
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDrug repurposing of fluoroquinolones as anticancer agents in 2023
Asmaa E. Kassab *a,
Rania M. Gomaabc and
Ehab M. Gedawyac
*a,
Rania M. Gomaabc and
Ehab M. Gedawyac
aDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Cairo University, Kasr El-Aini Street, P. O. Box 11562, Cairo, Egypt
bDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Mansoura University, P. O. Box 35516, Mansoura, Egypt
cDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy and Pharmaceutical Industries, Badr University in Cairo (BUC), Badr City, P. O. Box 11829, Cairo, Egypt. E-mail: asmaa.kassab@pharma.cu.edu.eg; Fax: +2023635140; Tel: +2023639307
First published on 20th November 2024
Abstract
Drug developers are currently focusing on investigating alternative strategies, such as “drug repositioning”, to address issues associated with productivity, regulatory obstacles, and the steadily rising cost of pharmaceuticals. Repositioning is the best strategy to stop searching for new drugs because it takes less time and money to investigate new indications for already approved or unsuccessful drugs. Although there are several potent Topo II inhibitors available on the market as important drugs used in the therapy of many types of cancer, more may be required in the future. The current inhibitors have drawbacks including acquired resistance and unfavorable side effects such as cardiotoxicity and subsequent malignancy. A substantial body of research documented the cytotoxic potential of experimental fluoroquinolones (FQs) on tumor cell lines and their remarkable efficacy against eukaryotic Topo II in addition to optimized physical and metabolic characteristics. The FQ scaffold has a unique ability to potentially resolve every major issue associated with traditional Topo II inhibitors while maintaining a highly desirable profile in crucial drug-likeness parameters; therefore, there is a significant chance that FQs will be repositioned as anticancer candidates. This review offers a summary of the most recent research on the anticancer potential of FQs that was published in 2023. Along with discussing structural activity relationship studies and the mechanism underlying their antiproliferative activity, this review aims to provide up-to-date information that will spur the development of more potent FQs as viable cancer treatment candidates.
1. Introduction
The uneven trade-off between investment in research and development and new product output has left the biopharmaceutical industry in a difficult position and significantly contributed to the current down market. It is estimated that the cost of the development of a novel drug can reach up to US $2.6 billion, and it can take 13–15 years for it to be commercialized, with a large percentage of drugs known to fail during clinical trials.1 To deal with the problem of productivity, regulatory barriers, and the ever-rising cost of pharmaceuticals, drug developers are now concentrating on developing alternative approaches, such as “drug repositioning”.2–4 Drug repositioning is the process of finding a new use for an existing approved or ineffective medication outside of its intended usage. It is also sometimes referred to as repurposing, reprofiling, retasking, or redirection. Although drug repositioning was serendipitously discovered, more recent developments in the domains of proteomics, genomics, and bioinformatics have allowed for a more thorough investigation of this field. Drug repositioning offers a feasible substitute for the traditional drawn-out drug discovery process in order to quickly bring more affordable, faster-acting drugs to market by taking pharmacokinetic data into account. The field of drug repositioning is expanding, and many biotechnology and pharmaceutical companies have included repositioning programmes into their main drug research agendas.5,6 One of the world's most dangerous and potentially fatal diseases is cancer. Many anticancer treatments are available on the market, but the development of acquired drug resistance and the severe side effects of these clinically prescribed anticancer drugs are significant obstacles to efficient chemotherapeutic treatments.7 Nearly 14.1 million cancer cases and 8.2 million cancer-related fatalities have been reported to date, according to the Cancer Society. This means that one in every seven deaths is attributable to cancer, which is more than the combined mortality from TB, AIDS, and protozoal infections. On this trajectory, 21.6 million new cancer diagnoses and roughly 13.0 million cancer-related deaths are predicted by 2030.8 Reports on experimental antibacterial fluoroquinolones (FQs) that showed cytotoxic activity against tumor cell lines and notable potency against eukaryotic type II topoisomerases (Topo II) surfaced in the late 1980s.FQs have the ability to combat cancer in a variety of ways. Primarily, they work by inhibiting Topo II, which causes DNA fragmentation and prevents DNA synthesis.9–11 Additional modes of action of FQs include suppression of the mitochondria, which is linked to endosymbiosis because of the structural resemblance between mitochondria and bacterial cells.12 Some FQs exerted their anticancer potential via kinase inhibition.13,14 Additionally, FQs cause cell death by apoptosis and necrosis or stop the cell cycle in the S and G2/M stages.15–18
As a result, several pharmaceutical companies started FQ anticancer programs to explore the potential of this class as an alternative to conventional human Topo II inhibitory antitumor drugs like doxorubicin and etoposide.19–26
FQs offer several advantageous characteristics for treating cancer, including lower toxicity, a decreased risk of resistance emerging, a decreased propensity for drug-induced secondary tumor development, greater potency in comparison to other Topo II inhibitors, and advantageous physicochemical and pharmacokinetic profiles.27 In addition, the growing prevalence of bacterial resistance to FQs seems to represent a threat to their continued use as antibiotics. Ciprofloxacin (CP) has been included in clinical trials for the treatment of acute myeloid leukemia (https://www.clinicaltrials.gov/study/NCT02773732) and bladder cancer (https://www.clinicaltrials.gov/study/NCT00003824). These developments have made them potential candidates for drug repositioning from antibiotics to anticancer drugs. Furthermore, the versatility and ease of synthesis of FQ derivatives utilizing various techniques and building blocks have prompted researchers to prepare a wide range of chemical structures, which have been documented in several works of literature.28,29 FQs largely impede DNA synthesis by interfering with the proper operation of DNA topoisomerase enzymes (particularly type II).30–32 This family of enzymes is conserved and necessary for all living organisms, prokaryotes and eukaryotes alike.33,34 Throughout a cell's life, DNA topoisomerases are in charge of maintaining the correct DNA topology.35,36 During the processes of transcription, recombination, replication, and repair, they control the degree of DNA supercoiling.31,37 These enzymes function by creating type I or type II single- or double-strand breaks, which are then reintegrated when other DNA strands pass through them.30,36 These processes are required to relax and separate DNA strands so that they can copy and replicate; to separate the two identical copies of the entire genome in each of the two daughter cells following replication; and, lastly, to supercoil and wind the genomes so that they can be compacted in the nuclei of the cells.37,38 Type II DNA topoisomerases are vital enzymes for cell survival in addition to cell replication.32,39 Targeting the mammalian equivalents of prokaryotic DNA topoisomerases can be a useful approach in the therapy of cancer.40,41
According to a large bulk of publications, FQs have significant anticancer potential, exerting their activity via different mechanisms such as induction of cell cycle arrest at different phases or induction of apoptosis through targeting the cancer suppressor gene P53, the apoptotic gene B cell lymphoma-2 (BCL-2), the apoptotic pro-BCL-2-associated-x (Bax) or caspase-3/8/9,10,17,42–60 inhibition of migration, invasion and metastasis61,62 or inhibition of proliferation via promoting miRNA processing, impairment of telomerase activity or suppressing DNA synthesis.63–70
This review provides an overview of FQ derivatives as potential anticancer agents reported in 2023, highlighting structural activity relationships and the mechanism of their antiproliferative activity to open the opportunity for further development of novel and potent FQ derivatives for cancer treatment.
2. Ciprofloxacin derivatives
A novel CP (1) derivative 2 (Fig. 1) was developed and evaluated against four human endometrial cancer cell lines, either alone or in conjunction with taxanes, by Alhaj-Suliman et al.71 According to in vitro research, paclitaxel (PTX) and CP derivative 2 (Table 1) together exhibited synergistic lethal effects against type-II human endometrial cancer cells that expressed multidrug resistance mutation 1 gene (MDR1) and had loss-of-function p53 (Hec50co LOFp53). Significant increases in caspase-3 expression, cell population changes towards the G2/M phase, and a decrease in cdc2 phosphorylation all supported the enhanced antitumor effects. It was discovered that compound 2 inhibited MDR1, Topo I, and Topo II in addition to amplifying the effects of PTX on microtubule assembly. When PTX and CP derivative 2 were administered in vivo together, there was a notable decrease in tumor growth and a marked increase in PTX accumulation in tumors (as opposed to CP derivative 2 alone). At all tested dosages and in a concentration-dependent manner, the caspase-9 activity in the cells treated with CP derivative 2 was considerably higher than that of the untreated group. The enhanced in vivo cytotoxic effects on tumor tissues were verified through histological and immunohistochemical analysis. Blood biochemistry and complete blood count data verified that there was no obvious off-target toxicity. Therefore, combination therapy that included CP derivative 2 and PTX targeted several pathways and may be more effective and tolerated by patients with type-II endometrial cancer who have p53 mutations and the MDR1 gene.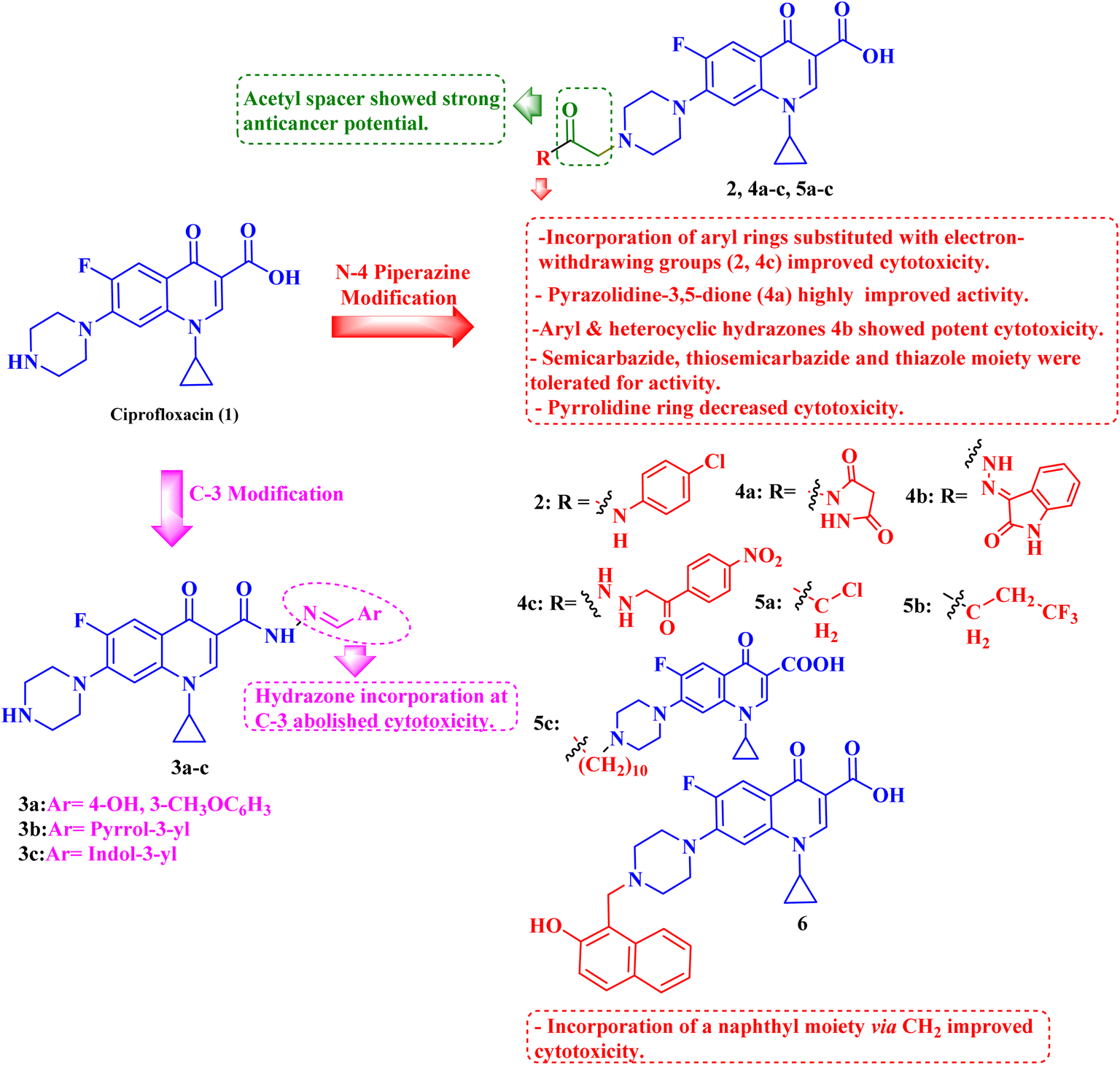 | ||
| Fig. 1 Structures and structure–activity relationships of CP derivatives 2–6 as antiproliferative agents. | ||
| Compound | Structure | Activity |
|---|---|---|
| 2 | 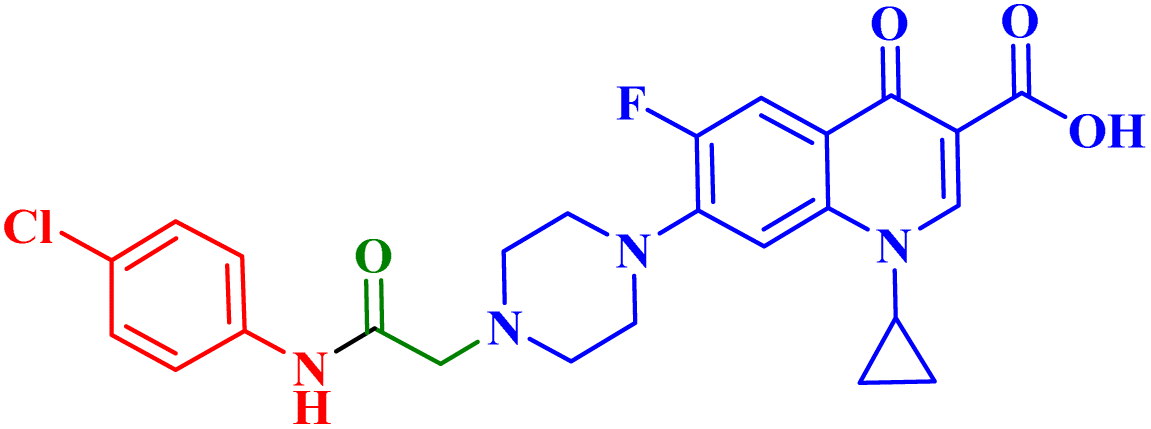 |
Synergistic augmentation of PTX cytotoxicity and augmented cell cycle arrest at G2/M phase |
| 3a | 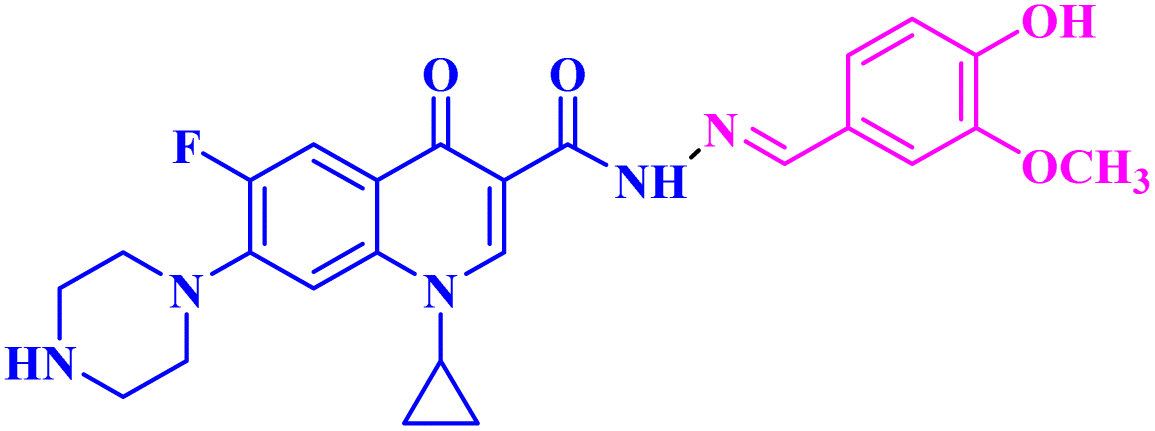 |
Mean GI% = 2.49% against NCI-60 human tumour cell lines |
| 3b | 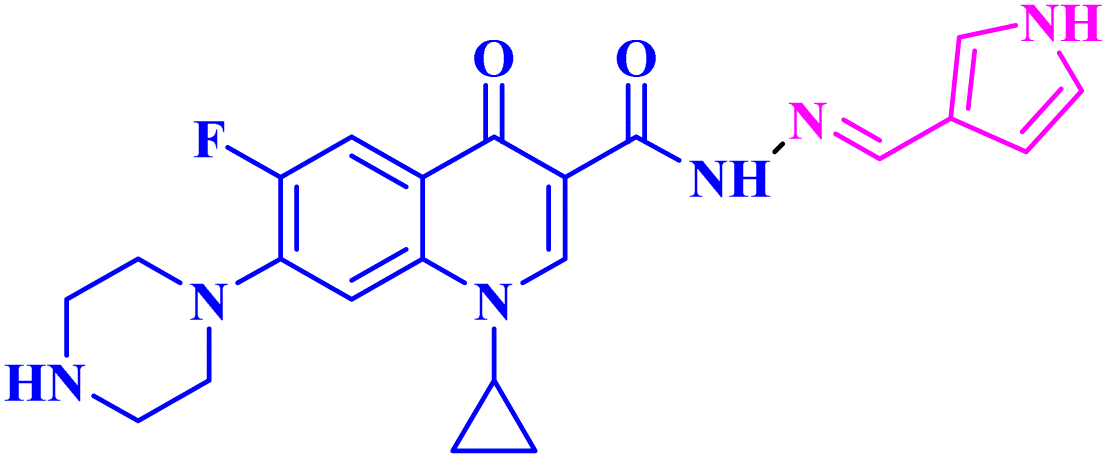 |
Mean GI% = 0.26% against NCI-60 human tumour cell lines |
| 3c | 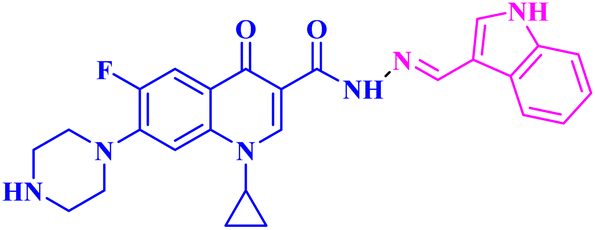 |
Mean GI% = 1.65% against NCI-60 human tumour cell lines |
| 4a | 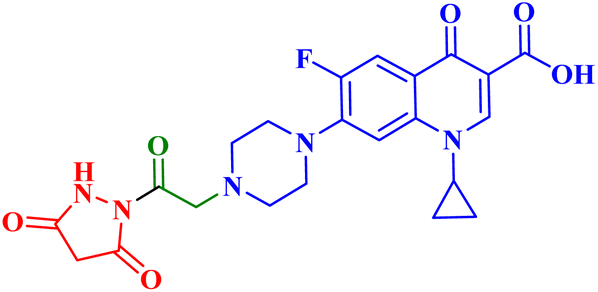 |
IC50 against T-24 = 5.68 μM; PC-3 IC50 = 92.16 μM; induced apoptosis 16.8-folds the control; increased apoptotic caspase-3 by 5.23-folds the control and caused G1-phase cell cycle arrest |
| 4b | 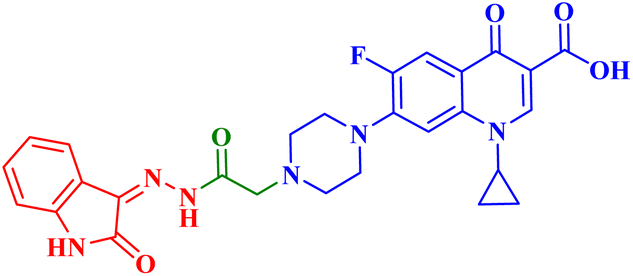 |
IC50 against T-24 = 3.36 μM; PC-3 IC50 = 10.95 μM; induced apoptosis 20.1-folds the control; increased apoptotic caspase-3 by 7.60-folds the control and caused G1-phase cell cycle arrest |
| 4c | 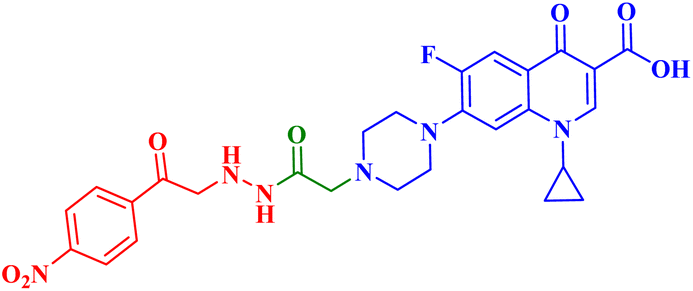 |
IC50 against T-24 = 10.08 μM; PC-3 IC50 = 3.25 μM |
| 5a | 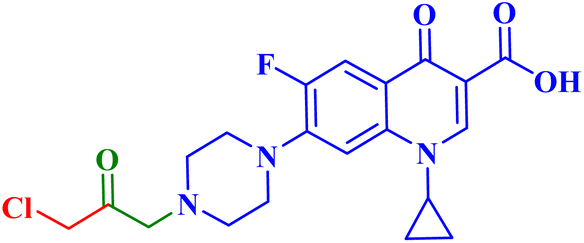 |
IC50 against PC-3 = 2.02 μM; SI = 17.60 towards PC-3 cells; induced apoptosis/necrosis in PC-3 cells 78.22%; reduced level of interleukin by 3.50 times the control |
| 5b | 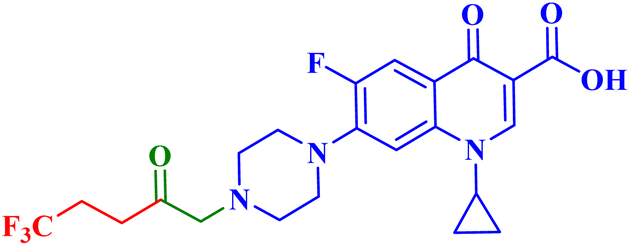 |
IC50 against PC-3 = 15.70 μM; SI = 5.40 towards PC-3 cells; reduced level of interleukin by 1.90 times the control |
| 5c | 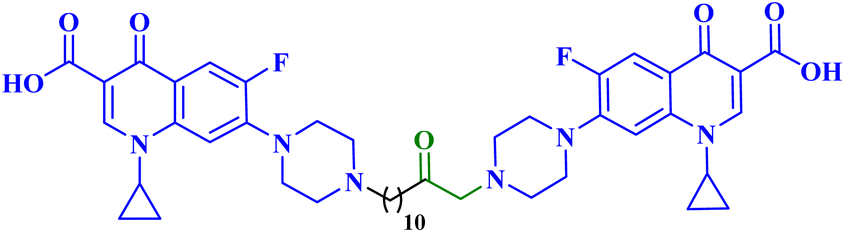 |
IC50 against PC-3 = 4.80 μM; SI = 15.20 towards PC-3 cells; induced apoptosis/necrosis in PC3 cells 69.29% |
| 6 | 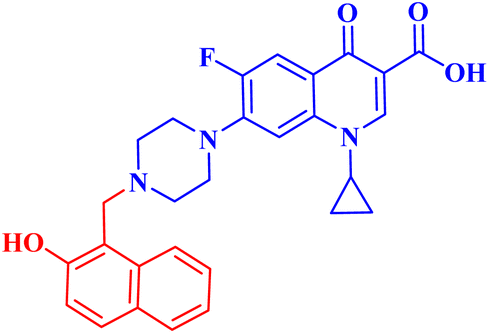 |
IC50 against OVCAR-3 = 44.34 μM; A-549 IC50 = 67.65 μM; increased the proportion of total apoptotic cells by 14.41-folds in OVCAR-3 and 19.65-folds in A-549 |
The same research group demonstrated that CP derivative 2 (Fig. 1) exhibited the ability to work in concert with PTX to increase its effectiveness by causing cell death in tests conducted on human endometrial cancer (EC) cell lines. CP derivative 2 was both safe and efficient, working in concert with PTX to inhibit the growth of LOF p53 type II EC both in vivo and in vitro. PEGylated polymeric nanoparticles (NP) encapsulating 3 reduced off-target effects while also enhancing the overall anticancer efficacy of PTX. These results suggested that treatment of type II EC may benefit from a multimodal therapy approach. Hec50co, a cell line generated from metastatic type II endometrial carcinoma, displayed chemo-resistance in prior research evaluating the efficacy of PTX in treating LOFp53 EC. To ascertain if 2 and PTX work in concert to modify EC cell growth, 2 and PTX were evaluated against LOFp53 EC before cytotoxicity was assessed. The results showed that when CP derivative 2 or PTX was given separately, their efficacy was low. However, while adding CP with PTX showed no advantage over PTX alone, the combined treatment of 2 and PTX had a dose-dependent synergistic impact that decreased the viability of Hec50co cells. Furthermore, the PTX and 2 pair was recently evaluated against three other EC cell lines (Ishikawa-H wild-type p53, Hec50co gain-of-function p53, and KLE gain-of-function p53), with the results showing that its cytotoxic properties were cell line-dependent. For instance, when doses of the PTX and 2 pair were applied to Ishikawa-H wild-type p53 cells, the cytotoxic activity was only slightly more than when the drug combination was applied to the other EC cell lines. Combining varying doses of CP derivative 2 (1, 10, 25, 50, and 100 μM) with 5 nM PTX resulted in a synergistic augmentation of PTX cytotoxicity, as all combinations tested had estimated Combination Index (CI) values less than one (Compusyn). The rapid decrease in relative cell viability from 80% and 95% to less than 20% after applying 5 nM PTX and 10 μM 2 (p < 0.001) provided additional evidence for this synergy. Comparably, adding 5 nM PTX and 25 μM CP derivative 2 together decreased cell survival to 14% (p < 0.001). This suggested synergism was validated by the 2.8–4-fold reduction in IC50 values for PTX paired with 2 over PTX alone and the 7–18-fold reduction in IC50 values for 2 combined with PTX over 2 alone. Hec50co cells were subjected to cell cycle analysis, which showed that adding 2 to PTX elevated the percentage of cells in the G2/M phase. Specifically, at 5 nM PTX alone, this percentage of cells elevated to 46%; however, at 10 μM 2, there was no discernible effect on the distribution of Hec50co cells in the cycle. All these investigations showed that treating Hec50co with PTX and 2 together reduced its survival, partly through changing the cell distribution over the cycle. Recent research on the main G2/M phase regulators discovered that the combination of PTX and 2 activated cdc2 by decreasing phosphorylation at Tyr15. On the other hand, 2-loaded nanoparticles (NPs) enhanced PTX's anticancer efficacy synergistically. The release of 2 from the NPs in vitro was gradual, with a release assay on day 10 reaching approximately 22%. Since none of the treated mice showed any obvious toxicity, in vivo data suggested a strong safety profile for CP derivative 2 NPs with or without PTX. Interestingly, compared to when 2 was administered in solution, IV delivery of CP derivative 2-loaded NPs greatly enhanced the tumor accumulation of 2, which probably amplified the action of PTX at the tumor site. In line with this, animals given PTX and 2 pair-loaded NPs had substantially reduced tumor growth. By allowing the use of lower PTX doses, this suggested therapeutic approach may help to decrease adverse effects associated with PTX use.72
Three CP derivatives 3a–c (Fig. 1) were designed and synthesized. They were first examined at a single dose of 10 μM in the NCI-60 Human Tumour Cell Lines Screen by The Developmental Therapeutic Programme (DTP) at the National Cancer Institute, USA (NCI). For every CP derivative, the mean growth inhibition percent was determined, which is the average of the growth inhibition percentage for all 60 cell lines. The mean GI% for CP derivatives 3a–c (Table 1) was 2.49%, 0.26%, and −1.65%, respectively.73
A set of new CP derivatives were designed and synthesized, with biologically active moieties introduced in the piperazine scaffold at the N-4 position. The study focused on their capacity to suppress the growth of bladder (T-24) and prostate (PC-3) cancer cell lines. Fourteen CP derivatives were 1.02 to 8.66 times more potent than doxorubicin against T-24 cancer cells, with IC50 values ranging from 3.36 to 28.55 μM. IC50 values ranging from 3.24 to 19.33 μM demonstrated the potency of ten compounds against PC-3 cancer cells, which were 1.2–7.1 times more potent than doxorubicin. Significant Topo II inhibitory activity was demonstrated by the most promising compounds (83–90% at 100 μM concentration). The potency of three CP derivatives, 4a–c (Fig. 1), was 1.01–2.32 times greater than that of doxorubicin (Table 1). The most promising CP derivatives, 4a and 4b, were studied in more detail to see how they affected the levels of active caspase-3, induction of apoptosis, and cell cycle progression in T-24 and PC-3 cell lines. In T-24 cells, both derivatives caused apoptosis (16.80- and 20.10-fold, respectively, in comparison to the control). An increase in the level of apoptotic caspase-3 (5.23- and 7.6-fold) corroborated this finding. In the S phase, compounds 4a and 4b halted the cell cycle. As for the induction of apoptosis in the PC-3 cell line, at respective IC50 concentrations, compounds 4a and 4b both produced G1-phase cell cycle arrest. The rise in the proportion of cells in the G1 phase (1.16 and 1.27-fold, respectively) and the corresponding decrease in the proportion of cells in the G2/M phase (1.50 and 20.10-fold, respectively) supported this finding. When PC-3 cells were treated with compounds 4a and 4b at their IC50 values, the proportion of viable cells decreased. The results demonstrated that both CP derivatives caused early and late apoptosis. These compounds' strong Topo II inhibitory activity can be explained by the molecular docking studies conducted with the Topo II protein, which identified more advantageous binding modes than merbarone. Different potencies were obtained by altering the CP scaffold at the piperazinyl N-4 position, according to the structure–activity correlation of the newly synthesized CP derivatives. CP compounds with an acetyl spacer-containing pyrazole ring (4a) exhibited strong anticancer properties. When it came to their ability to inhibit bladder cancer cell lines, the CP derivative with the pyrazolidine-3,5-dione moiety was the most potent. Considering CP hydrazones, the CP derivative with a cyclohexylidene moiety showed significant efficacy against the cell lines of bladder and prostate cancer. Subsequent investigation of these compounds showed that the hydrazones with substituted phenyl rings had more potency than those with thiophene rings. Among CP aryl hydrazones, an intriguing finding is that the CP hydrazone with an ortho hydroxyl group on the phenyl ring had the strongest antiproliferative action against prostate cancer. For both cell lines, the CP derivative with the acylhydrazone scaffold and a benzene ring containing a nearby NH demonstrated strong antiproliferative action. It was evident that the anticancer activity was negatively impacted by the bromine atom added to the indoline scaffold. Derivatives of CP semicarbazide and thiosemicarbazide showed strong action. The most effective CP derivative was found to have a 4-chloro-3-trifluoromethylphenyl moiety. When the phenyl moiety was substituted with an electron-withdrawing group, the anticancer activity against the two cell lines was markedly enhanced. Lower anticancer activity was seen in CP derivatives when monocyclic pyrrolidine or pyrrolidine fused with benzene was added. It is noteworthy that the antiproliferative effect was significantly enhanced by grafting the thiazole moiety.74
Struga M. et al.75 developed a novel series of N-acylated CP conjugates 5a–c (Fig. 1) and screened them for antiproliferative potential. Against prostate PC3 cells, CP derivatives 5a–c (Table 1) exhibited substantial antiproliferative activity (IC50 values of 2.02, 15.70, and 4.80 μM, respectively), up to 6.50–2.75 times stronger than cisplatin (IC50 = 13.20 μM). They entirely lowered the growth and proliferation rates of these cells. They did not have any cytotoxic effects on the normal HaCaT cell line with selectivity indices (SIs) of 17.6 and 5.4, sequentially. Additionally, derivatives 5a and 5c induced 78.22 and 69.29% apoptosis/necrosis in PC3 cells, respectively, most likely by raising the level of ROS inside the cell. Conjugates 5a and 5b reduced the level of interleukin by 3.50 and 1.90 times, which had an impact on PC3 cell proliferation.
Fawzy et al.76 aimed to evaluate the anticancer efficacy of a recently synthesized CP Mannich base 6 (Fig. 1) on lung cancer (A-549) and ovarian cancer (OVCAR-3) cell lines, as well as to investigate the underlying molecular pathways. The cytotoxic and pro-apoptotic effects of CP derivative 6 on the two cell lines were examined employing the MTT assay, annexin V assay, cell cycle analysis, and caspase-3 activation. While qRT-PCR was utilized to assess the gene expression pattern of the p53/Bax/BCL-2 pathway, western blotting was utilized to assess the downstream targets of the MAPK pathway. The results showed that OVCAR-3 cells had a lower IC50 value (44.34 μM) than A-549 cells (67.65 μM), indicating that they were more susceptible to 6 (Table 1). The proportion of total apoptotic cells increased in OVCAR-3 cells from 2.92% (control cells) to 36.26% and in A-549 cells from 1.71% (control cells) to 33.61%. In comparison to control cells, the cell proportion in the S phase of OVCAR-3 and A-549 cells rose to 57.69% and 51.44%, respectively. Treatment of OVCAR-3 cells with CP derivative 6 for 24 or 48 hours significantly boosted the expression of the p53 gene in a time-dependent manner as compared to untreated control cells, resulting in a 4.50- or 7.01-fold increase, respectively (p < 0.001). Furthermore, exposing A-549 cells to derivative 6 for 24 or 48 hours markedly increased the transcription of the p53 gene in a time-dependent manner, resulting in increases of 3.20 or 5.10-fold, respectively, in comparison to untreated control cells. In contrast to the relative expression of the p21 gene in untreated control cells, the results showed a significant time-dependent elevation in p21 gene expression in OVCAR-3 cells in response to treatment with 6 for 24 or 48 hours, with an 8- or a 5-fold increase, respectively (p < 0.001). In contrast to the relative expression of the p21 gene in untreated control cells, p21 was considerably and time-dependently elevated in A-549 cells, with increases of up to 3 and 6 times, respectively. After subjecting OVCAR-3 cells to treatment with CP derivative 6 for either 24 or 48 hours, there was a significant increase in Bax mRNA levels (3.80- or 5.06-fold, respectively) compared to the untreated control cells (p < 0.05). Additionally, A-549 cells treated with CP derivative 6 for 24 or 48 hours had a significant elevation of Bax gene expression (2.25- or 3.63-fold increase, respectively) compared to untreated control cells (p < 0.05). In addition, OVCAR-3 cancer cells treated with compound 6 for 24 or 48 hours showed a significant reduction in BCL-2's relative gene expression, to 0.70 or 0.33, respectively, in comparison to untreated control cells. When CP derivative 6 was applied to A-549 cells for either 24 or 48 hours, it was found that the expression of the BCL-2 gene was drastically reduced to 0.81 and 0.51, respectively, in comparison to untreated control cells. In OVCAR-3 cells, CP derivative 6 treatment for 24 or 48 hours resulted in strong activation of caspase-3, which was reflected in a time-dependent increase in active caspase-3 expression of 1.80 or 2.60- fold, respectively, relative to untreated control cells. Similar findings were observed in A-549 cells. There was a notable time-dependent increase in the level of active caspase-3 in A-549, as indicated by 1.50 and 2.40-fold increases when compared to untreated control cells. This finding indicated that CP derivative 6 activated caspase-3 to cause apoptosis in OVCAR-3 and A-549 cancer cells. Additionally, via inhibiting the MAPK pathway, CP derivative 6 arrested cell proliferation.
Given the biological characteristics of CP, it was possible to carry out amidification and esterification of CP, and the resultant compounds were tested in vitro for their antiproliferative potential against breast cancer cells (MCF-7). The in vitro studies exhibited that the synthesized derivatives revealed potent anticancer properties. The results of the antiproliferative assay of CP derivatives 7–9 (Table 2) showed that, in comparison to the control, the cell proliferation and viability for abemaciclib and CP derivatives 7–9 (Fig. 2) were in the range of 34.86% to 44.44% after 24 hours and at a concentration of 50 μg mL−1. The viability and proliferation of cells for abemaciclib and CP derivatives 7–9 were found to be in the range of 27.12% to 38.37% after 48 hours and at a concentration of 50 μg mL−1, in comparison to the control. Abemaciclib and CP derivatives were more potent after 48 hours than they were after 24 hours, as indicated by the proliferation and vitality of cells. The maximum viability and proliferation of cells in abemaciclib and synthetic CP derivatives occurred at a concentration of 50 μg mL−1 at both times (24 and 48 hours). Abemaciclib and CP derivatives 7–9 were found to have IC50 values in the range of 17.34 to 60.55 μM after 48 hours. The results indicated that derivative 8 exhibited the most potent anticancer potential. Furthermore, the anticancer activities of CP esterification/amidification derivatives (9a–c) were shown to be superior to those of CP derivatives (7a–d). It can be inferred that the addition of glycerol via esterification boosted the anticancer property of CP. It was discovered that the anticancer effects of compound 8 and the esterification/amidification products of CP were similar. Furthermore, the antitumor efficacy of CP amidification is nearly identical. Therefore, it may be inferred that the groups that were attached to the piperazine scaffold were not crucial in determining the anticancer capabilities of the compounds.77
| Compound | Structure | Activity |
|---|---|---|
| 7a | 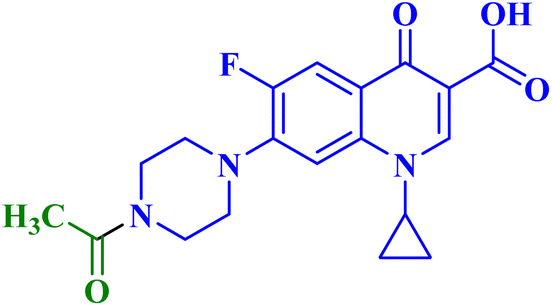 |
MCF-7 IC50 = 60.55 μM; decreased MCF-7 cells proliferation and viability to 42.94%, and 37.53% after 24 h and 48 h, respectively |
| 7b | 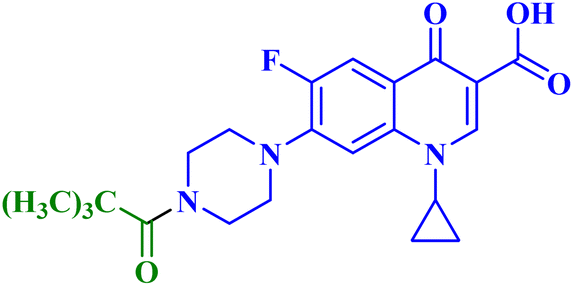 |
MCF-7 IC50 = 53.84 μM; decreased MCF-7 cells proliferation and viability to 41.67%, and 35.76% after 24 h and 48 h, respectively |
| 7c | 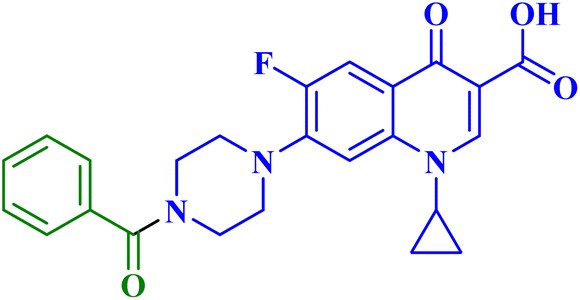 |
MCF-7 IC50 = 60.05 μM; decreased MCF-7 cells proliferation and viability to 44.44%, and 38.37% after 24 h and 48 h, respectively |
| 7d | 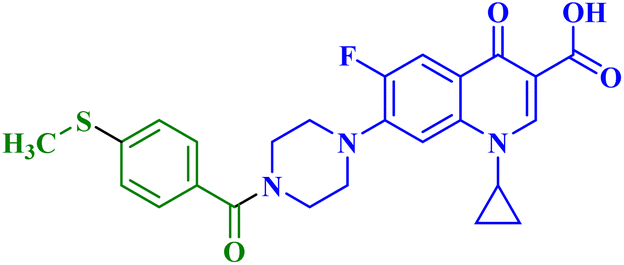 |
MCF-7 IC50 = 49.90 μM; decreased MCF-7 cells proliferation and viability to 40.51%, and 37.01% after 24 h and 48 h, respectively |
| 8 | 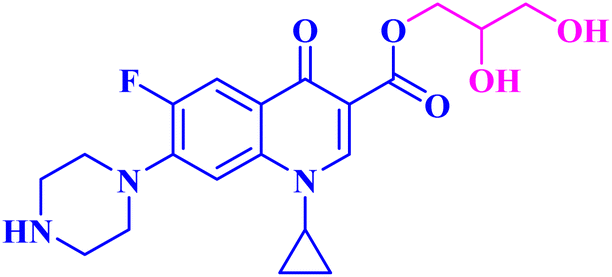 |
MCF-7 IC50 = 19.31 μM; decreased MCF-7 cells proliferation and viability to 34.86%, and 27.12% after 24 h and 48 h, respectively |
| 9 | 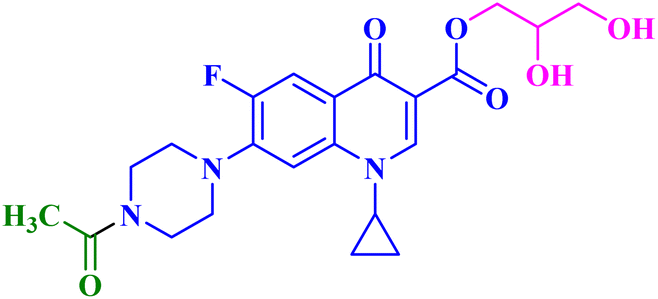 |
MCF-7 IC50 = 18.68 μM; decreased MCF-7 cells proliferation and viability to 34.91%, and 27.34% after 24 h and 48 h, respectively |
| 9b | 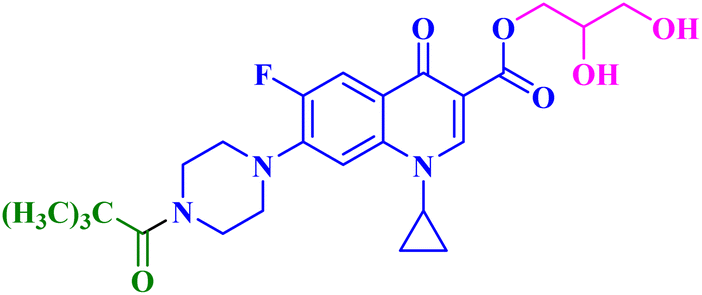 |
MCF-7 IC50 = 17.34 μM; decreased MCF-7 cells proliferation and viability to 35.01%, and 29.18% after 24 h and 48 h, respectively |
| 9c | 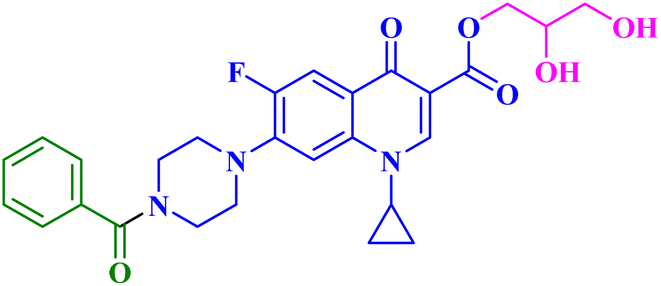 |
MCF-7 IC50 = 17.58 μM; decreased MCF-7 cells proliferation and viability to 35.99%, 27.19% after 24 h and 48 h, respectively |
| 10a | 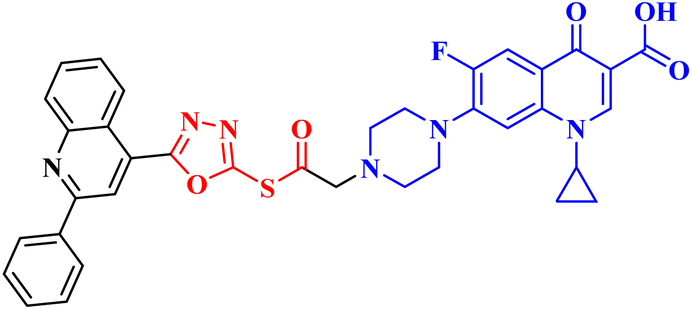 |
Induced GI in SR-leukemia cell line (33.25%), CAKI-1 renal cancer (26.92%), UO-31 renal carcinoma (64.19%), and LOX IMVI melanoma cancer (39.14%) |
| 10b | 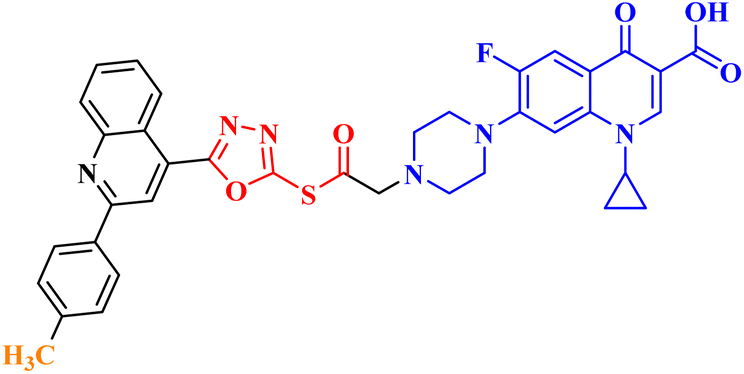 |
Induced GI in SR-leukemia cell line (52.62%), CAKI-1 renal cancer (39.81%), UO-31 renal carcinoma (55.49%), and LOX IMVI melanoma cancer (36.64%) |
| 10c | 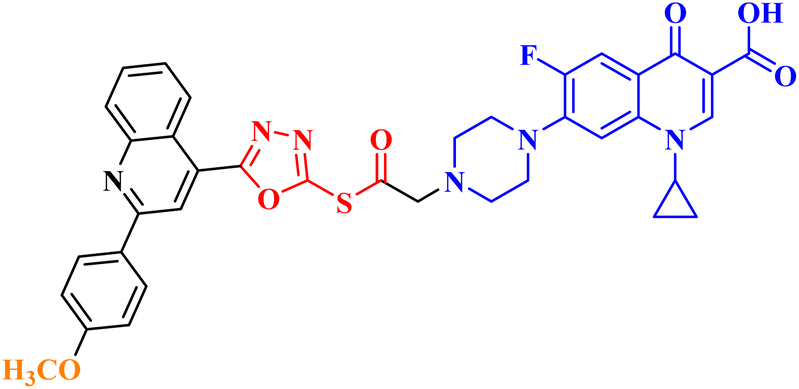 |
Induced GI in CAKI-1 renal cancer (27.31%) and UO-31 renal carcinoma (40.15%) |
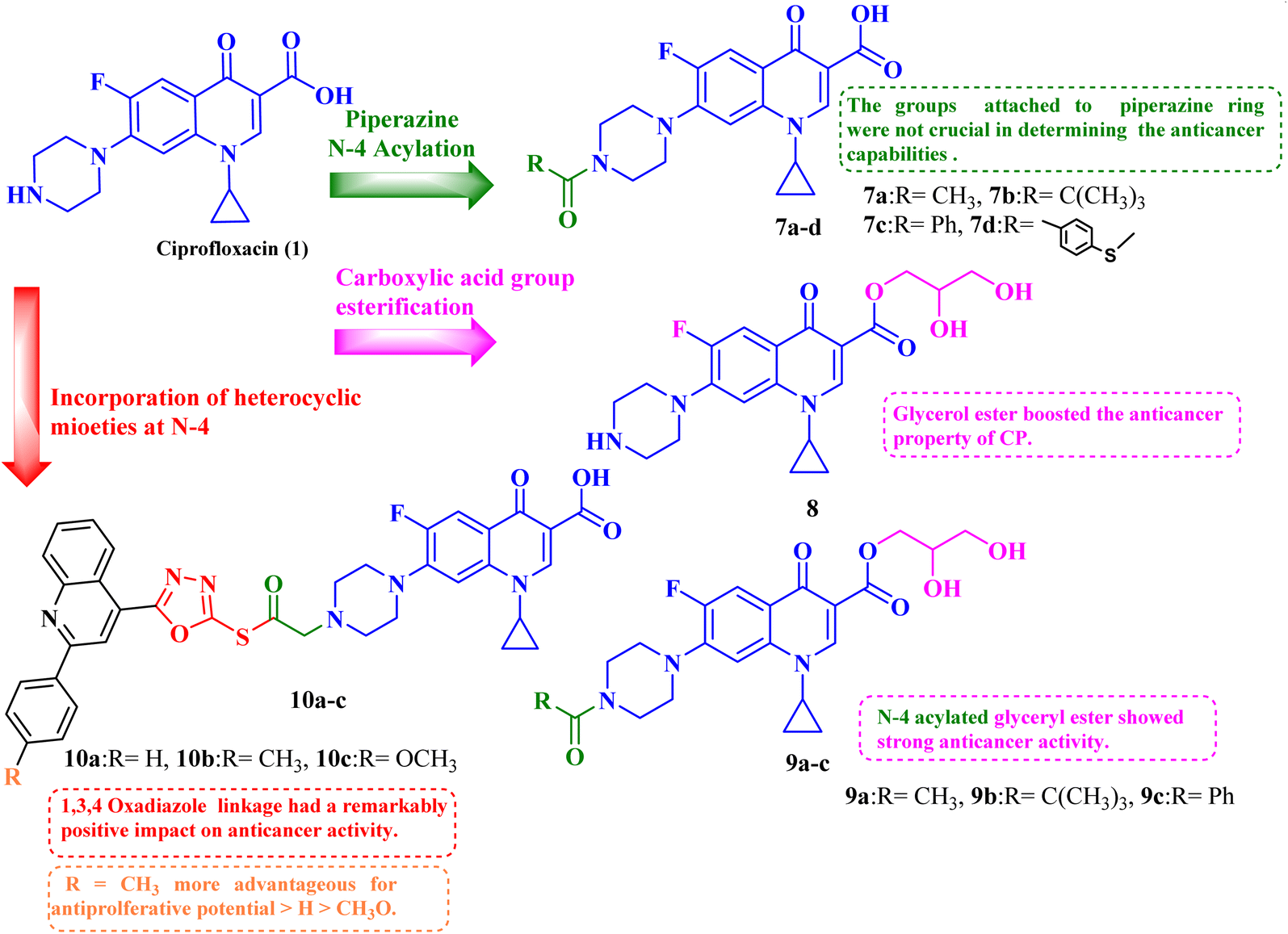 | ||
| Fig. 2 Structures and structure–activity relationships of CP derivatives 7–10 as antiproliferative agents. | ||
Molecular hybridization is an intriguing strategy that involves combining two or more biologically active compounds to create novel structures that can target many pharmacological targets. New CP-linked quinoline hybrids were designed and synthesized using a quinolin-4-yl-1,3,4-oxadiazole linker or a quinoline-4-carbonyl linker. The purpose of the study was to investigate the effect of incorporating such moieties in the C-7-piperazine moiety on the anticancer properties. The NCI findings of the novel heterocyclic compounds demonstrated that hybrids 10a–c (Fig. 2) exhibited superior antiproliferative properties compared to other hybrids (Table 2). CP hybrids 10a and 10b exhibited notable antiproliferative activity against SR-leukemia cell lines, resulting in growth inhibition percentages of 33.25% and 52.62%, respectively. The results demonstrated that the presence of an electron-donating substituent, such as a p-methyl group, on the phenyl ring (as in compound 10b) was more advantageous compared to having an unsubstituted phenyl ring at the same position (as in compound 10a). Conversely, substituting the p-methyl group with a p-methoxy group abolished the antiproliferative activity, as observed in compound 10c. For CAKI-1 renal cancer, CP hybrid 10b showed a superior anticancer effect compared to hybrids 10a and 10c, with growth inhibition percentages of 39.81%, 26.92%, and 27.31%, respectively. Hybrids 10a–c demonstrated significant antiproliferative effects against UO-31 renal carcinoma, with growth inhibition percentages of 64.19%, 55.49%, and 40.15%, respectively. In addition, compounds 10a and 10b exhibited strong anticancer efficacy against LOX IMVI melanoma cancer cells, with percentages of growth inhibition of 39.14% and 36.64%, respectively. Compound 10c, on the other hand, demonstrated negligible anticancer efficacy with a growth inhibition percentage of 8.95%. The anticancer effect of CP hybrid 10c was negatively impacted by the introduction of a stronger electron-donating group, such as the methoxy group at position 2 of the phenyl ring of the quinoline scaffold, when compared to compounds 10a and 10b. Consequently, compound 10a with an unsubstituted phenyl ring at this position showed higher antiproliferative potential against LOX IMVI melanoma, and the same reasoning was reflected in the growth inhibition percentages of compounds 10a–c on A498 renal cancer cell lines. Overall, rather than carbonyl linkage, the introduction of the 1,3,4 oxadiazole ring as a linker in hybrids 10a–c might have a remarkably positive impact on these hybrids' anticancer potential. This can be attributed to the well-known basic antiproliferative activity of 1,3,4 oxadiazole, which has various mechanisms that have been previously published in numerous literature research publications.78
3. Moxifloxacin derivatives
Chrzanowska et al.79 studied the antitumor activities of novel various fatty acids-moxifloxacin (MXF, 11) conjugates with different fatty acid chain lengths and different degrees of unsaturation. MXF conjugates with oleic and stearic acid (12a and b, respectively, Fig. 3) revealed more pronounced activities against colorectal cancer cell lines (SW480 and SW620) as well as prostate cancer cell line (PC3) than towards the control normal cell line HaCaT, with SIs ranging from 12.50 to 71.00 (Table 3). Moreover, the MXF conjugate with oleic acid 12a exhibited IC50 values in micromolar concentration (4.60, 3.40, and 1.30 μM) against SW480, SW620, and PC-3 cell lines, respectively, while stearic acid conjugate 12b showed IC50 values of 2.70, 6.50 and 2.40 μM against the three cancer cell lines, respectively. As for the interleukin-6 (IL-6) release inhibitory activity, both MXF conjugates showed more significant inhibitory activity than the unconjugated MXF, where the oleic acid conjugate 12a suppressed the level of this cytokine in SW480, SW620, and PC3 cell lines by 38%, 20%, and 15%, respectively. The stearic acid conjugate 12b decreased the IL-6 release in the three colorectal and prostate cancer cell lines by 40%, 16%, and 24%, respectively.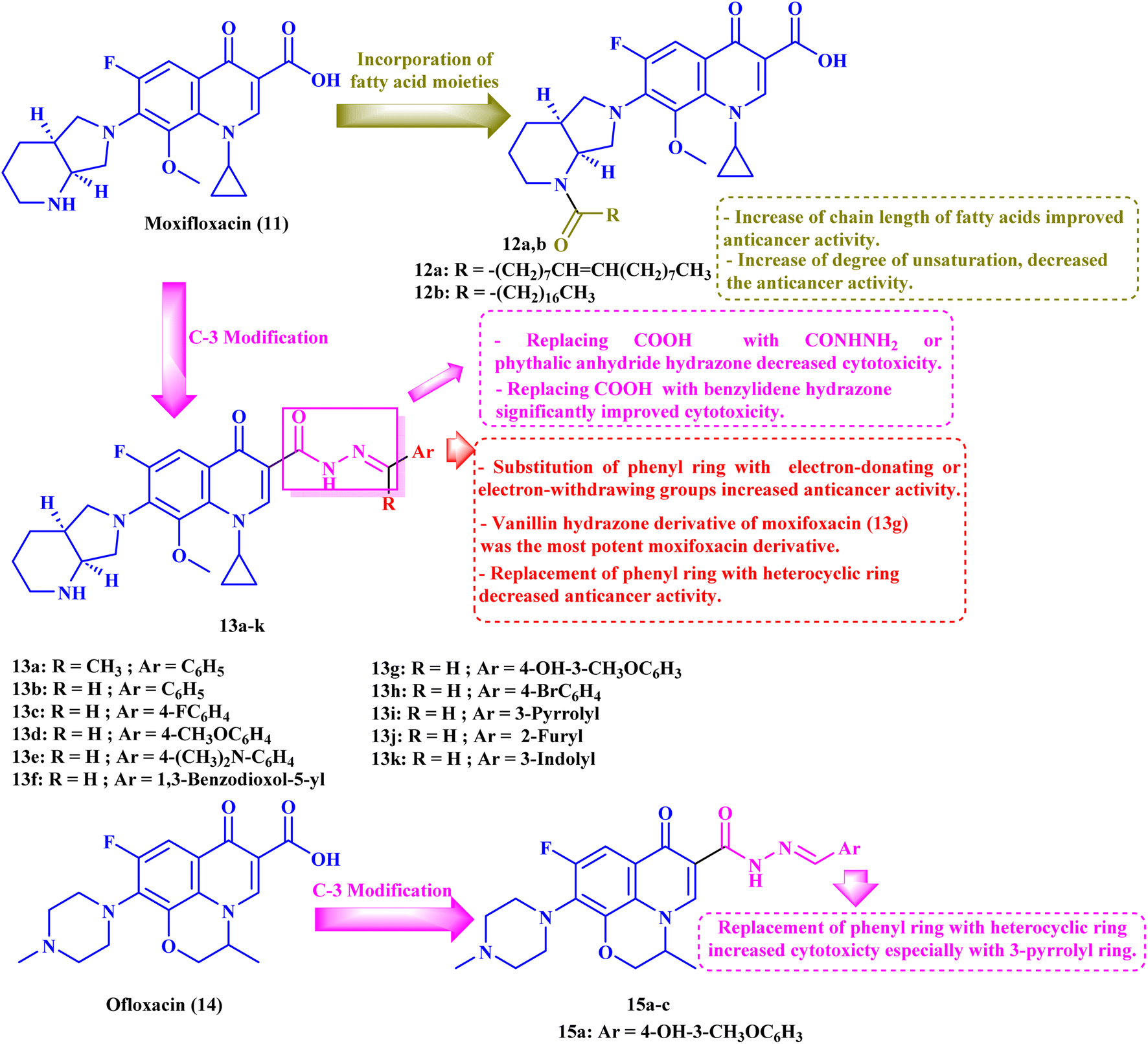 | ||
| Fig. 3 Structures and structure–activity relationships of MXF derivatives 12 and 13 and OFX derivatives 15a–c as antiproliferative agents. | ||
| Compound | Structure | Activity |
|---|---|---|
| 12 | 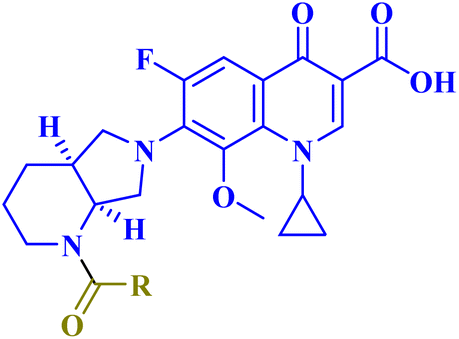 |
|
| 12a | R = –(CH2)7CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)7CH3 CH(CH2)7CH3 |
SW480 IC50 = 4.60 μM; SW620 IC50 = 3.40 μM; PC-3 IC50 = 4.60 μM. Showed IL-6 release inhibitory activity percentage (38%, 20%, 15%) in SW480, SW620, and PC-3 cells, respectively |
| 12b | R = –(CH2)16CH3 | SW480 IC50 = 2.70 μM; SW620 IC50 = 6.50 μM; PC-3 IC50 = 2.40 μM. Showed IL-6 release inhibitory activity percentage (40%, 16%, 24%) in SW480, SW620, and PC-3 cells, respectively |
| 13 | 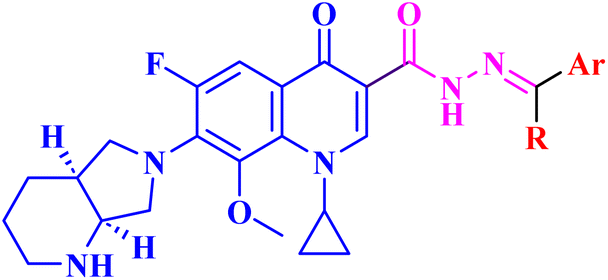 |
|
| 13a | R = CH3; Ar = C6H5 | Mean GI% against NCI-60 human tumor cell lines screen at 10 μM = 7.29% |
| 13b | R = H; Ar = C6H5 | Mean GI% against NCI-60 human tumor cell lines screen at 10 μM = 8.59% |
| 13c | R = H; Ar = 4-FC6H4 | Mean GI% against NCI-60 human tumor cell lines screen at 10 μM = 17.97% |
| 13d | R = H; Ar = 4-CH3OC6H4 | Mean GI% against NCI-60 human tumor cell lines screen at 10 μM = 17.67% |
| 13e | R = H; Ar = 4-(CH3)2N–C6H4 | Mean GI% against NCI-60 human tumor cell lines screen at 10 μM = 29.15% |
| 13f | R = H; Ar = 1,3-benzodioxol-5-yl | Mean GI% against NCI-60 human tumor cell lines screen at 10 μM = 39.24% |
| 13g | R = H; Ar = 4-OH-3-CH3OC6H3 | GI% NCI-60 human tumor cell lines screen ranging from 1.48% to 183.25% with mean GI% = 64.43% and mean GI50 of 1.78 μM; with high SI of 53.71 against cancer cells; increased in the overall apoptotic cells percentage about 33.75-fold; elevated Bax/BCL-2 ratio by 3-folds and a 3-fold rise in caspase-9 and a 2-fold increase in caspase-8 |
| 13h | R = H; Ar = 4-BrC6H4 | Mean GI% against NCI-60 human tumor cell lines screen at 10 μM = 25.87% |
| 13i | R = H; Ar = 3-pyrrolyl | Mean GI% against NCI-60 human tumor cell lines screen at 10 μM = 0.03% |
| 13j | R = H; Ar = 2-furyl | Mean GI% against NCI-60 human tumor cell lines screen at 10 μM = 1.85% |
| 13k | R = H; Ar = 3-indolyl | Mean GI% against NCI-60 human tumor cell lines screen at 10 μM = 5.48% |
| 15 | 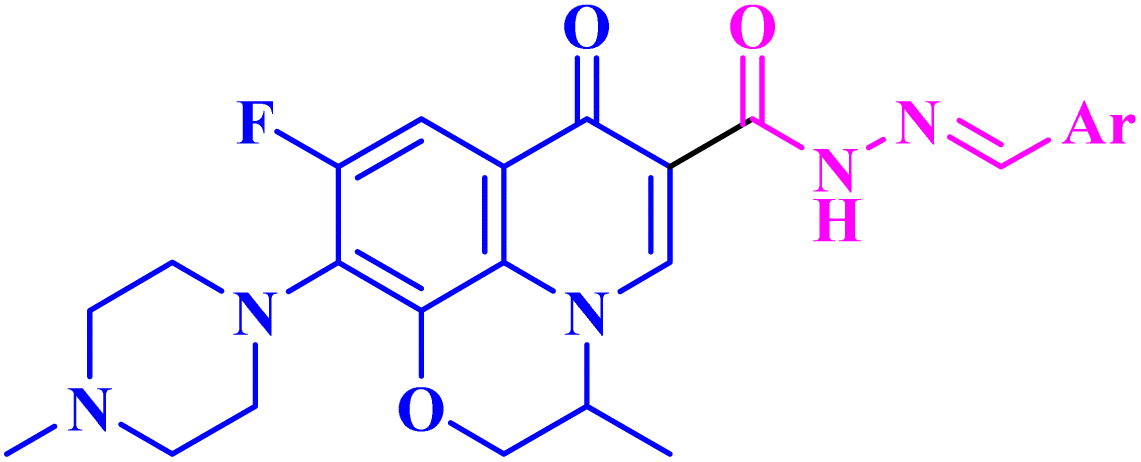 |
|
| 15a | Ar = 4-OH-3-CH3OC6H3 | Mean GI% against NCI-60 human tumor cell lines screen at 10 μM = 4.41% |
| 15b | Ar = 3-pyrrolyl | GI% NCI-60 human tumor cell lines screen ranging from 5.80% to 188.50% with mean GI% = 74.29% and mean GI50 of 1.45 μM |
| 15c | Ar = 3-indolyl | Mean GI% against NCI-60 human tumor cell lines screen at 10 μM = 0.35% |
Furthermore, the tested hybrids showed a significant effect on the induction of apoptosis and necrosis in SW480, SW620, and PC3 cell lines, where oleic acid MXF conjugate 12a showed an elevation of the percentage of the late apoptotic and necrotic cells by ranges of 2.70–68.60% and 3–18%, respectively. On the other hand, stearic acid MXF conjugate 12b revealed apparent induction of late apoptosis and necrosis in the three cancer cell lines by ranges of 16.30–78.30% and 6.00–19.20%, respectively.
In addition, the effect of the newly synthesized compounds on caspase 3/7 level was evaluated. The stearic acid conjugate 12b exhibited a significant increase in caspase 3/7 level by 109% relative to the control after 12 hours of incubation with SW480 at its IC50 and by 131% after 48 hours of incubation with the SW620 cancer cell line. For the cell line PC3, 12b revealed a 109% elevation in the caspase 3/7 levels after 24 hours of incubation. However, the oleic acid conjugate 12a revealed a moderate increase in the caspase level.
The authors also studied the cell cycle changes induced by MXF conjugates 12a and 12b at a molar concentration equivalent to their IC50 and half IC50 since changes in the cell cycle distribution profile frequently precede apoptosis induction. Both conjugates 12a and 12b revealed a dramatic increase in the cell percentage in the G0/G1 and sub-G1 phases in the SW480 as well as SW620 cell lines. On the other hand, treatment of PC3 cells with MXF conjugates 12a and 12b showed a maximal rise in the percentage of cells at the sub-G1 phase as well as cell cycle arrest in the G0/G1 phase.
Finally, MXF conjugates 12a and 12b showed more potent inhibition of NF-κB activation in colorectal and prostate cancer cell lines than MXF. The most potent inhibitory activities were recorded after treatment of SW480 cells with MXF hybrids 12a and 12b, where NF-κB inhibitory activity was decreased by 51% and 54%, respectively. While in the case of the SW620 cell line the decrease in the NF-κB inhibitory activity was 38% (12a) and 49% (12b), it was 28% (12a) and 40% (12b) in PC3 cells.
Elanany et al.73 reported the development and evaluation of the antitumor activity of several MXF derivatives 13a–k (Fig. 3). The results (Table 3) revealed that substituting the carboxylic acid group of MXF with benzylidene hydrazone significantly improved cytotoxicity. Substitution on the phenyl ring of benzylidene with either electron-donating or withdrawing groups improved the antitumor activity. Furthermore, the antitumor activity was significantly reduced when the phenyl ring was replaced by a heterocyclic ring such as 3-pyrrolyl, 2-furyl, or 3-indolyl. Among these derivatives, the vanillin hydrazone derivative of MXF 13g showed the most potent activity against a panel of 60 cancer cell lines with growth inhibition (GI%) ranging from 1.48% to 183.25% and a mean GI50 of 1.78 μM, whereas the assay of its cytotoxicity using the VERO normal cell line revealed a high SI of 53.71 against cancer cells. MXF derivative 13g caused apparent cell arrest for MCF-7 cells at G1/S after incubation for 24 hours at its GI50 concentration. Also, compound 13g resulted in a marked increase in the overall apoptotic cell percentage, about 33.75-fold that of the control. Moreover, the authors found that compound 13g promoted apoptosis via an intrinsic pathway, as indicated by an elevated ratio of Bax/BCL-2 by 3-fold and a threefold rise in caspase-9 and a two-fold increase in caspase-8 compared to the control, which approved the ability of the MXF derivatives to promote both intrinsic and extrinsic apoptotic pathways. Finally, compound 13g showed significant Topo II inhibitory activity with an inhibition percentage (83.54%) that was comparable to that of etoposide (96.84%); additionally, it showed apparent selectivity over Topo I.
4. Ofloxacin derivatives
To further investigate the antitumor activity of the repurposed FQ derivatives, the same authors73 designed ofloxacin (OFX, 14) analogs 15a–c (Fig. 3) and evaluated their antitumor activity. The authors found that the MXF hydrazine derivatives 13g and 13k showed more potent activity than the corresponding OFX derivatives (vanillin hydrazone and 3-indole hydrazone) 15a and 15c (Table 3). Compound 15b (3-pyrrolyl hydrazone) revealed the strongest antitumor activity against a panel of 60 cancer cell lines (Table 3), with GI% ranging from 5.80% to 188.50% and mean GI% equivalent to 74.29%; the results of its five-dose assay revealed potent antitumor activity with a mean GI50 of 1.45 μM with broad spectrum activity against various cell lines, which reached sub-micromolar GI50 values in breast cancer cell lines (MDA-MB-468 and MCF-7), NSCLC cell line HOP-92, and CNS cell lines (SNB-19 and U-251) (0.41, 0.42, 0.50, 0.51 and 0.61 μM, respectively). Furthermore, the OFX analog 15b exhibited an exceptional selectivity in the most sensitive MCF-7 cell line that exceeded 100-fold selectivity in the VERO normal cell line, as observed through cytotoxicity assay.Additionally, compound 15b elicited potent Topo II inhibitory activity with an inhibition percentage of 85.29%, which was nearly similar to that of etoposide (96.84%); on the other hand, it showed considerable activity against Topo I with an inhibition percentage of 41.87%. The tested OFX derivative 15b caused apparent cell arrest for MCF-7 cells at G1 as demonstrated by an elevation in G1 (64.76%) and a decrease in both S (27.15%) and G2/M (8.09%), compared to the control. Moreover, compound 15b significantly increased the overall percentage of apoptotic cells by 49.80-fold compared to that of the control. Also, compound 15b caused apparent elevation in the Bax level over two-fold as well as decreased BCL-2 to less than half, resulting in an increase of the Bax/BCL-2 ratio by 6-fold compared to β-actin. The previous results indicated the induction of apoptosis through an intrinsic pathway. Finally, the tested OFX derivative 15b showed a three-fold increase in caspase-9 and a two-fold increase in caspase-8 compared to the control, indicating that the OFX derivative might stimulate both intrinsic and extrinsic apoptotic pathways.
5. Levofloxacin derivatives
Ahadi et al.80 synthesized and evaluated the anticancer activity of a novel series of novel hybrids of levofloxacin (LVX, 16), with various substituted 1,3,4-thiadiazole moieties 17a–h (Fig. 4). The in vitro cytotoxicity assay on cancer cells, MCF-7, A549, and SKOV3, showed that the majority of the screened compounds possessed potent activity with IC50 ranging from 1.69 to 18.55 μM (Table 4).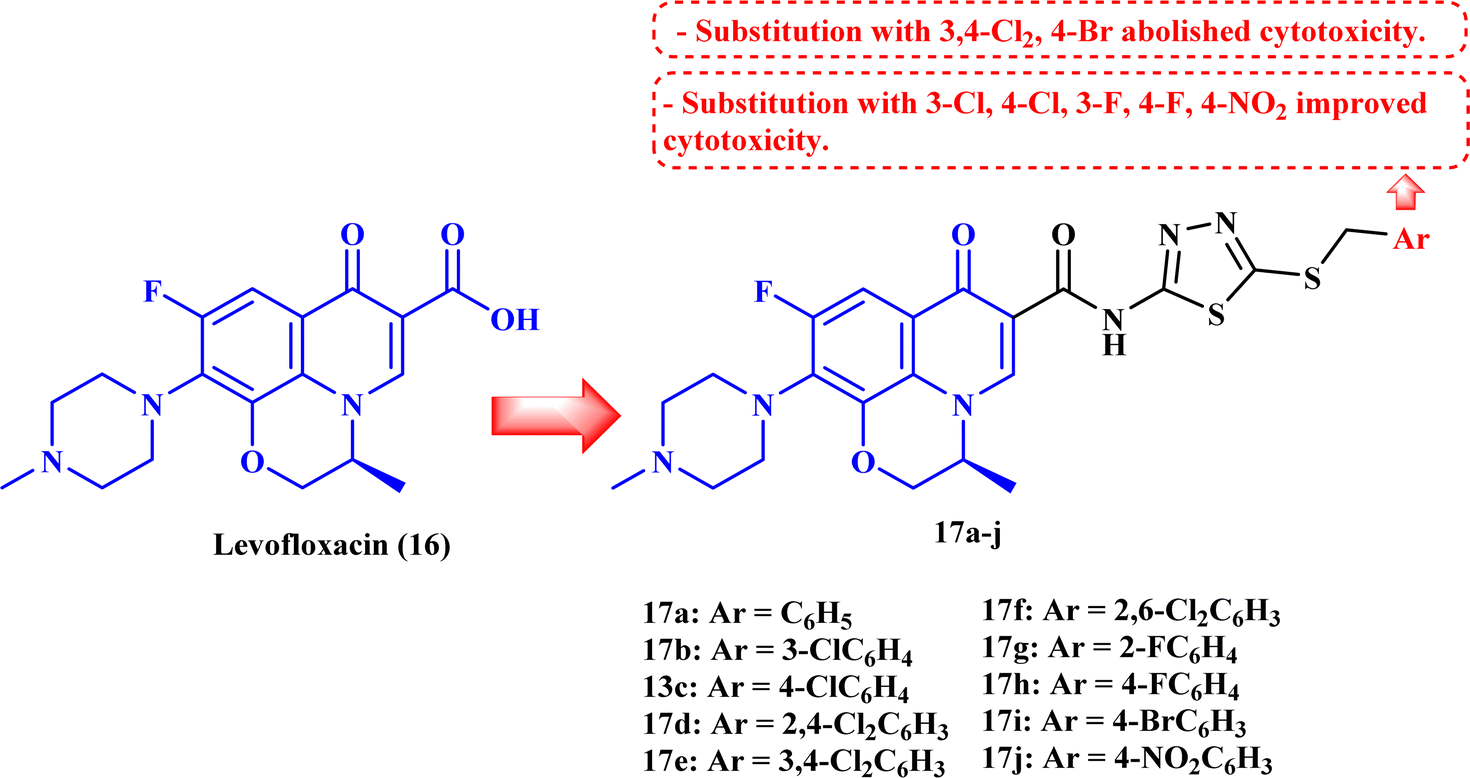 | ||
| Fig. 4 Structures and structure–activity relationships of LVX derivatives 17a–j as antiproliferative agents. | ||
| Compound | Structure | Activity |
|---|---|---|
| 17 | 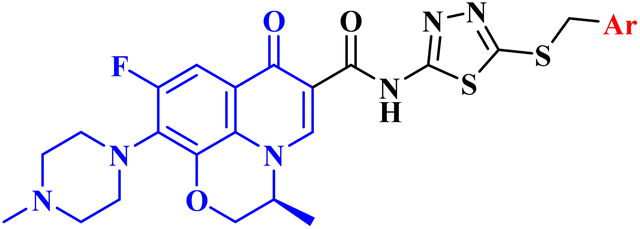 |
|
| 17a | Ar = C6H5 | MCF-7 IC50 = 4.82 μM |
| 17b | Ar = 3-ClC6H4 | MCF-7 IC50 = 2.82 μM; A549 IC50 = 3.81 μM; SKOV3 IC50 = 4.76 μM |
| 17c | Ar = 4-ClC6H4 | MCF-7 IC50 = 5.99 μM; SKOV3 IC50 = 3.38 μM |
| 17d | Ar = 2,4-Cl2C6H3 | MCF-7 IC50 = 10.86 μM; A549 IC50 = 2.65 μM; SKOV3 IC50 = 28.50 μM |
| 17e | Ar = 3,4-Cl2C6H3 | MCF-7 IC50 > 50 μM; A549 IC50 > 50 μM; SKOV3 IC50 > 50 μM |
| 17f | Ar = 2,6-Cl2C6H3 | MCF-7 IC50 = 8.42 μM; A549 IC50 = 15.50 μM; SKOV3 IC50 = 18.55 μM |
| 17g | Ar = 2-FC6H4 | MCF-7 IC50 = 6.08 μM; A549 IC50 = 10.23 μM; SKOV3 IC50 = 8.84 μM |
| 17h | Ar = 4-FC6H4 | MCF-7 IC50 = 1.69 μM; A549 IC50 = 2.62 μM; SKOV3 IC50 = 1.92 μM |
| 17bi | Ar = 4-BrC6H3 | MCF-7 IC50 > 50 μM; A549 IC50 > 50 μM; SKOV3 IC50 > 50 μM |
| 17j | Ar = 4-NO2C6H3 | MCF-7 IC50 = 4.11 μM; A549 IC50 = 11.48 μM; SKOV3 IC50 = 14.95 μM |
The structure–activity relationship studies exhibited that the phenyl ring substitution pattern of the benzyl moiety greatly affected the cytotoxicity, where anticancer activity was enhanced by the substitution with mono-chloro, 2,4-dichloro, 2,6-dichloro, fluoro, and nitro substituents. On the other hand, substitution with 3,4-dichloro and 4-bromo groups abolished the cytotoxicity. The most potent derivatives among the synthesized LVX analogs were 17b (3-chloro) and 17h (4-fluoro), which were found to be of comparable activity or 1.86-fold more potent compared to doxorubicin against the MCF-7 cell line with IC50 values of 2.82 and 1.69 μM, respectively. LVX derivatives 17b and 17h showed significant anticancer potential against the A549 cell line with IC50 values of 3.81 and 2.62 μM, respectively, while in the case of the SKOV3 cell line, they exhibited IC50 values of 4.76 and 1.92 μM, respectively.
Cell cycle assay results showed that LVX analog 17b induced cell cycle arrest in the G1 phase, which was proved by the significant drop in the percentage of G1 phase cells in MCF-7 cells from 59.06% to 46.19%; according to the results, LVX analog 17b could therefore stop cell division in the G1 phase. However, in the case of compound 17h, the proportion of cells in the G2/M phase rose noticeably from 9.59% to 16.18%, suggesting that this compound stopped the cell cycle in the G2/M phase. Both LVX derivatives 17b and 17h markedly elevated the percentage of cells in the sub-G1 phase, indicating the induction of apoptosis. The previous results encouraged the authors to examine the apoptotic effects of the tested compounds in MCF-7 cells. Compounds 17b and 17h induced a significant increase in the percentage of late apoptotic cells, from 3.62% to 52.30% and 37.50%, respectively, in MCF-7 cells at their IC50 values.
6. A recent patent filed
A recent patent81 has been filed for the usage of FQ-diphenyl ether hybrids as anticancer agents. The FQs prepared by this invention had the application of antitumor cell activity. The tumor cells comprised human breast cancer cells (MCF-7), human liver cancer cells (HepG 2), glioma cancer cells (U87 mg), and human lung cancer cells (A549). The results of in vitro cytotoxicity evaluation revealed that FQs 18a–c (IC50 values of 11.30, 10.40, and 12.90 μM, respectively) (Fig. 5) had better inhibitory activity against Umg87 cells at the same level as cisplatin (IC50 = 16.80 μM).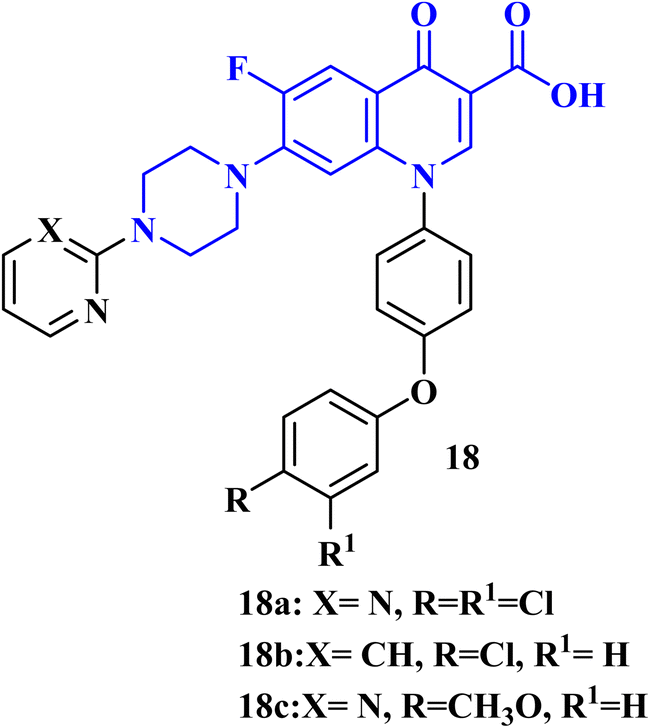 | ||
| Fig. 5 Structure of patented FQs 18a–c. | ||
7. Conclusion
Pharmaceutical companies appear to have a viable opportunity in drug repositioning. Biopharmaceutical businesses have already been influenced globally by the success stories of previously established repositioned drugs to adopt similar strategies and typologies to achieve success. FQs have dual benefit in medicine owing to their exceptional ability to suppress cancer cell development both in vivo and in vitro. Given that FQs are non-genotoxic and have a greater bioavailability rate than current anticancer medicines, their prospects as anticancer medications appear promising. FQs are underappreciated but important drugs with the potential to completely transform how people fight cancer. It is anticipated that continued advancements in the structure–activity relationship studies of FQs will result in the production of increasingly potent candidates soon. Collectively, in this review, we have updated the novel FQs reported in 2023 that have antiproliferative potential. An efficient approach to the SAR-based investigations of FQs as anticancer candidates has been adopted. Several FQs were reported as multitarget antiproliferative agents, exerting their antiproliferative effect via different mechanistic pathways such as Topo I and/or Topo II inhibition, apoptosis induction via intrinsic and/or extrinsic pathways, and cell cycle arrest. The combination of FQ derivatives with other anticancer drugs exhibited synergistic lethal effects against cancer cells. Several FQ analogs exhibited an exceptional selectivity towards cancer cells rather than normal cells, proving their safety. The SAR investigations of anticancer activity of FQ derivatives revealed that hybridization with other biologically active entities such as N-acylarylhydrazone, oxadiazole, semicarbazide, or quinoline greatly impacted their antiproliferative potential. A key factor in the efficacy of the FQ derivatives was the type and position of substituents on the incorporated entity. Amidification and esterification of FQs improved their anticancer activity. These compilations could be useful for medicinal chemists as a reference when developing FQ derivatives that target cancer and may eventually be approved in the future. Medicinal chemists may be inspired by this review to explore novel chemical entities with the FQ motif that have enhanced antiproliferative potential, which could provide new avenues for drug research and discovery.8. Future aspects
The promising biological results of CP derivatives 4b, 4c, and 5a, MXF derivative 12b, and LVX derivative 17h (Fig. 6) highlighted in this review emphasize the importance of FQ cores, especially CP, MXF, and LVX, as prominent scaffolds in cancer treatment. They showed significant anticancer potential against T-24, PC-3, SW480, MCF-7, and A549 human cancer cell lines with IC50 values in the range of 1.69 to 3.36 μM, which surpassed those of the standard anticancer drugs doxorubicin and cisplatin. CP derivatives 4b, 4c, and 5a were reported as multitarget antiproliferative agents, exerting their activity via Topo II inhibition, induction of apoptosis, and cell cycle progression arrest. As for the most potent MXF derivative 12b, it decreased the IL-6 release and inhibited NF-κB activation in cancer cells, induced apoptosis, and halted cell cycle progression. LVX derivative 17h stopped the cell cycle in the G2/M phase and elevated the percentage of cells in the sub-G1 phase, indicating the induction of apoptosis. The investigation of these FQ derivatives revealed that their prominent activity may be rationalized by the presence of a carbonyl group that can form hydrogen bonds with the amino acids in Topo I and Topo II active sites. They warrant further investigations such as modifications at various positions, which can further enhance the anticancer potential. Incorporation of a thioxo or an imino group as an isostere to the carbonyl group may enhance the anticancer activity of these FQ derivatives. Hybridizing these derivatives with different heterocyclic systems or varying the length of the embedded linkers may contribute to enhanced anticancer potency of these FQ derivatives. These compounds provide more opportunities for accelerated development of novel FQ derivatives with potential anticancer activity through the application of structure-based drug design or artificial intelligence software. By using these strategies, FQs may be repositioned as advantageous candidates for cancer treatment.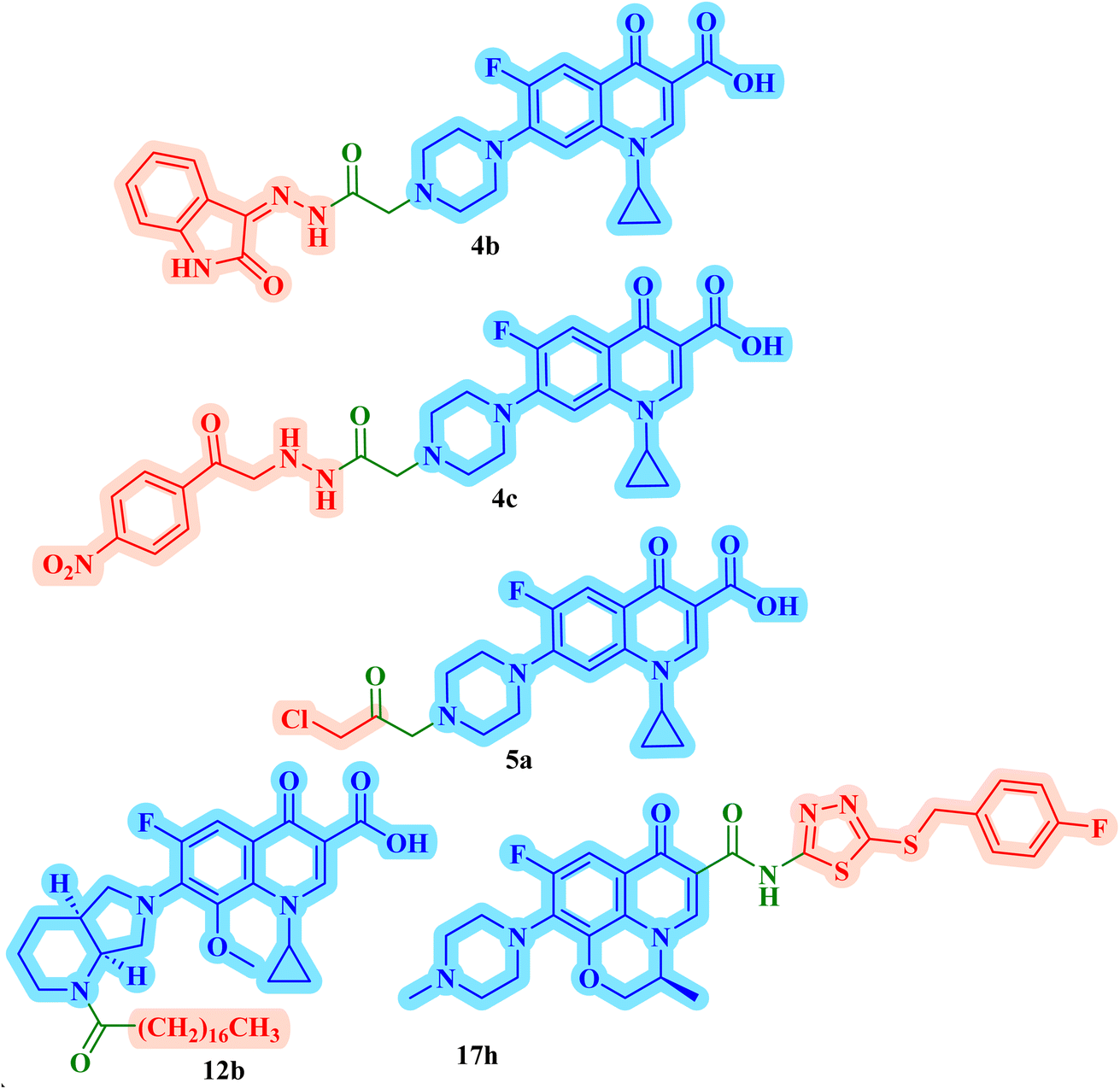 | ||
| Fig. 6 Structure of potent antiproliferative FQ derivatives 4b, 4c, 5a, 12b, and 17h. | ||
Data availability
No primary research results, software, or code have been included and no new data were generated or analyzed as part of this review.Author contributions
Asmaa E. Kassab: data collection, scientific writing, revision, and conceptualization Rania M. Gomaa: scientific writing and conceptualization, and Ehab M. Gedawy: scientific writing, revision, and conceptualization.Conflicts of interest
According to the authors, there are no conflicting financial interests or personal relationships that they are aware of that could affect the work reported in this article.Acknowledgements
The authors express their gratitude to the Faculty of Pharmacy, Cairo University.References
- J. W. Scannell, A. Blanckley, H. Boldon and B. Warrington, Diagnosing the decline in pharmaceutical R& D efficiency, Nat. Rev. Drug Discovery, 2012, 11(3), 191–200, DOI:10.1038/nrd3681
.
- V. Yadav and P. Talwar, Repositioning of fluoroquinolones from antibiotic to anti-cancer agents: An underestimated truth, Biomed. Pharmacother., 2019, 111, 934–946, DOI:10.1016/j.biopha.2018.12.119
- D. M. Klug, M. H. Gelb and M. P. Pollastri, Repurposing strategies for tropical disease drug discovery, Bioorg. Med. Chem. Lett., 2016, 26(11), 2569–2576, DOI:10.1016/j.bmcl.2016.03.103
- B. Singh, A. Sharma and N. Gunaganti, et al., Chemical Optimization of CBL0137 for Human African Trypanosomiasis Lead Drug Discovery, J. Med. Chem., 2023, 66(3), 1972–1989, DOI:10.1021/acs.jmedchem.2c01767
- T. T. Ashburn and K. B. Thor, Drug repositioning: identifying and developing new uses for existing drugs, Nat. Rev. Drug Discovery, 2004, 3(8), 673–683, DOI:10.1038/nrd1468
- S. Murteira, A. Millier, Z. Ghezaiel and M. Lamure, Drug reformulations and repositioning in the pharmaceutical industry and their impact on market access: regulatory implications, Journal of Market Access & Health Policy, 2014, 2(1), 22813, DOI:10.3402/jmahp.v2.22813
- R. Brown, E. Curry, L. Magnani, C. S. Wilhelm-Benartzi and J. Borley, Poised epigenetic states and acquired drug resistance in cancer, Nat. Rev. Cancer, 2014, 14(11), 747–753, DOI:10.1038/nrc3819
- F. Bray, A. Jemal, N. Grey, J. Ferlay and D. Forman, Global cancer transitions according to the Human Development Index (2008–2030): a population-based study, Lancet Oncol., 2012, 13(8), 790–801, DOI:10.1016/S1470-2045(12)70211-5
- M. K. Kathiravan, M. M. Khilare, K. Nikoomanesh, A. S. Chothe and K. S. Jain, Topoisomerase as target for antibacterial and anticancer drug discovery, J. Enzyme Inhib. Med. Chem., 2013, 28(3), 419–435, DOI:10.3109/14756366.2012.658785
- H. K. Swedan, A. E. Kassab, E. M. Gedawy and S. E. Elmeligie, Design, synthesis, and biological evaluation of novel ciprofloxacin derivatives as potential anticancer agents targeting topoisomerase II enzyme, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 118–137, DOI:10.1080/14756366.2022.2136172
- A. E. Kassab and E. M. Gedawy, Novel ciprofloxacin hybrids using biology oriented drug synthesis (BIODS) approach: Anticancer activity, effects on cell cycle profile, caspase-3 mediated apoptosis, topoisomerase II inhibition, and antibacterial activity, Eur. J. Med. Chem., 2018, 150, 403–418, DOI:10.1016/j.ejmech.2018.03.026
- J. W. Lawrence, S. Darkin-Rattray, F. Xie, A. H. Neims and T. C. Rowe, 4-Quinolones cause a selective loss of mitochondrial DNA from mouse L1210 leukemia cells, J. Cell. Biochem., 1993, 51(2), 165–174, DOI:10.1002/jcb.240510208
- K. Seo, R. Holt, Y. S. Jung, C. O. Rodriguez, X. Chen and R. B. Rebhun, Fluoroquinolone-Mediated Inhibition of Cell Growth, S-G2/M Cell Cycle Arrest, and Apoptosis in Canine Osteosarcoma Cell Lines, PLoS One, 2012, 7(8), e42960, DOI:10.1371/journal.pone.0042960
- S. Sha, H. W. Han and F. Gao, et al., Correction: Discovery of fluoroquinolone derivatives as potent, selective inhibitors of PI3Kγ, MedChemComm, 2015, 6(12), 2233, 10.1039/C5MD90050F
- T. Kloskowski, K. Szeliski and Z. Fekner, et al., Ciprofloxacin and Levofloxacin as Potential Drugs in Genitourinary Cancer Treatment—The Effect of Dose–Response on 2D and 3D Cell Cultures, Int. J. Mol. Sci., 2021, 22(21), 11970, DOI:10.3390/ijms222111970
- B. El-Rayes, R. Grignon, N. Aslam, O. Aranha and F. Sarkar, Ciprofloxacin inhibits cell growth and synergises the effect of etoposide in hormone resistant prostate cancer cells, Int. J. Oncol., 2002, 21(1), 207–211, DOI:10.3892/ijo.21.1.207
- A. Beberok, D. Wrześniok, J. Rok, Z. Rzepka, M. Respondek and E. Buszman, Ciprofloxacin triggers the apoptosis of human triple-negative breast cancer MDA-MB-231 cells via the p53/Bax/Bcl-2 signaling pathway, Int. J. Oncol., 2018, 52(5), 1727–1737, DOI:10.3892/ijo.2018.4310
- A. Beberok, Z. Rzepka, M. Respondek, J. Rok, M. Stradowski and D. Wrześniok, Moxifloxacin as an inducer of apoptosis in melanoma cells: A study at the cellular and molecular level, Toxicol. in Vitro, 2019, 55, 75–92, DOI:10.1016/j.tiv.2018.12.002
- M. J. Robinson, B. A. Martin, T. D. Gootz, P. R. McGuirk and N. Osheroff, Effects of novel fluoroquinolones on the catalytic activities of eukaryotic topoisomerase II: Influence of the C-8 fluorine group, Antimicrob. Agents Chemother., 1992, 36(4), 751–756, DOI:10.1128/AAC.36.4.751
- S. H. Elsea, N. Osheroff and J. L. Nitiss, Cytotoxicity of quinolones toward eukaryotic cells. Identification of topoisomerase II as the primary cellular target for the quinolone CP-115,953 in yeast, J. Biol. Chem., 1992, 267(19), 13150–13153, DOI:10.1016/S0021-9258(18)42185-0
- S. J. Froelich-Ammon, P. R. McGuirk, T. D. Gootz, M. R. Jefson and N. Osheroff, Novel 1-8-bridged chiral quinolones with activity against topoisomerase II: stereospecificity of the eukaryotic enzyme, Antimicrob. Agents Chemother., 1993, 37(4), 646–651, DOI:10.1128/AAC.37.4.646
- S. H. Elsea, P. R. McGuirk, T. D. Gootz, M. Moynihan and N. Osheroff, Drug features that contribute to the activity of quinolones against mammalian topoisomerase II and cultured cells: correlation between enhancement of enzyme-mediated DNA cleavage in vitro and cytotoxic potential, Antimicrob. Agents Chemother., 1993, 37(10), 2179–2186, DOI:10.1128/AAC.37.10.2179
- M. J. Robinson, S. H. Elsea, B. A. Martin, et al., A Novel Quinolone with Potent Activity against Eukaryotic DNA Topoisomerase II, Molecular Biology of DNA Topoisomerases and its Application to Chemotherapy, 1992, p. 189 Search PubMed
- T. D. Gootz, P. R. McGuirk, M. S. Moynihan and S. L. Haskell, Placement of alkyl substituents on the C-7 piperazine ring of fluoroquinolones: dramatic differential effects on mammalian topoisomerase II and DNA gyrase, Antimicrob. Agents Chemother., 1994, 38(1), 130–133, DOI:10.1128/AAC.38.1.130
- V. Hills and A. Park, US Pat., US4996355A, 1991 Search PubMed
- W. E. Kohlbrenner, N. Wideburg, D. Weigl, A. Saldivar and D. T. W. Chu, Induction of calf thymus topoisomerase II-mediated DNA breakage by the antibacterial isothiazoloquinolones A-65281 and A-65282, Antimicrob. Agents Chemother., 1992, 36(1), 81–86, DOI:10.1128/AAC.36.1.81
- G. S Bisacchi and M. R Hale, A “double-edged” scaffold: antitumor power within the antibacterial quinolone, Curr. Med. Chem., 2016, 23(6), 520–577 CrossRef PubMed
- M. A. A. Abdel-Aal, S. A. Abdel-Aziz, M. S. A. Shaykoon and G. E. D. A. Abuo-Rahma, Towards anticancer fluoroquinolones: A review article, Arch. Pharm., 2019, 352(7), e1800376, DOI:10.1002/ardp.201800376
- H. K. Swedan, A. E. Kassab, E. M. Gedawy and S. E. Elmeligie, Bioorganic Chemistry Topoisomerase II inhibitors design : Early studies and new perspectives, Bioorg. Chem., 2023, 136, 106548, DOI:10.1016/j.bioorg.2023.106548
- Y. Pommier, DNA Topoisomerases and Cancer, ed. Y. Pommier, Springer, New York, 2012, DOI:10.1007/978-1-4614-0323-4
- J. C. Wang, Cellular roles of DNA topoisomerases: a molecular perspective, Nat. Rev. Mol. Cell Biol., 2002, 3(6), 430–440, DOI:10.1038/nrm831
- C. R. Barker, Inhibition of Hsp90 acts synergistically with topoisomerase II poisons to increase the apoptotic killing of cells due to an increase in topoisomerase II mediated DNA damage, Nucleic Acids Res., 2006, 34(4), 1148–1157, DOI:10.1093/nar/gkj516
- V. Uivarosi, Metal Complexes of Quinolone Antibiotics and Their Applications: An Update, Molecules, 2013, 18(9), 11153–11197, DOI:10.3390/molecules180911153
- K. J. Marians and H. Hiasa, Mechanism of Quinolone Action, J. Biol. Chem., 1997, 272(14), 9401–9409, DOI:10.1074/jbc.272.14.9401
- M. S. Luijsterburg, M. F. White, R. van Driel and R. T. Dame, The Major Architects of Chromatin: Architectural Proteins in Bacteria, Archaea and Eukaryotes, Crit. Rev. Biochem. Mol. Biol., 2008, 43(6), 393–418, DOI:10.1080/10409230802528488
- V. Visone, A. Vettone and M. Serpe, et al., Chromatin Structure and Dynamics in Hot Environments: Architectural Proteins and DNA Topoisomerases of Thermophilic Archaea, Int. J. Mol. Sci., 2014, 15(9), 17162–17187, DOI:10.3390/ijms150917162
- J. J. Champoux, DNA Topoisomerases: Structure, Function, and Mechanism, Annu. Rev. Biochem., 2001, 70(1), 369–413, DOI:10.1146/annurev.biochem.70.1.369
- Y. Pommier, E. Leo, H. Zhang and C. Marchand, DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs, Chem. Biol., 2010, 17(5), 421–433, DOI:10.1016/j.chembiol.2010.04.012
- P. Bohley, Molecular Cell Biology, Biochem. Educ., 1987, 15(2), 99, DOI:10.1016/0307-4412(87)90114-2
- K. Kondaka and I. Gabriel, Targeting DNA Topoisomerase II in Antifungal Chemotherapy, Molecules, 2022, 27(22), 7768, DOI:10.3390/molecules27227768
- B. S. Dwarakanath, D. Khaitan and R. Mathur, Inhibitors of topoisomerases as anticancer drugs: problems and prospects, Indian J. Exp. Biol., 2004, 42(7), 649–659 CAS
- A. Beberok, Z. Rzepka, M. Respondek, J. Rok, D. Sierotowicz and D. Wrześniok, GSH depletion, mitochondrial membrane breakdown, caspase-3/7 activation and DNA fragmentation in U87MG glioblastoma cells: New insight into the mechanism of cytotoxicity induced by fluoroquinolones, Eur. J. Pharmacol., 2018, 835, 94–107, DOI:10.1016/j.ejphar.2018.08.002
- A. Beberok, D. Wrześniok and A. Minecka, et al., Ciprofloxacin-mediated induction of S-phase cell cycle arrest and apoptosis in COLO829 melanoma cells, Pharmacol. Rep., 2018, 70(1), 6–13, DOI:10.1016/j.pharep.2017.07.007
- K. Nishi, M. Kato, S. Sakurai, A. Matsumoto, Y. Iwase and N. Yumita, Enoxacin with UVA Irradiation Induces Apoptosis in the AsPC1 Human Pancreatic Cancer Cell Line Through ROS Generation, Anticancer Res., 2017, 37(11), 6211–6214, DOI:10.21873/anticanres.12071
- A. Beberok, D. Wrześniok and M. Szlachta, et al., Lomefloxacin Induces Oxidative Stress and Apoptosis in COLO829 Melanoma Cells, Int. J. Mol. Sci., 2017, 18(10), 2194, DOI:10.3390/ijms18102194
- S. Nakai, T. Imaizumi and T. Watanabe, et al., Photodynamically-induced Apoptosis Due to Ultraviolet A in the Presence of Lomefloxacin in Human Promyelocytic Leukemia Cells, Anticancer Res., 2017, 37(11), 6407–6413, DOI:10.21873/anticanres.12094
- M. Yu, R. Li and J. Zhang, Repositioning of antibiotic levofloxacin as a mitochondrial biogenesis inhibitor to target breast cancer, Biochem. Biophys. Res. Commun., 2016, 471(4), 639–645, DOI:10.1016/j.bbrc.2016.02.072
- M. Song, H. Wu and S. Wu, et al., Antibiotic drug levofloxacin inhibits proliferation and induces apoptosis of lung cancer cells through inducing mitochondrial dysfunction and oxidative damage, Biomed. Pharmacother., 2016, 84, 1137–1143, DOI:10.1016/j.biopha.2016.10.034
- V. Yadav, P. Varshney, S. Sultana, J. Yadav and N. Saini, Moxifloxacin and ciprofloxacin induces S-phase arrest and augments apoptotic effects of cisplatin in human pancreatic cancer cells via ERK activation, BMC Cancer, 2015, 15(1), 581, DOI:10.1186/s12885-015-1560-y
- P. Perucca, M. Savio and O. Cazzalini, et al., Structure–activity relationship and role of oxygen in the potential antitumour activity of fluoroquinolones in human epithelial cancer cells, J. Photochem. Photobiol., B, 2014, 140, 57–68, DOI:10.1016/j.jphotobiol.2014.07.006
- I. Jemel-Oualha, J. Elloumi-Mseddi, A. Beji, B. Hakim and S. Aifa, Controversial effect on Erk activation of some cytotoxic drugs in human LOVO colon cancer cells, J. Recept. Signal Transduction, 2016, 36(1), 21–25, DOI:10.3109/10799893.2014.975246
- E. J. Sousa, I. Graça and T. Baptista, et al., Enoxacin inhibits growth of prostate cancer cells and effectively restores microRNA processing, Epigenetics, 2013, 8(5), 548–558, DOI:10.4161/epi.24519
- J. H. Gong, X. J. Liu, B. Y. Shang, S. Z. Chen and Y. S. Zhen, HERG K+ channel related chemosensitivity to sparfloxacin in colon cancer cells, Oncol. Rep., 2010, 23(6), 1747–1756, DOI:10.3892/or_00000820
- D. J. Smart, H. D. Halicka, F. Traganos, Z. Darzynkiewicz and G. M. Williams, Ciprofloxacin-induced G2 arrest and apoptosis in TK6 lymphoblastoid cells is not dependent on DNA double-strand break formation, Cancer Biol. Ther., 2008, 7(1), 113–119, DOI:10.4161/cbt.7.1.5136
- P. S. Devi, M. S. Kumar and S. M. Das, Evaluation of Antiproliferative Activity of Red Sorghum Bran Anthocyanin on a Human Breast Cancer Cell Line (MCF-7), Int. J. Breast Cancer, 2011, 2011(1), 1–6, DOI:10.4061/2011/891481
- A. Gürbay, M. Osman, A. Favier and F. Hincal, Ciprofloxacin-Induced Cytotoxicity and Apoptosis in HeLa Cells, Toxicol. Mech. Methods, 2005, 15(5), 339–342, DOI:10.1080/153765291009877
- E. R. Mondal, S. K. Das and P. Mukherjee, Comparative evaluation of antiproliferative activity and induction of apoptosis by some fluoroquinolones on a human non-small cell lung cancer cell line in culture, Asian Pac. J. Cancer Prev., 2004, 5(2), 196–204 CAS
- O. Aranha, R. Grignon, N. Fernandes, T. McDonnell, D. Wood and F. Sarkar, Suppression of human prostate cancer cell growth by ciprofloxacin is associated with cell cycle arrest and apoptosis, Int. J. Oncol., 2003, 22(4), 787–794, DOI:10.3892/ijo.22.4.787
- O. Aranha, Jr D. P. Wood and F. H. Sarkar, Ciprofloxacin mediated cell growth inhibition, S/G2-M cell cycle arrest, and apoptosis in a human transitional cell carcinoma of the bladder cell line, Clin. Cancer Res., 2000, 6(3), 891–900 CAS
- A. E. Kassab and E. M. Gedawy, Novel ciprofloxacin hybrids using biology oriented drug synthesis (BIODS) approach: Anticancer activity, effects on cell cycle profile, caspase-3 mediated apoptosis, topoisomerase II inhibition, and antibacterial activity, Eur. J. Med. Chem., 2018, 150, 403–418, DOI:10.1016/j.ejmech.2018.03.026
- T. C. Chen, Y. L. Hsu, Y. C. Tsai, Y. W. Chang, P. L. Kuo and Y. H. Chen, Gemifloxacin inhibits migration and invasion and induces mesenchymal–epithelial transition in human breast adenocarcinoma cells, J. Mol. Med., 2014, 92(1), 53–64, DOI:10.1007/s00109-013-1083-4
- J. Y. Kan, Y. L. Hsu, Y. H. Chen, T. C. Chen, J. Y. Wang and P. L. Kuo, Gemifloxacin, a Fluoroquinolone Antimicrobial Drug, Inhibits Migration and Invasion of Human Colon Cancer Cells, BioMed Res. Int., 2013, 2013, 1–11, DOI:10.1155/2013/159786
- G. Valianatos, B. Valcikova and K. Growkova, et al., A small molecule drug promoting miRNA processing induces alternative splicing of MdmX transcript and rescues p53 activity in human cancer cells overexpressing MdmX protein, PLoS One, 2017, 12(10), e0185801, DOI:10.1371/journal.pone.0185801
- T. Kloskowski, N. Gurtowska and M. Nowak, et al., The influence of ciprofloxacin on viability of A549, HepG2, A375. S2, B16 and C6 cell lines in vitro, Acta Pol. Pharm., 2011, 68(6), 859–865 CrossRef CAS PubMed
- A. M. Kamat and D. L. Lamm, Antitumor activity of common antibiotics against superficial bladder cancer, Urology., 2004, 63(3), 457–460, DOI:10.1016/j.urology.2003.10.038
- A. M. Kamat, J. I. DeHaven and D. L. Lamm, Quinolone antibiotics: a potential adjunct to intravesical chemotherapy for bladder cancer, Urology, 1999, 54(1), 56–61, DOI:10.1016/S0090-4295(99)00064-3
- S. Ebisuno, T. Inagaki, Y. Kohjimoto and T. Ohkawa, The cytotoxic effects of fleroxacin and ciprofloxacin on transitional cell carcinoma in Vitro, Cancer, 1997, 80(12), 2263–2267, DOI:10.1002/(SICI)1097-0142(19971215)80:12<2263::AID-CNCR7>3.0.CO;2-V
- T. M. Seay, S. J. Peretsman and P. S. Dixon, Inhibition of Human Transitional Cell Carcinoma in Vitro Proliferation by Fluoroquinolone Antibiotics, J. Urol., 1996, 155(2), 757–762, DOI:10.1016/S0022-5347(01)66516-9
- E. Somekh, D. Douer, N. Shaked and E. Rubinstein, In vitro effects of ciprofloxacin and pefloxacin on growth of normal human hematopoietic progenitor cells and on leukemic cell lines, J. Pharmacol. Exp. Ther., 1989, 248(1), 415–418 CAS
- H. K. Swedan, A. E. Kassab, E. M. Gedawy and S. E. Elmeligie, Topoisomerase II inhibitors design: Early studies and new perspectives, Bioorg. Chem., 2023, 136, 106548, DOI:10.1016/j.bioorg.2023.106548
- S. O. Alhaj-Suliman, Y. W. Naguib and E. I. Wafa, et al., A ciprofloxacin derivative with four mechanisms of action overcomes paclitaxel resistance in p53-mutant and MDR1 gene-expressing type II human endometrial cancer, Biomaterials, 2023, 296, 122093, DOI:10.1016/j.biomaterials.2023.122093
- Y. W. Naguib, S. O. Alhaj-Suliman and E. I. Wafa, et al., Ciprofloxacin Derivative-Loaded Nanoparticles Synergize with Paclitaxel Against Type II Human Endometrial Cancer, Small, 2023, 2302931, DOI:10.1002/smll.202302931
- M. A. Elanany, E. E. A. Osman, E. M. Gedawy and S. M. Abou-Seri, Design and synthesis of novel cytotoxic fluoroquinolone analogs through topoisomerase inhibition, cell cycle arrest, and apoptosis, Sci. Rep., 2023, 13(1), 4144, DOI:10.1038/s41598-023-30885-5
- H. K. Swedan, A. E. Kassab and E. M. Gedawy, et al., Design , synthesis , and biological evaluation of novel ciprofloxacin derivatives as potential anticancer agents targeting topoisomerase II enzyme, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 118–137, DOI:10.1080/14756366.2022.2136172
- M. Struga, P. Roszkowski and A. Bielenica, et al., N-Acylated Ciprofloxacin Derivatives: Synthesis and In Vitro Biological Evaluation as Antibacterial and Anticancer Agents, ACS Omega, 2023, 8(21), 18663–18684, DOI:10.1021/acsomega.3c00554
- M. A. Fawzy, R. H. Abu-baih, G. E. D. A. Abuo-Rahma, I. M. Abdel-Rahman, A. A. K. El-Sheikh and M. H. Nazmy, In Vitro Anticancer Activity of Novel Ciprofloxacin Mannich Base in Lung Adenocarcinoma and High-Grade Serous Ovarian Cancer Cell Lines via Attenuating MAPK Signaling Pathway, Molecules, 2023, 28(3), 1137, DOI:10.3390/molecules28031137
- G. M. Alasadi and Z. Al-Obaidi, Synthesis of novel acylated and esterified ciprofloxacin derivatives as efficient anticancer and antimicrobial agents, Front. Mater., 2023, 10, 1255955, DOI:10.3389/fmats.2023.1255955
- H. A. A. Ezelarab, H. A. Hassan, G. E. D. A. Abuo-Rahma and S. H. Abbas, Design, synthesis, and biological investigation of quinoline/ciprofloxacin hybrids as antimicrobial and anti-proliferative agents, J. Iran. Chem. Soc., 2023, 20(3), 683–700, DOI:10.1007/s13738-022-02704-7
- A. Chrzanowska, D. Kurpios-Piec and B. Żyżyńska-Granica, et al., Anticancer activity and metabolic alteration in colon and prostate cancer cells by novel moxifloxacin conjugates with fatty acids, Eur. J. Pharmacol., 2023, 940, 175481, DOI:10.1016/j.ejphar.2022.175481
- H. Ahadi, M. Shokrzadeh, Z. Hosseini-khah, N. Ghassemi Barghi, M. Ghasemian and S. Emami, Conversion of antibacterial quinolone drug levofloxacin to potent cytotoxic agents, J. Biochem. Mol. Toxicol., 2023, 37(6), 1–10, DOI:10.1002/jbt.23334
- P. Zhang and W. X. Wanmei Li, Quinolone derivative and preparation method and application thereof, Chinese Pat., CN116444491A, 2023 Search PubMed
| This journal is © The Royal Society of Chemistry 2024 |