
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMitigating matrix effects in oil and gas wastewater analysis: LC-MS/MS method for ethanolamines†
Glen Andrew D.
de Vera
 *a,
Loredana
Caldiero
b,
Giovanni
Conte
b and
Desirée L.
Plata
a
*a,
Loredana
Caldiero
b,
Giovanni
Conte
b and
Desirée L.
Plata
a
aMassachusetts Institute of Technology, Department of Civil and Environmental Engineering, Parsons Laboratory, 15 Vassar Street, Cambridge, Massachusetts 02139, USA. E-mail: gdevera@mit.edu
bEni SpA – Upstream Technical Services, Via F. Maritano, 26 – 20097 San Donato M.se, MI, Italy
First published on 26th December 2024
Abstract
The high salinity and organic content in oil and gas wastewaters can cause ion suppression during liquid chromatography mass spectrometry (LC/MS) analysis, diminishing the sensitivity and accuracy of measurements in available methods. This suppression is severe for low molecular weight organic compounds such as ethanolamines (e.g., monoethanolamine (MEA), diethanolamine (DEA), triethanolamine (TEA), N-methyldiethanolamine (MDEA), and N,N-ethyldiethanolamine (EDEA)). Here, we deployed solid phase extraction (SPE), mixed-mode LC, triple quadrupole MS with positive electrospray ionization (ESI), and a suite of stable isotope standards (i.e., one per target compound) to correct for ion suppression by salts and organic matter, SPE losses, and instrument variability. The method was evaluated in produced water samples from Italy (NaCl salinity from 8110–18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 mg L−1; diesel range organic compounds ranging from 5.1–7.9 mg L−1). After correcting for matrix effects, ethanolamines in produced water samples were quantified. The first batch of samples (March 2019) had 37–646 μg L−1 total ethanolamines. The second batch of samples (September 2019) had greater ethanolamine content of 77–3976 μg L−1 which was attributed to a reduced water cut during oil production, enhancing the proportionate abundance of these compounds in the aqueous phase. In all samples, DEA and MEA were the dominant ethanolamine species. Possible sources (e.g., corrosion inhibitor and biotransformation) and natural attenuation potential during storage (e.g., at different temperatures, acidification, and addition of sodium azide) were investigated. The developed analytical method enables further investigation of the fate of low molecular weight organic additives in oil and gas development and provides an enhanced ability to evaluate risks associated with chemical release to the environment.
100 mg L−1; diesel range organic compounds ranging from 5.1–7.9 mg L−1). After correcting for matrix effects, ethanolamines in produced water samples were quantified. The first batch of samples (March 2019) had 37–646 μg L−1 total ethanolamines. The second batch of samples (September 2019) had greater ethanolamine content of 77–3976 μg L−1 which was attributed to a reduced water cut during oil production, enhancing the proportionate abundance of these compounds in the aqueous phase. In all samples, DEA and MEA were the dominant ethanolamine species. Possible sources (e.g., corrosion inhibitor and biotransformation) and natural attenuation potential during storage (e.g., at different temperatures, acidification, and addition of sodium azide) were investigated. The developed analytical method enables further investigation of the fate of low molecular weight organic additives in oil and gas development and provides an enhanced ability to evaluate risks associated with chemical release to the environment.
Environmental significanceOil and gas wastewaters pose a threat to drinking water sources due to presence of hazardous chemicals. Of particular concern are ethanolamines, which are extensively used within the industry and have the potential for leaching into the environment. The lack of standardized methods for measuring ethanolamines exacerbates the challenge of assessing and mitigating these risks. This study developed a robust LCMS method for accurately quantifying ethanolamines in produced waters. It comprehensively addressed matrix interferences through the use of solid-phase extraction and compound-specific isotopes. Finally, this study highlights inaccuracies in semi-quantitative approaches that lack authentic standards and careful matrix analysis; this has broad implications to the field. |
Introduction
Wastewaters from oil and gas industries including produced waters – water brought to the surface during extraction – pose a significant risk of contaminating drinking water sources due to the various chemicals involved throughout the extraction process. These chemicals, which include a complex mixture of hydrocarbons, volatile organic compounds, and other halogenated byproducts, among others,1,2 can be released from spills from pipelines or during transport, leaks from wastewater storage ponds/facilities, or subsurface migration from fluids through failed well casings.3 Several compounds in this wastewater are mobile in the environment and capable of leaching into the ground and fresh water sources, negatively impacting water quality and ecosystems. Understanding and addressing potential drinking water contamination would require rigorous analysis and monitoring of oil and gas wastewaters, but several challenges impede comprehensive evaluation of these complex mixtures.Quantitative analysis of individual organic compounds in oil and gas wastewaters are scarce due to instrumental limitations caused by the variable nature of the water samples and wide range of hydrogeochemical parameters. Particularly for liquid chromatography and mass spectrometry (LC-MS), the high salinity and organic matter content in oil and gas wastewaters can cause ion suppression with electrospray ionization. Such effect can be caused by a (a) decrease of the evaporation efficiency of the analyte due to increase in viscosity and surface tension of droplets caused by the sample matrix, (b) coprecipitation of analytes with non-volatile materials (e.g., macromolecules), (c) competition between analytes and interfering compounds for ionization energy that impacts efficiency of the technique, and (d) neutralization in the gas phase.4,5 All of these can affect the amount of charged ion in the gas phase ultimately reaching the detector. The matrix components, especially salts, can also accumulate in the capillary, increasing electric resistance and preventing ions from transfer into the MS. These processes can diminish accuracy and sensitivity of the method (e.g., non-detection of an analyte causing false negatives when ions are suppressed). Thus, a robust method that can correct for these matrix effects is needed for oil and gas wastewaters to be able to assess the risks of organic chemicals that can be inadvertently transported to groundwater sources.6 The method is particularly important for compounds that can partition easily from soil to water and readily transported to downstream drinking water sources.
Ethanolamines (Table 1) comprise a group of compounds that fall under this category. In oil and gas operations, these may be components of corrosion inhibitors, breakers, cross-linkers, or complexing agents of Zr(IV) to control the rate and timing of guar gum crosslinking,7 or components in the removal of acid gases (e.g., H2S)8 and CO2 capture9 during refining. These compounds are miscible in water, not volatile (low vapor pressures of 0.4 torr (for monoethanolamine) and <0.01 torr at 20 °C for di- and tri-ethanolamine),10 and do not adsorb significantly in organic carbon in soil (low organic carbon/water partition coefficient, Koc = log −0.2 to −0.3 for monoethanolamine and diethanolamine).11 These properties increase ethanolamines' likelihood of leaching and subsequent groundwater contamination. Considering the possible carcinogenic effects, as well as the potential for ethanolamine exposure to cause respiratory irritation, liver and kidney damage,12–14 it is important to have methods for accurate measurements of these compounds to better assess health and environmental risks. Ethanolamines can also act as precursors to other hazardous compounds, such as nitrosamines, which may form during water disinfection.15,16 As highlighted by the US Environmental Protection Agency,13 there is no standard method for ethanolamines despite their extensive use in the oil and gas industry. Methods that are available in the literature (e.g., Dionex Themo Fisher Note 271,17 ASTM D-7599 (ref. 18)) were developed using surface water samples and are not directly transferrable to oil and gas wastewater matrices. Matrix effects could result from the samples' high salinities (2000–30000 mg L−1), total suspended solids (TSS ∼ 200 mg L−1), petroleum hydrocarbon residues (around 60 mg L−1 on average, defined as nC10–nC40 or “C10–C40” hydrocarbons), and production chemical additives (e.g., corrosion inhibitors, scale inhibitors, and biocides). These matrix components could give rise to interferences leading to both false positive and false negative results. The inaccurate determination of ethanolamine concentrations may then lead to misplaced regulatory actions if the method does not correct for matrix interferences.
| Unlabelled (labelled) structure | Compound name | Precursor ion, m/z | Quantifier (qualifier) ion, m/z | t R, min | Collision energy, eV | MDL,b μg L−1 |
|---|---|---|---|---|---|---|
| a In parentheses are isotope labelled atoms, column: Acclaim Trinity P1. b MDL = standard deviation (n = 7, 1 μg L−1) × Student's t-value at 99% confidence interval. | ||||||
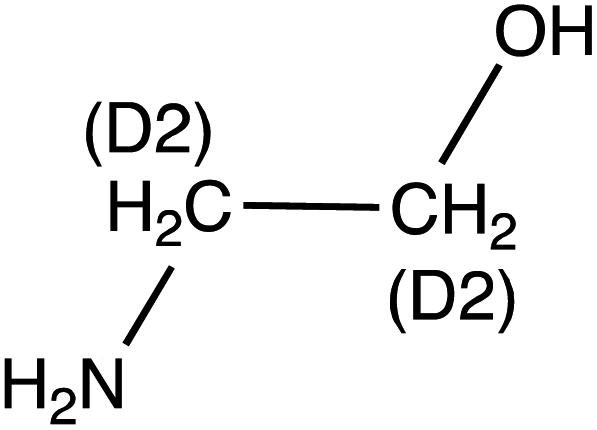
|
Ethanolamine (MEA) | 62.1 | 45 (44.2) | 5.4 | 9 (9) | 0.1 |
| d 4-Ethanolamine (d4-MEA) | 66.1 | 48.1 (30.1) | 5.4 | 9 (33) | ||
|
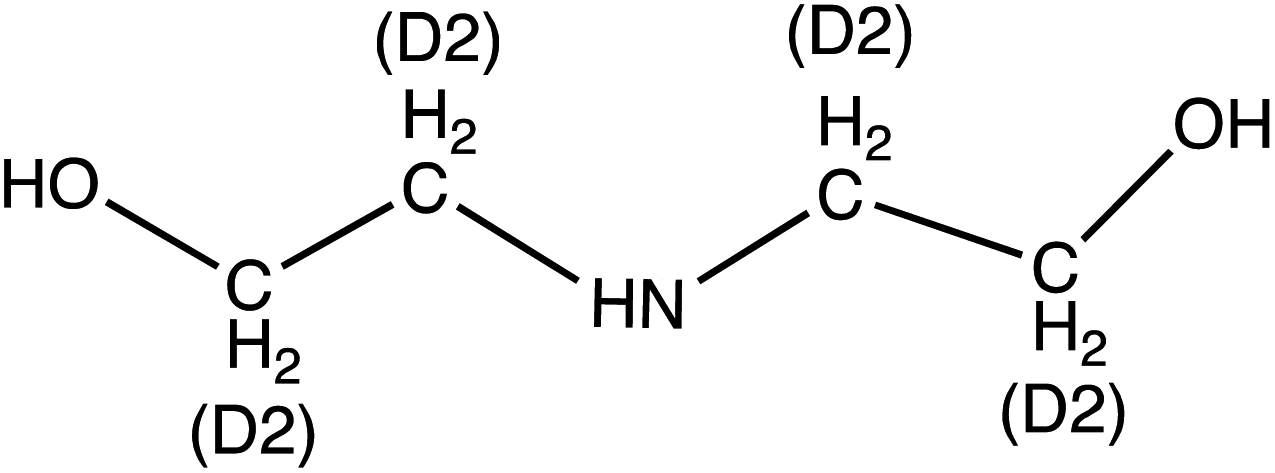
Diethanolamine (DEA) | 106.1 | 88 (69.9) | 4.6 | 9 (13) | 0.1 |
| d 8-Diethanolamine (d8-DEA) | 114.14 | 95.9 (78) | 4.6 | 9 (13) | ||
|
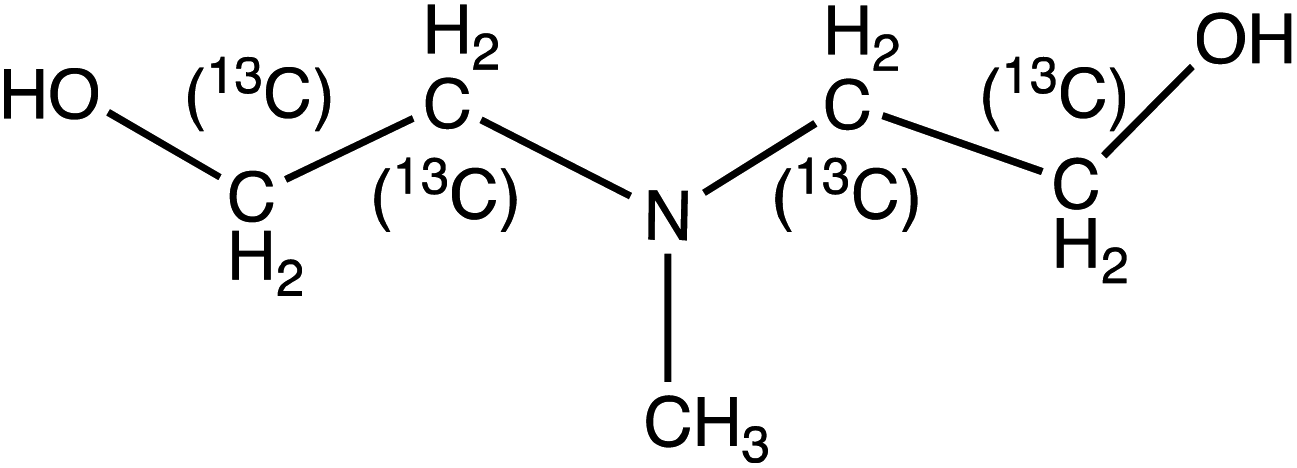
N-Methyldiethanolamine (MDEA) | 120.1 | 101.9 (58.1) | 2.9 | 13 (21) | 0.1 |
| 13C4-N-Methyldiethanolamine (13C4-MDEA) | 124.1 | 105.9 (60) | 2.9 | 13 (21) | ||
|
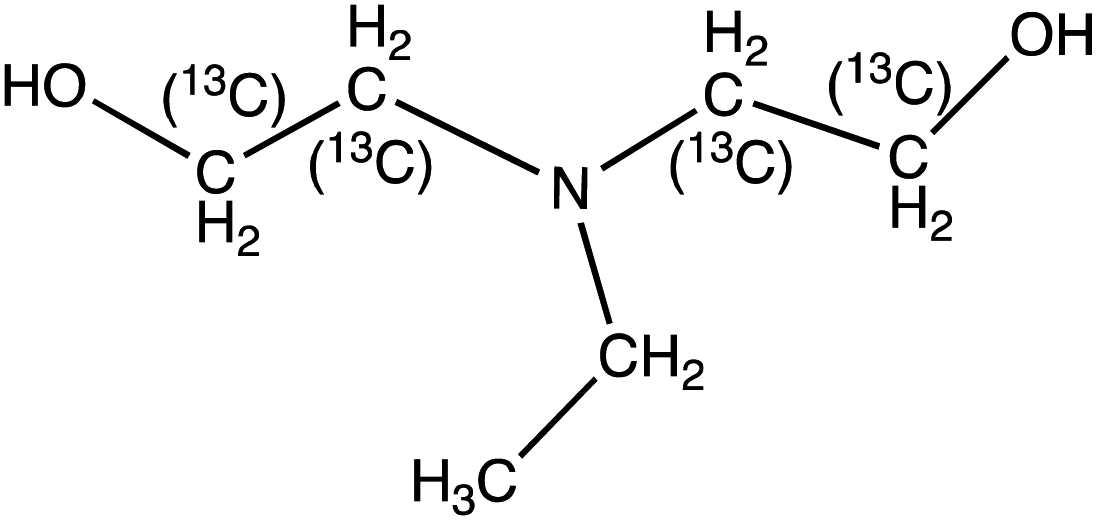
N,N-Ethyldiethanolamine (EDEA) | 134.1 | 115.9 (72, 44) | 2.2 | 13 (21, 25) | 0.2 |
| 13C4-N-Ethyldiethanolamine (13C4-EDEA) | 138.12 | 74 (46) | 2.2 | 17 (25) | ||
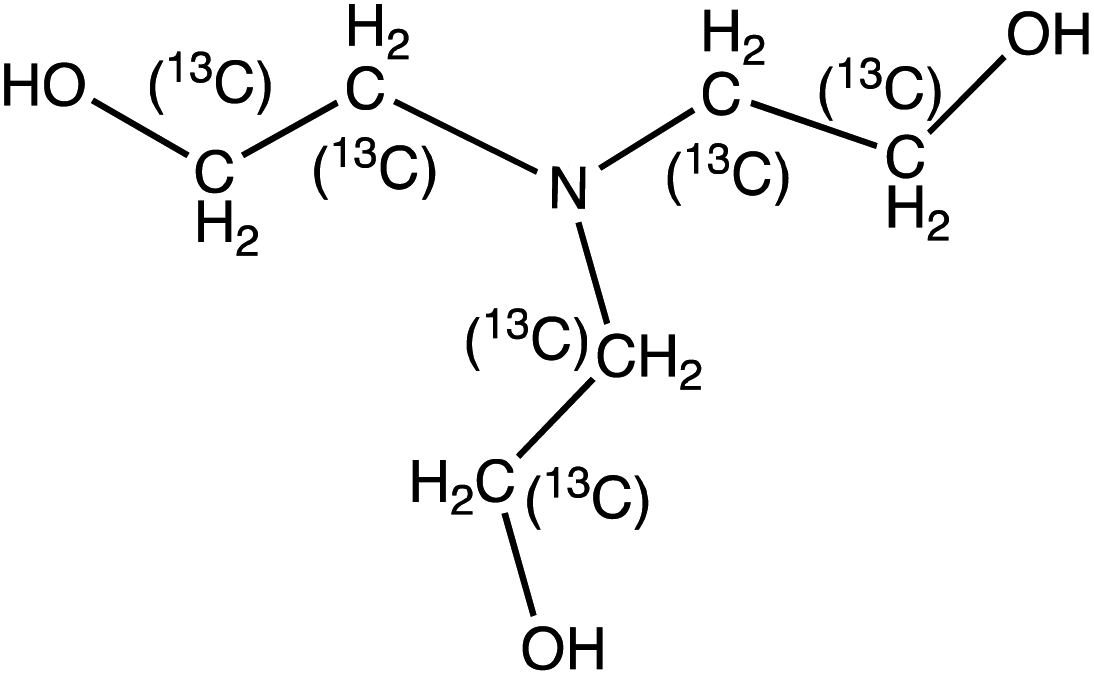
|
Triethanolamine (TEA) | 150.1 | 132 (88, 70) | 3.3 | 13 (18, 21) | 0.2 |
| 13C6-Triethanolamine (13C6-TEA) | 156.15 | 138.1 (47.2) | 3.3 | 12 (28) | ||
This work presents a robust LC-MS/MS method that accurately measures ethanolamines such as monoethanolamine (MEA), diethanolamine (DEA), methyldiethanolamine (MDEA), ethyldiethanolamine (EDEA), and triethanolamine (TEA) in various produced waters from an Italian oil and gas extraction facility. We evaluated interferences across a range of real produced water chemistries and identified ways to correct for these interferences by desalting via solid-phase extraction and use of multiple compound-specific isotopic standards in both synthetic and real water samples. Using the method, matrix effects were negligible. The method in this study was compared to the method proposed by a regional authority for the analysis of ethanolamines (Eni SpA, personal communication, March 2019) to highlight the importance of accounting for matrix interferences in implementing methods that have regulatory implications.
Experimental methods
Reagents and materials
The reagents and materials used for sample preparation and LC–MS analysis are described in Text S1 (ESI†). Methanol, acetonitrile, ammonium formate, formic acid, sodium hydroxide, unlabeled and labelled ethanolamine standards and other chemicals used for extraction and matrix effects evaluation (salts, Aldrich humic acid) were of reagent grade or higher. Solutions were prepared using Milli-Q ultrapure water (18.2 MΩ cm, Millipore).520 mg L−1 salinity (Table S2†) by diluting stock solutions of sodium chloride (50000 mg L−1), sodium bromide (2500 mg L−1), sodium sulfate (5000 mg L−1), and calcium carbonate (5000 mg L−1). A humic acid solution (Aldrich, WI, USA) was prepared to achieve total organic carbon (TOC) concentrations of 0–295 mgC L−1, simulating organic matter levels found in produced waters.20 Each solution was spiked with ethanolamine mixture (20 μg L−1 for each compound). Another set of samples was generated from the bulk organic matter extracted from PW-March samples (following solid-phase extraction, SPE). These extracts were spiked with isotopically labeled ethanolamines (10 μg L−1 per compound) to assess suppression caused by co-extracted organic compounds (Table S3†).
Mass spectrometry was performed using an Agilent 6495 iFunnel triple quadrupole system in positive ESI mode, and spectral data were acquired by multiple reaction monitoring (Table 1). Analysis was done using optimized MS ion source conditions (capillary = 2000 V, ion funnel high pressure RF = 150 V, low pressure RF = 50 V, nebulizer pressure = 50 psi, nozzle voltage = 0 V, sheath gas heater = 330 °C, sheath gas flow = 11 L min−1, drying gas temperature = 150 °C, drying gas flow = 18 L min−1). MEA was the compound with the weakest response, and optimization of compound- and source-dependent parameters was performed to maximize sensitivity for this target molecule. The most abundant product ions (Text S3, Fig. S4†) were used for quantification and confirmation, and were similar to those reported in Dionex and ASTM methods.
The concentrations of ethanolamines were determined by the relative response of each target ethanolamine to its isotopically labelled internal standard. Qualifier ions (Table 1) were used for peak confirmation, with close retention times as a quantification criterion. A laboratory-fortified sample (15 μg L−1 ethanolamine mixture spiked into LCMS-grade water) was analyzed during each SPE run to verify recovery, and sample matrix spikes were assessed during method development. To account for potential contamination, laboratory reagent blanks were included in every batch, and field blanks were analyzed alongside samples. Fresh calibration solutions were prepared for each analysis, and bracket standards were run after every 6–8 sample injections as part of quality control to ensure consistent system performance throughout the run.
Results and discussion
Method sensitivity, linearity, accuracy, and precision
The method detection limit (MDL) was determined by measuring ethanolamine concentrations in seven pure water samples fortified with 1 μg L−1 authentic standards and 10 μg L−1 isotope standards. The MDL was calculated by multiplying the standard deviation of the replicate measurements by the student's t-value (n − 1 degrees of freedom).22 The MDLs (Table 1) ranged from 0.1 μg L−1 (MEA, DEA, MDEA) to 0.2 μg L−1 (EDEA, TEA). The reporting limit was chosen to be 1.0 μg L−1 (1.0 ppb, level 1 calibration standard), which is 5 to 10 times greater than the calculated MDL. At this concentration, all compounds had acceptable signal-to-noise ratios (>100), were resolved from one another, and clearly detectable. The LC/MS method was calibrated with each analyte at 1.0–40.0 μg L−1 concentrations and an isotopically labelled version of each analyte was added to each calibration point (10.0 μg L−1) to generate a relative response ratio (Fig. S5†). Linear regression with 1/x weighting was used for all ethanolamines, where x is concentration, following the Dionex and ASTM methods.17,18 Coefficients of determination were required to be at least 0.990. In order to verify precision, replicate measurements of standard solutions were carried out. Precision was assessed in terms of repeatability of concentration (expressed as percent relative standard deviation, % RSD) and accuracy was evaluated in terms of recovery. Calculated ethanolamine concentrations were found to be reproducible with RSD below 10% (Table S6†) and recoveries in ultrapure water of 92–107% were observed (Table 2).| Level, μg L−1 | MEA | DEA | TEA | MDEA | EDEA |
|---|---|---|---|---|---|
| 1 | 92 (2) | 100 (7) | 103 (8) | 107 (2) | 97 (3) |
| 5 | 102 (2) | 100 (4) | 99 (3) | 98 (2) | 98 (2) |
| 10 | 103 (3) | 101 (5) | 100 (3) | 98 (2) | 101 (2) |
| 15 | 100 (3) | 99 (2) | 100 (4) | 101 (3) | 103 (5) |
| 20 | 99 (2) | 100 (2) | 100 (9) | 100 (6) | 98 (0) |
| 40 | 100 (1) | 103 (9) | 104 (4) | 100 (3) | 95 (2) |
Ion suppression and enhancement in ESI
Ion suppression can diminish accuracy and sensitivity of the method (e.g., non-detection of an analyte causing false negatives when ions are suppressed). Critically, we note that matrix effects can lead to overestimation if the internal standard experiences more severe suppression, resulting in disproportionate signal enhancement of the analyte (Fig. 1). Dionex 271 and ASTM D7599 methods are susceptible to these effects as they rely on the use of only d8-DEA as an internal standard. In this study, we investigated the impact of produced water matrix on the sensitivity of the method by monitoring the peak areas of the added isotopically labeled ethanolamine standards (constant at 10 μg L−1 level) for each sample. Due to the variable characteristics of the samples, the extent of ion suppression or enhancement was highly variable and compound dependent (Fig. 1). For example, the signals of d4-MEA in all produced water samples decreased by more than 90% and were hardly detectable (Fig. S6†). Similarly, d8-DEA, the recommended standard in Dionex 271 and ASTM D7599 exhibited high degrees of ion suppression (up to 80%; Fig. 1a). These results are consistent with other studies23 showing that smaller compounds (e.g., d4-MEA and d8-DEA) were more susceptible to ion suppression due to possible ion–ion interaction with more massive and highly charged molecules (e.g., bulk organic matter). Thus, without correcting for ion suppression, LC/MS analysis of ethanolamines in produced water samples could yield false negative results. Conversely, false positive results or overestimations would be obtained in the event that an internal standard used for other ethanolamines, such as d8-DEA, (i.e., following the Dionex 271 (proposed by a regional authority) and ASTM D7599 recommendations) is impacted significantly by matrix effects. For example, signal enhancement in MDEA (added at 10 μg L−1 consistently) was observed in three of the four PW samples and in one sample for TEA (Fig. 1a) while DEA signals were dramatically suppressed in those same waters. Consequently, the relative abundance of MDEA would be augmented by both the enhanced MDEA sensitivity and the diminished DEA response, generating an overestimation of the real MDEA abundance. A strategy that relies on reducing the salt content of the sample through the use of SPE resins and leveraging compound-specific isotopically labeled authentic standards is proposed in this study, and utilization of this method could help eliminate the inaccurate determinations of ethanolamines provided by the Dionex 271 and related methods.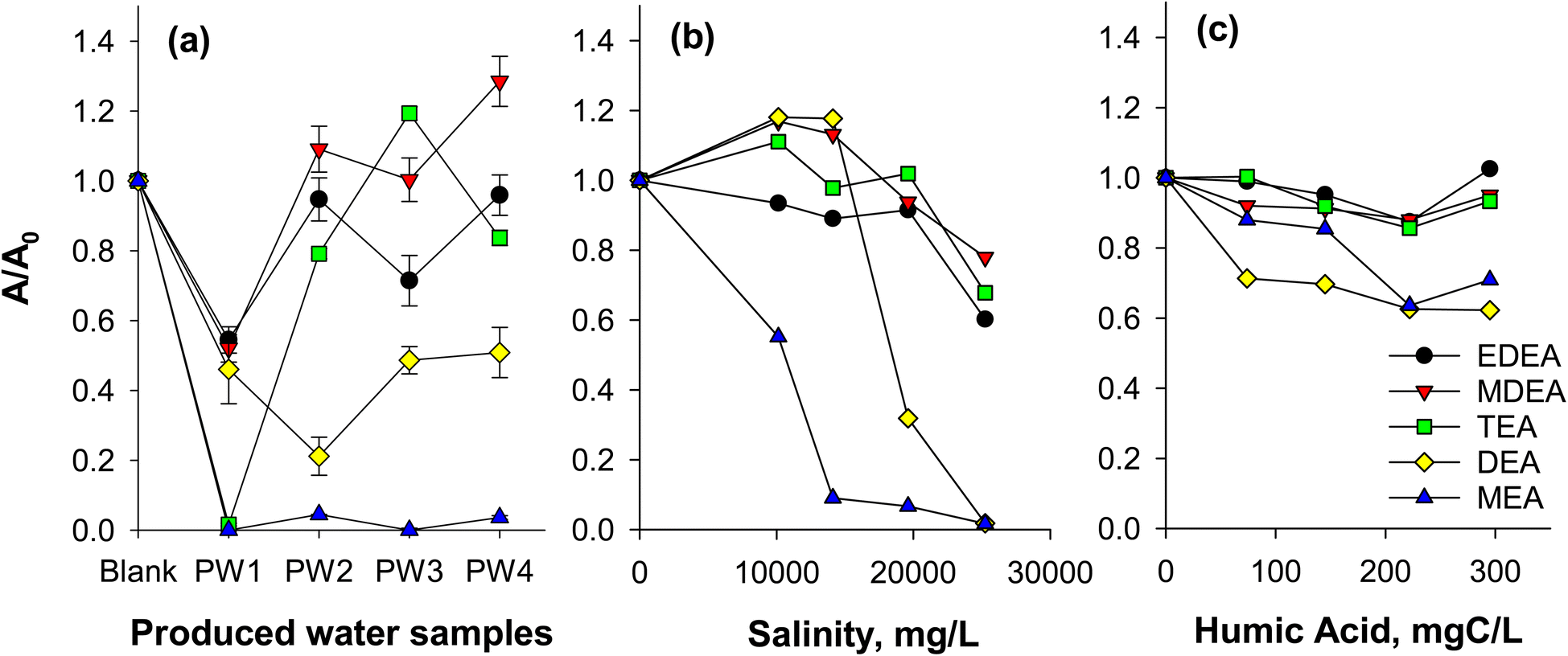 | ||
| Fig. 1 Ion suppression in (a) produced water samples (PW-March), (b) synthetic brine solutions, and (c) humic acid solutions. A/A0 corresponds to the area ratio of ethanolamines with matrix effects to ethanolamines without matrix effects (e.g., in ultrapure water). Area ratios in (a) were obtained from isotopic standards (10 μg L−1) of each ethanolamines, and are independent of unlabelled ethanolamines present in the sample. Error bars are standard errors (n = 3). For (b) and (c), area ratios were from unlabelled ethanolamines (20 μg L−1) spiked to synthetic brine and humic acid-containing solutions. Note that lines are only used to guide the eyes. | ||
To further understand the effects of salts and bulk organic matter, experiments using ethanolamine-spiked synthetic brine and humic acid solutions were conducted (Fig. 1b and c). DEA and MEA experienced a high degree of ion suppression (up to more than 90%) at salinities of over 20000 mg L−1 (Fig. 1b), confirming the contribution of salts for reduced analyte response. In addition to diminished mass spectral sensitivity, salts also caused significant retention time shifts (Fig. S7†) using the Acclaim Trinity P1 column. Analytes eluted earlier as salt concentration was increased. For example, retention time for MEA changed from 5.2 min to 3.6 min when salinity was increased from 0 to 25250 mg L−1, respectively (Fig. S8†). The shift in retention time was caused by the modified ion exchange properties of the column, where salts from the sample compete with ethanolamines for retention to the stationary phase. Large shifts in retention times are not acceptable, as they affect repeatability and may lead to non-detects (i.e., false negatives) when quantification criteria require retention times consistent with the calibration standards. In some cases, false positives would also be possible if the retention time of an interfering compound shifted into the response window of the ethanolamine and produced an interfering diagnostic mass fragment ion.
To investigate the effect of bulk organic matter on ion suppression, synthetic humic acid solutions (0–295 mgC L−1) were spiked with 15 μg L−1 ethanolamines. Note that the humic acid is representative of dissolved organic matter and may indicate an effect that could occur in the presence of hydrocarbons as well (i.e., in produced water). These synthetic mixtures were directly injected to the LC/MS to observe organic matter-induced ion suppression. Bulk organic matter was also found to cause ion suppression (Fig. 1c). Consistent with earlier results, DEA and MEA were the most impacted ethanolamines, with suppression of 30–40% at 295 mgC L−1 humic acid.
Correction of matrix effects
Sample preparation procedures were next investigated to correct for ion suppression by salts and organic compounds, as well as retention time shifts caused by high salt content. Sample clean-up by SPE was tested for improved ethanolamine detection and quantitation. Optimization experiments involved determination of (a) appropriate SPE cartridge, (b) conditions for analyte retention, (c) eluent for enhanced recovery, and (d) correction of losses using isotope-labelled internal standards.Solid-phase extraction
The SPE procedure was optimized by first evaluating appropriate SPE cartridge material and elution procedure. Oasis HLB and Agilent PPL cartridges were assessed since they were reported in previous studies for clean-up of produced water samples.24,25 For Oasis HLB, all ethanolamines except EDEA had poor adsorption (Fig. S9†). In contrast, Agilent PPL cartridge was able to retain most ethanolamines (Fig. S10†). The compounds were not observed in PPL's filtrate during loading and washing steps but were recovered during elution with methanol (bottom chromatogram in Fig. S10†). Therefore, the Agilent PPL cartridge was used to optimize the SPE method and no further experiments were conducted with the HLB cartridge.Although the recoveries and precision of SPE demonstrated good consistency across the pH range of 2.6 to 11.0 (Fig. S11†), sample pH was adjusted to pH 11 prior to extraction. This step ensured that ethanolamines predominantly existed in their deprotonated form (>97%, Fig. S12†), while also standardizing the initial pH of all samples, regardless of their source and composition. The suspended particles and precipitates that form at this pH require filtration through glass fiber filters (0.7 μm, pre-combusted at 450 °C for 12 h) prior to SPE. Pre-combusted glass fiber filters are preferred due to possible contamination from binders in plastic syringe filters (Fig. S13†).
In addition to the type of cartridge, the eluent composition also significantly affected ethanolamine recoveries (Fig. S14†). Using only methanol as the eluent, ethanolamine recoveries were at 2–55%. This recovery increased to 80–113% when the eluent was acidified with 2% formic acid, likely due to the decreased affinity of ethanolamines with the SPE material when converted back to their charged state via pH-dependent protonation.
Overall, the final SPE procedure in this study employed the Agilent PPL cartridge and the following steps: conditioning with 3 mL of methanol twice (flow rate 2 mL min−1), equilibration with 3 mL of pure water twice (flow rate 2 mL min−1), loading of 3 mL of pH 11 samples twice (at 1 mL min−1), washing/desalting with 3 mL of ultrapure water five times, 15 min cartridge drying under vacuum, and final elution with 3 mL of 2% formic acid in 90/10 methanol/water twice (flow rate 1 mL min−1). This procedure was tested against a synthetic brine solution (salinity of 25250 mg L−1, Fig. S15†) and the produced water samples (Fig. 2). The method was successful in desalting and minimizing ion suppression. Note that there was no concentration factor applied in this procedure, as target concentrations were achievable even without a concentration step. If future analysis requires increased sensitivity (e.g., below 1 μg L−1 (1 ppb)), the SPE extracts can be rotary evaporated to obtain the desired concentration factor. The total ion chromatograms of ethanolamines after SPE of the brine solutions and samples resulted in improved peak shapes, sensitivity, resolution, and repeatability of retention times across samples (Fig. 2).
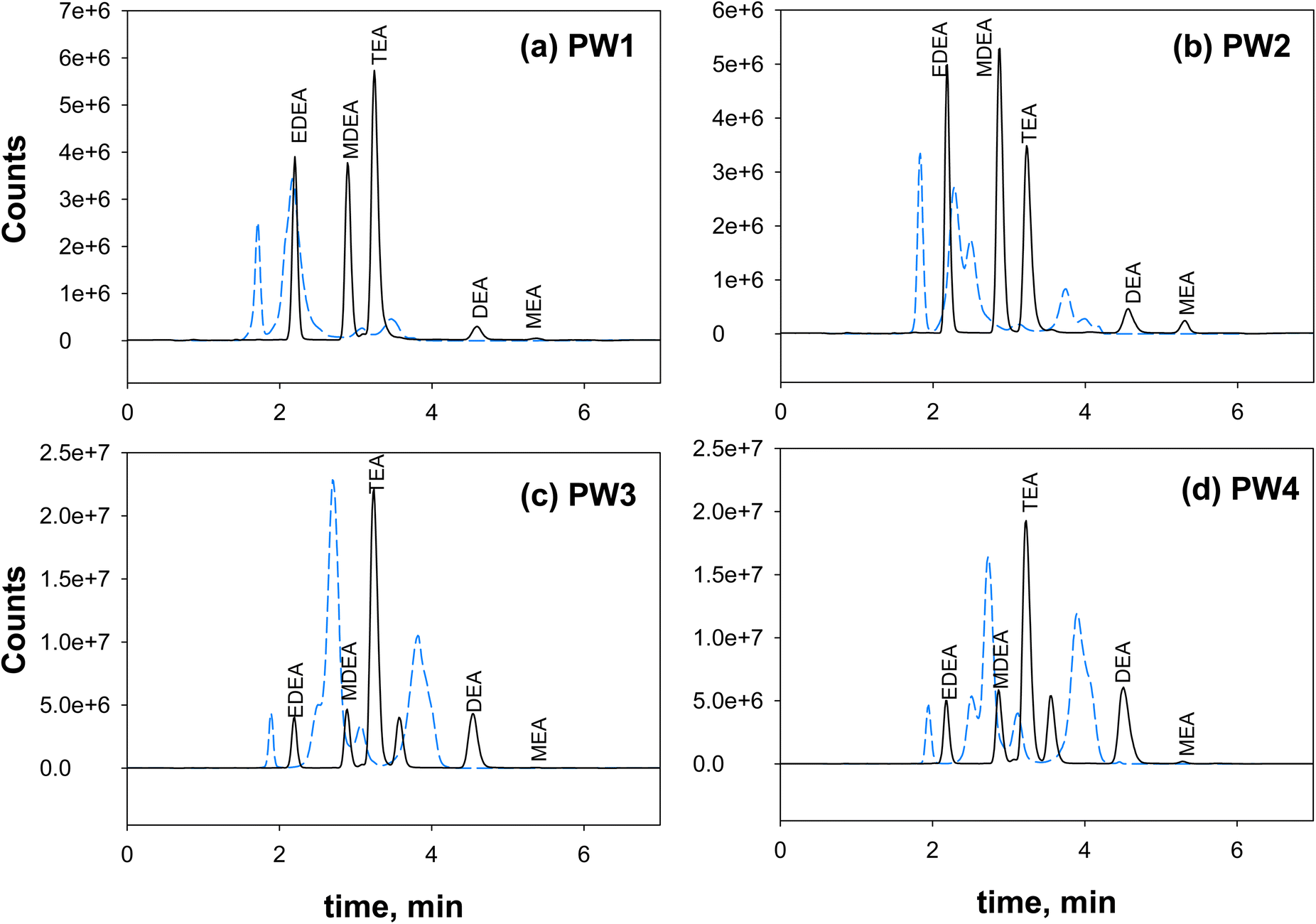 | ||
| Fig. 2 Total ion chromatograms (TIC) of ethanolamines (15 μg L−1) in PW-Mar samples ((a) PW1, (b) PW2, (c) PW3, (d) PW4) showing improved method sensitivity and peak resolution using the proposed SPE procedure. X-axis is time (min) and y-axis is counts/signal intensities. Peaks from left to right: EDEA, MDEA, TEA, DEA, MEA. Black line: chromatogram with SPE, Blue dashed line: chromatogram without SPE. | ||
Use of compound-specific stable isotopes
While SPE results in improved detection capability, it still suffers from interferences from co-eluting organic matter from the sample matrix, ion suppression effects, as well as losses from the sample loading and elution procedures. Recovery of different ethanolamines vary by type of sample (Fig. S16†), and this can be attributed to the varying water quality. We consistently observed that isotopically labelled DEA and MEA in produced water samples have low recoveries (e.g., less than 30% in produced water samples versus more than 50% in ultrapure water). This can be due to ion suppression caused by other matrix constituents (e.g., organic matter) not removed during desalting by SPE.The impact of the organic matter from produced waters was investigated by spiking isotopic standards to sample SPE extracts (Fig. S17†). This means that the samples have already been desalted, and SPE extracts only contained organic matter and ethanolamines from the sample. Compared to other ethanolamines, EDEA and MDEA were not significantly affected by the co-extracted organic matter. The opposite was observed for DEA and MEA. By comparing these results to samples spiked with isotopic standards followed by SPE, we could attribute the overall loss of ethanolamines from the SPE procedure and ion suppression caused by the bulk organic matter (Fig. S18†). Thus, to correct for these effects, isotope-labelled standards specific for each ethanolamine are required to accurately measure ethanolamine concentrations (Fig. S19†). Measuring concentrations of ethanolamines using only one internal standard (e.g., d8-DEA, i.e., indicated in the Dionex and ASTM methods) will not provide accurate results due to varying degrees of ion suppression per analyte (Fig. 3). Indeed, underrecovery or suppression of d8-DEA would lead to artificially enhanced other ethanolamine measurements in the absence of compound specific, isotopically labelled authentic standards.
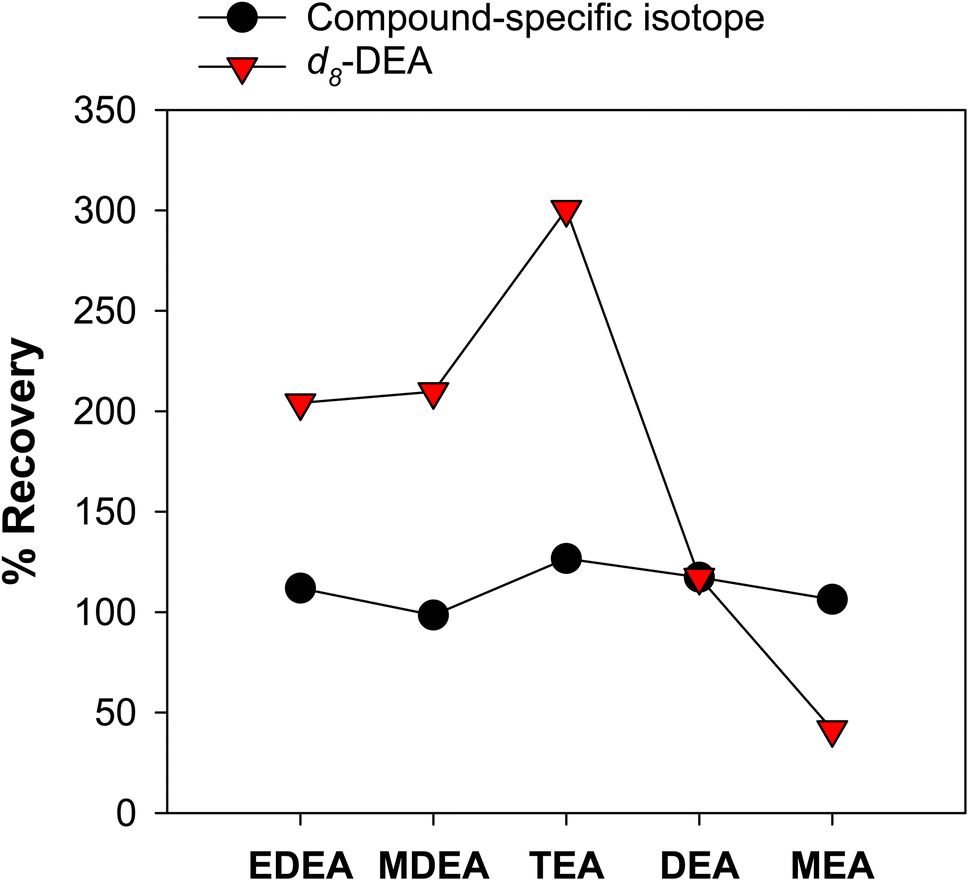 | ||
| Fig. 3 Impact on accuracy of using a single (d8-DEA) and compound-specific internal standards. Black symbols are ethanolamine recoveries using compound-specific isotopic standards. Red symbols are ethanolamine recoveries determined using d8-DEA alone, as recommended in the Dionex (proposed by a regional authority) and ASTM methods. % Recovery = internal standard response ratios before SPE/internal standard response ratio after SPE; calculated from 20 μg L−1 ethanolamine standards spiked in pure water; response ratio refers to the peak area of the target ethanolamine/peak area of the internal standard. | ||
Quantification of ethanolamines in produced water samples
Using the SPE procedure and multiple isotopic internal standards to correct for matrix effects, ethanolamines in produced water samples (PW-Mar, PW-Sept) were measured to prove practicality of the developed method in real applications. As previously discussed (Fig. 2), without SPE, ethanolamine peaks co-eluted at inconsistent retention times. With desalting by SPE, ethanolamines in produced waters were magnified with good resolution and eluted at repeatable retention times as the calibration standards resulting in more robust, sensitive, and accurate measurements. The addition of multiple isotopic internal standards further corrected for losses during the SPE procedure, instrument variability, and possible ion suppression from other matrix constituents. This study shows that the proposed method is applicable to water samples across a wide range of salinity and matrix compositions. Using this approach, matrix spike recoveries (spike level = 15 μg L−1) averaged (±standard deviation) 118 (±17) for MEA, 108% (±2) for DEA, 104% (±1) for MDEA, 108 (±1) for EDEA, and 107% (±1) for TEA across all PW samples (Table 3).| Compound | Ultrapure water | PW1 | PW2 | PW3 | PW4 |
|---|---|---|---|---|---|
| MEA | 103 (1) | 133 (4) | 97 (4) | 130 (3) | 111 (3) |
| DEA | 105 (1) | 108 (1) | 105 (2) | 110 (10) | 108 (10) |
| MDEA | 106 (2) | 106 (2) | 103 (1) | 104 (2) | 104 (2) |
| EDEA | 108 (4) | 109 (5) | 107 (4) | 107 (1) | 108 (2) |
| TEA | 103 (2) | 109 (6) | 107 (1) | 107 (4) | 107 (3) |
All produced water samples were found to contain some level of a variety of ethanolamines (Fig. 4). Among the samples, PW1 had the least amount of ethanolamines in both sampling events. In March 2019 samples, PW3 and PW4 had the highest total ethanolamine concentration, with DEA being the most abundant compound (432–465 μg L−1). PW3 and PW4 had relatively similar ethanolamine concentrations since both were collected from locations before reinjection to wells. PW4 was collected from a stock tank of all produced waters that were delivered to PW3 where reinjection occurs. On the other hand, PW1 and PW2 samples were taken from different oil separators. PW1 and PW2 samples (March 2019) had low ethanolamine content and were predominantly MEA (26–153 μg L−1). When samples were collected in September 2019, the concentration of ethanolamines was notably different. PW3 and PW4 still had similar concentration profile, but with DEA reaching over 1000 μg L−1. For these samples, MEA also increased to more than double its initial concentration, while TEA was relatively consistent. The biggest jump in ethanolamine concentrations occurred with PW2, which can be explained by different sampling location and water cuts of the well in March and September 2019. “Water cut” refers to the mass percentage of water compared to the total liquids from an oil well. The March 2019 sample was taken with 40% water cut while the samples from September 2019 only had 20%. Since the water cut decreased, the abundance of DEA in the aqueous phase was enhanced (i.e., DEA partitioned as it usually does, but into a smaller water volume). This resulted in a 1000-fold increase in DEA concentration, where PW2-Sept contained 3830 μg L−1 DEA. The proportional change in water cut did not correlate to the proportional change in ethanolamine concentrations, likely due to differences in composition of the produced waters over the hydrocarbon extraction that occurred in a 6 month time span or variations in utilized additives as well as transformations that may have transpired. In addition to changes in DEA, the shift to a lower water cut in PW2-Sept also led to the detection of MDEA (12.5 μg L−1) compared to PW2-Mar (below 1 μg L−1).
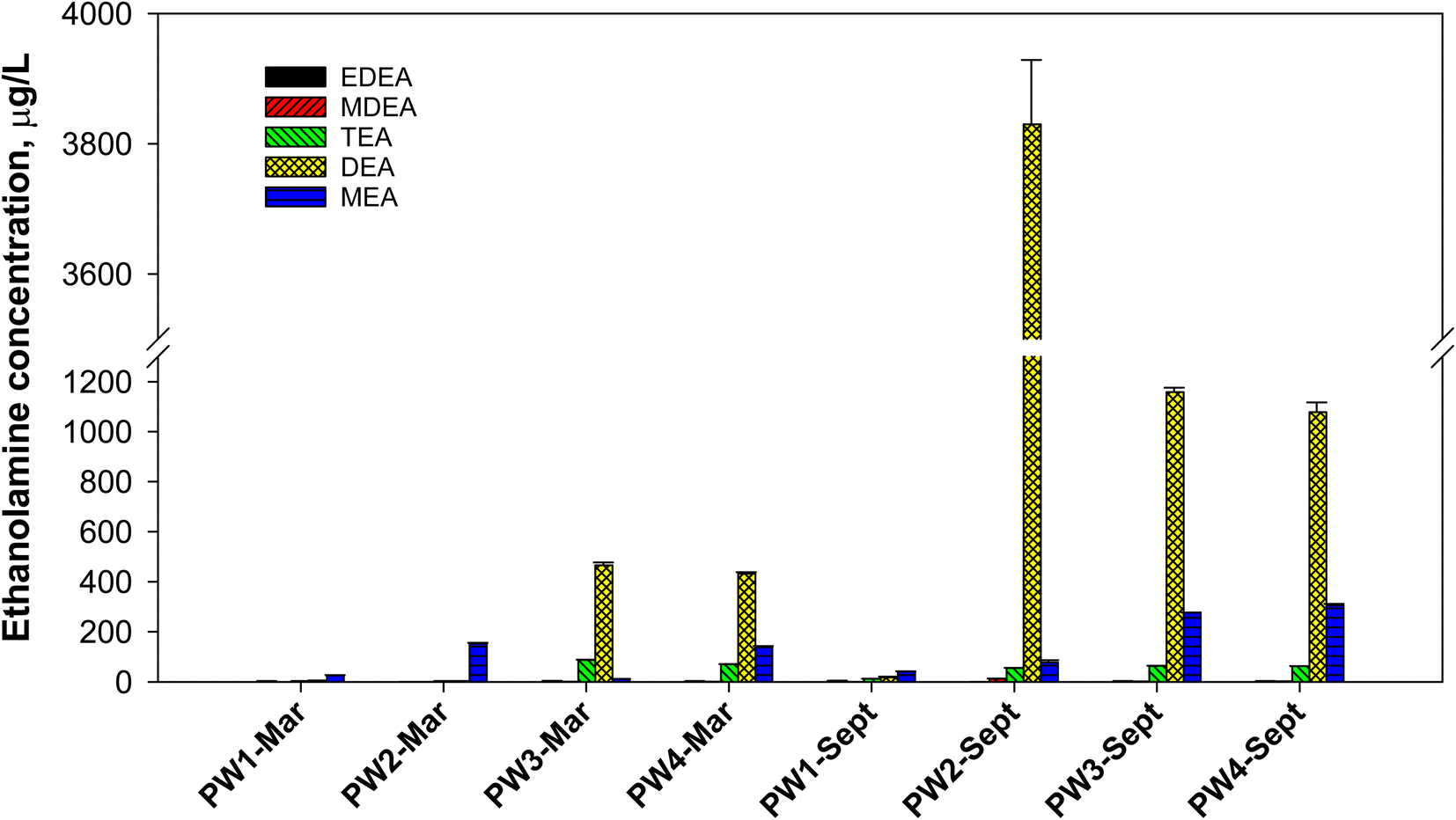 | ||
| Fig. 4 Concentrations of ethanolamines in produced water (PW) samples quantified using the proposed analytical method that involves SPE and compound-specific isotopic internal standards. Field blanks < 1 μg L−1 ethanolamines. Samples were diluted appropriately to fit the linear range. Error bars are standard deviation for PW-Mar, absolute deviation for PW-Sept. | ||
Microbial transformations can significantly influence ethanolamine concentrations in produced waters. For example, DEA can be produced from ethoxylated fatty amines in surfactants. For ethoxylated fatty amines, microbial degradation can cleave the Calkyl–N bond through a hydrogenation reaction forming DEA as a breakdown product. A closed-bottle test by van Ginkel et al. (1993) reported that alkylbis(2-hydroxyethyl)amine was rapidly biodegradable via an initial oxidation of the alkyl chain.26 The secondary amine byproducts did not biodegrade readily, contributing to the persistence of the amine byproducts. The same research group also reported that biodegradation of octadecylbis(2-hydroxyethyl)amine catalyzed by tertiary amine dehydrogenase could form DEA as one of the products.27 Thus, long chain ethanolamines could serves as potential sources of DEA. In all PW samples, m/z 190 consistently appeared at about 1 min (Fig. S20†). Although preliminary, m/z of 190 may correspond to N-hexyldiethanolamine and its biodegradation could offer a potential route for DEA formation via a pathway described in Fig. S21.† Further studies are warranted to confirm if these biotransformation routes can contribute towards the abundance of DEA in produced waters.
It is important to note that concentrations discussed in this section were determined using multiple isotopic internal standards and not d8-DEA alone, as recommended in the Dionex and ASTM methods. Use of one compound-specific, isotopically labelled internal standard per compound is necessary for accurate quantitation, as each ethanolamine can undergo ion suppression at varying degrees (Fig. 1). Critically, utilizing only d8-DEA as the internal standard can lead to overestimation of EDEA, MDEA, TEA via signal suppression of d8-DEA (via mechanisms described above) (Table S7†). MEA was underestimated when only d8-DEA was employed as a standard, as ion suppression for this compound was more severe compared to DEA. The use of a suite of internal standards and SPE extraction is necessary for accurate determination of ethanolamines.
Natural attenuation during storage and incubation
To check for formation or disappearance from biological or abiotic degradation pathways, several preservation techniques, such as acidification (pH 2), azide sterilization, freezing, and refrigeration were evaluated for 56 days. The spiked ethanolamines had varying trends when exposed to different storage conditions (Fig. S22†). EDEA and MDEA were relatively stable at all storage conditions (less than 20% decrease) while TEA and MEA decreased in 56 days. DEA was found to degrade by 50% after 7 days of storage and remained relatively at same concentration after a week. The decrease in DEA was accompanied by a slight increase in MEA which suggests possible biodegradation of DEA to MEA.28 Note that the loss of DEA at all storage conditions indicates that the use of d8-DEA as an internal standard (as recommended by the Dionex 271 and ASTM D7599 methods) would lead to artificial enhancement of ethanolamine quantitation if the samples are not analysed in a timely fashion (i.e., less than 7 days).For MEA, a significant decrease (70%) was observed for PW2 samples stored at room temperature (20 °C), while that for PW1 remained unchanged. This suggests differences in abundance and characteristics of microbial population between samples. Since PW2 samples showed biological activity (as loss of MEA at 20 °C in Fig. S22†), this sample was used as source of seed bacteria to induce biodegradation of ethanolamines in dilute solution of Versalis E-cori corrosion inhibitor (1.7 mg mL−1, contained 6.2 mg L−1 MEA). A synthetic mixture of two mL of PW2 samples were added to the corrosion inhibitor solution (60 mL) and changes in ethanolamine concentrations were monitored at 20 °C (Fig. 5). MDEA and EDEA in this solution were below 1 μg L−1 and remained so throughout the experiment. The results for DEA and TEA (over 75% and complete mineralization, respectively, after 56 days) suggest that occurrence of biodegradation in corrosion inhibitors could occur in natural microbial populations in or derived from produced water. Thus, ethanolamine transformations should be an expected source of inter-well variability and sample preservation techniques must be carefully designed. Further studies could explore the use of recovery standards added in the field to determine the influence of this process on key ethanolamine analytes.
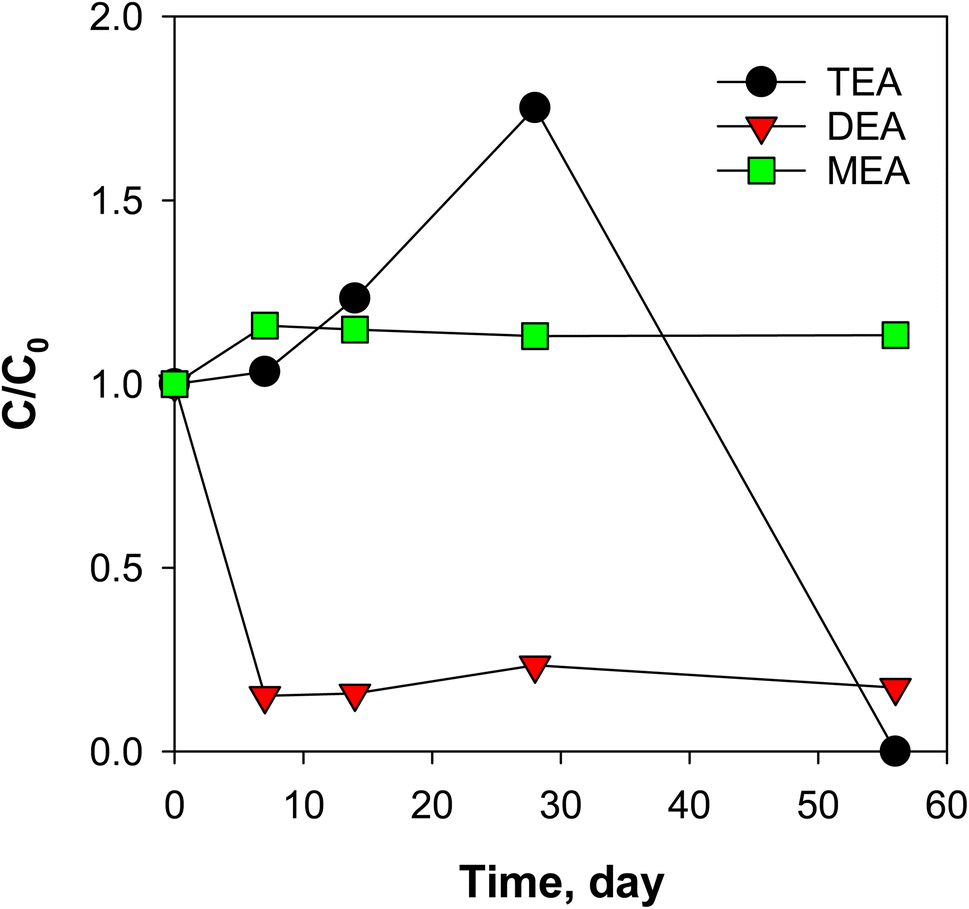 | ||
| Fig. 5 Inoculation of Versalis E-cori corrosion inhibitor (1.7 mg mL−1) with PW2-Mar sample (2 mL) suggesting microbial transformations of ethanolamines. EDEA and MDEA were below the quantification limit of 1 μg L−1. Total volume = 60 mL, temperature = 21 °C. | ||
Overall, the results of these stability tests show that acidification and azide addition or low-temperature preservation (i.e., freezing) can preserve the samples well. To minimize hazards associated with the use of acid and biocide, storing samples at cold conditions is strongly recommended. Freezing samples are preferred to limit biological activity even for long-term storage of samples (e.g., 56 days).
Conclusions
This study presents a reliable analytical method for the accurate quantification of ethanolamines in highly saline and organic-rich produced waters from oil and gas operations. A detailed step-by-step procedure is outlined in Text S4 and Fig. S23.† By utilizing SPE with Bondesil PPL cartridges and compound-specific isotopic standards, followed by triple quadrupole LC/MS, this method offers a robust and adaptable approach for analyzing ethanolamines in a wide range of water types. It effectively accounts for varying matrix constituents, correcting for significant ion suppression caused by salts and organic compounds. The method demonstrated its reliability across different sample conditions, providing detailed insights into ethanolamine concentrations and their variation with operational parameters. DEA and MEA were the most prevalent species identified. Further research is necessary to identify other additives that could contribute to ethanolamines in oil and gas operations and to further investigate compounds that can be biotransformed to produce ethanolamines. Inter-laboratory studies are also warranted to allow for comparison of results and standardize this method for use in the energy sector.This work emphasized that matrix effects should be rigorously evaluated in future LC/MS methods involving complex water chemistries, not only for ethanolamines but also for other low molecular weight compounds. The proposed method offers a valuable tool for further investigations into the environmental fate and risks of chemical additives in oil and gas production, contributing to improved environmental management and regulatory assessments.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Eni SpA through the MIT Energy Initiative. The authors are grateful to Barathkumar Baskaran for reviewing the manuscript and the anonymous reviewers for the constructive comments.References
- G. J. Getzinger, M. P. O'Connor, K. Hoelzer, B. D. Drollette, O. Karatum, M. A. Deshusses, P. L. Ferguson, M. Elsner and D. L. Plata, Natural Gas Residual Fluids: Sources, Endpoints, and Organic Chemical Composition after Centralized Waste Treatment in Pennsylvania, Environ. Sci. Technol., 2015, 49(14), 8347–8355, DOI:10.1021/acs.est.5b00471.
- D. G. Abraham, H. K. Liberatore, Md. T. Aziz, D. B. Burnett, L. H. Cizmas and S. D. Richardson, Impacts of Hydraulic Fracturing Wastewater from Oil and Gas Industries on Drinking Water: Quantification of 69 Disinfection by-Products and Calculated Toxicity, Sci. Total Environ., 2023, 882, 163344, DOI:10.1016/j.scitotenv.2023.163344.
- L. Konkel, Salting the Earth: The Environmental Impact of Oil and Gas Wastewater Spills, Environ. Health Perspect., 2016, 124(12), A230–A235, DOI:10.1289/ehp.124-A230.
- P. Panuwet, R. E. Hunter, P. E. D'Souza, X. Chen, S. A. Radford, J. R. Cohen, M. E. Marder, K. Kartavenka, P. B. Ryan and D. B. Barr, Biological Matrix Effects in Quantitative Tandem Mass Spectrometry-Based Analytical Methods: Advancing Biomonitoring, Crit. Rev. Anal. Chem., 2016, 46(2), 93–105, DOI:10.1080/10408347.2014.980775.
- J.-P. Antignac, K. de Wasch, F. Monteau, H. De Brabander, F. Andre and B. Le Bizec, The Ion Suppression Phenomenon in Liquid Chromatography–Mass Spectrometry and Its Consequences in the Field of Residue Analysis, Anal. Chim. Acta, 2005, 529(1), 129–136, DOI:10.1016/j.aca.2004.08.055.
- US EPA and C. Ridley, Hydraulic Fracturing for Oil and Gas: Impacts from the Hydraulic Fracturing Water Cycle on Drinking Water Resources in the United States (Final Report), https://cfpub.epa.gov/ncea/hfstudy/recordisplay.cfm?deid=332990, accessed 2023-10-18.
- M. Elsner and K. Hoelzer, Quantitative Survey and Structural Classification of Hydraulic Fracturing Chemicals Reported in Unconventional Gas Production, Environ. Sci. Technol., 2016, 50(7), 3290–3314, DOI:10.1021/acs.est.5b02818.
- U. Shoukat, D. D. D. Pinto and H. K. Knuutila, Study of Various Aqueous and Non-Aqueous Amine Blends for Hydrogen Sulfide Removal from Natural Gas, Processes, 2019, 7(3), 160, DOI:10.3390/pr7030160.
- B. Lv, B. Guo, Z. Zhou and G. Jing, Mechanisms of CO2 Capture into Monoethanolamine Solution with Different CO2 Loading during the Absorption/Desorption Processes, Environ. Sci. Technol., 2015, 49(17), 10728–10735, DOI:10.1021/acs.est.5b02356.
- S. Kim, C. Yoon and D. Park, Vaporization and Conversion of Ethanolamines Used in Metalworking Operations, Saf. Health Work, 2010, 1(2), 175–182, DOI:10.5491/SHAW.2010.1.2.175.
- Alberta Environment, Soil and Groundwater Remediation Guidelines for Monoethanolamine and Diethanolamine, 2010, https://open.alberta.ca/dataset/94cc43cc-7840-495c-9b1f-df268a767206/resource/36db3e20-bafe-4db8-991f-2262705e681e/download/soilgroundwaterremguideethanolamine-2010.pdf.
- H.-W. Leung, L. M. Kamendulis and W. T. Stott, Review of the Carcinogenic Activity of Diethanolamine and Evidence of Choline Deficiency as a Plausible Mode of Action, Regul. Toxicol. Pharmacol., 2005, 43(3), 260–271, DOI:10.1016/j.yrtph.2005.08.001.
- US EPA and J. Frithsen, Study of the Potential Impacts of Hydraulic Fracturing on Drinking Water Resources: Progress Report, (Dec 2012), https://cfpub.epa.gov/ncea/hfstudy/recordisplay.cfm?deid=320611, accessed 2023-10-18.
- CDC NIOSH, Ethanolamine National Institute for Occupational Safety and Health, 2011, https://Www.Cdc.Gov/Niosh/Pel88/141-43.Html, accessed 10 Oct 2024.
- N. Dai, A. D. Shah, L. Hu, M. J. Plewa, B. McKague and W. A. Mitch, Measurement of Nitrosamine and Nitramine Formation from NOx Reactions with Amines during Amine-Based Carbon Dioxide Capture for Postcombustion Carbon Sequestration, Environ. Sci. Technol., 2012, 46(17), 9793–9801, DOI:10.1021/es301867b.
- K. Yu, W. A. Mitch and N. Dai, Nitrosamines and Nitramines in Amine-Based Carbon Dioxide Capture Systems: Fundamentals, Engineering Implications, and Knowledge Gaps, Environ. Sci. Technol., 2017, 51(20), 11522–11536, DOI:10.1021/acs.est.7b02597.
- Thermo Fisher Scientific, Quantitative Analysis of Nitrogen Mustard Hydrolysis Products as Ethanolamines. Application Note 271, https://assets.fishersci.com/TFS-Assets/CMD/Application-Notes/109792-AN271-IC-NitrogenMustard-Ethanolamines-19Jan2011-LPN2684.pdf.
- ASTM, Standard Test Method for Determination of Diethanolamine, Triethanolamine, N-Methyldiethanolamine and N-Ethyldiethanolamine in Water by Single Reaction Monitoring Liquid Chromatography/Tandem Mass Spectrometry, https://www.astm.org/d7599-16r17.html, accessed 2023-10-19.
- B. Xiong, M. A. Soriano, K. M. Gutchess, N. Hoffman, C. J. Clark, H. J. Siegel, G. A. D. De Vera, Y. Li, R. J. Brenneis, A. J. Cox, E. C. Ryan, A. J. Sumner, N. C. Deziel, J. E. Saiers and D. L. Plata, Groundwaters in Northeastern Pennsylvania near Intense Hydraulic Fracturing Activities Exhibit Few Organic Chemical Impacts, Environ. Sci.: Processes Impacts, 2022, 24(2), 252–264, 10.1039/D1EM00124H.
- B. McDevitt, A. M. Jubb, M. S. Varonka, M. S. Blondes, M. A. Engle, T. J. Gallegos and J. L. Shelton, Dissolved Organic Matter within Oil and Gas Associated Wastewaters from U.S. Unconventional Petroleum Plays: Comparisons and Consequences for Disposal and Reuse, Sci. Total Environ., 2022, 838, 156331, DOI:10.1016/j.scitotenv.2022.156331.
- J. Owens, A. Vu and C. Koester, Analysis of Ethanolamines: Validation of Semi-Volatile Analysis by HPLC-MS/MS by EPA Method MS888; LLNL-TR-408180, 945589, 2008, p LLNL-TR-408180, 945589, DOI:10.2172/945589.
- US EPA, Definition and Procedure for the Determination of the Method Detection Limit, Revision 2, https://www.epa.gov/cwa-methods/procedures-detection-and-quantitation-documents, accessed 2023-11-17.
- R. George, A. Haywood, S. Khan, M. Radovanovic, J. Simmonds and R. Norris, Enhancement and Suppression of Ionization in Drug Analysis U.: Therapeutic Drug Monitoring, Ther. Drug Monit., 2018, 40, 1–8 CAS.
- J. L. Luek, P. Schmitt-Kopplin, P. J. Mouser, W. T. Petty, S. D. Richardson and M. Gonsior, Halogenated Organic Compounds Identified in Hydraulic Fracturing Wastewaters Using Ultrahigh Resolution Mass Spectrometry, Environ. Sci. Technol., 2017, 51(10), 5377–5385, DOI:10.1021/acs.est.6b06213.
- K. A. Sitterley, K. G. Linden, I. Ferrer and E. M. Thurman, Identification of Proprietary Amino Ethoxylates in Hydraulic Fracturing Wastewater Using Liquid Chromatography/Time-of-Flight Mass Spectrometry with Solid-Phase Extraction, Anal. Chem., 2018, 90(18), 10927–10934, DOI:10.1021/acs.analchem.8b02439.
- C. G. Van Ginkel, C. A. Stroo and A. G. M. Kroon, Biodegradability of Ethoxylated Fatty Amines: Detoxification through a Central Fission of These Surfactants, Sci. Total Environ., 1993, 134, 689–697, DOI:10.1016/S0048-9697(05)80072-5.
- C. G. Van Ginkel and A. G. M. Kroon, Metabolic Pathway for the Biodegradation of Octadecylbis(2-Hydroxyethyl)Amine, Biodegradation, 1992, 3(4), 435–443, DOI:10.1007/BF00240365.
- S. Wyman, Triethanolamine Pathway, https://eawag-bbd.ethz.ch/tea/tea_map.html, accessed 2023-11-17.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4em00716f |
| This journal is © The Royal Society of Chemistry 2025 |