DOI:
10.1039/D4DT03153A
(Paper)
Dalton Trans., 2025,
54, 3027-3038
Red and NIR light-triggered enhancement of anticancer and antibacterial activities of dinuclear Co(II)-catecholate complexes†
Received
11th November 2024
, Accepted 6th January 2025
First published on 10th January 2025
Abstract
Photoactive complexes of bioessential 3d metals, activable within the phototherapeutic window (650–900 nm), have gained widespread interest due to their therapeutic potential. Herein, we report the synthesis, characterization, and light-enhanced anticancer and antibacterial properties of four new dinuclear Co(II) complexes: [Co(phen)(cat)]2 (Co-1), [Co(dppz)(cat)]2 (Co-2), [Co(phen)(esc)]2 (Co-3), and [Co(dppz)(esc)]2 (Co-4). In these complexes, phen (1,10-phenanthroline) and dppz (dipyrido[3,2-a:2′,3′-c]phenazine) act as neutral N,N-donor ligands, while cat2− and esc2− serve as O,O-donor catecholate ligands derived from catechol (1,2-dihydroxybenzene) and esculetin (6,7-dihydroxy coumarin). Their high-spin paramagnetic nature and dimeric identity in solution were confirmed by magnetic susceptibility, UV-visible, emission, and mass spectral data. Co-1–Co-4 exhibited an absorption band within the 600–850 nm range, originating from a charge transfer transition. The electrically neutral complexes demonstrated sufficient solution stability both in the dark and under irradiated conditions. The dppz complexes Co-2 and Co-4 exhibited notable toxicity towards A549 lung carcinoma cells, with potency increasing significantly under brief (5 min) exposure to 660 nm (red) and 808 nm (NIR) laser light (IC50 ∼ 8.9 to 14.9 μM). Notably, their toxicity towards normal NIH-3T3 fibroblast cells was minimal. Cellular assays highlighted that the induced cell death followed an apoptotic pathway, primarily due to mitochondrial damage. Co-2 and Co-4 also demonstrated significant antibacterial potency against Gram-(+) S. aureus and Gram-(−) P. aeruginosa, with effectiveness significantly enhanced upon 808 nm laser irradiation (MIC ∼ 15–142 μM). The increase in the anticancer and antibacterial efficacies was attributed to the generation of cytotoxic singlet oxygen (1O2) species upon red/NIR light exposure. Notably, 808 nm NIR irradiation produced more pronounced effects compared to 660 nm. This study is the first to report on cobalt complexes exhibiting red and NIR light-triggered enhancement of antibacterial and anticancer activities, illuminating the path for the development of long-wavelength absorbing cobalt complexes with enhanced therapeutic efficacy.
Introduction
Phototherapy using transition metal complexes of non-macrocyclic ligands activatable by light for achieving anticancer activity has garnered significant interest in recent years.1–5 The promise shown by the Ru(II) complex TLD1433 in clinical trial studies for the photodynamic therapy (PDT) of bladder cancer has spurred substantial interest in designing non-macrocyclic metal complexes for cancer PDT applications.6 The rise of multi-drug-resistant (MDR) bacteria has become a serious global concern, presenting challenges in treating infections as these superbugs evade most clinical antibiotics.7,8 This highlights the pressing need for strategies to effectively combat these resilient pathogens. Antibacterial PDT (aPDT) has emerged as a paradigm shift to treat bacterial infections that normally do not respond to clinical antibiotics.9–11 In PDT and aPDT, cancer or bacterial cells are selectively destroyed by cytotoxic reactive oxygen species (ROS) formed by the photoactive drug following light irradiation.11,12 The advantages of aPDT include its broad-spectrum efficacy and rapid action upon irradiation, along with its capability to target MDR strains through a multitargeted mechanism.9–11
Drugs that operate through a multimodal mechanism of action offer distinct advantages in terms of enhanced therapeutic outcomes compared to monotherapies alone.13,14 For example, combining PDT with chemotherapy can lead to synergistic effects and improved therapeutic efficacy.15–17 Similarly, aPDT is effective against multidrug-resistant (MDR) infections, but photodynamic bacterial inactivation is often incomplete, leading to a potential relapse of infections.18 Therefore, combining PDT or aPDT with conventional chemotherapy or antibacterial therapy is considered a superior approach in terms of therapeutic effectiveness.
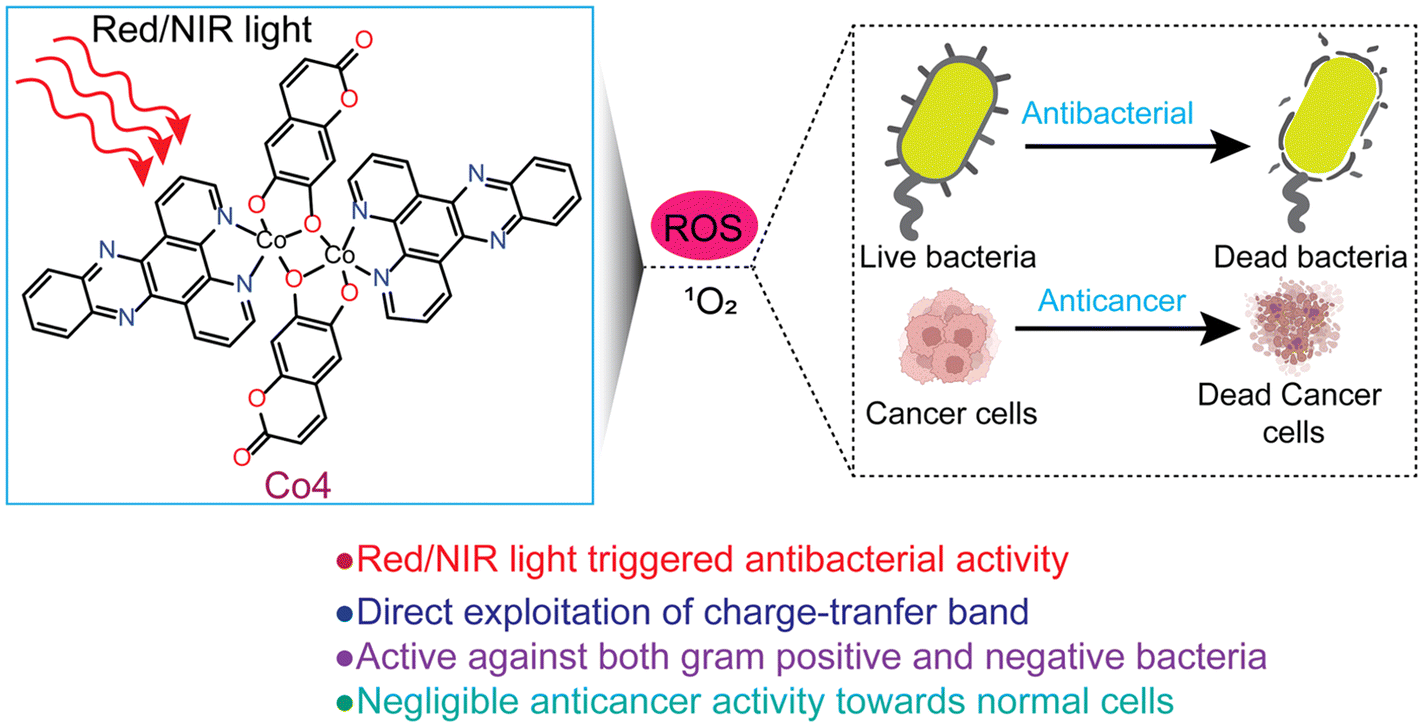
There is currently an escalating interest in designing photoactive anticancer metal complexes using first-row transition metal ions due to their endogenous presence, enhanced biocompatibility, and cost-effectiveness.19–22 While Co(II) and Co(III) complexes have been studied as cytotoxic agents using visible light, no studies are known for Co complexes showing red or NIR light-triggered cytotoxicity against cancer cells.23–28 In a maiden report, Chakravarty et al. studied a Co(III)-BODIPY complex as a phototoxic agent using red light.29 However, the observed phototoxicity was because of the released BODIPY ligand from the complex following light irradiation. Similarly, among the 3d metal complexes, there are only a few reports on Fe, Co, Cu, and Zn complexes as aPDT agents, but all utilize visible light.30–34 However, reports on 3d metal complexes showing aPDT activity using red or NIR light are unknown. Notably, photosensitizers that can be activated within the clinical PDT window (650–900 nm) are highly desirable due to their enhanced ability to penetrate and access deep tissue regions, making them beneficial for cancer PDT in treating deeply located tumors.3–5 Additionally, these red/NIR photosensitizers are valuable in aPDT for treating non-superficial infections.35–37 Herein, we report the synthesis, characterization, and light-enhanced anticancer and antibacterial properties of four newly developed dinuclear Co(II) complexes: [Co(phen)(cat)]2 (Co-1), [Co(phen)(esc)]2 (Co-2), [Co(dppz)(cat)]2 (Co-3), and [Co(dppz)(esc)]2 (Co-4). In these complexes, phen (1,10-phenanthroline) and dppz (dipyrido[3,2-a:2′,3′-c]phenazine) act as bidentate N,N-donor heterocyclic ligands, while cat2− and esc2− act as bidentate O,O-donor catecholate ligands derived from catechol (1,2-dihydroxybenzene) and esculetin (6,7-dihydroxycoumarin) (Chart 1). The dppz as a α-diimine ligand is utilized for its property to form stable chelates with 3d metals and act as a photosensitizer, contributing to the phototherapeutic effect.23–25,38,39 At present, there is rising interest in investigating photoactive natural compounds for PDT and aPDT applications.40,41 Esculetin, a naturally derived coumarin with a catecholate metal-binding moiety, is known to serve as an anticancer and photosensitizing ligand.22,24,42 Key features of our study include: (a) the first report of a cobalt-based complex with red and NIR light-triggered apoptotic cytotoxicity; (b) negligible toxicity toward normal NIH-3T3 embryonic fibroblast cells; (c) the first 3d metal complex with antibacterial activity under red/NIR light; (d) effectiveness against both Gram-(+) and Gram-(−) bacteria; and (e) direct exploitation of the charge transfer (CT) band to achieve biological activity with superior PDT and aPDT activity under NIR light compared to red light (Fig. 1).
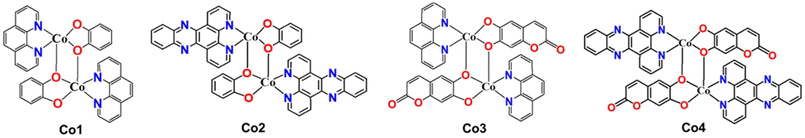 |
| | Chart 1 Chemical representations of Co-1–Co-4. | |
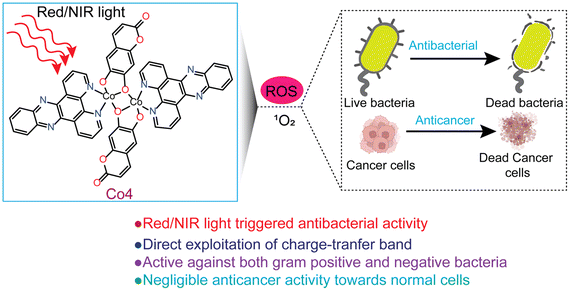 |
| | Fig. 1 A diagrammatic illustration of the light-triggered PDT and aPDT activity of Co-4. | |
Results and discussion
Synthesis and characterization
Co(II) complexes Co-1–Co-4 were synthesized and isolated as green solids by reacting anhydrous CoCl2 with stoichiometric amounts of the α-diimine and deprotonated catecholate ligands, using anhydrous THF as the solvent (Scheme S1, ESI†). The solid-state purity and solution-state identity of the compounds were confirmed through elemental (CHN) and high-resolution mass spectral analyses (ESI-HRMS), respectively. While CHN analysis showed an excellent match with the theoretical values, the HRMS spectra in H2O/MeOH [1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19 (v/v)] revealed a prominent peak for the [M + H]+ species (Fig. S1–S4, ESI†). The FT-IR spectra of Co-2 and Co-4, which contain the esc2− ligand, exhibited a prominent peak at 1663 cm−1, attributed to the C
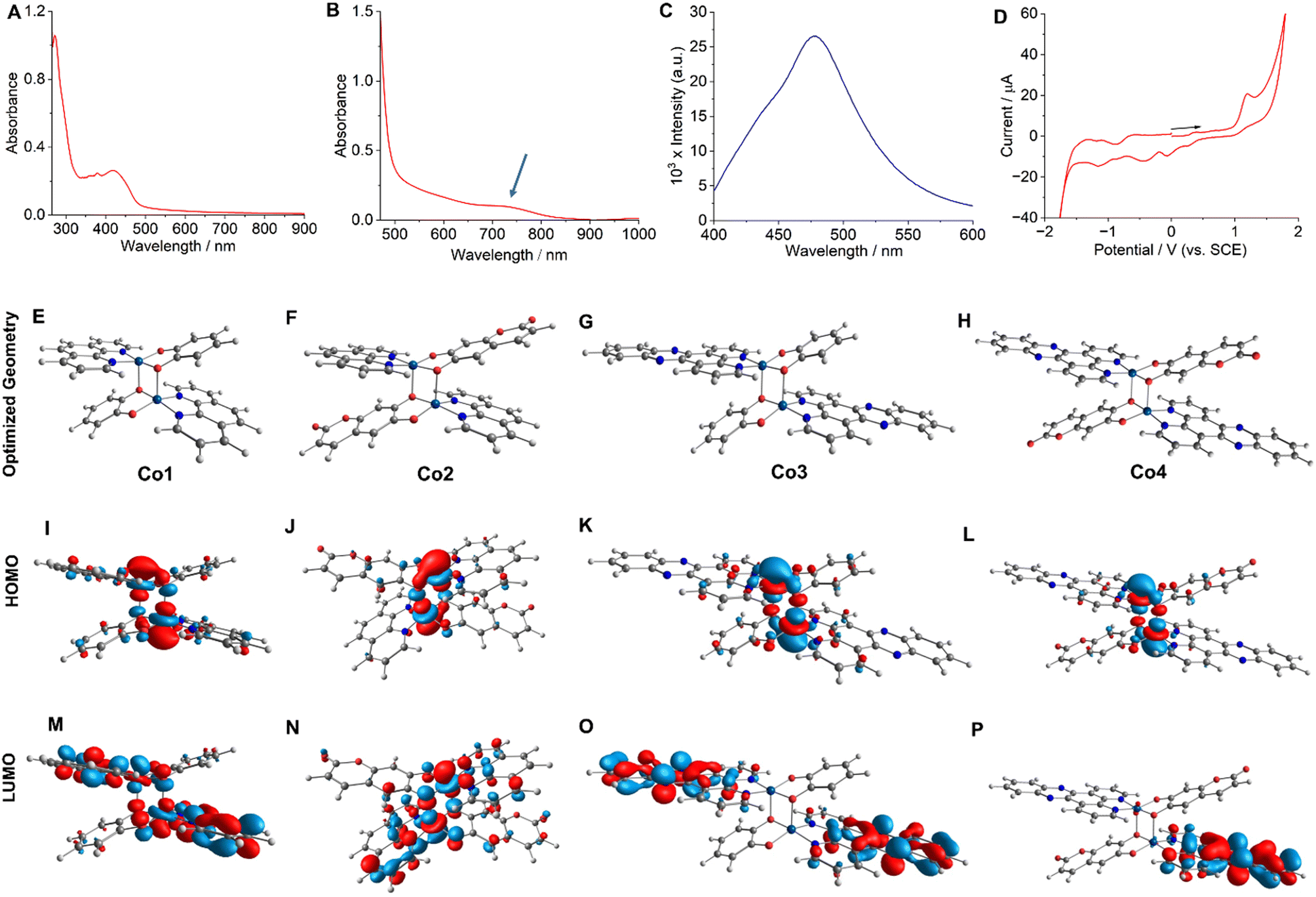
:19 (v/v)] revealed a prominent peak for the [M + H]+ species (Fig. S1–S4, ESI†). The FT-IR spectra of Co-2 and Co-4, which contain the esc2− ligand, exhibited a prominent peak at 1663 cm−1, attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration. Additionally, the characteristic peaks assignable to the aromatic moieties in all complexes were observed within their anticipated frequency ranges (Fig. S5–S8, ESI†). Molar conductivity (ΛM = 10–14 S cm2 mol−1) studies in dimethylformamide (DMF) suggested that the compounds were charge-neutral.43 Solution magnetic susceptibility measurements at room temperature using the Evans method yielded μeff values ranging from 6.36 to 6.45 BM.22 These values are suggestive of two isolated high-spin paramagnetic Co(II) centers with a quartet spin state (S = 3/2), as reported previously.44 The μeff values obtained were consistent with the value recently reported in the solid state for a structurally related dinuclear Co(II) complex with α-diimine and catecholate ligands.44 The UV-visible spectra of Co-1–Co-4 acquired at 25 °C in a 5:1 (v/v) phosphate buffered saline (PBS)/DMF solution (pH = 7.2) displayed a broad (600–850 nm) and moderately intense (M + L)L′CT absorption band (Fig. 2A, B and S9–S12, ESI†).44 While Co-1 and Co-3 did not yield any emission, Co-2 and Co-4 showed respective emission bands around 490 nm (λex = 405 nm) and 477 nm (λex = 420 nm) in PBS/DMF solvent mixture (pH = 7.2) with an emission quantum yield (Φf) value of 0.03 (Fig. 2C and S13, ESI†). Co-2 exhibited a larger Stokes shift of 85 nm compared to Co-4, which displayed a shift of 57 nm. Electrochemical (cyclic voltammetry, CV) studies of Co-1–Co-4 performed in PBS/DMF solvent mixture indicated an irreversible Co(III)/Co(II) redox response around −0.18 V, −0.11 V, −0.21 V, and −0.10 V respectively (vs. SCE) (Fig. 2D and S14–S16, ESI†). Two catecholate ligand-based responses at +1.12 V and −0.42 V and one α-diimine-based response at −1.52 V (for phen) and −1.17 V (for dppz) were additionally observed.21,24,25 The energy-optimized geometries obtained through the density functional theory (DFT) calculations showed a distorted square pyramidal (SPY) coordination environment for the complexes (Fig. 2E–H and Tables S1–S4, ESI†). One O-donor atom from the cat2− or esc2− ligand acts as a bridging ligand, connecting the two Co(II) centers to form the dimeric geometry. A previously reported X-ray single-crystal structure of a structurally similar Co(II) complex further corroborates the predicted geometry.44 The HOMOs of the complexes exhibited contributions from the catecholate and metal orbitals while the LUMOs were predominantly centered on the α-diimine ligands (Fig. 2I–P). Furthermore, the theoretical electronic transitions obtained for Co-4 from the time-dependent (TD) DFT calculations aligned well with the experimentally observed transitions in the visible, red, and NIR regions (Table S5, ESI†).
O stretching vibration. Additionally, the characteristic peaks assignable to the aromatic moieties in all complexes were observed within their anticipated frequency ranges (Fig. S5–S8, ESI†). Molar conductivity (ΛM = 10–14 S cm2 mol−1) studies in dimethylformamide (DMF) suggested that the compounds were charge-neutral.43 Solution magnetic susceptibility measurements at room temperature using the Evans method yielded μeff values ranging from 6.36 to 6.45 BM.22 These values are suggestive of two isolated high-spin paramagnetic Co(II) centers with a quartet spin state (S = 3/2), as reported previously.44 The μeff values obtained were consistent with the value recently reported in the solid state for a structurally related dinuclear Co(II) complex with α-diimine and catecholate ligands.44 The UV-visible spectra of Co-1–Co-4 acquired at 25 °C in a 5:1 (v/v) phosphate buffered saline (PBS)/DMF solution (pH = 7.2) displayed a broad (600–850 nm) and moderately intense (M + L)L′CT absorption band (Fig. 2A, B and S9–S12, ESI†).44 While Co-1 and Co-3 did not yield any emission, Co-2 and Co-4 showed respective emission bands around 490 nm (λex = 405 nm) and 477 nm (λex = 420 nm) in PBS/DMF solvent mixture (pH = 7.2) with an emission quantum yield (Φf) value of 0.03 (Fig. 2C and S13, ESI†). Co-2 exhibited a larger Stokes shift of 85 nm compared to Co-4, which displayed a shift of 57 nm. Electrochemical (cyclic voltammetry, CV) studies of Co-1–Co-4 performed in PBS/DMF solvent mixture indicated an irreversible Co(III)/Co(II) redox response around −0.18 V, −0.11 V, −0.21 V, and −0.10 V respectively (vs. SCE) (Fig. 2D and S14–S16, ESI†). Two catecholate ligand-based responses at +1.12 V and −0.42 V and one α-diimine-based response at −1.52 V (for phen) and −1.17 V (for dppz) were additionally observed.21,24,25 The energy-optimized geometries obtained through the density functional theory (DFT) calculations showed a distorted square pyramidal (SPY) coordination environment for the complexes (Fig. 2E–H and Tables S1–S4, ESI†). One O-donor atom from the cat2− or esc2− ligand acts as a bridging ligand, connecting the two Co(II) centers to form the dimeric geometry. A previously reported X-ray single-crystal structure of a structurally similar Co(II) complex further corroborates the predicted geometry.44 The HOMOs of the complexes exhibited contributions from the catecholate and metal orbitals while the LUMOs were predominantly centered on the α-diimine ligands (Fig. 2I–P). Furthermore, the theoretical electronic transitions obtained for Co-4 from the time-dependent (TD) DFT calculations aligned well with the experimentally observed transitions in the visible, red, and NIR regions (Table S5, ESI†).
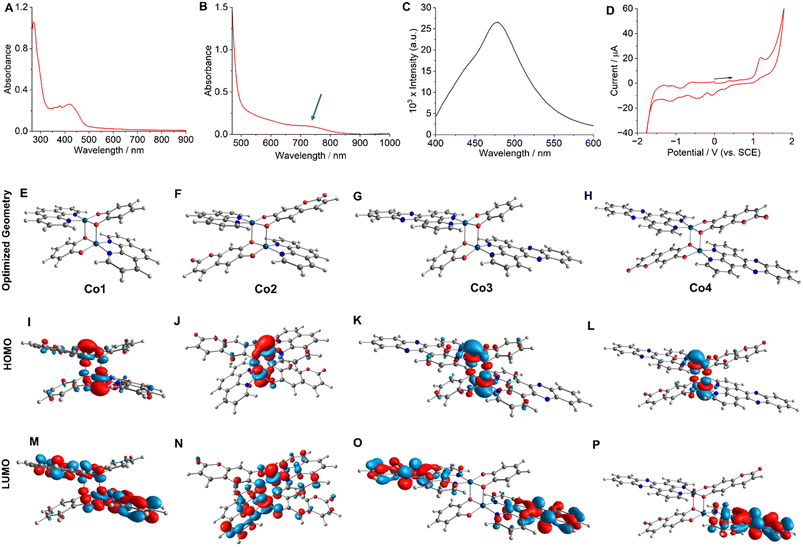 |
| | Fig. 2 UV-visible (in A and B) and emission (in C) spectra of Co-4 (11 μM) in PBS/DMF (5:1 v/v, pH = 7.2) at 25 °C. The spectrum shown in B was recorded at a higher concentration (55 μM) to observe the charge transfer band. For emission measurement, λex = 410 nm. Cyclic voltammogram (in D) of Co-4 in 1:9 (v/v) PBS/DMF. DFT optimized structures (E to H), HOMOs (I to L), and LUMOs (M to P) of Co-1–Co-4, respectively. | |
Solution stability, photostability and lipophilicity
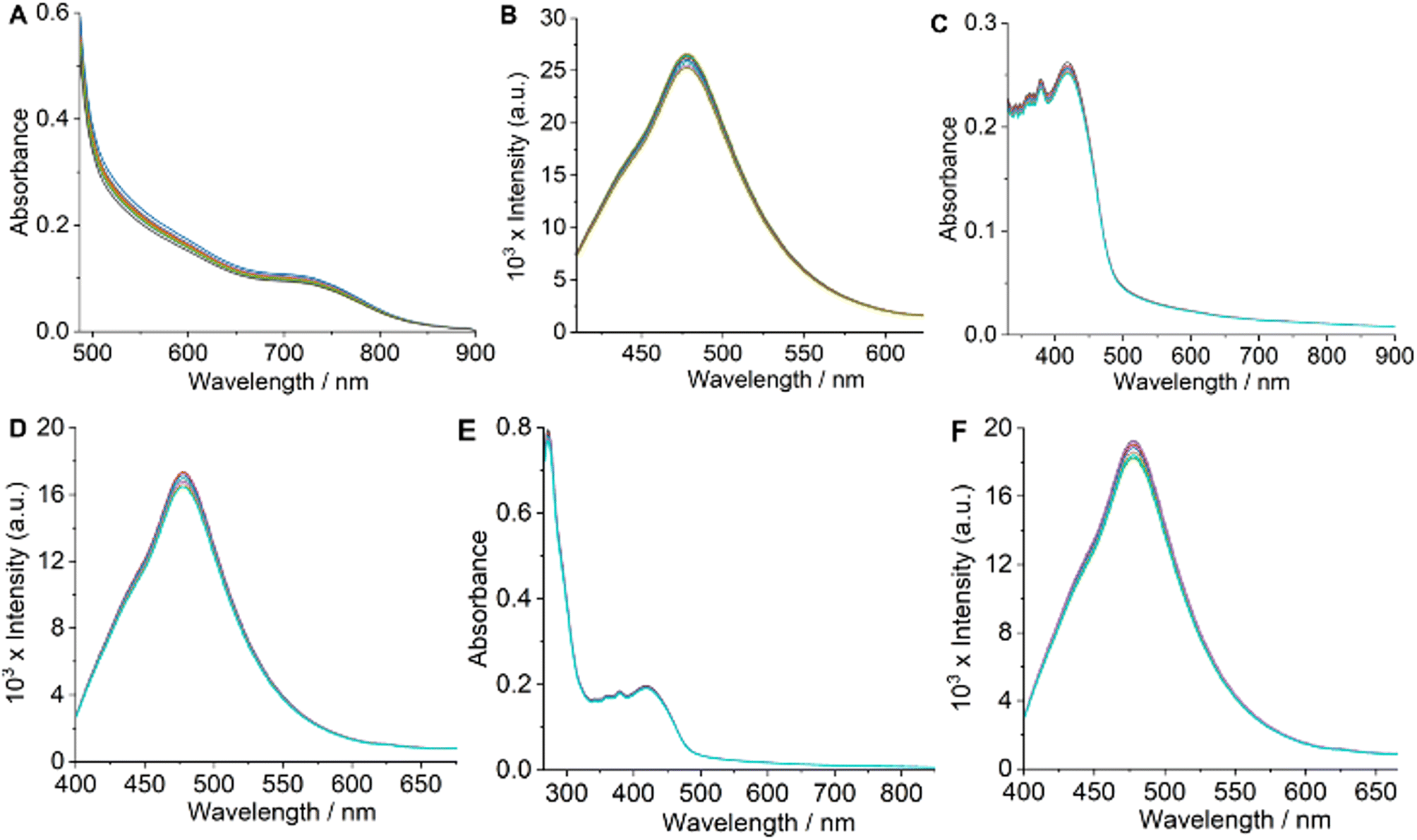
For therapeutic effectiveness, a prospective drug must demonstrate adequate thermodynamic stability in aqueous solution. The ESI-HRMS (+) spectra obtained for the complexes in H2O/MeOH (1:19 v/v) solvent mixture displayed a prominent molecular ion peak corresponding to the [M + H]+ species (Fig. S1–S4†). This observation confirms the dimeric structures of the complexes in solution. Additionally, solution magnetic susceptibility measurements indicated that the μeff are consistent with the presence of two distinct Co(II) centers. If the complexes were monomeric, we would expect μeff values characteristic of a single Co(II) center, thereby reinforcing their dimeric nature. To further judge the solution stability of Co-1–Co-4, we used UV-visible and emission spectroscopy employing a PBS/DMF [5:1 (v/v), pH = 7.2, 25 °C] solution of Co-4. UV-visible and emission spectra of Co-4 showed no discernible changes during periodic monitoring over 48 h, indicating prolonged stability (Fig. 3A and B).21,24 Additionally, HRMS spectral analysis conducted after 48 h using an H2O/MeOH [1:19 (v/v)] solution showed no extra peaks besides the molecular [M + H]+ ion peak at m/z = 1035.0758, indicating that the dimeric structure of the complex stayed intact (Fig. S17, ESI†). Additionally, the colorimetric test to detect any free Co(II) species in the solution gave negative results.45 These results unequivocally establish the fact that the synthesized complexes are sufficiently stable in an aqueous phase and retain their dimeric structures. The preservation of the dimeric structure in solution is further corroborated by a recent study on a similar mixed-ligand dinuclear Co(II) complex featuring α-diimine and catecholate ligands.44 Photostability is an essential aspect of any photosensitizer intended for photodynamic applications. To study whether the synthesized complexes meet this criterion, we monitored the UV-visible and emission spectra of Co-4 under continuous laser irradiation as a representative example. The results demonstrated no significant alternations in the UV-visible and emission spectra throughout 1 h, with measurements taken every 10 minutes during continuous laser irradiation (808 nm, 450 mW), indicating their photostability (Fig. 3C and D).21,24 Additionally, no spectral changes were noticed for Co-4 treated with reduced glutathione (GSH, 1.0 mM), revealing that Co-1–Co-4 maintained their identity under the reducing conditions typically found inside cells (Fig. 3E and F). Drug's lipophilicity is a key factor that influences the efficiency of drug delivery at the cellular level. The experimental determination of lipophilicity involved measuring the octanol/water (o/w) partition coefficient (Po/w) and expressing the results as logPo/w. The Po/w measurements gave the respective logPo/w values of 0.91 ± 0.04, 1.25 ± 0.03, 1.07 ± 0.02, and 1.43 ± 0.04 for Co-1–Co-4, correlating with the lipophilicity order of the α-diimine and catecholate ligands.23,24 Positive logPo/w values suggest a diffusion-mediated cellular uptake mechanism for the complexes.
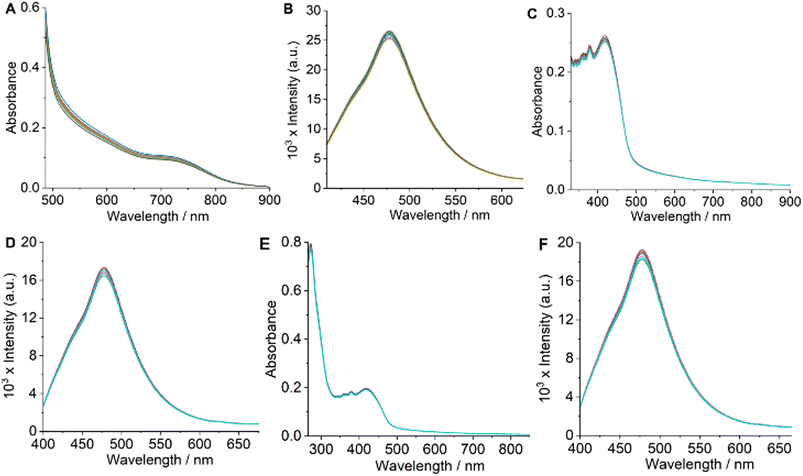 |
| | Fig. 3 Time-dependent UV-visible (in A) and emission (in B) spectra of Co-4 monitored over 48 h. Time-dependent UV-visible (in C) and emission (in D) spectra of Co-4, taken at 10 min intervals (up to 1 h) during continuous laser irradiation at 808 nm (450 mW). Time-dependent UV-visible (in E) and emission (in F) spectra of Co-4 with reduced GSH (1.0 mM), taken at 8 h intervals up to 48 h. All UV-visible and emission spectra were acquired at 25 °C in a 5:1 (v/v) PBS/DMF solvent mixture (pH = 7.2). [Complex] = 8–11 μM. | |
Anticancer activity (cytotoxicity)
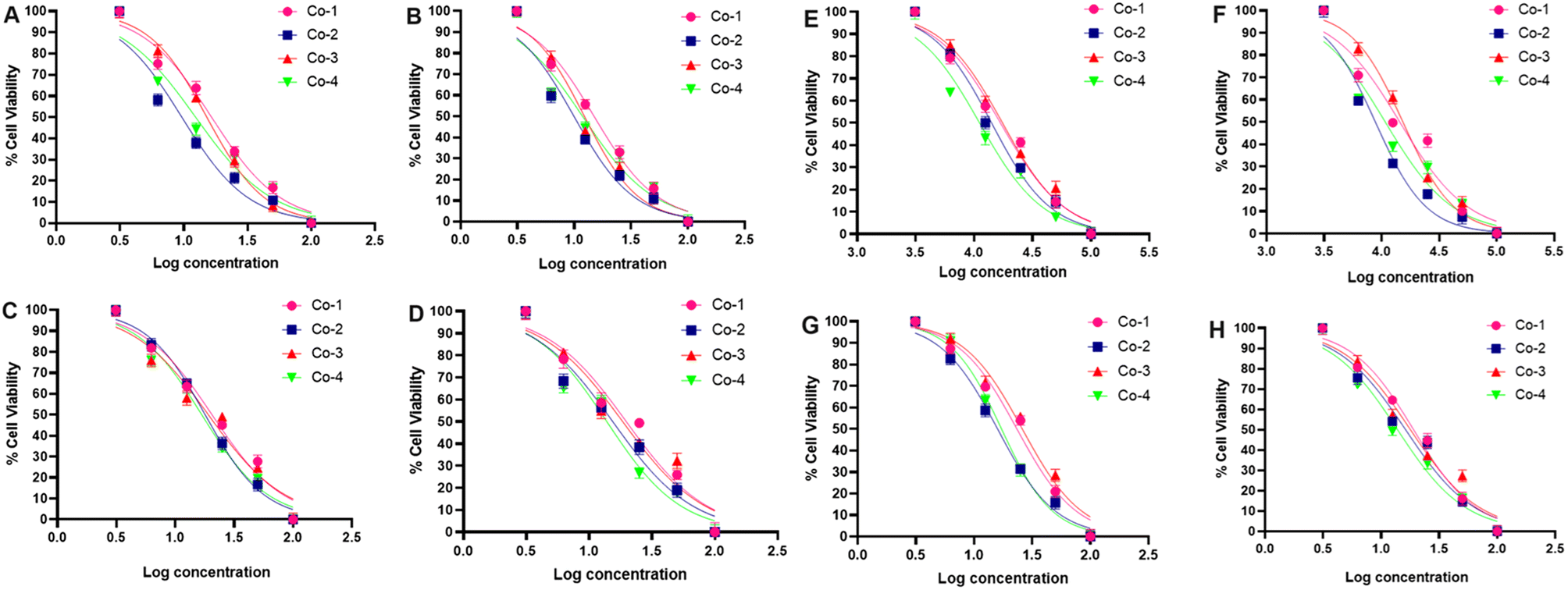
As the next step in our investigation, we assessed the cytotoxic potential of Co-1–Co-4 utilizing the monolayer A549 cancerous lung cell line and conducting the colorimetric MTT assay. The viability of the cells was studied under the dark and light-irradiated (red light, 660 nm; NIR light, 808 nm, 1.0 W cm−2, 5 min) conditions after 24 h and 48 h of drug treatment.46 The choice of these wavelengths aligns with the absorption characteristics of the complexes, facilitating optimal photodynamic activation. Under dark conditions, the phen complexes Co-1 and Co-3 displayed notable cytotoxicity giving IC50 values of 22.90 and 19.47 μM at 24 h and 19.73 and 18.48 μM at 48 h. For the dppz complexes Co-2 and Co-4, the respective IC50 values were 22.17 and 18.76 μM at 24 h and 18.54 and 15.52 μM at 48 h. With 660 nm laser irradiation, the IC50 values for Co-1 and Co-3 decreased to 17.85 and 19.42 at 24 h and 14.88 and 13.92 μM at 48 h. For the dppz complexes Co-2 and Co-4, the respective values were 14.93 and 12.22 μM at 24 h and 10.27 and 11.82 μM at 48 h (Fig. 4 and Table 1). In another experiment designed for 808 nm laser irradiation, Co-1 and Co-3 yielded IC50 values of 23.17 and 21.69 μM in the dark (at 24 h) and 19.32 and 18.26 μM (at 48 h). Under 808 nm laser irradiation, the IC50 values decreased to 16.81 and 17.80 μM (at 24 h) and 14.37 and 15.20 μM (at 48 h). The dppz complexes Co-2 and Co-4 yielded respective IC50 values of 17.92 and 16.46 μM (at 24 h) and 16.44 and 14.11 μM (at 48 h) under dark conditions. Interestingly, the values decreased to 14.12 and 11.24 μM at 24 h and 8.82 and 10.13 μM at 48 h with 808 nm laser irradiation (Fig. 4 and Table 1). Our data reveal that the compounds caused cancer cell death through a mechanism of chemotherapy and PDT (Chemo-PDT), showing enhanced effectiveness with 808 nm light over 660 nm light. The observed activity in the dark may result from the joint influence of the metal and ligand under dark and irradiated conditions. Similarly, the increased light-triggered activity is due to the photoactivity of the α-diimine (dppz > phen) and esc2− ligands. The dppz complexes Co-2 and Co-4 had better cytotoxic effects than the corresponding phen complexes Co-1 and Co-3. The esculetin ligand gave IC50 values of 58.4 ± 2.5 μM (24 h) and 42.76 ± 1.8 μM (48 h) in the dark which changed negligibly under laser irradiation due to lack of any absorption in the red and NIR regions. Our results are important since they identify cobalt complexes with increased cytotoxic effects upon activation by long-wavelength red and NIR light, not previously reported for cobalt complexes within the clinically relevant range for PDT. A drug intended for chemo-PDT applications should not exhibit notable toxicity toward normal cells. To assess the impact of Co-2 and Co-4 on healthy cells, we tested them on the NIH-3T3 embryonic fibroblast cell line, using the MTT assay under comparable conditions. The IC50 values of CO-2 and Co-4 were >100 μM (48 h, 808 nm, 5 min, Fig. S18, ESI†). Thus, the results suggested that these compounds exhibited a negligible effect on the survival of normal fibroblast cells, highlighting their potential for selectively targeting cancer cells.
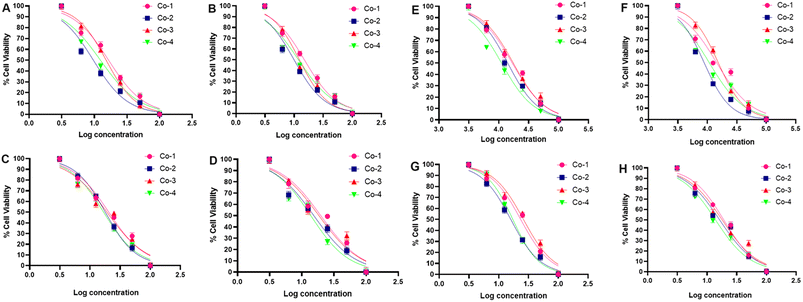 |
| | Fig. 4 IC50 graphs (in A to D) of A549 cells treated with Co-1, Co-2, Co-3, or Co-4 with/without 660 nm laser irradiation (1.0 W cm−2, 5 min) for 24 h and 48 h. With laser 24 h (in A), with laser 48 h (in B), without laser 24 h (in C), and without laser 48 h (in D). IC50 graphs (in E to H) of A549 cells treated with Co-1, Co-2, Co-3, or Co-4 with/without 808 nm laser irradiation (1.0 W cm−2, 5 min) for 24 h and 48 h. With laser 24 h (in E), with laser 48 h (in F), without laser 24 h (in G), and without laser 48 h (in H). | |
Table 1 The IC50 (in μM units) values of Co-1–Co-4 as determined from the colorimetric MTT assay
| Compound |
24 (−light) |
48 h (−light) |
24 h (+660 nm) |
48 h (+660 nm) |
24 (−light) |
48 h (−light) |
24 h (+808 nm) |
48 h (+808 nm) |
|
Co-1
|
22.90 ± 1.31 |
19.73 ± 1.54 |
17.85 ± 1.37 |
14.88 ± 1.02 |
23.17 ± 1.28 |
19.32 ± 1.4 |
16.81 ± 1.23 |
14.37 ± 1.05 |
|
Co-2
|
22.17 ± 1.12 |
18.54 ± 1.23 |
14.93 ± 0.3 |
10.27 ± 1.42 |
17.92 ± 1.11 |
16.44 ± 0.9 |
14.12 ± 0.35 |
8.9 ± 1.30 |
|
Co-3
|
19.47 ± 1.35 |
18.48 ± 1.19 |
19.42 ± 1.22 |
13.92 ± 1.28 |
21.69 ± 1.30 |
18.26 ± 1.2 |
17.80 ± 1.32 |
15.20 ± 1.33 |
|
Co-4
|
18.76 ± 1.04 |
15.52 ± 0.8 |
12.22 ± 1.52 |
11.82 ± 1.25 |
16.46 ± 0.6 |
14.11 ± 1.1 |
11.24 ± 0.78 |
10.0 ± 1.23 |
Apoptosis detection
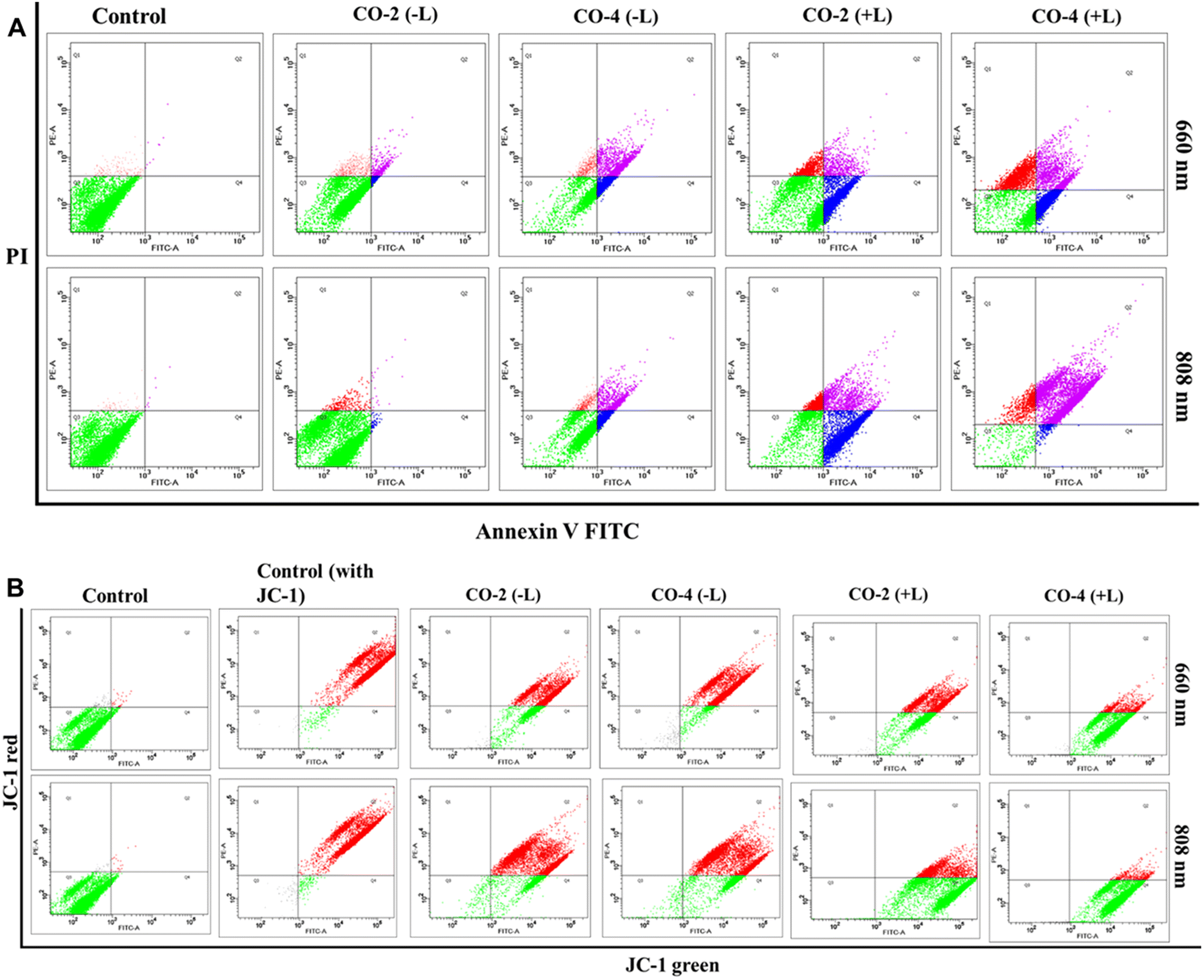
To explore the mode of cell death, we performed the Annexin-V-FITC assay using Co-2 and Co-4 (15 μM) under both dark and light-irradiated (red light, 660 nm; NIR light, 808 nm; 1.0 W cm−2; 5 min) conditions with drug treatment (24 h).46 The assay results indicated that the cell death was apoptotic, with a notable rise in cell death observed under 660 nm and 808 nm light irradiation, in contrast to non-irradiated conditions (Fig. 5A). The impact was stronger for Co-4 (∼20% early apoptosis and ∼47% late apoptosis) than for Co-2 (∼15% early apoptosis and ∼30% late apoptosis).
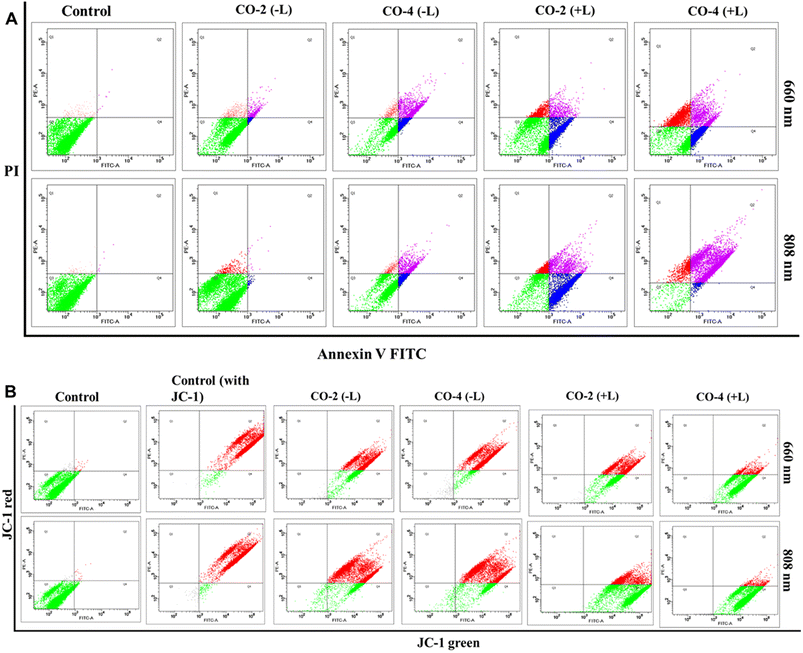 |
| | Fig. 5 Annexin-V-assay (A) and MMP assay using JC-1 dye (B) conducted in A549 cells treated with Co-2 and Co-4 (15 μM) under conditions with (+L) and without (–L) laser irradiation at 660 nm or 808 nm (1.0 W cm−2) for 5 min. | |
Mitochondrial damage
Excessive production of drug-induced ROS can cause harm to mitochondrial membranes, leading to a loss of mitochondrial membrane potential (MMP) and cellular apoptosis. We used the reliable JC-1 assay to assess changes in MMP under both dark and light-irradiated (red light, 660 nm; NIR light, 808 nm; 1.0 W cm−2; 5 min) conditions, using Co-2 and Co-4 (15 μM).46 In normal cells with a high MMP, JC-1 dye forms red-fluorescent J-aggregates, while decreased MMP increases green fluorescent J-monomers. Findings from our assay revealed that control groups exhibited strong red fluorescence and minimal green fluorescence, indicating high MMP. In contrast, Co-2 and Co-4 caused a substantial reduction in MMP under irradiation at 660 nm and 808 nm, with Co-4 having a more pronounced effect than Co-2 (Fig. 5B).
ROS generation
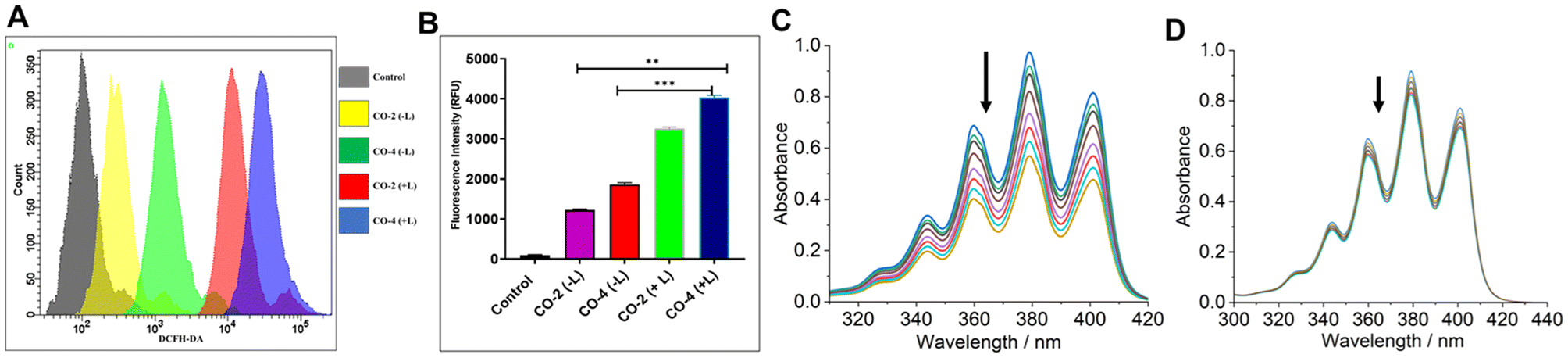
To detect ROS generation by Co-2 and Co-4 leading to apoptotic death of A549 cancer cells, we employed DCFH-DA assay under both dark and light-irradiated (red light, 660 nm; NIR light, 808 nm; 1.0 W cm−2; 5 min) conditions.21 The findings indicated that the compounds triggered ROS production at 24 h of treatment and this effect was significantly amplified under irradiated conditions (Fig. 6A and B). Next, we used singlet oxygen (1O2)-specific probe 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA) to detect the actual identity of the ROS generated. Treatment of ABDA (7 μM) with Co-4 (15 μM) and exposed to NIR laser light (808 nm, 450 mW) led to a gradual decrease in absorbance of the π → π* absorption band at 378 nm (Fig. 6C).47 In contrast, no marked spectral alternation was noticed when the 1O2 quencher NaN3 was used (Fig. 6D). These findings suggest the photo-triggered production of 1O2 as the cell-killing ROS in a type-II PDT process. The quantum yield (ϕΔ) of 1O2 generation was determined to be 0.08 for Co-2 and 0.13 for Co-4 (Fig. S19 and S20, ESI†).15
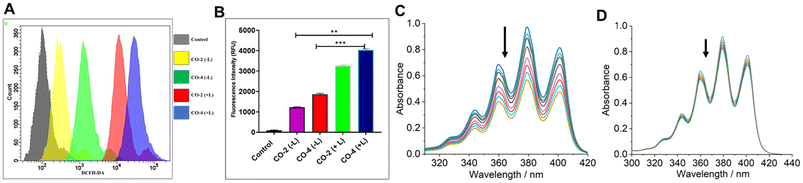 |
| | Fig. 6 (A) DCFH-DA assay to detect ROS generation (A549 cells) using Co-2 and Co-4 (15 μM, 24 h) in the dark and with laser irradiation. Light source: 808 nm, 1.0 W cm−2, 5 min. (B) Bar diagram showing the variation of the emission intensity of DCFH in A549 cells treated with Co-2 and Co-4 (15 μM, 24 h). (C) Time-dependent UV-visible spectral traces of ABDA (7 μM) at 378 nm treated with Co-4 (15 μM) and recorded at 1 min intervals of continuous NIR laser irradiation (808 nm, 450 mW). (D) The UV-visible spectra of ABDA (6 μM) treated with Co-4 (15 μM), in the presence of NaN3, and acquired every 1 min during continuous laser irradiation (808 nm, 450 mW). | |
Antibacterial activity
Significant research is currently focused on understanding how bacteria contribute to the initiation, growth, and advancement of cancer.48,49 Patients undergoing cancer therapy are at increased risk of bacterial infections, a condition worsened by the prescription of immunosuppressive medications during treatment.50 The increasing challenge posed by MDR bacterial infections, coupled with this scenario, spurred our exploration of the antibacterial effectiveness of Co-2 and Co-4. We assessed the antibacterial potency of Co-2 and Co-4 using the broth microdilution method.51 The Gram-(+) S. aureus (SA) and Gram-(−) P. aeruginosa (PA) bacteria were treated with Co-2 and Co-4 for 15 min within the concentration range of 0.03125 to 512 μg mL−1. The antibacterial effect was evaluated in the dark and under 808 nm laser irradiation (1.0 W cm−2, 5 min). The MIC (minimum inhibitory concentration) values for Co-2 were determined to be 128 μg mL−1 (142 μM) in the dark for both SA and PA which decreased to 32 μg mL−1 (35 μM) and 64 μg mL−1 (71 μM) respectively with laser irradiation. In contrast, the MIC values for Co-4 under dark conditions were 32 μg mL−1 (31 μM) and 64 μg mL−1 (62 μM), which were further reduced to 16 μg mL−1 (15 μM) and 32 μg mL−1 (31 μM) for SA and PA respectively with laser irradiation (Table 2 and Fig. S21, ESI†). Thus, Co-4 was more effective in killing both types of bacteria than Co-2.
Table 2 The MIC (in μg mL−1 and μM units) values of Co-2 and Co-4
| Complex |
S. Aureus (dark) |
S. Aureus (+808 nm) |
P. aeruginosa (dark) |
P. aeruginosa (+808 nm) |
|
Co-2
|
128 μg mL−1 (142 μM) |
32 μg mL−1 (35 μM) |
128 μg mL−1 (123 μM) |
64 μg mL−1 (71 μM) |
|
Co-4
|
32 μg mL−1 (31 μM) |
16 μg mL−1 (15 μM) |
64 μg mL−1 (62 μM) |
32 μg mL−1 (31 μM) |
Live/dead staining of bacteria
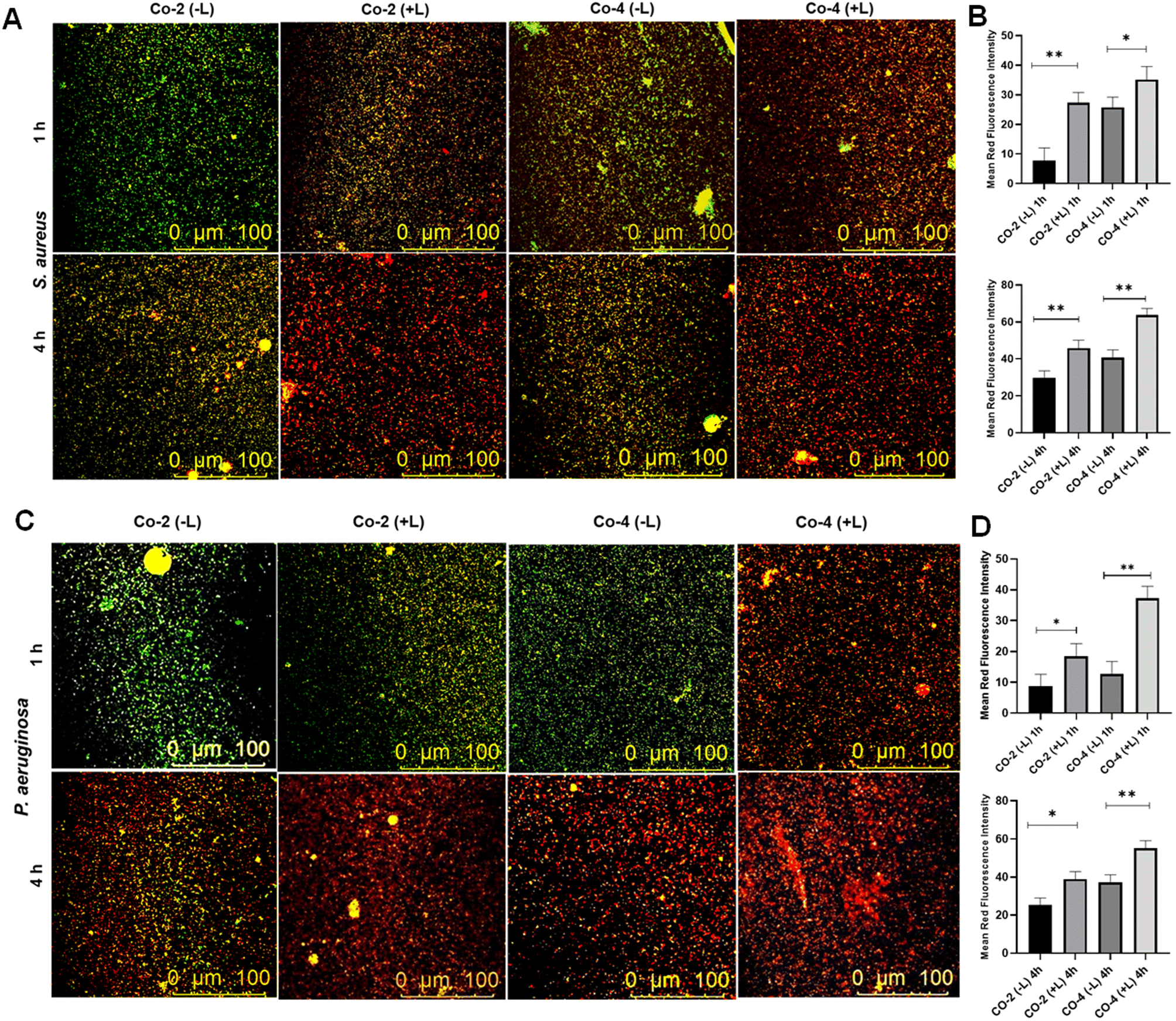
Next, we conducted the live-dead analysis of planktonic bacteria using SYTO9 and PI staining.52 SYTO9 stains both viable and non-viable bacterial cells, emitting green fluorescence, whereas PI selectively stains cells with compromised membranes, emitting red fluorescence. Our results showed a higher proportion of bacterial death induced by Co-4 than Co-2, both with and without laser irradiation (Fig. 7A and B). The red fluorescence intensities of dead populations of S. aureus and P. aeruginosa cultures have been quantified by using ImageJ software. The mean red fluorescence intensity increased in cultures incubated after laser irradiation compared to the cultures incubated in the dark conditions in both bacterial strains, indicating increased bacterial killing efficiency of the complexes after light exposure (Fig. 7B and D). The bacteria-killing ability of the complexes was considerably more pronounced under irradiation compared to darkness. Additionally, Co-4 exhibited a more pronounced effect on ROS generation compared to Co-2 under similar conditions as evidenced by the greater fluorescence of DCFH in cells treated could stem from membrane integrity disruption, potentially due to pore formation caused by the compound's interaction, leading to bacterial death.
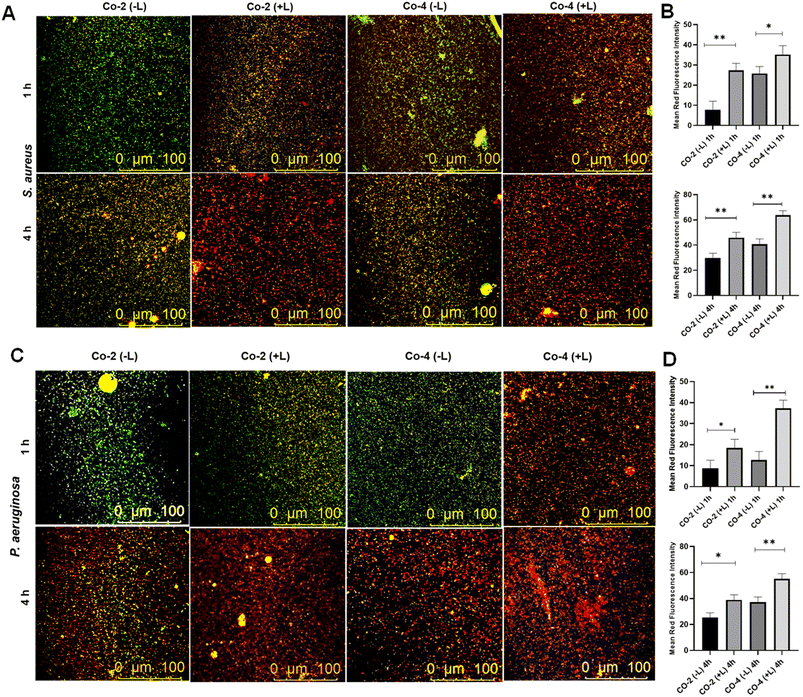 |
| | Fig. 7 Live/dead staining assay using S. aureus (in A) and P. aeruginosa (in C) cultures after incubating (1 h and 4 h) with Co-2 and Co-4 under dark and with laser irradiation. The bar graphs represent the mean red fluorescence intensity of dead cells of S. aureus (B) and P. aeruginosa (D) quantified by ImageJ software. Light source: 808 nm, 1.0 W cm−2, 5 min. | |
Conclusions
In conclusion, we have reported four mixed-ligand dinuclear Co(II) complexes featuring α-diimine (phen, dppz) and catecholate (cat2− and esc2−) ligands which demonstrate significantly enhanced PDT activity under red and NIR light against cancerous A549 cells giving IC50 values in the micromolar range. These complexes had marginal toxicity towards the NIH-3T3 healthy embryonic fibroblast cells. The dppz complexes Co-2 and Co-4 also demonstrated strong antibacterial efficacy against Gram (+) SA and Gram (−) PA, with significant enhancement observed upon red and NIR light irradiation. The markedly enhanced activity exhibited by Co-2 and Co-4 against cancer and bacterial cells is attributed to the generation of cytotoxic singlet oxygen species via a type-II PDT process, facilitated by the photoactive dppz and esculetin moieties. To our knowledge, this represents the first study on cobalt complexes directly utilizing its charge transfer transition to boost toxicity towards cancer cells and pathogenic bacteria under red and NIR light irradiation. Our findings illuminate the path for developing cobalt-based coordination compounds capable of addressing cancer and bacterial infections through dual chemo-PDT using long-wavelength red and NIR lights.
Experimental section
Synthesis of complexes Co-1–Co-4
A common synthesis pathway was employed for the complexes, as detailed below: under an argon atmosphere, anhydrous CoCl2 (0.13 g, 1.0 mmol) was dissolved in anhydrous THF (20 mL) and subsequently, 1.0 mmol of anhydrous phen or dppz (phen, 0.198 g; dppz, 0.282 g) solubilized in anhydrous THF (20 mL) was added dropwise. In the case of dppz, the THF was heated to solubilize the ligand before adding it to the reaction. After 30 minutes of stirring at room temperature, 1.0 mmol of catechol (1,2-dihydroxybenzene, H2cat, 0.11 g, 1.0 mmol) or esculetin (6,7-dihydroxycoumarin, H2esc, 0.178 g) completely solubilized in a mixture of anhydrous THF (10 mL), anhydrous CHCl3 (5 mL), and sodium hydroxide (0.08 g, 2.0 mmol) was slowly introduced into the reaction by maintaining the argon atmosphere. The reaction mixture was left to stir at room temperature for 2 h, during which a green precipitate formed. Filtration allowed for the recovery of a dark green solid which was washed with cooled anhydrous THF (3 × 10 mL) followed by Et2O (2 × 15 mL). Finally, the solid was meticulously dried overnight in a vacuum desiccator using P4O10 as the desiccant.
Characterization data
[Co(phen)(cat)]2 (Co-1).
Yield (%): ∼63. Anal. calcd for C36H24N4O4Co2: C, 66.26; H, 3.48; N, 8.07; observed: C, 66.34; H, 3.51; N, 8.10. HRMS (ESI+) in H2O/MeOH [1:19 (v/v)]. Calcd for [M + H]+: m/z 695.0540. Observed: m/z 695.0557. FT–IR (cm−1): 1633 s, 1521 s, 1472 s, 1424 s, 1393 m, 1335 w, 1253 s, 1224 w, 1141 w, 1098 w, 1034 w, 855 s, 713 s, 660 w, 636 w. (vs, very strong; s, strong; m, medium; w, weak). UV-visible [5:1 (v/v) PBS/DMF; λmax, nm (ε, M−1 cm−1)]: 270 (46600), 674 (1070). μeff = 6.45 B.M. at 25 °C. Molar conductivity (ΛM) in DMF at 25 °C [S cm2 mol−1]: 14.
[Co(dppz)(cat)]2 (Co-2).
Yield (%): ∼58. Anal. calcd for C48H28N8O4Co2: C, 64.15; H, 3.14; N, 12.47; observed: C, 64.26; H, 3.13; N, 12.43. HRMS (ESI+) in H2O/MeOH [1:19 (v/v)]. Calcd for [M + H]+: m/z 899.0976. Observed: m/z 899.0992. FT–IR (cm−1): 2793 w, 2718 w 1625 m, 1489 m, 1412 w, 1360 w, 1276 w, 1180 w, 1150 w, 1077 m, 1036 m, 827 m, 729 m, 648 w, 586 w. UV-visible [5:1 (v/v) PBS/DMF; λmax, nm (ε, M−1 cm−1)]: 273 (36700), 360 (7050), 377 (6800), 680 (1360). μeff = 6.39 B.M. at 25 °C. Molar conductivity (ΛM) in DMF at 25 °C [S cm2 mol−1]: 12.
[Co(phen)(esc)]2 (Co-3).
Yield (%): ∼52. Anal. calcd for C42H24N4O8Co2: C, 60.74; H, 2.91; N, 6.75; observed: C, 60.81; H, 2.89; N, 6.73. HRMS (ESI+) in H2O/MeOH [1:19 (v/v)]. Calcd for [M + H]+: m/z 831.0336. Observed: m/z 831.0345. FT–IR (cm−1): 1661 s, 1584 s, 1535 s, 1476 s, 1428 s, 1389 s, 1360 w, 1341 w, 1288 s, 1253 s, 1224 w, 1191 m, 1161 w, 1140 w, 1092 w, 1041 w, 926 w, 843 m, 812 w, 771 w, 750 w, 729 m, 657 w, 626 w. UV-visible [5:1 (v/v) PBS/DMF; λmax, nm (ε, M−1 cm−1)]: 271 (50500), 405 (21300), 674 (1120). Emission [5:1 (v/v) PBS/DMF]: λem = 490 nm (λex = 405 nm). μeff = 6.41 B.M. at 25 °C. Molar conductivity (ΛM) in DMF at 25 °C [S cm2 mol−1]: 10.
[Co(dppz)(esc)]2 (Co-4).
Yield (%): ∼66. Anal. calcd for C54H28N8O4Co2: C, 62.68; H, 2.73; N, 10.83; observed: C, 62.74; H, 2.72; N, 10.86. HRMS (ESI+) in H2O/MeOH [1:19 (v/v)]. Calcd for [M + H]+: m/z 1035.0772. Observed: m/z 1035.0789. FT–IR (cm−1): 2793 m, 2740 w, 1662 s, 1570 w, 1469 m, 1396 m, 1262 s, 1190 w, 1150 m, 1060 m, 1037 w, 957 w, 830 m, 726 m, 648 w. UV-visible [5:1 (v/v) PBS/DMF; λmax, nm (ε, M−1 cm−1)]: 272 (95400), 361 (19800), 380 (23700), 420 (26200), 730 (1920). Emission [5:1 (v/v) PBS/DMF]: λem = 477 nm (λex = 420 nm). μeff = 6.36 B.M. at 25 °C. Molar conductivity (ΛM) in DMF at 25 °C [S cm2 mol−1]: 11.
Cell viability
A549 cells were added to a 96-well plate (10000 cells per well) and allowed to incubate overnight. The next day, cells were treated with Co-1–Co-4. After 12 h of treatment, they were subjected to NIR laser irradiation at 660 nm and 808 nm (1.0 W cm−2) for 5 min per well, according to a method described earlier.46 The cells underwent further incubation for 24 h and 48 h. After incubation, MTT reagent (50 μL, 5 mg mL−1) was added to each well and left to incubate for 4 hours. To dissolve the purple formazan crystals that formed, 150 μL of DMSO was then added. After one hour, absorbance readings were taken at 570 nm and 630 nm using a SpectraMax Multi-Mode Microplate Reader (Molecular Devices, USA). The outcomes of laser-exposed cells were compared with those of cells that were treated but not exposed to laser irradiation.
Apoptosis assay
Flow cytometry was performed using PI/Annexin V-FITC staining to evaluate apoptosis levels. Following a 24 h exposure to Co-2 and Co-4, cells underwent treatment. Laser exposure at 660 nm and 808 nm wavelengths lasted for 5 min per well after 12 h of treatment, followed by an additional 12 h incubation as reported previously earlier.46 Post-treatment, the cells were trypsinized, washed with PBS, and resuspended in 500 μL of 1× binding buffer. Following the addition of Annexin V-FITC (4 μL) and PI (10 μL), the cells were incubated in the dark at 25 °C for 15 min. Flow cytometric analysis was conducted with a BD-FACS instrument, and the results were processed using Facs Diva Software. The apoptotic cell percentages were calculated after accounting for autofluorescence, and results are expressed as mean ± SEM.
Mitochondrial damage assay
JC-1 dye was utilized to assess mitochondrial depolarization. This cationic dye selectively accumulates in mitochondria in a potential-dependent manner, with a shift in fluorescence emission from red to green indicating mitochondrial damage and membrane potential loss. After exposing cells to Co-2 and Co-4 at 15 μM concentrations for 24 h, NIR laser irradiation (660 and 808 nm, 1.0 W cm−2) was applied for 5 minutes following 12 h of incubation, according to the method outlined previously.46 Subsequently, the cells underwent washing with PBS and incubation with 10 μg mL−1 JC-1 dye for 15 min at 37 °C. The examination was conducted using a confocal microscope (Leica, Germany). Quantitative analysis of mitochondrial membrane potential (MMP) was carried out using a flow cytometer (BD FACS Area III), with fluorescence measured at 485 nm (excitation) and 590 nm (emission). The instrument was carefully calibrated to ensure accuracy, with appropriate compensation and threshold settings applied.
ROS generation assay
Reactive oxygen species (ROS) levels were quantified using the DCFH-DA dye, which permeates the cell and reacts with ROS to produce dichlorofluorescein (DCF), a green fluorescent compound. ROS levels were measured by flow cytometry, following the protocol detailed earlier.21 A stock solution of DCFH-DA (10 mM) was first diluted to a working concentration of 100 μM. Cancerous A549 lung cells were exposed to 15 μM concentrations of Co-2 and Co-4, followed by exposure to 660 nm and 808 nm laser radiation (1.0 W cm−2, 5 min) for 24 h. After treatment, cells underwent washing with PBS and incubation with 100 μM DCFH-DA for 30 minutes at 37 °C. The quantification of the ROS levels was done using a flow cytometer (BD FACS Area III).
Antibacterial assay (MIC determination)
The MICs of Co-2 and Co-4 were measured through the broth microdilution method, following a modified version of the procedure described by Soares et al.51 Overnight cultures of PA and SA in LB broth were adjusted to a concentration of 1 × 105 CFU mL−1. A 100 μL aliquot of this suspension was then dispensed into each well of a sterile 96-well plate. Various concentrations of Co-2 and Co-4 (ranging from 0.03125 to 512 μg mL−1) were prepared and added in equal volumes to the bacterial suspensions. Sterile PBS was used in the control wells. Optical density (OD) measurements of the cultures were performed at different time intervals (over 24 h) using a SpectraMax UV spectrophotometer. To determine how laser irradiation impacts bacterial growth treated with complex, bacterial suspensions were exposed to varying concentrations of Co-2 and Co-4 for 15 min to allow for internalization. The suspensions were then exposed to a laser light (1.0 W cm−2, 808 nm) for 5 minutes per well. OD measurements were taken at specified time points over 24 h using the SpectraMax UV spectrophotometer. The MIC was established as the minimum concentration that resulted in the inhibition of bacterial growth.
Live/dead staining
Live/dead staining was employed to evaluate the impact of Co-2 and Co-4 on bacterial viability. Bacterial cultures at a density of 1 × 106 CFU mL−1 were exposed to MIC concentrations of the compounds, both with and without laser irradiation, and subsequently incubated for 1 h and 4 h. Post incubation, the bacteria were collected by centrifugation, washed with sterile PBS, and resuspended in 100 μL of PBS. A staining solution (5 μL) containing SYTO9 and PI was added to the suspension and incubated under dark conditions for 15 min. A 10 μL portion of the stained bacterial suspension was transferred onto a glass slide and then covered with a coverslip. Bacterial viability was examined using a confocal microscope.52
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
A. H. thanks the Department of Biotechnology (DBT) for providing financial assistance through Grant No. BT/PR25668/NER/95/1278/2017. J. D. acknowledges the financial support received from DBT as a research fellow. The Department of Chemistry at Handique Girls’ College acknowledges financial assistance from the Department of Science and Technology (DST), Government of India, under the DST-CURIE (WISE KIRAN Division) scheme for Women PG Colleges (Ref. No. DST/CURIE-PG/2022/88(G)). T. S. is thankful to CSIR-Indian Institute of Chemical Technology, Hyderabad.
References
-
(a) H. Shi and P. J. Sadler, Br. J. Cancer, 2020, 123, 871–873 CrossRef PubMed
 ;
(b) C. Imberti, P. Zhang, H. Huang and P. J. Sadler, Angew. Chem., Int. Ed., 2020, 59, 61–73 CrossRef CAS PubMed .
;
(b) C. Imberti, P. Zhang, H. Huang and P. J. Sadler, Angew. Chem., Int. Ed., 2020, 59, 61–73 CrossRef CAS PubMed .
-
(a) Y. Wu, S. Li, Y. Chen, W. He and Z. Guo, Chem. Sci., 2022, 13, 5085–5106 RSC ;
(b) N. P. Toupin, S. J. Steinke, M. K. Herroon, I. Podgorski, C. Turro and J. J. Kodanko, Photochem. Photobiol., 2022, 98, 378–388 CrossRef CAS PubMed .
-
(a) J. Karges, Angew. Chem., Int. Ed., 2022, 61, e202112236 CrossRef CAS PubMed ;
(b) H. Zhou, D. Tang, Y. Yu, L. Zhang, B. Wang, J. Karges and H. Xiao, Nat. Commun., 2023, 14, 5350 CrossRef CAS PubMed .
-
(a) J. Shum, L. C.-C. Lee, M. W.-L. Chiang, Y.-W. Lam and K. K.-W. Lo, Angew. Chem., Int. Ed., 2023, 62, e202303931 CrossRef CAS PubMed ;
(b) E. Ortega-Forte, A. Rovira, M. López-Corrales, A. Hernández-García, F. J. Ballester, E. Izquierdo-García, M. Jordà-Redondo, M. Bosch, S. Nonell, M. D. Santana, J. Ruiz, V. Marchán and G. Gasser, Chem. Sci., 2023, 14, 7170–7184 RSC ;
(c) J. Karges, F. Heinemann, M. Jakubaszek, F. Maschietto, C. Subecz, M. Dotou, R. Vinck, O. Blacque, M. Tharaud, B. Goud, E. V. Zahínos, B. Spingler, I. Ciofini and G. Gasser, J. Am. Chem. Soc., 2020, 142, 6578–6587 CrossRef CAS PubMed .
-
(a) F. J. Ballester, A. Hernández-García, M. D. Santana, D. Bautista, P. Ashoo, E. Ortega-Forte, G. Barone and J. Ruiz, Inorg. Chem., 2024, 63, 6202–6216 CrossRef CAS PubMed ;
(b) A. Mani, T. Feng, A. Gandioso, R. Vinck, A. Notaro, L. Gourdon, P. Burckel, B. Saubaméa, O. Blacque, K. Cariou, J.-E. Belgaied, H. Chao and G. Gasser, Angew. Chem., Int. Ed., 2023, 62, e202218347 CrossRef CAS PubMed .
-
(a) L. C.-C. Lee and K. K.-W. Lo, J. Am. Chem. Soc., 2022, 144, 14420–14440 CrossRef CAS PubMed ;
(b) S. Monro, K. L. Colon, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed .
- A. Parmanik, S. Das, B. Kar, A. Bose, G. R. Dwivedi and M. M. Pandey, Curr. Microbiol., 2022, 79, 388 CrossRef CAS PubMed .
- A. J. Kunz Coyne, A. El Ghali, D. Holger, N. Rebold and M. J. Rybak, Infect. Dis. Ther., 2022, 11, 661–682 CrossRef PubMed .
-
(a) N. Soliman, V. Sol, T.-S. Ouk, C. M. Thomas and G. Gasser, Pharmaceutics, 2020, 12, 961 CrossRef CAS PubMed ;
(b) S. P. Songca and Y. Adjei, Int. J. Mol. Sci., 2022, 23, 3209 CrossRef CAS PubMed ;
(c) Y. Liu, R. Qin, S. A. J. Zaat, E. Breukink and M. Heger, J. Clin. Transl. Res., 2015, 1, 140 Search PubMed .
-
(a) J. Ghorbani, D. Rahban, S. Aghamiri, A. Teymouri and A. Bahador, Laser Ther., 2018, 27, 293–302 CrossRef PubMed ;
(b) X. Y. Ng, K. W. Fong, L. V. Kiew, P. Y. Chung, Y. K. Liew, N. Delsuc, M. Zulkefeli and M. L. Low, J. Inorg. Biochem., 2024, 250, 112425 CrossRef CAS PubMed .
-
(a) M. Kolarikova, B. Hosikova, H. Dilenko, K. Barton-Tomankova, L. Valkova, R. Bajgar, L. Malina and H. Kolarova, Med. Res. Rev., 2023, 43, 717 CrossRef CAS PubMed ;
(b) R. Youf, M. Müller, A. Balasini, F. Thétiot, M. Müller, A. Hascoët, U. Jonas, H. Schönherr, G. Lemercier, T. Montier and T. Le Gall, Pharmaceutics, 2021, 13, 1995 CrossRef CAS PubMed ;
(c) Y. Jao, S.-J. Ding and C.-C. Chen, J. Dent. Sci., 2023, 18, 1453–1466 CrossRef PubMed .
-
(a) J. H. Correia, J. A. Rodrigues, S. Pimenta, T. Dong and Z. Yang, Pharmaceutics, 2021, 13, 1332 CrossRef CAS PubMed ;
(b) C. P. Sabino, M. S. Ribeiro, M. Wainwright, C. dos Anjos, F. P. Sellera, M. Dropa, N. B. Nunes, G. T. P. Brancini, G. U. L. Braga, V. E. Arana-Chavez, R. O. Freitas, N. Lincopan and M. S. Baptista, J. Photochem. Photobiol., 2023, 99, 742–750 CrossRef CAS PubMed .
- S. A. McFarland, A. Mandel, R. Dumoulin-White and G. Gasser, Curr. Opin. Chem. Biol., 2020, 56, 23–27 CrossRef CAS PubMed .
- T. Zhong, J. Yu, Y. Pan, N. Zhang, Y. Qi and Y. Huang, Adv. Healthcare Mater., 2023, 12, 2300253 CrossRef CAS PubMed .
- X. Lin, F. Chen, X. Yu, H. Wang, H. Qiu, Y. Li, S. Yin and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203994119 CrossRef CAS PubMed .
- C. Shi, H. Huang, X. Zhou, Z. Zhang, H. Ma, Q. Yao, K. Shao, W. Sun, J. Du, J. Fan, B. Liu, L. Wang and X. Peng, ACS Appl. Mater. Interfaces, 2021, 13, 45259–45268 CrossRef CAS PubMed .
- W. Abuduwaili, X. Wang, A.-T. Huang, J.-L. Sun, R.-C. Xu, G.-C. Zhang, Z.-Y. Liu, F. Wang, C.-F. Zhu, T.-T. Liu, L. Dong, J.-M. Zhu, S.-Q. Weng, Y. Li and X.-Z. Shen, ACS Appl. Mater. Interfaces, 2022, 14, 37356–37368 CrossRef CAS PubMed .
- Y. Feng, C. C. Tonon, S. Ashraf and T. Hasan, Adv. Drug Delivery Rev., 2021, 177, 113941 CrossRef CAS PubMed .
-
(a) M. Pröhl, U. Schubert, W. Weigand and M. Gottschaldt, Coord. Chem. Rev., 2016, 307, 32–41 CrossRef ;
(b) S. Banerjee and A. R. Chakravarty, Acc. Chem. Res., 2015, 48, 2075–2083 CrossRef CAS PubMed .
- L. Gourdon, K. Cariou and G. Gasser, Chem. Soc. Rev., 2022, 51, 1167–1195 RSC .
- T. Sarkar, S. Sahoo, S. Neekhra, M. Paul, S. Biswas, B. N. Babu, R. Srivastava and A. Hussain, Eur. J. Med. Chem., 2023, 261, 115816 CrossRef CAS PubMed .
- T. Sarkar, A. Bhattacharyya, S. Banerjee and A. Hussain, Chem. Commun., 2020, 56, 7981–7984 RSC .
- J. Dutta, A. Bera, A. Upadhyay, A. K. Yadav, S. Banerjee, T. Sarkar and A. Hussain, ChemBioChem, 2024, 25, e202400484 Search PubMed .
- T. Sarkar, A. Kumar, S. Sahoo and A. Hussain, Inorg. Chem., 2021, 60, 6649–6662 CrossRef CAS PubMed .
- T. Sarkar, S. Banerjee and A. Hussain, RSC Adv., 2015, 5, 16641–16653 RSC .
-
(a) A. Garai, I. Pant, S. Banerjee, B. Banik, P. Kondaiah and A. R. Chakravarty, Inorg. Chem., 2016, 55, 6027–6035 CrossRef CAS PubMed ;
(b) A. Jana, P. Kundu, S. Paul, P. Kondaiah and A. R. Chakravarty, Inorg. Chem., 2022, 61, 6837–6851 CrossRef CAS PubMed .
- A. K. Renfrew, N. S. Bryce and T. C. Hambley, Chem. – Eur. J., 2015, 21, 15224–15234 CrossRef CAS PubMed .
-
(a) D. Das, A. Banaspati, N. Das, B. Bora, M. K. Raza and T. K. Goswami, Dalton Trans., 2019, 48, 12933–12942 RSC ;
(b) D. Das, A. Noor, M. K. Raza and T. K. Goswami, J. Biol. Inorg. Chem., 2021, 26, 881–893 CrossRef CAS PubMed .
- A. Jana, S. Sahoo, S. Paul, S. Sahoo, C. Jayabaskaran and A. R. Chakravarty, Inorg. Chem., 2024, 63, 6822–6835 CrossRef CAS PubMed .
- C. N. Sudhamani, H. S. Bhojya Naik, K. R. Sangeetha Gowda, M. Giridhar, D. Girija and P. N. Prashanth Kumar, Med. Chem. Res., 2017, 26, 1160–1169 CrossRef CAS .
- C. N. Sudhamani, H. S. Bhojya Naik, K. R. Sangeetha Gowda, D. Girija and M. Giridhar, Nucleosides, Nucleotides Nucleic Acids, 2018, 37, 546–562 CrossRef CAS PubMed .
- C. N. Sudhamani, H. S. Bhojya Naik, K. R. Sangeetha Gowda, M. Giridhar, D. Girija and P. N. Prashanth Kumar, Spectrochim. Acta, Part A, 2015, 138, 780–788 CrossRef CAS PubMed .
- A. Mandal, R. Rai, S. Saha, R. Kushwaha, L. Wei, H. Gogoi, A. Mandal, A. Yadav, H. Huang, A. Dutta, P. Dhar and S. Banerjee, Dalton Trans., 2023, 52, 17562–17572 RSC .
- R. Kushwaha, R. Rai, V. Gawande, V. Singh, A. K. Yadav, B. Koch, P. Dhar and S. Banerjee, ChemBioChem, 2024, 25, e202300652 CrossRef CAS PubMed .
- C. Gourlot, A. Gosset, E. Glattard, C. Aisenbrey, S. Rangasamy, M. Rabineau, T.-S. Ouk, V. Sol, P. Lavalle, C. Gourlaouen, B. Ventura, B. Bechinger and V. Heitz, ACS Infect. Dis., 2022, 8, 1509–1152 CrossRef CAS PubMed .
- M. Wegener, M. J. Hansen, A. J. M. Driessen, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2017, 139, 17979–17986 CrossRef CAS PubMed .
- M. Piksa, C. Lian, I. C. Samuel, K. J. Pawlik, D. W. Samuel and K. Matczyszyn, Chem. Soc. Rev., 2023, 52, 1697–1722 RSC .
- B. A. Albani, B. Peña, N. A. Leed, N. A. B. G. de Paula, C. Pavani, M. S. Baptista, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2014, 136, 17095–17101 CrossRef CAS PubMed .
- A. M. Angeles-Boza, P. M. Bradley, P. K.-L. Fu, S. E. Wicke, J. Bacsa, K. R. Dunbar and C. Turro, Inorg. Chem., 2004, 43, 8510–8519 CrossRef CAS PubMed .
- A. Mansoori, A. Mohammadi, M. D. Amin, F. Mohammadnejad, F. Kamari, M. F. Gjerstorff, B. Baradaran and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2019, 26, 395–404 CrossRef PubMed .
- E. Polat and K. Kang, Biomedicines, 2021, 9, 584 CrossRef CAS PubMed .
- Y. J. Jeon, J. Y. Jang, J. H. Shim, P. K. Myung and J. I. Chae, Cancer Prev. Res., 2015, 20, 106–112 CrossRef PubMed .
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS .
- K. I. Pashanova, I. V. Ershova, O. Y. Trofimova, R. V. Rumyantsev, G. K. Fukin, A. S. Bogomyakov, M. V. Arsenyev and A. V. Piskunov, Molecules, 2022, 27, 8175 CrossRef CAS PubMed .
- R. E. Kitson, Anal. Chem., 1950, 22, 664–667 CrossRef CAS .
- M. Paul, H. Bhatt, S. Kumbham, B. Ghosh and S. Biswas, ACS Appl. Nano Mater., 2023, 6, 13385–13399 CrossRef .
- Y. Yuan, C.-J. Zhang, S. Xu and B. Liu, Chem. Sci., 2016, 51, 1862–1866 RSC .
- G. El Tekle and W. S. Garrett, Nat. Rev. Cancer, 2023, 23, 600–618 CrossRef CAS PubMed .
- S. Eyvazi, M. Asghari Vostakolaei, A. Dilmaghani, O. Borumandi, M. S. Hejazi, H. Kahroba and V. Tarhriz, Microb. Pathog., 2020, 141, 104019 CrossRef CAS PubMed .
- D. Ron and P. Walter, Nat. Rev. Mol. Cell Biol., 2007, 8, 519–529 CrossRef CAS PubMed .
- J. M. Soares, F. E. G. Guimarães, V. V. Yakovlev, V. S. Bagnato and K. C. Blanco, Sci. Rep., 2022, 12, 1 CrossRef PubMed .
- T. G. Deligeorgiev, S. Kaloyanova and J. J. Vaquero, Recent Pat. Mater. Sci., 2009, 2, 1–26 CrossRef CAS .
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, methods, additional experimental details, schemes, and figures. See DOI: https://doi.org/10.1039/d4dt03153a |
| ‡ These authors contributed equally to the work. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b and
Akhtar
Hussain
*b and
Akhtar
Hussain