
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-intensity dependence of visible-light CO2 reduction over Ru(II)-complex/Ag/polymeric carbon nitride hybrid photocatalysts†
Ryuichi
Nakada
a,
Chao
Zhang
b,
Jo
Onodera
a,
Toshiya
Tanaka
a,
Megumi
Okazaki
 a,
Guigang
Zhang
*b and
Kazuhiko
Maeda
*ac
a,
Guigang
Zhang
*b and
Kazuhiko
Maeda
*ac
aDepartment of Chemistry, School of Science, Institute of Science Tokyo, 2-12-1-NE-2 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: maeda@chem.sci.isct.ac.jp
bState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou 350108, P. R. China. E-mail: guigang.zhang@fzu.edu.cn
cResearch Center for Autonomous Systems Materialogy (ASMat), Institute of Science Tokyo, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8501, Japan
First published on 2nd December 2024
Abstract
Photocatalytic CO2 reduction over Ru(II)-complex/Ag/polymeric carbon nitride (PCN) was studied with respect to light intensity and the type of Ru(II) complex. In experiments using two different Ru(II) complex cocatalysts, the reduction potential of the Ru complex was found to balance efficient CO2 reduction on the Ru complex with electron transfer from Ag/PCN. This balance avoided the formation of H2 as a byproduct, minimized charge accumulation in Ag/PCN, and maximized the apparent quantum yield for CO2-to-HCOOH conversion.
Introduction
CO2 is an important source of C1 chemicals in organic synthesis1–3 and biosynthesis.4 Photocatalytic CO2 reduction is another promising approach because it produces value-added chemicals (e.g., HCOOH and CO) using inexhaustible sunlight. Metal-complex (photo)catalysts are highly active toward the reduction of CO2 with high selectivity. However, the photo-oxidative ability of the metal-complex photocatalysts is insufficient to achieve artificial photosynthetic reactions using water as an electron donor. By contrast, semiconductor photocatalysts generally exhibit good photo-oxidation power but low selectivity for CO2 reduction. To further exploit the advantages of both metal-complex and semiconductor photocatalysts while minimizing their weakness, researchers have intensively studied metal-complex/semiconductor hybrid photocatalysts over the past decade.5–8One of the most studied metal-complex/semiconductor hybrid photocatalysts is based on a polymeric carbon nitride (PCN) absorber modified with a mononuclear metal complex cocatalyst (Scheme 1).9–16 For example, PCNs functionalized with trans(Cl)-[RuII(bpyX2)(CO)2Cl2] (bpyX2 = 2,2′-bipyridine with substituents X in the 4-positions; X = PO3H2, CH2PO3H2), represented as RuP and RuCP hereafter, can reduce CO2 into HCOOH with 80–90% selectivity under visible light in the presence of triethanolamine (TEOA) as an electron donor.9,11 The activity of the Ru(II) complex/PCN hybrid is further enhanced through modification of the PCN component with Ag nanoparticles, which promote charge separation followed by electron transfer to the Ru complex.17,18
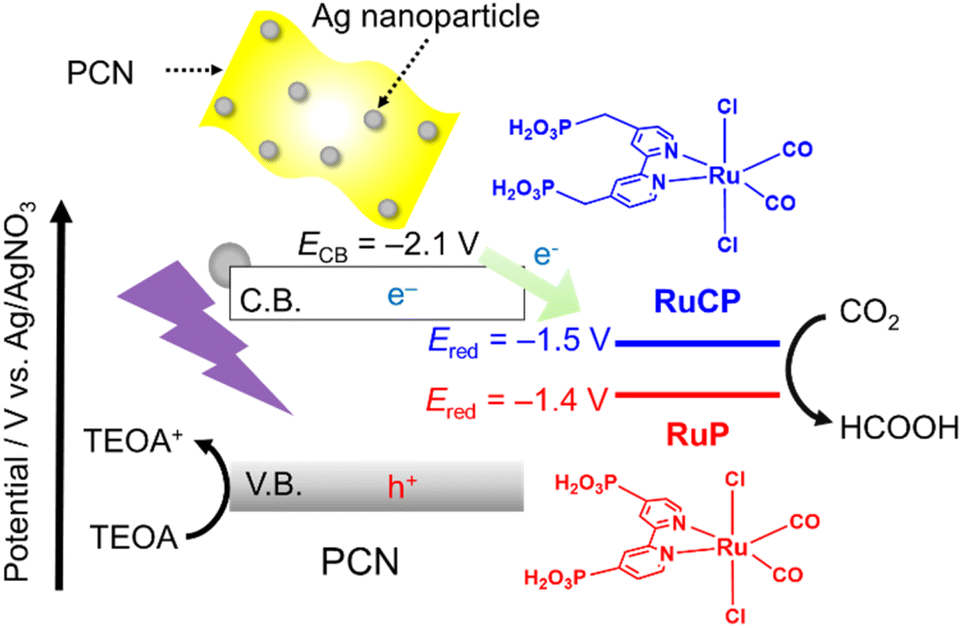 | ||
| Scheme 1 Schematic of photocatalytic CO2 reduction over Ru complex/Ag/PCN. | ||
For heterogeneous photocatalysts, especially those for water splitting or H2 evolution reactions, investigating the dependence of the photocatalytic activity on the light intensity has been shown to provide mechanistic insights into the bottlenecks in the associated photocatalytic reactions.19–24 By contrast, such studies involving heterogeneous photocatalysts for CO2 reduction are rare. In the conversion of CO2 to CH4 by a P25 TiO2 photocatalyst, the rate of CH4 formation has been reported to increase in proportion to the square root of the light intensity; however, the origin of the CH4 remains unclear because of a lack of 13CO2 isotope experiments.25 Shangguan et al. have reported that Au nanoparticles photocatalyze the reduction of CO2 to CO with the aid of H2O as a result of interband transitions of Au driven by visible light and that the rate of CO production increases almost linearly with increasing light intensity.26 For homogeneous systems, Ishida et al. have reported the light-intensity dependence of photocatalytic CO2 reduction using a mixed system of trans(Cl)-Ru(bpy)(CO)2Cl2 and [Ru(bpy)3]2+ as a catalyst and a photosensitizer, respectively, with the aid of 1-benzyl-1,4-dihydronicotinamide as an electron donor.27 According to their study, the ratio of CO/HCOOH products increased with increasing light intensity. Because some derivatives of trans(Cl)-Ru(bpy)(CO)2Cl2 have been used as active molecular cocatalysts for CO2 reduction with PCN, studying the light-intensity dependence of visible-light CO2 reduction over Ru complex/PCN hybrid photocatalysts, which has not been reported thus far, would be intriguing.
In the present study, we focused on the three-component hybrid photocatalysts RuP (or RuCP)/Ag/PCN and investigated the light-intensity dependence of the CO2 photoreduction quantum yield and the product selectivity. The significant effect of the reduction potential of the Ru complexes on CO2 photoreduction quantum yield and product selectivity with respect to the incident light intensity is reported.
Results and discussion
PCN and Ag/PCN were synthesized according to a previously reported method.17 Some essential characterization results, including the results of X-ray diffraction, UV-visible diffuse-reflectance spectroscopy, electron microscopy, and X-ray photoelectron spectroscopy experiments, are shown in the ESI (Fig. S1–S3†). The syntheses of RuP and RuCP were conducted in accordance with a previous report.28 As reported elsewhere, PCN absorbs visible light with wavelengths as long as ∼420 nm,17 whereas RuP and RuCP basically do not absorb visible light.11 The detailed procedures and materials characterization results can be found in the corresponding literature.First, the CO2 reduction reaction over RuP/Ag/PCN was carried out in a CO2-saturated mixed solution of N,N-dimethylacetamide (DMA) and TEOA (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) under 400 nm monochromatized light with different light-intensity conditions. In this work, the wavelength of 400 nm was employed because PCN absorbs 400 nm light, while RuP and RuCP do not.11 Here, the apparent quantum yield (AQY) for HCOOH generation was calculated using an equation adopted from a previous report:24
:1 v/v) under 400 nm monochromatized light with different light-intensity conditions. In this work, the wavelength of 400 nm was employed because PCN absorbs 400 nm light, while RuP and RuCP do not.11 Here, the apparent quantum yield (AQY) for HCOOH generation was calculated using an equation adopted from a previous report:24
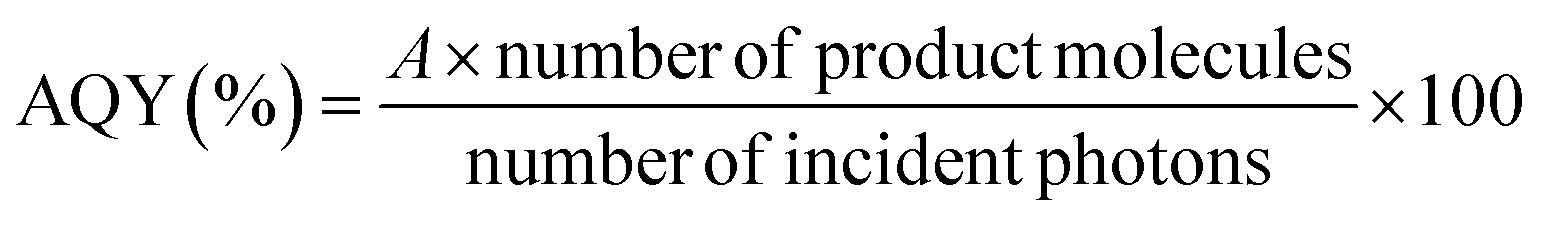
| Number of product molecules [h−1] = v[μmol h−1] × NA [mol−1] × 10−6 |
where I, S, h, and c are the wavelength of the incident light, the light intensity (measured by a spectrophotometer; LS-100, Eko Instruments), the irradiation area, Planck's constant, and the speed of light, respectively. In this research, the irradiation area was fixed at 4.4 cm2, and the light intensity was controlled by adjusting the output power of the Xe lamp. The maximum light intensity employed in this work was ∼10 mW cm−2 due to the power limit of the light source.
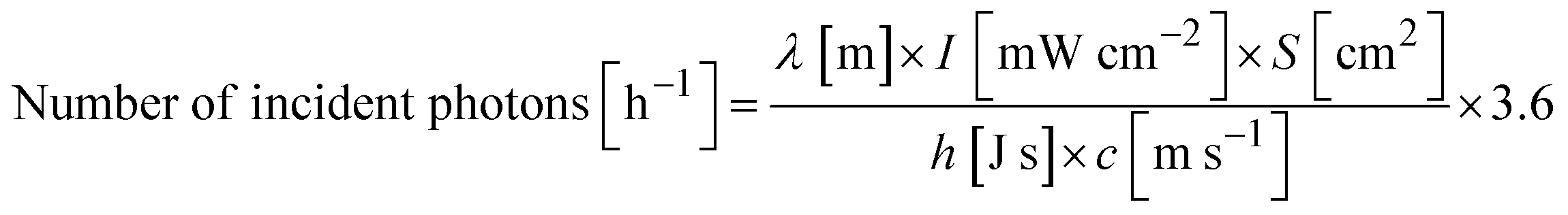
Fig. 1a shows the AQY for visible-light-driven HCOOH production over RuP/Ag/PCN as a function of the incident-light intensity. The AQY recorded in the light-intensity range ≤5.0 mW cm−2 was ∼2.5% and almost independent of the light intensity. However, the AQY decreased sharply as the light intensity was increased beyond 5.0 mW cm−2. The results confirmed that the turnover number for HCOOH formation (TONHCOOH) was much larger than 1 in all cases (Table S1†), indicating that HCOOH formation was catalytically cycled. In previous work, we demonstrated that the generation of HCOOH over the RuP/Ag/PCN hybrid photocatalyst is stable until the TONHCOOH reaches ∼5800.17 In the present work, the recorded TONHCOOH was at most ∼300 because the reactions were conducted under relatively low-intensity visible light for 2 h. Therefore, the degradation of the activity of the RuP/Ag/PCN hybrid photocatalyst during the CO2 reduction can be regarded as negligible.
 | ||
| Fig. 1 (a) Apparent quantum yield, (b) HCOOH production rate, (c) HCOOH selectivity, and (d) rate of gas-phase product formation from RuP/Ag/PCN as function of the incident-light intensity. Reaction conditions: catalyst, 14 mg; reactant solution, DMA:TEOA mixture solvent (4:1 v/v, 14 mL); wavelength of light, 400 nm, Ag: 2.0 wt%, adsorbed amount of RuP: 2 μmol g−1. | ||
The relationship between the rate of HCOOH production (vHCOOH) and the incident-light intensity (I) is displayed in Fig. 1b. The data plot in lower-light-intensity regions could be fitted by the equation:
| vHCOOH = aIb |
The HCOOH production rate (vHCOOH) was almost proportional to the light intensity (I0.97) in the region ≤5.0 mW cm−2 (Table S2†). However, in the region >5.0 mW cm−2, the HCOOH rate remained almost unchanged. The HCOOH selectivity was also found to decrease (Fig. 1c), similar to the AQY results. The decrease in the HCOOH selectivity is attributed to the reaction order for the main gas-phase byproduct, H2, not changing with increasing light intensity (Fig. 1d). The corresponding H2 evolution AQY was ∼0.2%, independent of the light intensity.
The reduction of CO2 over RuCP/Ag/PCN was similarly tested; the results are shown in Fig. 2 and Table S3.† Unlike the AQY achieved with RuP/Ag/PCN, that achieved with RuCP/Ag/PCN decreased gradually with increasing light intensity (Fig. 2a). From Fig. 2b, the reaction order for light the intensity of RuCP/Ag/PCN was determined to be 0.72, which was lower than that for the RuP loading in the light-intensity regime of ≤5.0 mW cm−2 (0.97). The CO2-to-HCOOH selectivity was 90–95% irrespective of the light intensity (Fig. 2c). The rate of H2 byproduct formation tended to increase with increasing light intensity (Fig. 2d). No CO was detected in the products generated over RuCP/Ag/PCN. The trend of CO2 reduction selectivity for RuCP/Ag/PCN with respect to light intensity was thus different from that for RuP/Ag/PCN.
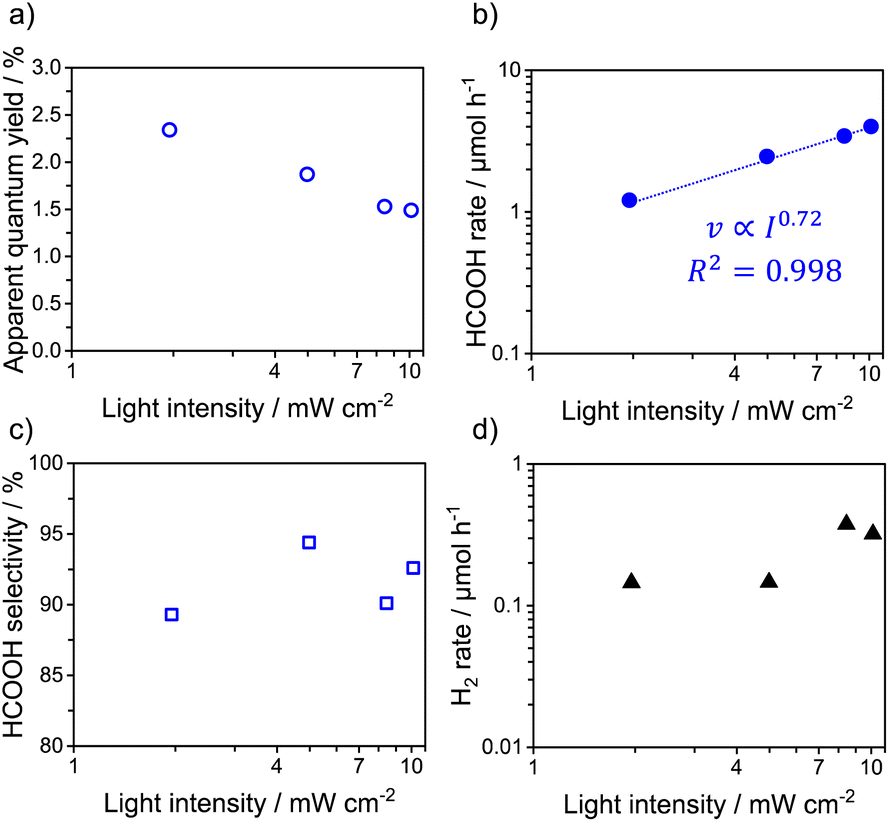 | ||
| Fig. 2 (a) Apparent quantum yield, (b) HCOOH production rate, (c) HCOOH selectivity, and (d) rate of gas-phase product formation from RuCP/Ag/PCN as function of the incident-light intensity. Reaction conditions: catalyst, 14 mg; reactant solution, DMA:TEOA mixture solvent (4:1 v/v, 14 mL); wavelength of light, 400 nm, Ag: 2.0 wt%, adsorbed amount of RuCP: 2 μmol g−1. | ||
The results thus demonstrated that the behavior of Ru complex/Ag/PCN ternary hybrid photocatalysts toward CO2 reduction was dependent on the type of the Ru complex and the incident light intensity. In the light-intensity range ≤5.0 mW cm−2, RuP/Ag/PCN gave a HCOOH generation reaction order close to unity with respect to light intensity (Fig. 1b). This result indicates that the overall reaction rate was determined by light absorption of the PCN component, as reported for other heterogeneous photocatalysts for water splitting.19,21,23,24 Of course, at least ∼90% of the photogenerated carriers were unused for the surface reaction because of recombination. When the light intensity was greater than 5.0 mW cm−2, light absorption by PCN was no longer the rate-determining step. Increasing the light intensity above 5.0 mW cm−2 resulted in a monotonic decrease in the AQY for HCOOH generation and a concomitant increase in H2 evolution, suggesting that there is a critical CO2-reduction bottleneck other than light absorption and charge migration in PCN.
The RuCP/Ag/PCN photocatalyst showed a different light-intensity dependence of photocatalytic CO2 reduction, giving a dependence of I∼0.7 in the entire examined light-intensity range (Fig. 2b). We speculate that the difference in the light-intensity dependence of CO2 reduction originates from the difference in the reduction potential of the Ru complexes. The reduction potential of RuCP has been shown to be 0.1 V more negative than that of RuP.11 Therefore, compared with RuP, RuCP exhibits greater difficulty in receiving an electron from Ag/PCN from the viewpoint of thermodynamics, but has a higher driving force for CO2 reduction. It should be also noted that the catalytic Ru center in RuCP is more distant from Ag/PCN than RuP because of the presence of methylene spacer, which would adversely affect the efficiency of electron transfer.29
That is, RuP is disadvantageous over RuCP in terms of the driving force for CO2 reduction, which could become the rate-determining step under high-light-intensity conditions. For a homogeneous CO2 photoreduction system composed of trans-(Cl)-[Ru(5,5′-diamide-2,2′-bipyridine)(CO)2Cl2]-type complex catalysts and [Ru(bpy)3]2+ as a photosensitizer, Ishida et al. have reported that the turnover frequency for CO2 reduction increases exponentially with a negative shift of the potential of the catalyst complex until the potential reaches the reduction potential of [Ru(bpy)3]2+.30 Notably, the rate of H2 byproduct formation during the CO2 reduction over RuP/Ag/PCN increased with a reaction order of ∼0.9. Because the Ag on the PCN can act as a H2 evolution cocatalyst,31 we speculate that the electrons in Ag/PCN, which had nowhere else to go because RuP no longer functioned properly, participated in H2 generation on the loaded Ag. We also note that Ag/PCN alone (i.e., without RuP or RuCP) gave AQYs of ∼0.2% and <0.1% for H2 and HCOOH generation, respectively, at 5.0 mW cm−2. That is, the selectivity of Ag/PCN to HCOOH was at most ∼35%, which is much lower than the selectivities recorded using the RuP- or RuCP-loaded analogues. Thus, when the outlet for CO2 reduction is blocked, the HCOOH generation AQY decreases drastically, whereas H2 production becomes relatively dominant (although the H2-evolution AQY remains approximately the same).
Because of the greater CO2 reduction driving force for RuCP, the CO2 reduction process on the RuCP is not a critical bottleneck. However, a certain degree of charge accumulation in Ag/PCN occurred even in combination with RuCP because electron transfer from Ag/PCN to RuCP is less efficient than that to RuP,11 as previously mentioned. That is, the greater the difficulty of the terminal reduction reaction, the more pronounced the charge accumulation in the PCN and the lower the reaction quantum yield.
In this regard, we conducted the H2 evolution reaction under the same reaction conditions as CO2 reduction but with PCN modified with a Pt cocatalyst, which is one of the most effective promoters of H2 evolution for PCN.32 The characterization data for Pt/PCN are included in Fig. S1–S3.† Fig. S4† shows the H2 evolution AQY and rate as functions of the light intensity. As expected, nearly twofold higher AQY (4.5–5.5%) was obtained compared with that recorded in CO2 reduction (∼2.5% at most, see Fig. 1a and 2a). The Pt/PCN gave a H2 evolution reaction order of ∼0.8 for light intensity, which was higher than the CO2 reduction order recorded using RuCP/Ag/PCN (∼0.7). From this result, one might expect that further improving the activity for CO2 reduction over metal-complex/PCN hybrid photocatalysts could be realized if a suitable metal-complex cocatalyst with a more negative reduction potential not exceeding the conduction-band minimum of PCN is developed.
Conclusions
In conclusion, we observed unique light-intensity-dependent CO2 reduction over Ru complex/Ag/PCN hybrid photocatalysts under 400 nm visible light. Using RuP, which features a more positive reduction potential, we found two operational regimes: One regime is ≤5.0 mW cm−2, where the HCOOH production rate increased in proportion with the light intensity while a high CO2-to-HCOOH selectivity (∼95%) was maintained; that is, the AQY was independent of the light intensity. The other regime is >5.0 mW cm−2, where the reaction order for the HCOOH rate decreased dramatically, accompanied by more pronounced byproduct formation of H2. However, a RuCP hybrid photocatalyst with a more negative reduction potential showed an I∼0.7 dependence of CO2 reduction to HCOOH with 90–95% selectivity in the entire examined light-intensity range. On the basis of these results, we concluded that, in RuP/Ag/PCN, light absorption dominates the overall CO2 reduction rate at I ≤ 5.0 mW cm−2, whereas CO2 reduction over RuP is the rate-determining step at I ≥ 5.0 mW cm−2. In RuCP/Ag/PCN, electron transfer from PCN to RuCP is the bottleneck that determines the overall reaction rate. These results suggest that, under lower light-intensity conditions, molecular catalysts with a more positive reduction potential function better and give a greater reaction quantum yield than those with a more negative potential, although the absolute amount of CO2 reduction product becomes inevitably low.Thus far, the development of metal-complex/semiconductor hybrid photocatalysts has relied on the individual refinement of both the metal-complex and semiconductor parts, where CO2 reduction occurs more quickly/selectively and charge recombination in relation to any back electron transfer reaction is minimal.6,8 The results of the present work based on the light-intensity dependence clearly provide more general insights into improving the CO2 reduction performance of metal-complex/semiconductor hybrid photocatalysts.
Experimental
Purification of solvent
DMA and TEOA were distilled before use. The distillation was performed according to our previous work.31 DMA was dried over 4 Å molecular sieves for several days and distilled under reduced pressure (10–20 torr). TEOA was distilled under reduced pressure (<1 torr). Distilled DMA and TEOA were kept under an Ar atmosphere before use. All other reagents were commercially available and used without further purification.Synthesis of PCN
PCN samples were synthesized according to a previous method33 by heating 12.0 g of urea (>99.0%, FUJIFILM Wako Pure Chemical) in air at a ramp rate of 2.2 K min−1 to 823 K, maintaining that temperature for 4 h, then cooling without temperature control. Approximately 0.58 g of the PCN powder was obtained.Synthesis of Ag/PCN
The Ag was loaded in the same manner as in our previous study.17 Ag species as promoters were loaded onto the surface of PCN by an impregnation method using AgNO3 (>99.8%, Wako Pure Chemicals) as the precursor. PCN was dispersed in an aqueous AgNO3 solution (10 mL). The water content was slowly removed under reduced pressure at room temperature (∼298 K) using an evaporator in the dark. The resultant solid sample was heated under a H2 stream (20 mL min−1) at 473 K for 1 h. The Ag loading amount was 2.0 wt% (nominal value).Synthesis of RuP and RuCP
Ru complexes (RuP and RuCP) were synthesized according to the procedures reported by Anderson et al.28 Briefly, Ru polymer [Ru(CO)2Cl2]n was refluxed in ethanol with (PO3Et2)2bpy (for RuP) or (PO3Et2CH2)2bpy (for RuCP) ligands and the obtained powders were refluxed in HCl aqueous solution (4 M) for the hydrolysis to obtain phosphonic (or methylphosphonic) acid anchors.Adsorption of Ru complex by Ag/PCN
The adsorption of RuP (or RuCP) was conducted in a manner similar to that previously reported.17,31,33 PCN was dispersed in methanol containing dissolved RuP. The suspension was stirred for 24 h at room temperature (∼298 K) in the dark under an Ar atmosphere to allow for adsorption/desorption equilibrium, followed by filtration. The amount of adsorbed RuP (or RuCP) was calculated on the basis of the UV-vis spectrum of the filtrate. In the present report, the amount of adsorbed Ru complexes was 2 μmol g−1. Typical UV-visible absorption spectra of the solutions before and after the adsorption procedure are shown in Fig. S5.†Synthesis of Pt/PCN
Pt loading was carried out by impregnation followed by H2 reduction, similar to the aforementioned method of Ag loading. Pt was loaded onto the PCN using H2PtCl6·6H2O (>98.5%, Kanto Chemical) as the precursor. The water was subsequently removed slowly by heating. The resultant solid sample was heated under a H2 stream (20 mL min−1) at 473 K for 1 h. The Pt loading amount was 2.0 wt% (nominal value).Photocatalytic reaction
Photocatalytic reactions were performed in a 30 mL Pyrex cuvette containing 14 mL of a mixed solution of DMA and TEOA (4:1 v/v) and 14 mg of photocatalyst, which was kept at room temperature using a water jacket. In this study, CO2 reduction was performed by RuP/Ag/PCN (or RuCP/Ag/PCN) and H2 evolution by Pt/PCN. Before irradiation, the suspension was purged with CO2 (>99.995%, Taiyo Nippon Sanso) for 25–30 min. The light source was a 400 W Xe lamp (Asahi Spectra, MAX-400D) equipped with a bandpass filter (λ = 400 nm) that allowed the light intensity to be controlled. Some spectral irradiance data are displayed in Fig. S6.† The gaseous reaction products were analyzed using a gas chromatograph equipped with a thermal conductivity detector (GL Science, GC32 or Shimadzu, GC-2014; carrier gas, Ar). The HCOOH generated in the liquid phase was analyzed using a capillary electrophoresis system (Otsuka Electronics, Agilent-7100).
The turnover number (TON) was calculated by the following equation:
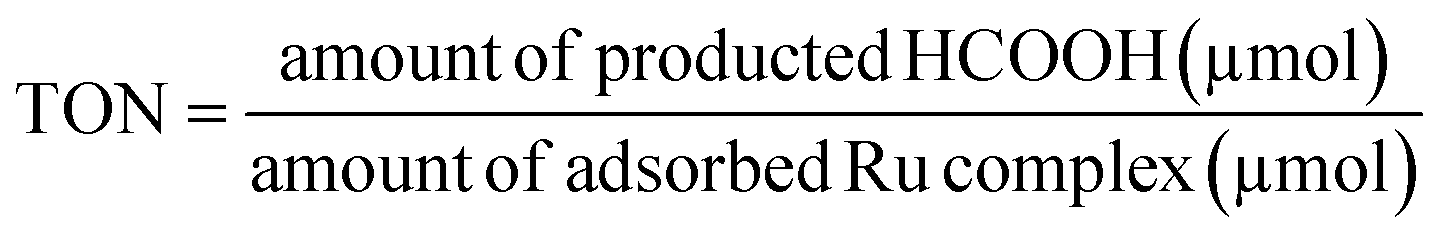
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
K. M. designed/supervised the project and wrote the manuscript draft with R. N. and M. O. R. N. conducted photocatalytic reactions and main characterization. J. O. and T. T. synthesized Ru complexes with R. N. C. Z. and G. Z. performed TEM and XPS measurements. All authors reviewed the manuscript and approved its submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Transformative Research Areas (A) “Supra-ceramics” (JP22H05148) and a bilateral collaboration program (JPJSBP120237406) (JSPS). Acknowledgement is also extended to a grant from the National Natural Science Foundation of China (22311540011).Notes and references
- J. H. Ye, T. Ju, H. Huang, L. L. Liao and D. G. Yu, Acc. Chem. Res., 2021, 54, 2518–2531 CrossRef CAS PubMed.
- M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2022, 61, e202112835 CrossRef CAS.
- L. Wang, C. Qi, W. Xiong and H. Jiang, Chin. J. Catal., 2022, 43, 1598–1617 CrossRef CAS.
- Y. Amao, Sustain. Energy Fuels, 2018, 2, 1928–1950 RSC.
- S. Sato, T. Arai and T. Morikawa, Inorg. Chem., 2015, 54, 5105–5113 CrossRef CAS PubMed.
- K. Maeda, Adv. Mater., 2019, 31, e1808205 CrossRef PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- A. Nakada, H. Kumagai, M. Robert, O. Ishitani and K. Maeda, Acc. Mater. Res., 2021, 2, 458–470 CrossRef CAS.
- K. Maeda, K. Sekizawa and O. Ishitani, Chem. Commun., 2013, 49, 10127–10129 RSC.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2013, 2, 353–358 CrossRef.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS.
- J. J. Walsh, C. Jiang, J. Tang and A. J. Cowan, Phys. Chem. Chem. Phys., 2016, 18, 24825–24829 RSC.
- G. Zhao, H. Pang, G. Liu, P. Li, H. Liu, H. Zhang, L. Shi and J. Ye, Appl. Catal., B, 2017, 200, 141–149 CrossRef CAS.
- C. Cometto, R. Kuriki, L. Chen, K. Maeda, T. C. Lau, O. Ishitani and M. Robert, J. Am. Chem. Soc., 2018, 140, 7437–7440 CrossRef CAS.
- S. Roy and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 12180–12184 CrossRef CAS.
- B. Ma, G. Chen, C. Fave, L. Chen, R. Kuriki, K. Maeda, O. Ishitani, T. C. Lau, J. Bonin and M. Robert, J. Am. Chem. Soc., 2020, 142, 6188–6195 CrossRef CAS.
- K. Maeda, D. An, C. S. Kumara Ranasinghe, T. Uchiyama, R. Kuriki, T. Kanazawa, D. Lu, S. Nozawa, A. Yamakata, Y. Uchimoto and O. Ishitani, J. Mater. Chem. A, 2018, 6, 9708–9715 RSC.
- N. Sakakibara, K. Kamogawa, A. Miyoshi, K. Maeda and O. Ishitani, Energy Fuels, 2024, 38, 2343–2350 CrossRef CAS.
- S. Tabata, H. Ohnishi, E. Yagasaki, M. Ippommatsu and K. Domen, Catal. Lett., 1994, 28, 417–422 CrossRef CAS.
- T. Torimoto, Y. Aburakawa, Y. Kawahara, S. Ikeda and B. Ohtani, Chem. Phys. Lett., 2004, 392, 220–224 CrossRef CAS.
- T. Hisatomi, K. Maeda, K. Takanabe, J. Kubota and K. Domen, J. Phys. Chem. C, 2009, 113, 21458–21466 CrossRef CAS.
- P. Ketwong, S. Yoshihara, S. Takeuchi, M. Takashima and B. Ohtani, J. Chem. Phys., 2020, 153, 124709 CrossRef CAS.
- R. Han, M. A. Melo, Z. Zhao, Z. Wu and F. E. Osterloh, J. Phys. Chem. C, 2020, 124, 9724–9733 CrossRef CAS.
- K. Maeda, T. Maeda, C. Suppaso, S. Nishioka, Y. Kamakura, S. Yasuda and T. Yokoi, Sustain. Energy Fuels, 2024, 8, 36–42 RSC.
- M. Dilla, A. Mateblowski, S. Ristig and J. Strunk, ChemCatChem, 2017, 9, 4345–4352 CrossRef CAS.
- W. Shangguan, Q. Liu, Y. Wang, N. Sun, Y. Liu, R. Zhao, Y. Li, C. Wang and J. Zhao, Nat. Commun., 2022, 13, 3894 CrossRef CAS PubMed.
- Y. Kuramochi, J. Itabashi, K. Fukaya, A. Enomoto, M. Yoshida and H. Ishida, Chem. Sci., 2015, 6, 3063–3074 RSC.
- P. A. Anderson, G. B. Deacon, K. H. Haarmann, F. R. Keene, T. J. Meyer, D. A. Reitsma, B. W. Skelton, G. F. Strouse and N. C. Thomas, Inorg. Chem., 1995, 34, 6145–6157 CrossRef CAS.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- Y. Kuramochi, K. Fukaya, M. Yoshida and H. Ishida, Chem.–Eur. J., 2015, 21, 10049–10060 CrossRef CAS.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940–4947 CrossRef CAS.
- M. Shizuno, K. Kato, S. Nishioka, T. Kanazawa, D. Saito, S. Nozawa, A. Yamakata, O. Ishitani and K. Maeda, ACS Appl. Energy Mater., 2022, 5, 9479–9486 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization and reaction data. See DOI: https://doi.org/10.1039/d4se01488j |
| This journal is © The Royal Society of Chemistry 2025 |