
Enhancement of carbamazepine photodegradation using hybrid of phosphorescent carbon dots coupled with highly porous TiO2 photocatalyst†
Daniela
Kujawa
 *a,
Anna
Grzegórska
b,
Anna
Zielińska-Jurek
b,
Marzena
Fandzloch
a,
Oleksii
Bezkrovnyi
a and
Paweł
Głuchowski
*a
*a,
Anna
Grzegórska
b,
Anna
Zielińska-Jurek
b,
Marzena
Fandzloch
a,
Oleksii
Bezkrovnyi
a and
Paweł
Głuchowski
*a
aInstitute of Low Temperature and Structural Research, Polish Academy of Sciences, Okólna 2, Wrocław, 50-422, Poland. E-mail: d.kujawa@intibs.pl; p.gluchowski@intibs.pl
bDepartment of Process Engineering and Chemical Technology, Faculty of Chemistry, Gdańsk University of Technology, G. Narutowicza 11/12, Gdańsk, 80-233, Poland
First published on 27th November 2024
Abstract
This study focuses on harnessing the synergistic effects between titanium oxide (TiO2) and carbon dots (CDs) to enhance the photocatalytic performance of TiO2. The work describes the synthesis of carbon dots exhibiting fluorescence (CDs) or phosphorescence (PhCDs), as well as the preparation of the TiO2 hybrid. Structural and textural measurements (X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), N2 sorption isotherms) were conducted to elucidate the compositional and morphological changes induced by the incorporation of CDs into a matrix. Spectroscopic analyses revealed a shift of the absorption edge and range into the visible region of the CDs@TiO2 hybrid compared with pure TiO2. Infrared (IR) and Raman spectroscopy revealed the presence of diverse bonds associated with functional groups on the surface of the dots, enabling control over the spectroscopic properties of the resulting hybrids. Photocatalytic assessments demonstrated an enhancement in the PhCDs@TiO2 hybrid activity compared with pure TiO2. The proposed mechanism for the increase in photocatalytic activity in PhCDs@TiO2 is based on the slowdown of carrier recombination, which is linked to the confinement of electrons within traps located below the conduction band. The demonstrated enhancement in photocatalytic activity holds promise for the more effective decomposition of organic compounds in water, while the utilization of carbon dots unveils new avenues for modifying existing photocatalysts.
1. Introduction
Photocatalysis phenomena on TiO2 and zinc oxide semiconductors have been studied since the 1960s. During these studies, it was found that some compounds, such as O2 or H2O, were adsorbed or desorbed on the surface of the solid under the influence of UV light, depending on the surface conditions. This phenomenon was explained by the band theory of semiconductors which explains how electrons behave in such materials. It highlights two key energy bands: the valence band (VB), where electrons are tightly bound to atoms, and the conduction band (CB), where electrons can move freely. The energy gap between these bands, called the band gap, determines the conductivity properties of a semiconductor. When electrons absorb energy, they are transported from the VB to the CB, and when they return to the VB, the excess energy is released.1–4 The first important research on the photocatalysis process on a TiO2 photocatalyst using UV light was described by Masuo in 1964.5 Honda and Fujishima6 discovered the photosensitization effect on TiO2 electrodes in 1972 and used it to split water, which was performed at a much lower bias voltage compared with normal electrolysis. These processes were performed using Pt electrodes as the cathode and TiO2 as the photoanode. Further research was focused on improving photolysis using TiO2.7,8 This material is a very good photocatalyst, which works with high efficiency, but the main disadvantage is that it absorbs only UV light and cannot work in the visible range. That is why researchers focus on finding new materials or using TiO2 coupled with other materials that have similar or better efficiency and can also absorb visible light.Materials that in recent years focused the research on adapting photocatalysis to the visible range are CDs, which can play different roles in this process. CDs themselves can be used as photocatalysts because of their strong ability to absorb light, excite electrons and generate holes. These materials can also act as electron donors and/or acceptors. The nature of the dots is defined by the amount and type of functional groups on the surface.9 They have the ability to absorb energy and emit photons with energy higher than that absorbed. It means that they can be used as up-converters.10 This property is useful during photocatalysis for many hybrid systems consisting of dots attached to the surface of semiconductors with a wide bandgap. The CDs can absorb long-wavelength visible or infrared light and emit a photon in the ultraviolet range, which then excites electrons in a wide-band semiconductor. This allows such materials to work indirectly in visible light.11
For photocatalytic applications, the CDs are embedded in porous materials. This prevents the aggregation of nanoparticles and improves their optical performance.12 In this case, many researchers are using metal–organic frameworks (MOFs), which are one-, two- or three-dimensional structures. MOFs are formed by metal ions or metal clusters and organic ligands.13,14 This material is a good example of a scaffold for CDs that can be used in photocatalytic applications. MOF with encapsulated dots exhibits high photostability, tunable photoluminescence, rapid electron transfer, and up-conversion luminescence.15–18 Raw MOF is not often used as a photocatalyst because it does not have sufficient ability to absorb light and recombine the charges which are generated by light. CDs can absorb light with higher efficiency. Moreover, their structure provides faster transmission of photogenerated electrons and transforms dots into electron acceptors. Hybrids of MOFs and CDs are used as photocatalysts because they form a structure with many active sites, similar to semiconductors. Each active site in MOFs, such as a metal–oxygen cluster, can be considered a single quantum dot, acting as a light absorber, charge generator, and catalytic site, much like a small semiconductor. Additionally, organic linkers in MOFs can act as antennas, absorbing additional ultraviolet (UV) light and transferring energy to active sites via ligand–metal charge transfer (LMCT).14 Adding carbon dots to that structure could shift the absorption edge to the visible light and slow electron–hole recombination, improving the photocatalytic process.19–22
CDs have also been introduced into commonly known and used photocatalysts to improve their photocatalysis process. The TiO2 semiconductor is used in photocatalysis due to its possible double-walled structure, characterized by a large specific surface area and porosity.22 It has been shown that titanium oxide in the anatase structure exhibits better photocatalytic activity compared with rutile or brookite. Augustynski23 investigated all TiO2 structures and found that the higher photocatalytic performance depends on the presence of surface-bound peroxy species, the formation of which is promoted by the lattice plane of anatase TiO2. To extend the absorption range of anatase, carbon dots can be used as they absorb light mainly in the visible range.24 Yin et al.25 created an N,S-CQDs-TiO2 hybrid (CQDS – carbon quantum dots, which are a subgroup of CDs always showing a spherical structure and crystalline structure with functional groups on the surface) that was able to generate H2 when exposed to light. After preparing such a hybrid, they obtained 77% better results in generating H2 during light exposure than for pure CDs. The conduction band of TiO2 acts as an electron intermediate that efficiently transfers electrons from the inert to the active band of CDs (responsible for H2 generation). Zhang et al.22 obtained CQD/TiO2 nanotubes (NT), which utilized up-conversion emission from carbon dots to degrade rhodamine B in the visible light range. Zhang et al.24 prepared hybrids of CQD with TiO2 NT, which can absorb visible light and near-infrared radiation, promoting charge separation and electron transfer. That hybrid exhibits better photocatalytic properties in reducing CO2 than pure TiO2 NT. Mozdbar et al.26 developed a hybrid material by combining carbon quantum dots with TiO2 and investigated its photocatalytic activity under both visible and UV light. They found that the formation of this hybrid resulted in a shift of the absorption edge towards visible light and led to higher photocatalytic activity in methyl blue removal under visible light irradiation compared with pure TiO2. It was demonstrated that carbon dots can be prepared to exhibit phosphorescence by embedding them in various matrices. The phosphorescence mechanism relies on the transfer of an electron from the singlet to the triplet state via intersystem crossing (ISC), a process in which spin multiplicity changes from a singlet state (paired electron spins) to a triplet state (unpaired electron spins). Once in the triplet state, the electron experiences a slower rate of decay, allowing for long-lasting phosphorescence to occur due to the forbidden nature of triplet–singlet transitions. The matrix in which the carbon dots are embedded plays a critical role in stabilizing the triplet state, often by limiting molecular vibrations or interactions that would otherwise quench the triplet state, thus extending the lifetime of the excited state. Phosphorescent carbon dots, where electrons are maintained in the excited triplet state for extended periods, can effectively reduce the recombination of electron–hole pairs, a significant limiting factor in photocatalytic processes. By slowing down recombination, phosphorescent dots improve the overall efficiency of photocatalytic reactions. In contrast, non-phosphorescent carbon dots, which do not benefit from this stabilization of excited states, exhibit faster recombination rates, which limits their potential in such applications.27–29
The aim of this work was to increase the photocatalytic activity of anatase TiO2 in the degradation of carbamazepine by incorporation of CDs into its pores. This was achieved by impregnating two types of TiO2 with two types of CD. TiO2 was obtained in two different ways: the well-known solvothermal reaction of TBT and acetic acid, and the unconventional route of thermal decomposition of Ti-based MOF. Both materials are in the anatase phase but have different sizes, shapes, porosities, and therefore different properties, so both were used to synthesize new hybrids. Two types of carbon dot were used to show the impact of their different spectroscopic properties on the photocatalytic activity. Luminescent CDs were added to shift the absorption edge of the TiO2 to enable the use of the hybrid in a wider range of light (including the visible range). Phosphorescent CDs also shift the absorption range, but additionally due to their longer emission decay (slower carrier recombination) allow an increase in the photocatalytic activity of the hybrid. It was assumed that the presence of CDs in the TiO2 pores would increase the absorption of light in the visible range, and improve energy transfer in the semiconductor, and that by using dots exhibiting phosphorescence, the carrier recombination process will be slowed down. All the above changes will result in an increase in photocatalytic activity. A number of studies have been conducted to verify our thesis. For pure and hybrid materials, their structure, morphology, spectroscopic properties, textural properties, and photocatalytic activity were examined.
2. Experimental
2.1. Materials
2.2. Equipment
The XRD data were collected between 10 and 80 2Q degrees. The measurements were carried out at room temperature using an X'PERT PRO PANalytical (Malvern Panalytical, Malvern, UK) diffractometer with CuKα1 radiation (1.5406 Å) and step 0.03°. The samples were examined using a Philips CM20 Super Twin operating at 160 kV and also a Tecnai G2 20 X-TWIN transmission electron microscope (TEM) equipped with a LaB6 cathode electron gun, an FEI Eagle 2K CCD camera, and a high-angle annular dark-field (HAADF) detector for scanning transmission electron microscopy (STEM). Measurements were conducted at an accelerating voltage of 200 kV in both bright field and scanning and transmission modes. Additionally, measurements were taken with a Hitachi S-3400N scanning electron microscope (SEM), which featured a tungsten cathode gun, a secondary electron (SE) detector, and a backscattered electron detector. The pure TiO2 samples were sputter-coated with gold using a Cressington 108A gold sputter coater. Imaging was carried out at an accelerating voltage of 10 kV. The N2 adsorption–desorption isotherms were obtained at 77 K using a Micromeritics 3Flex Surface Characterization Analyzer. Prior to isotherm acquisition, the materials were activated and outgassed (150 °C, 1.3 kPa) for 12 h. MicroActive software was used to determine the specific surface area according to the Brunauer–Emmett–Teller (BET) method and total pore volume at a relative pressure (p/p0) = 0.9.The mid-IR spectra of all samples suspended in KBr pellets were measured using a Nicolet iSTM50 FT-IR spectrometer. The Raman spectra were measured using the micro-Raman apparatus (inVia™ Renishaw) with a 514 nm excitation line in the range 100–3200 cm−1. Absorption spectra were measured with a Varian Model 5E UV–VIS–NIR spectrophotometer at room temperature in the range 200–800 nm in reflectance mode. The spectral bandwidth of the instrument was set to 0.1 nm. Based on the absorption spectra, the band gap of materials was calculated by using the Kubelka–Munk theory34,35 which is based on the measurement of diffuse reflection and describes the behaviour of light inside a sample that scatters light. This theory is applicable to particles that are comparable in size to or smaller than the wavelength of the incident light. When the thickness of the samples does not exceed a certain limit, it does not influence the reflectance. It can then be described by the Kubelka–Munk function:
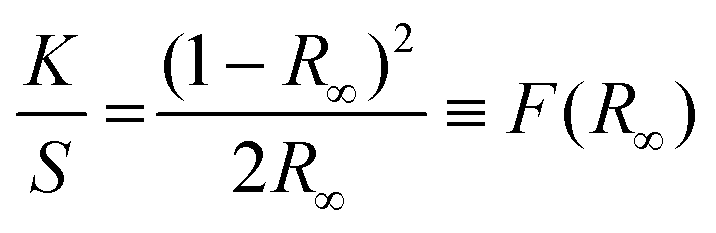 | (1) |
Here, K and S are the absorption and scattering coefficients respectively, R∞ represents the diffuse reflectance, and (R∞) is referred to as the Kubelka–Munk function. In materials with a parabolic band structure, the absorption coefficient and energy gap (Eg) are related through the well-known Tauc36 relation. For different band gap materials, the Tauc relation is given by:
| αhv = A(hv − Eg)n | (2) |
| [F(R∞)hv]2 = A(hv − Eg) | (3) |
In the next step, a graph of (αhv)2vs. hv was plotted, which facilitated the determination of the energy gap.
The emission spectra of TiO2, carbon dots, and hybrid materials were measured using the FLS980 Fluorescence Spectrometer from Edinburgh Instruments (Livingston, UK), equipped with 1800 lines per mm holographic grating and 300 mm focal length monochromators in a Czerny–Turner configuration. To obtain the spectra, a 450 W xenon lamp was employed, and a Hamamatsu R928P side window photomultiplier tube (Hamamatsu Photonics, Shizuoka, Japan) served as the detector. The decay time was measured by using a femtosecond Libra laser (Coherent, USA) and Streak Camera (Hamamatsu, Japan) as a detector.
2.3. Photocatalytic analysis
The photocatalytic activities of the prepared materials were examined in the reaction of carbamazepine (CBZ) degradation. A CBZ solution concentration of 14 mg dm−3 (initial TOC value of 10.67 mg dm−3) was used, while the photocatalyst content was 2 g dm−3. The photocatalytic processes were performed in a glass reactor with a quartz window equipped in air supply mode and placed on a magnetic stirrer. In a typical experiment, 50 mg of the photocatalyst was dispersed in 25 cm3 of the CBZ solution. The irradiation source was a 300 W xenon lamp (LOT Quantum Design) with a light intensity in the UV range (310–380 nm) equal to 30 W m−2. The spectrum of the lamp is presented in Fig. S1.† Before irradiation, the dark reaction was performed for 30 min to reach the adsorption–desorption equilibrium. Experiments were conducted at a constant temperature of 20 °C. Samples (2 cm3) were collected at 0, 20, 40, and 60 min during the photocatalysis process. The suspension was filtered using a 0.2 μm syringe filter to separate the photocatalyst from the solution. The progress of CBZ degradation was monitored using reverse-phase high-performance liquid chromatography (Shimadzu UFLC LC-20AD Kyoto, Japan) with a photodiode array detector (Shimadzu SPD-M20A). The parameters of the HPLC measurement are listed in Table S1.† The mineralization efficiency, measured as the total organic carbon (TOC) removal, was measured using a Shimadzu TOC analyzer.Further experiments were conducted to investigate the influence of the photocatalyst dosage (0.5, 1 and 2 g dm−3) and carbamazepine (CBZ) solution concentration (8.5, 14 and 19.5 mg dm−3) on the efficacy of the photocatalytic process. Additionally, trapping experiments were performed using scavengers (10 mM) to elucidate the reactive species involved in pharmaceutical degradation. Ammonium oxalate was employed as a hole scavenger, tert-butanol as a hydroxyl radical scavenger, ascorbic acid for trapping superoxide anion radicals, and sodium azide as a singlet oxygen scavenger.
3. Results and discussion
3.1. Structure and morphology
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (ICSD #52290), which shows the presence of a polycrystalline boric acid matrix. The CDs show a broad band centred at around 23 2Q degrees that is characteristic of the graphene structure.37 Hybrid materials with PhCDs and CDs show a structure assigned to the anatase phase of the TiO2 framework. The hybrid materials change colour from white to grey after impregnation (Fig. S2†), indicating that the PhCDs/CDs are incorporated into the pores of the TiO2. On the other hand, in the XRD of the hybrid materials, no peaks/bands from the graphene material can be seen, which leads to the conclusion that the content of carbon dots in the TiO2 pores is less than 5%. For hybrid structures, an increase of FWHM (full width at half maximum) of the peaks can be observed compared with the diffractogram for TiO2 (Table 1). The increasing FWHM peaks could result from increasing a structural disorder38 or increasing the tensile stress in the material.39 To test both theories, the strain was calculated from the XRD diffraction pattern. It was shown that after impregnation, the strain increased for almost all samples except PhCDs@TiO2(1), where strain decreased.
space group (ICSD #52290), which shows the presence of a polycrystalline boric acid matrix. The CDs show a broad band centred at around 23 2Q degrees that is characteristic of the graphene structure.37 Hybrid materials with PhCDs and CDs show a structure assigned to the anatase phase of the TiO2 framework. The hybrid materials change colour from white to grey after impregnation (Fig. S2†), indicating that the PhCDs/CDs are incorporated into the pores of the TiO2. On the other hand, in the XRD of the hybrid materials, no peaks/bands from the graphene material can be seen, which leads to the conclusion that the content of carbon dots in the TiO2 pores is less than 5%. For hybrid structures, an increase of FWHM (full width at half maximum) of the peaks can be observed compared with the diffractogram for TiO2 (Table 1). The increasing FWHM peaks could result from increasing a structural disorder38 or increasing the tensile stress in the material.39 To test both theories, the strain was calculated from the XRD diffraction pattern. It was shown that after impregnation, the strain increased for almost all samples except PhCDs@TiO2(1), where strain decreased.
 | ||
| Fig. 1 XRD diffractogram for raw TiO2, carbon dots and hybrid materials. | ||
| Sample | Strains, % | FWHM, ° |
|---|---|---|
| TiO2(1) | 0.071 | 0.3711 |
| PhCDs@TiO2(1) | 0.052 | 0.5188 |
| CDs@TiO2(1) | 0.083 | 0.5274 |
| TiO2(2) | 0.071 | 0.5292 |
| PhCDs@TiO2(2) | 0.091 | 0.7201 |
| CDs@TiO2(2) | 0.082 | 0.7426 |
 | ||
| Fig. 2 TEM (a and b) and SEM (c and d) images of TiO2(1) (a and c) and TiO2(2) (b and d). | ||
TEM images of hybrid materials show that the morphology and size of the grains do not change after impregnation (Fig. 3). It can be observed that after impregnation, the TiO2(1) (Fig. 3a and c) structure shows a lower porosity than before impregnation. In the TEM image of the TiO2(2) hybrid, small grains can be seen on the surface of the titanium dioxide (Fig. 3e and g). This indicates that carbon dots are located partially in the pores but also on the surface of the photocatalysts. The CDs can be observed as nanostructures with a diameter of less than 10 nm (Fig. 3b and f). It can also be observed that the hybrids have planes with an interplanar spacing of 0.33–0.35 nm, which is a characteristic value for a graphene multilayer structure present in CDs as well as TiO2 for the (101) plane of the anatase phase.40,41 Therefore, it is not possible to determine from the TEM images if the smallest particles on the surface are TiO2 or CDs, but based on the spectroscopic results presented later, it can be concluded that they are CDs.
 | ||
| Fig. 3 TEM images of (a and b) CDs@TiO2(1), (c and d) PhCDs@TiO2(1), (e and f) CDs@TiO2(2), and (g and h) PhCDs@TiO2(2). | ||
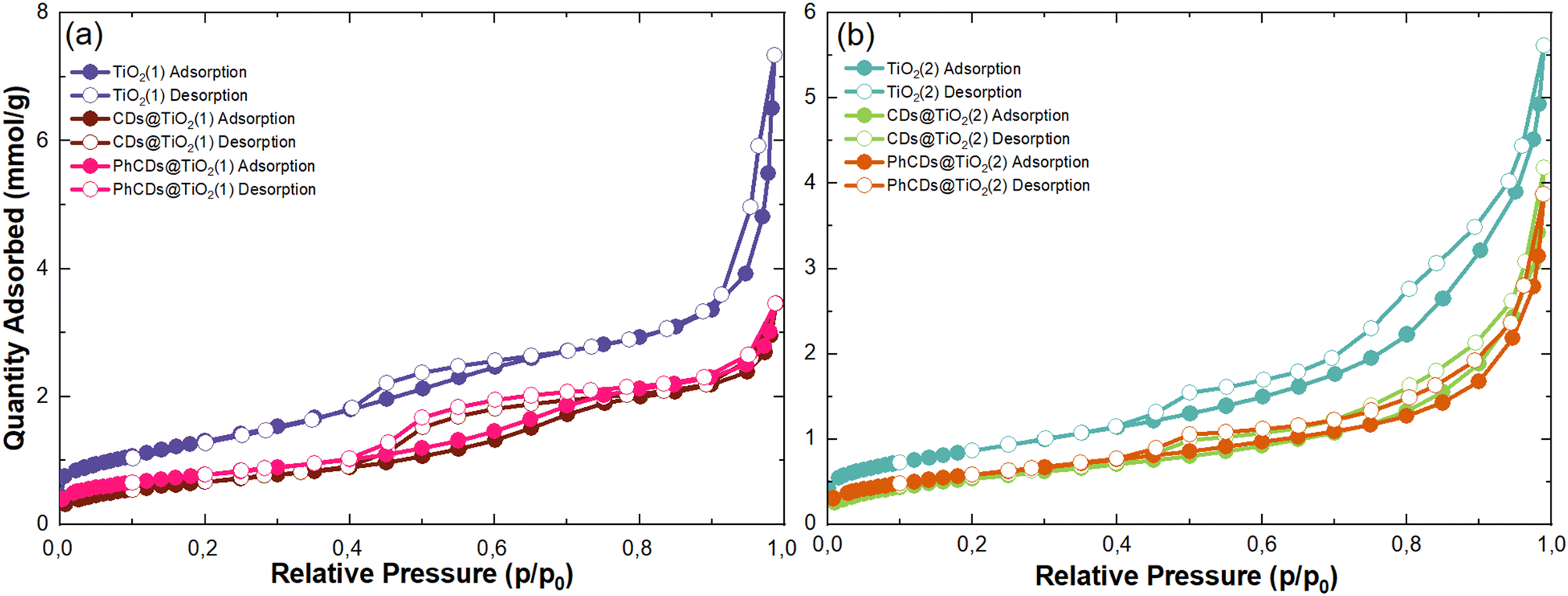 | ||
| Fig. 4 N2 adsorption–desorption isotherms (77 K) of raw TiO2(1) (a) TiO2(2) (b) and their hybrids. | ||
| Sample | S BET, m2 g−1 | V total, cm3 g−1 |
|---|---|---|
| TiO2(1) | 106 | 0.12 |
| CDs@TiO2(1) | 54 | 0.08 |
| PhCDs@TiO2(1) | 62 | 0.08 |
| TiO2(2) | 68 | 0.11 |
| CDs@TiO2(2) | 42 | 0.07 |
| PhCDs@TiO2(2) | 47 | 0.06 |
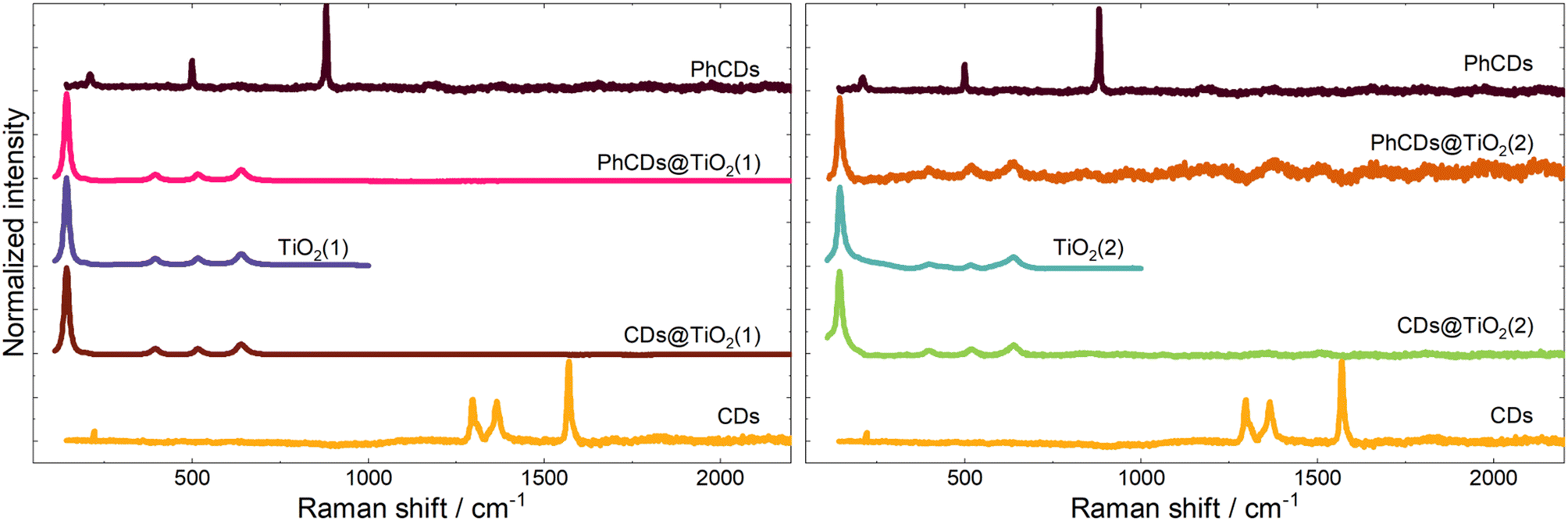 | ||
| Fig. 5 Raman spectra of CDs, TiO2, and hybrid materials. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C can be observed at around 750 and 1500 cm−1 respectively.55 On the surface of carbon dots, carbon atoms are connected with oxygen and nitrogen, as seen in the results. The peaks at around 1700, 1300, and 1150 cm−1 are assigned to the CO,56 C–N–C saturated57 and C–O58 bonds, respectively. The hybrid materials yield similar results, and the characteristic peak of bond Ti–C cannot be observed at about 460 cm−1 (ref. 59) which means that the carbon dots are not connected with TiO2 by metallic or covalent bonds.
C can be observed at around 750 and 1500 cm−1 respectively.55 On the surface of carbon dots, carbon atoms are connected with oxygen and nitrogen, as seen in the results. The peaks at around 1700, 1300, and 1150 cm−1 are assigned to the CO,56 C–N–C saturated57 and C–O58 bonds, respectively. The hybrid materials yield similar results, and the characteristic peak of bond Ti–C cannot be observed at about 460 cm−1 (ref. 59) which means that the carbon dots are not connected with TiO2 by metallic or covalent bonds.
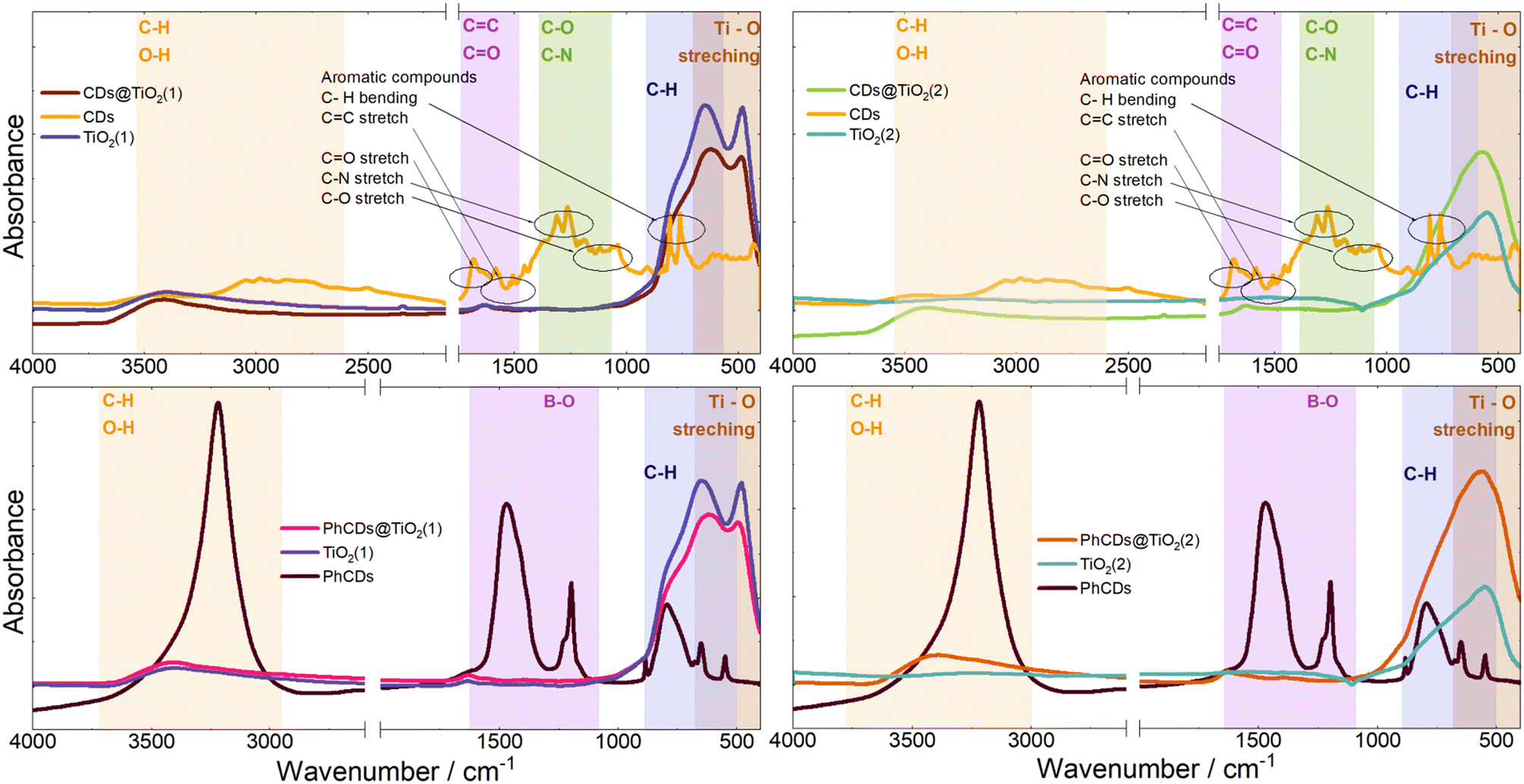 | ||
| Fig. 6 IR spectra of CDs, TiO2, and hybrid materials. | ||
3.2. Spectroscopic properties of CDs@TiO2
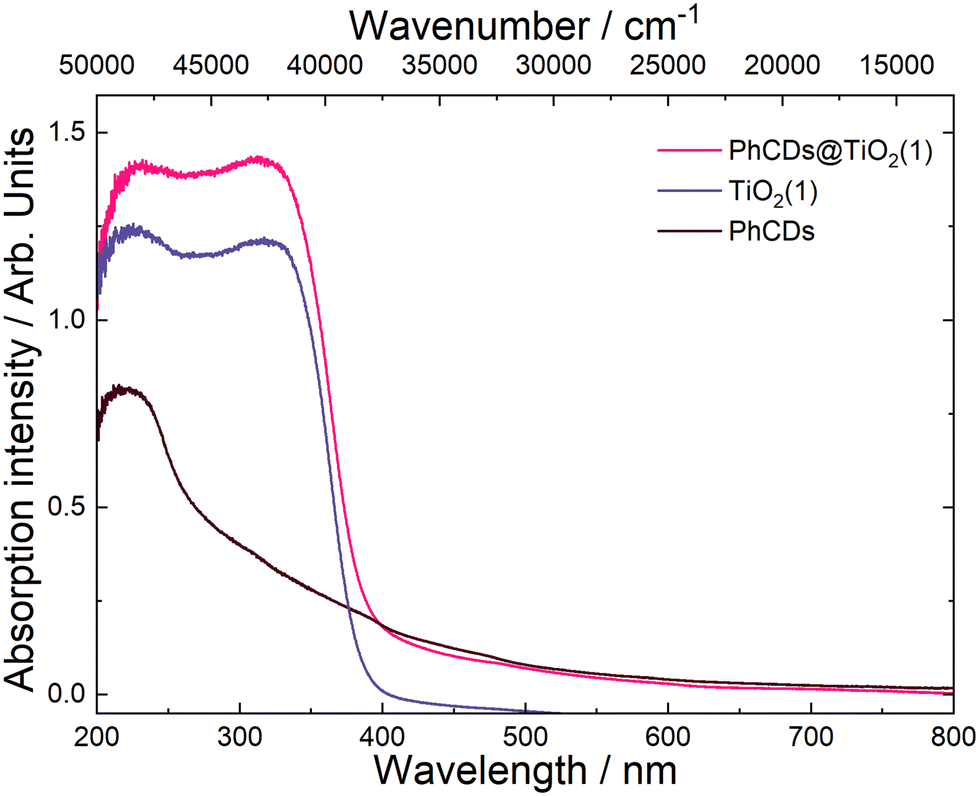 | ||
| Fig. 7 Absorption spectrum of PhCDs@TiO2(1), PhCDs and TiO2(1) compounds. | ||
The spectrum of PhCDs (Fig. 7) shows a wide band of absorption from UV to visible light with the highest intensity in the UV range. The maximum absorption at 220 nm is assigned to the p–p* transition of the CC bond.54 The CDs (Fig. S4†) absorb very well in the UV and blue range, and in the spectrum two separated bands are present, each consisting of two peaks. Absorption peaks at 225 and 253 nm can be also assigned to the p–p* transition of the CC bond, while peaks at 420 and 444 nm can be attributed to the surface states in which free electron pairs can overlap with n–p* transitions.
The hybrid of CDs@TiO2(1) shows a significant increase in the absorption intensity in the UV compared with pure TiO2(1) (Fig. S4†) and also results in a broader absorption range extending from UV to VIS. After the addition of CDs to TiO2(2) (Fig. S4†), a band assigned to 450 nm of CDs can be seen in the absorption spectrum, proving that CDs are embedded in TiO2 pores.
TiO2 exhibited the highest emission intensity in the 350–500 nm range when excited at wavelengths of 320, 350, and 370 nm (Fig. 8 and Fig. S5†). The maximum emission intensity for TiO2(2) is shifted to the red compared with the TiO2(1) when the excitation wavelength was 320 or 350 nm. At 390 nm (Fig. S5†) excitation, it was observed that TiO2(1) emitted effectively in the 400–600 nm range, while TiO2(2) emitted within the narrower range of 400–500 nm. When the samples were excited by a 420 nm (Fig. 8) or 450 nm (Fig. S5†) wavelength, both types of TiO2 displayed a similar broad emission band in the 450–800 nm range.
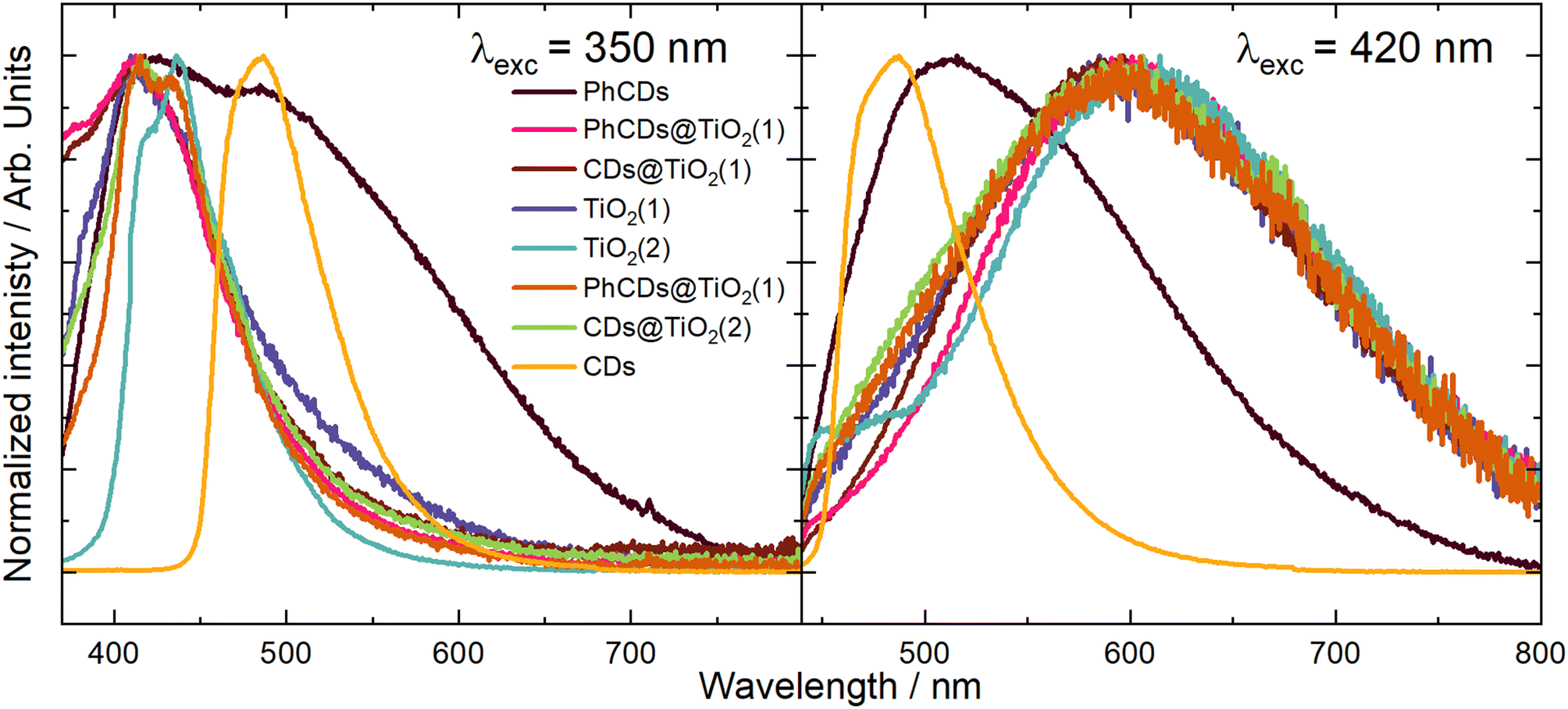 | ||
| Fig. 8 The emission spectrum of CDs, TiO2, and hybrid materials under excitation wavelengths of 350 and 420 nm. | ||
The nature of the emission can be explained by the mechanisms observed for TiO2. It is based on the presence of oxygen vacancies present in the structure of the material. They occur when an oxygen atom is missing from its designated position in the lattice. This absence can occur due to various reasons, such as structural irregularities within the lattice. For instance, there may be titanium atoms without complete oxygen coordination in certain regions of the lattice, such as at the edges. These vacancies can lead to the creation of free electrons which can migrate to other locations within the lattice, where they may occupy Ti4+. This process results in the formation of Ti3+ and F+ centers, which are categorized as shallow and deep trap states, respectively. The Ti3+ trap is located at 0.51 or 0.8 eV below the CB. The energy gap calculated based on the absorption spectra was 3.37 and 3.34 eV for TiO2(1) and TiO2(2), respectively. So shallow traps are located at 2.87 and 2.57 eV in TiO2(1) and 2.84 and 2.54 eV in TiO2(2).
To precisely understand the emission mechanism, deconvolution operations were performed to extract the component peaks that constitute the emission band visible in the obtained spectra. The obtained results allowed for a better understanding of the energy structure of materials. Calculations showed that at an excitation wavelength of 350 nm for TiO2(1), the emission band consisted of three components corresponding to the energy states of this structure. The most effective emission is observed from states of 3.05 eV, which is visible on the graph as the maximum emission. The second level is at 2.83 eV, and the third is at 2.6 eV, which are close to the theoretical positions of the shallow traps described earlier. Both of these states are visible in the spectrum through a broad tail for wavelengths higher than the emission maximum. In the spectrum of TiO2(2), three emission intensity maxima can be seen, corresponding to energy states of 2.99, 2.87 and 2.6 eV. As in the case of TiO2(1), the energy values of the second and third traps are close to the theoretical values. The emission in TiO2(2) is most intense for the second state. The emission bandwidth of TiO2(1) is wider than that of TiO2(2), which indicates that the energy states are more separated for the former than for the latter. The energy states isolated based on the deconvolution process showed that the two highest states in TiO2(1) are separated more widely than in TiO2(2). Because the excitation is more energetic than the bandgap of titanium oxide, electrons are transferred from the VB to above the CB, creating an additional state located just below the CB to which electrons pass non-radiatively. These additional states appear in the emission spectra as the most energetic emissions. At an excitation wavelength of 420 nm, the emission spectra for both TiO2 appear as a broad band. The deconvolution operation resulted in the separation of two band components, which correspond to two energy states. The distinction between the levels distance in TiO2(1) and TiO2(2) is similar to that in the previous excitation. The states in TiO2(1) are located at 2.2 and 1.9 eV, and for TiO2(2) at 2.14 and 1.94 eV. In this case, the amount of energy supplied with the excitation, which corresponds to 2.95 eV, can be considered the energy gap. According to the theory of traps in TiO2 described above, the traps should be located 0.51 and 0.8 eV below the energy gap. This means that states with energies of 2.44 and 2.15 eV should be created for this excitation. Only the second trap can be observed in the emission spectra. This means that electrons excited below the CB are trapped in the first defect (∼2.44 eV) and non-radiatively pass to the second one (∼2.15 eV), creating an additional virtual energy state of ∼1.9 eV. Emission occurs in both of these energy states. For this excitation, the difference in bandwidth for TiO2(1) and TiO2(2) is also visible. The emission band of TiO2(1) is much broader than that of TiO2(2), which means that the energy difference of the states is also larger. The broader distribution of states in TiO2(1) compared with TiO2(2) results in more effective electron transfer between levels. In TiO2(2), where the energy states are closer, non-radiative transitions may occur more easily. Therefore, the photocatalysis process should be more effective for the first type of titanium dioxide than the second one, which was proved later.
The emission spectra of carbon dots were measured under six excitation wavelengths, the same as in the case of TiO2. PhCDs are carbon dots placed in a polycrystalline matrix. The emission in this structure depends on the excitation wavelength, which, according to a leading article, can be attributed to the existence of many triplet excited states in the hybrid (Fig. 8 and Fig. S5†).25 The polycrystalline matrix is responsible for stabilizing the triplet levels in the carbon dots, resulting in a phosphorescence. In these studies, all the spectra (Fig. 8) show a wide emission band. For these types of carbon dot, the emission spectra are based on two factors. The first is fluorescence (FL), during which electrons move from the singlet state to the VB of the carbon dots and emit photons. The second factor is phosphorescence, which involves the transfer of electrons via an intersystem transition (ISC) to the triplet state and then transfer to the VB while emitting photons (Fig. 9). Both of these processes can be observed in the emission spectra as one broad band consisting of a narrow peak corresponding to fluorescence and a broad peak at higher wavelengths corresponding to phosphorescence. It can also be observed that different excitation wavelengths result in differences in the emission spectra. The use of a higher energy excitation source results in the presence of two maxima in the emission spectra. However, after performing the deconvolution operation, three components of a given band can be isolated. When the excitation has a lower energy, it results in reduced band separation in the spectrum, narrower bands, and the extraction of only two components. This observation leads to the conclusion that lower excitation energy results in fewer electrons transferred through the inter-system transition to the triplet state, and phosphorescence is observed with lower efficiency. As expected, CDs show narrower bands (Fig. 8 and Fig. S5†) because these types of carbon dot only show fluorescence. Therefore, no additional bands resulting from phosphorescence can be observed.
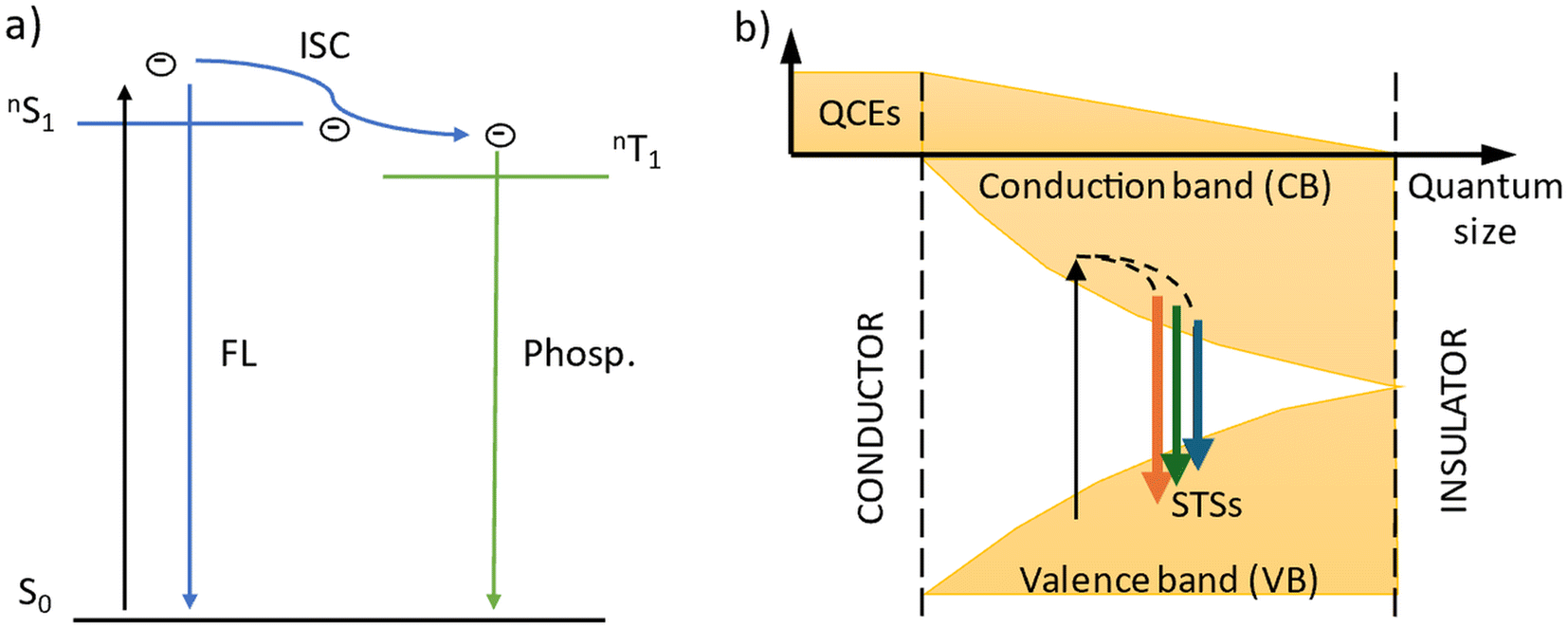 | ||
| Fig. 9 Scheme of the emission process in (a) PhCDs and (b) CDs. | ||
For carbon dots, the emission depends on the quantum confinement effects (QCEs) or surface trap states (STSs) (Fig. 9). The size of the carbon dots influences the width between the CB and VB, and surface defects introduce metastable energy levels in the bandgap. By adjusting this metastable level, it is possible to slightly change the color of the emission in carbon dots of the same size. For these studies, solutions containing a mixture of carbon dots of various sizes and surface functionalizations were used. This diversity of material is visible on the spectrum as a narrow band with a wide base. This band consists of four components, which is confirmed by the presence of carbon dots of different sizes and surface groups. The spectra show that the location of the maximum emission intensity and even the shape of the bands do not depend on the excitation wavelength. This means that CDs always emit photons of the same energy. The only change when using a different excitation wavelength is the change in emission intensity.
In the case of hybrid materials, the emission spectra have a shape and maximum intensity location similar to the results obtained for pure TiO2. This result was expected due to the small amount of carbon dots in the hybrid materials, resulting in a higher response from pure TiO2 compared with carbon dots. It has been observed that some levels occurring in pure materials are not active in the emission process in hybrids (Fig. 10). Such states were called inactive states. Hybrid materials with TiO2(1) show a higher intensity (Fig. S6†) of emission than pure TiO2(1). This is because electrons are transferred from inactive states in separate materials to levels from which emission occurs in hybrid materials, causing more radiative transitions to occur in the hybrid than in the titanium dioxide (1). Conversely, opposite results can be observed in hybrid materials with TiO2(2) where the intensity of emission is lower than for pure materials (Fig. S7†). This indicates that electrons are transferred from emission states to inactive states, resulting in non-radiative energy loss. What is worth emphasizing is that using 350 nm excitation in the case of TiO2(1) hybrids, emission can be observed both from the CB (2) and from defects located in the energy gap (3). In the case of the TiO2(1) hybrid, the transfer of electrons from inactive states to emission states results in an additional influx of electrons that can be used for the photocatalysis process (Fig. 10). If the electrons occupying the energy levels are used for photocatalysis, supplementary electrons from the carbon dots become available. Conversely, in TiO2(2) hybrids (Fig. S8†), electron transfer occurs from the hybrids to states within carbon dots where electron energy is dissipated non-radiatively (4). These electrons are not utilized as a substitute for others involved in photocatalysis.
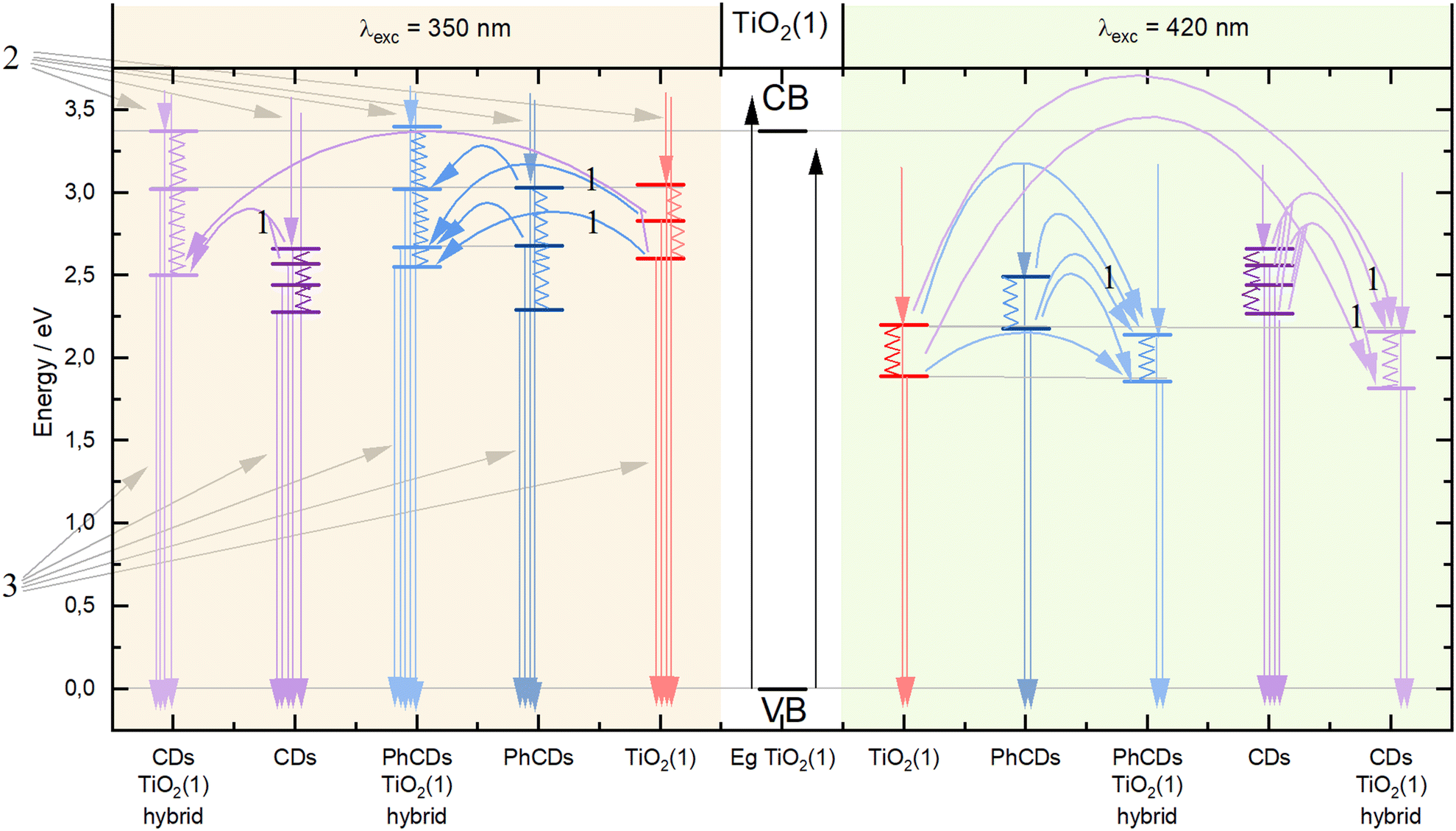 | ||
| Fig. 10 Mechanism of energy transfer in TiO2(1), PhCDs, CDs, and hybrid materials. | ||
| Sample | TiO2(1) | TiO2(2) | CDs | CDs@TiO2(2) | PhCDs | PhCDs@TiO2(1) | PhCDs@TiO2(2) | |
|---|---|---|---|---|---|---|---|---|
| λ exc (nm) | λ em (nm) | τ (ns) | ||||||
| 350 | 440 | — | 3.29 | — | — | 10.2 | — | 1.81 |
| 470 | — | 3.31 | 12.5 | 2.6 | — | — | — | |
| 480 | — | 3.33 | — | — | 12.1 | — | 1.77 | |
| 485 | — | 3.31 | 12.7 | 2.51 | — | — | — | |
| 490 | — | 3.37 | 12.7 | 2.46 | — | — | — | |
| 500 | — | 3.42 | — | — | 13.3 | — | 1.72 | |
| 520 | — | 3.47 | 12.6 | 2.38 | 14.1 | — | 1.76 | |
| 390 | 460 | 2.68 | — | — | — | 16.5 | 6.39 | — |
| 470 | 2.70 | — | — | — | 17.1 | 6.56 | — | |
At 350 nm excitation, the maximum emission occurred at wavelengths of 440 and 480 nm for PhCDs, 520 nm for TiO2(2), and 500 nm for their hybrid. Therefore, to compare the lifetimes of these materials, calculations were performed for all four wavelengths (440, 480, 500, and 520 nm). For CDs, maximum emission values occurred at wavelengths of 470 and 485 nm, and for the CDs@TiO2(2) hybrids at 490 nm. Consequently, lifetimes for dots, TiO2(2), and hybrids were calculated for four wavelengths (470, 485, 490, and 520 nm). For excitation at 390 nm, PhCDs had maximum emission at 470 nm, TiO2(1) at 460 nm, and the hybrid, like PhCDs, at 470 nm. Thus, lifetimes for these materials were calculated for two wavelengths (460 and 470 nm).
Based on the obtained luminescence kinetics measurements, it can be concluded that for the 350 nm excitation, the lifetime of electrons in TiO2(2) in the excited state is approximately 3.3 ns. When excited with a higher wavelength of 390 nm, the lifetime of electrons in TiO2(1) is shorter, at approximately 2.7 ns.
The lifetime of electrons in PhCDs emitting in the range of 440–520 nm depends on the maximum emission value for which they were measured. For lower wavelengths, the lifetime is shorter, around 10 ns, and increases to 14 ns as the emission wavelength increases. This is expected due to the phosphorescence properties of these dots. As described in the emission spectra analysis, the emission shortly after excitation is responsible for the maximum emission at shorter wavelengths. However, the tail in the emission spectrum at longer wavelengths reflects phosphorescence, where electrons are held for a period before relaxing, accounting for their longer lifetime. For the 390 nm excitation, where the measured times were for closely located maxima, the lifetime was 16.5–17 ns, indicating that with lower energy excitation, electrons remain in the excited state longer.
CDs excited at a wavelength of 350 nm emit in a narrow range which is in alignment with the maximum wavelengths determined during luminescence kinetics measurements. This means that in these dots, electron relaxation occurs shortly after excitation without additional storage. Electron lifetimes in the emission range of 470–520 nm were similar, around 12.7 ns.
The addition of PhCDs/CDs to TiO2(2) significantly reduced electron lifetimes compared with pure TiO2(2). The addition of PhCDs nearly halved the lifetime. In the hybrid, electrons have a lifetime of about 1.8 ns. Notably, for the PhCDs@TiO2(2) hybrid, the lifetimes for various maximum emissions were similar, unlike in pure PhCDs. For CDs, whose lifetime did not depend on the emission wavelength, their addition to TiO2(2) resulted in a hybrid material with electrons of different lifetimes. Electrons in CDs@TiO2(2) have the longest lifetime of 2.6 ns for the shortest emission wavelength, decreasing to 2.38 ns as the wavelength increases. For TiO2(1), adding PhCDs almost tripled the lifetimes (6.39 and 6.56 ns). The lifetime extension indicates the positive impact of adding PhCDs to the photocatalyst on the photocatalysis process. A longer lifetime means a slower recombination of electron–hole pairs, increasing the efficiency of the photocatalytic process.
The measurements were conducted to compare the decay times of CDs, PhCDs, TiO2, and their respective hybrids. All measurements were taken at specific emission wavelengths that excluded the phosphorescence emission maximum (observed at about 560 nm), and therefore, the resulting decay times are not fully representative of this process. To investigate the phosphorescence phenomenon in more detail, an additional experiment was performed. Emission spectra of PhCDs were collected under UV irradiation and at 100 ms intervals after ceasing excitation (Fig. 11a). The spectra were recorded over a broader wavelength range (300–700 nm) to capture phosphorescence, which exhibits a maximum at approximately 560 nm (Fig. 11a). It was observed that the phosphorescence maximum is red-shifted relative to the fluorescence emission (inset in Fig. 11a). From the recorded spectra, the decay of phosphorescence was calculated (Fig. 11b) to be approximately 970 ms, which is consistent with the characteristic behavior of the phosphorescence process and is consistent with the data presented in the source article on which the synthesis was based, as well as with results obtained by other research groups (Table 4).
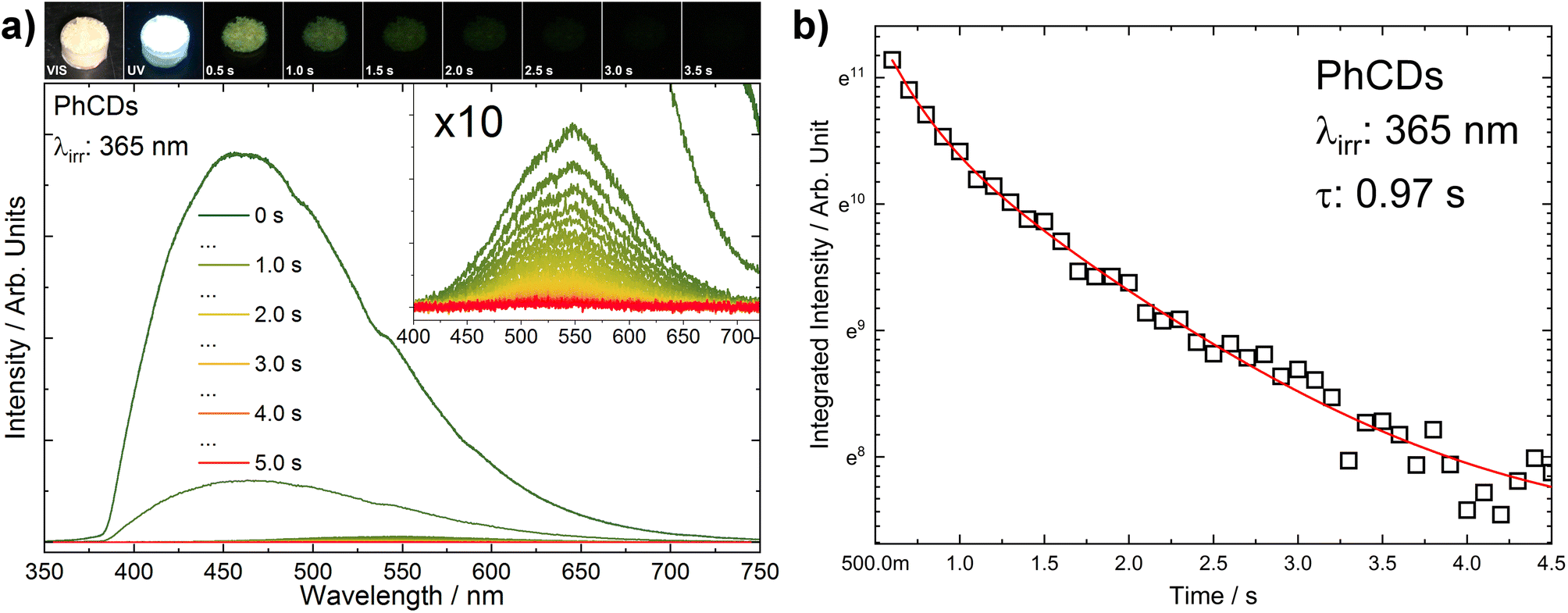 | ||
| Fig. 11 (a) PhCDs luminescence spectrum as a function of time (the inset shows the enlarged range in which phosphorescence is observed). Above the graph, PhCDs photos are presented under VIS and UV irradiation, and after UV ceasing with a step of 0.5 s. (b) The decay of PhCDs phosphorescence. | ||
3.3. Photocatalytic activity
The photocatalytic activity of the prepared materials was evaluated based on the degradation process of carbamazepine, a pharmaceutical commonly detected in water that is non-susceptible to biodegradation. As can be seen in Fig. 12, TiO2(1) showed improved photocatalytic activity compared with the reaction of CBZ degradation using TiO2(2). For TiO2(1), the removal efficiency reached nearly 96% within 60 min of irradiation under simulated solar light. Furthermore, for this material, the reduction of total organic carbon (TOC) was 51%, which indicates the potential for efficient photodegradation and mineralization of persistent organic pollutants. The enhanced photocatalytic activity may be correlated with the larger surface area of the TiO2(1) (106 m2 g−1) photocatalyst than that of TiO2(2) (68 m2 g−1). Generally, a photocatalyst with a higher surface area possesses more reactive sites and is characterized by a shorter migration distance; therefore, the recombination rate may be reduced. For TiO2(1), modification of the surface with CDs did not affect the CBZ degradation efficiency. However, in the case of TiO2(1) modified with PhCDs, enhancement in the photocatalytic activity was observed. The CBZ degradation rate constant increased from 5.31 × 10−2 min−1 to 6.59 × 10−2 min−1, and TOC removal increased to 60%. The opposite effect was observed for TiO2(2), where the modification of the TiO2 surface was not accompanied by an improvement in the photocatalytic activity. In particular, the presence of luminescent carbon dots caused a decrease in the degradation efficiency. Moreover, none of these samples showed the ability to mineralize organic pollutant (Table 5).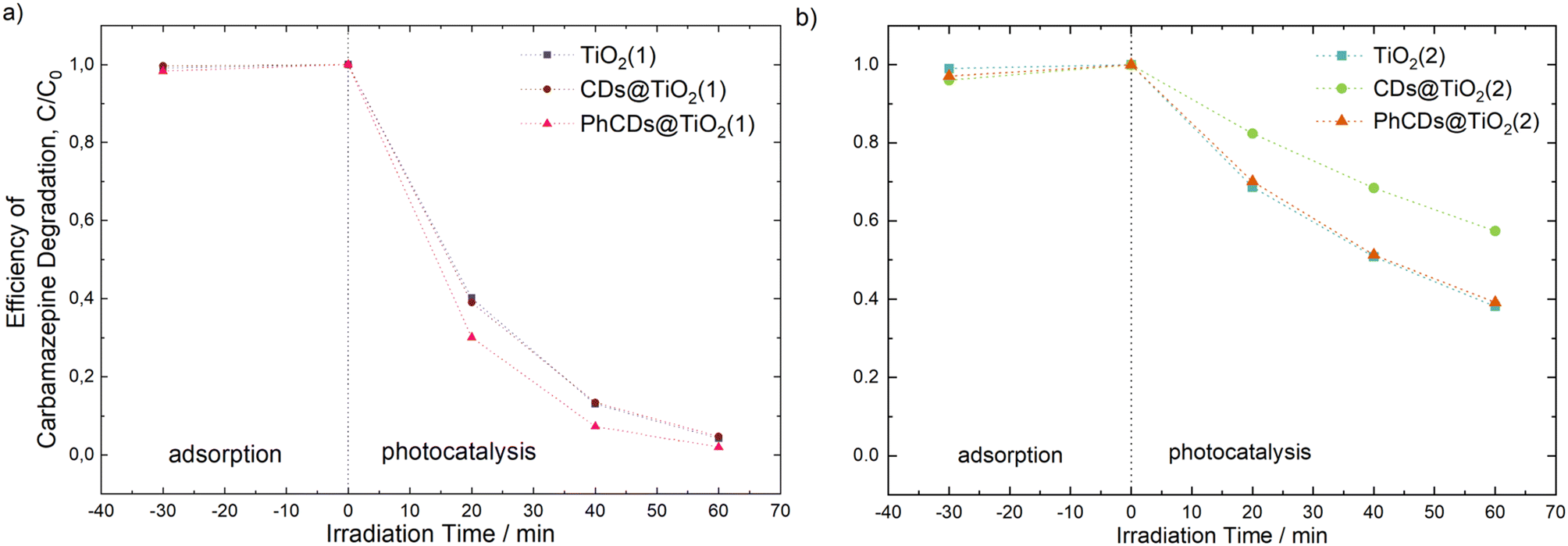 | ||
| Fig. 12 Efficiency of carbamazepine degradation for (a) pure TiO2(1) and hybrid materials PhCDs@TiO2(1) and CDs@TiO2(1), and (b) pure TiO2(2) and hybrid materials PhCDs@TiO2(2) and CDs@TiO2(2). | ||
| Sample name | Carbamazepine degradation efficiency, % | Degradation rate constant k × 10−2, min−1 | Efficiency of TOC reduction, % |
|---|---|---|---|
| TiO2(1) | 95.8 | 5.31 | 50.7 |
| CDs@TiO2(1) | 95.3 | 5.11 | 2.4 |
| PhCDs@TiO2(1) | 98.0 | 6.59 | 60.2 |
| TiO2(2) | 61.8 | 1.60 | 0 |
| CDs@TiO2(2) | 42.6 | 0.93 | 0 |
| PhCDs@TiO2(2) | 60.8 | 1.56 | 0 |
The above results and the results obtained from emission and lifetime measurements show a correlation between the efficiency of photocatalytic activity and the energy transfer mechanism. The addition of PhCDs and CDs to TiO2(1) resulted in an increase in emission intensity, which was explained as the non-radiative electron transfer between the excited states of TiO2 and CDs to the valence band and participating in the luminescence observed in the hybrid. During photocatalysis these electrons take part in the production of radicals that participate in the decomposition of pollutants. The phosphorescence phenomenon occurring in PhCDs helps to slow down the recombination of the charges due to trapping electrons and participation of the holes in the generation of radicals, also involved in the degradation of carbamazepine. The addition of PhCDs to TiO2(1) resulted in a prolonging of the hybrid luminescence lifetime compared with pure TiO2(1), which confirms the positive effect of this combination. Due to this process, an improvement in the PhCDs@TiO2(1) photocatalysis process can be observed. In the case of TiO2(2) hybrids, CDs/PhCDs are located in large amounts on the surface of the photocatalyst, not in its pores. Based on the emission spectra, it was found that the electron transfer is reverse. Electrons move from hybrid states to states in carbon dots, where non-radiative transitions occur. This results in a limited number of electrons that can take part in the formation of radicals. Also in the lifetime measurements, a negative effect of the addition of carbon dots to TiO2(2) could be noticed. This is the reason why a decrease in photocatalysis efficiency is observed in the case of TiO2(2) hybrids.
Additional experiments were conducted for the PhCDs@TiO2(1) sample exhibiting the highest photocatalytic activity in the degradation of CBZ under UV-vis irradiation. As illustrated in Fig. 13a and Table 6, even a two- and four-fold reduction in the photocatalyst concentration within the reaction medium facilitated a high degradation efficiency. The optimal photocatalyst concentration was determined as 1 g dm−3. In the case of 2 g dm−3, the increased turbidity may have resulted in light scattering and screening effects, consequently decreasing the photocatalyst activation. The effect of the CBZ solution concentration on the photocatalytic activity was also investigated. As shown in Fig. 13b and Table 6, a decrease in the CBZ concentration to 8.5 mg dm−3 resulted in an increase in the degradation rate constant from 7.33 × 10−2 min−1 to 9.94 × 10−2 min−1. Almost complete degradation was observed within 40 min of irradiation. Conversely, increasing the CBZ concentration to 19.5 mg dm−3 produced the opposite effect: the degradation rate constant decreased to 5.22 × 10−2 min−1. When the concentration of the pollutant increases, the number of intermediates, which may compete for active sites because of the non-selective nature of the photocatalytic process, also increases. This may have led to a decrease in the overall degradation rate.65 Additionally, higher initial concentrations cause the pollutant to occupy more active sites, thereby hindering the production of oxidants. Furthermore, higher initial CBZ concentrations lead to greater photon absorption. As a result, fewer photons are available to activate the photocatalyst, causing diminished photocatalytic activity because the catalyst surface becomes inactive.66
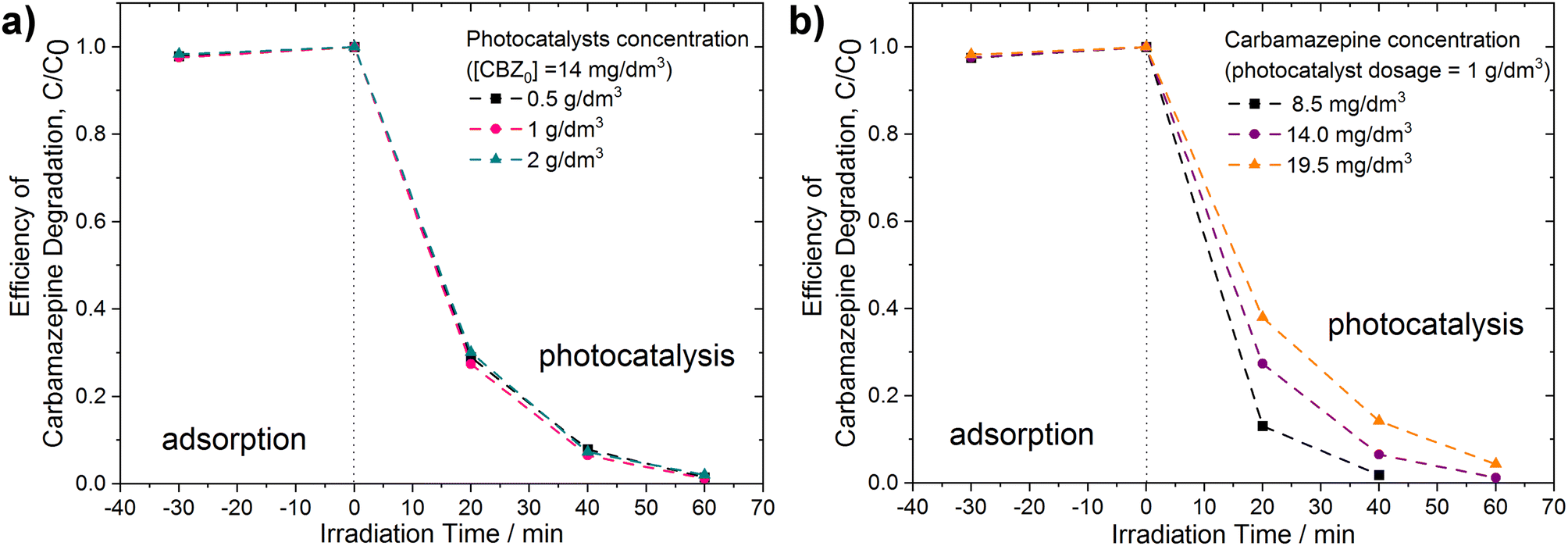 | ||
| Fig. 13 Efficiency of carbamazepine degradation (a) at various dosages of PhCDs@TiO2(1) photocatalyst, [CBZ0] = 14 mg dm−3, and (b) using PhCDs@TiO2(1) photocatalyst at various concentrations of CBZ solution, photocatalyst dosage = 1 g dm−3. | ||
| Dosage of | Degradation rate constant k × 10−2 (min−1) | Efficiency of TOC reduction (%) | |
|---|---|---|---|
| photocatalyst (g dm−3) | CBZ (mg dm−3) | ||
| 0.5 | 14 | 7.01 | 63.1 |
| 1 | 14 | 7.33 | 61.9 |
| 2 | 14 | 6.59 | 60.2 |
| 1 | 8.5 | 9.94 | 67.0 |
| 1 | 14 | 7.33 | 61.9 |
| 1 | 19.5 | 5.22 | 65.6 |
To identify the primary reactive species involved in the carbamazepine degradation process under UV-vis light using PhCDs@TiO2(1) photocatalyst, trapping experiments were conducted in the presence of scavengers. As illustrated in Fig. 14, the most significant inhibition of CBZ degradation was observed following the addition of ascorbic acid. In this instance, the degradation efficiency decreased to approximately 25% within 60 minutes of irradiation, indicating that superoxide anion radicals play a crucial role in CBZ removal. Furthermore, ˙OH and 1O2 may also be attributed a significant role in the degradation process. The addition of tert-butanol and sodium azide resulted in a reduction of photocatalytic activity, with 57% and 67% degradation within 60 minutes, respectively. Upon the addition of ammonium oxalate, a scavenger of h+, the degradation efficiency decreased to 93%, indicating that these species have a minor contribution to the reaction pathway of CBZ.
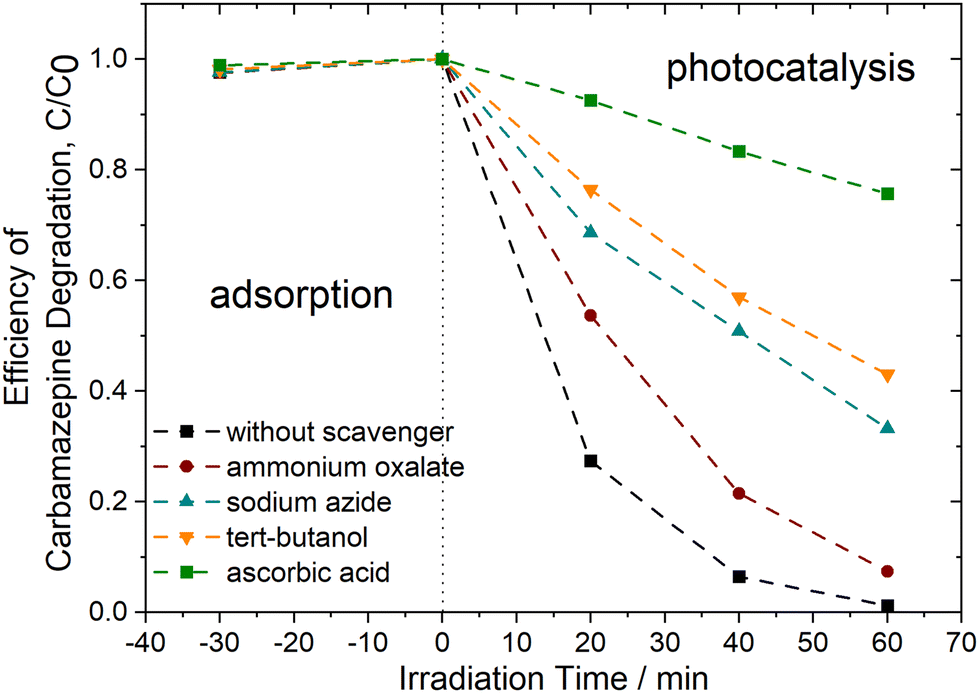 | ||
| Fig. 14 Efficiency of carbamazepine degradation of PhCDs@TiO2(1) photocatalyst in the presence of scavengers, [CBZ0] = 14 mg dm−3, photocatalyst dosage = 1 g dm−3. | ||
4. Conclusions
The investigated hybrid materials consisted of titanium oxide impregnated with carbon dots. Four hybrids were created consisting of two types of TiO2 and two types of carbon dot. Structural tests showed no changes in the structure of TiO2 after the addition of carbon dots, which was confirmed by SEM, TEM and BET analysis. In Raman and IR spectroscopy, no significant differences were observed between the hybrid materials and pure titanium oxide due to the small number of CDs. Absorption measurements showed that adding CDs to TiO2(1) causes the absorption edge to shift towards visible light and increases the absorption intensity across the entire range for all hybrids. Analysing the emission spectra of the raw and hybrid materials allowed for understanding the mechanism of luminescence in hybrid materials. It has been shown that in the case of TiO2(1), adding carbon dots increases the emission intensity. This means that emission in hybrid materials occurs from the electrons of TiO2 and carbon dots which enhance the photocatalytic process. In the case of TiO2(2), the opposite relationship was observed, namely, the addition of carbon dots to titanium oxide resulted in a reduction in emission intensity. The location of CDs mostly on the TiO2 surface probably leads to non-radiative electron energy loss, which negatively impacts the photocatalysis process. Photocatalytic activity tests confirm this thesis. Thus, in the case of TiO2(1), the addition of phosphorescent dots improved the photocatalytic process. The optimal photocatalyst concentration was determined as 1 g dm−3. The addition of CDs to this type of titanium oxide did not cause major differences in this process. In the case of TiO2(2), the addition of both types of carbon dot resulted in a reduction in the efficiency of this process.To summarize, the thesis regarding the improvement of the photocatalysis process by adding phosphorescent carbon dots to titanium oxide has been confirmed. Additionally, it was found that the type of titanium oxide and the properties of the carbon dots have a significant impact on the photocatalysis process. It has been proved that to improve this process, it is important to create a material that can hold electrons in the excited state. This will slow down the recombination of electron–hole pairs, which has a significant impact on the photocatalysis process.
Data availability
Data for this article, including raw data are available at Zenodo at https://doi.org/10.5281/zenodo.13832245.Conflicts of interest
There are no conflicts to declare.References
- D. R. Kennedy, M. Ritchie and J. Mackenzie, Trans. Faraday Soc., 1958, 54, 119–129 RSC
.
- T. I. Barry, F. S. Stone and F. C. Tompkins, Proc. R. Soc. London, Ser. A, 1997, 255, 124–144 Search PubMed
- Y. Fujita and T. Kwan, Bull. Chem. Soc. Jpn., 1958, 31, 379b–3380 CrossRef
- W. Doerffler and K. Hauffe, J. Catal., 1964, 3, 156–170 CrossRef CAS
- F. Masuo and K. Kato, Kogyo Kagaku Zasshi, 1964, 67, 1136–1144 CrossRef
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS
- S. N. Frank and A. J. Bard, J. Am. Chem. Soc., 1977, 99, 303–304 CrossRef CAS
- Y. Wang and A. Hu, J. Mater. Chem. C, 2014, 2, 6921–6939 RSC
- A. P. Sunitha, K. Sandeep, P. Praveen and K. J. Saji, Mater. Today: Proc., 2020, 33, 1298–1300 CAS
- K. Akbar, E. Moretti and A. Vomiero, Adv. Opt. Mater., 2021, 9, 2100532 CrossRef CAS
- J. Ren, L. Stagi and P. Innocenzi, Prog. Solid State Chem., 2021, 62, 100295 CrossRef CAS
- P. Tong, J. Liang, X. Jiang and J. Li, Crit. Rev. Anal. Chem., 2019, 50, 376–392 CrossRef PubMed
- M. Szymczak, A. Mauri, S. Galli, L. Marciniak and M. Fandzloch, Adv. Funct. Mater., 2024, 34, 2313045 CrossRef CAS
- Q. Liu, B. Wu, M. Li, Y. Huang, L. Li, Q. Liu, Y. Huang, B. Wu, L. Li and M. Li, Adv. Sci., 2022, 9, 2103911 CrossRef CAS
- D. Yang, G. Yang, S. Gai, F. He, C. Li and P. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 6829–6838 CrossRef CAS PubMed
- L. Xu, G. Fang, J. Liu, M. Pan, R. Wang and S. Wang, J. Mater. Chem. A, 2016, 4, 15880–15887 RSC
- Y. Gao, M. Hilbers, H. Zhang and S. Tanase, Eur. J. Inorg. Chem., 2019, 2019, 3925–3932 CrossRef CAS
- G. Zhang, G. Kim and W. Choi, Energy Environ. Sci., 2014, 7, 954–966 RSC
- F. X. Llabrés i Xamena, A. Corma and H. Garcia, J. Phys. Chem. C, 2007, 111, 80–85 CrossRef
- R. Lin, S. Li, J. Wang, J. Xu, C. Xu, J. Weng, C. Li and Z. Li, Inorg. Chem. Front., 2018, 5, 3170–3177 RSC
- J. Pan, Y. Sheng, J. Zhang, J. Wei, P. Huang, X. Zhang and B. Feng, J. Mater. Chem. A, 2014, 2, 18082–18086 RSC
- J. Augustynski, Electrochim. Acta, 1993, 38, 43–46 CrossRef CAS
- J. Zhang, J. Xu and F. Tao, ACS Appl. Energy Mater., 2021, 4, 13120–13131 CrossRef CAS
- F. Zhao, F. Zhang, D. Han, K. Huang, Y. Yang and H. Yin, Phys. Chem. Chem. Phys., 2021, 23, 10448–10455 RSC
- A. Mozdbar, A. Nouralishahi, S. Fatemi and F. S. Talatori, J. Water Process Eng., 2023, 51, 103465 CrossRef
- W. Li, W. Zhou, Z. Zhou, H. Zhang, X. Zhang, J. Zhuang, Y. Liu, B. Lei and C. Hu, Angew. Chem., Int. Ed., 2019, 58, 7278–7283 CrossRef CAS
- C. Lin, Y. Zhuang, W. Li, T. L. Zhou and R. J. Xie, Nanoscale, 2019, 11, 6584–6590 RSC
- K. Jiang, Y. Wang, Z. Li and H. Lin, Mater. Chem. Front., 2020, 4, 386–399 RSC
- Z. Liu, H. Zou, N. Wang, T. Yang, Z. Peng, J. Wang, N. Li and C. Huang, Sci. China: Chem., 2018, 61, 490–496 CrossRef CAS
- Y. Ding, X. Wang, M. Tang and H. Qiu, Adv. Sci., 2022, 9, 2103833 CrossRef CAS
- J. Ye, W. Liu, J. Cai, S. Chen, X. Zhao, H. Zhou and L. Qi, J. Am. Chem. Soc., 2011, 133, 933–940 CrossRef CAS PubMed
- A. W. Augustyniak, M. Sadakiyo, J. A. R. Navarro and A. M. Trzeciak, ChemistrySelect, 2018, 3, 7934–7939 CrossRef CAS
-
P. Kubelka and F. Munk, An Article on Optics of Paint Layers, 1931 Search PubMed
-
A. E. Morales, E. S. Mora, A. E. Morales, E. S. Mora and U. Pal, Use of Diffuse Reflectance Spectroscopy for Optical Characterization of Un-Supported Nanostructures, 2007, vol. 53 Search PubMed
- G. P. Joshi, N. S. Saxena, R. Mangal, A. Mishra and T. P. Sharma, Bull. Mater. Sci., 2003, 26, 387–389 CrossRef CAS
- P. Głuchowski, R. Tomala, A. Jeżowski, D. Szewczyk, B. Macalik, I. Smolina, T. Kurzynowski and W. Stręk, Sci. Rep., 2020, 10, 11121 CrossRef
- J. M. Kim and H. T. Chung, J. Power Sources, 2003, 115, 125–130 CrossRef CAS
- S. Rai, B. K. Choudhary, T. Jayakumar, K. B. S. Rao and B. Raj, Int. J. Pressure Vessels Piping, 1999, 76, 275–281 CrossRef CAS
- T. E. Thompson, Phys. Today, 1978, 31, 36–45 CrossRef
- S. Dai, Y. Wu, T. Sakai, Z. Du, H. Sakai and M. Abe, Nanoscale Res. Lett., 2010, 5, 1829–1835 CrossRef CAS PubMed
- W. Su, J. Zhang, Z. Feng, T. Chen, P. Ying and C. Li, J. Phys. Chem. C, 2008, 112, 7710–7716 CrossRef CAS
- T. Ohsaka and Y. Fujiki, Raman Spectrum of Anatase, TiO, J. Raman Spectrosc., 1978, 7, 321–324 CrossRef
- M. A. Boda and M. A. Shah, Mater. Res. Express, 2017, 4(7), 075908 CrossRef
- A. Erdemir, M. Halter and G. R. Fenske, Wear, 1997, 205, 236–239 CrossRef CAS
- K. Krishnan, The Raman Spectrum of Boric Acid, Proc. Indian Acad. Sci, 1963, 57, 103–108 CrossRef CAS
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS
- P. D. Moran, G. A. Bowmaker, R. P. Cooney, K. S. Finnie, J. R. Bartlett and J. L. Woolfrey, Inorg. Chem., 1998, 37, 2741–2748 CrossRef CAS
- Y. Gao, Y. Masuda, W.-S. Seo, H. Ohta and K. Koumoto, Ceram. Int., 2004, 30, 1365–1368 CrossRef CAS
- A. B. Younis, V. Milosavljevic, T. Fialova, K. Smerkova, H. Michalkova, P. Svec, P. Antal, P. Kopel, V. Adam, L. Zurek and K. Dolezelikova, BMC Microbiol., 2023, 23, 207 CrossRef CAS PubMed
- P. Praveen, G. Viruthagiri, S. Mugundan and N. Shanmugam, Spectrochim. Acta, Part A, 2014, 117, 622–629 CrossRef CAS
- L. Chougala, M. Yatnatti, R. Linganagoudar, R. Kamble and J. Kadadevarmath, J. Nano- Electron. Phys., 2017, 9, 4001–4005 Search PubMed
- D. E. Bethell and N. Sheppard, Trans. Faraday Soc., 1955, 51, 9–15 RSC
- O. M. Moon, B.-C. Kang, S.-B. Lee and J.-H. Boo, Thin Solid Films, 2004, 464–465, 164–169 CrossRef CAS
- B. C. Smith, Spectroscopy, 2021, 36, 22–25 CAS
- B. C. Smith, Spectroscopy, 2022, 37, 10–13 CAS
- B. C. Smith, Spectroscopy, 2019, 34, 22–26 Search PubMed
- B. C. Smith, Spectroscopy, 2017, 32, 14–21 CAS
- N. Chang, Y. Ji, J. Liu, J. Liu and Y. Chen, Ind. Eng. Chem. Res., 2023, 62, 17428–17439 CrossRef
- J. Joseph and A. A. Anappara, Phys. Chem. Chem. Phys., 2017, 19, 15137–15144 RSC
- Q. Li, M. Zhou, M. Yang, Q. Yang, Z. Zhang and J. Shi, Nat. Commun., 2018, 9, 734 CrossRef PubMed
- Q. Li, M. Zhou, Q. Yang, Q. Wu, J. Shi, A. Gong and M. Yang, Chem. Mater., 2016, 28, 8221–8227 CrossRef CAS
- W. Li, W. Zhou, Z. Zhou, H. Zhang, X. Zhang, J. Zhuang, Y. Liu, B. Lei and C. Hu, Angew. Chem., Int. Ed., 2019, 58, 7278–7283 CrossRef CAS
- J. Tan, J. Zhang, W. Li, L. Zhang and D. Yue, J. Mater. Chem. C, 2016, 4, 10146–10153 RSC
- P. Roushenas, Z. C. Ong, Z. Ismail, Z. Majidnia, B. C. Ang, M. Asadsangabifard, C. C. Onn and J. H. Tam, Desalin. Water Treat., 2018, 120, 109–118 CrossRef CAS
- M. A. Al-Nuaim, A. A. Alwasiti and Z. Y. Shnain, Chem. Pap., 2023, 77, 677–701 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt02196g |
| This journal is © The Royal Society of Chemistry 2025 |