
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Delocalization error poisons the density-functional many-body expansion†
Dustin R.
Broderick
 and
John M.
Herbert
*
and
John M.
Herbert
*
Department of Chemistry & Biochemistry, The Ohio State University, 151 W. Woodruff Ave., Columbus, Ohio 43210, USA. E-mail: herbert@chemistry.ohio-state.edu
First published on 30th October 2024
Abstract
The many-body expansion is a fragment-based approach to large-scale quantum chemistry that partitions a single monolithic calculation into manageable subsystems. This technique is increasingly being used as a basis for fitting classical force fields to electronic structure data, especially for water and aqueous ions, and for machine learning. Here, we show that the many-body expansion based on semilocal density functional theory affords wild oscillations and runaway error accumulation for ion–water interactions, typified by F−(H2O)N with N ≳ 15. We attribute these oscillations to self-interaction error in the density-functional approximation. The effect is minor or negligible in small water clusters, explaining why it has not been noticed previously, but grows to catastrophic proportion in clusters that are only moderately larger. This behavior can be counteracted with hybrid functionals but only if the fraction of exact exchange is ≳50%, whereas modern meta-generalized gradient approximations including ωB97X-V, SCAN, and SCAN0 are insufficient to eliminate divergent behavior. Other mitigation strategies including counterpoise correction, density correction (i.e., exchange–correlation functionals evaluated atop Hartree–Fock densities), and dielectric continuum boundary conditions do little to curtail the problematic oscillations. In contrast, energy-based screening to cull unimportant subsystems can successfully forestall divergent behavior. These results suggest that extreme caution is warranted when the many-body expansion is combined with density functional theory.
1 Introduction
Ostensibly, the many-body expansion (MBE) offers a method-agnostic way to apply electronic structure theory to large molecular systems,1–4 avoiding the steep nonlinear scaling of ab initio quantum chemistry by partitioning a supersystem into small fragments. The total energy is then approximated as a sum of n-body interactions between these fragments:5 | (1) |
The MBE in eqn (1) forms the basis of a wide variety of fragment-based approximation schemes.5–8 These have been used in hybrid quantum mechanics/molecular mechanics approaches,10–14 as a basis for developing classical force fields for water–water and ion–water interactions,15–31 as a means to elucidate the nature of intermolecular interactions,30–38 and as a way to overcome the dimensionality problem in machine learning.39–43 In principle, correlated wave function models can be used for the electronic structure, since only small n-body subsystem calculations are required, but density functional theory (DFT) has also been suggested for general use in force-field development.44 One can imagine the MBE as a means to accelerate DFT-based ab initio molecular dynamics simulations,45–47 and the low cost of DFT calculations make this an attractive choice for generating the enormous data sets that are necessary for machine learning applications. It is these DFT-based applications (and potential applications) that concern us.
DFT has become the dominant tool for electronic structure calculations due to its combination of favorable scaling and quantitative or semi-quantitative accuracy for a wide range of chemical problems.48–50 Nevertheless, it is not without systemic problems. Among these, perhaps the most pernicious and pervasive is self-interaction error (SIE),51–54 also known as delocalization error.55–57 Although SIE is most often associated with exaggerated delocalization of unpaired spins,58–71 including fractional atomic charges at stretched bond lengths,51,72–74 SIE also produces a driving force to delocalize charge in closed-shell cases. It is especially problematic for solvated and condensed-phase ions.75–80 In the present work, we demonstrate how delocalization error interacts with the MBE to create a feedback loop leading to runaway error accumulation. The problem is most pronounced for semilocal functionals derived within the generalized gradient approximation (GGA), but is still serious for hybrid functionals such as B3LYP or PBE0, and meta-GGAs such as ωB97X-V81 and SCAN.82 It significantly impairs the applicability of the DFT-based MBE. That alone should give pause as this method is considered for qualitative analysis, machine learning, or force-field development.
2 Results
In what follows, we consider errors in DFT-based MBE(n) calculations, meaning that eqn (1) is truncated at n-body interactions. Explicit expressions for the n-body corrections (ΔEIJ, ΔEIJK, etc.) can be found elsewhere.2 Calculations were performed with the FRAGME∩T code83,84 interfaced to Q-CHEM,85 as described in Section 5. For calculations on ion–water clusters X±(H2O)N, the property of interest is the ion's interaction energy with the water cluster, ΔEint. Errors in MBE(n) approximations are defined with respect to a counterpoise (CP) corrected,86 supramolecular calculation of ΔEint at the same level of theory that is used for the n-body calculations. The CP correction in the supersystem benchmark is useful for comparison to CP-corrected MBE(n) results but it amounts to a constant offset and does not affect the oscillations that are the primary focus of this work.2.1 Fluoride–water clusters
We first consider a data set consisting of ten F−(H2O)15 clusters, with calculations performed using either the PBE functional or else Hartree–Fock (HF) theory. We examine basis sets aug-cc-pVXZ (abbreviated “aXZ”) for X = D, T, and Q. Errors in MBE(n) approximations up to n = 6 are plotted in Fig. 1. Ratios of the MBE(n) approximation, ΔEint[MBE(n)], to the CP-corrected supramolecular result (ΔEint) are plotted in Fig. S1† (analogous ratios are provided in the ESI† for other error plots provided in this work). | ||
| Fig. 1 Errors in MBE(n) approximations for ΔEint in a set of ten F−(H2O)15 clusters, computed at (a)–(c) the PBE/aXZ level (top, for X = D, T, and Q) and (d)–(f) the HF/aXZ level (bottom). In each case, error is defined with respect to a CP-corrected, supramolecular calculation at the indicated level of theory. | ||
The HF-based MBE(n) interaction energies converge as expected to the reference supersystem value, with five-body terms that are negligible in what is effectively the basis-set limit, aQZ. Higher-order n-body terms can sometimes be artifacts of basis-set superposition error (BSSE),87,88 which likely explains the diminished importance of the six-body terms at the HF/aTZ and HF/aQZ levels, relative to HF/aDZ results.
In contrast, PBE-based MBE(n) calculations are subject to wild oscillations that grow worse as n increases; the expansion appears to be divergent for this and other semilocal functionals that we have tested. Histograms of the various interaction terms ΔEIJ⋯ are plotted in Fig. 2 for the HF/aQZ and PBE/aQZ calculations, and summary statistics are provided in Table S1.† The histograms are separated into fluoride-containing subsystems, which afford larger interactions on average, and those that contain only water molecules. Even for the seemingly divergent PBE-based expansions, the magnitude of the n-body corrections does decrease with n. However, for the PBE calculations the net contribution from the fluoride-containing subsystems increases as a function of n, leading to the observed divergence. For PBE, the total contribution from the fluoride-containing terms is −115.9 kcal mol−1 for n = 4 and +193.0 kcal mol−1 for n = 5. These values are unmatched in the water-only terms, which sum to −4.0 kcal mol−1 (n = 4) and +1.8 kcal mol−1 (n = 5). Even those values are still significantly larger than the water-only terms obtained at the HF level, which sum to −0.6 kcal mol−1 (n = 4) and +0.1 kcal mol−1 (n = 5).
 | ||
| Fig. 2 Histograms of all n-body interactions ΔEIJ⋯ up to n = 5, for MBE(n) applied to (a)–(d) F−(H2O)15 at the HF/aQZ level (left) and (e)–(h) the PBE/aQZ level (right). The interaction terms ΔEIJ⋯ that involve F− are shown in blue while those that only involve water are plotted in gold. The overall magnitude of the n-body corrections ΔEIJ⋯ decreases with n for both methods but for high-order interactions computed using PBE, the fluoride-containing terms are significantly larger than the water-only terms. | ||
Thus, the divergent behavior is exacerbated by the presence of an anion. A combinatorial increase in the number of n-body terms results in divergence for PBE-based MBE(n) despite the fact that individual ΔEIJ⋯ corrections decrease order-by-order. Our data are consistent with previous studies that observed a marked increase in errors for MBE-based energy decomposition analyses (based on two-body terms only) when GGAs were employed.89,90 Those studies, however, were limited to (H2O)6 clusters that do not engender the rapid divergence that we observe using F−(H2O)15.
Four- and five-body terms computed using PBE also show systematic negative and positive biases, respectively, indicating exaggerated magnitude for the higher-order n-body corrections. No such systematic error is observed in the HF results, where the n = 4 and n = 5 histograms are much more symmetric about zero. Assuming oscillating errors in the total energy, consistent with the data in Fig. 1, then for MBE(n) to converge it must be the case that
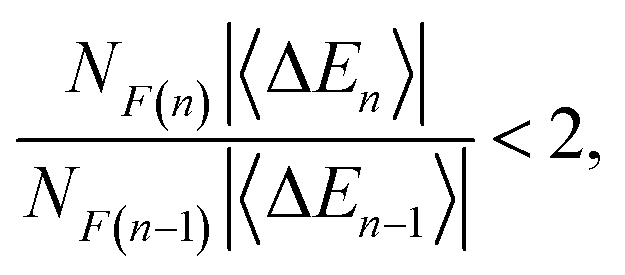 | (2) |
If divergence of MBE(n) calculations is indeed driven by combinatorial error accumulation, then smaller clusters with fewer fragments should exhibit improved convergence properties. To examine this hypothesis, we extracted clusters F−(H2O)N with N = 5–25 from a molecular dynamics simulation of F−(H2O)128. The absolute error per monomer in ΔEint, computed at the PBE/aDZ level, is plotted in Fig. 3 up to N = 15 and plots up to N = 25 can be found in Fig. S5–S7,† for both PBE/aDZ and HF/aDZ calculations. Normalizing the errors by the number of monomers accounts for overall errors that are expected to be size-extensive (i.e., a roughly constant error per hydrogen bond),91 and indeed the normalized HF errors in Fig. S5–S7† are independent of cluster size. In contrast, PBE errors diverge as N increases, for MBE(3), MBE(4), and MBE(5). We posit that SIE-induced error accumulation explains divergent behavior in DFT-MBE(n) calculations that has been documented previously by our group.1–3
 | ||
| Fig. 3 Error per monomer for the F− interaction energy in F−(H2O)N clusters, for calculations at the PBE/aDZ level. | ||
Examining the PBE-MBE(n) results in Fig. 3 and moving up the ladder from n = 2 to n = 5, we observe an order-by-order reduction in the error for small clusters, up to F−(H2O)8. For larger clusters, however, MBE(4) affords a larger error than MBE(3) and by F−(H2O)16, the two-body expansion affords the smallest error per monomer. For larger clusters, higher-order n-body terms are actually detrimental to the accuracy! Considering the product NF(n)〈ΔEn〉 in eqn (2) suggests two strategies for improving convergence of MBE(n): either reduce the per-fragment error via strategies to mitigate SIE, or else reduce the number of fragments via screening. Both strategies are pursued in Section 3.
2.2 Neutral and cationic clusters
SIE is especially pernicious for anions,92–100 so we next consider whether spurious oscillations in MBE(n) are limited to hydrated anions. To do so, we extracted a set of (H2O)15 and Na+(H2O)14 clusters from molecular dynamics simulations. MBE(n) results for these systems are plotted in Fig. 4, using the functionals PBE, PBE0, and HF, corresponding to fractions of Hartree–Fock exchange αhfx = 0, 0.25, and 1.0, respectively. We also examine results for the long-range corrected LRC-ωPBE functional101 that switches between αhfx = 0 at short range and αhfx = 1 at long range.101,102 Results for F−(H2O)14 are also plotted in Fig. 4 to facilitate side-by-side comparison to results from the previous section.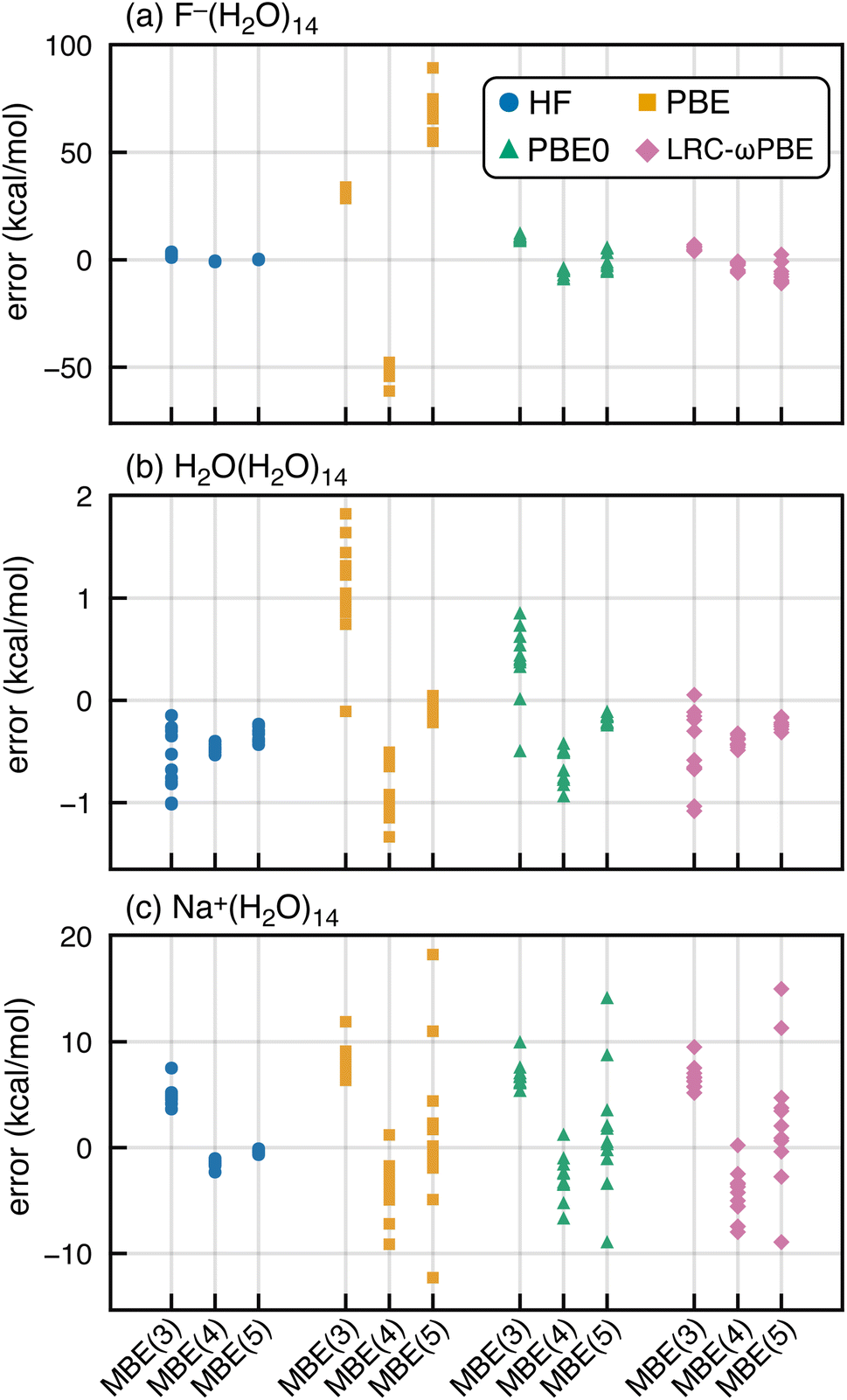 | ||
| Fig. 4 MBE(n) errors in ΔEint for (a) F−(H2O)14, (b) H2O(H2O)14, and (c) Na+(H2O)14, computed using DFT/aTZ with the functionals indicated. Each data set contains 11 structures extracted from a simulation. In (b), ΔEint is defined as the energy to remove a single, central H2O molecule whereas in (a) and (c) it is the energy required to remove the ion. | ||
Oscillations in the n-body interactions are quite small for the charge-neutral water clusters, albeit still largest with the PBE functional. These oscillations are reduced in magnitude, though not eliminated, by the hybrid functionals. A previous study of hydrated ions using SIE-corrected functionals concluded that SIE is important in F−(H2O)N but not for Na+(H2O)N,103 although the systems examined were limited to N ≤ 2. For Na+(H2O)14, we find that MBE(n) diverges using any of the aforementioned functionals except for HF. These exaggerated many-body effects are much larger than what is observed at the three-body level for Na+(H2O)2 or F−(H2O)2,36,37 even with semilocal functionals. This difference between microhydrated systems considered in previous studies, and the full solvation-shell clusters examined here, may explain why problems with DFT-based MBE(n) calculations have not been reported previously.
In what follows, we will focus on fluoride–water clusters where the problem is most severe but cationic systems are clearly not immune to the SIE problems documented herein. Semilocal functionals also exaggerate many-body effects even in charge-neutral clusters.
3 Discussion
3.1 SIE exacerbates n-body BSSE
A large basis set is vital for minimizing both BSSE and basis-set incompleteness error. Oscillatory convergence of MBE(n) towards the supersystem energy is sometimes mitigated in larger basis sets,104 but the data in Fig. 1 show little change as the basis set approaches completeness. This suggests that basis-set incompleteness is not primarily responsible for the oscillations that we observe.BSSE can be eliminated from the MBE(n) calculations by performing all subsystem calculations in the full-cluster basis set.104 This is somewhat expensive and was pursued using a small basis set, with HF/6-31G and PBE/6-31G results shown in Fig. 5 alongside conventional MBE(n) results that use only subsystem basis functions. The latter approach engenders significantly large errors and a −50 kcal mol−1 shift in the magnitude of the n-body corrections, for both HF and PBE. The CP-corrected HF/6-31G data (Fig. 5c) converge by n = 4. For PBE/6-31G, use of the full-cluster basis significantly dampens the magnitude of the oscillations yet they remain quite large, with errors of ∼20 kcal mol−1 at the n = 3 and n = 5 level. The errors that are comparable in magnitude, but opposite in sign, for n = 4 and n = 6.
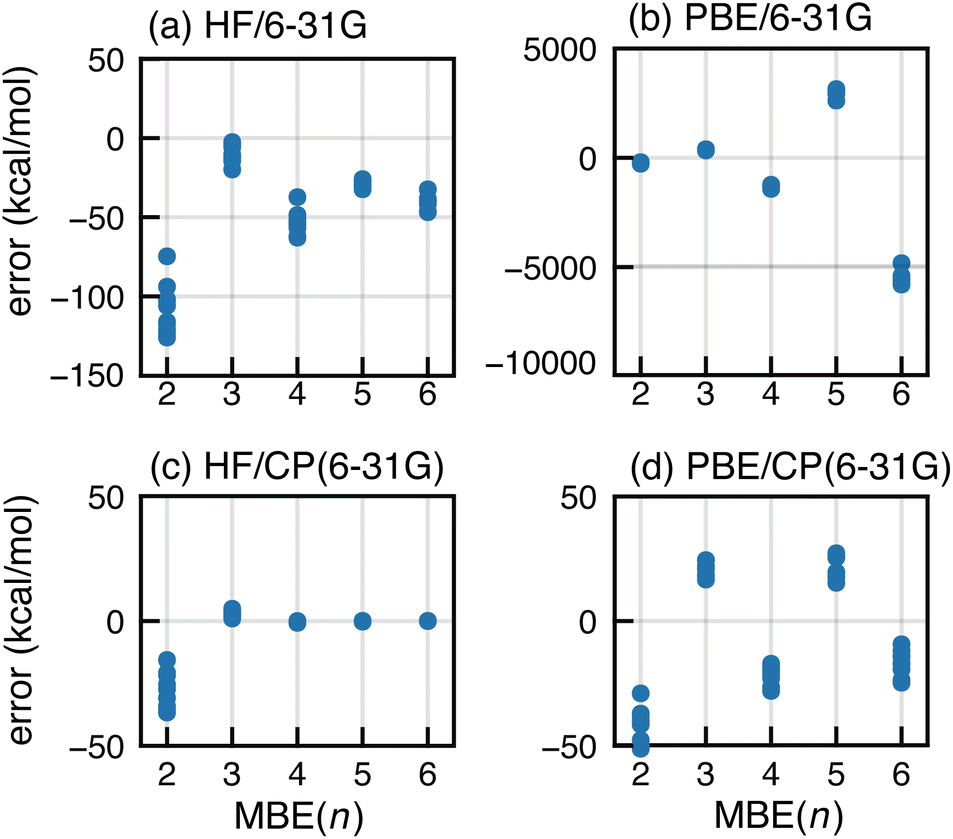 | ||
| Fig. 5 Errors in MBE(n) interaction energies for F− in a set of ten F−(H2O)15 clusters at (a) the HF/6-31G level and (b) the PBE/6-31G level. Also shown are results using a full-cluster CP correction, again at the (c) HF/6-31G and (d) PBE/6-31G levels. | ||
The difference between full-cluster HF and PBE results points to the interplay between SIE and BSSE. These effects are coupled because the loss of neighboring basis functions (when the full-cluster basis is replaced by a subsystem basis) confines electron density to a small number of monomers, preventing it from delocalizing throughout space. Similar artificial localization has been observed for anions, where SIE in semilocal functionals leads to an unbound electron that may become artificially bound in a finite basis set.95–100 Examples of the same phenomenon include fractional charges on well-separated moieties72–74 and charge-sloshing leading to oscillations when MBE(n) is applied to proteins with ionic side chains.105
As a result of SIE, the mere presence of a distant (therefore, non-interacting) subsystem has a stabilizing effect on the total energy.56 In MBE(n), both proximate and distant systems are added and removed as n changes, resulting in dramatic overstabilization of higher-order n-body correction terms when GGA functionals are used. This imbalance is somewhat mitigated by CP correction because the anion's charge can delocalize to other ghost atom sites in each of the subsystem calculations. Absent CP correction, aQZ basis functions extend only about 3 Å beyond the nuclei,106 so cannot support a delocalized electron beyond the monomers that are present in the subsystem.
3.2 Strategies to reduce SIE
The most common strategy to mitigate SIE or delocalization error is to incorporate a fraction of exact Fock exchange, with a coefficient 0 ≤ αhfx ≤ 1. We examine this approach by comparing results for a sequence of functionals: BLYP (with αhfx = 0),107,108 B3LYP (αhfx = 0.2),108,109 BH&H-LYP (αhfx = 0.5),110 and HF-LYP (αhfx = 1). As shown in Fig. 6, divergent behavior for F−(H2O)15 persists using B3LYP but results appear to converge for BH&H-LYP, and the oscillations disappear completely for HF-LYP.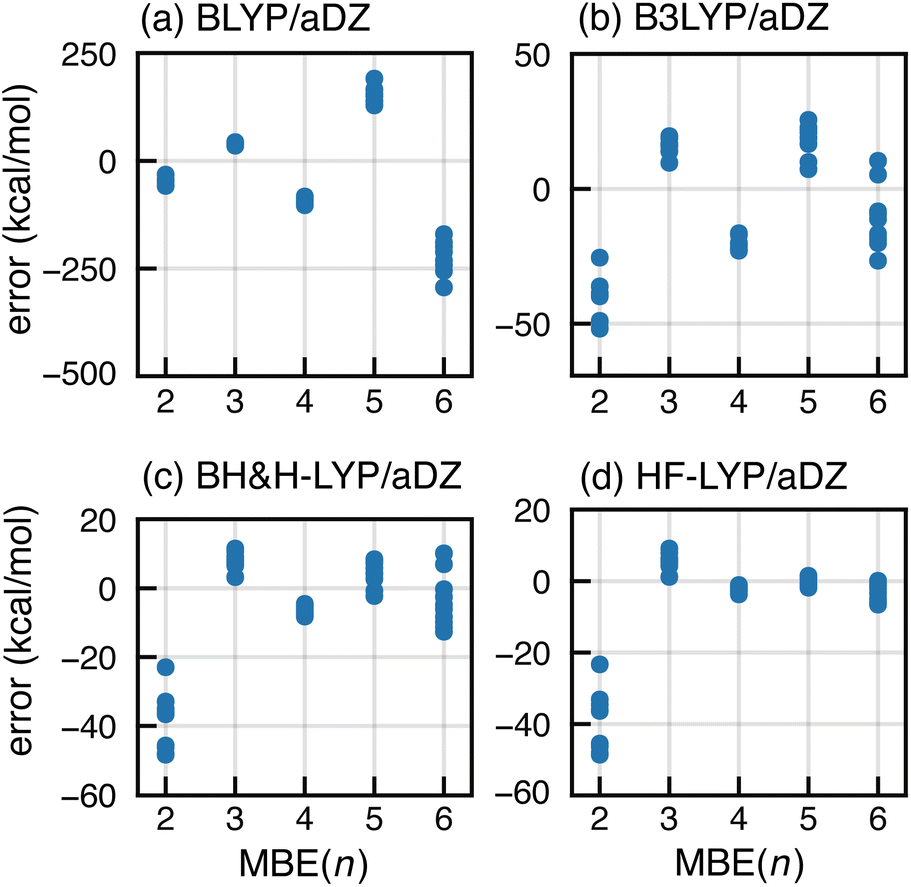 | ||
| Fig. 6 Errors in MBE(n) interaction energies for ten configurations of F−(H2O)15 computed using BLYP-based functionals with different fractions of exact exchange: (a) semilocal BLYP functional with αhfx = 0, (b) B3LYP hybrid functional with αhfx = 0.2, (c) half-and-half functional (BH&H-LYP) with αhfx = 0.5, and (d) HF-LYP with αhfx = 1. All calculations use the aDZ basis set. | ||
Apparently, the BH&H-LYP functional can be used to obtain convergent n-body expansions and it is interesting that the same functional often works well in problematic cases of ground- or excited-state charge transfer,69,70,111–118 whereas functionals such as B3LYP and PBE0 (the latter with αhfx = 0.25) often substantially underestimate charge-transfer energies.119–124 In the early days of molecular DFT, the BH&H-LYP functional was assessed as unfit for general-purpose calculations,72,125 at least in comparison to B3LYP. Indeed, errors for atomization energies109,125 and for reaction energies126 are somewhat larger as compared to B3LYP, yet BH&H-LYP is superior to B3LYP for barrier heights.126,127 In Table S2,† we compare BH&H-LYP side-by-side with B3LYP (including an empirical dispersion correction for both functionals),128 using the GMTKN55 data set.129 The overall performance of BH&H-LYP is only marginally worse than that of B3LYP, so the former may be a sensible alternative in cases where standard functionals exhibit SIE problems, including applications of MBE(n).
As an alternative to BH&H-LYP, one might consider using newer meta-GGA functionals. Based on tests for (H2O)6, it has been suggested that the many-body SIE is small for the semilocal SCAN functional.89 For F−(H2O)15, however, SCAN exhibits divergent behavior in MBE(n) calculations as shown in Fig. 7a. The same is true for the hybrid SCAN0 functional,130 which uses αhfx = 0.25 (Fig. 7b). We also tried ωB97X-V, which sets αhfx = 0.167 for short-range exchange81 and is a very good all-around density functional,50 but it also exhibits serious oscillations for F−(H2O)15 as shown in Fig. S8b.† Moreover, each of these meta-GGAs is inferior to BH&H-LYP for the SIE-dominated “SIE4 × 4” subset of GMTKN55,131 suggesting that these functionals exhibit larger all-around SIE as compared to BH&H-LYP.
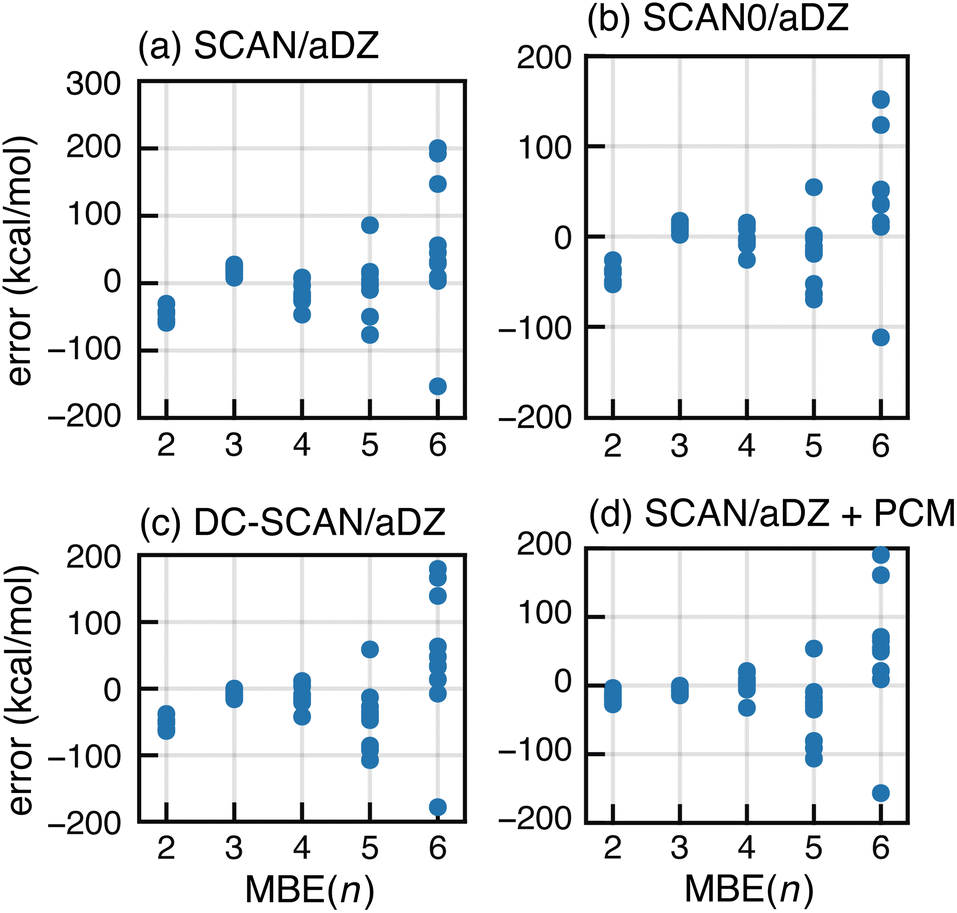 | ||
| Fig. 7 Errors in MBE(n) interaction energies for ten configurations of F−(H2O)15, computed using (a) the SCAN functional, (b) SCAN0 (with 25% exact exchange), (c) DC-SCAN, and (d) SCAN in conjunction with dielectric boundary conditions (ε = 4). All calculations used the aDZ basis set. | ||
Next, we consider the density-corrected132–135 (DC-)SCAN approach, also known as “SCAN@HF”,136 in which the SCAN exchange–correlation functional is evaluated non-self-consistently atop a self-consistent HF density. This procedure has been shown to reduce density delocalization across hydrogen bonds,90,136 and DC-SCAN has been used to generate many-body force fields.28–30 In the present calculations, however, DC-SCAN fails to forestall the runaway behavior of MBE(n); see Fig. 7c. This observation suggests that promising preliminary results for DC-SCAN applied to small clusters28–30,136 do not probe the full extent of problems that are exposed in calculations on a hydrated anion with a complete solvation shell.
Finally, we consider incorporation of low-dielectric boundary conditions as a means to mitigate charge delocalization. In previous work,105 we showed that a continuum solvation model with a dielectric constant ε = 4 eliminates oscillatory behavior in MBE(n) as applied to a large enzyme model with individual amino acids as fragments. The boundary conditions were implemented using a polarizable continuum model (PCM),137 not as a model of solvation but rather to reduce charge delocalization that can lead to a vanishing gap and concomitant convergence problems in large-molecule DFT calculations.138–141 These problems are sometimes ameliorated by electrostatic stabilization of the molecular surface.105,140,142 Notably, convergence is also improved using the BH&H-LYP functional rather than B3LYP or GGAs.138,139
MBE(n)-SCAN results with boundary conditions corresponding to ε = 4 are shown in Fig. 7d but the PCM fails to mitigate the oscillations. As compared to our calculations on proteins,105 the fragments used here are rather small and hydrogen bonds may fit within the molecular surface used by the solvent model.143 In other words, intermolecular charge delocalization across hydrogen bonds remains possible, and divergent results for MBE(n) suggests that this behavior is not mitigated by the low-dielectric PCM.
The same error mitigation strategies that are tested for SCAN in Fig. 7 are examined for PBE in Fig. S5.† The PBE0, DC-PBE, and PBE+PCM methods each significantly reduce (but do not eliminate) oscillatory behavior in MBE(n). This suggests that instabilities in MBE(n) calculations may manifest differently for GGA versus meta-GGA functionals. These functional-dependent differences will be examined in future work.
3.3 Screening
High-order MBE calculations quickly become intractable due to combinatorial growth in the number of subsystems, which creates follow-on difficulties for maintaining precision.1–3 Therefore, a screening mechanism to reduce the number of subsystems is vital to large-scale deployment of fragmentation, but simple distance-based screening can miss energetically important subsystems.4,144 We have shown that energy-based screening is superior in both accuracy and efficiency,145 and this type of screening is native to the FRAGME∩T software used here.83We next test the effects of screening on MBE(4) interaction energies for F−(H2O)15, computed at either the HF/aQZ level or the PBE/aQZ level. In either case, three-body fragments are screened using the semi-empirical GFN2-xTB model146 with an adjustable threshold τ3. Four-body subsystems are created from energetically important three-body subsystems, allowing for M = 1 missing parents.83 This means that tetramer IJKL is included if at least three of its four three-body sub-clusters is above threshold (e.g., |ΔEIJK| > τ3). This procedure has previously been shown to afford energetically converged four-body expansions.83
HF/aQZ results in Fig. 8a exhibit minimal error in ΔEint when all three-body terms are retained (τ3 = 0). Errors increase as τ3 increases and the screening becomes more aggressive, but they remain acceptably small for τ3 ∼ 0.1 kcal mol−1, a value that also affords good results for neutral water clusters.83 In Fig. 8b, the same HF/aQZ errors are plotted as a function of the number of subsystems included in the calculation. As expected, errors decrease as the calculation becomes more complete, up to a certain point (around 500 subsystems) when all energetically important terms have been incorporated. Beyond that point there is no further benefit to tightening τ3, and perhaps some noise introduced as the number of subsystems increases.
 | ||
| Fig. 8 MBE(4) results for ten configurations of F−(H2O)15. (a) Errors in ΔEint for calculations at the HF/aQZ level, plotted as a function of the three-body screening threshold τ3. (b) Same data as in (a), plotted as a function of the number of subsystems. (c) Errors in ΔEintversus τ3 for calculations at the PBE/aQZ level. (d) Same data as in (c), versus the number of subsystems. | ||
These sensible trends are inverted in the PBE/aQZ data, for which the τ3 = 0 limit engenders catastrophic error accumulation as documented in Section 2.1. Increasing τ3, which more aggressively removes subsystems from the calculation, dramatically reduces the error in ΔEint; see Fig. 8c. These errors are plotted as a function of the number of subsystems in Fig. 8d. The first ca. 250 subsystems do reduce the error, but beyond that the additional subsystems lead to error accumulation. Our interpretation is that a relatively small number of terms is required to get the gross electronic structure correct (meaning that it roughly represents the solvation environment of F− in the full cluster), but once that is achieved any fine details are washed out by cumulative SIE. For large systems, screening not only reduces the cost but also keeps error accumulation in check.
3.4 Repercussions and outlook
Paesani and co-workers have suggested DFT-based MBE(n) as a tool for generating “data-driven” classical force fields,44 and reasonable results have been obtained for neat liquid water and for ion–water interactions using DC-SCAN.28–30 In our view, this approach succeeds by limiting the expansion to three-body terms, using a classical polarization model to replace ab initio four-body interactions,29 and incorporating conservative distance cutoffs.19–23,30,31 The generality of this approach is questionable, however, in view of the results presented above. Although four-body terms appear to be sufficient for both neat water83,88 and monovalent ion–water interactions,147 higher-order interactions are sizable in divalent ion–water clusters.148Our results do suggest there is a “sweet spot” where just enough neighbors are included for accuracy but not so many as to cause significant accumulation of delocalization error; this is exemplified by PBE results in Fig. 8d. However, this may not be sufficient to salvage all ion–water interactions, or for water–solute interactions involving larger, asymmetric solute molecules. A safer strategy is to retreat to HF theory, leveraging the efficiency of the energy-screened MBE to apply post-HF, correlated wave function models. The BH&H-LYP functional so far appears to be a satisfactory workaround with moderate accuracy and DFT cost.
4 Conclusions
Pairing MBE(n) with DFT results in slow convergence of the n-body interactions for neutral systems and rapid divergence for hydrated ions, using a variety of common GGA and meta-GGA functionals. Hybrid functionals with 20–25% Fock exchange also exhibit unphysical oscillations in the n-body interactions, and functionals such as BH&H-LYP (with 50% Fock exchange) are required in order to eliminate SIE-induced divergence. The latter functional may be a useful workaround. A DC-DFT correction scheme132–134 has shown preliminary promise in small-cluster MBE(n) calculations,28–30,136 but it does not solve the aforementioned problem in a general way.These results may have important implications for the application of fragment-based quantum chemistry to study enzymatic reactions, especially for metalloenzymes149–152 where different oxidation states of a transition metal might be expected to exhibit varying degrees of SIE. Use of larger fragments may help to mitigate wild oscillations in MBE(n) that are documented here, as demonstrated previously for proteins (including those with ionic residues) using low-dielectric boundary conditions.105
That strategy is less straightforward in aqueous systems, where the simplest choice is single-H2O fragments, although methods with overlapping fragments have been used for calculations on various molecular clusters.5,153–161 Screening and culling the subsystem interactions can also mitigate oscillations in MBE(n) while simultaneously reducing cost. In that regard, there seems to exist a “Goldilocks point” at which enough solvating water molecules have been included to describe the solute's environment with reasonable accuracy, yet not so many that delocalization error overwhelms the result. Whether this balance can be codified in an unambiguous way remains an important issue for future study.
At present, the safest approach is to rely on MBE(n) as an efficient means to apply post-HF correlated wave function methods to large systems, starting from a SIE-free HF calculation and aggressively screening the correlated calculations.83,145,162–164 In this way, second-order Møller–Plesset (MP2) calculations have been demonstrated in which a small-basis HF calculation for the entire system is used to recover long-range polarization, with short-range MBE(3) calculations to describe electron correlation, such that the total cost is dramatically reduced with respect to conventional MP2 calculations.145 In future work, we will extend this approach to coupled-cluster calculations that can achieve benchmark accuracy for thermochemistry and non-covalent interactions.
5 Methods
We have previously reported a “bottom-up” algorithm to implement MBE(n) using low-level (typically semi-empirical) energy screening to construct a representation of the n-body interactions in the form of a directed acyclic graph.83 This is vastly more efficient than order-by-order screening, which allows us to extend the expansion to unprecedented orders (n) and systems sizes (N).83,105 This technique is implemented in an open-source code called FRAGME∩T,84 which is used for all of the calculations reported here. Electronic structure calculations are performed by interfacing FRAGME∩T with Q-CHEM.85Interaction energies are computed using the supramolecular approach,
| ΔEint(AB) = E(AB) − E(A) − E(B). | (3) |
The MBE(n) approximation to ΔEint can be computed at a cost that is greatly reduced as compared to naive application of MBE(n) to all three terms in eqn (3). This builds on previous work using a generalization of the MBE that can handle overlapping fragments.169,170 In this approach, a fragment F is simply a subset of the nuclei Ai,83
| F = {A1, A2, …, An}, | (4) |
| S(x) = {F1, F2, … , FN} | (5) |
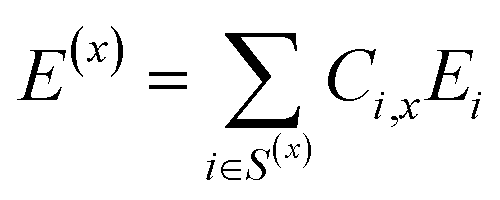 | (6) |
To calculate ΔEint for a system FAB, we define two subsystems FA = {A1, A2, …} and FB = {B1, B2, …}, such that FAB = FA ∪ FB and FA ∩ FB = ∅. The interaction energy for any fragmentation scheme S(x)AB of the system FAB can be computed as
 | (7) |
In Section 3.3, low-level screening is performed using the GFN2-xTB method,146 eliminating terms with
| |ΔEIJK| < τ3, | (8) |
Data availability
The FRAGME∩T code is available at the URL specified in ref. 84. All molecular structures are provided in the ESI.†Author contributions
J. M. H. conceived the project. D. R. B. wrote the code, designed and executed the computational experiments, and performed the analysis. The manuscript was written by both authors.Conflicts of interest
J. M. H. is part owner of Q-Chem Inc. and serves on its board of directors.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences under Award No. DE-SC0008550. Calculations were performed at the Ohio Supercomputer Center.171 We thank Montgomery Gray for providing thermochemical benchmarks for BH&H-LYP + D3 and B3LYP + D3 (Table S2†), using protocols from ref. 172.Notes and references
- R. M. Richard, K. U. Lao and J. M. Herbert, Aiming for benchmark accuracy with the many-body expansion, Acc. Chem. Res., 2014, 47, 2828–2836 CrossRef
.
- R. M. Richard, K. U. Lao and J. M. Herbert, Understanding the many-body expansion for large systems. I. Precision considerations, J. Chem. Phys., 2014, 141, 014108 CrossRef PubMed
- K. U. Lao, K.-Y. Liu, R. M. Richard and J. M. Herbert, Understanding the many-body expansion for large systems. II. Accuracy considerations, J. Chem. Phys., 2016, 144, 164105 CrossRef
- K.-Y. Liu and J. M. Herbert, Understanding the many-body expansion for large systems. III. Critical role of four-body terms, counterpoise corrections, and cutoffs, J. Chem. Phys., 2017, 147, 161729 CrossRef PubMed
- J. M. Herbert, Fantasy versus reality in fragment-based quantum chemistry, J. Chem. Phys., 2019, 151, 170901 CrossRef PubMed
- M. S. Gordon, D. G. Fedorov, S. R. Pruitt and L. V. Slipchenko, Fragmentation methods: a route to accurate calculations on large systems, Chem. Rev., 2012, 112, 632–672 CrossRef
- M. A. Collins and R. P. Bettens, Energy-based molecular fragmentation methods, Chem. Rev., 2015, 115, 5607–5642 CrossRef
-
Fragmentation: Toward Accurate Calculations on Complex Molecular Systems, ed. M. S. Gordon, John Wiley & Sons, Hoboken, 2017 Search PubMed
- V. Gavini, S. Baroni, V. Blum, D. R. Bowler, A. Buccheri, J. R. Chelikowsky, S. Das, W. Dawson, P. Delugas, M. Dogan, C. Draxl, G. Galli, L. Genovese, P. Giannozzi, M. Giantomassi, X. Gonze, M. Govoni, F. Gygi, A. Gulans, J. M. Herbert, S. Kokott, T. D. Kühne, K.-H. Liou, T. Miyazaki, P. Motamarri, A. Nakata, J. E. Pask, C. Plessl, L. E. Ratcliff, R. M. Richard, M. Rossi, R. Schade, M. Scheffler, O. Schütt, P. Suryanarayana, M. Torrent, L. Truflandier, T. L. Windus, Q. Xu, V. W.-Z. Yu and D. Perez, Roadmap on electronic structure codes in the exascale era, Modell. Simul. Mater. Sci. Eng., 2023, 31, 063301 CrossRef
- G. J. O. Beran, Approximating quantum many-body intermolecular interactions in molecular clusters using classical polarizable force fields, J. Chem. Phys., 2009, 130, 164115 CrossRef
- A. Sebetci and G. J. O. Beran, Spatially homogeneous QM/MM for systems of interacting molecules with on-the-fly ab initio force-field parameterization, J. Chem. Theory Comput., 2010, 6, 155–167 CrossRef CAS PubMed
- S. Wen and G. J. O. Beran, Accurate molecular crystal lattice energies from a fragment QM/MM approach with on-the-fly ab initio force field parametrization, J. Chem. Theory Comput., 2011, 7, 3733–3742 CrossRef CAS PubMed
- S. Wen, K. Nanda, Y. Huang and G. J. O. Beran, Practical quantum mechanics-based fragment methods for predicting molecular crystal properties, Phys. Chem. Chem. Phys., 2012, 14, 7578–7590 RSC
- T. L. Teuteberg, M. Eckhoff and R. A. Mata, A full additive QM/MM scheme for the computation of molecular crystals with extension to many-body expansions, J. Chem. Phys., 2019, 150, 154118 CrossRef PubMed
- Y. Wang, X. Huang, B. C. Shepler, B. J. Braams and J. M. Bowman, Flexible, ab initio potential, and dipole moment surfaces for water. I. Tests and applications for clusters up to the 22-mer, J. Chem. Phys., 2011, 134, 094509 CrossRef
- A. Nandi, C. Qu, P. L. Houston, R. Conte, Q. Yu and J. M. Bowman, A CCSD(T)-based 4-body potential for water, J. Phys. Chem. Lett., 2021, 12, 10318–10324 CrossRef CAS PubMed
- Q. Yu, C. Qu, P. L. Houston, R. Conte, A. Nandi and J. M. Bowman, q-AQUA: a many-body CCSD(T) water potential, including four-body interactions, demonstrates the quantum nature of water from clusters to the liquid phase, J. Phys. Chem. Lett., 2022, 13, 5068–5074 CrossRef CAS PubMed
- C. Qu, Q. Yu, R. Conte, P. L. Houston, A. Nandi and J. M. Bowman, A Δ-machine learning approach for force fields, illustrated by a CCSD(T) 4-body correction to the MB-pol water potential, Digital Discovery, 2022, 1, 658–664 RSC
- V. Babin, C. Leforestier and F. Paesani, Development of a “first principles” water potential with flexible monomers: dimer potential energy surface, VRT spectrum, and second virial coefficient, J. Chem. Theory Comput., 2013, 9, 5395–5403 CrossRef CAS PubMed
- V. Babin, G. R. Medders and F. Paesani, Development of a “first principles” water potential with flexible monomers. II. Trimer potential energy surface, third Virial coefficient, and small clusters, J. Chem. Theory Comput., 2014, 10, 1599–1607 CrossRef CAS PubMed
- P. Bajaj, A. W. Götz and F. Paesani, Toward chemical accuracy in the description of ion–water interactions through many-body representations. I. Halide–water dimer potential energy surfaces, J. Chem. Theory Comput., 2016, 12, 2698–2705 CrossRef CAS
- M. Riera, N. Mardirossian, P. Bajaj, A. W. Götz and F. Paesani, Toward chemical accuracy in the description of ion–water interactions through many-body representations. Alkali–water dimer potential energy surfaces, J. Chem. Phys., 2017, 147, 161715 CrossRef PubMed
- A. Caruso and F. Paesani, Data-driven many-body models enable a quantitative description of chloride hydration from clusters to bulk, J. Chem. Phys., 2021, 155, 064502 CrossRef CAS PubMed
- F. Paesani, P. Bajaj and M. Riera, Chemical accuracy in modeling halide ion hydration from many-body representations, Adv. Phys.: X, 2019, 4, 1631212 CAS
-
F. Paesani, Water: many-body potential from first principles (from the gas to the liquid phase), in Handbook of Materials Modeling, ed. W. Andreoni and S. Yip, Springer International Publishing, Cham, 2020, pp. 635–660 Search PubMed
- E. Lambros, S. Dasgupta, E. Palos, S. Swee, J. Hu and F. Paesani, General many-body framework for data-driven potentials with arbitrary quantum mechanical accuracy: water as a case study, J. Chem. Theory Comput., 2021, 17, 5635–5650 CrossRef CAS
- X. Zhu, M. Riera, E. F. Bull-Vulpe and F. Paesani, MB-pol(2023): sub-chemical accuracy for water simulations from the gas to the liquid phase, J. Chem. Theory Comput., 2023, 19, 3551–3566 CrossRef CAS
- E. Palos, A. Caruso and F. Paesani, Consistent density functional theory-based description of ion hydration through density-corrected many-body representations, J. Chem. Phys., 2023, 159, 181101 CrossRef CAS PubMed
- S. Dasgupta, E. Lambros, J. P. Perdew and F. Paesani, Elevating density functional theory to chemical accuracy for water simulations through a density-corrected many-body formalism, Nat. Commun., 2021, 12, 6359 CrossRef CAS
- E. Lambros, J. Hu and F. Paesani, Assessing the accuracy of the SCAN functional for water through a many-body analysis of the adiabatic connection formula, J. Chem. Phys., 2021, 17, 3739–3749 CAS
- M. Riera, E. Lambros, T. T. Nguyen, A. W. Götz and F. Paesani, Low-order many-body interactions determine the local structure of liquid water, Chem. Sci., 2019, 10, 8211–8217 RSC
- J. M. Pedulla, K. Kim and K. D. Jordan, Theoretical study of the n-body interaction energies of the ring, cage and prism forms of (H2O)6, Chem. Phys. Lett., 1998, 291, 78–84 CrossRef CAS
- A. D. Kulkarni, V. Ganesh and S. R. Gadre, Many-body interaction analysis: algorithm development and application to large molecular clusters, J. Chem. Phys., 2004, 121, 5043–5050 CrossRef CAS PubMed
-
R. A. Christie and K. D. Jordan, n-Body decomposition approach to the calculation of interaction energies of water clusters, in Intermolecular Forces and Clusters II, ed. D. Wales and R. A. Christie, Structure and Bonding, Springer-Verlag, Heidelberg, 2005, vol. 116, pp. 27–41 Search PubMed
- L. Rincón, R. Almeida and D. G. Aldea, Many-body energy decomposition analysis of cooperativity in hydrogen fluoride clusters, Int. J. Quantum Chem., 2005, 102, 443–453 CrossRef
- B. B. Bizzarro, C. K. Egan and F. Paesani, Nature of halide–water interactions: insights from many-body representations and density functional theory, J. Chem. Theory Comput., 2019, 15, 2983–2995 CrossRef CAS PubMed
- C. K. Egan, B. B. Bizzarro, M. Riera and F. Paesani, Nature of ion–water interactions: insights from many-body representations and density functional theory. II, J. Chem. Theory Comput., 2020, 16, 3055–3072 CrossRef CAS PubMed
- J. P. Heindel, K. M. Herman and S. S. Xantheas, Many-body effects in aqueous systems: synergies between interaction analysis techniques and force field development, Annu. Rev. Phys. Chem., 2023, 74, 337–360 CrossRef CAS PubMed
- K. Yao, J. E. Herr and J. Parkhill, The many-body expansion combined with neural networks, J. Chem. Phys., 2017, 146, 014106 CrossRef PubMed
- T. T. Nguyen, E. Székely, G. Imbalzano, J. Behler, G. Csányi, M. Ceriotti, A. W. Götz and F. Paesani, Comparison of permutationally invariant polynomials, neural networks, and Gaussian approximation potentials in representing water interactions through many-body expansions, J. Chem. Phys., 2018, 148, 241725 CrossRef
- M. Shiranirad, C. J. Burnham and N. J. English, Machine-learning-based many-body energy analysis of argon clusters: fit for size?, Chem. Phys., 2022, 552, 111347 CrossRef CAS
- A. M. Maldonado, I. Poltavsky, V. Vassilev-Galindo, A. Tkatchenko and J. A. Keith, Modeling molecular ensembles with gradient-domain machine learning force fields, Digital Discovery, 2023, 2, 871–880 RSC
- S. R. Xie, M. Rupp and R. G. Hennig, Ultra-fast interpretable machine-learning potentials, npj Comput. Mater., 2023, 9, 162 CrossRef
- E. Palos, S. Dasgupta, E. Lambros and F. Paesani, Data-driven many-body potentials from density functional theory for aqueous phase chemistry, Chem. Phys. Rev., 2023, 4, 011301 CrossRef CAS
- E. E. Dahlke and D. G. Truhlar, Electrostatically embedded many-body expansions for simulations, J. Chem. Theory Comput., 2008, 4, 1–6 CrossRef CAS
- T. C. Ricard, A. Kumar and S. S. Iyengar, Embedded, graph-theoretically defined many-body approximations for wavefunction-in-DFT and DFT-in-DFT: applications to gas- and condensed-phased ab initio molecular dynamics, and potential surfaces for quantum nuclear effects, Int. J. Quantum Chem., 2020, 120, e26244 CrossRef CAS
- T. C. Ricard and S. S. Iyengar, Efficient and accurate approach to estimate hybrid functional and large basis-set contributions to condensed-phase systems and molecule–surface interactions, J. Chem. Theory Comput., 2020, 16, 4790–4812 CrossRef CAS
- P. J. Hasnip, K. Refson, M. I. J. Probert, J. R. Yates, S. J. Clark and C. J. Pickard, Density functional theory in the solid state, Philos. Trans. R. Soc., A, 2014, 372, 20130270 CrossRef
- R. Peverati and D. G. Truhlar, Quest for a universal density functional: the accuracy of density functionals across a broad spectrum of databases in chemistry and physics, Philos. Trans. R. Soc., A, 2014, 372, 20120476 CrossRef
- N. Mardirossian and M. Head-Gordon, Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS
- P. Mori-Sánchez, A. J. Cohen and W. Yang, Many-electron self-interaction error in approximate density functionals, J. Chem. Phys., 2006, 125, 201201 CrossRef
- T. Tsuneda and K. Hirao, Self-interaction corrections in density functional theory, J. Chem. Phys., 2014, 140, 18A513 CrossRef PubMed
- J. P. Perdew, A. Ruzsinszky, J. Sun and M. R. Pederson, Paradox of self-interaction correction: how can anything so right be so wrong?, Adv. At., Mol., Opt. Phys., 2015, 64, 1–14 Search PubMed
- B. G. Janesko, Replacing hybrid density functional theory: motivation and recent advances, Chem. Soc. Rev., 2021, 50, 8470–8495 RSC
- A. J. Cohen, P. Mori-Sanchez and W. Yang, Insights into current limitations of density functional theory, Science, 2008, 321, 792–794 CrossRef CAS
- A. J. Cohen, P. Mori-Sanchez and W. Yang, Challenges for density functional theory, Chem. Rev., 2012, 112, 289–320 CrossRef CAS PubMed
- K. R. Bryenton, A. A. Adeleke, S. G. Dale and E. R. Johnson, Delocalization error: the greatest outstanding challenge in density-functional theory, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2023, 13, e1631 CAS
- B. Braïda, P. C. Hiberty and A. Savin, A systematic failing of current density functionals: overestimation of two-center three-electron bonding energies, J. Phys. Chem. A, 1998, 102, 7872–7877 CrossRef
- M. Sodupe, J. Bertran, L. Rodríguez-Santiago and E. J. Baerends, Ground state of the (H2O)2+ radical cation: DFT versus post-Hartree–Fock methods, J. Phys. Chem. A, 1999, 103, 166–170 CrossRef CAS
- J. M. Herbert and M. Head-Gordon, Calculation of electron detachment energies for water cluster anions: an appraisal of electronic structure methods, with application to (H2O)20− and (H2O)24−, J. Phys. Chem. A, 2005, 109, 5217–5229 CrossRef CAS PubMed
- Y. A. Mantz, F. L. Gervasio, T. Laino and M. Parrinello, Charge localization in stacked radical cation DNA base pairs and the benzene dimer studied by self-interaction corrected density-functional theory, J. Phys. Chem. A, 2007, 111, 105–112 CrossRef CAS PubMed
- E. Livshits, R. S. Granot and R. Baer, A density functional theory for studying ionization processes in water clusters, J. Phys. Chem. A, 2011, 115, 5735–5744 CrossRef CAS PubMed
- P. R. Tentscher and J. S. Arey, Binding in radical-solvent binary complexes: benchmark energies and performance of approximate methods, J. Chem. Theory Comput., 2013, 9, 1568–1579 CrossRef CAS
- E. R. Johnson, M. Salamone, M. Bietti and G. A. DiLabio, Modeling noncovalent radical–molecule interactions using conventional density-functional theory: beware erroneous charge transfer, J. Phys. Chem. A, 2013, 117, 947–952 CrossRef CAS
- E. R. Johnson, A. Otero-de-la-Roza and S. G. Dale, Extreme density-driven delocalization error for a model solvated-electron system, J. Chem. Phys., 2013, 139, 184116 CrossRef PubMed
- M.-C. Kim, E. Sim and K. Burke, Ions in solution: density corrected density functional theory (DC-DFT), J. Chem. Phys., 2014, 140, 18A528 CrossRef PubMed
- M.-C. Kim, H. Park, S. Son, E. Sim and K. Burke, Improved DFT potential energy surfaces via improved densities, J. Phys. Chem. Lett., 2015, 6, 3802–3807 CrossRef CAS PubMed
- J. VandeVondele and M. Sprik, A molecular dynamics study of the hydroxyl radical in solution applying self-interaction-corrected density functional methods, Phys. Chem. Chem. Phys., 2005, 7, 1363–1367 RSC
- B. Rana and J. M. Herbert, Role of hemibonding in the structure and ultraviolet spectroscopy of the aqueous hydroxyl radical, Phys. Chem. Chem. Phys., 2020, 22, 27829–27844 RSC
- B. Rana and J. M. Herbert, Hidden hemibonding in the aqueous hydroxyl radical, J. Phys. Chem. Lett., 2021, 12, 8053–8060 CrossRef CAS PubMed
- B. Rana, M. P. Coons and J. M. Herbert, Detection and correction of delocalization errors for electron and hole polarons using density-corrected DFT, J. Phys. Chem. Lett., 2022, 13, 5275–5284 CrossRef CAS PubMed
- Y. Zhang and W. Yang, A challenge for density functionals: self-interaction error increases for systems with a noninteger number of electrons, J. Chem. Phys., 1998, 109, 2604–2608 Search PubMed
- A. Ruzsinszky, J. P. Perdew, G. I. Csonka, O. A. Vydrov and G. E. Scuseria, Spurious fractional charge of dissociated atoms: pervasive and resilient self-interaction error of common density functionals, J. Chem. Phys., 2006, 125, 194112 Search PubMed
- O. A. Vydrov, G. E. Scuseria and J. P. Perdew, Tests of functionals for systems with fractional electron number, J. Chem. Phys., 2007, 126, 154109 CrossRef
- S. Grimme, W. Hujo and B. Kirchner, Performance of dispersion-corrected density functional theory for the interactions in ionic liquids, Phys. Chem. Chem. Phys., 2012, 14, 4875–4883 RSC
- I. Lage-Estebanez, A. Ruzanov, J. M. García de la Vega, M. V. Fedorov and V. B. Ivaništštev, Self-interaction error in DFT-based modelling of ionic liquids, Phys. Chem. Chem. Phys., 2016, 18, 2175–2182 Search PubMed
- Q. Zhao and H. J. Kulik, Where does the density localize in the solid state? Divergent behavior for hybrids and DFT+U, J. Chem. Theory Comput., 2018, 14, 670–683 CrossRef CAS
- B. Janesko, Coupled alkali halide color centers: fractional charge errors, fractional spin errors, and a failure of spin symmetry breaking produce challenging tests for condensed-phase electronic structure calculations, J. Chem. Phys., 2019, 151, 064109 Search PubMed
- J. A. Gauthier, L. D. Chen, M. Bajdich and K. Chan, Implications of the fractional charge of hydroxide at the electrochemical interface, Phys. Chem. Chem. Phys., 2020, 22, 6964–6969 RSC
- M. R. Pederson, K. P. K. Withanage, Z. Hooshmand, A. I. Johnson, T. Baruah, Y. Yamamoto, R. R. Zope, D.-Y. Kao, P. B. Shukla, J. K. Johnson, J. E. Peralta and K. A. Jackson, Use of FLOSIC for understanding anion-solvent interactions, J. Chem. Phys., 2023, 159, 154112 CrossRef CAS
- N. Mardirossian and M. Head-Gordon,
ωB97X-V: a 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy, Phys. Chem. Chem. Phys., 2014, 16, 9904–9924 RSC
- J. Sun, A. Ruzsinszky and J. P. Perdew, Strongly constrained and appropriately normed semilocal density functional, Phys. Rev. Lett., 2015, 115, 036402 Search PubMed
- D. R. Broderick and J. M. Herbert, Scalable generalized screening for high-order terms in the many-body expansion: algorithm, open-source implementation, and demonstration, J. Chem. Phys., 2023, 159, 174801 CrossRef CAS PubMed
- FRAGME∩T, https://gitlab.com/fragment-qc/fragment, accessed 2024-10-08.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z.-Q. You, Y. Zhu, B. Alam, B. J. Albrecht, A. Aldossary, E. Alguire, J. H. Andersen, V. Athavale, D. Barton, K. Begam, A. Behn, N. Bellonzi, Y. A. Bernard, E. J. Berquist, H. G. A. Burton, A. Carreras, K. Carter-Fenk, R. Chakraborty, A. D. Chien, K. D. Closser, V. Cofer-Shabica, S. Dasgupta, M. de Wergifosse, J. Deng, M. Diedenhofen, H. Do, S. Ehlert, P.-T. Fang, S. Fatehi, Q. Feng, T. Friedhoff, J. Gayvert, Q. Ge, G. Gidofalvi, M. Goldey, J. Gomes, C. E. González-Espinoza, S. Gulania, A. O. Gunina, M. W. D. Hanson-Heine, P. H. P. Harbach, A. Hauser, M. F. Herbst, M. Hernández Vera, M. Hodecker, Z. C. Holden, S. Houck, X. Huang, K. Hui, B.
C. Huynh, M. Ivanov, A. Jász, H. Ji, H. Jiang, B. Kaduk, S. Kähler, K. Khistyaev, J. Kim, G. Kis, P. Klunzinger, Z. Koczor-Benda, J. H. Koh, D. Kosenkov, L. Koulias, T. Kowalczyk, C. M. Krauter, K. Kue, A. Kunitsa, T. Kus, I. Ladjánszki, A. Landau, K. V. Lawler, D. Lefrancois, S. Lehtola, R. R. Li, Y.-P. Li, J. Liang, M. Liebenthal, H.-H. Lin, Y.-S. Lin, F. Liu, K.-Y. Liu, M. Loipersberger, A. Luenser, A. Manjanath, P. Manohar, E. Mansoor, S. F. Manzer, S.-P. Mao, A. V. Marenich, T. Markovich, S. Mason, S. A. Maurer, P. F. McLaughlin, M. F. S. J. Menger, J.-M. Mewes, S. A. Mewes, P. Morgante, J. W. Mullinax, K. J. Oosterbaan, G. Paran, A. C. Paul, S. K. Paul, F. Pavošević, Z. Pei, S. Prager, E. I. Proynov, A. Rák, E. Ramos-Cordoba, B. Rana, A. E. Rask, A. Rettig, R. M. Richard, F. Rob, E. Rossomme, T. Scheele, M. Scheurer, M. Schneider, N. Sergueev, S. M. Sharada, W. Skomorowski, D. W. Small, C. J. Stein, Y.-C. Su, E. J. Sundstrom, Z. Tao, J. Thirman, G. J. Tornai, T. Tsuchimochi, N. M. Tubman, S. P. Veccham, O. Vydrov, J. Wenzel, J. Witte, A. Yamada, K. Yao, S. Yeganeh, S. R. Yost, A. Zech, I. Y. Zhang, X. Zhang, Y. Zhang, D. Zuev, A. Aspuru-Guzik, A. T. Bell, N. A. Besley, K. B. Bravaya, B. R. Brooks, D. Casanova, J.-D. Chai, S. Coriani, C. J. Cramer, G. Cserey, A. E. DePrince III, R. A. DiStasio Jr., A. Dreuw, B. D. Dunietz, T. R. Furlani, W. A. Goddard III, S. Hammes-Schiffer, T. Head-Gordon, W. J. Hehre, C.-P. Hsu, T.-C. Jagau, Y. Jung, A. Klamt, J. Kong, D. S. Lambrecht, W. Liang, N. J. Mayhall, C. W. McCurdy, J. B. Neaton, C. Ochsenfeld, J. A. Parkhill, R. Peverati, V. A. Rassolov, Y. Shao, L. V. Slipchenko, T. Stauch, R. P. Steele, J. E. Subotnik, A. J. W. Thom, A. Tkatchenko, D. G. Truhlar, T. Van Voorhis, T. A. Wesolowski, K. B. Whaley, H. L. Woodcock III, P. M. Zimmerman, S. Faraji, P. M. W. Gill, M. Head-Gordon, J. M. Herbert and A. I. Krylov, Software for the frontiers of quantum chemistry: an overview of developments in the Q-Chem 5 package, J. Chem. Phys., 2021, 155, 084801 Search PubMed
- M. Gray, P. E. Bowling and J. M. Herbert, Systematic examination of counterpoise correction in density functional theory, J. Chem. Theory Comput., 2022, 18, 6742–6756 Search PubMed
- R. M. Richard, K.
U. Lao and J. M. Herbert, Approaching the complete-basis limit with a truncated many-body expansion, J. Chem. Phys., 2013, 139, 224102 Search PubMed
- J. P. Heindel and S. S. Xantheas, The many-body expansion for aqueous systems revisited: I. Water–water interactions, J. Chem. Theory Comput., 2020, 16, 6843–6855 CrossRef CAS
- K. Sharkas, K. Wagle, B. Santra, S. Akter, R. R. Zope, T. Baruah, K. A. Jackson, J. P. Perdew and J. E. Peralta, Self-interaction error overbinds water clusters but cancels in structural energy differences, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 11283–11288 Search PubMed
- E. Palos, E. Lambros, S. Swee, J. Hu, S. Dasgupta and F. Paesani, Assessing the interplay between functional-driven and density-driven errors in DFT models of water, J. Chem. Theory Comput., 2022, 18, 3410–3426 CrossRef CAS
- L. D. Jacobson and J. M. Herbert, An efficient, fragment-based electronic structure method for molecular systems: self-consistent polarization with perturbative two-body exchange and dispersion, J. Chem. Phys., 2011, 134, 094118 CrossRef
- K. Schwarz, Instability of stable negative ions in the Xα method or other local density functional schemes, Chem. Phys. Lett., 1978, 57, 605–607 Search PubMed
- A. A. Jarȩcki and E. R. Davidson, Density functional theory calculations for F−, Chem. Phys. Lett., 1999, 300, 44–52 CrossRef
- J. M. Galbraith and H. F. Schaefer III, Concerning the applicability of density functional methods to atomic and molecular ions, J. Chem. Phys., 1996, 105, 862–864 CrossRef CAS
- N. Rösch and S. B. Trickey, Comment on ‘Concerning the applicability of density functional methods to atomic and molecular negative ions’, J. Chem. Phys., 1997, 106, 8940–8941 Search PubMed
- D. Lee and K. Burke, Finding electron affinities with approximate density functionals, Mol. Phys., 2010, 108, 2687–2701 CrossRef CAS
- D. Lee, F. Furche and K. Burke, Accuracy of electron affinities of atoms in approximate density functional theory, J. Phys. Chem. Lett., 2010, 1, 2124–2129 CrossRef CAS
- F. Jensen, Describing anions by density functional theory: fractional electron affinity, J. Chem. Theory Comput., 2010, 6, 2726–2735 CrossRef CAS PubMed
-
J. M. Herbert, The quantum chemistry of loosely-bound electrons, in Reviews in Computational Chemistry, ed. A. L. Parill and K. Lipkowitz, Wiley-VCH, Hoboken, 2015, ch. 8, vol. 28, pp. 391–517 Search PubMed
- M. J. G. Peach, A. M. Teale, T. Helgaker and D. J. Tozer, Fractional electron loss in approximate DFT and Hartree–Fock theory, J. Chem. Theory Comput., 2015, 11, 5262–5268 CrossRef CAS
- M. A. Rohrdanz, K. M. Martins and J. M. Herbert, A long-range-corrected density functional that performs well for both ground-state properties and time-dependent density functional theory excitation energies, including charge-transfer excited states, J. Chem. Phys., 2009, 130, 054112 Search PubMed
- B. Alam, A. F. Morrison and J. M. Herbert, Charge separation and charge transfer in the low-lying excited states of pentacene, J. Phys. Chem. C, 2020, 124, 24653–24666 CrossRef CAS
- K. Wagle, B. Santra, P. Bhattarai, C. Shahi, M. R. Pederson, K. A. Jackson and J. P. Perdew, Self-interaction correction in water–ion clusters, J. Chem. Phys., 2021, 154, 094302 CrossRef CAS
- J. F. Ouyang, M. W. Cvitkovic and R. P. A. Bettens, Trouble with the many-body expansion, J. Chem. Theory Comput., 2014, 10, 3699–3707 CrossRef CAS PubMed
- P. E. Bowling, D. R. Broderick and J. M. Herbert, Fragment-based calculations of enzymatic thermochemistry require dielectric boundary conditions, J. Phys. Chem. Lett., 2023, 14, 3826–3834 Search PubMed
- Y. Zhu and J. M. Herbert, High harmonic spectra computed using time-dependent Kohn–Sham theory with Gaussian orbitals and a complex absorbing potential, J. Chem. Phys., 2022, 156, 204123 CrossRef CAS
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 Search PubMed
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B, 1988, 37, 785–789 Search PubMed
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- A. D. Becke, A new mixing of Hartree–Fock and local density-functional theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS
- E. Ruiz, D. R. Salahub and A. Vela, Charge-transfer complexes: stringent tests for widely used density functionals, J. Phys. Chem., 1996, 100, 12265–12276 CrossRef CAS
- M.-S. Liao, Y. Lu and S. Scheiner, Performance assessment of density-functional methods for study of charge-transfer complexes, J. Comput. Chem., 2003, 24, 623–631 CrossRef CAS
- A. Nakata, Y. Imamura, T. Otsuka and H. Nakai, Time-dependent density functional theory calculations for core-excited states: assessment of standard exchange-correlation functionals and development of a novel hybrid functional, J. Chem. Phys., 2006, 124, 094105 Search PubMed
- R. J. Magyar and S. Tretiak, Dependence of spurious charge-transfer excited states on orbital exchange in TDDFT: large molecules and clusters, J. Chem. Theory Comput., 2007, 3, 976–987 Search PubMed
- D. M. Chipman, Absorption spectrum of OH radical in water, J. Phys. Chem. A, 2008, 112, 13372–13381 CrossRef CAS PubMed
- N. A. Besley, M. J. G. Peach and D. J. Tozer, Time-dependent density functional theory calculations of near-edge X-ray absorption fine structure with short-range corrected functionals, Phys. Chem. Chem. Phys., 2009, 11, 10350–10358 RSC
- A. Otero-de-la-Roza, E. R. Johnson and G. A. DiLabio, Halogen bonding from dispersion-corrected density-functional theory: the role of delocalization error, J. Chem. Theory Comput., 2014, 10, 5436–5447 CrossRef CAS
- S. Schwalbe, K. Trepte and S. Lehtola, How good are recent density functionals for ground and excited states of one-electron systems?, J. Chem. Phys., 2022, 157, 174113 Search PubMed
- A. Dreuw, J. L. Weisman and M. Head-Gordon, Long-range charge-transfer excited states in time-dependent density functional theory require non-local exchange, J. Chem. Phys., 2003, 119, 2943–2946 CrossRef CAS
- A. Lange and J. M. Herbert, Simple methods to reduce charge-transfer contamination in time-dependent density-functional calculations of clusters and liquids, J. Chem. Theory Comput., 2007, 3, 1680–1690 CrossRef CAS
- A. W. Lange, M. A. Rohrdanz and J. M. Herbert, Charge-transfer excited states in a π-stacked adenine dimer, as predicted using long-range-corrected time-dependent density functional theory, J. Phys. Chem. B, 2008, 112, 6304–6308 CrossRef CAS PubMed
- A. W. Lange and J. M. Herbert, Both intra- and interstrand charge-transfer excited states in B-DNA are present at energies comparable to, or just above, the 1ππ* excitonic bright states, J. Am. Chem. Soc., 2009, 131, 3913–3922 CrossRef CAS
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, Excitation energies in density functional theory: an evaluation and a diagnostic test, J. Chem. Phys., 2008, 128, 044118 CrossRef PubMed
- J. M. Herbert and A. Mandal, Importance of orbital invariance in quantifying electron–hole separation and exciton size, J. Chem. Theory Comput., 2024 DOI:10.1021/acs.jctc.4c01085
- C. Adamo and V. Barone, Toward reliable adiabatic connection models free from adjustable parameters, Chem. Phys. Lett., 1997, 274, 242–250 CrossRef CAS
- B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, Adiabatic connection for kinetics, J. Phys. Chem. A, 2000, 104, 4811–4815 CrossRef CAS
- B. Chan and J. M. Simmie, Barriometry—an enhanced database of accurate barrier heights for gas-phase reactions, Phys. Chem. Chem. Phys., 2018, 20, 10732–10740 RSC
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef
- L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry kinetics, and noncovalent interactions, Phys. Chem. Chem. Phys., 2017, 19, 32184–32215 RSC
- K. Hui and J.-D. Chai, SCAN-based hybrid and double-hybrid density functionals from models without fitted parameters, J. Chem. Phys., 2016, 144, 044114 CrossRef PubMed
- D. R. Lonsdale and L. Goerigk, The one-electron self-interaction error in 74 density functional approximations: a case study on hydrogenic mono- and dinuclear systems, Phys. Chem. Chem. Phys., 2020, 22, 15805–15830 RSC
- S. Vuckovic, S. Song, J. Kozlowski, E. Sim and K. Burke, Density functional analysis: the theory of density-corrected DFT, J. Chem. Theory Comput., 2019, 15, 6336–6346 Search PubMed
- E. Sim, S. Song, S. Vuckovic and K. Burke, Improving results by improving densities: density-corrected density functional theory, J. Am. Chem. Soc., 2022, 144, 6625–6639 CrossRef CAS
- S. Song, S. Vuckovic, E. Sim and K. Burke, Density-corrected DFT explained: questions and answers, J. Chem. Theory Comput., 2022, 18, 817–827 CrossRef CAS PubMed
- B. Rana, G. J. O. Beran and J. M. Herbert, Correcting π-delocalisation errors in conformational energies using density-corrected DFT, with application to crystal polymorphs, Mol. Phys., 2023, 121, e2138789 CrossRef
- S. Dasgupta, C. Shahi, P. Bhetwal, J. P. Perdew and F. Paesani, How good is the density-corrected SCAN functional for neutral and ionic aqueous systems, and what is so right about the Hartree–Fock density?, J. Chem. Theory Comput., 2022, 18, 4745–4761 CrossRef CAS PubMed
- A. W. Lange and J. M. Herbert, Polarizable continuum reaction-field solvation models affording smooth potential energy surfaces, J. Phys. Chem. Lett., 2010, 1, 556–561 CrossRef CAS
- E. Rudberg, E. H. Rubensson and P. Sałek, Kohn–Sham density functional theory electronic structure calculations with linearly scaling computational time and memory usage, J. Chem. Theory Comput., 2011, 7, 340–350 CrossRef CAS PubMed
- E. Rudberg, Difficulties in applying pure Kohn–Sham density functional theory electronic structure methods to protein molecules, J. Phys.: Condens. Matter, 2012, 24, 072202 CrossRef PubMed
- G. Lever, J. D. Cole, N. D. M. Hine, P. D. Haynes and M. C. Payne, Electrostatic considerations affecting the calculated HOMO–LUMO gap in protein molecules, J. Phys.: Condens. Matter, 2013, 25, 152101 CrossRef
- S. A. Slattery, J. C. Yon and E. F. Valeev, Revisiting artifacts of Kohn–Sham density functionals for biosimulation, J. Chem. Theory Comput., 2024, 20, 6652–6660 CrossRef CAS PubMed
- F. Ren and F. Liu, Impacts of polarizable continuum models on the SCF convergence and DFT delocalization error of large molecules, J. Chem. Phys., 2022, 157, 184106 CrossRef CAS
- J. M. Herbert, Dielectric continuum methods for quantum chemistry, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1519 CAS
- J. F. Ouyang and R. P. A. Bettens, When are many-body effects significant?, J. Chem. Theory Comput., 2016, 12, 5860–5867 CrossRef CAS PubMed
- K.-Y. Liu and J. M. Herbert, Energy-screened many-body expansion: a practical yet accurate fragmentation method for quantum chemistry, J. Chem. Theory Comput., 2020, 16, 475–487 CrossRef
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB—an accurate and broadly parameterized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed
- J. P. Heindel and S. S. Xantheas, The many-body expansion for aqueous systems revisited: II. Alkali metal and halide ion–water interactions, J. Chem. Theory Comput., 2021, 17, 2200–2216 CrossRef CAS
- K. M. Herman, J. P. Heindel and S. S. Xantheas, The many-body expansion for aqueous systems revisited: III. Hofmeister ion–water interactions, Phys. Chem. Chem. Phys., 2021, 23, 11196–11210 RSC
- E. K. Kurbanov, H. R. Leverentz, D. G. Truhlar and E. A. Amin, Electrostatically embedded many-body expansion for neutral and charged metalloenzyme model systems, J. Chem. Theory Comput., 2012, 8, 1–5 CrossRef CAS PubMed
- M. Xu, X. He, T. Zhu and J. Z. H. Zhang, A fragment quantum mechanical method for metalloproteins, J. Chem. Theory Comput., 2019, 15, 1430–1439 CrossRef CAS
- J. Hellmers, E. D. Hedegård and C. König, Fragmentation-based decomposition of a metalloenzyme–substrate interaction: a case study for a lytic polysaccharide monooxygenase, J. Phys. Chem. B, 2022, 126, 5400–5412 CrossRef CAS
- R. Paciotti, N. Re and L. Storchi, Combining the fragment molecular orbital and GRID approaches for the prediction of ligand–metalloenzyme binding affinity: the case study of hCA II inhibitors, Molecules, 2024, 29, 3600 CrossRef CAS
- L. D. Jacobson, R. M. Richard, K. U. Lao and J. M. Herbert, Efficient monomer-based quantum chemistry methods for molecular and ionic clusters, Annu. Rep. Comput. Chem., 2013, 9, 25–58 CAS
- K. Wang, W. Li and S. Li, Generalized energy-based fragmentation CCSD(T)-F12a method and application to the relative energies of water clusters (H2O)20, J. Chem. Theory Comput., 2014, 10, 1546–1553 CrossRef CAS PubMed
- D. Yuan, X. Shen, W. Li and S. Li, Are fragment-based quantum chemistry methods applicable to medium-sized water clusters?, Phys. Chem. Chem. Phys., 2016, 18, 16491–16498 RSC
- D. Yuan, Y. Li, Z. Ni, P. Pulay, W. Li and S. Li, Benchmark relative energies for large water clusters with the generalized energy-based fragmentation method, J. Chem. Theory Comput., 2017, 13, 2696–2704 CrossRef CAS
- L. Zhang, W. Li, T. Fang and S. Li, Accurate relative energies and binding energies of large ice–liquid water clusters and periodic structures, J. Phys. Chem. A, 2017, 121, 4030–4038 CrossRef CAS
- G. Singh, A. Nandi and S. R. Gadre, Breaking the bottleneck: use of molecular tailoring approach for the estimation of binding energies at MP2/CBS limit for large water clusters, J. Chem. Phys., 2016, 144, 104102 CrossRef
- N. Sahu, G. Singh, A. Nandi and S. R. Gadre, Toward an accurate and inexpensive estimation of CCSD(T)/CBS binding energies of large water clusters, J. Phys. Chem. A, 2016, 120, 5706–5714 CrossRef CAS PubMed
- S. S. Khire, N. D. Gurav, A. Nandi and S. R. Gadre, Enabling rapid and accurate construction of CCSD(T)-level potential energy surface of large molecule using molecular tailoring approach, J. Phys. Chem. A, 2022, 126, 1458–1464 CrossRef CAS
- N. Sahu and S. R. Gadre, Molecular tailoring approach: a route for ab initio treatment of large clusters, Acc. Chem. Res., 2014, 47, 2739–2747 CrossRef CAS PubMed
- S. S. Khire and S. R. Gadre, Pragmatic many-body approach for economic MP2 energy estimation of molecular clusters, J. Phys. Chem. A, 2019, 123, 5005–5011 CrossRef CAS PubMed
- S. S. Khire and S. R. Gadre, Development and testing of an algorithm for efficient MP2/CCSD(T) energy estimation of molecular clusters with the 2-body approach, J. Comput. Chem., 2023, 44, 261–267 CrossRef CAS
- S. S. Khire, T. Nakajima and S. R. Gadre, REAlgo: rapid and efficient algorithm for estimating MP2/CCSD energy gradients for large molecular systems, J. Chem. Phys., 2023, 159, 184109 CrossRef CAS PubMed
- M. Kamiya, S. Hirata and M. Valiev, Fast electron correlation methods for molecular clusters without basis set superposition errors, J. Chem. Phys., 2008, 128, 074103 CrossRef
- R. M. Richard, K. U. Lao and J. M. Herbert, Achieving the CCSD(T) basis-set limit in sizable molecular clusters: counterpoise corrections for the many-body expansion, J. Phys. Chem. Lett., 2013, 4, 2674–2680 CrossRef CAS PubMed
- J. F. Ouyang and R. P. A. Bettens, Many-body basis set superposition effect, J. Chem. Theory Comput., 2015, 11, 5132–5143 CrossRef CAS
- R. M. Richard, B. W. Bakr and C. D. Sherrill, Understanding the many-body basis set superposition error: beyond Boys and Bernardi, J. Chem. Theory Comput., 2018, 14, 2386–2400 CrossRef CAS PubMed
- R. M. Richard and J. M. Herbert, A generalized many-body expansion and a unified view of fragment-based methods in electronic structure theory, J. Chem. Phys., 2012, 137, 064113 CrossRef
- R. M. Richard and J. M. Herbert, The many-body expansion with overlapping fragments: analysis of two approaches, J. Chem. Theory Comput., 2013, 9, 1408–1416 CrossRef CAS
- Ohio Supercomputer Center, https://www.osc.edu/ark:/19495/f5s1ph73, accessed 2024-10-06.
- M. Gray, P. E. Bowling and J. M. Herbert, Comment on “Benchmarking basis sets for density functional theory thermochemistry calculations: why unpolarized basis sets and the polarized 6-311G family should be avoided”, J. Phys. Chem. A, 2024, 128, 7739–7745 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05955g |
| This journal is © The Royal Society of Chemistry 2024 |