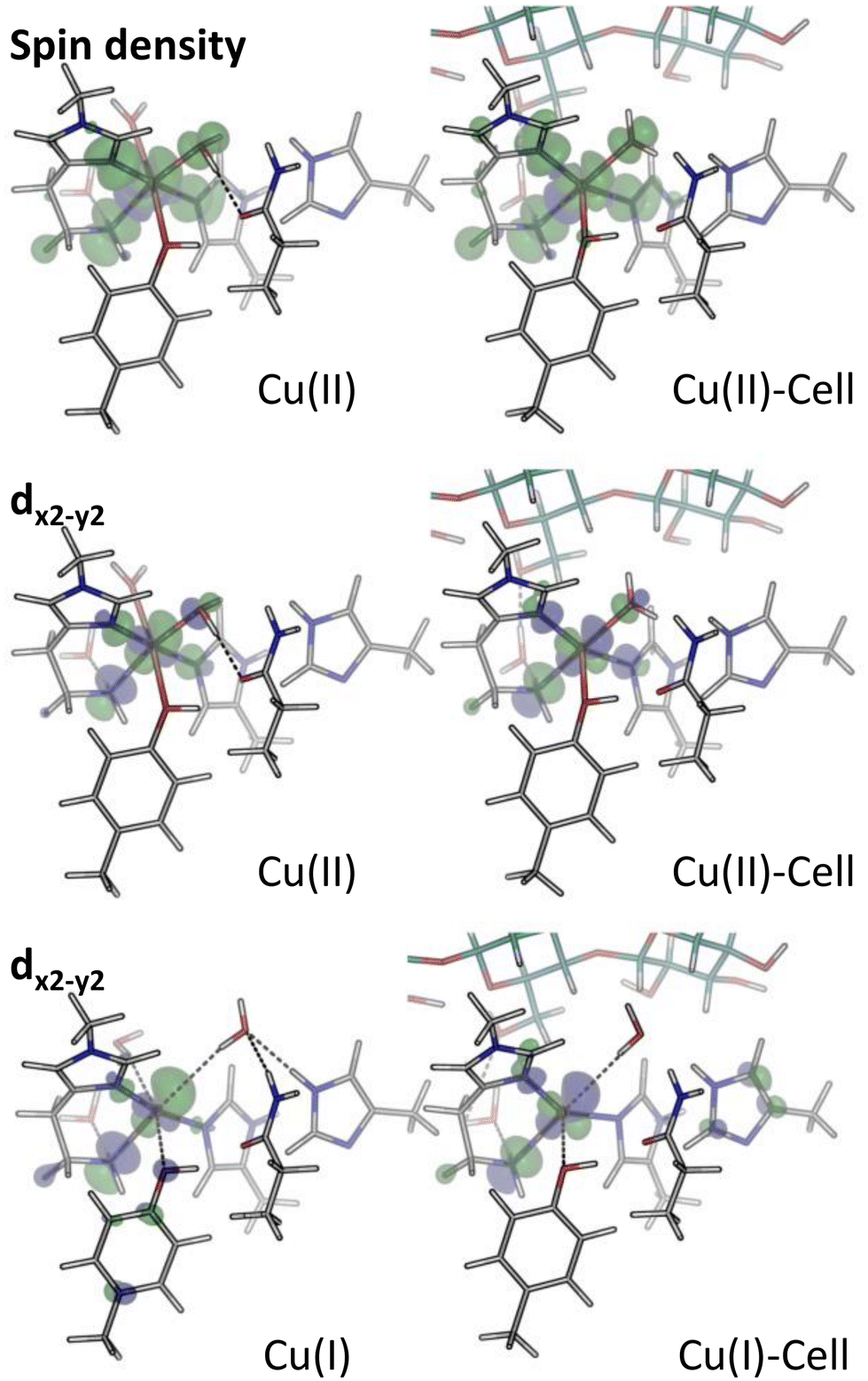 | ||
| Fig. 4 Spin density (isovalue 0.001 e bohr−3) and dx2−y2-orbitals (isovalue 0.05 e bohr−3) for the structures in blue boxes in Fig. 1, obtained from B3LYP/def2-TZVPP calculations. In both cases, green colour corresponds to positive values, and blue to negative ones. The spin density is defined as the difference in the densities contributed by the α and β-electrons. Shown is the LUMO for Cu(II) and Cu(II)-Cell, as well as the highest occupied orbital of copper d-character for Cu(I) and Cu(I)-Cell, in all cases corresponding to a dx2−y2-orbital. | ||
Moving to the orbital energies of the Cu(I) structure (see Fig. 3), the frontier MOs are dominated by orbitals of copper d-character. HOMO−1, HOMO−7, and HOMO−9 to HOMO−11 are mainly orbitals of copper d-character (40–90%) with some smaller contributions from the surrounding ligands (see Table S8†).
Hence, the priming electron brings the occupied orbitals of copper d-character to the frontier, where they are available for incoming small molecules. This provides a simple frontier-orbital-based explanation for the “priming reduction” requirement for activity.13,33,34,43
Comparison of substrate-free and substrate-bound systems
Having established the effect of the priming electron on the frontier orbitals, we next analyse the influence of substrate binding (corresponding to reactions (2a) and (2b) in Fig. 1), comparing the Cu(II) and Cu(II)-Cell structures as well as the Cu(I) and Cu(I)-Cell structures shown in Fig. 2.Tandrup et al. report two correlated changes to the active site geometries upon substrate binding: first, binding of the substrates induced a loss of planarity in the copper and the equatorial ligands that are almost coplanar in the Cu(II) structure without substrate. Second, the distance between copper and tyrosine was reduced by ca. 0.2 Å upon binding of the substrate to the Cu(II) structure (according to reaction (1b) in Fig. 1, see Table 1 for distances).22 A third change already described by Frandsen et al. is that the axial water molecule is displaced by a hydroxymethyl group of the substrate (see Fig. 2).21 In the following, we will describe how these structural changes influence the electronic structure.
For the individual oxidation states, the frontier orbital splittings are qualitatively similar before and after the addition of substrate (see Fig. 3 and S10†): the Cu(II)/Cu(II)-Cell systems display occupied frontier MOs mainly of ligand character for Cu(II) and ligand or substrate character for Cu(II)-Cell. The β-LUMOs are in both cases metal-based orbitals of d-character (dx2−y2 as depicted in Fig. 4). Thus, even if the substrate binds to the Cu(II) state, i.e. before reduction, the reduction of Cu(II)-Cell will likely result in the population of the same dx2−y2-orbital as in Cu(II).
Moving to the reduced Cu(I) and Cu(I)-Cell structures, we see from Fig. 3 (and Fig. S10†) that the orbitals of copper d-character are now in both cases close to the frontier – and that the binding of the substrate does not change this fact. The Cu(I) HOMO−1 is the dx2−y2-orbital shown in Fig. 4. Consistent with Lim et al.,51 we observe that the dx2−y2-orbital in the Cu(I) structure is isolated and 1.3–1.6 eV higher in energy than the other copper d-orbitals (see Table S8†). However, we also observe that after the substrate binds, the copper orbitals of d-character are closer in energy (see Cu(I)-Cell in Fig. 3 and S10†): the energy difference between the dx2−y2-orbital and the next lower copper d-orbital is reduced more than three-fold upon substrate binding, from 1.3 eV in the substrate-free Cu(I) structure to 0.4 eV in the substrate-bound Cu(I)-Cell structure. This was not seen by Lim et al. since they did not include a substrate. However, they did investigate the effect of the co-substrate binding (in this case H2O2) on these energy differences, and we therefore further analyse these differences in more detail in the following section.
Before explicitly including an oxidizing species, we note that the proposal with an isolated dx2−y2-orbital high in energy was also suggested by Courtade et al.56 They argued that substrate binding induced the relative increase of the dx2−y2-orbital. Their study was based on EPR spectra measured for BlAA10A with and without chitin56 and thus inherently based on the Cu(II) states. For the Cu(II) and Cu(II)-Cell structures, the spin density (Fig. 4) shows that for both substrate-bound and unbound states, the spin density is mostly localized on copper. The spin density closely mimics the dx2−y2-orbital, showing that it is reasonable to interpret this orbital as the single-occupied molecular orbital (SOMO), as done by Courtade et al. However, the increase in relative energy of the dx2−y2-orbital cannot be unequivocally confirmed. This may be due to large differences between the LsAA9A LPMO we study here and the AA10 LPMO studied by Courtade et al. or it is a consequence of the different methods employed to model the substrate (Courtade et al. did not include the substrate explicitly in their calculations but used a four-coordinate species to mimic a substrate-bound intermediate).
We further note that in all four structures (Cu(II), Cu(I), Cu(II)-Cell and Cu(I)-Cell), the HOMO is a tyrosine orbital (see Tables S7–S10†), but its character barely changes upon substrate binding despite the shortening of the Cu–OTyr distance and it is not involved in the reaction with the substrate (or co-substrate as we will see in the next section). Nevertheless, a small part of the spin density is redistributed to Tyr164 (involving both the oxygen atom and the π-system) upon substrate binding: essentially no spin density is located on tyrosine in Cu(II) (Löwdin spin population of 0.00 for tyrosine, see Table S3†), which increases to 0.02 in Cu(II)-Cell, reflecting the closer distance between copper and tyrosine in the substrate-bound structures.
In summary, the impact of substrate binding on the electronic structure is smaller compared to the impact of the reduction: the reduction brings the occupied copper d-orbitals to the frontier, and this impact of the reduction is not influenced significantly by the presence of the substrate. However, before substrate binding, there is a significant energy difference between the highest occupied copper dx2−y2-orbital and the remaining copper d-orbitals. This difference is reduced by a factor of three when the substrate binds.
Impact of an oxidizing species
For LPMOs in general (and particularly for LsAA9A), H2O2 has shown to be the (only) co-substrate.12,23,24 Therefore, we start by discussing substrate-free and substrate-bound structures with H2O2 replacing the equatorial water or chloride in Cu(I) and Cu(I)-Cell-Cl−, respectively (note that neither the water nor the chloride are coordinated in these structures). We aim to obtain the same pre-bound state as Lim et al.51 (indicated as Cu(I)⋯H2O2 in Fig. 1), but in one case additionally include a substrate (Cu(I)-Cell⋯H2O2 in Fig. 1). In the pre-bound state, H2O2 is not coordinated to Cu(I), but instead located in the pocket between the substrate and active site, where it is stabilized by interactions with a second-sphere histidine and glutamine (see His147 and Gln162 in Fig. 2).18–20 We observe that substrate binding brings H2O2 closer to copper (Cu–O distance of 2.54 Å and 2.39 Å in Cu(I)⋯H2O2 and Cu(I)-Cell⋯H2O2, respectively) and increases the O–O bond in H2O2 from 1.50 Å to 1.72 Å (see Table 1). QM/MM calculations of the pre-bound intermediate in LsAA9A (including the substrate) obtained a Cu–O distance of 2.77–2.98 Å and an O–O distance of 1.44–1.46 Å. In the caged intermediates the electron has been transferred from copper to the σ*-orbital of H2O2 (reaction (6b) in Fig. 1). These intermediates are in an open-shell singlet state, and they are characterized by long O–O distances of 1.97–2.14 Å and shorter Cu–O distances of approximately 1.9 Å.18–20 Hence, our Cu(I)-Cell⋯H2O2 intermediate is geometrically somewhat in between a pre-bound and a caged species.Indeed, upon binding of hydrogen peroxide, a small charge transfer occurs from copper to H2O2 (see Table S4†): the positive charge on copper increases from 0.61 in Cu(I) to 0.68 in Cu(I)⋯H2O2 and from 0.64 in Cu(I)-Cell to 0.77 in Cu(I)-Cell⋯H2O2. The net charge of the equatorial water (not in coordination distance) is close to neutral (−0.02 in Cu(I) and −0.01 in Cu(I)-Cell), whereas the net charge of H2O2 is more negative with −0.09 in Cu(I)⋯H2O2 and −0.25 in Cu(I)-Cell⋯H2O2. The larger magnitude of the charge transfer in the presence of substrate is consistent with the copper and H2O2 being closer in the structures with bound substrate, and supports that Cu(I)-Cell⋯H2O2 is somewhere in between a pre-bound and a caged species. Comparison with results for amyloid β-peptide models coordinated with copper confirms this observation.78
Analysing the orbital energy splittings for Cu(I)⋯H2O2 and Cu(I)-Cell⋯H2O2 in Fig. 5 (see Fig. S11† for TPSS results), we observe a similar character and splitting of the occupied frontier orbitals as in Cu(I) and Cu(I)-Cell, with the occupied orbitals of copper d-character close to the frontier. For Cu(I)⋯H2O2, we obtain an energy difference of 1.2–1.7 eV between the highest occupied dx2−y2-orbital and the remaining MOs of copper d-character (see Table S11†), which is consistent with the difference of 1.1–1.4 eV reported by Lim et al.51 We also find that the dx2−y2-orbital (see Fig. 6) is the HOMO in Cu(I)⋯H2O2. When the substrate binds, the HOMO is a tyrosine orbital but the dx2−y2-orbital (see Fig. 6) remains the highest-lying orbital with d-character, while the LUMO is always an orbital of co-substrate (H2O2) character. Each LUMO is shown in Fig. 6 and has the shape of a σ*-orbital. This agrees well with the LUMO described by Lim et al.51 However, as in the Cu(I) systems, we observe that substrate binding brings the remaining orbitals of copper d-character closer to the dx2−y2-orbital, decreasing the energy difference between them to 0.8–1.8 eV. Additionally, the substrate lowers the energy difference between the dx2−y2-orbital and the H2O2 σ*-orbital (from 3.9 eV in Cu(I)⋯H2O2 to 3.1 eV in Cu(I)-Cell⋯H2O2, see Tables S11 and S12†), making the electron transfer (reaction (6b) in Fig. 1) more favourable with the bound substrate. This is in line with QM/MM and QM-cluster calculations that show that this reaction is overall favourable in LsAA9A with and without substrate.19,20,51
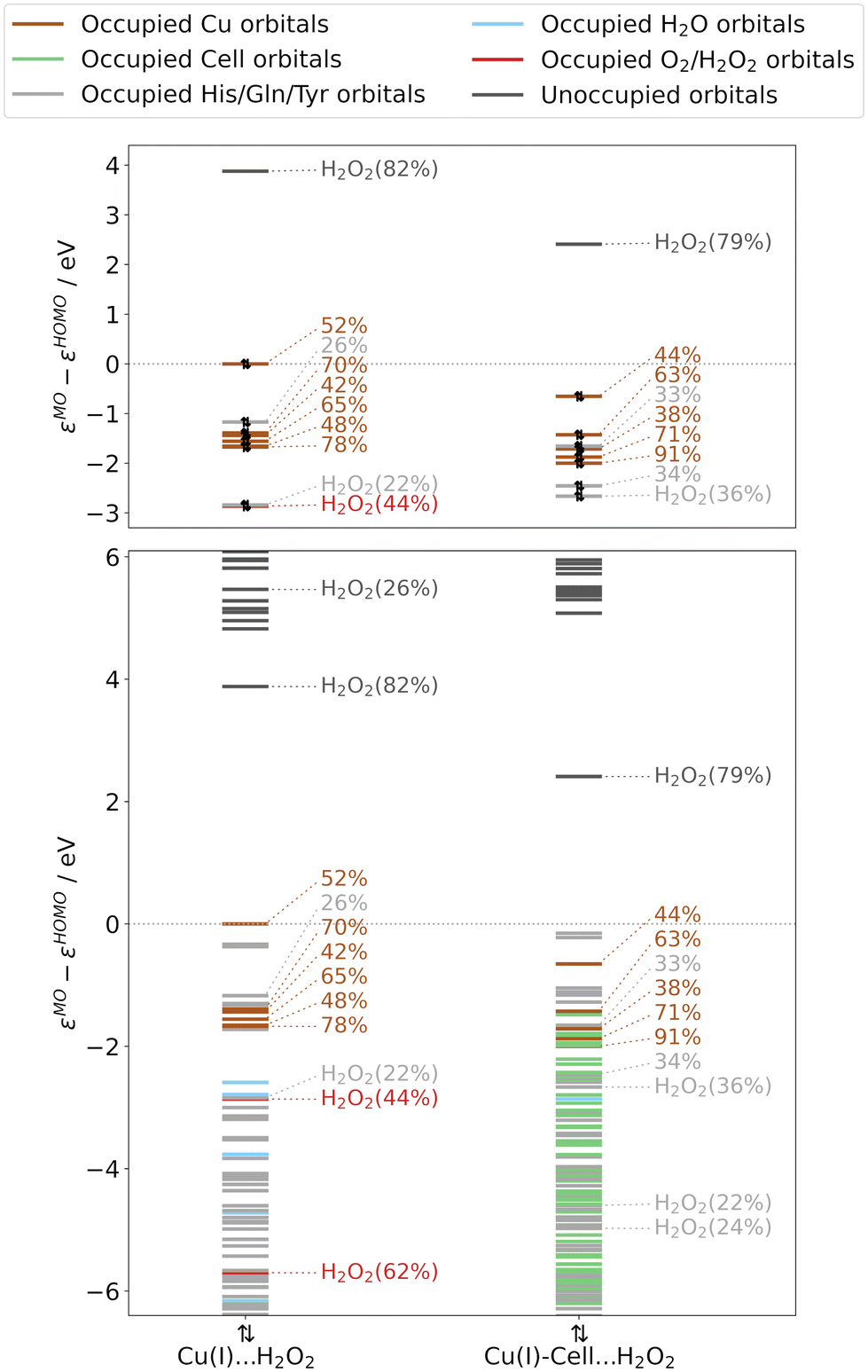 | ||
| Fig. 5 Normalized orbital energies and Löwdin reduced orbital populations per molecular orbital for substrate-free and substrate-bound structures with pre-bound H2O2, obtained from B3LYP/def2-TZVPP calculations (and obtained from adding H2O2 to the crystal structures, see right green boxes in Fig. 1). The colour indicates the residue with the largest Löwdin reduced orbital population (see legend). Löwdin reduced orbital population are reported if the population of the copper d-orbitals (here only numbers are given) and H2O2 orbitals is larger than 20%. Only these orbitals are shown in the top panels, whereas the bottom panels include all orbitals. The orbital energies are normalized to the HOMO energy. | ||
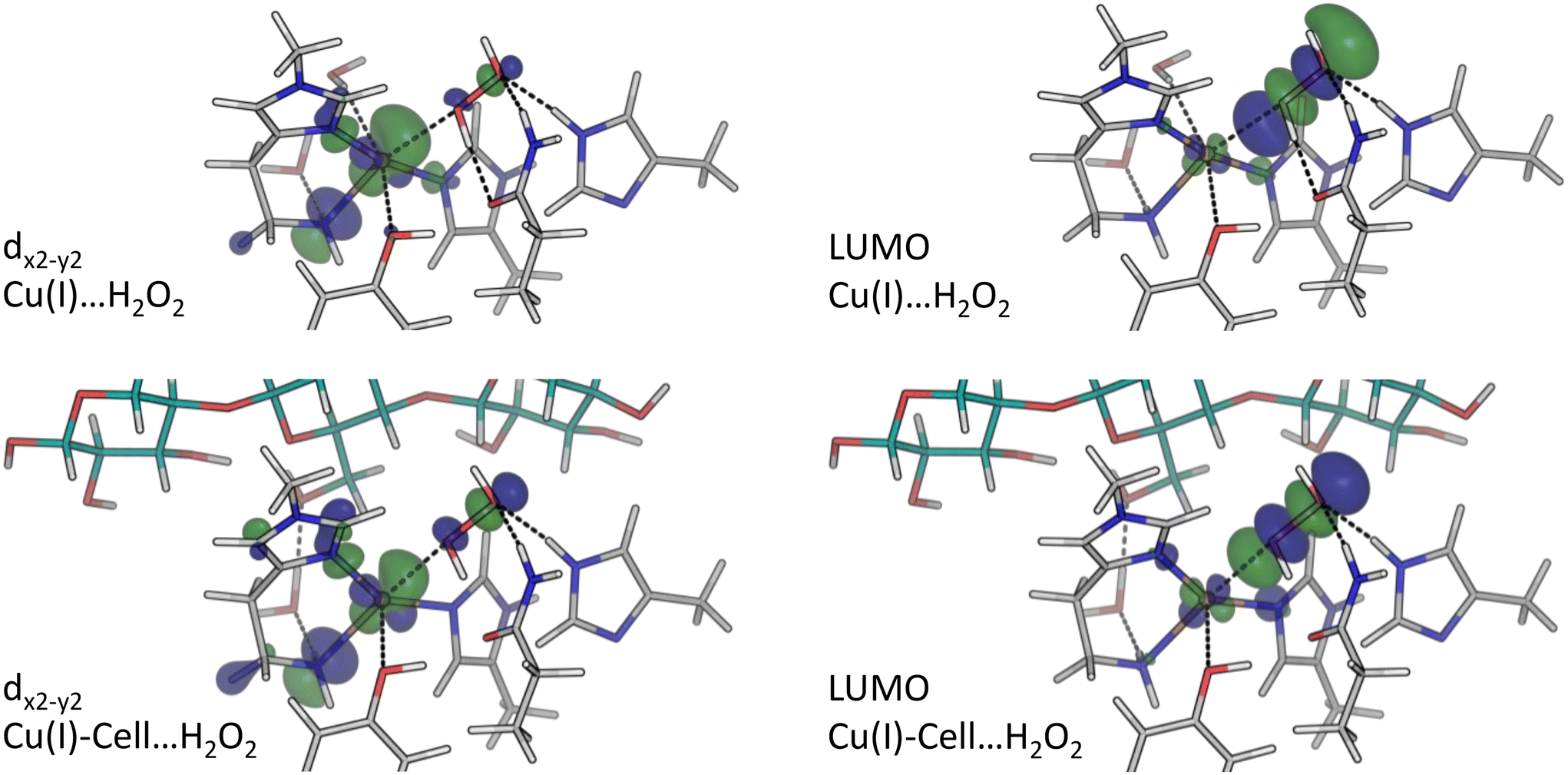 | ||
| Fig. 6 dx2−y2-orbitals and LUMO (isovalue 0.05 e bohr−3) for the structures with pre-bound H2O2 obtained from B3LYP/def2-TZVPP calculations (see Cu(I)⋯H2O2 and Cu(I)-Cell⋯H2O2 structures in Fig. 1). Green colour corresponds to positive values, and blue to negative ones. | ||
For comparison with older LPMO papers and to investigate if the conclusions for H2O2 were independent of the co-substrate, we additionally carried out the same experiment with O2 (see section S1 in the ESI† for further details). However, since several independent studies23,24 showed that the former is not a co-substrate of LsAA9A, we will discuss these results in the ESI.†
Conclusions
Starting from four different crystal structures of the LPMO LsAA9A, we investigated the influence of reduction and substrate binding on the electronic structure of LPMOs.Our results show that reduction from Cu(II) to Cu(I) has a significant influence on the orbital energies, bringing the occupied orbitals of copper d-character close to the frontier, where they can engage in a reaction. This is independent of the presence or absence of substrate binding and explains why the unique priming reduction is required for LPMO activity.
Further, we observed a similar orbital splitting in Cu(I) as a previous study51 in pre-bound Cu(I)⋯H2O2, where the highest occupied copper d-orbital (the dx2−y2) is raised by more than 1 eV in energy compared to the other occupied copper d-orbitals. Yet, this energy difference decreases by a factor of three upon substrate binding. Previous studies suggest that a relative rise in energy of the dx2−y2-orbital could be favourable for LPMO reactions, bringing the dx2−y2-orbital closer to the anti-bonding σ*- or π*-orbitals in hydrogen peroxide or dioxygen, respectively.51,56 Our observation seems to contradict this suggestion. However, the relative energy of the anti-bonding orbitals should also be considered as well as substrate binding. Therefore, we additionally investigated how pre-bound H2O2 (and O2) impact the electronic structure of substrate-free and substrate-bound LsAA9A. For the substrate-free H2O2 structure, we find that the dx2−y2-orbital is more than 1 eV higher in energy than the lower-lying copper d-orbitals, which is consistent with previous results.51 This energy difference is decreased by one-third upon substrate binding, which is consistent with our findings without co-substrate. However, substrate binding additionally causes the H2O2 σ*-orbital to be lower in energy, bringing it closer to the copper dx2−y2-orbital. Based on our calculations, it is this lowering of the H2O2 σ*-orbital upon substrate binding that facilitates the electron transfer from copper to H2O2. This does not occur in calculations with O2 or superoxide (see ESI†).
The above trends are independent of the employed functionals (B3LYP and TPSS). To further generalize our findings, and to compare to EPR data of AA10 LPMOs, substrate-bound structures of this and other LPMO families would be a valuable starting point.
Our findings have significant biological significance, as they explain why the reduction plays an important role in activating LsAA9A, independently of the presence or absence of substrate. Rather, the substrate binding has a more subtle impact on the electronic structure, facilitating the reaction between LPMO and H2O2. From an electronic structure perspective, it could thus be more important to control the supply of reductant and H2O2 than the substrate supply in experimental settings, since substrate binding alone does not seem to activate LsAA9A.
Author contributions
E. K. W.: conceptualization, investigation, formal analysis, validation, writing – original draft, writing – review & editing, visualization, project administration. L. L. L.: conceptualization, formal analysis, validation, writing – review & editing, supervision, funding acquisition, project administration. E. D. H.: conceptualization, formal analysis, validation, writing – review & editing, supervision, funding acquisition, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
EDH thanks The Villum Foundation, Young Investigator Program (grant no. 29412), the Swedish Research Council (grant no. 2019-04205), and Independent Research Fund Denmark (grant no. 2064-00002B) for support. LLL acknowledges funding from the Independent Research Fund Denmark (grant no. 8021-00273B). All of the computing for this project was performed on the GenomeDK cluster. The authors would like to thank GenomeDK and Aarhus University for providing computational resources and support that contributed to these research results.References
- R. J. Quinlan, M. D. Sweeney, L. Lo Leggio, H. Otten, J. C. Poulsen, K. S. Johansen, K. B. Krogh, C. I. Jorgensen, M. Tovborg, A. Anthonsen, T. Tryfona, C. P. Walter, P. Dupree, F. Xu, G. J. Davies and P. H. Walton, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15079–15084 CrossRef CAS PubMed.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sorlie and V. G. Eijsink, Science, 2010, 330, 219–222 CrossRef CAS PubMed.
- P. V. Harris, D. Welner, K. C. McFarland, E. Re, J. C. Navarro Poulsen, K. Brown, R. Salbo, H. Ding, E. Vlasenko, S. Merino, F. Xu, J. Cherry, S. Larsen and L. Lo Leggio, Biochemistry, 2010, 49, 3305–3316 CrossRef CAS PubMed.
- V. V. Vu, W. T. Beeson, E. A. Span, E. R. Farquhar and M. A. Marletta, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13822–13827 CrossRef CAS PubMed.
- L. Lo Leggio, T. J. Simmons, J. C. Poulsen, K. E. Frandsen, G. R. Hemsworth, M. A. Stringer, P. von Freiesleben, M. Tovborg, K. S. Johansen, L. De Maria, P. V. Harris, C. L. Soong, P. Dupree, T. Tryfona, N. Lenfant, B. Henrissat, G. J. Davies and P. H. Walton, Nat. Commun., 2015, 6, 5961 CrossRef CAS PubMed.
- M. Couturier, S. Ladeveze, G. Sulzenbacher, L. Ciano, M. Fanuel, C. Moreau, A. Villares, B. Cathala, F. Chaspoul, K. E. Frandsen, A. Labourel, I. Herpoel-Gimbert, S. Grisel, M. Haon, N. Lenfant, H. Rogniaux, D. Ropartz, G. J. Davies, M. N. Rosso, P. H. Walton, B. Henrissat and J. G. Berrin, Nat. Chem. Biol., 2018, 14, 306–310 CrossRef CAS PubMed.
- F. Sabbadin, G. R. Hemsworth, L. Ciano, B. Henrissat, P. Dupree, T. Tryfona, R. D. S. Marques, S. T. Sweeney, K. Besser, L. Elias, G. Pesante, Y. Li, A. A. Dowle, R. Bates, L. D. Gomez, R. Simister, G. J. Davies, P. H. Walton, N. C. Bruce and S. J. McQueen-Mason, Nat. Commun., 2018, 9, 756 CrossRef PubMed.
- C. Filiatrault-Chastel, D. Navarro, M. Haon, S. Grisel, I. Herpoel-Gimbert, D. Chevret, M. Fanuel, B. Henrissat, S. Heiss-Blanquet, A. Margeot and J. G. Berrin, Biotechnol. Biofuels, 2019, 12, 55 CrossRef PubMed.
- F. Sabbadin, S. Urresti, B. Henrissat, A. O. Avrova, L. R. J. Welsh, P. J. Lindley, M. Csukai, J. N. Squires, P. H. Walton, G. J. Davies, N. C. Bruce, S. C. Whisson and S. J. McQueen-Mason, Science, 2021, 373, 774–779 CrossRef CAS PubMed.
- J. O. Ipsen, M. Hallas-Moller, S. Brander, L. Lo Leggio and K. S. Johansen, Biochem. Soc. Trans., 2021, 49, 531–540 CrossRef CAS PubMed.
- K. K. Meier, S. M. Jones, T. Kaper, H. Hansson, M. J. Koetsier, S. Karkehabadi, E. I. Solomon, M. Sandgren and B. Kelemen, Chem. Rev., 2018, 118, 2593–2635 CrossRef CAS PubMed.
- Z. Forsberg, M. Sorlie, D. Petrovic, G. Courtade, F. L. Aachmann, G. Vaaje-Kolstad, B. Bissaro, A. K. Rohr and V. G. Eijsink, Curr. Opin. Struct. Biol., 2019, 59, 54–64 CrossRef CAS PubMed.
- T. M. Vandhana, J. L. Reyre, D. Sushmaa, J. G. Berrin, B. Bissaro and J. Madhuprakash, New Phytol., 2022, 233, 2380–2396 CrossRef CAS PubMed.
- S. Garcia-Santamarina, C. Probst, R. A. Festa, C. Ding, A. D. Smith, S. E. Conklin, S. Brander, L. N. Kinch, N. V. Grishin, K. J. Franz, P. Riggs-Gelasco, L. Lo Leggio, K. S. Johansen and D. J. Thiele, Nat. Chem. Biol., 2020, 16, 337–344 CrossRef CAS PubMed.
- F. Askarian, S. Uchiyama, H. Masson, H. V. Sorensen, O. Golten, A. C. Bunaes, S. Mekasha, A. K. Rohr, E. Kommedal, J. A. Ludviksen, M. O. Arntzen, B. Schmidt, R. H. Zurich, N. M. van Sorge, V. G. H. Eijsink, U. Krengel, T. E. Mollnes, N. E. Lewis, V. Nizet and G. Vaaje-Kolstad, Nat. Commun., 2021, 12, 1230 CrossRef CAS PubMed.
- K. S. Johansen, Biochem. Soc. Trans., 2016, 44, 143–149 CrossRef CAS PubMed.
- G. Muller, A. Varnai, K. S. Johansen, V. G. Eijsink and S. J. Horn, Biotechnol. Biofuels, 2015, 8, 187 CrossRef PubMed.
- E. Drula, M. L. Garron, S. Dogan, V. Lombard, B. Henrissat and N. Terrapon, Nucleic Acids Res., 2022, 50, D571–D577 CrossRef CAS PubMed.
- C. H. Kjaergaard, M. F. Qayyum, S. D. Wong, F. Xu, G. R. Hemsworth, D. J. Walton, N. A. Young, G. J. Davies, P. H. Walton, K. S. Johansen, K. O. Hodgson, B. Hedman and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8797–8802 CrossRef CAS PubMed.
- W. T. Beeson, V. V. Vu, E. A. Span, C. M. Phillips and M. A. Marletta, Annu. Rev. Biochem., 2015, 84, 923–946 CrossRef CAS PubMed.
- B. Bissaro and V. G. H. Eijsink, Essays Biochem., 2023, 67, 575–584 CrossRef PubMed.
- B. Bissaro, A. K. Rohr, G. Muller, P. Chylenski, M. Skaugen, Z. Forsberg, S. J. Horn, G. Vaaje-Kolstad and V. G. H. Eijsink, Nat. Chem. Biol., 2017, 13, 1123–1128 CrossRef CAS PubMed.
- S. Brander, R. Tokin, J. Ø. Ipsen, P. E. Jensen, C. Hernández-Rollán, M. H. H. Nørholm, L. Lo Leggio, P. Dupree and K. S. Johansen, ACS Catal., 2021, 11, 13848–13859 CrossRef CAS.
- H. Chang, N. Gacias Amengual, A. Botz, L. Schwaiger, D. Kracher, S. Scheiblbrandner, F. Csarman and R. Ludwig, Nat. Commun., 2022, 13, 6258 CrossRef CAS PubMed.
- P. H. Walton and G. J. Davies, J. Biol. Inorg. Chem., 2022, 27, 705–713 CrossRef CAS PubMed.
- T. Isaksen, B. Westereng, F. L. Aachmann, J. W. Agger, D. Kracher, R. Kittl, R. Ludwig, D. Haltrich, V. G. Eijsink and S. J. Horn, J. Biol. Chem., 2014, 289, 2632–2642 CrossRef CAS PubMed.
- R. Kittl, D. Kracher, D. Burgstaller, D. Haltrich and R. Ludwig, Biotechnol. Biofuels, 2012, 5, 79 CrossRef CAS PubMed.
- V. G. H. Eijsink, D. Petrovic, Z. Forsberg, S. Mekasha, A. K. Rohr, A. Varnai, B. Bissaro and G. Vaaje-Kolstad, Biotechnol. Biofuels, 2019, 12, 58 CrossRef PubMed.
- B. Bissaro, E. Kommedal, A. K. Rohr and V. G. H. Eijsink, Nat. Commun., 2020, 11, 890 CrossRef CAS PubMed.
- A. A. Stepnov, V. G. H. Eijsink and Z. Forsberg, Sci. Rep., 2022, 12, 6129 CrossRef CAS PubMed.
- A. A. Stepnov, Z. Forsberg, M. Sorlie, G. S. Nguyen, A. Wentzel, A. K. Rohr and V. G. H. Eijsink, Biotechnol. Biofuels, 2021, 14, 28 CrossRef CAS PubMed.
- O. Golten, I. Ayuso-Fernandez, K. R. Hall, A. A. Stepnov, M. Sorlie, A. K. Rohr and V. G. H. Eijsink, FEBS Lett., 2023, 597, 1363–1374 CrossRef CAS PubMed.
- D. Kracher, S. Scheiblbrandner, A. K. Felice, E. Breslmayr, M. Preims, K. Ludwicka, D. Haltrich, V. G. Eijsink and R. Ludwig, Science, 2016, 352, 1098–1101 CrossRef CAS PubMed.
- M. Frommhagen, A. H. Westphal, W. J. H. van Berkel and M. A. Kabel, Front. Microbiol., 2018, 9, 1080 CrossRef PubMed.
- D. Kracher, M. Andlar, P. G. Furtmuller and R. Ludwig, J. Biol. Chem., 2018, 293, 1676–1687 CrossRef CAS PubMed.
- P. H. Walton, G. J. Davies, D. E. Diaz and J. P. Franco-Cairo, FEBS Lett., 2023, 597, 485–494 CrossRef CAS PubMed.
- K. D. Dubey and S. Shaik, Acc. Chem. Res., 2019, 52, 389–399 CrossRef PubMed.
- S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949–1017 CrossRef CAS PubMed.
- T. M. Hedison, E. Breslmayr, M. Shanmugam, K. Karnpakdee, D. J. Heyes, A. P. Green, R. Ludwig, N. S. Scrutton and D. Kracher, FEBS J., 2021, 288, 4115–4128 CrossRef CAS PubMed.
- M. M. Hagemann and E. D. Hedegard, Chemistry, 2023, 29, e202202379 CrossRef CAS PubMed.
- B. Wang, E. M. Johnston, P. Li, S. Shaik, G. J. Davies, P. H. Walton and C. Rovira, ACS Catal., 2018, 8, 1346–1351 CrossRef CAS.
- E. D. Hedegard and U. Ryde, Chem. Sci., 2018, 9, 3866–3880 RSC.
- K. E. Frandsen, T. J. Simmons, P. Dupree, J. C. Poulsen, G. R. Hemsworth, L. Ciano, E. M. Johnston, M. Tovborg, K. S. Johansen, P. von Freiesleben, L. Marmuse, S. Fort, S. Cottaz, H. Driguez, B. Henrissat, N. Lenfant, F. Tuna, A. Baldansuren, G. J. Davies, L. Lo Leggio and P. H. Walton, Nat. Chem. Biol., 2016, 12, 298–303 CrossRef CAS PubMed.
- T. Tandrup, S. J. Muderspach, S. Banerjee, G. Santoni, J. O. Ipsen, C. Hernandez-Rollan, M. H. H. Norholm, K. S. Johansen, F. Meilleur and L. Lo Leggio, IUCrJ, 2022, 9, 666–681 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
- P. Chylenski, B. Bissaro, M. Sorlie, Å. K. Rohr, A. Várnai, S. J. Horn and V. G. H. Eijsink, ACS Catal., 2019, 9, 4970–4991 CrossRef CAS.
- O. Caldararu, E. Oksanen, U. Ryde and E. D. Hedegard, Chem. Sci., 2019, 10, 576–586 RSC.
- B. Wang, Z. Wang, G. J. Davies, P. H. Walton and C. Rovira, ACS Catal., 2020, 10, 12760–12769 CrossRef CAS.
- C. Laurent, E. Breslmayr, D. Tunega, R. Ludwig and C. Oostenbrink, Biochemistry, 2019, 58, 1226–1235 CrossRef CAS PubMed.
- Z. Wang, S. Feng, C. Rovira and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 2385–2392 CrossRef CAS PubMed.
- H. Lim, M. T. Brueggemeyer, W. J. Transue, K. K. Meier, S. M. Jones, T. Kroll, D. Sokaras, B. Kelemen, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2023, 145, 16015–16025 CrossRef CAS PubMed.
- A. Paradisi, E. M. Johnston, M. Tovborg, C. R. Nicoll, L. Ciano, A. Dowle, J. McMaster, Y. Hancock, G. J. Davies and P. H. Walton, J. Am. Chem. Soc., 2019, 141, 18585–18599 CrossRef CAS PubMed.
- T. J. Simmons, K. E. H. Frandsen, L. Ciano, T. Tryfona, N. Lenfant, J. C. Poulsen, L. F. L. Wilson, T. Tandrup, M. Tovborg, K. Schnorr, K. S. Johansen, B. Henrissat, P. H. Walton, L. Lo Leggio and P. Dupree, Nat. Commun., 2017, 8, 1064 CrossRef CAS PubMed.
- L. Bertini, R. Breglia, M. Lambrughi, P. Fantucci, L. De Gioia, M. Borsari, M. Sola, C. A. Bortolotti and M. Bruschi, Inorg. Chem., 2018, 57, 86–97 CrossRef CAS PubMed.
- A. S. Borisova, T. Isaksen, M. Dimarogona, A. A. Kognole, G. Mathiesen, A. Varnai, A. K. Rohr, C. M. Payne, M. Sorlie, M. Sandgren and V. G. Eijsink, J. Biol. Chem., 2015, 290, 22955–22969 CrossRef CAS PubMed.
- G. Courtade, L. Ciano, A. Paradisi, P. J. Lindley, Z. Forsberg, M. Sorlie, R. Wimmer, G. J. Davies, V. G. H. Eijsink, P. H. Walton and F. L. Aachmann, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 19178–19189 CrossRef CAS PubMed.
- B. Bissaro, I. Isaksen, G. Vaaje-Kolstad, V. G. H. Eijsink and A. K. Rohr, Biochemistry, 2018, 57, 1893–1906 CrossRef CAS PubMed.
- P. Chen and E. I. Solomon, J. Inorg. Biochem., 2002, 88, 368–374 CrossRef CAS PubMed.
- R. E. Cowley, J. Cirera, M. F. Qayyum, D. Rokhsana, B. Hedman, K. O. Hodgson, D. M. Dooley and E. I. Solomon, J. Am. Chem. Soc., 2016, 138, 13219–13229 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- S. E. Bowman, J. Bridwell-Rabb and C. L. Drennan, Acc. Chem. Res., 2016, 49, 695–702 CrossRef CAS PubMed.
- M. Gudmundsson, S. Kim, M. Wu, T. Ishida, M. H. Momeni, G. Vaaje-Kolstad, D. Lundberg, A. Royant, J. Stahlberg, V. G. Eijsink, G. T. Beckham and M. Sandgren, J. Biol. Chem., 2014, 289, 18782–18792 CrossRef CAS PubMed.
- Schrödinger Release 2021-2, Maestro, Schrödinger, LLC, New York, NY, 2021.
- S. Banerjee, S. J. Muderspach, T. Tandrup, K. E. H. Frandsen, R. K. Singh, J. O. Ipsen, C. Hernandez-Rollan, M. H. H. Norholm, M. J. Bjerrum, K. S. Johansen and L. Lo Leggio, Biomolecules, 2022, 12, 194 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- A. D. Becke, Phys. Rev. A: Gen. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- E. D. Hedegard and U. Ryde, ACS Omega, 2017, 2, 536–545 CrossRef PubMed.
- Schrödinger, The PyMOL Molecular Graphics System, Version 2.6, 2015 Search PubMed.
- T. A. Keith, TK Gristmill Software, Overland Park KS, USA, 2019 Search PubMed.
- T. Prosdocimi, L. De Gioia, G. Zampella and L. Bertini, J. Biol. Inorg. Chem., 2016, 21, 197–212 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Selected distances and angles for investigated structures. Löwdin spin populations for all open-shell systems. QTAIM atomic charges for copper and summed for all residues. Section S1. Impact of pre-bound oxygen vs. superoxide. Section S2. Impact of chloride. Section S3. Results with TPSS. Section S4. Selected orbital energies and Löwdin reduced orbital populations. See DOI: https://doi.org/10.1039/d3dt04275h |
| This journal is © The Royal Society of Chemistry 2024 |