
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in heavier group 15 (P, As, Sb, Bi) radical chemistry – frameworks, small molecule reactivity, and catalysis
Deana L. G. Symes
 and
Jason D. Masuda
*
and
Jason D. Masuda
*
Department of Chemistry, Saint Mary's University, Halifax, Nova Scotia B3H 3C3, Canada. E-mail: jason.masuda@smu.ca
First published on 25th February 2025
Abstract
Main group radical chemistry has been a targeted research area for several decades. With growing examples of phosphorus radicals, even heavier pnictogen radicals including arsenic, antimony, and bismuth have also become important targets. A diverse framework of group 15 radicals has been reported in the 21st century and is covered herein. Reactivity and applications of selected radicals and future directions for this field are discussed.
 Deana L. G. Symes | Deana Symes completed her BSc with an Honours in Chemistry (2024) from Saint Mary's University with a thesis on substituted cyclopentadienyl main group complexes. Since 2024 she is pursuing an MASc at the same institution under the continued supervision of Dr Jason Masuda as an NSERC CGS-M recipient. Her research focuses on the generation and stabilization of group 15 and group 16 element-centered radicals. |
 Jason D. Masuda | Jason Masuda completed his BSc (2000) and MSc (2002) in Chemistry at the University of Lethbridge. After receiving his PhD in 2005 from the University of Windsor, Jason was an NSERC Postdoctoral Fellow at the University of California, Riverside and a Director's Postdoctoral Fellow at Los Alamos National Lab. He started his independent career in 2008 at Saint Mary's University and is currently a Full Professor and an Associate Dean of Science. His research is focused on ligand development/coordination chemistry, the stabilization of new main group atom combinations, and trapping highly reactive small molecules using carbenes and related molecules. |
Introduction
Radical-based molecules are popular in modern chemistry but they are often unstable and short-lived due to their high reactivity. To generate and isolate main-group radicals while preventing dimerization or polymerization, these systems generally require delocalization of the unpaired electron through a π-system and the introduction of sterically hindering ligands.1,2 The first organic radical to be isolated, Ph3C˙, was reported in 19003 and is stable in the solution as a free radical, but exists in the solid state as a dimer.4 The persistence of this radical is suggested to arise from a combination of resonance contributions and steric protection from the surrounding phenyl rings.5 Since this discovery, various radical categories have been developed including categorization by the number of unpaired electrons, electronic charge, and relative stabilities.1A specific class of main group radicals that have gained popularity are phosphorus-centered radicals. These can be further divided into three general categories of neutral phosphorus radicals including phosphinyl (R2P˙), phosphonyl (R2PO˙), and phosphoranyl (R4P˙) radicals,5,6 however, other forms such as phosphoniumyl radical cations (R3P˙+),7,8 carbene stabilized phosphorus radical ions (NHC) → P˙+(R),2,9 and phosphidyl radical anions (R3P˙−)9,10 are examples of charged phosphorus radical species that have been investigated. Nonetheless, these radicals and radical ions are arranged into three classes based on their kinetic and thermodynamic stability: stable, persistent, and fleeting or transient.1,5,9,11 Stable radicals are long-lived, and can be isolated and stored in an inert atmosphere for extended periods without decomposing. These species are sufficiently stable to be analyzed by single crystal X-ray diffraction.1,9,12 Persistent radicals have a shorter relative lifetime and can be characterized by standard spectroscopic methods such as electron paramagnetic resonance (EPR) or UV-vis, but cannot be isolated under standard conditions.1,9,12 In the case of fleeting or transient radicals, these species are very short-lived due to their high reactivity and instability, resulting in unselective products, dimerization, and difficulty with characterization.1,9
More recently, the development of heavy group 15-based radicals including arsenic, antimony, and bismuth has begun to emerge. Examples of the heavy group 15 radical analogues are much more limited, however, there has been increased interest as of late.13–15 The trend of increasing difficulty in radical stabilization moving down the group 15 elements can be rationalized with a few fundamental chemistry concepts. From an atomic orbital perspective, as the principle quantum number increases, the valence atomic orbitals have a larger radial extension and are more diffuse.16 Additionally, with larger valence atomic orbitals, the energy of the highest occupied molecular orbital (HOMO) generally increases while simultaneously decreasing the energy difference between the singly occupied molecular orbital (SOMO) and the HOMO from P → Bi, making the heavier radical species easier to generate (e.g. milder reaction conditions or reagents). However, the heavier pnictogen-centered radical species are more reactive and therefore increasingly challenging to isolate.17 This effect is presented throughout the literature, where a significant number of phosphorus radicals have been reported, along with many arsenic analogues,13 however, there are a limited number of persistent or isolable antimony radicals and an even shorter list of bismuth radicals.13,18
The physical and chemical properties of pnictogens have a reported secondary periodicity, where an irregular change is observed throughout the group.16,19,20 A “sawtooth” shaped pattern is observed in various electronic structure traits of pnictogens, generally arising from the large relativistic effects present in heavier atoms, and the scandide (d-block) and lanthanide (f-block) contractions of the valence shells of antimony and bismuth, respectively.20 As previously mentioned, the valence atomic orbitals increase going down the group; however, it has a varying impact on the electronic structure, including size, pnictogen bonding, and the valence orbital ionization energy.16 This sawtooth trend is clear when looking at the van der Waals radii of the group 15 elements, where phosphorus and arsenic radii are clustered (1.80 Å and 1.85 Å, respectively), followed by a larger jump in size to the clustered antimony and bismuth radii (2.05 Å and 2.07 Å, respectively).16 The trend in atomic size throughout group 15 further promotes a sawtooth-like pattern with the increase in Pn–C bond lengths when comparing within the PnPh3 series,16 where the P–C (1.93 Å)21 and As–C (1.96 Å)22 bond lengths demonstrate a small increase but are noticeably shorter than the Sb–C (2.15 Å)23 and Bi–C (2.25 Å)24 bond lengths. When looking at the single electron ionization energies of the pnictogen atomic orbitals, a uniform increase of the valence p orbital energy is observed going down the group, contrarily to the s orbital ionization energy, which displays another sawtooth-like trend.16 The energy required to remove one electron from the As 4s orbital is marginally increased compared to removing an electron from the P 3s orbital. For the heavier pnictogens Sb and Bi, a significant increase in the orbital ionization energy of the Sb 5s orbital to the bismuth 6s orbital is observed due to the significant relativistic stabilization of the latter.16
From the considerably impactful relativistic effects experienced by heavier elements, bismuth compounds have distinctive properties compared to their lighter pnictogen congeners (N, P, As, Sb), such as the inert pair effect observed for the 6s2 electron pair which remains formally unoxidized, however, can be stereochemically active.14,24–28 Relativistic effects are responsible for or influence certain properties of heavy elements such as their colour, the geometries of heavy element-containing molecules, and their reactivity behaviour.24 When looking at the s and p orbitals going down the group 15 elements, there is a significant decrease in the spatial overlap of the 6s and 6p orbitals due to their increasing size difference, resulting in less s/p mixing to give essentially non-hybridized orbitals for bismuth.14,16 With the large and diffuse atomic orbitals of bismuth, this leads to inefficient overlap with the orbitals of other bound atoms, resulting in weaker and longer Bi–E bonds.14,16,29 This effect has been observed with the Pn–H bond dissociation enthalpies for the PnH3 series (Pn = P: 81.4; As: 74.6; Sb: 63.3; Bi: 51.8 kcal mol−1),30,31 where the valence s-character of the heavier elements going down the group tends to accumulate in a non-bonding lone pair type orbital. This leads to the Pn–H bonds being made essentially by unhybridized p-orbitals.14,16,32
In the 21st century, there are a few reviews on the recent developments of pnictogen-centered radicals.1,5,9,10,13,18,33 Additional reviews in the literature describe specific areas of group 15 radical chemistry including the use of carbene ligands for effective stabilization,2,34 synthesis and reactivity of biradicals,35 as well as the applications of EPR spectroscopy36 and frustrated radical pairs.37,38 group 15 radical species possess the potential for stoichiometric and catalytic transformations including small molecule activation and conversion, as well as thermal and photochemical coupling reactions, which are areas that transition metals have otherwise dominated. Herein, we aim to highlight recent developments within group 15-based radicals and the direction we envision as the next potential steps moving forward in new and related research. Our goal is to contextualize the significance of these developments and discuss the challenges still faced in the area of group 15-based radicals while putting this area in a new perspective.
Phosphorus-centered radicals
Phosphinyl radicals
Phosphinyl radicals are neutral two-coordinate radicals with an unpaired electron and a lone pair, commonly generated by homolytic cleavage of a P–P single bond.9,39–41 The homolytic bond dissociation of P–P bonds is generally a non-spontaneous process, with the parent diphosphine, H2P–PH2 having a bond dissociation energy (BDE) of 61.2 kcal mol−1.42,43 Although this energy barrier is much lower than its carbon analogue, it remains sufficiently high, preventing the homolytic cleavage of the P–P bond.42,44 This facile dimerization can create challenges in the utility of these phosphorus radicals, necessitating the development of an approach to stabilize these species. By tuning the steric and electronic properties of the substituents, homolytic cleavage of the P–P bond is encouraged in solution through steric crowding of the radical center, which elongates and weakens the central P–P bond. Additionally, dimerization can be minimized through the resonance stabilization of the unpaired electron by delocalization through a conjugated system.42,45–47Other factors that determine the dissociation equilibrium are the system's dispersion forces and entropic effects. When dispersion forces are neglected, it is calculated that many diphosphanes would be energetically more favourable as their respective monomeric radical species in the solid state rather than their dimeric forms.48 Since this is not the case for experimentally observed results,17,39–41,45,47,49–52 it reinforces the significance of dispersion force effects in the theoretical calculations of sterically crowded molecules.48 Increased steric bulk of the substituents gives rise to an increase in strain energies and steric repulsion between the substituents, thus the attractive dispersion forces prevail over the susceptibility of the dimer to dissociate.48
Methods for generating diphosphines that homolytically cleave to give phosphinyl radicals, or the direct formation of phosphinyl radicals typically involve the reduction of R2P-X (X = halide). Generally this is done with common reducing reagents such as magnesium metal,51,53 potassium on graphite,1,54,55 or metallocences such as [Cp2TiCl]2 and [Cp2Ti(btmsa)] (btmsa = bis(trimethylsilyl)acetylene).53 In other approaches, one electron oxidation of an amino phosphalkene56 or a cationic phosphanide57 and reduction of a fluorenyl-based phosphalkenes with potassium or lithium metal58 are a few additional redox approaches for generating phosphinyl radicals. Other phosphinyl radical generating processes include photochemical dehydrocoupling of N-heterocyclic phosphanes to form the related diphosphines that can homolytically cleave to form the radical59–61 as well as thermochemical generation in the presence of radical initiators.62
Two popular frameworks for phosphinyl radicals are the dialkyl-substituted and the bis-amido-phosphorus derivatives. The first report on persistent phosphinyl radicals was published in 1976 by Lappert et al.,49 where a radical for each framework was generated (1a–b), containing bis(trimethylsilyl) groups on the atoms adjacent to the phosphorus center (Scheme 1).49 The substantial steric strain of these bulky substituents accumulates significant potential energy, which induces the parent diphosphines to dissociate into the respective phosphinyl radicals when released from the solid state.47–50,63 This specific design is known in the literature as a molecular “jack-in-the-box” as the substituents behave similarly to stored energy in a compressed spring.47,48,63
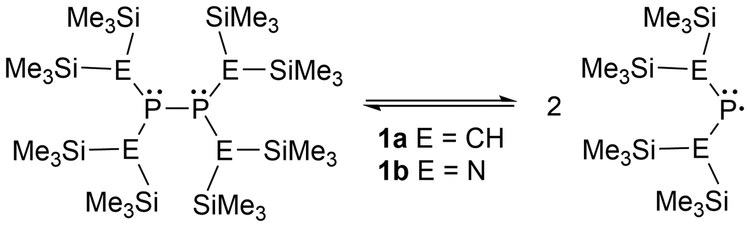 | ||
| Scheme 1 Equilibrium of the diphosphine and its respective phosphinyl radical in solution. | ||
Furthermore, the substituents adjacent to the phosphorus radical center can easily be modified to explore an array of steric environments. Many examples have been described in the literature including diphosphines incapable of homolytic cleavage (2),64 persistent asymmetric acyclic (3–7g),50,65–67 and symmetric acyclic (7h)66 or cyclic (8–11)17,40,41,45,51,52,55 radicals in the solution state (Fig. 1). Although 2 contains a bulkier backbone than 9–11, this species does not dissociate into the monomeric radical form as the isopropyl substituents have minimal steric repulsion.64 The examples 3–10 are dimeric in the solid state, however, in solution, they exist in an equilibrium between the diphosphine and the phosphinyl monomer, which gives radical character when in solution. Compound 11 was the first example of a phosphinyl monomer in the solid state.55 Compounds 8a–c contain a unique phosphinyl radical framework where a ferrocene fragment is incorporated into the structure by linking it to the terminal heteroatoms (P or N) in the P–P–P and N–P–N systems, providing a rigid cyclic structure beneficial for selective stereochemistry.52
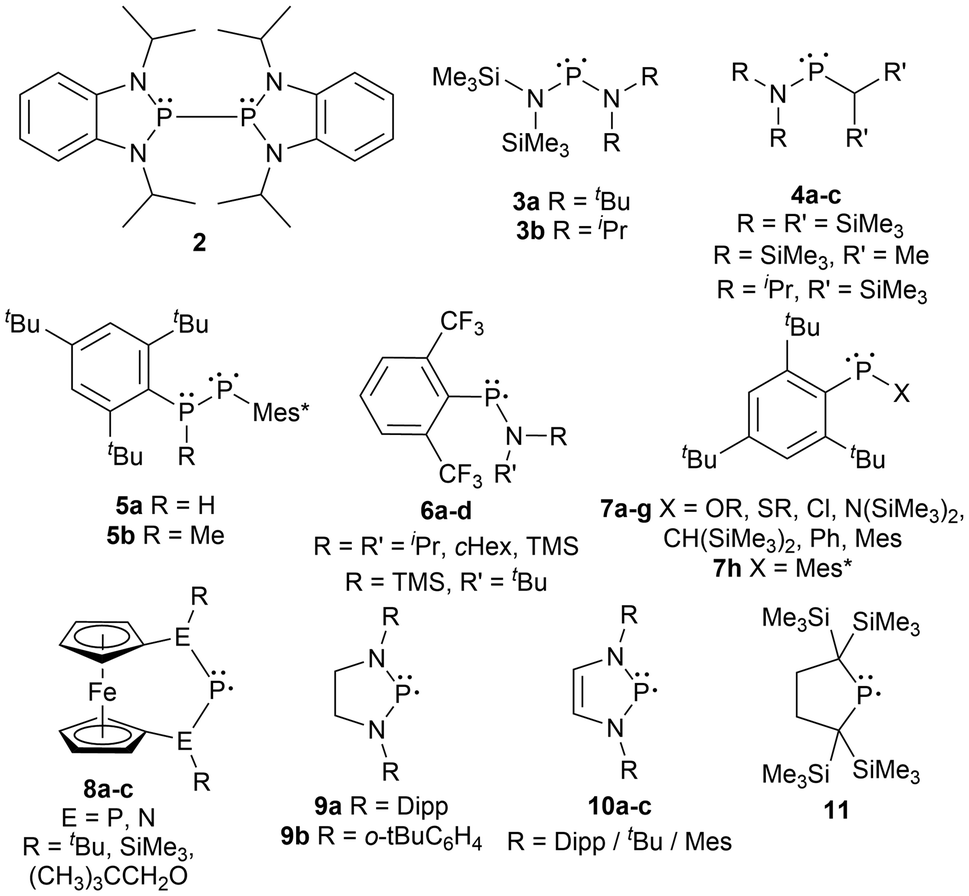 | ||
| Fig. 1 A diphospine (2) and phosphinyl radicals (3–11). | ||
Phosphorus-centered radical ions
In the literature, there are both phosphorus radical cations and phosphorus radical anions, with the former being more prevalent (Fig. 2).1,33 Although this observation has been addressed in the literature,1,33 the underlying reasons were left to the reader's interpretation. When comparing a phosphorus radical cation to a phosphorus radical anion, the former has an overall positive charge, making the unpaired electron less susceptible to reactivity as it is held more tightly by the phosphorus radical center. Additionally, most phosphorus radical ions are generated from P(III) species, where the resulting radical ion has two or three substituents: in many cases, carbon-based groups. These substituents inductively donate electron density to the radical center, stabilizing cationic species but de-stabilizing anionic species, making them more challenging to isolate.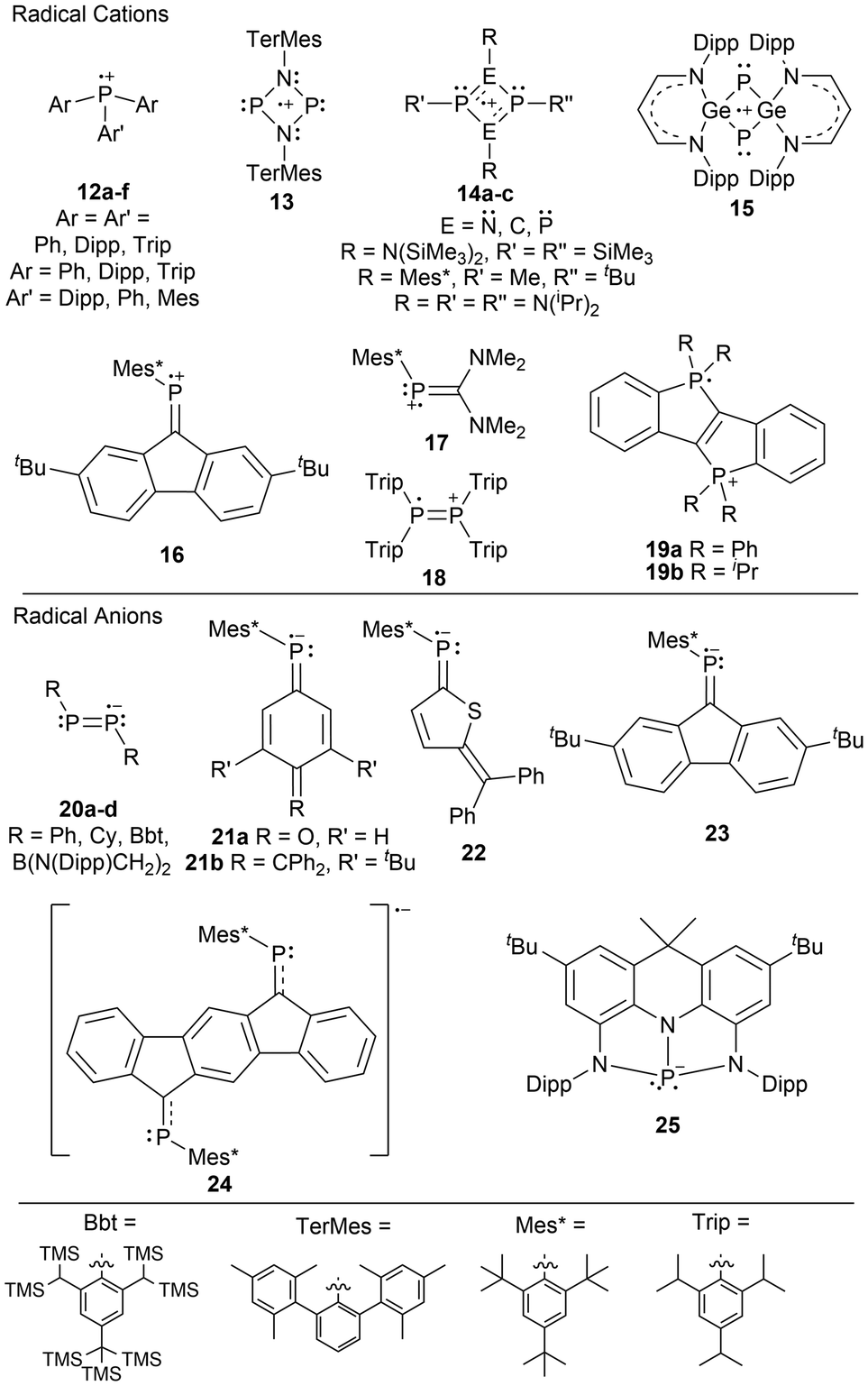 | ||
| Fig. 2 Phosphorus radical cations and radical anions. | ||
The generation of stable phosphorus radical cations continues to develop by incorporating sterically bulky substituents allowing for the delocalization of the positive charge across a larger area.10,33 These radical species are typically generated through chemical oxidation with silver, trityl, or ferrocenium salts.9 Stable phosphorus radical anions are typically generated by reducing phosphorus-containing unsaturated bonds, such as those found in phosphalkenes10,33 or diphosphenes,68,69 with strong reducing agents such as alkali metals.
Phosphorus radical cation examples in the literature generally consist of tricoordinate aryl-substituted phosphorus radical centers (12),7,8,70 or 4-membered ring systems of dimeric phosphorus species; N2P2 (13, 14a),71,72 C2P2 (14b)73 P2P2 (14c),71 and Ge2P2 (15).74 Other examples of phosphorus radical cations are those containing phosphaalkenes (16, 17),75 a tetraaryldiphosphine (18),76 and diphosphadibenzo[a,e]pentalenes (19a–b) (Fig. 2).77 For phosphorus radical anions, the examples are more limited, however, they contain dicoordinate aryl-substituted diphosphenes (20a–c),68,69 boryl-substituted diphosphenes (20d),78 phosphaalkenes (21–24),79–83 and the rigid chelating diamidodihydroacridinide ligand (25), where the radical precursor contains a vacant p-orbital suitable as an acceptor orbital for one electron reduction reactions (Fig. 2).84 Many of these radical ionic species have been previously discussed in detail in recent review-type articles.9,10
Carbene stabilized phosphorus radicals
Innovative designs for stabilizing radicals are often used and over the last two decades, singlet carbenes have proven their utility in this manner.82,85–92 The use of N-heterocyclic carbenes (NHCs) and cyclic(alkyl)(amino) carbenes (CAACs) has allowed for the generation, and occasionally the isolation of radical-containing species, which otherwise could not be detected spectroscopically.2 Carbenes effectively stabilize inherently highly energetic radical centers due to their tunable, typically bulky substituents, and a low-lying empty carbon p-orbital available to accept and delocalize electron density of the unpaired electron.85,93–95 A minireview of carbene stabilized main group radicals and radical ions was published2 in 2013 and includes pnictogen-centered derivatives, however, additional carbene-stabilized pnictogen-based radicals have since been reported.87,88,90,90–92There are a few variations of phosphorus-centered radicals and radical ions stabilized by carbenes including single phosphorus atoms (26, 27),56,57 phosphorus-phosphorus systems (28a–c),87,89 nitrogen–phosphorus systems (29),96 and systems containing multiple nitrogen and or multiple phosphorus atoms N–P–N (30a),97 P–P–P (30b),98 P–N–P (31),91 and P–N–P–N (32a–b)92 (Fig. 3). In most cases, the species generated is a radical cation56,87,89,92,96 or a radical dication,57 except for the neutral tripnictogen systems,91,97,98 or in a more recent development that includes a neutral diphosphene radical stabilized by a carbene and a Dipp group (33).90 Other radicals incorporating a carbene functionality include a diphosphene stabilized by an N-heterocyclic vinyl (NHV) scaffold (34),88 dicarbondiphosphide-based radical cations stabilized by NHCs (35, 36),99,100 and an α-radical phosphine species (37)101 generated by reducing a bicyclic(alkyl)(amino)carbene (BICAAC) stabilized phosphenium cation (Fig. 3).
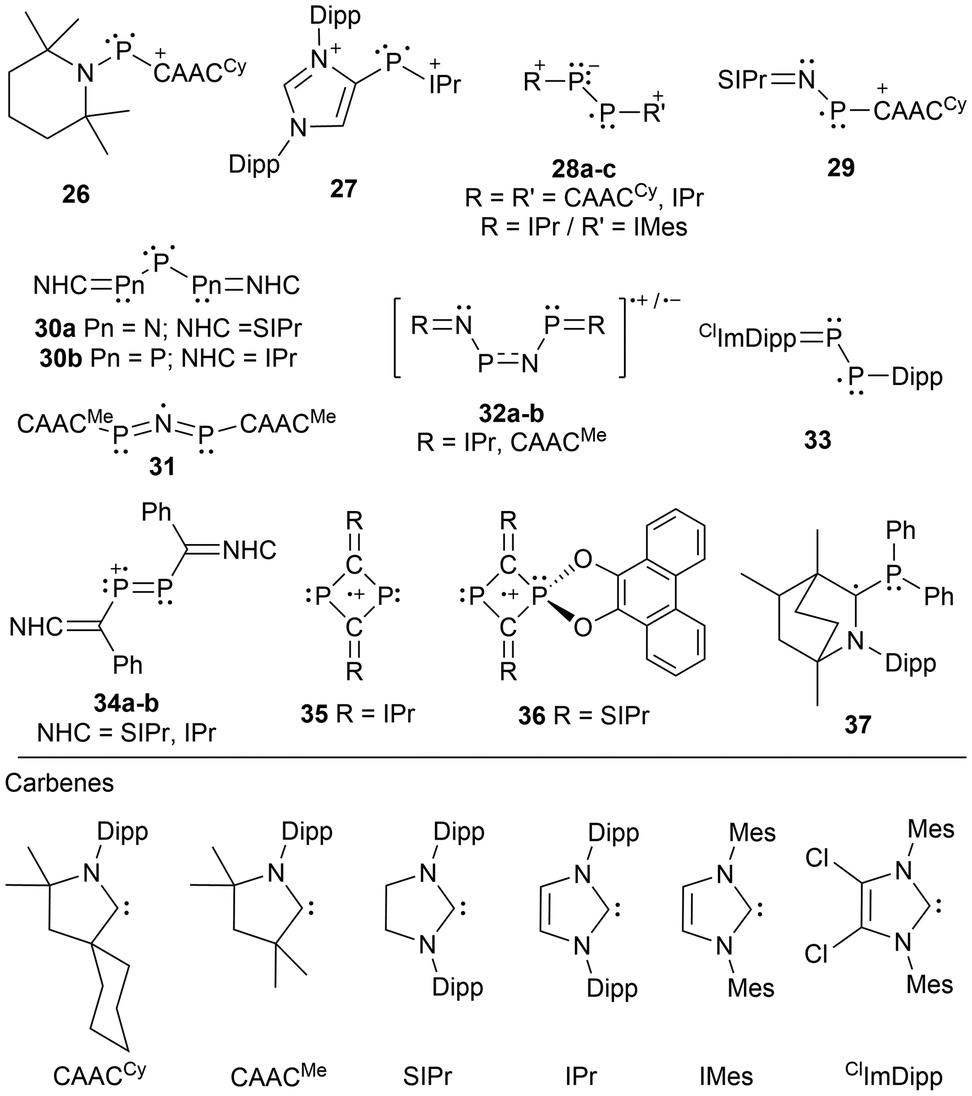 | ||
| Fig. 3 Carbene stabilized phosphorus and phosphorus–nitrogen based radicals. | ||
Fig. 3 shows that these radical systems can also be categorized by whether they contain two identical or different NHCs, two CAACs, one NHC and one CAAC, or a single CAAC. When comparing five-membered NHCs with saturated or unsaturated backbones, the former is typically calculated to be the stronger σ-donor and stronger π-acceptor. However, when considering CAACs the σ-donor and π-acceptor properties are further strengthened, making CAACs a better choice for radical stabilization.102 This difference in donor and acceptor abilities can be recognized as a consequence of the energy difference between the highest occupied molecular orbital (HOMO) and the lowest occupied molecular orbital (LUMO), where the HOMO-LUMO gap of an NHC is typically larger compared to those of CAACs.102,103
Heavy group 15 based radicals
Arsenic-centered radicals
The first arsenic paramagnetic species to be structurally characterized in the solid state was an arsenic radical cation stabilized by two NHCs in 2013 (38).104 Only a few years after this initial discovery, a minireview on long-lived heavier Group 15 radicals was published13 in 2020, listing a variety of arsenic-centered radicals. Similar to the frameworks listed for phosphorus-centered radicals, arsenic derivatives of dicoordinate bis-trimethyl silyl substituted acyclic49,50 and cyclic17 neutral radicals (39, 40), tricoordinate aryl-substituted radical cations (41),105 tricoordinate tris-amide substituted radical anions (42),84 tripnictogen-centered radicals (P–As–P) stabilized by two NHCs (43),98 radical dications stabilized by two CAACs (44),106 and dicoordinate organoarsenic radical anions (45, 46a)107 and biradical dianions (46b, 47)107 have been reviewed (Fig. 4).13 | ||
| Fig. 4 Arsenic-centered radical analogues of phosphorus-based systems. | ||
Newer radical systems have also been reported for arsenic radical derivatives where some have been previously discussed in reviews.108 Phosphaarsene (P–As) radical cations (48)109 and radical anions (49)110 stabilized by carbenes and aryl groups respectively have been generated, with the former being isolated. There have also been reports of dicoordinate radical cations stabilized by a single CAAC, where the other coordinating group is an NHV scaffold (50),111 and radical cations within 4-membered ring systems of arsenic dimers, As2N2 (51a),72 and mixed pnictogens, AsNPN (51b).72 The newer systems not covered by previous reviews include dicoordinate radical cations stabilized by a CAAC and a gallium substituent (52),112 or arsenic radical centers stabilized by two gallium substituents (53)113 (Fig. 5).
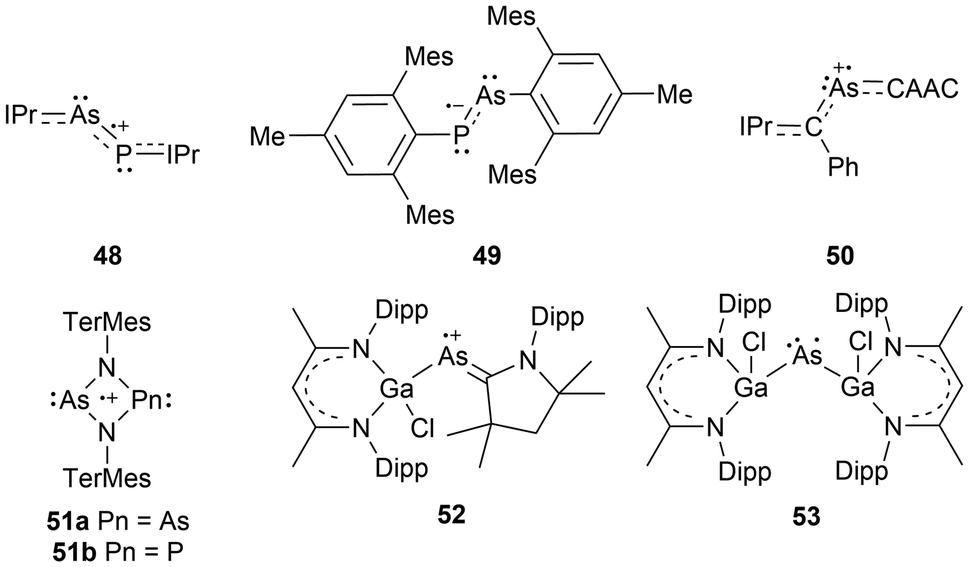 | ||
| Fig. 5 Arsenic-centered radicals generated with new frameworks. | ||
Antimony and bismuth-centered radicals
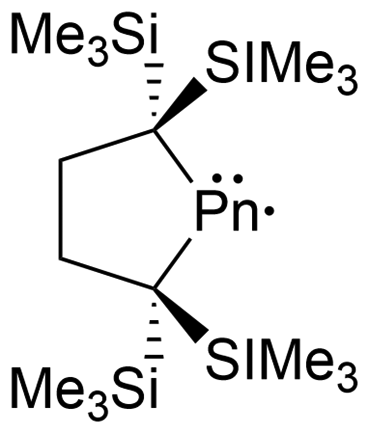
The heavy pnictogen-centered radicals of antimony and bismuth have limited examples in the literature. As mentioned in the introduction, the list of persistent or isolable bismuth radicals has significantly fewer examples than that for antimony, which may be attributed to the higher tendency for bismuth radicals to undergo decomposition and dimerization reactions.13,18 Nonetheless, these radicals can be generated under a few conditions similarly to phosphorus-centered derivatives. The homolytic cleavage of dipnictogen bonds17,114 or Pn–E bonds (E = C, O, transition metal fragment)29,115–120 in reversible equilibrium reactions114 or non-reversible conditions115 respectively is a standard strategy that employs mild reaction conditions for the generation of some antimony and bismuth radicals.18 Similarly, methods including photochemically induced homolytic bond dissociation have been investigated for these pnictogens, with no success in isolating the radical species generated, however, their implementation as radical intermediates in stoichiometric and catalytic reactions has been reported.15As anticipated, analogues to the phosphorus and arsenic radicals previously discussed have been generated for antimony and bismuth. These include dicoordinate bis-trimethylsilyl substituted neutral radicals (54a–b),114 dicoordinate radical cations stabilized by a single CAAC and a gallium-based substituent (55a–b),112 and bis-gallium-based substituted neutral radicals (56a–d)113,121,122 (Fig. 6). Unlike the phosphorus and arsenic analogues, the stibinyl and bismuthinyl radicals 54a and 54b are reported as dimers in the solid state.17 With the small covalent radii of phosphorus and arsenic relative to antimony and bismuth, the lighter pnictogens are more sensitive to steric influences, in this case, the steric repulsion of the bulky trimethylsilyl substituents.16,17 The influence of changing the pnictogen radical center from phosphorus to bismuth is displayed by the varying Pn–C bond lengths and C–Pn–C bond angles throughout this series (Table 1).
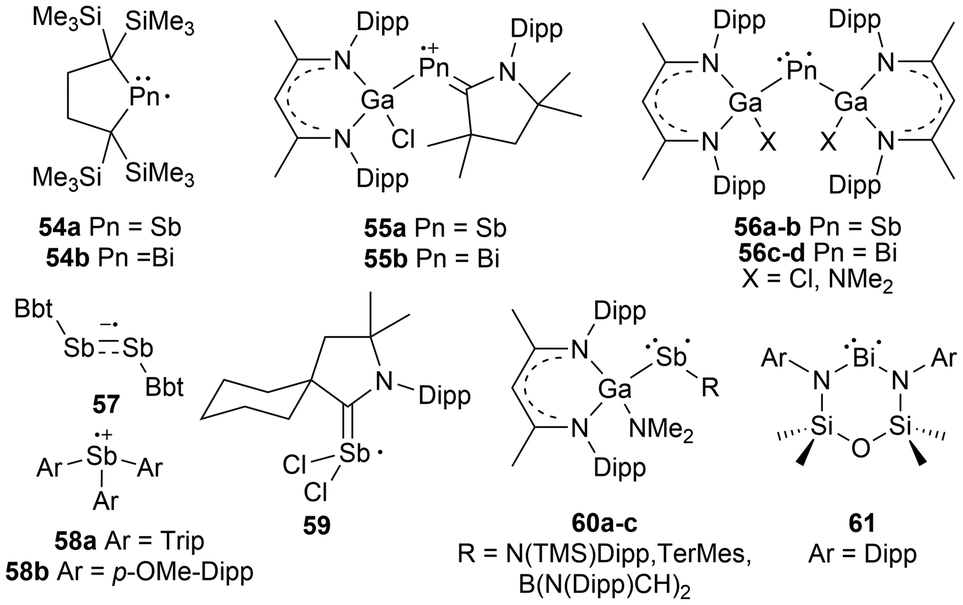 | ||
| Fig. 6 Antimony and bismuth-centered radicals described in the literature. | ||
|
||||
|---|---|---|---|---|
| Pn | P | As | Sbb | Bib |
| a Optimized at (U)M06-2X/def2-SVP level of theory.b X-ray bond lengths and angles are from the Pn–Pn dimer. | ||||
| Pn–C X-ray | 1.869(1) | 2.006(2), 2.007(2) | 2.252(2) | 2.371(3), 2.370(3) |
| C–Pn–C X-ray | 96.16(5) | 92.98(7) | 86.92(7) | 84.74(9) |
| Pn–C calca | 1.877, 1.878 | 2.005 | 2.217, 2.218 | 2.318, 2.323 |
| C–Pn–C calca | 95.120 | 91.816 | 86.557 | 84.131 |
For antimony, this list can be expanded to include a dicoordinate aryl substituted distibene radical anion (57),69,121 triarylstibine radical cations (58a–b),123 and a carbene-stabilized neutral dichlorostibanyl radical (59);124 the first antimony-centered radical to be characterized in solution. In addition to the pre-existing systems, some systems have been altered to generate unique antimony and bismuth radicals. New radical systems incorporating a gallium-based substituent and either an amine (60a), aryl group (60b), or N-heterocyclic borane (60c) have been explored for antimony.116 For unique bismuth radicals, a diamidobismuth(II) radical has been isolated and characterized (61) (Fig. 6).125
Another example of trends descending through Group 15 is the tricoordinate aryl-substituted radical cations. For the [Trip3Pn]˙+ (Pn = P, As, Sb) series, a decreasing trend in the sum of the C–Pn–C bond angles has been reported going down the group (359.99° (P),8,105 354.35° (As),105,126 348.54° (Sb)123,126). As a comparison, for the neutral and sterically comparable Dipp3Pn series the sum of the C–Pn–C bond angles also decreases from P to Bi 335.01° (P),70,127 328.83° (As),127,128 320.88° (Sb),127 318.50° (Bi).129 The phosphorus radical cation species is planar, whereas a slight pyramidalization is observed for the arsenic and antimony derivatives. The structural differences observed in this series can be rationalized by the increase in atomic radius going down the group, creating longer Pn–C bond lengths and a reduced degree of steric shielding from the bulky substituents.8,105,123
Biradicals
Biradicals are an even-electron molecular species containing two radical centers in two degenerate (or nearly degenerate) orbitals that are nearly independent of each other.35,53,130–133 In terms of biradicaloids, this refers to biradicals in which the two radical centers interact significantly.35,134,135 These two systems can be further differentiated by the spin of the two unpaired electrons. For biradicals, the spin of the two electrons is parallel, resulting in a triplet state, whereas in biradicaloids, the spin of the two electrons is antiparallel, forming an open-shell singlet state.53,130,132 Furthermore, another type of biradical includes disbiradicals, which are biradicals that display negligibly small to no coupling between the two unpaired electrons and give, in spectroscopic terms, two-doublet radical species.35,131,132,135 For simplicity, since there is no defined measurable degree of interaction, the term biradical is used synonymously for biradicaloid in this review, as similarly done in previous publications.35,135Heteroatom-based biradical systems have recently been extensively reviewed by Schulz,135 as well as a more specific Frontier article on cyclobutane-1,3-diyl and cyclopentane1,3-diyl group 15 biradical analogues by Schulz and coworkers.35 To avoid significant repetition of this work, a brief background of biradicals and some highlights of group 15 biradicals will be discussed, followed by an overview of newer developments within this field. To begin, after Gomberg reported the trityl radical as the first observed organic radical species in 19003 the report of the first biradical 62 was published by Schlenk and Brauns in 1915.136 This biradical species was generated by the reduction of two C–Cl bonds of 1,3-(diphenylchloro)phenyl with copper bronze (CuSn) (Scheme 2).136 Studies of heteroatom-containing biradicals were initiated by the study of the four-membered sulfur-nitrogen heterocycle S2N2 (63)137 and later were reinvigorated by the Niecke-type biradicals (64) (Scheme 2).138,139
 | ||
| Scheme 2 Pioneering research for main group biradicals. | ||
In-depth studies of the P2C2 heterocyclic system, 1,3-diphospha-cyclobutane-2,4-diyl, have been done by the Niecke group138 as reviewed by Schoeller139 which facilitated the experimental and theoretical investigations of cyclobutane1,3-diyl analogues of the type [E(μNTerMes)E′]2 (E = E′ = P, As, Sb, Bi; E = P, E′ = As)130,140–142 (65a–e) (Scheme 3). For the biradicals 65a–e, the phosphorus and arsenic derivatives have been reported in good yields, however, the heavy analogues, antimony and bismuth, have only been observed in situ due to their higher relative reactivity.135 Based on theoretical calculations, when going down group 15 (P → Bi) the biradical character significantly increases in the [E(μNTerMes)E′]2 (E = E′ = P, As, Sb, Bi) series as observed in the elongation of the trans-annular E–E′ distances in a sawtooth-like pattern (E = E′ = P: 2.669 Å; As: 2.896 Å; Sb: 3.250 Å, Bi: 3.386 Å).130,140,141
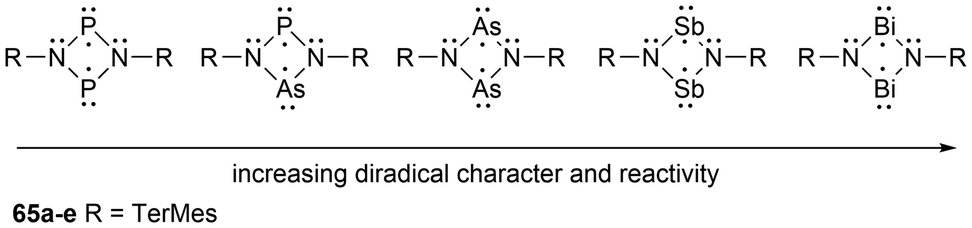 | ||
| Scheme 3 Altering properties in four-membered biradical ring systems by changing the pnictogen atom(s). | ||
For newer developments in this field, improvements in the synthesis of four-membered cyclic biradical systems (66) have been recently reported from the reduction of acyclic materials134 alternatively to the classic reduction of the halogenated heterocyclic precursors (Scheme 4).35 Additionally, as theorized in the review by Hinz et al.135 the first phosphorus-centered dis-biradical, linked by a hexyl ligand was synthesized (67).132 Recently as well, Schulz et al. reported a zirconocene-bridged phosphorus-centered bis-biradical (68), the first bis-biradical-based molecular switch attached to a transition metal fragment (Fig. 7).143 Additionally, in 2023, monocoordinated antimony and bismuth complexes in a triplet ground state were isolated incorporating sterically demanding hydrindacene-based ligands to obtain a stibinidene (69)144 and bismuthinidene (70).145 Further discussion of these species or analogous derivatives can be found in a later section; light-induced reactivity.
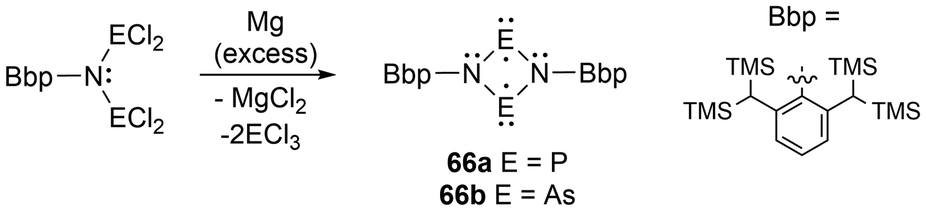 | ||
| Scheme 4 Reduction acyclic materials to generate four-membered cyclic biradicals. | ||
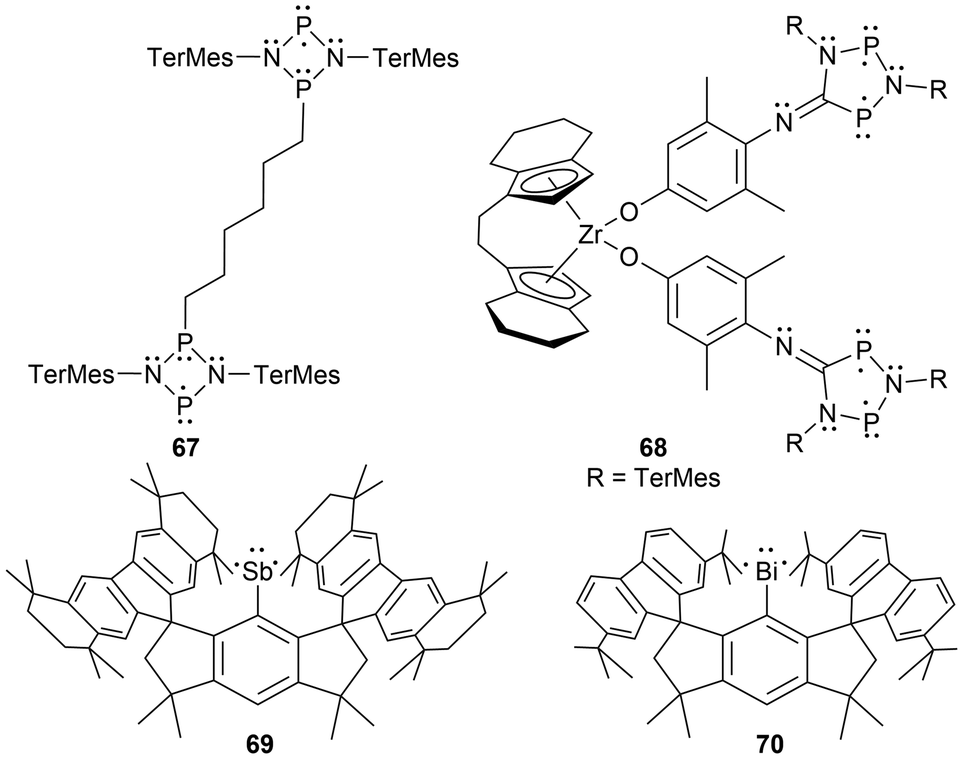 | ||
| Fig. 7 Recent developments of multi-radical centered pnictogens. | ||
Radical reactivity studies
Radical coupling reactions
Main group element-centered radicals are highly chemically reactive species due to the presence of an unpaired electron.10,108 Historically, these species undergo poorly controlled reactions, however, this research field is rapidly developing with their importance in theoretical, biological, and synthetic chemistry, as well as material science.1,10,108 The utilization of sterically encumbering substituents to create kinetic2,10,108 and thermodynamic barriers146 and π-conjugated systems to delocalize the unpaired electron density are applied strategies to stabilize radical-containing species.Cross-radical coupling reactions are a frequently performed reactivity experiment that supports the generation of a radical species. A cross-radical coupling reaction involves the combination of two radicals to generate a new bond, which can capture the radical species of interest in a more stable form. A readily available reagent used in many cases is 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO). Cross-radical coupling products of TEMPO with the pnictogens discussed in this paper have been reported in various studies.17,39,114,147 Another reagent investigated for cross-radical coupling reactions is 2-azaadamantane-N-oxyl (AZADO), another stable aminoxyl radical.39 When the phosphinyl radical 11 is reacted with TEMPO, the rearrangement product 71 is observed, however, when reacted with AZADO, three rearrangement products are observed (71, 72, 73). The heavier pnictogen radical analogues (40, 54a, and 54b), have been investigated in radical cross-coupling reactions with TEMPO, yielding the anticipated cross-coupling products (74a–c) (Fig. 8).17 Another unique phosphorus-centered radical system investigated in similar radical trapping reactions includes a square pyramidal phosphoranyl radical.148 This radical is very unstable and readily dimerizes, however could be trapped with benzophenone and (C6F5)2CO as radical adducts (75a–b) (Fig. 8).148
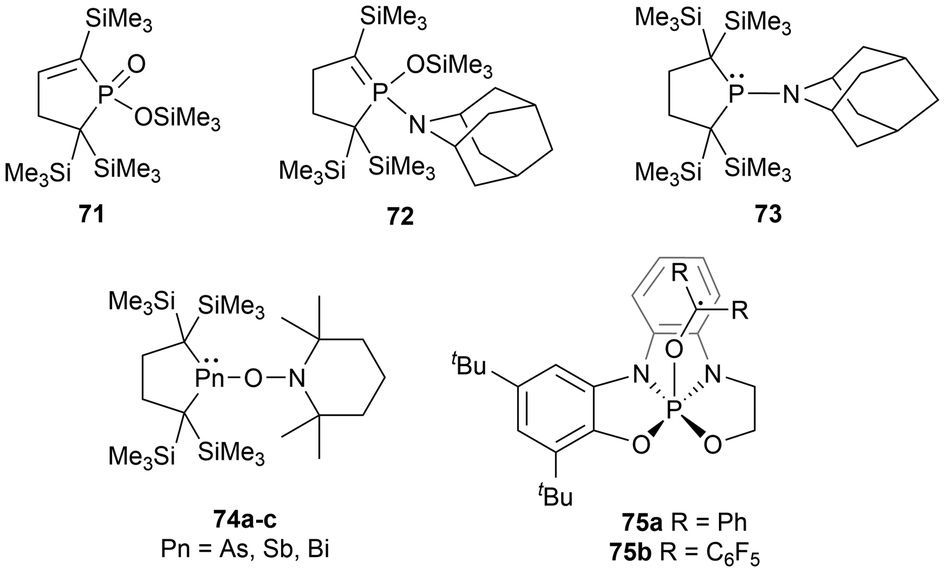 | ||
| Fig. 8 Radical cross-coupling products for group 15 radicals. | ||
The reaction of the stable phosphinyl radical 11 with AZADO generated the anticipated radical coupling product 76 at −40 °C as an intermediate species.39 When warmed to room temperature, the aminoxyphosphine 76 decomposes to the silyl phosphinate 71, the cyclic phosphorane 72, and the aminophosphine 73 in 7%, 82%, and 5% yields, respectively, with the remaining trace amounts being unidentified products. This distribution of products is drastically changed when the reaction is conducted at room temperature, giving a more even distribution of 27% (71), 28% (72), and 35% (73).39 Based on the products observed for the decomposition of the aminoxyphopshine intermediate 76, a proposed mechanism was suggested by Iwamoto et al., as seen in Scheme 5.39 The first suspected step of this mechanism involves the homolytic cleavage of the N–O bond to give the aminyl (77) and phosphinoyl (78) radicals, where the latter radical intermediate (78) undergoes a 1,3-silyl migration to generate the more energetically favourable radical 77. Next, there are three variations of radical coupling reactions as seen in Scheme 5 that give 72, 73, and 80. To generate the third observed product 71, a second N–O bond cleavage of 80 followed by hydrogen abstraction is the suggested formation mechanism.39
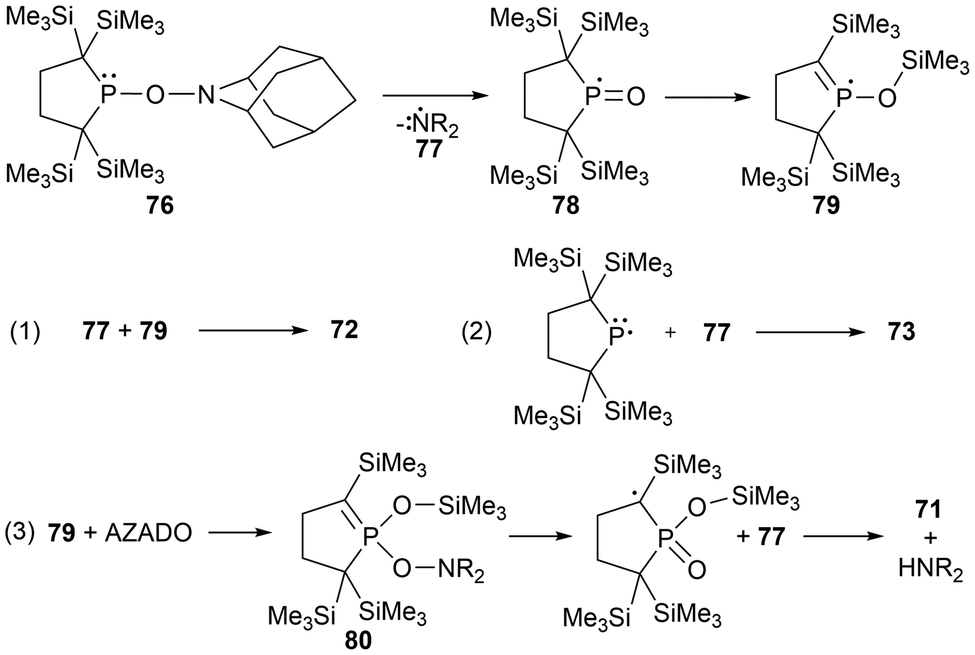 | ||
| Scheme 5 Proposed formation mechanism for 71–73. | ||
For the arsenic cross-coupling product 74a, two sets of signals were observed by EPR spectroscopy, suggesting a partially reversible dissociation of the product into the arsinyl radical 40 and TEMPO due to the shorter Pn–O bond distance, causing severe steric hindrance between the two subunits. For the antimony derivative, the Pn–O bond is longer, slightly reducing the steric strain, and therefore does not dissociate into the stibinyl radical 54a and TEMPO.17 This concept is further supported by the molecular structures of the cross-coupling products 74a and 74b, as the intramolecular distances of the methyl carbons of the trimethylsilyl substituents and TEMPO have a shorter average bond distance in the arsenic derivative (3.77 Å) compared to that observed in the antimony derivative (3.91 Å).17 Despite the longer Pn–O bond for the bismuth derivative 74c, which would further reduce the steric repulsion between the bulky alkyl ligand and TEMPO moieties, this cross-coupling product reversibly dissociates to the bismuthinyl radical and TEMPO. With significantly large and diffuse orbitals on bismuth, it is predicted that this reported observation is due to the weak Bi–O bond.17
Small molecule reactivity
A few of the Group 15 radicals listed have been investigated within small molecule reactivity studies. The phosphinyl radicals 1a and 9a have been investigated by adding various chalcogens (O2, S8, Se, and Te), resulting in chalcogen-bridged diphosphines (Scheme 6).45,149 In a subsequent report, 9a was reacted with carbon disulfide, phenyl isocyanate, and phenyl isothiocyanate.41 The radical species were added across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S and CO double bond to generate neutral diamagnetic species.41 Similar reactivity of 4-membered cyclic phosphorus (64a) and arsenic (64c) biradicals were also reported (Scheme 6).35,53,140,150,151
S and CO double bond to generate neutral diamagnetic species.41 Similar reactivity of 4-membered cyclic phosphorus (64a) and arsenic (64c) biradicals were also reported (Scheme 6).35,53,140,150,151
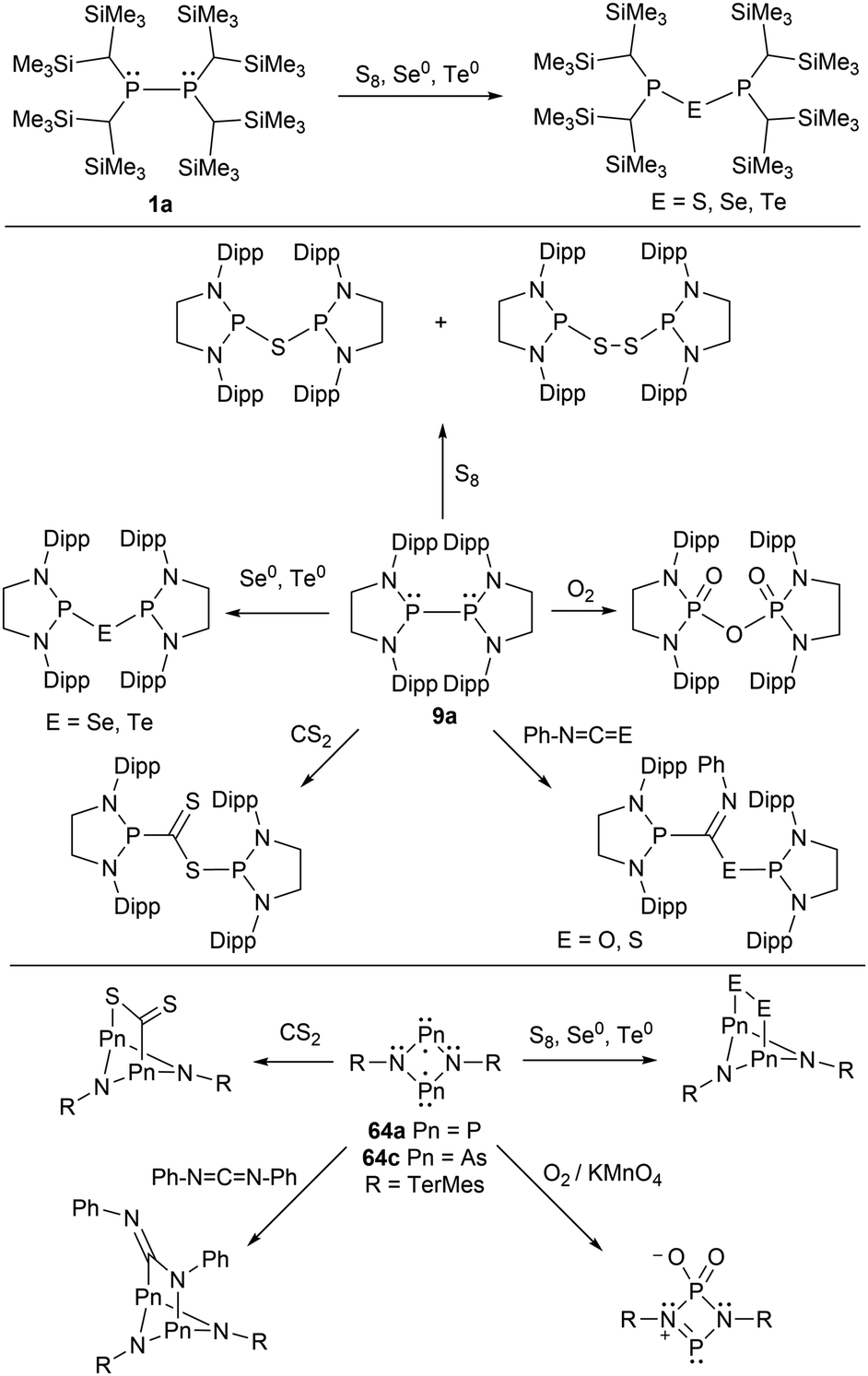 | ||
| Scheme 6 Reactivity of phosphinyl radicals (1a, 9a) and biradicals (64a, 64c) with chalcogens and other small molecules. | ||
Phosphorus radicals 3b,67 9a,45 and 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 90 have also been used for white phosphorus (P4) activation. In all cases, a butterfly-type core structure was observed, capped by two of the phosphorus radical fragments (Scheme 7).45,90 Further reactivity of radicals 9a and 33 was attempted with other small molecules including CO2, CO, and H2 however no reactions were observed. In other work, a tetraradical (81) made by Schulz et al., was successfully applied in the activation of two equivalents of H2 (Scheme 7).131
90 have also been used for white phosphorus (P4) activation. In all cases, a butterfly-type core structure was observed, capped by two of the phosphorus radical fragments (Scheme 7).45,90 Further reactivity of radicals 9a and 33 was attempted with other small molecules including CO2, CO, and H2 however no reactions were observed. In other work, a tetraradical (81) made by Schulz et al., was successfully applied in the activation of two equivalents of H2 (Scheme 7).131
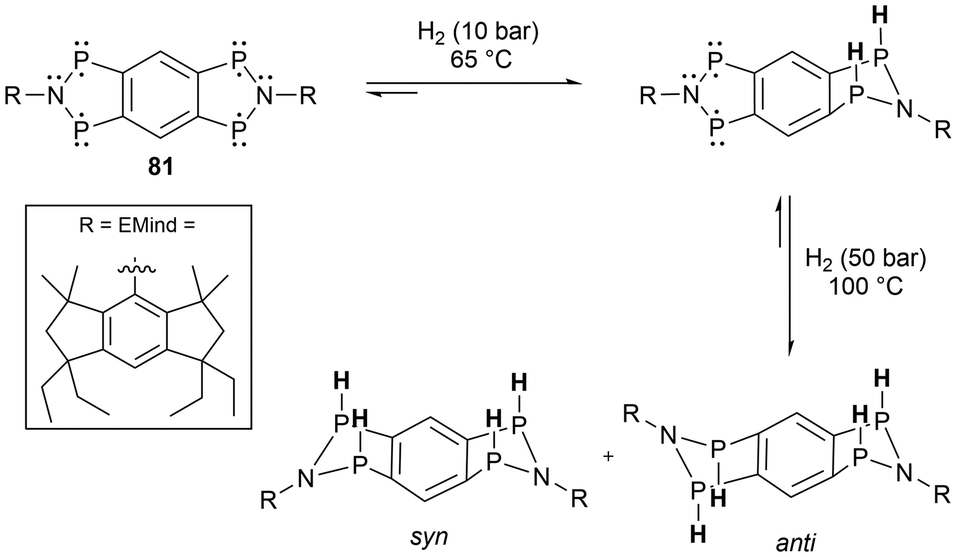 | ||
| Scheme 7 H2 activation by the tetraradical (81). | ||
For biradicals, the four-membered ring system [P(μNTerMes)]2 (64a) has undergone reactivity with CO, where the CO inserts into a P–N bond to generate the hetero cyclopentane-1,3-diyl biradical 82, which was subsequentially reacted with various small molecules (Scheme 8).152 Following this work, the four-membered biradical ring systems 64a and 64b were reacted with isonitriles CN-R′ (R′ = N(SiMe3)2, tBu, TerMes, DMP = 2,6-dimethylphenyl, and Mtp = 2,6-dimethyl-4-tert-butyl-phenyl) to generate new cyclopentane-1,3-diyl derivatives, 83a–d, and 84a–d, respectively, by once again, inserting the carbon atom into a P–N bond (Scheme 9).153,154 The biradicals 83d, was then further reacted in a later study by Schulz et al. to generate Staudinger-type adducts 85a–b.155
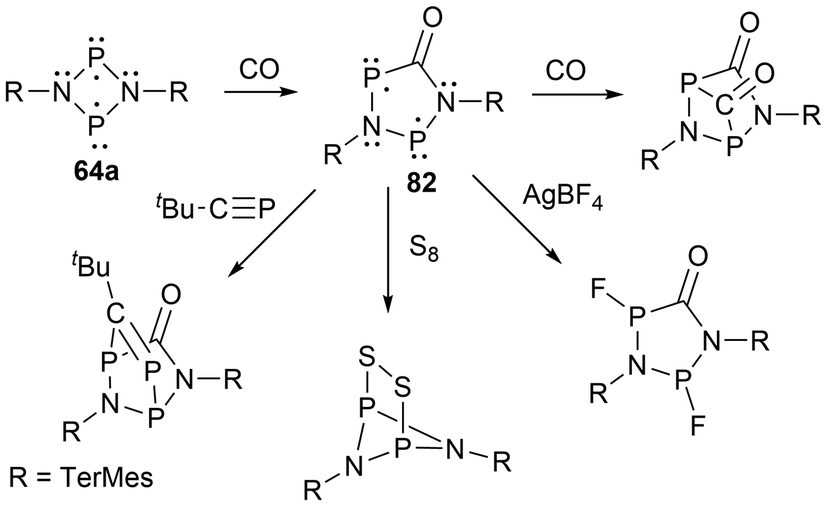 | ||
| Scheme 8 Generation of cyclopentane-1,3-diyl biradical 82 and its reactivity studies. | ||
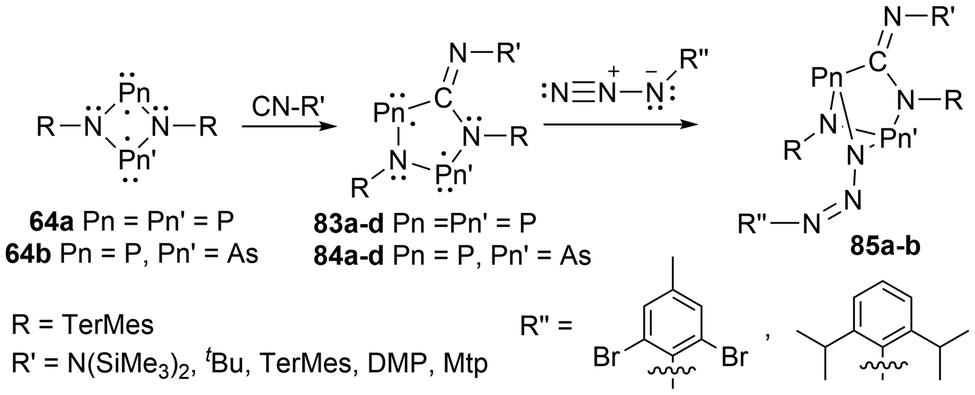 | ||
| Scheme 9 Reactivity of biradicals of the type [E(μNTerMes)E′]2 to generate cyclopentane-1,3-diyl derivatives (83a–d, 84a–d), and Staudinger-type adducts (85a–b). | ||
Light-induced reactivity
More recently, phosphorus-based radicals have been implicated in light-induced reactivity studies. The bimetallic Ni complex 86, was investigated with bond activation reactions using visible light, indicating that the diphosphine P–P bond behaves as an active site under these conditions, demonstrating radical-type reactivity.156 By irradiation with 536 nm light, a variety of bonds including H–H, N–H, N–N, and O–H were activated by the light-induced cleavage of the P–P bond of 86 (Scheme 10).156 Another study reported light-induced functionalization of diazaphospholenes with electrophiles via the phosphinyl radical 10b in the absence of radical initiators at room temperature (Scheme 11).157 The two products generated in these reactions could then easily be separated by their solubilities.157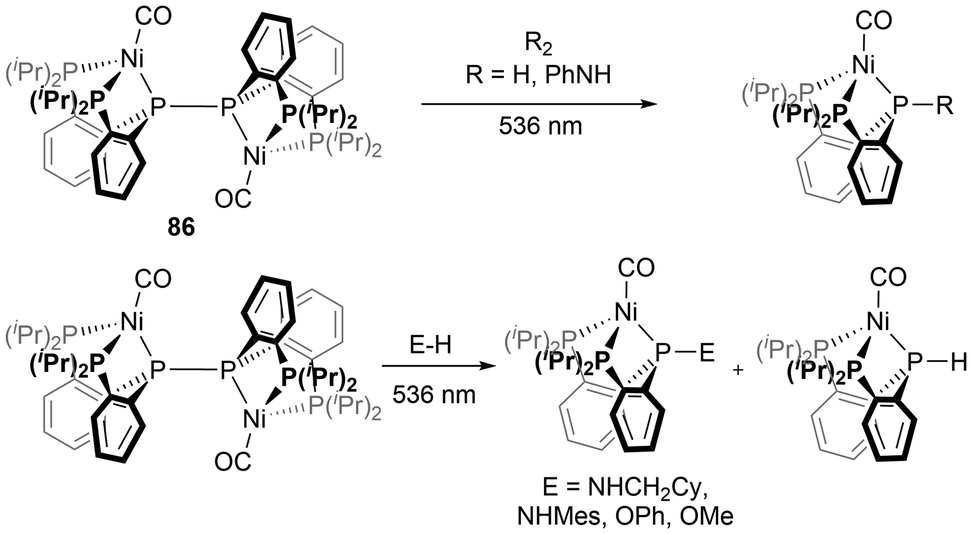 | ||
| Scheme 10 Photo-induced P–P bond cleavage of a rare metal-based diphosphine and reactions with small molecules. | ||
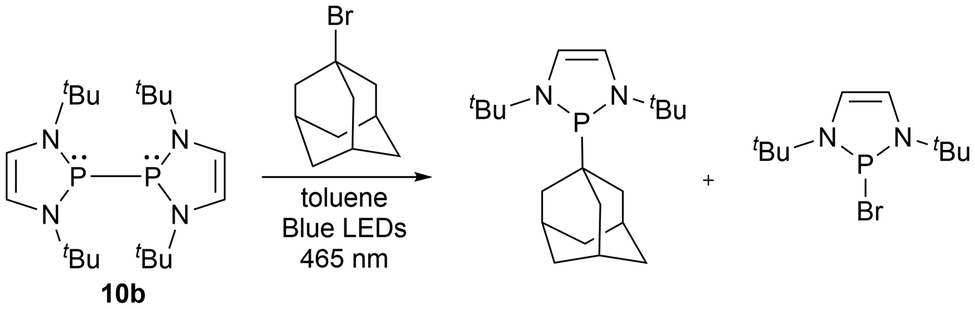 | ||
| Scheme 11 A selected example of photoinduced P–P bond cleavage and activation of an sp3 hybridized C–Br bond. | ||
Furthermore, in the recent literature, diazaphospholene-catalyzed radical reactions have been reported.60,61 Diazaphospholene-mediated cyclization of aryl halides was earlier reported using stoichiometric amounts at room temperature [Scheme 12, eqn (1)],157 followed by radical intramolecular annulations of aryl halides with stoichiometric diazaphosphinane at elevated temperatures in the presence of the radical initiator azobis(isobutyronitrile) (AIBN) [Scheme 12, eqn (2)].62 In this work it is suspected that the phosphinyl radical generated activates the carbon–halogen bond through a mechanism involving single electron transfer (SET).62,157 In 2022, the introduction of visible light irradiation significantly accelerated the annulation of aryl halides and requires as little as 5% mol catalyst loading of the phosphine pre-catalyst in a DBU/HBpin system [Scheme 12, eqn (3)]61 and 10% mol catalyst loading phosphine pre-catalyst when combined with phenylsilane, an alkali metal salt, and a base [Scheme 12, eqn (4)].60
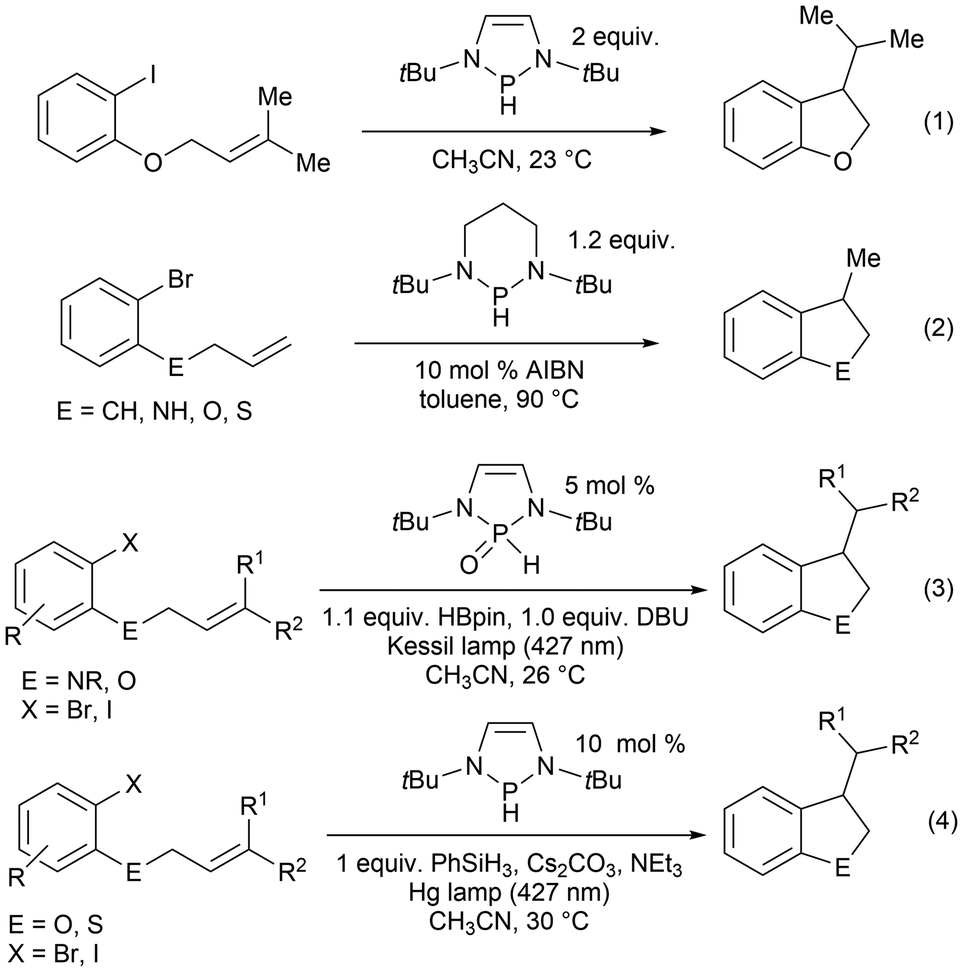 | ||
| Scheme 12 Stoichiometric and catalytic ring closure reactions that implicate phosphorus-based radicals as an active species in the reaction. | ||
The proposed catalytic cycles for the cyclization of aryl halides previously discussed (Scheme 12; eqn (3) and (4)) have been reported and feature equivalent key intermediates.60,61 In both cases, a pre-catalyst is introduced to the system and converted to the respective phosphinyl radical by light irradiation at 427 nm. However, for Cramer's approach, an additional step of reducing the phosphine oxide with HBpin must be done prior. The generation of the phosphinyl radicals initiates a radical chain process by abstracting the halide, resulting in the aryl radical (I) and a phosphine halide.60,61 The aryl radical then adds across the CC bond in a 5-exo-trig manner to generate an alkyl radical (II). Separately, the phosphine halide reacts with HBpin (a)61 or Cs2CO3 then PhSiH3 (b)60 to regenerate the pre-catalyst phosphine. The alkyl radical (II) then abstracts the hydrogen atom from this phosphine to generate the cyclization product and regenerate the phosphinyl radical (Scheme 13).60,61 Cramer et al. also explored using the diphosphine as the catalyst for the cyclization reactions to bypass the light activation step, which resulted in reduced yields in the dark. These results indicate the significance of the light, which can regenerate the phosphinyl radical and restore the catalytic cycle from radical chain terminations that occur before the completion of the reaction.61
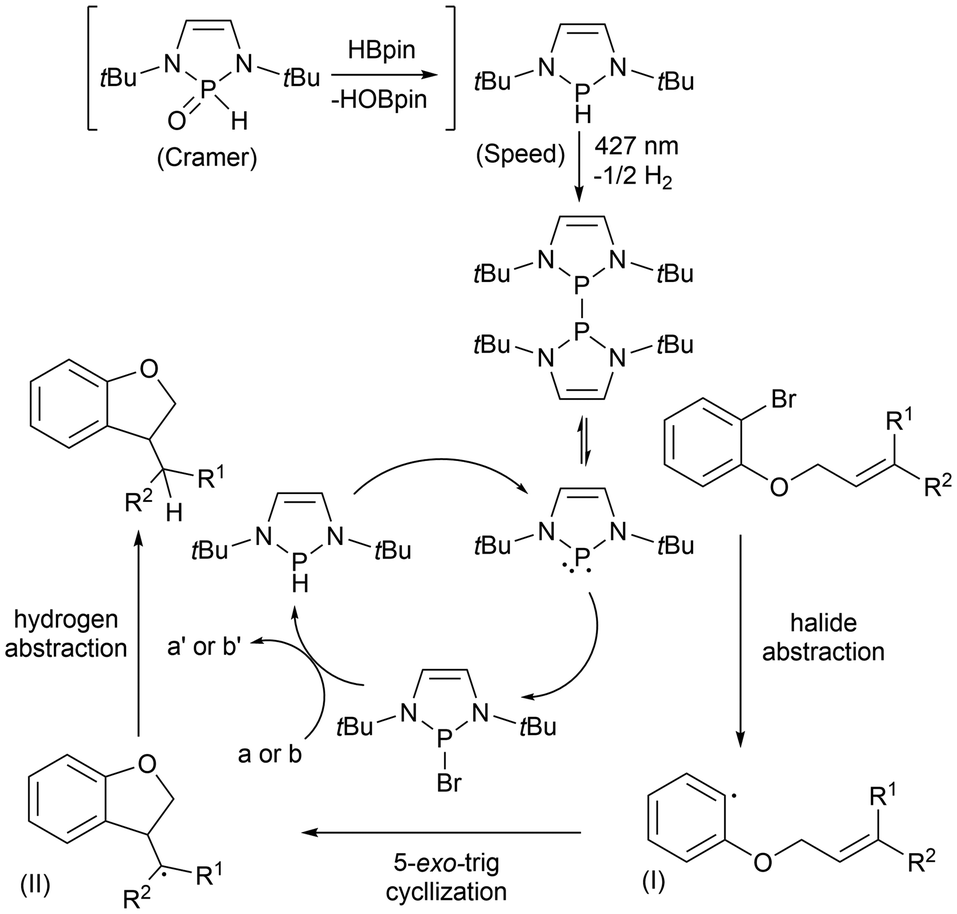 | ||
| Scheme 13 Proposed catalytic cycle of aryl halide cyclization reactions. | ||
One of the most well-known and early developed uses of bismuth in catalysis is in the heterogeneous Standard Oil of Ohio Company (SOHIO) process. In the SOHIO process, a bismuth molybdate catalyst such as Bi2O3·MoO3 is involved in the rate-determining hydrogen abstraction step of the ammoxidation or oxidation of propene to give acrylonitrile or acrolein respectively.158,159 Further application of bismuth in new catalytic radical reactions is of recent interest due to bismuth's low relative costs, non-carcinogenetic properties, low toxicity, potential recyclability, and its relativistic interaction of the large spin-orbit coupling, allowing for UV-vis transitions.14,15,29 In 2023, Cornella et al. published work applying a bismuthinidene (87) in catalytic amounts to promote C(sp3)–N bond formation in a unique manner without requiring a photo-redox system, a chemical oxidant, an external base, or an electrochemical set-up; the previously necessary tools or components for the successful transformation reaction.160 This bismuth-catalyzed cross-coupling reaction proceeds through a single-electron transfer from the Bi(I) complex 87, with an overall Bi(I/II) or Bi(I/II/III) radical incorporating redox cycle as seen in the general Scheme 14.160 Following a recent review focused on bismuth in radical chemistry and catalysis,14 the area of bismuth in photocatalysis continued to expand.161–163 In one instance, the application of stoichiometric organobismuth in photocatalytic arylations has been reported.161 This work generated aryl radicals through the light-mediated SET of organobismuth radical cation intermediates to generate aryl radicals, which further react to obtain several arylated products.161
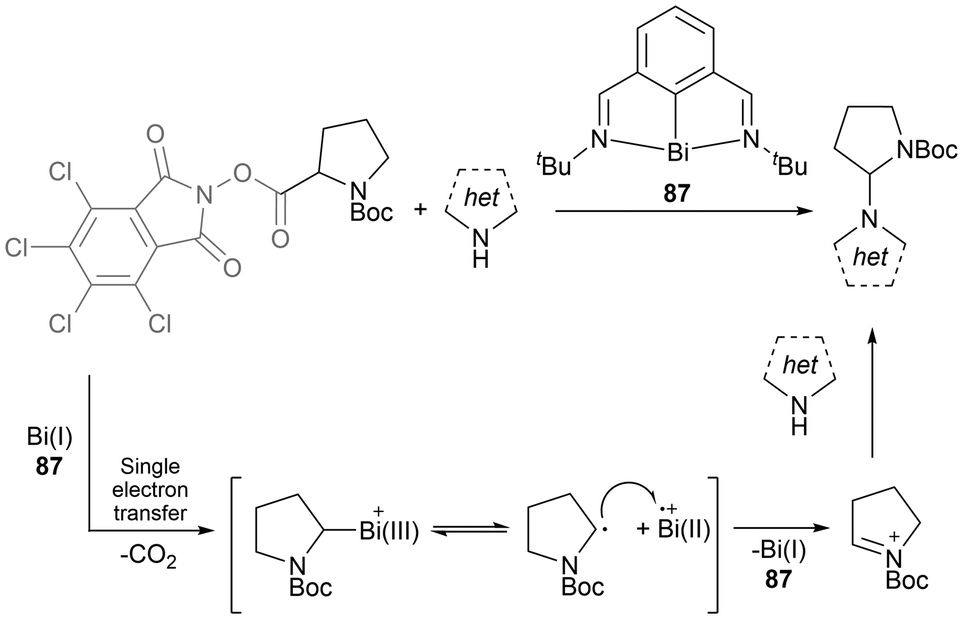 | ||
| Scheme 14 Bi(I)-catalyzed formal C(sp3)–N cross-coupling by radical activation of α-amino redox-active esters. | ||
Following this work, bismuth photocatalysis has also been recently explored in a publication by Cornella and coworkers using the same bismuthinidene.162,163 The activation and C–C coupling of aryl iodides was reported by the light-induced homolytic cleavage of bismuth–aryl bonds, generating a series of aryl–aryl coupling products [Scheme 15, eqn (1)].163 The bismuth catalyst (61) used in this work generates a bismuth radical intermediate, where then the product and bismuth catalyst are generated and regenerated respectively by a proton-coupled electron transfer step.163 Furthermore, using the same bismuth catalyst design (87, 88), Cornella et al. also reported the intermolecular [Scheme 15, eqn (2)] and intramolecular [Scheme 15, eqn (3)] cyclopropanation of double bonds under light irradiation.162 With these recent developments, the further expansion of organobismuth complexes in photocatalysis is a promising area for utilizing heavier Group 15 radicals.
 | ||
| Scheme 15 Light-induced catalytic reactions using bismuth-based catalysts. | ||
Recently, multi-radical centered pnictogens have been reported to demonstrate photoswitching behaviour. The bis-biradical 89 has been reported to experience light-induced on-off switching of H2 addition, where as seen in Scheme 16, the H2 addition is dependent on the irradiation/housane (89-H) formation.143 Furthermore, in other work reported by Schulz et al., an additional cyclopentane-1,3-diyl derivative (90) was generated and functions as a novel molecular double switch capable of performing either a constitutional or stereo-isomerization process when irradiated with different visible light frequencies (Scheme 17).164
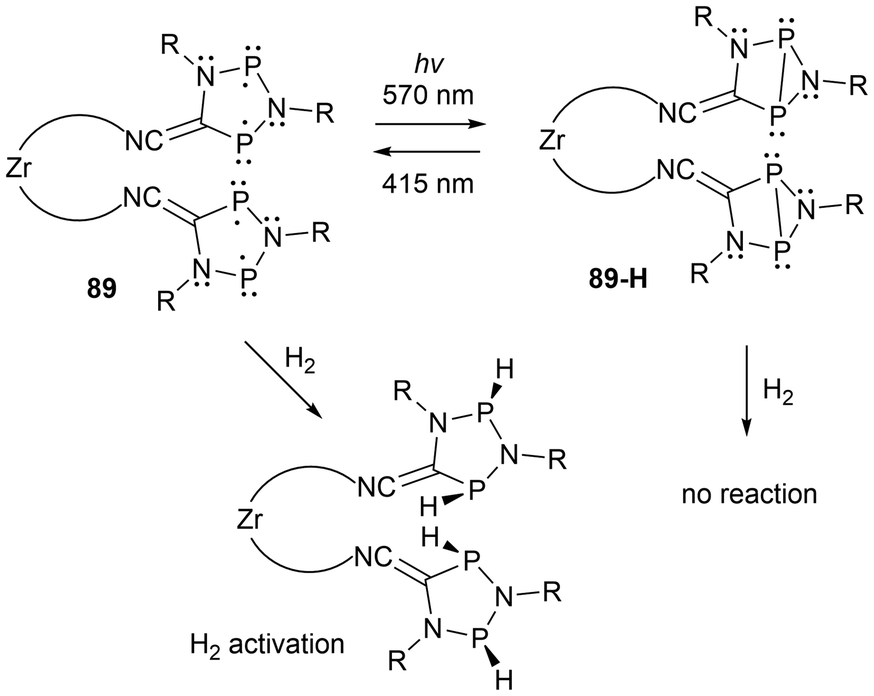 | ||
| Scheme 16 Photo-switching and H2 activation investigation of tetraradical 89 and its bis(housane) 89-H. | ||
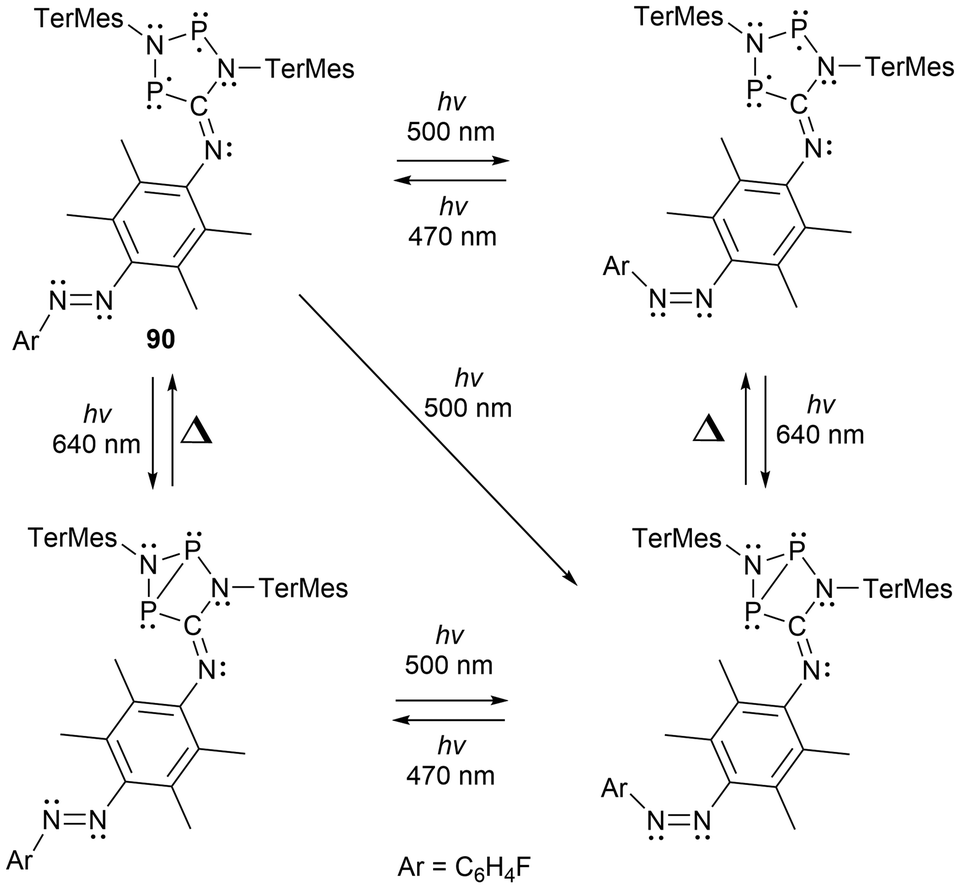 | ||
| Scheme 17 Novel molecular double switch undergoing constitutional and stereo-isomerization upon irradiation with visible light. | ||
Future implications and conclusions
Within the field of Group 15 radicals, there are potential directions this work could be directed. An area with promising potential for future work is the light-induced reactivity studies with the phosphinyl radical catalysts. Between the phosphinyl radical catalysis work done by Speed60 and Cramer61 as seen in Scheme 12, eqn (3) and (4), no examples of intermolecular cyclization, and only two examples of intramolecular cyclization reactions using aryl chlorides were successful. Tuning the phosphinyl radical through the N–R substituents and backbone to give the desired reactivity, while still maintaining sufficient stability for ease of pre-catalyst or catalyst handling could be explored for this application. For example, the N–P–N angle could be tuned by changing the size of the heterocycle to give 6- or 7-membered ring systems to change the energies of the frontier orbitals at the P atom. Additionally, in a patent by Speed, an N-heterocyclic phosphine similar to the phosphinyl pre-catalyst used in the annulation of aryl halides60 was used to reduce imines asymmetrically.165 The phosphine in this case incorporates chiral alkyl substituents attached to the nitrogen atoms, promoting the enantioselectivity of the products (Fig. 9).165 Future studies using chiral phosphinyl catalysts should be investigated for use in enantioselective catalysis.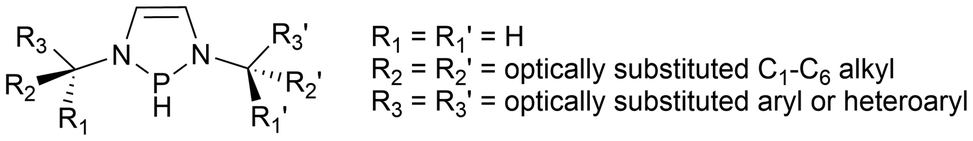 | ||
| Fig. 9 Chiral diazaphopholene framework developed by Speed.165 | ||
An additional area to further develop within group 15 mono-radicals includes mixed pnictogen systems stabilized by carbenes and/or aryl substituents. The current frameworks include a variety of bi-, tri-, and tetrapnictogen-centered radicals and radical ions for nitrogen and phosphorus (Fig. 3), as well as some examples of bi- and tripnictogen nitrogen-arsenic and phosphorus-arsenic examples. Expanding this list to include examples of antimony and bismuth in these systems should be the next step. In general, the next steps should focus on developing more stable or even air-stable group 15 radical species, which can be further investigated within these processes.
Work in the area of bismuth-based radicals is expected to be a continued hot area and changes to the ligand frameworks should provide new and exciting chemistry. For example, as we were writing this conclusion, Cornella et al. published a red-light activated bismuthinidene where oxidative addition of aryl iodides occurs at bismuth through a SET mechanism.166
In addition to the wide variety of known ligands and substituent combinations available, new ligand frameworks are always being developed. We anticipate the area of heavier group 15 radicals to continue to be a hot area in main group chemistry and it is our hope that further breakthroughs will direct the area towards discoveries that will be embraced by the general chemistry community.
Author contributions
Conceptualization, data curation, formal analysis, project administration, and writing (review/editing) was contributed by all authors. JDM contributed to funding acquisition, resources, and supervision. DLGS performed the investigation, visualization, and writing of the original draft.Data availability
No primary research results, software or code have been included, and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Natural Science and Engineering Council of Canada (NSERC) for operating funds (JDM) and the award of a Canadian Graduate Scholarship – Master's (DLGS). We also thank the reviewers for their insightful comments and suggestions.References
- Z. Feng, S. Tang, Y. Su and X. Wang, Chem. Soc. Rev., 2022, 51, 5930–5973 RSC.
- C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020 RSC.
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- L. N. Bochkarev, N. E. Molosnova, L. N. Zakharov, G. K. Fukin, A. I. Yanovsky and Y. T. Struchkov, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 1995, 51, 489–491 CrossRef.
- P. P. Power, Chem. Rev., 2003, 103, 789–810 CrossRef CAS PubMed.
- V. V. Pen'kovskii, Russ. Chem. Rev., 1975, 44, 449–467 CrossRef.
- J. P. Bullock, A. M. Bond, R. T. Boeré, T. M. Gietz, T. L. Roemmele, S. D. Seagrave, J. D. Masuda and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11205–11215 CrossRef CAS PubMed.
- X. Pan, X. Chen, T. Li, Y. Li and X. Wang, J. Am. Chem. Soc., 2013, 135, 3414–3417 CrossRef CAS PubMed.
- B. Das, A. Makol and S. Kundu, Dalton Trans., 2022, 51, 12404–12426 RSC.
- G. Tan and X. Wang, Chin. J. Chem., 2018, 36, 573–586 Search PubMed.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1976, 9, 13–19 CrossRef CAS.
- Y. Su and R. Kinjo, Coord. Chem. Rev., 2017, 352, 346–378 CrossRef CAS.
- C. Helling and S. Schulz, Eur. J. Inorg. Chem., 2020, 2020, 3209–3221 CrossRef CAS.
- M. Mato and J. Cornella, Angew. Chem., Int. Ed., 2024, 63, e202315046 CrossRef CAS PubMed.
- J. Ramler, I. Krummenacher and C. Lichtenberg, Chem. – Eur. J., 2020, 26, 14551–14555 CrossRef CAS PubMed.
- J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS PubMed.
- S. Ishida, F. Hirakawa and T. Iwamoto, Bull. Chem. Soc. Jpn., 2018, 91, 1168–1175 CrossRef CAS.
- C. Lichtenberg, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, Wiley, 2nd edn, 2020, pp. 1–12 Search PubMed.
- N. S. Imyanitov, Found. Chem., 2019, 21, 255–284 CrossRef CAS.
- M. Seth, M. Dolg, P. Fulde and P. Schwerdtfeger, J. Am. Chem. Soc., 1995, 117, 6597–6598 CrossRef CAS.
- B. J. Dunne and A. G. Orpen, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 1991, 47, 345–347 CrossRef.
- A. N. Sobolev, V. K. Belsky, N. Y. Chernikova and F. Y. Akhmadulina, J. Organomet. Chem., 1983, 244, 129–136 CrossRef CAS.
- E. A. Adams, J. W. Kolis and W. T. Pennington, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 1990, 46, 917–919 CrossRef.
- L. Bučinský, D. Jayatilaka and S. Grabowsky, J. Phys. Chem. A, 2016, 120, 6650–6669 CrossRef PubMed.
- R. J. F. Berger, D. Rettenwander, S. Spirk, C. Wolf, M. Patzschke, M. Ertl, U. Monkowius and N. W. Mitzel, Phys. Chem. Chem. Phys., 2012, 14, 15520 RSC.
- P. S. Bagus, Y. S. Lee and K. S. Pitzer, Chem. Phys. Lett., 1975, 33, 408–411 CrossRef CAS.
- P. Pyykko, Chem. Rev., 1988, 88, 563–594 CrossRef CAS.
- A. Kyono and M. Kimata, Am. Mineral., 2004, 89, 932–940 CrossRef CAS.
- J. Ramler, I. Krummenacher and C. Lichtenberg, Angew. Chem., Int. Ed., 2019, 58, 12924–12929 CrossRef CAS PubMed.
- K. Balasubramanian, Y. S. Chung and W. S. Glaunsinger, J. Chem. Phys., 1993, 98, 8859–8869 CrossRef CAS.
- D. Dai and K. Balasubramanian, J. Chem. Phys., 1990, 93, 1837–1846 CrossRef CAS.
- M. Kaupp, in The Chemical Bond, ed. G. Frenking and S. Shaik, Wiley, 1st edn, 2014, pp. 1–24 Search PubMed.
- Y. H. Budnikova, Dokl. Chem., 2022, 507, 229–259 CrossRef CAS.
- S. Kundu, S. Sinhababu, V. Chandrasekhar and H. W. Roesky, Chem. Sci., 2019, 10, 4727–4741 RSC.
- A. Schulz, Dalton Trans., 2018, 47, 12827–12837 RSC.
- G. E. Cutsail, Dalton Trans., 2020, 49, 12128–12135 RSC.
- A. Dasgupta, E. Richards and R. L. Melen, Angew. Chem., Int. Ed., 2021, 60, 53–65 CrossRef CAS PubMed.
- L. J. C. van der Zee, S. Pahar, E. Richards, R. L. Melen and J. C. Slootweg, Chem. Rev., 2023, 123, 9653–9675 Search PubMed.
- S. Ishida, F. Hirakawa and T. Iwamoto, Chem. Lett., 2015, 44, 94–96 CrossRef.
- F. Hirakawa, H. Ichikawa, S. Ishida and T. Iwamoto, Organometallics, 2015, 34, 2714–2716 CrossRef CAS.
- N. A. Giffin, A. D. Hendsbee and J. D. Masuda, Dalton Trans., 2016, 45, 12636–12638 RSC.
- A. Ott, P. R. Nagy and Z. Benkő, Inorg. Chem., 2022, 61, 16266–16281 Search PubMed.
- Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, Boca Raton, 2002 Search PubMed.
- C. Branfoot, T. A. Young, D. F. Wass and P. G. Pringle, Dalton Trans., 2021, 50, 7094–7104 Search PubMed.
- N. A. Giffin, A. D. Hendsbee, T. L. Roemmele, M. D. Lumsden, C. C. Pye and J. D. Masuda, Inorg. Chem., 2012, 51, 11837–11850 CrossRef CAS PubMed.
- M. Blum, O. Puntigam, S. Plebst, F. Ehret, J. Bender, M. Nieger and D. Gudat, Dalton Trans., 2016, 45, 1987–1997 Search PubMed.
- S. L. Hinchley, C. A. Morrison, D. W. H. Rankin, C. L. B. Macdonald, R. J. Wiacek, A. H. Cowley, M. F. Lappert, G. Gundersen, J. A. C. Clyburne and P. P. Power, Chem. Commun., 2000, 2045–2046 RSC.
- J.-D. Guo, S. Nagase and P. P. Power, Organometallics, 2015, 34, 2028–2033 CrossRef CAS.
- M. J. S. Gynane, A. Hudson, M. F. Lappert and P. P. Power, J. Chem. Soc., Chem. Commun., 1976, 623–624 RSC.
- M. J. S. Gynane, A. Hudson, M. F. Lappert, P. P. Power and H. Goldwhite, J. Chem. Soc., Dalton Trans., 1980, 2428–2433 RSC.
- O. Puntigam, D. Förster, N. A. Giffin, S. Burck, J. Bender, F. Ehret, A. D. Hendsbee, M. Nieger, J. D. Masuda and D. Gudat, Eur. J. Inorg. Chem., 2013, 2013, 2041–2050 Search PubMed.
- S. Isenberg, S. Weller, D. Kargin, S. Valić, B. Schwederski, Z. Kelemen, C. Bruhn, K. Krekić, M. Maurer, C. M. Feil, M. Nieger, D. Gudat, L. Nyulászi and R. Pietschnig, ChemistryOpen, 2019, 8, 1235–1243 CrossRef CAS PubMed.
- A. Hinz, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Chem. – Eur. J., 2014, 20, 14659–14673 Search PubMed.
- T. Taniguchi, Synthesis, 2017, 3511–3534 Search PubMed.
- S. Ishida, F. Hirakawa and T. Iwamoto, J. Am. Chem. Soc., 2011, 133, 12968–12971 Search PubMed.
- O. Back, M. A. Celik, G. Frenking, M. Melaimi, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 10262–10263 CrossRef CAS PubMed.
- K. Schwedtmann, S. Schulz, F. Hennersdorf, T. Strassner, E. Dmitrieva and J. J. Weigand, Angew. Chem., Int. Ed., 2015, 54, 11054–11058 Search PubMed.
- X. Pan, X. Wang, Y. Zhao, Y. Sui and X. Wang, J. Am. Chem. Soc., 2014, 136, 9834–9837 Search PubMed.
- O. Puntigam, L. Könczöl, L. Nyulászi and D. Gudat, Angew. Chem., Int. Ed., 2015, 54, 11567–11571 Search PubMed.
- R. D. Riley, B. S. N. Huchenski, K. L. Bamford and A. W. H. Speed, Angew. Chem., Int. Ed., 2022, 61, e202204088 CrossRef CAS PubMed.
- J. Klett, Ł. Woźniak and N. Cramer, Angew. Chem., Int. Ed., 2022, 61, e202202306 CrossRef CAS PubMed.
- J. Zhang, J.-D. Yang and J.-P. Cheng, Chem. Sci., 2020, 11, 4786–4790 RSC.
- S. L. Hinchley, C. A. Morrison, D. W. H. Rankin, C. L. B. Macdonald, R. J. Wiacek, A. Voigt, A. H. Cowley, M. F. Lappert, G. Gundersen, J. A. C. Clyburne and P. P. Power, J. Am. Chem. Soc., 2001, 123, 9045–9053 CrossRef CAS PubMed.
- D. M. C. Ould, A. C. Rigby, L. C. Wilkins, S. J. Adams, J. A. Platts, S. J. A. Pope, E. Richards and R. L. Melen, Organometallics, 2018, 37, 712–719 CrossRef CAS.
- A. Dumitrescu, V. L. Rudzevich, V. D. Romanenko, A. Mari, W. W. Schoeller, D. Bourissou and G. Bertrand, Inorg. Chem., 2004, 43, 6546–6548 CrossRef CAS PubMed.
- B.
![[C with combining dot below]](https://www.rsc.org/images/entities/char_0043_0323.gif) etinkaya, P. B. Hitchcock, M. F. Lappert, A. J. Thorne and H. Goldwhite, J. Chem. Soc., Chem. Commun., 1982, 691–693 Search PubMed.
etinkaya, P. B. Hitchcock, M. F. Lappert, A. J. Thorne and H. Goldwhite, J. Chem. Soc., Chem. Commun., 1982, 691–693 Search PubMed. - J.-P. Bezombes, P. B. Hitchcock, M. F. Lappert and J. E. Nycz, Dalton Trans., 2004, 499–501 Search PubMed.
- J. Geier, J. Harmer and H. Grützmacher, Angew. Chem., Int. Ed., 2004, 43, 4093–4097 Search PubMed.
- T. Sasamori, E. Mieda, N. Nagahora, K. Sato, D. Shiomi, T. Takui, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 12582–12588 Search PubMed.
- R. T. Boeré, A. M. Bond, S. Cronin, N. W. Duffy, P. Hazendonk, J. D. Masuda, K. Pollard, T. L. Roemmele, P. Tran and Y. Zhang, New J. Chem., 2008, 32, 214–231 Search PubMed.
- Y. Su, X. Zheng, X. Wang, X. Zhang, Y. Sui and X. Wang, J. Am. Chem. Soc., 2014, 136, 6251–6254 CrossRef CAS PubMed.
- A. Brückner, A. Hinz, J. B. Priebe, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 7426–7430 Search PubMed.
- M. Yoshifuji, A. J. Arduengo III, T. A. Konovalova, L. D. Kispert, M. Kikuchi and S. Ito, Chem. Lett., 2006, 35, 1136–1137 CrossRef CAS.
- H. Cui, D. Xiao, L. Zhang, H. Ruan, Y. Fang, Y. Zhao, G. Tan, L. Zhao, G. Frenking, M. Driess and X. Wang, Chem. Commun., 2020, 56, 2167–2170 RSC.
- X. Pan, X. Wang, Z. Zhang and X. Wang, Dalton Trans., 2015, 44, 15099–15102 RSC.
- X. Pan, Y. Su, X. Chen, Y. Zhao, Y. Li, J. Zuo and X. Wang, J. Am. Chem. Soc., 2013, 135, 5561–5564 CrossRef CAS PubMed.
- P. Federmann, H. K. Wagner, P. W. Antoni, J.-M. Mörsdorf, J. L. Pérez Lustres, H. Wadepohl, M. Motzkus and J. Ballmann, Org. Lett., 2019, 21, 2033–2038 Search PubMed.
- S. Asami, S. Ishida, T. Iwamoto, K. Suzuki and M. Yamashita, Angew. Chem., Int. Ed., 2017, 56, 1658–1662 CrossRef CAS PubMed.
- G. Tan, J. Li, L. Zhang, C. Chen, Y. Zhao, X. Wang, Y. Song, Y.-Q. Zhang and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 12741–12745 CrossRef CAS PubMed.
- C. Chen, Z. Hu, J. Li, H. Ruan, Y. Zhao, G. Tan, Y. Song and X. Wang, Inorg. Chem., 2020, 59, 2111–2115 CrossRef CAS PubMed.
- F. Murakami, S. Sasaki and M. Yoshifuji, J. Am. Chem. Soc., 2005, 127, 8926–8927 CrossRef CAS PubMed.
- S. Sasaki, F. Murakami and M. Yoshifuji, Angew. Chem., Int. Ed., 1999, 38, 340–343 CrossRef CAS PubMed.
- G. Tan, S. Li, S. Chen, Y. Sui, Y. Zhao and X. Wang, J. Am. Chem. Soc., 2016, 138, 6735–6738 CrossRef CAS PubMed.
- M. K. Mondal, L. Zhang, Z. Feng, S. Tang, R. Feng, Y. Zhao, G. Tan, H. Ruan and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 15829–15833 Search PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- J. Back, J. Park, Y. Kim, H. Kang, Y. Kim, M. J. Park, K. Kim and E. Lee, J. Am. Chem. Soc., 2017, 139, 15300–15303 CrossRef CAS PubMed.
- A. Doddi, D. Bockfeld, M.-K. Zaretzke, C. Kleeberg, T. Bannenberg and M. Tamm, Dalton Trans., 2017, 46, 15859–15864 RSC.
- M. K. Sharma, D. Rottschäfer, S. Blomeyer, B. Neumann, H.-G. Stammler, M. Van Gastel, A. Hinz and R. S. Ghadwal, Chem. Commun., 2019, 55, 10408–10411 Search PubMed.
- O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369–373 Search PubMed.
- J. Haberstroh, C. Taube, J. Fidelius, S. Schulz, N. Israel, E. Dmitrieva, R. M. Gomila, A. Frontera, R. Wolf, K. Schwedtmann and J. J. Weigand, Chem. Commun., 2024, 60, 8537–8540 RSC.
- E. A. LaPierre, L. K. Watanabe, B. O. Patrick, J. M. Rawson, H. M. Tuononen and I. Manners, J. Am. Chem. Soc., 2023, 145, 9223–9232 Search PubMed.
- E. A. LaPierre, B. O. Patrick and I. Manners, J. Am. Chem. Soc., 2024, 146, 6326–6335 CrossRef CAS PubMed.
- H. Kim and E. Lee, Bull. Korean Chem. Soc., 2022, 43, 1328–1341 CrossRef CAS.
- Y. Kim and E. Lee, Chem. – Eur. J., 2018, 24, 19110–19121 CrossRef CAS PubMed.
- Y. Song, H. Song, Y. Choi, J. Seo and E. Lee, Chem. Commun., 2024, 60, 8043–8046 RSC.
- R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930–5933 CrossRef CAS PubMed.
- O. Back, B. Donnadieu, M. Von Hopffgarten, S. Klein, R. Tonner, G. Frenking and G. Bertrand, Chem. Sci., 2011, 2, 858 RSC.
- A. M. Tondreau, Z. Benkő, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545–1554 RSC.
- Z. Li, X. Chen, D. M. Andrada, G. Frenking, Z. Benkö, Y. Li, J. R. Harmer, C.-Y. Su and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 5744–5749 CrossRef CAS PubMed.
- X. Chen, L. L. Liu, S. Liu, H. Grützmacher and Z. Li, Angew. Chem., Int. Ed., 2020, 59, 23830–23835 CrossRef CAS PubMed.
- R. Yadav, A. Sharma, B. Das, C. Majumder, A. Das, S. Sen and S. Kundu, Chem. – Eur. J., 2024, 30, e202401730 CrossRef CAS PubMed.
- D. Munz, Organometallics, 2018, 37, 275–289 CrossRef CAS.
- E. Welz, J. Böhnke, R. D. Dewhurst, H. Braunschweig and B. Engels, J. Am. Chem. Soc., 2018, 140, 12580–12591 CrossRef CAS PubMed.
- M. Y. Abraham, Y. Wang, Y. Xie, R. J. Jr Gilliard, P. Wei, B. J. Vaccaro, M. K. Johnson, H. F. I. Schaefer, P. V. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2013, 135, 2486–2488 CrossRef CAS PubMed.
- T. Li, G. Tan, C. Cheng, Y. Zhao, L. Zhang and X. Wang, Chem. Commun., 2018, 54, 1493–1496 RSC.
- K. M. Melancon, M. B. Gildner and T. W. Hudnall, Chem. – Eur. J., 2018, 24, 9264–9268 CrossRef CAS PubMed.
- Y. Fang, L. Zhang, C. Cheng, Y. Zhao, M. Abe, G. Tan and X. Wang, Chem. – Eur. J., 2018, 24, 3156–3160 CrossRef CAS PubMed.
- C. Helling and S. Schulz, Eur. J. Inorg. Chem., 2020, 3209–3221 CrossRef CAS.
- A. Doddi, D. Bockfeld, M.-K. Zaretzke, T. Bannenberg and M. Tamm, Chem. – Eur. J., 2019, 25, 13119–13123 CrossRef CAS PubMed.
- K. Tsuji, Y. Fujii, S. Sasaki and M. Yoshifuji, Chem. Lett., 1997, 26, 855–856 CrossRef.
- M. K. Sharma, S. Blomeyer, B. Neumann, H.-G. Stammler, A. Hinz, M. Van Gastel and R. S. Ghadwal, Chem. Commun., 2020, 56, 3575–3578 RSC.
- J. Krüger, J. Haak, C. Wölper, G. E. I. Cutsail, G. Haberhauer and S. Schulz, Inorg. Chem., 2022, 61, 5878–5884 CrossRef PubMed.
- C. Helling, C. Wölper, Y. Schulte, G. E. I. Cutsail and S. Schulz, Inorg. Chem., 2019, 58, 10323–10332 CrossRef CAS PubMed.
- S. Ishida, F. Hirakawa, K. Furukawa, K. Yoza and T. Iwamoto, Angew. Chem., Int. Ed., 2014, 53, 11172–11176 CrossRef CAS PubMed.
- C. Ganesamoorthy, C. Helling, C. Wölper, W. Frank, E. Bill, G. E. Cutsail and S. Schulz, Nat. Commun., 2018, 9, 87 CrossRef PubMed.
- C. Helling, G. E. Cutsail III, H. Weinert, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2020, 59, 7561–7568 CrossRef CAS PubMed.
- C. Helling, C. Wölper, G. E. Cutsail III, G. Haberhauer and S. Schulz, Chem. – Eur. J., 2020, 26, 13390–13399 CrossRef CAS PubMed.
- C. Lichtenberg, F. Pan, T. P. Spaniol, U. Englert and J. Okuda, Angew. Chem., Int. Ed., 2012, 51, 13011–13015 CrossRef CAS PubMed.
- D. P. Mukhopadhyay, D. Schleier, S. Wirsing, J. Ramler, D. Kaiser, E. Reusch, P. Hemberger, T. Preitschopf, I. Krummenacher, B. Engels, I. Fischer and C. Lichtenberg, Chem. Sci., 2020, 11, 7562–7568 RSC.
- T. A. Hanna, A. L. Rieger, P. H. Rieger and X. Wang, Inorg. Chem., 2002, 41, 3590–3592 CrossRef CAS PubMed.
- H. M. Weinert, C. Wölper, J. Haak, G. E. Cutsail and S. Schulz, Chem. Sci., 2021, 12, 14024–14032 RSC.
- J. Krüger, C. Wölper and S. Schulz, Inorg. Chem., 2020, 59, 11142–11151 CrossRef PubMed.
- T. Li, H. Wei, Y. Fang, L. Wang, S. Chen, Z. Zhang, Y. Zhao, G. Tan and X. Wang, Angew. Chem., 2017, 129, 647–651 CrossRef.
- R. Kretschmer, D. A. Ruiz, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 8176–8179 Search PubMed.
- R. J. Schwamm, J. R. Harmer, M. Lein, C. M. Fitchett, S. Granville and M. P. Coles, Angew. Chem., Int. Ed., 2015, 54, 10630–10633 CrossRef CAS PubMed.
- S. Sasaki, K. Sutoh, F. Murakami and M. Yoshifuji, J. Am. Chem. Soc., 2002, 124, 14830–14831 CrossRef CAS PubMed.
- J. S. Wenger, M. Weng, G. N. George and T. C. Johnstone, Nat. Chem., 2023, 15, 633–640 Search PubMed.
- M. Taghavikish, B. L. Price, T. L. Roemmele and R. T. Boeré, Aust. J. Chem., 2013, 66, 1226 Search PubMed.
- T. Dunaj, M. Egorycheva, A. Arebi, K. Dollberg and C. von Hänisch, Z. Anorg. Allg. Chem., 2023, 649, e202300004 Search PubMed.
- S. Demeshko, C. Godemann, R. Kuzora, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2013, 52, 2105–2108 CrossRef CAS PubMed.
- E. Zander, J. Bresien, V. V. Zhivonitko, J. Fessler, A. Villinger, D. Michalik and A. Schulz, J. Am. Chem. Soc., 2023, 145, 14484–14497 CrossRef CAS PubMed.
- J. Rosenboom, F. Taube, L. Teichmeier, A. Villinger, M. Reinhard, S. Demeshko, M. Bennati, J. Bresien, B. Corzilius and A. Schulz, Angew. Chem., Int. Ed., 2024, 63, e202318210 CrossRef CAS PubMed.
- H. Steffenfauseweh, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2022, 61, e202207415 Search PubMed.
- J. Bresien, A. Schulz, L. S. Szych, A. Villinger and R. Wustrack, Dalton Trans., 2019, 48, 11103–11111 RSC.
- A. Hinz, J. Bresien, F. Breher and A. Schulz, Chem. Rev., 2023, 123, 10468–10526 CrossRef CAS PubMed.
- W. Schlenk and M. Brauns, Ber. Dtsch. Chem. Ges., 1915, 48, 661–669 CrossRef CAS.
- M. Goehring and D. Voigt, Naturwissenschaften, 1953, 40, 482–482 Search PubMed.
- E. Niecke, A. Fuchs, F. Baumeister, M. Nieger and W. W. Schoeller, Angew. Chem., Int. Ed. Engl., 1995, 34, 555–557 Search PubMed.
- W. W. Schoeller, Eur. J. Inorg. Chem., 2019, 2019, 1495–1506 CrossRef CAS.
- T. Beweries, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2011, 50, 8974–8978 CrossRef CAS PubMed.
- J. Bresien, A. Hinz, A. Schulz and A. Villinger, Dalton Trans., 2018, 47, 4433–4436 RSC.
- A. Hinz, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 668–672 CrossRef CAS PubMed.
- P. Fritz, H. AlHamwi, A. Villinger, D. Michalik, J. Bresien, F. Reiß, T. Beweries and A. Schulz, Chem. – Eur. J., 2024, 30, e202402415 CrossRef CAS PubMed.
- M. Wu, H. Li, W. Chen, D. Wang, Y. He, L. Xu, S. Ye and G. Tan, Chem, 2023, 9, 2573–2584 CAS.
- Y. Pang, N. Nöthling, M. Leutzsch, L. Kang, E. Bill, M. van Gastel, E. Reijerse, R. Goddard, L. Wagner, D. SantaLucia, S. DeBeer, F. Neese and J. Cornella, Science, 2023, 380, 1043–1048 CrossRef CAS PubMed.
- N. Burford, J. A. C. Clyburne and M. S. W. Chan, Inorg. Chem., 1997, 36, 3204–3206 CrossRef CAS PubMed.
- J. Zhang, J.-D. Yang and J.-P. Cheng, Chem. Sci., 2020, 11, 3672–3679 RSC.
- S. Volodarsky, I. Malahov, D. Bawari, M. Diab, N. Malik, B. Tumanskii and R. Dobrovetsky, Chem. Sci., 2022, 13, 5957–5963 RSC.
- N. J. Hill, G. Reeske and A. H. Cowley, Main Group Chem., 2010, 9, 5–10 CAS.
- A. Hinz, A. Schulz and A. Villinger, Chem. – Eur. J., 2014, 20, 3913–3916 CrossRef CAS PubMed.
- S. Demeshko, C. Godemann, R. Kuzora, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2013, 52, 2105–2108 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 2776–2779 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, J. Am. Chem. Soc., 2015, 137, 9953–9962 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, Chem. Sci., 2016, 7, 745–751 RSC.
- Y. Pilopp, J. Bresien, K. P. Lüdtke and A. Schulz, Chem. – Eur. J., 2025, 31, e202403893 CrossRef CAS PubMed.
- Y. Kim and Y. Lee, Angew. Chem., Int. Ed., 2018, 57, 14159–14163 Search PubMed.
- B. S. N. Huchenski, K. N. Robertson and A. W. H. Speed, Eur. J. Org. Chem., 2020, 5140–5144 CrossRef CAS.
- T. Hanna, Coord. Chem. Rev., 2004, 248, 429–440 CrossRef CAS.
- Y. H. Jang and W. A. Goddard, J. Phys. Chem. B, 2002, 106, 5997–6013 Search PubMed.
- M. Mato, D. Spinnato, M. Leutzsch, H. W. Moon, E. J. Reijerse and J. Cornella, Nat. Chem., 2023, 15, 1138–1145 CrossRef CAS PubMed.
- N. D. Chiappini, E. P. Geunes, E. T. Bodak and R. R. Knowles, ACS Catal., 2024, 14, 2664–2670 CrossRef CAS.
- S. Ni, D. Spinnato and J. Cornella, J. Am. Chem. Soc., 2024, 146, 22140–22144 CrossRef CAS PubMed.
- M. Mato, A. Stamoulis, P. C. Bruzzese and J. Cornella, Angew. Chem., Int. Ed., 2025, 64, e202418367 CrossRef CAS PubMed.
- Y. Pilopp, H. Beer, J. Bresien, D. Michalik, A. Villinger and A. Schulz, Chem. Sci., 2025, 16, 876–888 RSC.
- A. W. H. Speed, US Pat., 11161863, 2021 Search PubMed.
- A. Stamoulis, M. Mato, P. C. Bruzzese, M. Leutzsch, A. Cadranel, M. Gil-Sepulcre, F. Neese and J. Cornella, J. Am. Chem. Soc., 2025, 147, 6037–6048 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |