
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombining photocontrolled-cationic and anionic-group-transfer polymerizations using a universal mediator: enabling access to two- and three-mechanism block copolymers†
Brandon M.
Hosford‡
 ,
William
Ramos‡
and
Jessica R.
Lamb
*
,
William
Ramos‡
and
Jessica R.
Lamb
*
Department of Chemistry, University of Minnesota—Twin Cities, 207 Pleasant Street SE, Minneapolis, MN 55455, USA. E-mail: jrlamb@umn.edu
First published on 30th July 2024
Abstract
An ongoing challenge in polymer chemistry is accessing diverse block copolymers from multiple polymerization mechanisms and monomer classes. One strategy to accomplish this goal without intermediate compatibilization steps is the use of universal mediators. Thiocarbonyl thio (TCT) functional groups are well-known mediators to combine radical with either cationic or anionic polymerization, but a sequential cationic-anionic universal mediator system has never been reported. Herein, we report a TCT universal mediator that can sequentially perform photocontrolled cationic polymerization and thioacyl anionic group transfer polymerization to access poly(ethyl vinyl ether)-block-poly(thiirane) polymers for the first time. Thermal analyses of these block copolymers provide evidence of microphase separation. The success of this system, along with the established compatibility of radical polymerization, enabled us to further chain extend the cationic-anionic diblock using radical polymerization of N-isopropylacrylamide. The resulting terpolymer represents the first example of a triblock made from three different monomer classes incorporated via three different mechanisms without any end-group modification steps. The development of this simple, sequential synthesis using a universal mediator approach opens up new possibilities by providing facile access to diverse block copolymers of vinyl ethers, thiiranes, and acrylamides.
Introduction
Block copolymers (BCPs) are important and versatile materials due to their unique properties and functions, which are dictated by their composition and microstructure.1–3 Most BCPs are made from a single polymerization mechanism where compatible monomers are added sequentially.4–6 However, this approach limits the accessible chemical space to similar monomer classes7–11 or to a prescribed addition order to maintain the active chain-end.6,12–16 Ideally, polymer chemists would not be confined to this paradigm, such that disparate monomers could be combined to access an expanded property space. To accomplish this goal, it is necessary to develop new ways to easily incorporate different polymerization mechanisms (e.g., cationic, anionic, and radical) into a single material. Combining cationic and anionic mechanisms has been particularly understudied,17 presumably due to the incompatibility of their reactive intermediates.Common methods to combine multiple mechanisms – such as homopolymer coupling,18–20 end-group modification,21–34 or the use of multi-functional initiators35–38 – require additional synthetic steps and/or are limited in the number and arrangement of blocks in the final material. Universal mediators – which are functional groups that can facilitate distinct mechanisms under different conditions – are an elegant solution to these challenges (Fig. 1A). Due to the fact that the mediator stays at the chain end, multi-mechanism BCPs can be made via simple sequential polymerizations, and the user is theoretically not limited in the number of blocks accessible. Thiocarbonyl thio (TCT) moieties are well-studied universal mediators to sequentially combine radical polymerization with either cationic39–44 or anionic group transfer45–50 polymerization, but they have never been reported to mediate cationic and anionic polymerizations (Fig. 1B), possibly due to the opposing natures of these two mechanisms resulting in vastly different reaction condition requirements. Bridging this gap would open new and exciting possibilities to access novel poly(vinyl ether)-b-poly(thiirane) materials and to change between all three (i.e., cationic, anionic, and radical) polymerizations without extra compatibilization steps – vastly expanding the available chemical space accessible via this streamlined synthetic method. On the basis of the rich literature of cationic40,51–58 and anionic45,59,60 homopolymerizations using TCTs, we hypothesized that proper selection of the monomers, R and Z groups of the TCT,61,62 and reaction conditions would successfully enable sequential cationic-anionic polymerizations using a universal mediator for the first time.
 | ||
| Fig. 1 (A) General scheme for the synthesis of BCPs using a universal mediator. (B) Previous TCT universal mediator reports for radical/cationic and radical/anionic polymerizations. (C) This work combining cationic and anionic polymerizations using a TCT universal mediator. | ||
Herein, we report a straightforward, multi-pot, sequential method that combines photocontrolled cationic polymerization (photo-CP) and thioacyl anionic group transfer polymerization (TAGT) to generate novel BCPs of poly(vinyl ether)s and poly(thiirane)s (Fig. 1C). We identified a viable TCT universal mediator and explored [monomer]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[TCT] achieving number-average molar mass (Mn) up to 53 kDa and monomer scope of the homopolymerizations. Next, poly(ethyl vinyl ether) (p(EVE)) was chain extended with a variety of thiiranes to form diblock copolymers which were characterized by NMR spectroscopy, size-exclusion chromatography (SEC), thermogravimetric analysis (TGA), and differential scanning calorimetry (DSC). For each BCP, we observed two glass transition temperatures (Tgs), which suggest microphase separation between the blocks.63 Capitalizing on the success of this method completing the trio of mechanisms using this universal mediator, we further chain extended the BCPs with N-isopropylacrylamide (NIPAM) via photocontrolled radical polymerization. This novel triblock terpolymer is produced from three different mechanisms and three monomer classes without any end-group manipulations using this universal mediator approach.
:[TCT] achieving number-average molar mass (Mn) up to 53 kDa and monomer scope of the homopolymerizations. Next, poly(ethyl vinyl ether) (p(EVE)) was chain extended with a variety of thiiranes to form diblock copolymers which were characterized by NMR spectroscopy, size-exclusion chromatography (SEC), thermogravimetric analysis (TGA), and differential scanning calorimetry (DSC). For each BCP, we observed two glass transition temperatures (Tgs), which suggest microphase separation between the blocks.63 Capitalizing on the success of this method completing the trio of mechanisms using this universal mediator, we further chain extended the BCPs with N-isopropylacrylamide (NIPAM) via photocontrolled radical polymerization. This novel triblock terpolymer is produced from three different mechanisms and three monomer classes without any end-group manipulations using this universal mediator approach.
Results and discussion
Universal mediator identification
We began our compatibilization efforts by adapting reported reaction conditions for photo-CP64 and TAGT.46 For the photo-CP of ethyl vinyl ether (EVE), we used the classic oxidizing photocatalyst 2,4,6-tri-(p-methoxyphenyl)pyrylium tetrafluoroborate (PC) in dichloromethane (DCM). For TAGT of 3-phenoxypropylene sulfide (POPS), we used tetraphenyl phosphonium chloride (TPPCl) as the nucleophilic catalyst in N,N-dimethylacetamide (DMAc).We explored a number of TCT mediators, varying the R and Z groups (Table 1) to determine the optimal mediator for both photo-CP and TAGT. We hypothesized that a thioacetal R group would be vital for controlled initiation of photo-CP44,65,66 because the thioacetal helps facilitate the C–S bond cleavage to generate the cationic chain end. On the other hand, the Z group will dictate the nucleophilic attack of the activated thiolate monomer in TAGT by controlling the electrophilicity of the thiocarbonyl carbon. If the TCT is not electrophilic enough, the thiolate can instead directly attack another thiirane monomer, leading to uncontrolled polymerization characterized by a large dispersity and an Mn much larger than the theoretical Mn. As expected, polymerizations with TCT167 and TCT2, which are common TCTs used for photo-CP64 and TAGT,46,48 respectively, resulted in well-controlled polymerizations for their reported mechanisms (Table 1, entries 1a and 2b), but had poor control for the other polymerization (entries 1b and 2a). The dithiocarbamate of TCT1 is not electrophilic enough, resulting in uncontrolled TAGT, and TCT2 lacks the thioacetal R group for efficient photo-CP initiation. Combining the two important features into TCT3 – a more electrophilic trithiocarbonate with a thioacetal R group – resulted in moderate control for both photo-CP and TAGT (entries 3a and 3b).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TCT | Entry | Cationic (photo-CP)a | Entry | Anionic (TAGT)b | ||||||
| Conv.c (%) | M n,theo (kDa) | M n (kDa) | Ð | Conv.d (%) | M n,theo (kDa) | M n (kDa) | Ð | |||
| a Photo-CP standard conditions: TCT (20 μmol, 1 eq.), EVE (50 eq.), PC (2.0 mol% relative to TCT), DCM (92 mM), RT, 456 nm LED, 6 h. b TAGT standard conditions: TCT (15 μmol, 1 eq.), POPS (100 eq.), TPPCl (0.33 eq.), DMAc (1.0 mL), 60 °C, 6 h. c Determined via1H NMR spectroscopy. d Determined via SEC in DMF with 0.025 M LiBr against PMMA standards. e Polymerization run with no PC. f Polymerization run with 1.0 mol% of PC relative to TCT. n.d. = not determined. | ||||||||||
| TCT1 | 1a | 95 | 3.7 | 3.8 | 1.13 | 1b | 92 | 15.8 | 97.2 | 3.55 |
| TCT2 | 2a | >99 | 3.9 | 6.2 | 2.51 | 2b | >99 | 17.4 | 16.5 | 1.22 |
| TCT3 | 3a | >99 | 3.8 | 3.8 | 1.34 | 3b | 88 | 15.6 | 19.5 | 1.35 |
| TCT3 | 4ae | <1 | — | — | — | 4b | n.d. | n.d. | n.d. | n.d. |
| — | 5a | 99 | — | 10.1 | 1.95 | 5b | 73 | — | 289.6 | 1.87 |
| TCT3 | 6af | 98 | 3.8 | 4.5 | 1.17 | 6bf | 60 | 9.92 | 14.1 | 1.67 |

Control experiments showed that no p(EVE) was made in the absence of PC (entry 4a) and that TCT is necessary for controlled polymerization in both cases (entries 5a and 5b). Successful polymerization of EVE without the presence of a mediator is evidence of direct oxidation of the vinyl ether monomer by PC, but previous studies have shown that TCT oxidation occurs much faster than direct monomer oxidation at low [vinyl ether]:[TCT] ratios.66
Factors affecting sequential polymerization
During the initial polymerizations, we discovered that p(EVE) is difficult to purify because it is highly soluble (>10 mg mL−1) in a wide range of solvents – hexanes, diethyl ether, tetrahydrofuran, DCM, methanol, and DMAc (see ESI, Section S1.A†). Therefore, it was necessary to explore the effect of the photo-CP components on TAGT. Even a small amount of DCM inhibits TAGT, but both residual monomer and DCM can be completely removed from the polymer by drying in a vacuum oven at 70 °C for 16 hours. Conversely, the PC is not volatile, which makes it more challenging to remove. Unfortunately, dialysis was unsuccessful, and precipitation into water resulted in co-precipitation of p(EVE) and PC; therefore, we were unable to effectively remove the PC from the polymer. Thus, we tested the effect of varying amounts of PC on the TAGT (see ESI, Section S2.A†). At high loadings, PC inhibits the anionic polymerization (13% conversion in 6 h at 4 mol% PC relative to TCT). However, at 1 mol% PC, most of the activity is retained (60% conversion in 6 h; Table 1, entry 6b). During this exploration, we discovered the photo-CP still achieved 98% conversion at half of our standard loading (entry 6a). Therefore, in order to minimize the amount of PC contamination for chain extensions, we continued using the halved PC loading moving forward.Homopolymerization scope
Next, we explored the scope and limitations of TCT3 as a mediator for both polymerizations. To determine control over a wide molar mass range, we performed a [monomer]:[TCT3] screen for photo-CP of EVE and TAGT of POPS (Fig. 2; for tabulated data, see Section S2.B†). Photo-CP performed well when [EVE]:[TCT] was 50:1 and 100:1 – resulting in a close match of Mn to Mn,theo and a low dispersity. However, at larger [EVE]:[TCT] (200–300:1), Mn deviates from Mn,theo, which is common for photo-CP due to the increased levels of direct monomer oxidation.66 This oxidation acts as a competing initiation pathway, leading to more chains and resulting in a lower Mn.68 TAGT performed well for all [POPS]:[TCT] screened and resulted in good agreement between Mn and Mn,theo, achieving molar masses up to 53 kDa while maintaining a low-to-moderate dispersity (<1.55).
 | ||
| Fig. 2 [Monomer]:[TCT3] screen of (A) photo-CP and (B) TAGT homopolymerizations. Black squares: Mn. Red circles: Đ. Black dashed line: Mn,theo. Photo-CP conditions: TCT (20 μmol, 1 eq.), vinyl ether, PC (0.02 mol% relative to EVE), DCM (92 mM), RT, 456 nm LED, 6 h. TAGT conditions: TCT3 (4 μmol, 1 eq.), thiirane, TPPCl (0.33 eq.), DMAc (0.27 mL), 60 °C, 6 h. | ||
While TCT3 has previously been reported for photo-CP of EVE, isobutyl vinyl ether (IBVE), and nbutyl vinyl ether (BVE);64,66 we attempted to expand the scope to high Tg poly(vinyl ethers)69,70 from cyclohexyl vinyl ether (CHVE) and 2,3-dihydrofuran (DHF) (Fig. 3A). These monomers achieved full conversion in 6 hours, but SEC analysis showed bimodal distributions for both polymers (Fig. S4 and S5†), which we hypothesize is due to both photocatalytic activation and direct monomer activation. Unfortunately, all poly(vinyl ethers) beyond p(EVE) were found to be insoluble in DMAc, and thus were not carried onto chain extension trials or pursued further.
 | ||
| Fig. 3 (A) Vinyl ether and (B) thiirane monomer scope. Mn and Ð determined via SEC in DMF with 0.025 M LiBr against PMMA standards. Photo-CP standard conditions: TCT (20 μmol, 1 eq.), vinyl ether (50 eq.), PC (2.0 mol% relative to TCT), DCM (92 mM), RT, 456 nm LED, 6 h. TAGT standard conditions: TCT3 (4 μmol, 1 eq.), thiirane (100 eq.), TPPCl (0.33 eq.), DMAc (0.27 mL), 60 °C, 6 h. aReaction run for 24 h. bEstimated conversion due to overlapping peaks. | ||
Unlike for photo-CP, TCT3 has never been reported for TAGT. Therefore, we tested common thiirane monomers46,48,49,59,60 POPS (vide supra), propylene sulfide (PS), and allyloxy propylene sulfide (AOPS) in addition to a new monomer, phenyl propylene sulfide (PPS), using this mediator (Fig. 3B). TCT3 was very effective for PS, reaching >99% conversion to form p(PS) (Mn = 11.9 kDa) with a narrow Ð (1.20). AOPS only achieved 17% conversion in 24 hours, resulting in a low molar mass (3.02 kDa), and a slightly elevated Ð (1.54). PPS proceeded with moderate conversion (66%), forming p(PPS) (Mn = 14.1 kDa) with a narrow Ð (1.39). Despite the variability in reactivity between the thiiranes, all were tested in the chain extensions with p(EVE). These results showing successful photo-CP and TAGT validate TCT3's promise as a universal mediator for these mechanisms.
Chain-extension of p(EVE)
Following photo-CP, the p(EVE) samples were used as macro-mediators for chain extension via TAGT. Initially, we utilized 50, 100, 200, and 300 monomer equivalents for both EVE and POPS to target p(EVE)-b-p(POPS) with a range of block lengths (see Section S2.E†). Excitingly, the 1H NMR spectra show the presence of both blocks (Fig. 4A) and the SEC peaks shift to higher molar masses, indicating the successful chain extension in every case (Fig. 4B and S9–S12†). Due to the challenge of accurately determining degrees of polymerization for BCPs, all polymers were named on the basis of monomer feed (e.g., p(EVE)50-b-p(POPS)50). For BCPs made from p(EVE)200 or p(EVE)300, bimodal peaks were observed in the SEC. We hypothesize this bimodality arises from the increased amount of direct monomer oxidation, which yields some p(EVE) chains that cannot be extended due to the lack of a TCT end group. Despite some loss of control at high molar masses, these chain extensions represent the first examples of sequential cationic-anionic polymerizations using a TCT universal mediator. | ||
| Fig. 4 (A) Stacked 1H NMR spectra of respective homopolymers and p(EVE)50-b-p(POPS)50. (B) Representative SEC traces showing chain extension from p(EVE)50 to p(EVE)50-b-p(POPS)50. | ||
Unfortunately, during the purification process via precipitation in methanol, we encountered fractionation of p(EVE)-b-p(POPS) on the basis of the relative composition of the different blocks (Section S2.F†). To avoid this fractionation from interfering with the polymer characterization, the chain extension screen was repeated – focusing on the block lengths that retained control (i.e., 50 and 100 monomer equivalents) – and the resulting BCPs were purified using preparative SEC (prepSEC) (Table 2). As the ratio of [POPS]:[p(EVE)] went up from 50 to 100, the TAGT conversion also went up (compare entries 1 and 2 or 3 and 4), which we hypothesize is due to the higher concentration of thiirane monomer resulting in a chemical environment more similar to a POPS homopolymerization. Additionally, we observe lower conversion of POPS when using p(EVE)100 (entries 3–4), possibly arising from more residual PC and/or mass transport limitations. Similar to our initial chain extension screen, all BCPs showed clean peak shifts to lower retention times in the SEC relative to p(EVE) homopolymer (see Section S2.G†).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Starting polymer | [POPS]:[p(EVE)] |
Conv.b (%) | M n (kDa) | M w (kDa) | Đ | T o (°C) | T g (°C) |
| a Standard reaction conditions: p(EVE) (4.0 µmol, 1 eq.), POPS (n eq.), TPPCl (0.33 eq.), DMAc (15 µM p(EVE)), 6 h, 60 °C. b Determined via1H NMR spectroscopy. c Determined via SEC in DMF with 0.025 M LiBr against PMMA standards. d Calculated from TGA thermograms. e Determined by DSC. p(EVE)50: Mn = 4.80 kDa, Đ = 1.27. p(EVE)100: Mn = 7.17 kDa, Đ = 1.43. | ||||||||
| 1 | p(EVE)50 | 50:1 |
55% | 10.3 | 14.7 | 1.43 | 283, 376 | −30, 18 |
| 2 | p(EVE)50 | 100:1 |
61% | 18.7 | 27.4 | 1.47 | 272, 375 | −32, 19 |
| 3 | p(EVE)100 | 50:1 |
36% | 10.4 | 15.2 | 1.46 | 271, 373 | −41, 4 |
| 4 | p(EVE)100 | 100:1 |
44% | 18.0 | 27.2 | 1.51 | 277, 379 | −49, 4 |
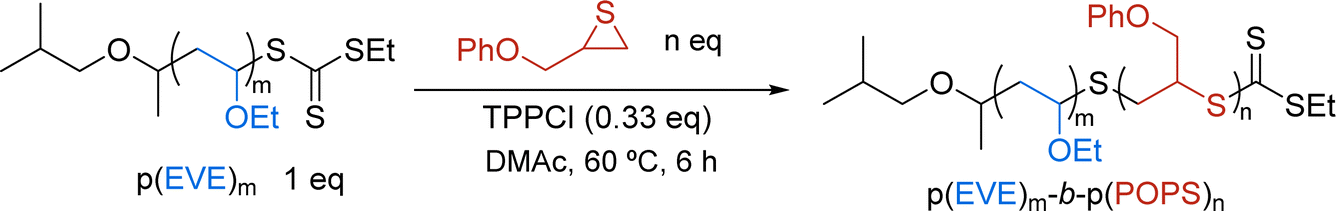
The expanded thiirane scope also cleanly extended p(EVE)50 under the standard TAGT conditions (Table 3 and Fig. S21–S23†). The polymerization of PS proceeded rapidly, reaching high conversion (94%) within 3 hours and maintaining a moderate Ð (1.45) (entry 1). On the basis of the lower reactivity of AOPS and PPS in the homopolymerization studies (Fig. 3B) we increased the polymerization time for the chain extension experiments using these monomers. In 18 hours, AOPS only achieved 45% conversion (entry 2), whereas PPS reached full conversion (entry 3). In both cases, the resulting BCP maintained a narrow Ð (1.35 and 1.39, respectively).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Thiirane | Conv.b (%) | M n (kDa) | Đ | T o (°C) | T g (°C) |
| a Standard reaction conditions: p(EVE) (4.0 μmol, 1 eq.), thiirane (50 eq.), TPPCl (0.33 eq.), DMAc (0.27 mL), 3 h, 60 °C. b Determined via1H NMR spectroscopy of the crude reaction mixture. c Determined via SEC in DMF with 0.025 M LiBr against PMMA standards. d Calculated from TGA thermograms. e Determined by DSC. f Starting from p(EVE)50: Mn = 4.45 kDa, Đ = 1.26. g 18 h reaction time. h Starting from p(EVE)50: Mn = 4.23 kDa, Đ = 1.29. | ||||||
| 1f | PS | 94 | 7.03 | 1.45 | 284, 403 | −39, −31 |
| 2f,g | AOPS | 45 | 6.90 | 1.35 | 284, 403 | −53, −34 |
| 3g,h | PPS | >99 | 11.7 | 1.39 | 288, 399 | −30, 7 |

End-group analysis
In order to verify the presence of the trithiocarbonate end-group, we conducted COSY, HSQC, and HMBC NMR spectroscopy experiments (see Section S2.H†). To increase the relative intensity of the end-group peaks, low [monomer]:[TCT3] polymers (p(EVE)10 and p(EVE)10-b-p(POPS)10) were synthesized. We utilized the 2D NMR spectra to assign the 1H and 13C peaks of each polymer.
The HMBC spectrum of p(EVE)10 (Fig. S26†) showed correlations between the thiocarbonyl carbon and both the methylene of the thiyl end-group and the methine of the adjacent repeat unit. Additionally, MALDI-TOF MS analysis validated the desired end-groups for p(EVE) (Table S7 and Fig. S30 and S31†). For p(EVE)10-b-p(POPS)10 (Fig. S29†), a similar HMBC correlation between the thiocarbonyl carbon with the methylene of the adjacent repeat unit was observed. These experiments, along with the successful chain extensions, provide evidence that the trithiocarbonate chain end has been retained through both homopolymerization and diblock formation.
Thermal analysis
Next, we investigated the thermal properties of the BCPs purified by prepSEC. Extrapolated onset temperatures of degradation (Tos) were calculated from TGA thermograms and compared to the corresponding homopolymers (see Section S2.I†). All p(EVE)-b-p(POPS) BCPs had two major mass-loss features: To = 271–283 °C and 373–379 °C, corresponding to the p(POPS) and p(EVE) blocks, respectively (Fig. 5A and S39–S42†). As expected, the extent of each mass loss event corresponds to the size of the associated block. For example, the p(POPS) To accounts for approximately 33%, 50%, and 67% of the total mass loss for p(EVE)100-b-p(POPS)50 (Fig. S41†), p(EVE)100-b-p(POPS)100 (Fig. 5A), p(EVE)50-b-p(POPS)100 (Fig. S40†), respectively. A small mass-loss event is seen for each BCP around 150 °C which was also observed in the multi-stage degradation of the p(EVE) homopolymer (Fig. S32†). | ||
| Fig. 5 (A) TGA and (B) DSC of p(EVE)100-b-p(POPS)100 with To and Tg features corresponding to p(POPS) (solid red box) and p(EVE) (dashed blue box) highlighted. | ||
Similarly, the PS-, AOPS-, and PPS-based BCPs displayed two major mass loss events with observed To values of 284–288 °C and 399–403 °C (Table 3 and Fig. S43–S45†). The intensities of these losses match well with the mass of each block.
DSC for each BCP revealed two Tg features (Fig. 5B, Tables 2 and 3, and Fig. S53–S59†), indicating that microphase separation between the two blocks has occurred.63 The Tgs of the diblocks match closely to their respective homopolymers (Fig. S46, S47, S50–S52, Table S8†): the Tg at −49 to 30 °C originates from the p(EVE) block, and the other Tg originates from the thiirane block (p(POPS): 4–19 °C, p(PS): −39 °C, p(AOPS): −53 °C, p(PPS): 7 °C).
BCPs from three-mechanisms and three-monomer classes
Now that a TCT universal mediator can successfully combine cationic and anionic polymerizations, we wanted to demonstrate the power of this approach by expanding to three-mechanism BCPs. Because the TCT remains on the chain end, we were able to take advantage of its generality to change between photo-CP, TAGT, and now radical polymerization without intermediate steps. To accomplish this, the newly synthesized p(EVE)50-b-p(POPS)50 was chain extended using photocontrolled radical polymerization46,71–80 of NIPAM (50 eq. relative to TCT) to make a novel terpolymer (Fig. 6). | ||
| Fig. 6 SEC traces showing chain extension to a three-mechanism, three-monomer-class BCP. Radical polymerization conditions: p(EVE)-b-p(POPS) (4.0 μmol, 1 eq.), NIPAM (50 or 100 eq.), DCM (3 mM), Ir(ppy)3 (0.045 mol% relative to NIPAM), RT, 456 nm LED, 6 h. | ||
Gratifyingly, we observed another significant shift of the SEC peak to a shorter retention time (Fig. 6), indicating successful triblock formation. Chain extension was also observed in another trial with block lengths of 100 for each block (see Section S2.J†). To the best of our knowledge, Kamigaito and coworkers reported81 the only other example of sequentially combining anionic, cationic, and radical polymerizations, but their system did not use a different monomer class for each mechanism and required end-group modification after the anionic step. Therefore, our protocol represents the first report combining cationic, anionic, and radical polymerizations without any intermediate compatibilization or end-group modification steps to synthesize a BCP of three different monomer classes.
Conclusions
In summary, we identified and applied TCT3 as a universal mediator for photo-CP and TAGT in a simple, sequential procedure. The proper selection of the R and Z groups of the TCT and determining the importance of p(EVE)'s solubility in DMAc enabled this approach. Clean shifts in the SEC traces support complete chain extension with a variety of thiiranes to form novel diblock copolymers. In particular, p(EVE)-b-p(POPS) BCPs were prepared with a range of block lengths. These materials exhibited two Tg features in the DSC thermograms, suggesting microphase separation into two distinct domains. The fidelity of the trithiocarbonate chain end was characterized by 2D NMR experiments and further demonstrated by a second chain extension using radical polymerization of NIPAM to form a three-monomer-class triblock terpolymer using three different mechanisms. Future work will focus on expanding the utility of this system as well as further characterization and application of these novel materials.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Credit: Brandon M. Hosford data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, and writing – review & editing; William Ramos data curation, formal analysis, investigation, methodology, visualization, writing – original draft, and writing – review & editing; Jessica R. Lamb conceptualization, formal analysis, funding acquisition, project administration, resources, supervision, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (NSF) Materials Research Science and Engineering Center (MRSEC) (DMR-2011401) and the University of Minnesota (UMN). W. R. was partially supported by the 3M Science and Technology Graduate Fellowship. We thank Prof. Aleksandr Zhukhovitskiy and Hilary Djomnang-Fokwa for assistance with running prepSEC. NMR analyses were done in the Nuclear Magnetic Resonance Laboratory and the Minnesota NMR Center, which are supported by the Research and Innovation Office (RIO), the Medical School, the College of Biological Science, College of Science and Engineering (CSE), and the Department of Chemistry at UMN, and the Office of the Director, National Institutes of Health (NIH, S10OD011952), the NSF, and the Minnesota Medical Foundation. Mass spectrometry analysis was performed at the UMN Department of Chemistry Mass Spectrometry Laboratory (MSL), supported by RIO, CSE, and the Department of Chemistry at UMN, as well as the NSF (CHE-1336940). Part of this work was carried out in the CSE Polymer Characterization Facility at UMN, which has received capital equipment funding from the National Science Foundation through the UMN MRSEC. The content of this paper is the sole responsibility of the authors and does not represent the official views of or endorsement by the NIH or NSF.Notes and references
- H. J. Harwood, Angew. Chem., Int. Ed. Engl., 1965, 4, 1051–1060 CrossRef.
- J.-F. Lutz, in Sequence-Controlled Polymers: Synthesis, Self-Assembly, and Properties, American Chemical Society, 2014, vol. 1170, pp. 1–11 Search PubMed.
- J. G. Baker, R. Zhang and C. A. Figg, J. Am. Chem. Soc., 2024, 146, 106–111 CrossRef CAS PubMed.
- R. B. Grubbs and R. H. Grubbs, Macromolecules, 2017, 50, 6979–6997 CrossRef CAS.
- G. K. K. Clothier, T. R. Guimarães, S. W. Thompson, J. Y. Rho, S. Perrier, G. Moad and P. B. Zetterlund, Chem. Soc. Rev., 2023, 52, 3438–3469 RSC.
- D. J. Keddie, Chem. Soc. Rev., 2013, 43, 496–505 RSC.
- K. Min, H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2005, 127, 3825–3830 CrossRef CAS PubMed.
- K. Matyjaszewski and J. Spanswick, Mater. Today, 2005, 8, 26–33 CrossRef CAS.
- Y. K. Chong, T. P. T. Le, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 2071–2074 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed.
- M. Ouchi and M. Sawamoto, Polym. J., 2018, 50, 83–94 CrossRef CAS.
- N. Ekizoglou and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2166–2170 CrossRef CAS.
- T. P. Lodge and P. C. Hiemenz, in Polymer Chemistry, CRC Press, Taylor and Francis Group, Boca Raton, 3rd edn, 2020 Search PubMed.
- A. Favier and M.-T. Charreyre, Macromol. Rapid Commun., 2006, 27, 653–692 CrossRef CAS.
- G. Moad, J. Chiefari, Y. K. Chong, J. Krstina, R. T. Mayadunne, A. Postma, E. Rizzardo and S. H. Thang, Polym. Int., 2000, 49, 993–1001 CrossRef CAS.
- M. Khan, T. R. Guimarães, K. Choong, G. Moad, S. Perrier and P. B. Zetterlund, Macromolecules, 2021, 54, 736–746 CrossRef CAS.
- A. Dey, U. Haldar and P. De, Front. Chem., 2021, 9, 644547 CrossRef CAS PubMed.
- X.-H. Guo, G.-G. Gu, T.-J. Yue and W.-M. Ren, Polym. Chem., 2023, 14, 5034–5039 RSC.
- S. Creutz, C. Vandooren, R. Jérôme and P. Teyssié, Polym. Bull., 1994, 33, 21–28 CrossRef CAS.
- A. Verma, A. Nielsen, J. E. McGrath and J. S. Riffle, Polym. Bull., 1990, 23, 563–570 CrossRef CAS.
- A. Puglisi, E. Murtezi, G. Yilmaz and Y. Yagci, Polym. Chem., 2017, 8, 7307–7310 RSC.
- M. U. Kahveci, G. Acik and Y. Yagci, Macromol. Rapid Commun., 2012, 33, 309–313 CrossRef CAS PubMed.
- S. Kumagai, K. Nagai, K. Satoh and M. Kamigaito, Macromolecules, 2010, 43, 7523–7531 CrossRef CAS.
- M. Schäfer, P. C. Wieland and O. Nuyken, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3725–3733 CrossRef.
- J. Feldthusen, B. Iván and A. H. E. Müller, Macromolecules, 1997, 30, 6989–6993 CrossRef CAS.
- E. Yoshida and A. Sugita, Macromolecules, 1996, 29, 6422–6426 CrossRef CAS.
- R. Nomura, M. Narita and T. Endo, Macromolecules, 1994, 27, 4853–4854 CrossRef CAS.
- Q. Liu, M. Konas, R. M. Davis and J. S. Riffle, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 1709–1717 CrossRef CAS.
- S. Nemes and J. P. Kennedy, J. Macromol. Sci., Chem., 1991, 28, 311–328 Search PubMed.
- T. Kitayama, T. Nishiura and K. Hatada, Polym. Bull., 1991, 26, 513–520 CrossRef CAS.
- T. Souel, F. Schué, M. Abadie and D. H. Richards, Polymer, 1977, 18, 1292–1294 CrossRef CAS.
- F. J. Burgess, A. V. Cunliffe, J. R. MacCallum and D. H. Richards, Polymer, 1977, 18, 726–732 CrossRef CAS.
- F. J. Burgess, A. V. Cunliffe, J. R. MacCallum and D. H. Richards, Polymer, 1977, 18, 719–725 CrossRef CAS.
- F. J. Burgess, A. V. Cunliffe, J. V. Dawkins and D. H. Richards, Polymer, 1977, 18, 733–740 CrossRef CAS.
- K. Zhang, T. Bai and J. Ling, Macromolecules, 2023, 56, 7389–7395 CrossRef CAS.
- H. Qiu, T. Shen, Z. Yang, F. Wu, X. Li, Y. Tu and J. Ling, Chin. J. Chem., 2022, 40, 705–712 CrossRef CAS.
- H. Dong, Y. Zhu, Z. Li, J. Xu, J. Liu, S. Xu, H. Wang, Y. Gao and K. Guo, Macromolecules, 2017, 50, 9295–9306 CrossRef CAS.
- Y. Hepuzer, Y. Yagci, T. Biedron and P. Kubisa, Angew. Makromol. Chem., 1996, 237, 163–171 CrossRef CAS.
- M. Guerre, M. Uchiyama, E. Folgado, M. Semsarilar, B. Améduri, K. Satoh, M. Kamigaito and V. Ladmiral, ACS Macro Lett., 2017, 6, 393–398 CrossRef CAS PubMed.
- M. Uchiyama, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2015, 54, 1924–1928 CrossRef CAS PubMed.
- Q. Ma, X. Zhang, Y. Jiang, J. Lin, B. Graff, S. Hu, J. Lalevée and S. Liao, Polym. Chem., 2022, 13, 209–219 RSC.
- V. Kottisch, Q. Michaudel and B. P. Fors, J. Am. Chem. Soc., 2017, 139, 10665–10668 CrossRef CAS PubMed.
- Y. Ma, V. Kottisch, E. A. McLoughlin, Z. W. Rouse, M. J. Supej, S. P. Baker and B. P. Fors, J. Am. Chem. Soc., 2021, 143, 21200–21205 CrossRef CAS PubMed.
- M. J. Supej, B. M. Peterson and B. P. Fors, Chem, 2020, 6, 1794–1803 CAS.
- A. Nagai, N. Koike, H. Kudo and T. Nishikubo, Macromolecules, 2007, 40, 8129–8131 CrossRef CAS.
- Z. Zhang, T.-Y. Zeng, L. Xia, C.-Y. Hong, D.-C. Wu and Y.-Z. You, Nat. Commun., 2018, 9, 2577 CrossRef PubMed.
- J.-W. Li, M. Chen, Z. Zhang, C.-Y. Pan, W.-J. Zhang and C.-Y. Hong, Polym. Chem., 2022, 13, 402–410 RSC.
- Z. Zhang, L. Xia, T.-Y. Zeng, D.-C. Wu, W.-J. Zhang, C.-Y. Hong and Y.-Z. You, Polym. Chem., 2019, 10, 2117–2125 RSC.
- C.-H. Wang, Y.-S. Fan, Z. Zhang, Q.-B. Chen, T.-Y. Zeng, Q.-Y. Meng and Y.-Z. You, Appl. Surf. Sci., 2019, 475, 639–644 CrossRef CAS.
- M. K. Gupta, T. A. Meyer, C. E. Nelson and C. L. Duvall, J. Controlled Release, 2012, 162, 591–598 CrossRef CAS PubMed.
- S. W. Spring, C. S. Cerione, J. H. Hsu, S. L. Shankel and B. P. Fors, Chin. J. Chem., 2023, 41, 399–404 CrossRef CAS.
- S. L. Shankel, T. H. Lambert and B. P. Fors, Polym. Chem., 2022, 13, 5974–5979 RSC.
- B. M. Peterson, S. Lin and B. P. Fors, J. Am. Chem. Soc., 2018, 140, 2076–2079 CrossRef CAS PubMed.
- J. Li, A. Kerr, Q. Song, J. Yang, S. Häkkinen, X. Pan, Z. Zhang, J. Zhu and S. Perrier, ACS Macro Lett., 2021, 10, 570–575 CrossRef CAS PubMed.
- M. Matsuda, M. Uchiyama, Y. Itabashi, K. Ohkubo and M. Kamigaito, Polym. Chem., 2022, 13, 1031–1039 RSC.
- X. Zhang, Y. Jiang, Q. Ma, S. Hu and S. Liao, J. Am. Chem. Soc., 2021, 143, 6357–6362 CrossRef CAS PubMed.
- S. Singha, S. Pan, S. S. Tallury, G. Nguyen, R. Tripathy and P. De, ACS Polym. Au, 2024, 3, 189–207 CrossRef PubMed.
- P. C. Knutson, A. J. Teator, T. P. Varner, C. T. Kozuszek, P. E. Jacky and F. A. Leibfarth, J. Am. Chem. Soc., 2021, 143, 16388–16393 CrossRef CAS PubMed.
- Z. Zhang, X. Nie, F. Wang, G. Chen, W.-Q. Huang, L. Xia, W.-J. Zhang, Z.-Y. Hao, C.-Y. Hong, L.-H. Wang and Y.-Z. You, Nat. Commun., 2020, 11, 3654 CrossRef CAS PubMed.
- A. Takahashi, S. Tsunoda, R. Yuzaki and A. Kameyama, Macromolecules, 2020, 53, 5227–5236 CrossRef CAS.
- M. R. Hill, R. N. Carmean and B. S. Sumerlin, Macromolecules, 2015, 48, 5459–5469 CrossRef CAS.
- M.-N. Antonopoulou, R. Whitfield, N. P. Truong and A. Anastasaki, Eur. Polym. J., 2022, 174, 111326 CrossRef CAS.
- H. Daimon, H. Okitsu and J. Kumanotani, Polym. J., 1975, 7, 460–466 CrossRef CAS.
- V. Kottisch, Q. Michaudel and B. P. Fors, J. Am. Chem. Soc., 2016, 138, 15535–15538 CrossRef CAS PubMed.
- B. M. Peterson, V. Kottisch, M. J. Supej and B. P. Fors, ACS Cent. Sci., 2018, 4, 1228–1234 CrossRef CAS PubMed.
- Q. Michaudel, T. Chauviré, V. Kottisch, M. J. Supej, K. J. Stawiasz, L. Shen, W. R. Zipfel, H. D. Abruña, J. H. Freed and B. P. Fors, J. Am. Chem. Soc., 2017, 139, 15530–15538 CrossRef CAS PubMed.
- A high degree of purity is needed to polymerize EVE with TCT1.66.
- V. Kottisch, M. J. Supej and B. P. Fors, Angew. Chem., Int. Ed., 2018, 57, 8260–8264 CrossRef CAS PubMed.
- S. W. Spring, J. H. Hsu, R. J. Sifri, S.-M. Yang, C. S. Cerione, T. H. Lambert, C. J. Ellison and B. P. Fors, J. Am. Chem. Soc., 2022, 144, 15727–15734 CrossRef CAS PubMed.
- H. Cramail and A. Deffieux, Macromol. Chem. Phys., 1994, 195, 217–227 CrossRef CAS.
- J. Xu, C. Fu, S. Shanmugam, C. J. Hawker, G. Moad and C. Boyer, Angew. Chem., Int. Ed., 2017, 56, 8376–8383 CrossRef CAS PubMed.
- M. Chen, S. Deng, Y. Gu, J. Lin, M. J. MacLeod and J. A. Johnson, J. Am. Chem. Soc., 2017, 139, 2257–2266 CrossRef CAS PubMed.
- J. Xu, S. Shanmugam, C. Fu, K.-F. Aguey-Zinsou and C. Boyer, J. Am. Chem. Soc., 2016, 138, 3094–3106 CrossRef CAS PubMed.
- S. Shanmugam, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55, 1036–1040 CrossRef CAS PubMed.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- S. Shanmugam, J. Xu and C. Boyer, Chem. Sci., 2015, 6, 1341–1349 RSC.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed.
- S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9988–9999 CrossRef CAS PubMed.
- M. Chen, M. J. MacLeod and J. A. Johnson, ACS Macro Lett., 2015, 4, 566–569 CrossRef CAS PubMed.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- K. Satoh, Y. Mori and M. Kamigaito, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 465–473 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02511c |
| ‡ B. M. H. and W. R. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |