
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent studies on protein kinase signaling inhibitors based on thiazoles: review to date
Manal S. Ebaida,
Hoda Atef Abdelsattar Ibrahim b,
Asmaa F. Kassemc and
Ahmed Sabt*d
b,
Asmaa F. Kassemc and
Ahmed Sabt*d
aDepartment of Chemistry, College of Science, Northern Border University, Arar, Saudi Arabia
bPediatric Department, Faculty of Medicine, Cairo University, Cairo, Egypt
cDepartment of Chemistry, College of Science and Humanities in Al-Kharj, Prince Sattam Bin Abdulaziz University, Al-Kharj, 11942, Saudi Arabia
dChemistry of Natural Compounds Department, Pharmaceutical and Drug Industries Research Institute, National Research Center, Dokki, Cairo 12622, Egypt. E-mail: sabt.nrc@gmail.com
First published on 19th November 2024
Abstract
Due to the important role of protein kinases in protein phosphorylation within vital cellular processes, their abnormal function, especially in cancer situations, has underscored their importance in therapy. Thiazole structures are versatile frameworks present in numerous bioactive compounds. Thiazole derivatives, as a highly favored structural motif, have garnered considerable interest from both industrial and medicinal researchers and have demonstrated notable success over past decades due to their diverse biological properties, including anticancer, antibacterial, antifungal, anti-HIV, antiulcer, and anti-inflammatory activities. Moreover, several thiazole-based drugs are widely recognized pharmaceuticals on the market. Due to their specific structural features, thiazole derivatives have a high potential for interacting with different protein kinases, leading researchers to investigate a variety of structural changes. This thorough review thoroughly examines the design and biological evaluations of small molecules utilizing thiazole as potential agents that target various kinases for anti-cancer applications. These compounds are categorized into two classes: inhibitors of serine/threonine and tyrosine kinases. The goal is to promote the development and progress of more effective, targeted compounds for cancer treatment by highlighting the potential of thiazole in inhibiting kinases.
1. Introduction
Heterocyclic chemistry is currently receiving increased focus due to its classification of molecules that have been utilized in medicinal chemistry applications.1–3 A primary objective within the realms of organic and medicinal chemistry is the creation, synthesis, and advancement of molecules that exhibit promise for application as pharmaceutical agents.4,5 Numerous biologically active molecules are characterized by the presence of five-membered rings containing two heteroatoms. Heterocyclic compounds containing nitrogen are particularly significant in the process of drug discovery,6 with the thiazole ring being a notable example of such compounds.Thiazole, while not occurring naturally, is present in various natural compounds like peptide alkaloids, metabolites, and cyclopeptides.7 For example, Cyclotheonaellazole A (1) and Oriamide (2) are cyclic peptides containing thiazolyl propenoic acid that have been found in the marine sponge Theonella sp.8,9 Apratoxin A (3), which is derived from Lyngbya majuscula, is a well-known marine product with anticancer properties.10,11 Dendroamide A (4), obtained from the cyanobacterium Stigonema dendroideum fremy, is used in the treatment of tumors that develop resistance.12 Argyrin A (5), a cyclic peptide from the myxobacterium Archangium gephyra, is known for its strong anti-tumor effects,13 Fig. 1.
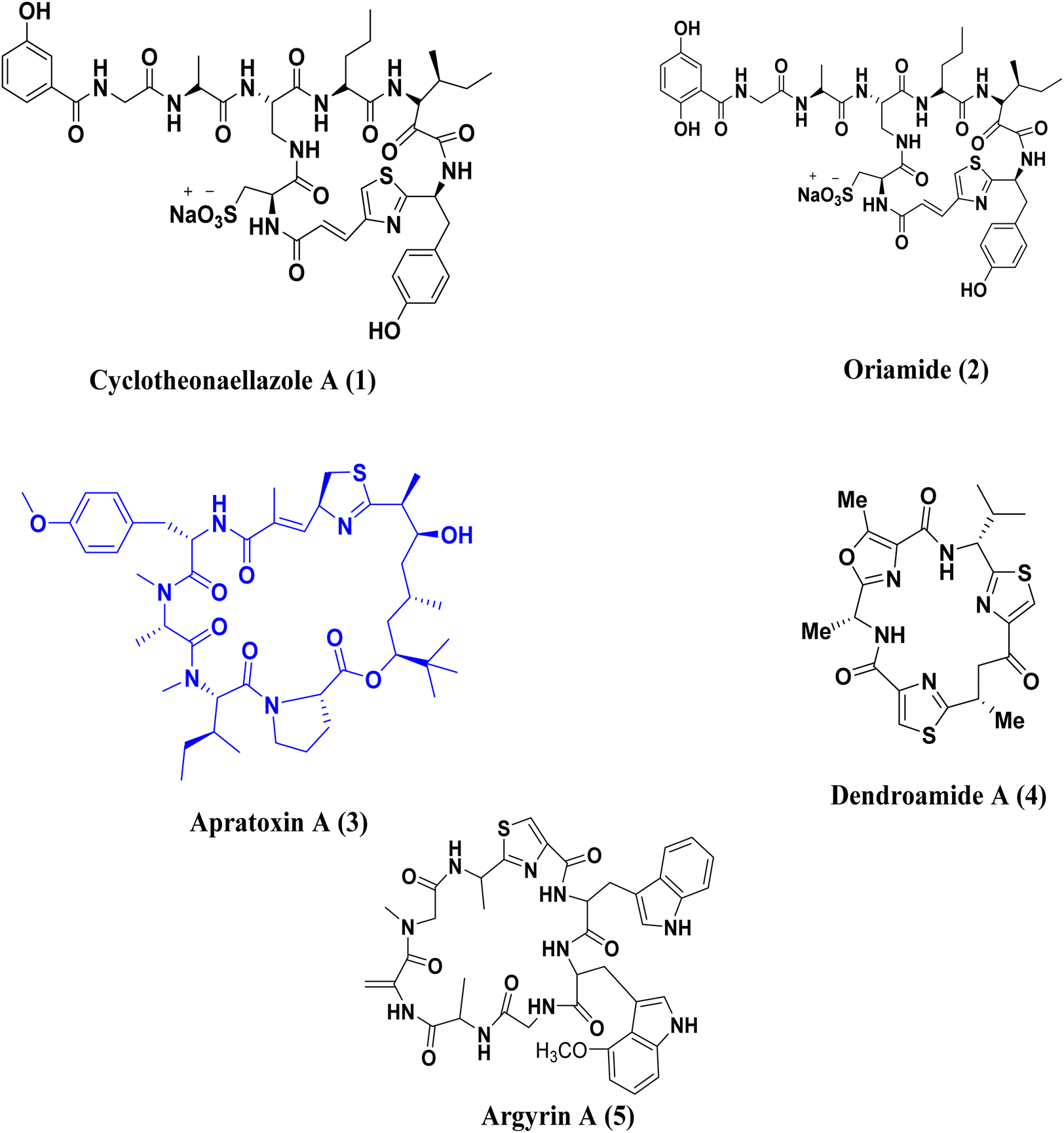 | ||
| Fig. 1 Various natural compounds containing thiazole ring. | ||
Furthermore, thiazole derivatives, including vitamin B1 (6), are naturally present in various foods and marine sources e.g. 7a–c and 8.14 Vitamin B1, also known as thiamin, is an essential water-soluble vitamin crucial for mitochondrial energetics, particularly in ATP formation. Moreover, it acts as a vital coenzyme in glucose and amino acid metabolic pathways for living organisms,15 Fig. 2.
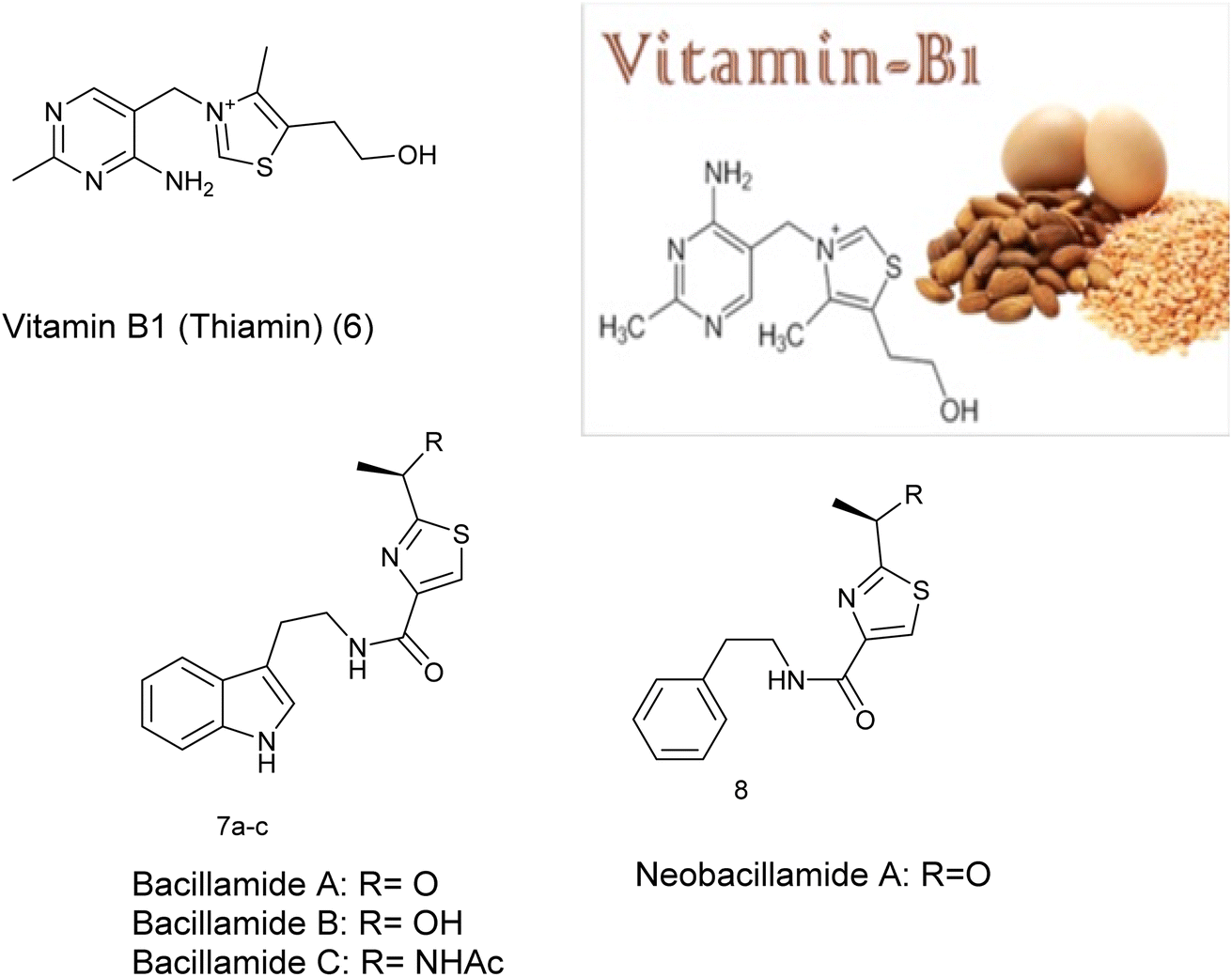 | ||
| Fig. 2 Thiazole ring present in many foods and marine source. | ||
In recent years, the thiazole ring, a heterocyclic structure composed of sulfur and nitrogen atoms, has demonstrated a wide range of biological properties.16 It functions as a significant pharmacophore core with extensive pharmaceutical applications, including carbonic anhydrase inhibitory, antimalarial, antiparasitic, anti-SARS-CoV-2, antimicrobial, anti-inflammatory, and anticancer activities.17–24 Moreover, the existence of thiazole rings in a variety of natural compounds, medications, and human enzymes has led to the development of numerous thiazole analogs that exhibit strong antitumor or cytotoxic properties. These analogs have been custom-made to focus on particular pathways in cancer cells.25 It is worth mentioning that a number of existing drugs that include thiazole have been authorized for use as anti-cancer treatments such as Vosaroxin (9),26 Dabrafenib (10),27 patellamide A (11),26–28 dasatinib (12), epothilones (13), ixabepilone (14), tiazofurin (15), Kud 773 (16), and bleomycin (17)29 Fig. 3 and 4.
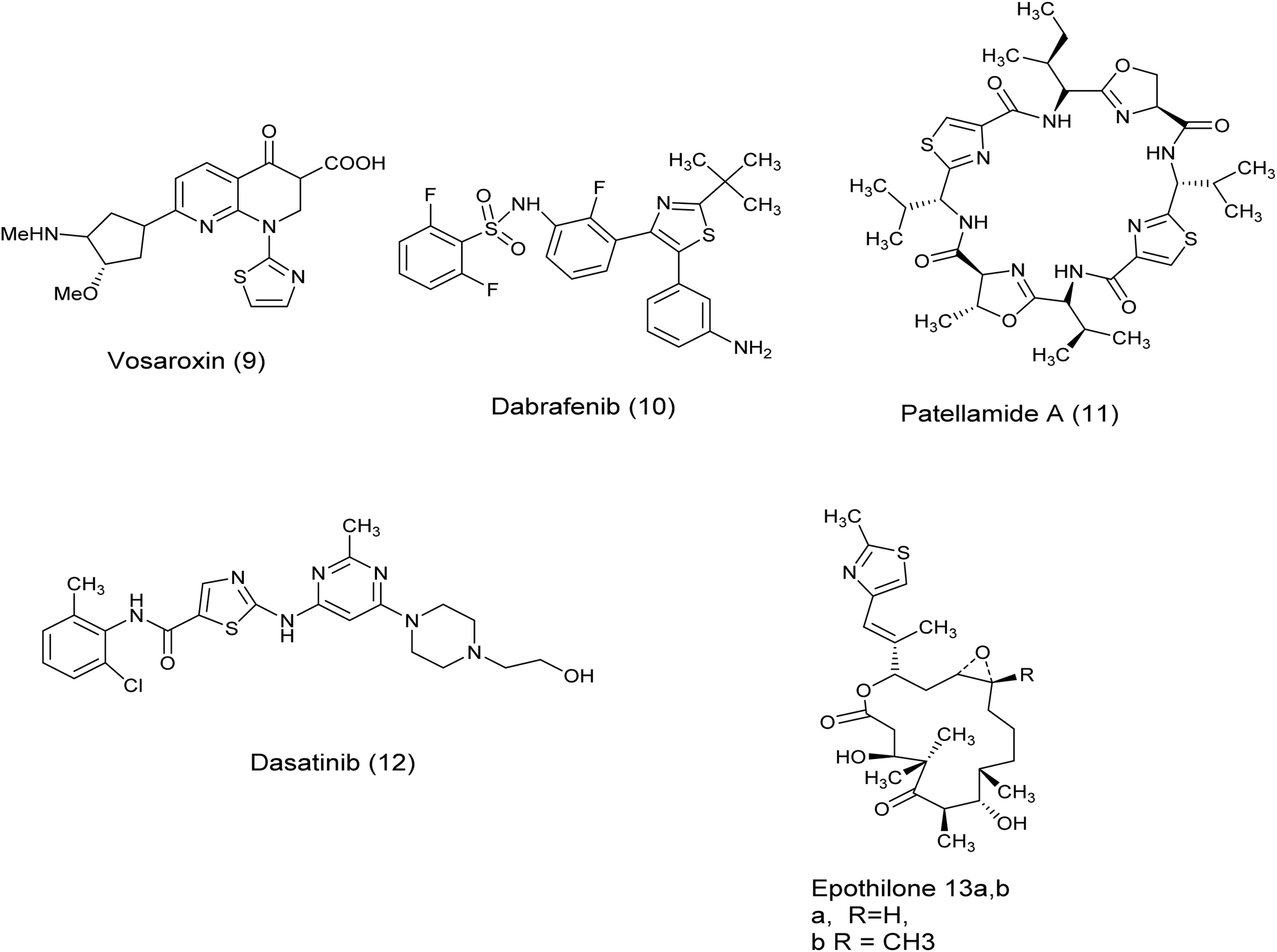 | ||
| Fig. 3 Some clinically available thiazole-containing anti-cancer drugs. | ||
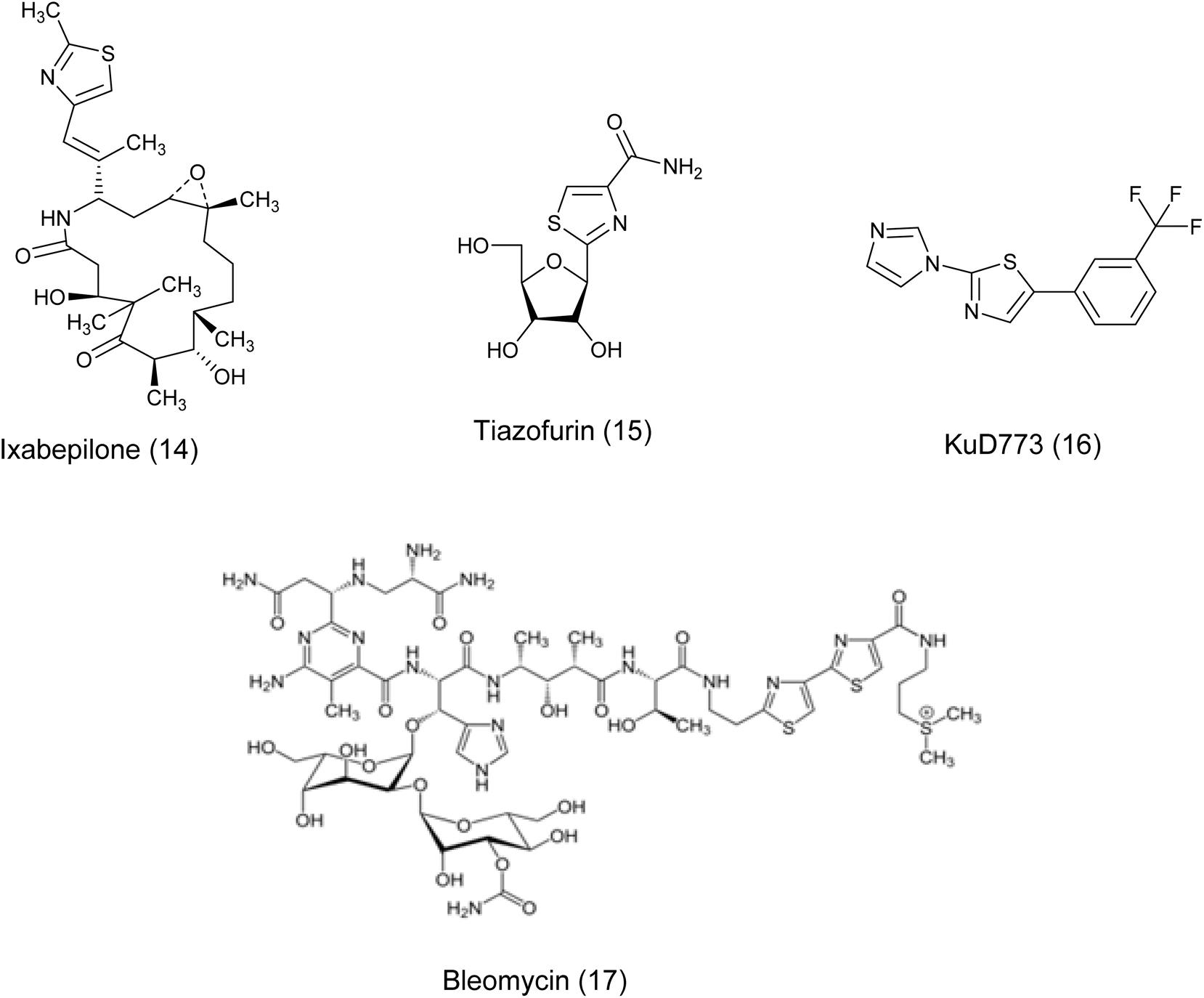 | ||
| Fig. 4 Continuous: some clinically available thiazole-containing anti-cancer drugs. | ||
Because of the various pharmaceutical effects linked to thiazole derivatives, they have been instrumental in the design and advancement of drugs. Research has shown that these structures have displayed significant anti-cancer properties, particularly through kinase inhibition. This review aims to summarize the studies on small molecules containing thiazole frameworks that have shown kinase inhibitory effects. The goal is to draw the interest of medicinal chemists towards identifying new leads that could potentially be developed into novel drugs in collaboration with pharmaceutical companies.
2. Chemistry of thiazole
Thiazole, also referred to as 1,3-thiazole, is a type of five-membered ring belonging to the azoles group. It contains a sulfur atom at position 1 and a nitrogen atom at position 3. The characteristics of thiazole are summarized and presented in Table 1.30| Chemical formula | C3H3NS |
| Color | Pale yellow |
| Odour | Like pyridine |
| Solubility | Soluble in alcohol, ether, acetone; slightly soluble in water and sparingly soluble on dyes and solvents of organic origin |
| Boiling point | 116–118 °C |
| Relative density | 1.1998 |
| Refractive index | 1.5969 |
The resonance investigation revealed an aromatic nature attributed to the delocalization of a lone pair of electrons originating from the sulfur atom, resulting in a 6π electron system, as illustrated in Fig. 5.
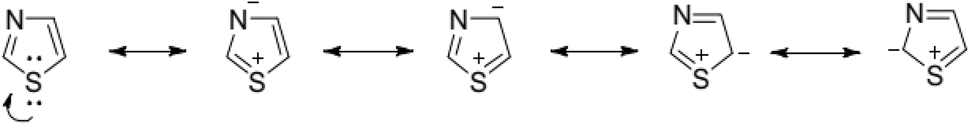 | ||
| Fig. 5 The resonance structure of thiazole. | ||
Thiazole has many analogues, such as isothiazole, thiazolone, 1,3,4-thiadiazole, hydrogenated thiazole (such as thiazoline and thiazolidine), and polycyclic or fused thiazoles (benzothiazole or naphthothiazole, etc.), as shown in Fig. 6.
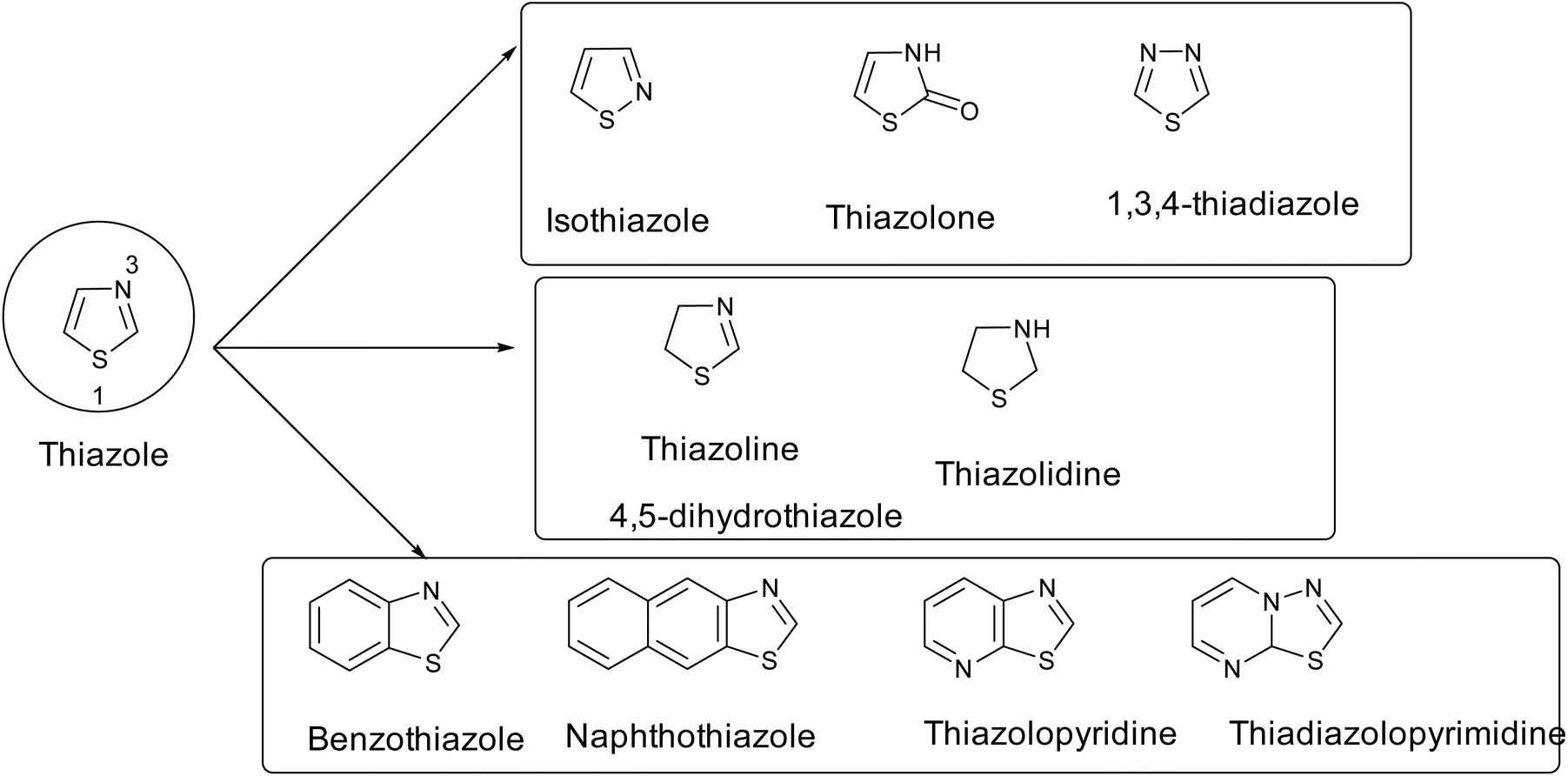 | ||
| Fig. 6 Analogues of thiazole. | ||
Based on the π electron density analysis, electrophilic substitution is primarily observed at the C-5 position, with the C-4 position being the next most reactive site, while nucleophilic substitution predominantly occurs at the C-2 position.31,32 (Fig. 7).
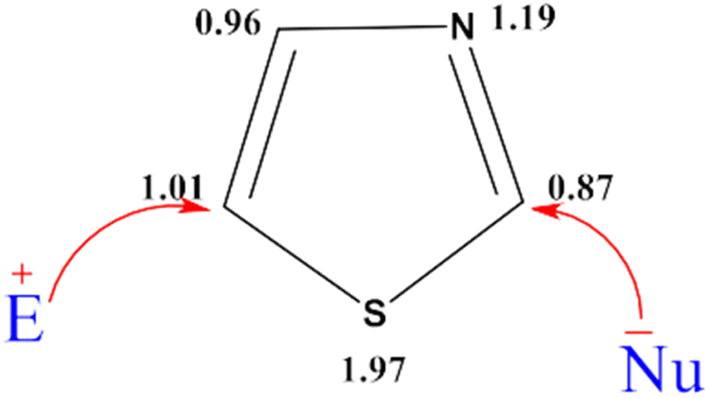 | ||
| Fig. 7 Calculated π-electron density of thiazole. | ||
2.1. Synthesis of thiazole derivatives
Hantzsch was the first great chemist and the pioneer of thiazole chemistry. The Hantzsch technique is the oldest and most widely used technique for the synthesis of thiazole rings. The procedure involves cyclizing α-halocarbonyl compounds with thioamide or thiourea to form thiazole.33 Earlier, historical synthesizes of thiazole derivatives have been listed previously in many reviews15,16,34–36 and summarized in Fig. 8.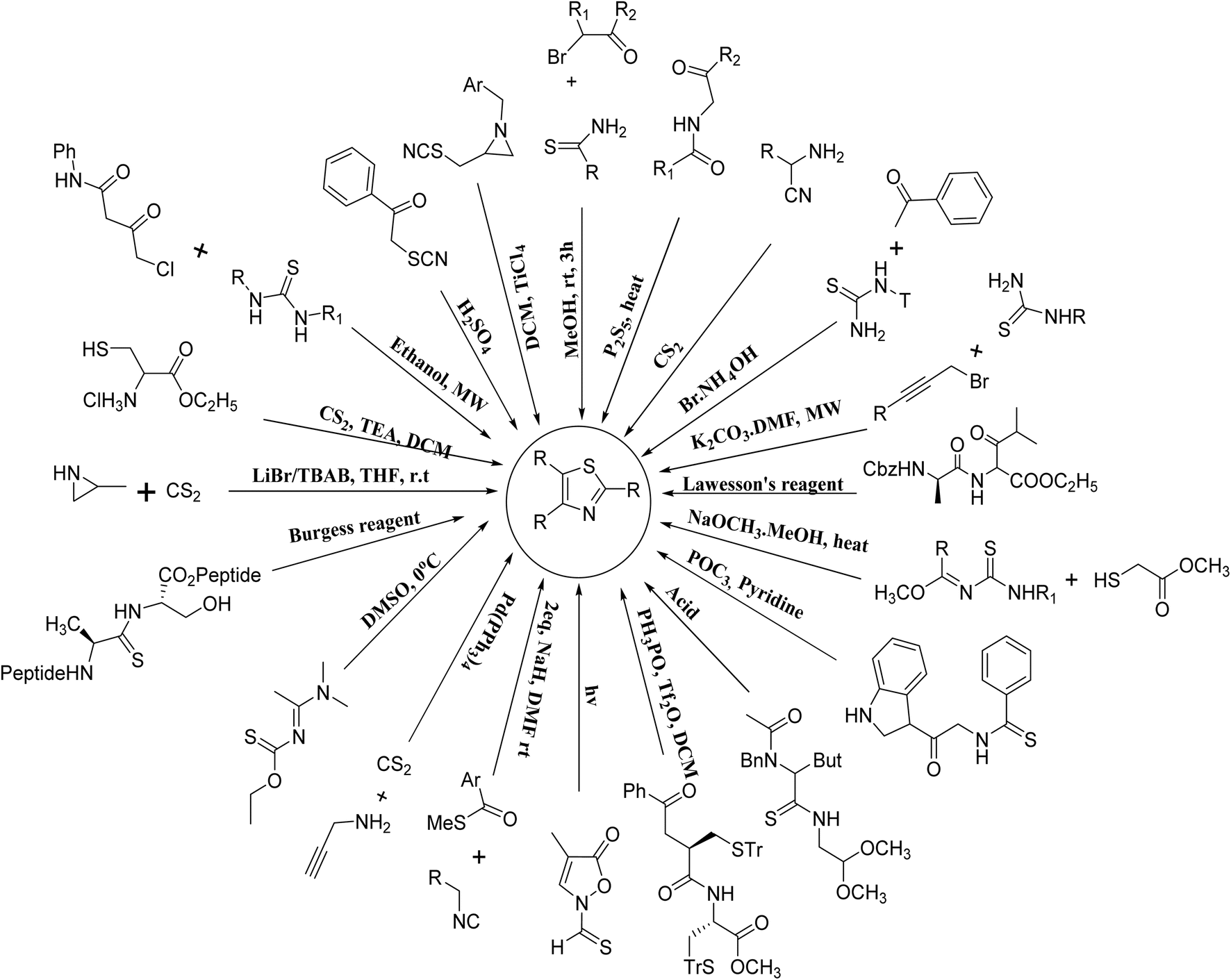 | ||
| Fig. 8 A variety synthesized techniques for thiazole synthesis. | ||
2.2. Recent advancements in the synthesis of thiazole derivatives have been observed
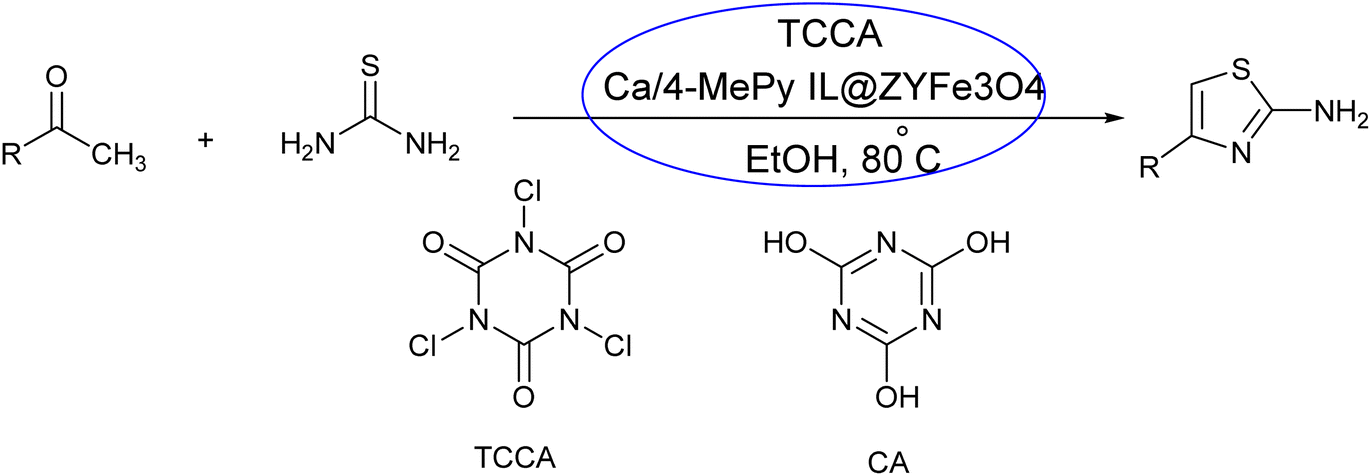 | ||
| Scheme 1 Synthesis of 2-aminothiazole using catalytic nanosystem. | ||
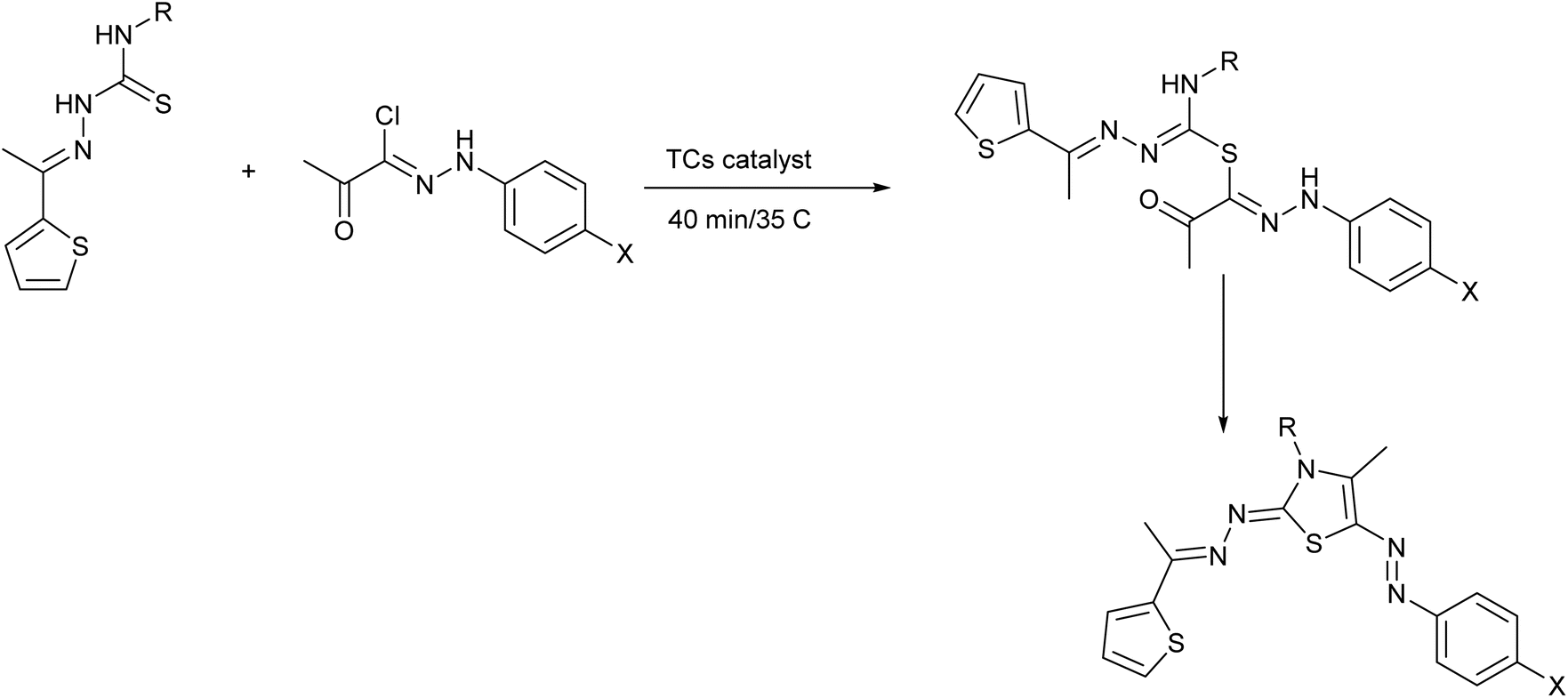 | ||
| Scheme 2 Synthesis of thiazole derivatives by TCs catalyst. | ||
Moreover, the application of Fe3O4 and SiO2-bipyridine-CuCl2 in ethanol under reflux conditions for the synthesis of thioamides and phenylbromide represents a distinctive and environmentally sustainable catalytic approach to produce thiazole derivatives with an excellent yield (95%), as demonstrated in Scheme 3.39
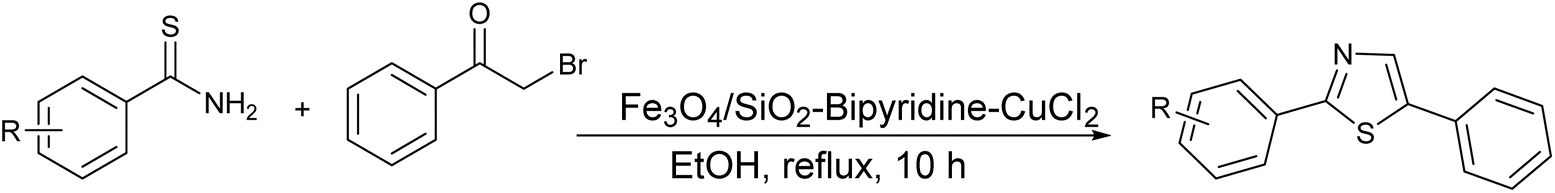 | ||
| Scheme 3 Synthesis of thiazole derivatives by ecofriendly catalyst. | ||
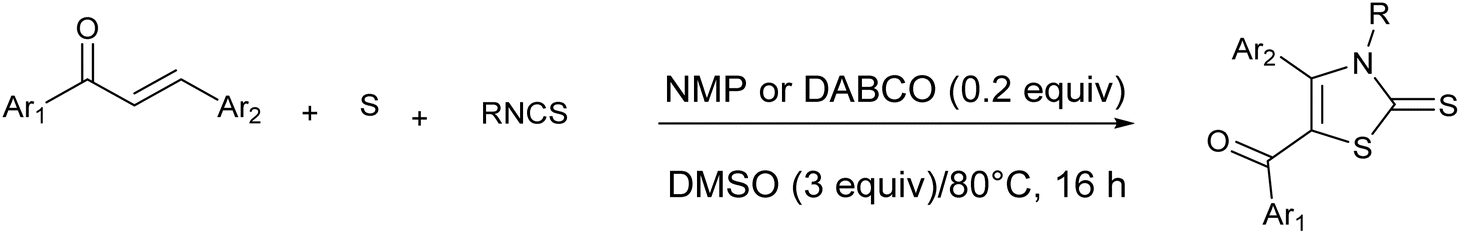 | ||
| Scheme 4 Synthesis of thiazole derivative by three component reactants. | ||
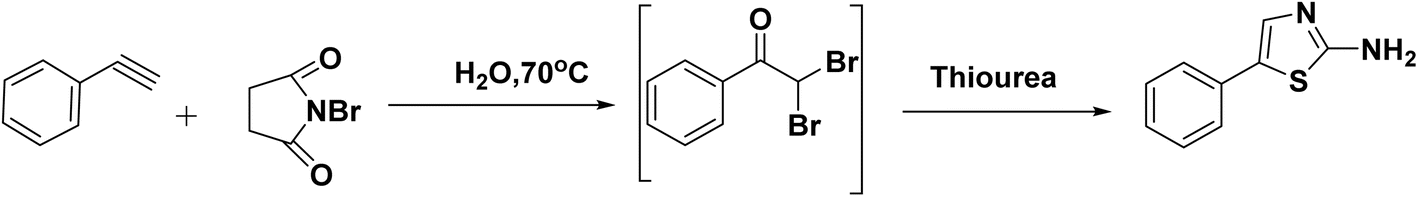 | ||
| Scheme 5 Synthesis of 2-aminothiazoles via one-pot multicomponent reaction. | ||
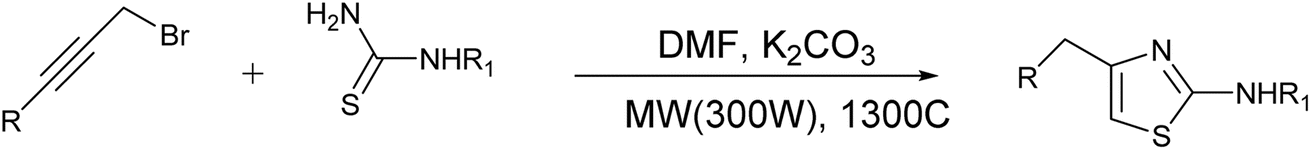 | ||
| Scheme 6 Synthesis of 2-aminothiazoles via domino alkylation – cyclization reaction. | ||
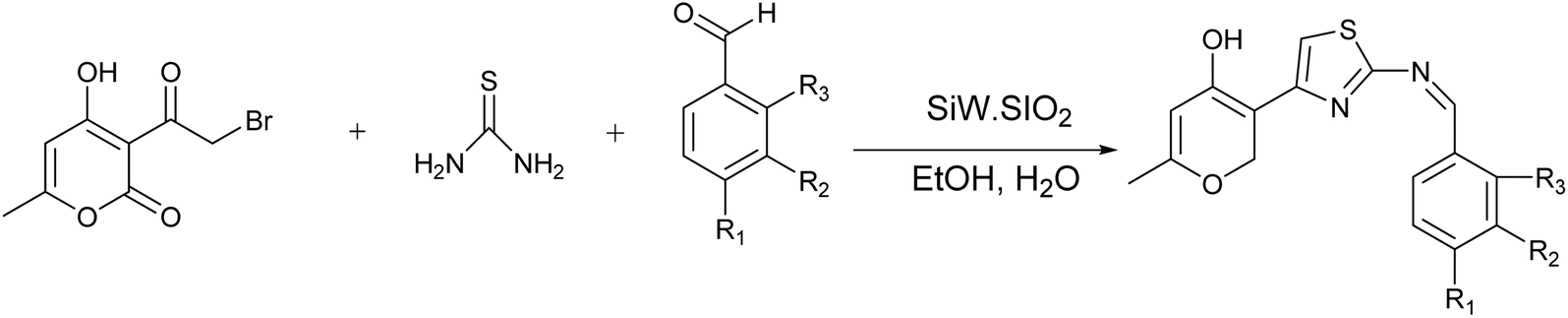 | ||
| Scheme 7 Utilizing silica-supported tungstosilicic acid for the synthesis of 2-aminothiazoles. | ||
3. Cancer disease
Cancer is a complex ailment resulting from a sophisticated interplay of external and internal factors. It ranks as the second leading cause of mortality globally, following cardiovascular diseases. The World Health Organization (WHO) has reported that in 2018, there were 9.6 million deaths attributed to cancer, a figure projected to rise to 13.1 million by 2030. Over the past decade, there has been a worrisome escalation in the incidence and fatality rates of cancer. The primary clinical modalities employed in cancer treatment include chemotherapy, surgery, and radiotherapy, with chemotherapy being the most commonly utilized method. Consequently, a plethora of chemotherapeutic agents have been extensively researched and developed to combat various cancer types through diverse mechanisms.44,45 Enzyme inhibition has emerged as a notable and viable target strategy for tumor treatment, with thiazole compounds demonstrating efficacy in inhibiting several such enzymes, particularly protein kinases.4. Protein kinases in cancer
The kinase inhibitors represent a diverse class of potent antineoplastic agents that selectively target protein kinases exhibiting aberrant activity in cancer cells, contributing to their uncontrolled proliferation. Protein kinases are a large group of enzymes with over 500 members that modify other proteins by adding a phosphate group from ATP, leading to changes in their function. This phosphorylation process typically controls signaling pathways crucial for cell processes like growth, differentiation, and survival.46 In recent years, researchers have extensively investigated the role of dysregulated kinases, particularly in cancer and other human diseases.47,48 Eukaryotic organisms have two main types of protein kinases: serine/threonine kinases (which add phosphate to serine/threonine residues) and tyrosine kinases (which add phosphate to tyrosine residues).49 Many protein kinases function as cell surface receptors, initiating intracellular signaling cascades upon ligand binding, often cytokines or growth factors. Inhibitors targeting these kinases are referred to as protein kinase receptor inhibitors.5. Thiazole as kinase inhibitors
A variety of heterocyclic small molecules have been created and advanced to inhibit these kinases. Out of these fragrant heterocyclic compounds, thiazole derivatives have shown notable inhibitory properties. Therefore, this article describes the main structural features of small molecule thiazoles and how they interact within the active sites of kinases to highlight the importance of this scaffold, which may be due to its unique physical and chemical properties. The review is organized based on categorizing kinases into serine/threonine and tyrosine kinases.5.1. Serine/threonine thiazole-based kinase inhibitors
Li et al., developed nineteen novel compounds derived from the thiazole scaffold and evaluated their efficacy in suppressing the proliferation of cancer cells in A549, MCF-7, U-87 MG, and HCT-116 cell lines. The majority of compounds exhibited favorable activity. Subsequently, these derivatives were further examined for their potential as inhibitors of the PI3K/AKT/mTOR pathway. Compound 18, Fig. 9, showed the most potent anticancer effects among all tested cell lines, with an IC50 value between 0.50–4.75 μM, outperforming the reference medication (BEZ235). Additionally, western blot analysis showed that derivative 18 successfully blocked the PI3K/AKT/mTOR signaling pathway and slowed down the advancement of the tumor.51
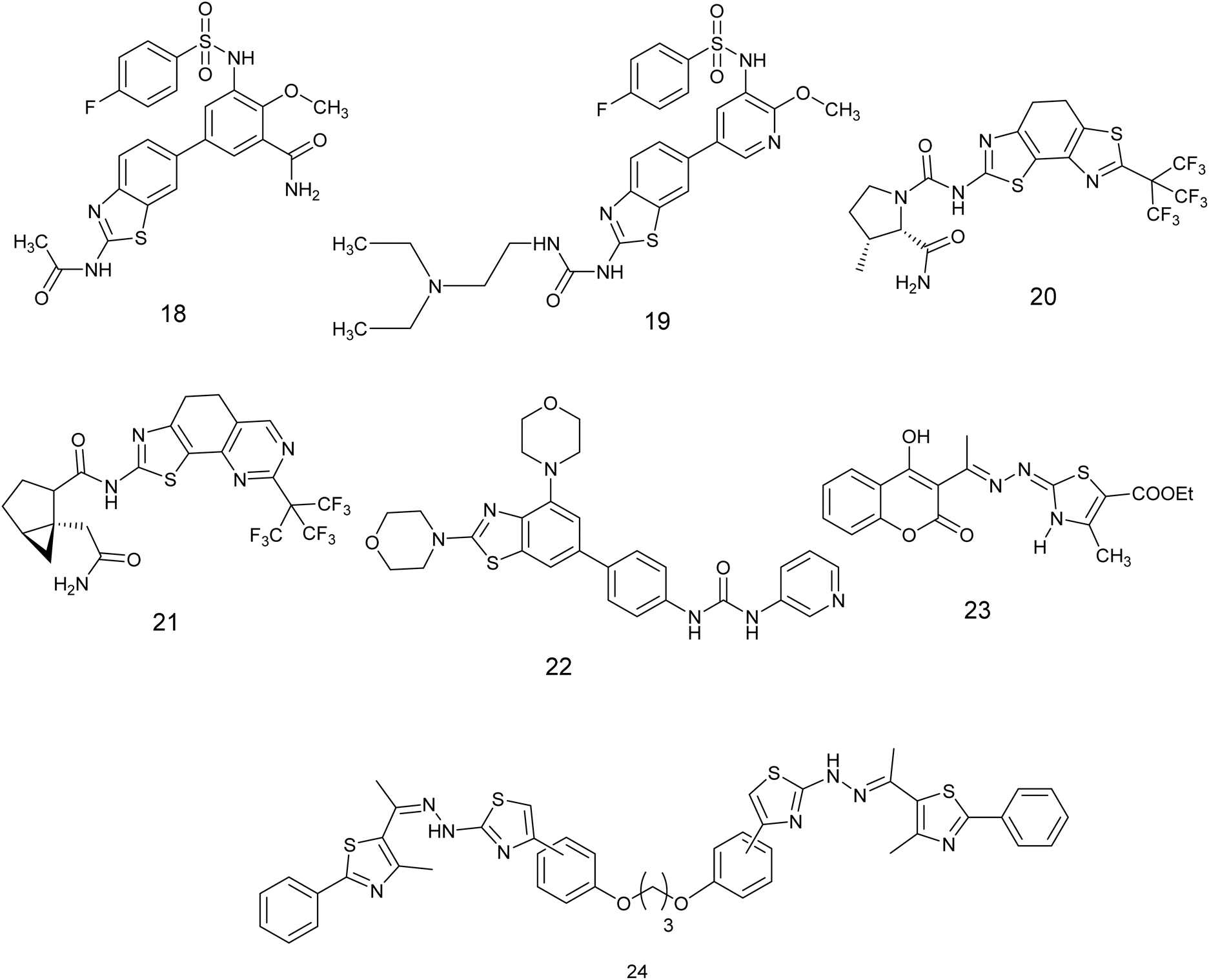 | ||
| Fig. 9 Thiazole derivatives as PI3K/AKT/mTOR signaling pathway inhibitors (18–24). | ||
Xie et al. conducted an investigation on benzothiazole derivatives containing pyridine to assess their inhibitory impacts on PI3Ks and mTORC1, along with their anti-proliferative characteristics against MCF-7, U87 MG, A549, and HCT116 cell lines. The results displayed that compound 19, Fig. 9 demonstrated the most potent antiproliferative activity across all tested cells (IC50 = 0.30–0.45 μM). Furthermore, findings from experiments conducted on cells showed that derivative 19 displayed notable inhibitory properties against PI3K and mTORC1. The inhibitory effects on PI3Ka, PI3Kb, and PI3Kg were discovered to be better than those of the standard medication BEZ235.52
Fairhurst et al. discovered a 4′,5-bisthiazole variant of an (S)-proline-amide aminothiazoleurea derivative that acts as a selective inhibitor of phosphatidylinositol-3 kinase alpha (PI3K). In their research, derivatives 20 and 21, Fig. 9 were discovered to be highly effective in blocking signaling via the PI3K pathway. They were identified as the most powerful and specific inhibitors of PI3Ka, with IC50 values varying from 9–290 nM.53
A novel set of inhibitors has been developed to target the PI3Kβ enzyme, a component of the beta subunit of phosphatidylinositol-3-kinases, has been developed and prepared using a benzothiazole scaffold structure. The researchers evaluated the inhibitory impacts of these substances on PI3Kα, β, γ, δ, and mTOR (Mammalian target of rapamycin). Compound 22, Fig. 9 exhibited the highest potency and selectivity against PI3Ks/mTOR, demonstrating potent activity PI3Kβ with an IC50 value of 0.02 μM. Additionally, Compound 22, the most favorable compound exhibited significant anti-proliferative effects and selectivity among different cancer cell types, with a particular emphasis on its efficacy in prostate cancer cells. Furthermore, the structure–activity relationship (SAR) analysis of compound 22, as illustrated in Fig. 10, indicates that the inclusion of a benzothiazole ring is critical for both the activity and selectivity of the inhibitors. Additionally, the presence of a morpholine group at the 2-position of the benzothiazole is required for significant antitumor efficacy. The carbamide group is also essential for activity; also the carbamide's substitutions were found to be intolerable, suggesting that the presence of a pyridine moiety is necessary for maintaining activity.54
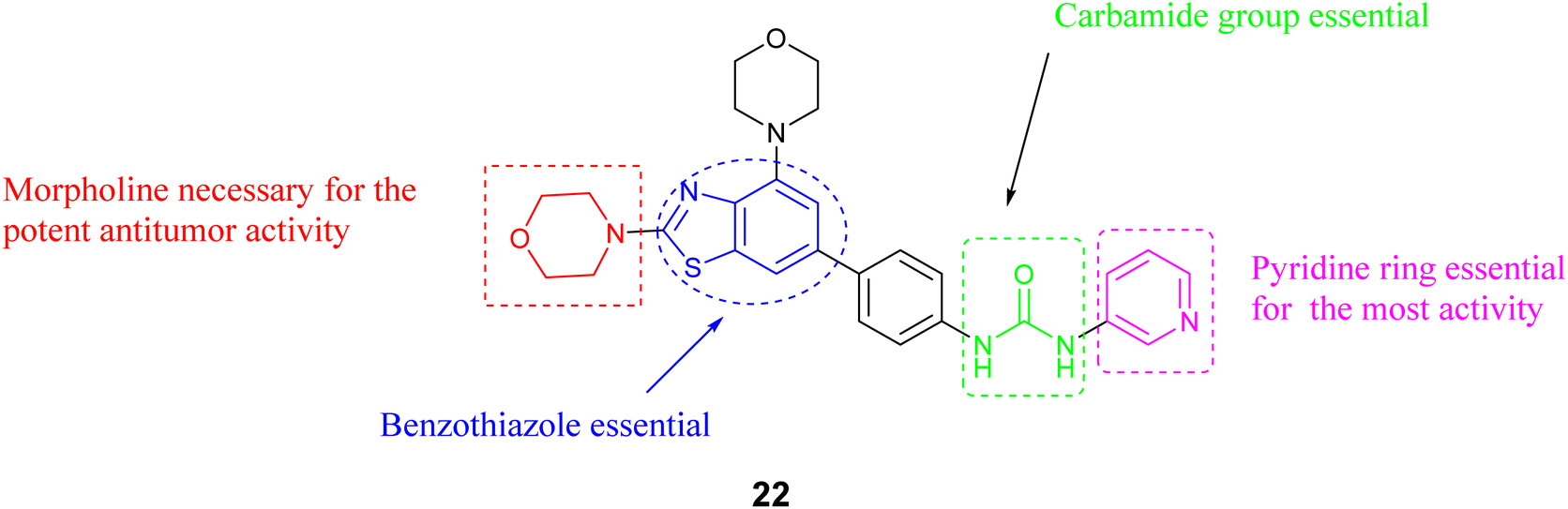 | ||
| Fig. 10 SAR for compound 22. | ||
A range of thiazoline and thiazolidinone-based derivatives of 4-hydroxycoumarin were synthesized through traditional methods as well as microwave-assisted techniques. These novel compounds exhibited notable efficacy and inhibition of proliferation in different cancer cell lines including HCT-116, HepG2, and MCF-7, while demonstrating no toxicity towards normal cells (BJ-1). The potential anti-proliferative agents were assessed for their capacity to hinder the activity of EGFR and PI3K/mTOR signaling pathways. Compound 23, Fig. 9 demonstrated the most notable inhibitory effect on the signaling pathway, the value of IC50; 0.184 ± 0.01, 0.719 ± 0.04, and 0.131 ± 0.007, respectively.55
In 2024, Salem and colleagues introduced a new class of bis-dithiazoles connected through aliphatic, aromatic, or heterocyclic frameworks and assessed their potential as anticancer agents targeting HT29 colon cancer cells. The most potent compounds were selected for further investigation as inhibitors of the PI3K enzyme. Compound 24, Fig. 9 exhibited notable enzyme suppression with an IC50 value of 2.33 nm, outperforming the standard drug alpelisib which displayed an IC50 of 4.96 nM. Furthermore, the inhibitory efficacy of this compound was qualitatively assessed using western blotting, revealing a significant reduction in PI3K gene expression to half of its maximum level.56
A group of researchers led by Shao H. synthesized and studied a group of pyrimidines with thiazole ring systems and tested their ability to inhibit cell proliferation and CDK activity. The researchers analyzed the relationship between the structure of the compounds and their activity, focusing on the selectivity for CDK9. Among the compounds tested, compound 25, Fig. 11 showed the highest potency as a CDK9 inhibitor and demonstrated strong anti-proliferative effects on various cancer cell lines, with IC50 values ranging from 0.64 to 2.01 μM.59
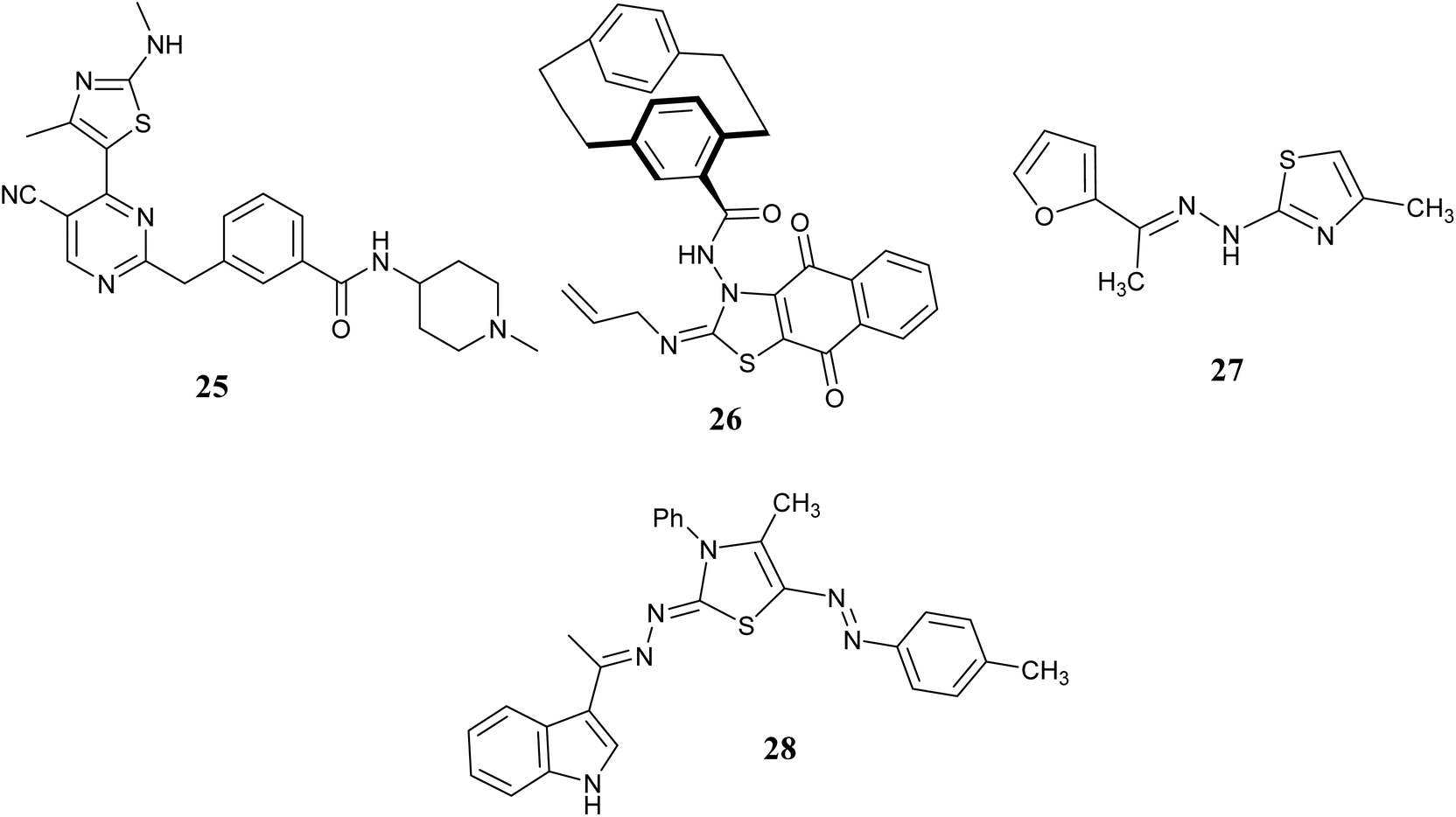 | ||
| Fig. 11 Thiazole derivatives as cyclin-dependent kinases (CDKs) inhibitors (25–28). | ||
In 2020, Abdelhafez et al. introduced three novel series of new compounds to evaluate their efficacy by conducting anti-cancer screening in vitro on 60 different cancer cell lines. Majority of the synthesized compounds exhibited significant activity, with particular emphasis on those containing 1,4-dihydronaphthoquinone which demonstrated the highest potency without affecting normal cells. Subsequently, the most promising compounds were assessed for their potential as cyclin-dependent kinase (CDK) inhibitors. Among these, compound 26, Fig. 11 stood out by inhibiting eight isoforms of CDK, notably displaying the lowest IC50 value of 54.8 nM against CDK1 compared to the reference compound Dinaciclib. Furthermore, compound 26 predominantly induced cell cycle arrest in the pre-G1 and G2/M phases upon examination of the SK-MEL-5 cell line. Notably, the sequential caspase-3 assay for compound 26 revealed a significant upregulation level.60
A. M. El-Naggar and colleagues identified arylidene-hydrazinyl-thiazole as potential CDK2 inhibitors through a one-pot synthesis method conducted under conditions that are both environmentally friendly and utilize ultrasound and microwave technology. Most of the newly created compounds showed strong inhibition of growth in laboratory tests against three types of cancer cells (MCF-7, HCT-116, and HepG2). Furthermore, the compounds that exhibited the most potent anti-proliferative effects were assessed for their activity against various kinase enzymes. The efficacy of CDK2 inhibitor 27, Fig. 11 was further examined to evaluate its effects on cell cycle progression and apoptosis in the HepG2 cell line. Results indicated that this compound led to inhibition in the G2/M phase of the cell cycle and demonstrated apoptotic properties.61
In 2023, S. M. Gomha and colleagues synthesized a new series of 3-thiazolyl-indoles and tested their impact on the growth of three types of human cancer cells (MCF-7, HCT-116, and HepG2) in a laboratory setting. The findings indicated that a large proportion of the synthesized compounds displayed notable cytotoxic effects. Furthermore, the inhibitory capacity of the most successful compounds was assessed using a laboratory enzyme test focused on CDK2. The findings showed that compound 28, Fig. 11 exhibited the strongest effectiveness, with an IC50 value of 0.35 ± 1.07 μM, in contrast to roscovitine's IC50 value of 0.39 ± 0.47 μM.62
Gray and colleagues have developed novel compounds utilizing aminothiazole as specific Aurora kinase inhibitors. The findings indicate that a significant number of the synthesized compounds exhibit notable activity. Notably, compounds 29 and 30, Fig. 12 were identified as the most potent inhibitors, demonstrating IC50 values of 79 and 140 nM, respectively, and displaying a high level of kinase selectivity against Aurora A. While these compounds were found to effectively inhibit histone H3 phosphorylation, impede the cell cycle, and induce siRNAs targeting Aurora A, their practical application in experiments is hindered by limited cellular penetration, necessitating further refinement through chemical optimization.65
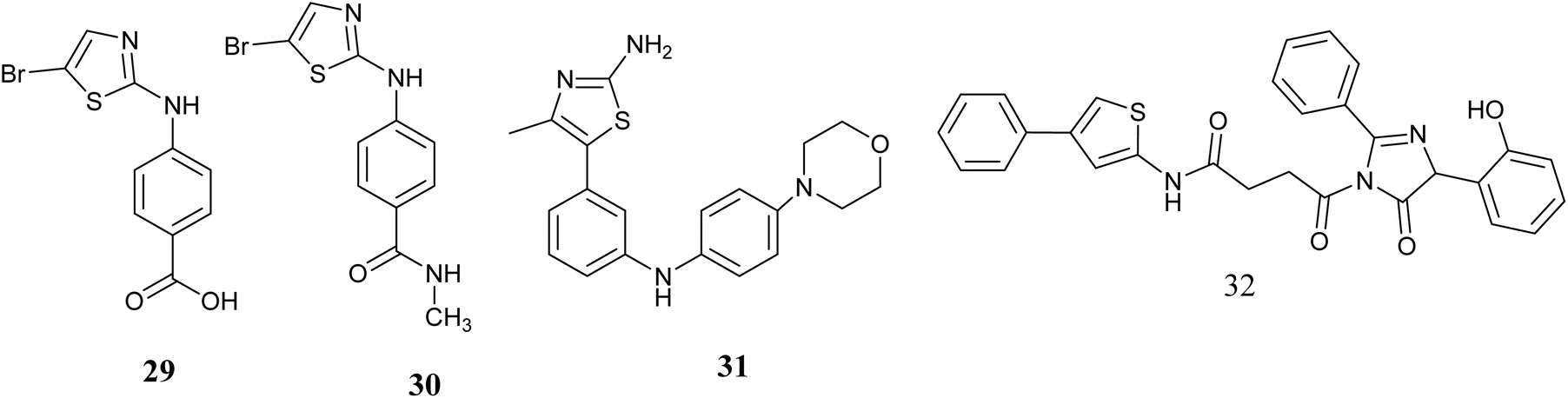 | ||
| Fig. 12 Thiazole derivatives as Aurora kinase inhibitors(29–32). | ||
Wang and colleagues reported the identification of a series of modified 4-(4-methylthiazol-5-yl)-N-phenylpyrimidin-2-amines with potential anticancer properties targeting aurora kinase inhibitors. Compound 31, Fig. 12 exhibited significant cytotoxic effects on cell lines derived from cancerous tissues, also the suppression of mitotic histone H3 phosphorylation was observed by this compound 31, leading to the manifestation of atypical mitotic activities. Subsequent investigations confirmed these compounds as strong blockers of aurora A and B enzymes. Compound 31 demonstrated anticancer efficacies with Ki values of 8.0 and 9.2 nM for aurora A and B, Subsequent in vivo assessments demonstrated that compound 30 displayed oral bioavailability and demonstrated efficacy in combating cancer.66
In 2023, Shaik and colleagues utilized QSARINS software to conduct quantitative structure–activity relationship (QSAR) analysis in order to identify potential anti-breast cancer agents with aurora kinase inhibitory properties. Following this analysis, the most promising compounds were further investigated through molecular docking studies against the aurora kinase protein (1MQ4). Compound 32, Fig. 12 emerged with the highest docking score (−9.67) and was subsequently subjected to molecular dynamic simulation for 100 nanoseconds to assess its stable binding with 1MQ4. The confirmation of compound 32's binding stability with 1MQ4 was validated through various analyses including RMSD, RMSF, RoG, H-bond interactions, molecular mechanics-generalized Born surface area (MM-GBSA) calculations, free binding energy assessments, and solvent-accessible surface area (SASA) evaluations. Additionally, the newly designed compound 32 demonstrated favorable ADMET properties. These results suggest that compound 32 could serve as a promising theoretical lead for future experimental investigations aimed at selectively inhibiting aurora kinase.67
In 2018, Yarmoluk et al. conducted a study aimed at discovering new CK2 inhibitors. Virtual screening experiments were carried out utilizing Autodock software to identify potential compounds. Subsequently, the top-scoring compounds were subjected to in vitro testing through P32 radioactive kinase assay. The findings revealed the presence of small-molecular inhibitors targeting protein kinase CK2 within the derivatives of 1,3-thiazole-5-carboxylic acid. Notably, compound 33, Fig. 13 exhibited the highest activity, displaying potent inhibition of CK2 IC50 = 0.4 μM.69
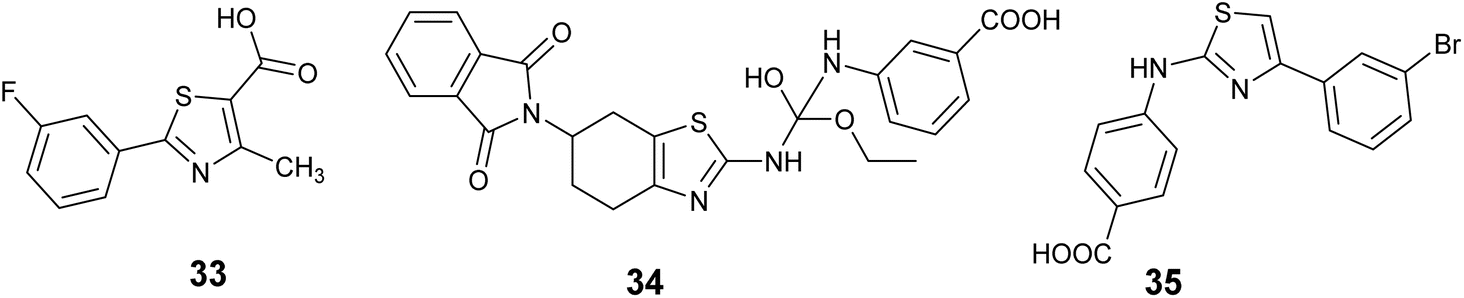 | ||
| Fig. 13 Thiazole derivatives as casein kinases II (CK2) inhibitors (33–35). | ||
Furthermore, Vasu and colleagues employed molecular docking techniques to discover and produce a new lead compound with comparable characteristics to Hit15 (ZINC20464516, a molecule sourced from the ZINC database) as dual kinase inhibitors targeting CK2 and GSK3β. The results revealed that compound 34, Fig. 13 demonstrated the highest efficacy in inhibiting dual kinases effects against CK2 and GSK3β in the in vitro kinase inhibition assay, with IC50 values of 1.9 ± 0.05 and 0.67 ± 0.27 μM, respectively.70
In the current investigation, a novel series of aryl 2-aminothiazoles was identified as a fresh category of CK2 inhibitors, showcasing a mode of action that is non-competitive with ATP and leading to the stabilization of an inactive conformation of CK2 in solution. Through a range of experimental methodologies including enzyme kinetics investigations, STD-NMR analysis, circular dichroism spectroscopy, and native mass spectrometry, it was demonstrated that these substances interact with an allosteric cavity located adjacent to, but distinct from, the ATP-binding region. Molecular docking studies further supported these findings, indicating that this newly identified binding site is positioned at the junction of the αC helix and the flexible glycine-rich loop. Significantly, compound 35, Fig. 13 exhibited the most potent activity against purified CK2α, as evidenced by an IC50 value of 3.4 μM, and displayed a favorable selectivity profile.71
In this context, Zhao et al. synthesized a series of 4,5-dihydropyrazole derivatives incorporating thiazole and thiophene groups, which were assessed for their potential as inhibitors of the V600E mutant B-RAF kinase enzymes implicated in tumor initiation and progression. The biological assessment indicated that most of the compounds displayed notable inhibitory effects against B- RAFV600E and showed antiproliferative properties on MCF-7 and WM266.4 cell lines. Significantly, Compound 36, Fig. 14 exhibited the highest antiproliferative efficacy against MCF-7 and WM266.4, demonstrating IC50 values of 0.16 μM and 0.12 μM, respectively, surpassing the efficacy of sorafenib. Compound 36 also exhibited strong bioactivity against V600E mutant B-RAF kinase IC50 = 0.05 μM. Furthermore, this compound triggered notable apoptosis in WM266.4 and MCF-7 cells in a manner that was dependent on the dosage administered.73
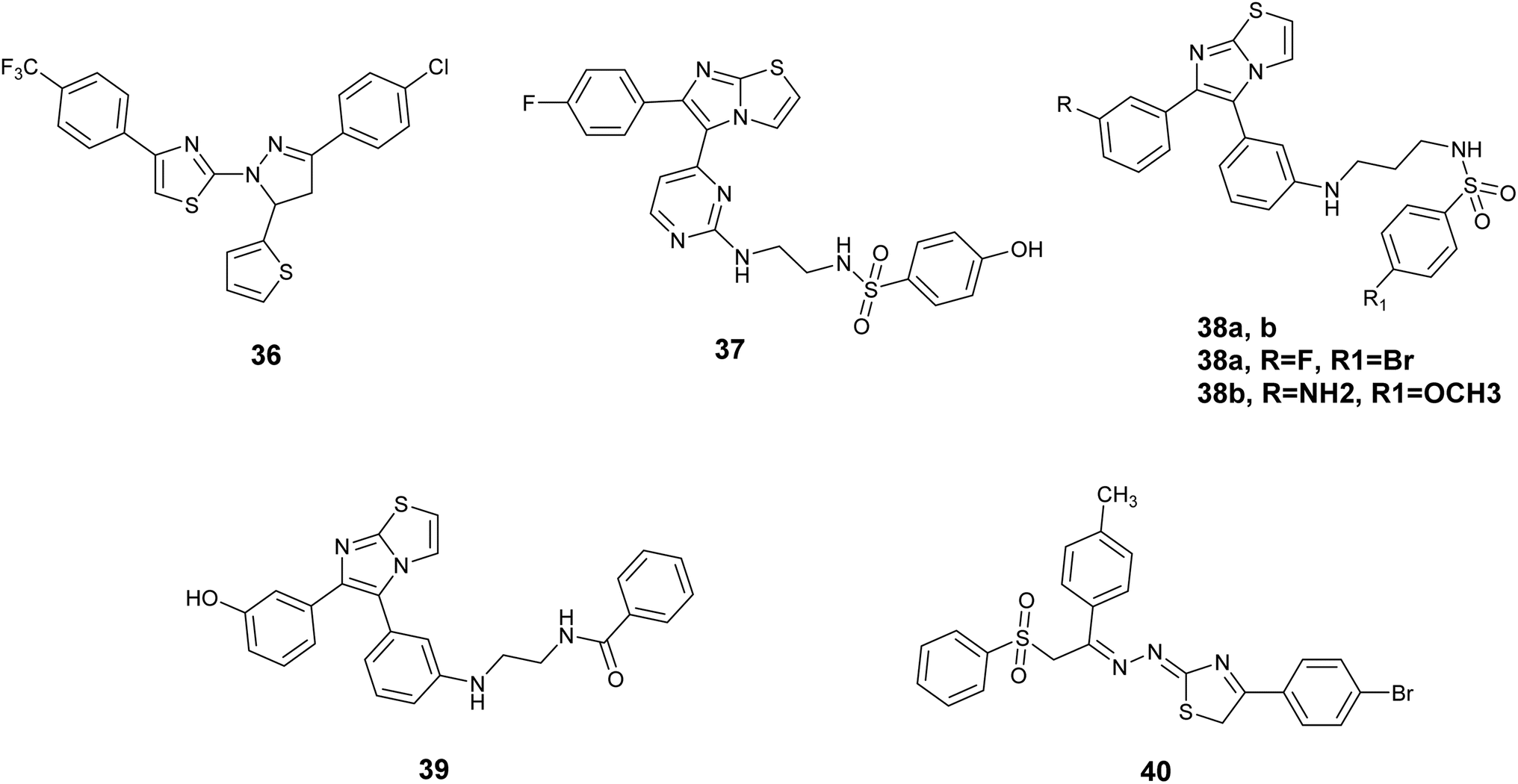 | ||
| Fig. 14 Thiazole derivatives as B-RAF inhibitors (36–40). | ||
Abdel-Maksoud and colleagues investigated a new group of compounds that included imidazo[2,1-b]thiazole for their effects on inhibiting cell growth in 57 different human cancer cell lines at the National Cancer Institute (NCI). Certain compounds demonstrated similar average inhibition percentages at a concentration of 10 μM compared to sorafenib. Compound 37, Fig. 14 demonstrated increased effectiveness against MCF-7 cells with a notably lower IC50 value of 0.475 μM compared to reference drug sorafenib with the value of IC50; 2.51 μM. Additionally, Compound 37 exhibited similar potency to sorafenib when tested against wild-type B-RAF, CRAF (IC50; 19 nM), V600E-B-RAF (IC50; 39 nM), ERK and MEK kinases. Furthermore, the structure–activity relationship (SAR) analysis for compound 37, Fig. 15 indicated that the presence of a 4-fluoro substituent on the phenyl ring is critical for biological activity. Additionally, the incorporation of an ethylene linker generally enhances the activity of most compounds in comparison to a propylene linker. Moreover, the para-hydroxyphenyl terminal ring exhibited superior activity relative to analogs such as trifluoromethyl, tosyl, and fused thiazole, which are also deemed essential for activity.74
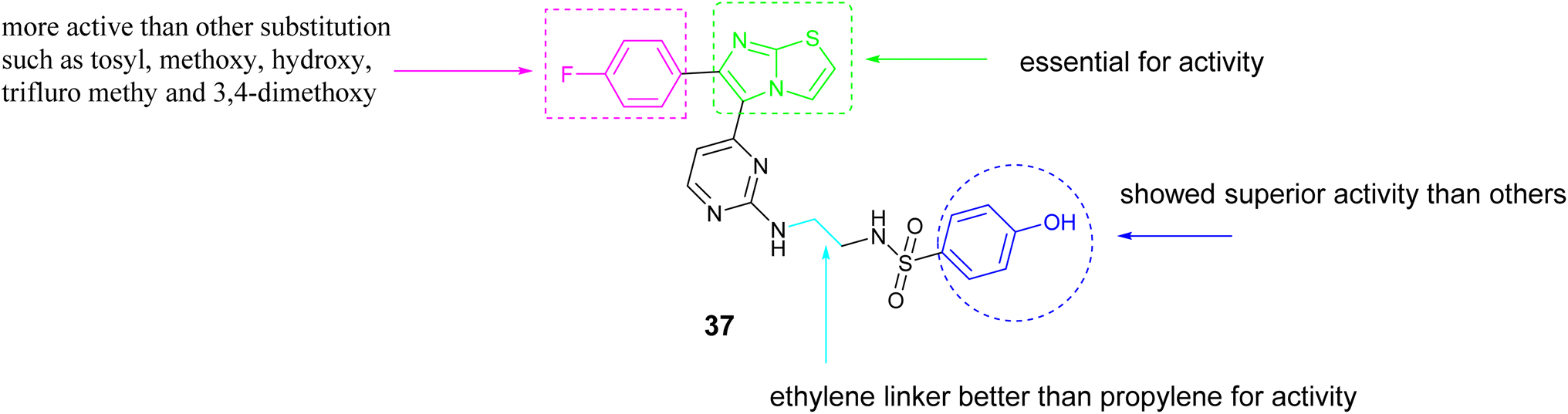 | ||
| Fig. 15 SAR of compound 37. | ||
Additionally, a novel set of imidazothiazole compounds incorporating pyrimidine were formulated and produced in order to design potential anticancer drugs. These compounds were studied on NCI 60 cell lines, with many showing notable inhibition percentages, especially against melanoma and colon cancer cell lines. The most successful substances were then subjected to additional testing on WTBRAF, V600EBRAF, and CRAF, revealing that Compound 38a, Fig. 14 exhibited the highest inhibitory effect on V600EBRAF at 9.30 nM.75 The identical research team synthesized additional derivatives utilizing imidazothiazole, with compound 38b, Fig. 14 demonstrating the highest activity against V600E B-Raf kinase, exhibiting an IC50 value of 1.20 nM.76
A study was conducted to investigate the molecular mechanism of action of a set of imidazothiazole derivatives that show promise in targeting melanoma cells. The compounds were evaluated for their effects on RAF1 and V600E-B-RAF kinases. Out of the compounds examined, 39, Fig. 14 exhibited the most significant efficacy against both kinases, displaying IC50 values of 8.2 and 0.978 nM, respectively. Notably, it exhibited a certain degree of selectivity towards the V600E mutant B-RAF kinase. Furthermore, compound 39 was evaluated for its activity against four melanoma cell lines, demonstrating enhanced effectiveness (IC50 = 0.18–0.59 μM) in comparison to the reference drug sorafenib (IC50 1.95–5.45 μM). Subsequent whole-cell kinase assays revealed that compound 39 effectively inhibited in-cell V600E-B-RAF kinase activity (IC50 = 0.19 μM). Moreover, it was noted that compound 39 triggered apoptosis instead of necrosis in the UACC-62 melanoma cell line, which exhibited the highest level of responsiveness.77
Thiazole derivatives containing a phenyl sulfonyl group were synthesized with the purpose of acting as inhibitors of the B-RAFV600E kinase. All synthesized compounds exhibited notable inhibition of the B-RAFV600E kinase enzyme at nanomolar concentrations. Particularly, Compound 40, Fig. 14 demonstrated exceptional inhibitory effects on B-RAFV600E with the value of IC50; 23.1 ± 1.2 nM, surpassing the standard drug dabrafenib which had an IC50 of 47.2 ± 2.5 nM. Furthermore, the most potent compounds were subjected to evaluation for their anticancer properties against both B-RAFV600E-mutated and wild-type melanoma cells. The findings indicated that the tested compounds effectively suppressed the proliferation of WM266.4 melanoma cells, with IC50 values ranging from 1.24 to 17.1 μM, in comparison to dabrafenib (IC50 = 16.5 ± 0.91 μM).78
The thiazole compound AR-A014418 (41), Fig. 16 created by Astra Zeneca, functions as a selective and ATP-competitive inhibitor of GSK3β. This compound has demonstrated notable efficacy as a selective competitor of the ATP binding site, with an IC50 value of 100 nM. Furthermore, it has shown inhibitory effects on tau phosphorylation, safeguarded neuronal cells against apoptotic triggers, and hindered β-amyloid-induced neurodegeneration.81
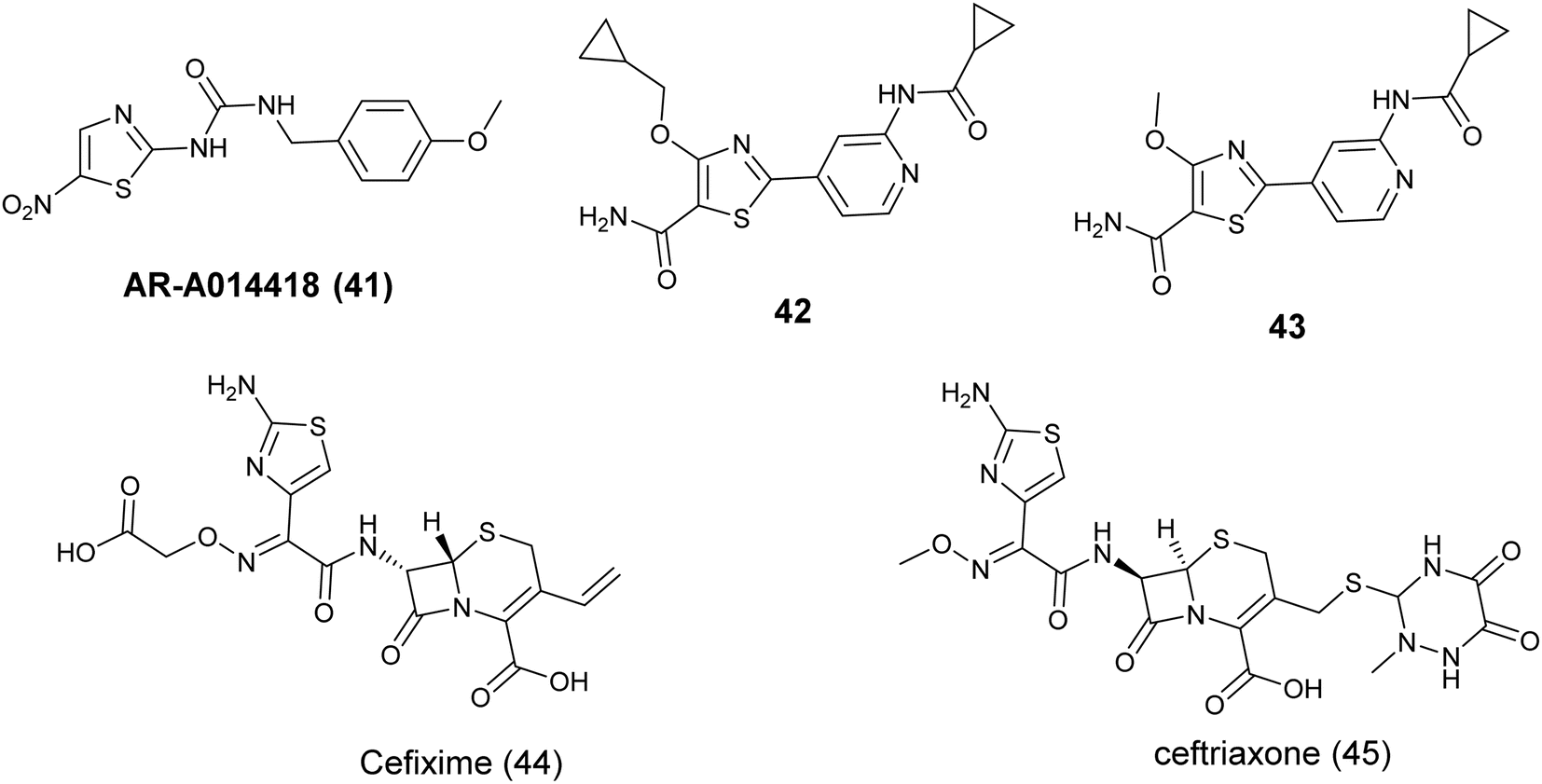 | ||
| Fig. 16 Thiazole derivatives as Glycogen synthase kinase 3 (GSK-3) inhibitors (41–45). | ||
Shivaprakasam and colleagues (2019) conducted a study on the structural optimization of acylaminopyridines to develop highly effective and specific inhibitors of GSK-3β. Their research demonstrated that incorporating a primary carboxamide group on the thiazole ring significantly improved the potency of compounds, with nanomolar activity observed. Specifically, compound 42, Fig. 16 exhibited the most potent inhibition of GSK-3β, with an IC50 value of 0.29 ± 0.01 nM, attributed to the presence of larger ether groups at the C-4 position. Furthermore, compound 43, Fig. 16 exhibited strong efficacy against Glycogen synthase kinase-3 (GSK-3) with an IC50 value of 1.1 ± 0.1 nM. Moreover, in investigations into the mechanism of action of compound 43, a notable decrease in pTau396 levels was observed when it was orally administered at a dosage of 30 mg kg−1 as a nano-suspension to LaFerla 3xTg-C57BL6 male mice.82
Recent research has highlighted the potential benefits of cephalosporin antibiotics in cancer treatment. In a study conducted in 2023 by Nassar et al., these antibiotics were examined for their capacity to suppress the activity of GSK3b. The study focused on four specific cephalosporins – cefixime, ceftriaxone, cephalexin, and cefadroxil – and their interaction with the GSK3b binding pocket through molecular docking. The findings indicated that cefixime (44) and ceftriaxone (45) showed the most favorable docking scores (−7.36 and −7.06) attributed to hydrogen bonding between their aminothiazole group and hinge residues of GSK3b. Subsequent in vitro experiments were conducted to measure the inhibitory effects of these cephalosporins on GSK3b. Consistent with the docking results, Cefixime (44), Fig. 16 demonstrated the most potent inhibitory activity, with IC50 values of 2.55 μM.83
5.2. Inhibitors of kinases that are based on tyrosine thiazole
A set of novel 4-phenoxyquinoline derivatives containing the benzo[d]thiazole-2-yl urea moiety were synthesized and evaluated for their cytotoxic properties on the MKN-45, H460, and HT-29 cell lines. The results indicated that the majority of these compounds demonstrated varying levels of efficacy against the three cell lines under investigation. Notably, compound 46, Fig. 17, exhibited the highest activity among the tested compounds, with IC50 values of 0.01, 0.06, and 0.18 μM against the H460, MKN-45, and HT-29 cell lines, respectively. Also, this compound emerged as a promising candidate against c-Met with nanomolar activity (IC50 = 17.6 nM).85
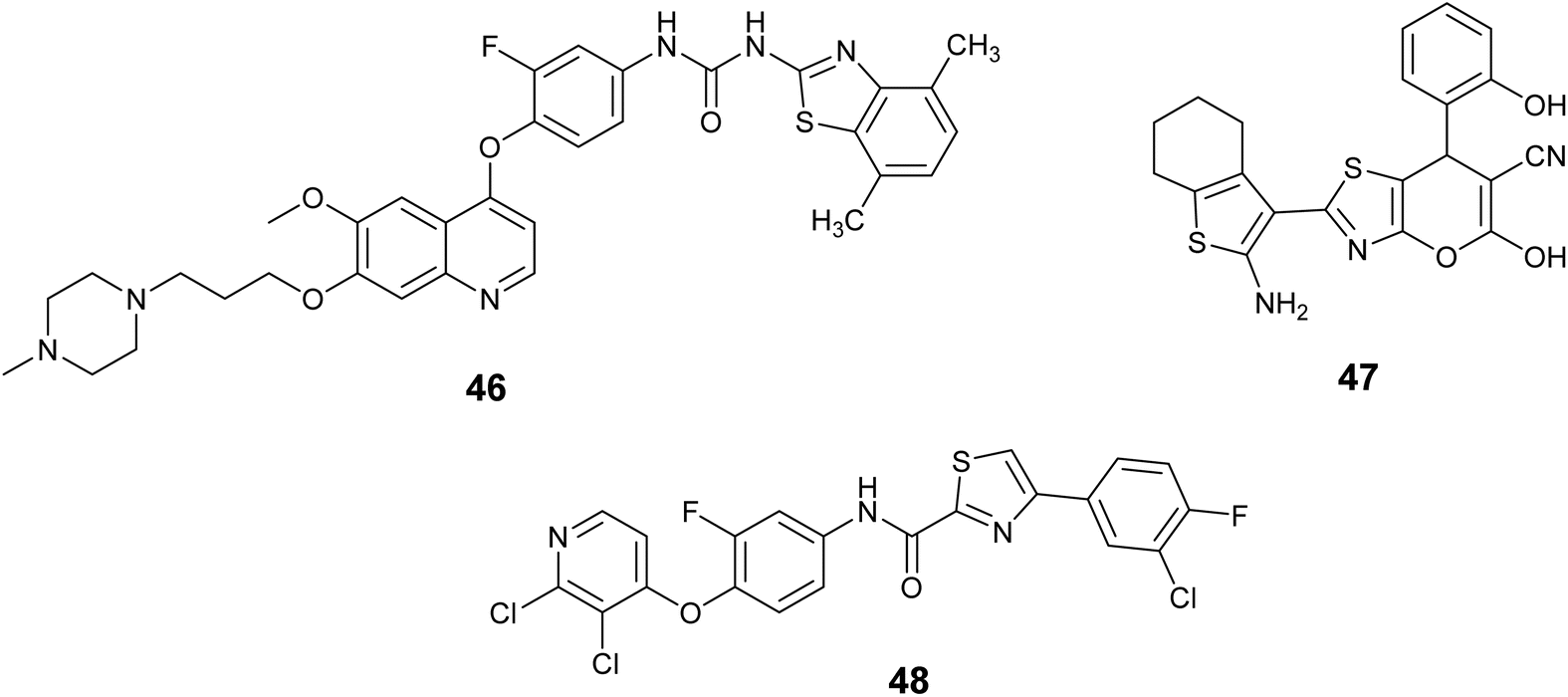 | ||
| Fig. 17 Thiazole derivatives as c-MET inhibitors (46–48). | ||
In 2018, Abdallah et al. employed multicomponent reactions (MCRs) for the synthesis of novel thiazole derivatives. These substances were tested in a laboratory setting to determine their effectiveness in fighting cancer, using six different types of cancer cells. The findings indicated that a significant number of compounds exhibited notable activity, leading to the selection of promising candidates for further investigation as enzymatic inhibitors of c-Met. Compound 47, Fig. 17 demonstrated the highest potency with nanomolar activity (IC50 = 0.06 ± 0.01 nM), surpassing the standard drug foretinib (IC50 = 1.16 ± 0.17 nM).86
In 2023, Nan et al. aimed to identify new c-Met inhibitors with potential antitumor properties by designing, synthesizing, and testing four series of thiazole/thiadiazole carboxamide-derived analogues against c-Met and four human cancer cell lines in vitro. Compound 48, Fig. 17 demonstrated significant potency toward c-Met, with the value of IC50 = 2.54 ± 0.49 nM. Moreover, compound 48 was found to cause cell cycle arrest and apoptosis in MKN-45 cells, suppress c-Met phosphorylation in cellular and cell-free environments, and exhibit advantageous pharmacokinetic properties in BALB/c mice.87
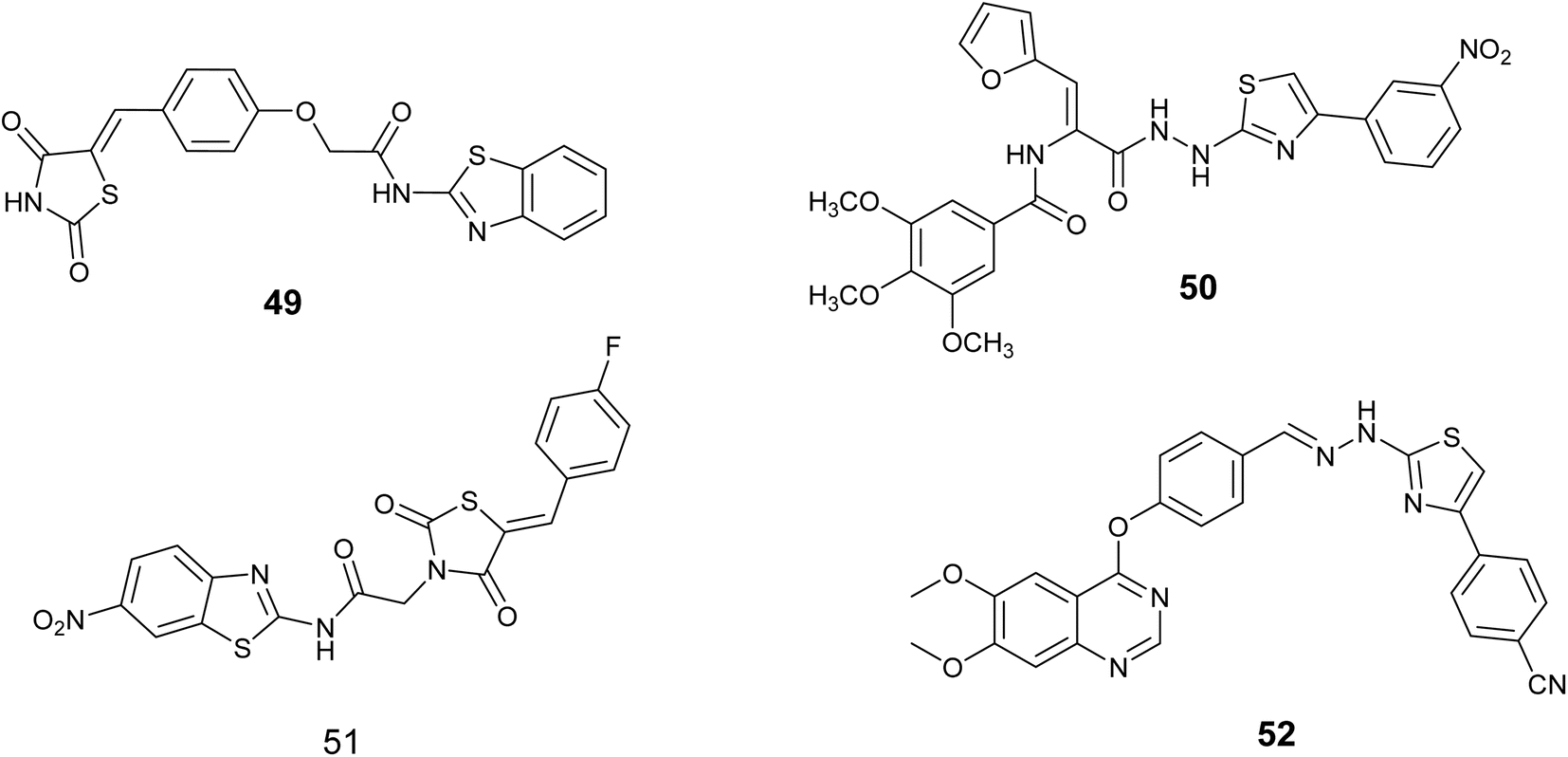
Bhanushali and colleagues conducted a screening of a new group of 5-benzylidene-2,4- thiazolidinediones and assessed their potential as inhibitors of VEGFR-2 kinase in vitro to investigate their anti-angiogenic properties. Compound 49, Fig. 18, exhibited significant inhibitory effects in both CAM and zebrafish assays, leading to its selection for further evaluation as a VEGFR kinase inhibitor. Subsequent findings indicated that this compound effectively inhibited the kinase with an IC50 value of 0.5 μM.89
 | ||
| Fig. 18 Thiazole derivatives as Vascular endothelial growth factor receptor (VEGFR) inhibitors (49–52). | ||
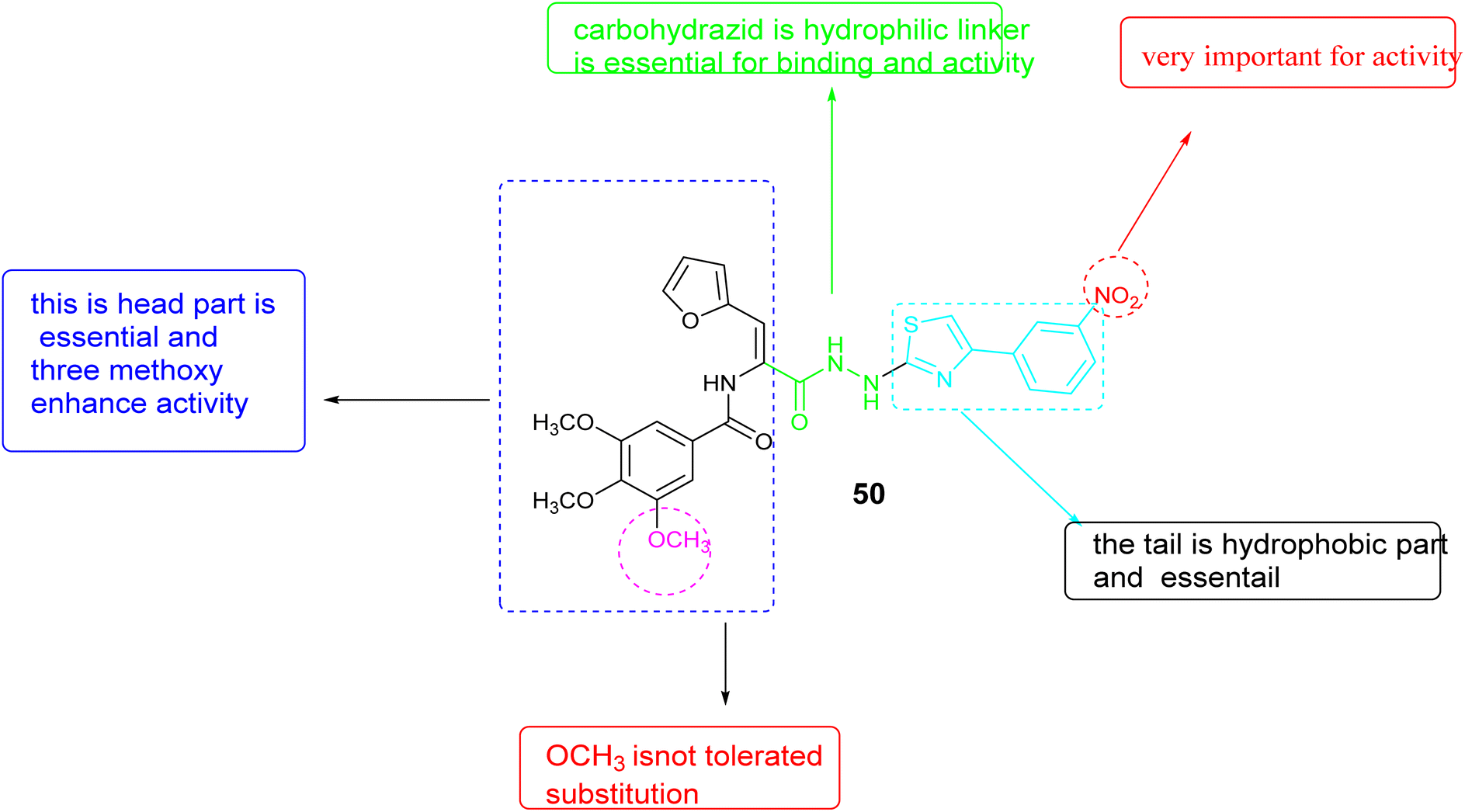
In 2022, Zaki and colleagues synthesized novel series based on thiazole core and assessed their activity as anticancer agent toward the breast cancer (MDA-MB-231) cell line. The results showed that derivative 50, Fig. 18, demonstrated notable effectiveness, with an IC50 value of 1.21 ± 0.09 μM. Furthermore, this compound demonstrated the ability to inhibit VEGFR-2, with an inhibition percentage of 85.72%. Following DNA flow cytometry analysis showed that compound 50 caused cells to stop at the G1 and G2/M stages of the cell cycle. Additionally, it triggered apoptosis, as shown by a rise in the percentage of cells in the pre-G1 phase. Furthermore, the structure–activity relationship (SAR) analysis for compound 50, Fig. 19 indicated that the carbohydrazide linker exhibits flexibility and hydrophilicity, which are advantageous for anti-VEGFR-2 activity. The presence of a 4-nitro group on the phenyl ring is critical for the compound's efficacy, while the combination of these two components with the thiazole ring functions as a tail segment. The head portion of the compound is characterized by trimethoxy substitutions on the phenyl ring, which is vital for its biological activity, and is connected to the furan moiety through an amide linker.90
 | ||
| Fig. 19 SAR for compound 50. | ||
A novel series of compounds derived from aminobenzothiazole was created and tested for their ability to inhibit tumor growth in laboratory settings using three different types of cancer cells (MCF-7, HCT-116, and HEPG-2). Compound 51, Fig. 18 exhibited the highest potency among the tested compounds, demonstrating IC50 values of 3.84, 5.61 and 7.92 μM against the respective cell lines, while also displaying favorable safety characteristics against noncancer WI-38 cells. Compound 51 significantly inhibited S phase cell proliferation in MCF-7 cells as determined by flow cytometry. Further analysis revealed that compound 51, with an IC50 value of 91 nanometers, was the most effective inhibitor of VEGFR-2.91
In 2024, Sandor and colleagues introduced a new set of quinazoline-thiazole hybrid compounds with potential antiproliferative and anti-angiogenic agents. The majority of these compounds exhibited enhanced antiproliferative activity (IC50 = 1.83–4.24 μM) against HepG2 cells compared to sorafenib (IC50 = 6.28 μM). The interaction with the VEGFR2 kinase domain was evaluated using computational methods such as molecular docking, molecular dynamics simulations, and MM-PBSA. The compound series demonstrated a notable resemblance to sorafenib in terms of binding orientation within the VEGFR2 active site, with compound 52, Fig. 18, forming the most stable complex with VEGFR2 in comparison to sorafenib. The highest free energy was observed for 52 (−71.23 ± 5.29 kcal mol−1), which closely approximated the value for sorafenib (–69.39 ± 3.63 kcal mol−1).92
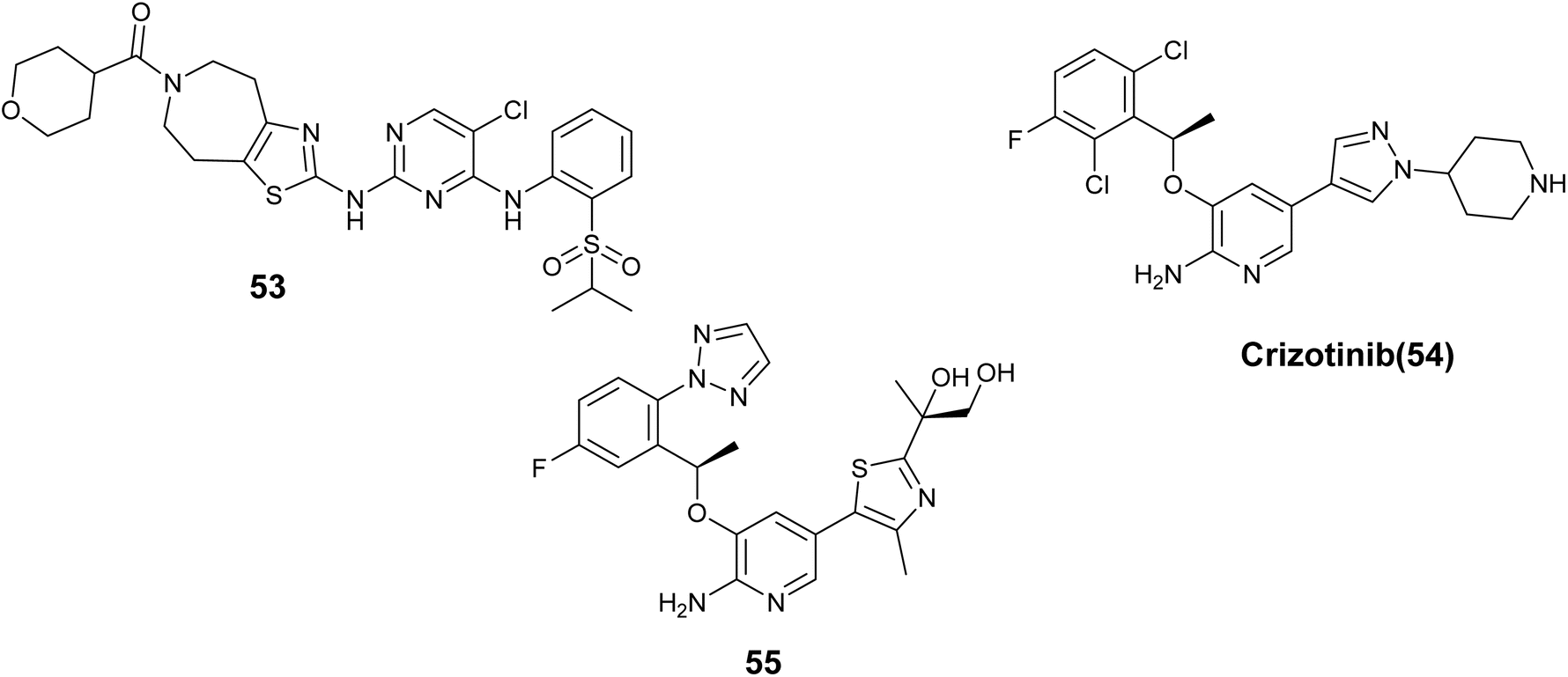
Z. Liu and colleagues introduced a novel series of 2-(thiazol-2-amino)-4-arylaminopyrimidines designed as inhibitors of Anaplastic lymphoma kinase (ALK). These compounds exhibited varying levels of effectiveness toward ALK kinase, Compound 53, Fig. 20 showed the strongest potency at 12.4 nM, although it had only moderate effectiveness in SUP-M2 cells with NPM-ALK. However, it displayed significant selectivity for kinases and effectively inhibited the ALK gatekeeper mutation L1196M (IC50 = 24.1 nM). Furthermore, derivative 53 dose-dependently inhibited ALK phosphorylation and its related signaling pathways.94
 | ||
| Fig. 20 Thiazole derivatives as anaplastic lymphoma kinase (ALK) inhibitors (53–55). | ||
Crizotinib (54), Fig. 20 is a drug that inhibits the ALK receptor tyrosine kinase and has shown effectiveness in patients who test positive for ALK and ROS. However, the development of point mutations in the kinase domain of ALK under crizotinib treatment leads to resistance and disease progression. To address this issue, Huang et al. conducted optimization studies to identify a novel drug candidate with enhanced lipophilic efficiency capable of overcoming resistance in cell lines. The results indicated that compound 55, Fig. 20 demonstrated significant efficacy against various engineered ALK mutant cell lines, displaying an IC50 value of 0.8 nM in contrast to 80 nM for Crizotinib. Additionally, compound 55 demonstrated high potency against ROS1 with a Ki value of 0.02 nM. Furthermore, it exhibited favorable preclinical pharmacokinetic properties and substantial suppression of tumor progression observed in a resistant cell line (H3122-L1196M) with an IC50 of 6.6 nM.95
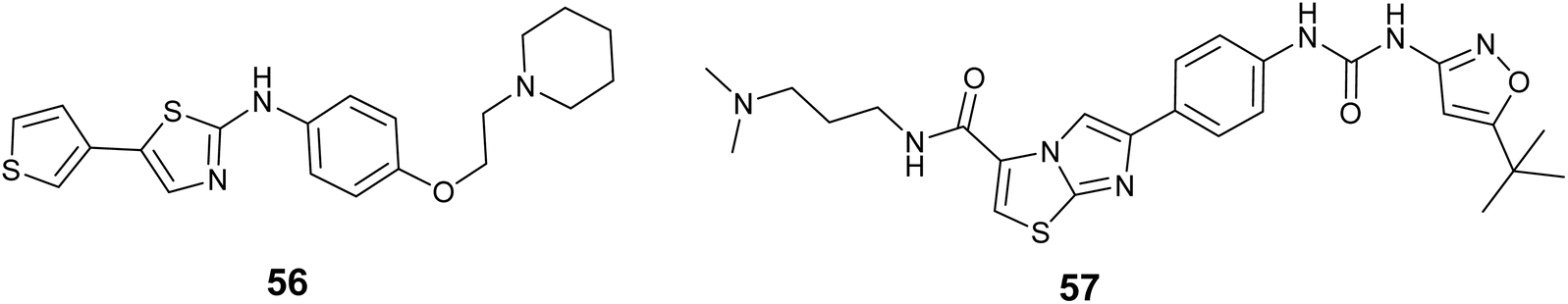
Novartis developed a group of thiazole derivatives that function as Flt3 inhibitors. Compound 56, Fig. 21 a representative of this series, Showed notable effectiveness with an IC50 value of 22 nM toward Flt3.97 Furthermore, a set of imidazothiazole derivatives was synthesized. By examining the relationship between the structure and activity of these compounds through cellular tests, Multiple compounds were found to be highly effective against FLT3-dependent human acute myeloid leukemia (AML) cell line MV4-11, but showed little to no impact on FLT3-independent human cervical cancer cell line HeLa. Following experiments were carried out on the most effective compounds to inhibit FLT3 kinase. Compound 57, Fig. 21 emerged as the most effective, demonstrating high potency in both cellular MV4-11 and enzymatic (FLT3) assays with the value of IC50; 0.002 μM and 0.022 μM, respectively.98
 | ||
| Fig. 21 Thiazole derivatives as FMS-like tyrosine kinase 3 (FLT3) inhibitors (56 and 57). | ||
In 2020, Groendyke B. J. has launched a new collection of small molecules based on a tricyclic pyrimidothiazolodiazepinone core, which demonstrates notable effectiveness and selectivity against FAK. BJG-03-025 (58), Fig. 22 was developed through the use of Structure–Activity Relationship (SAR) studies and modifications. This compound shows potent biochemical suppression of FAK (IC50 = 20 nM), high selectivity within the kinome, effectiveness in breast and gastric cancer models grown in 3D settings, and positive pharmacokinetic properties in mouse models. BJG-03-025(58) is a useful chemical tool for evaluating biological processes related to FAK.100
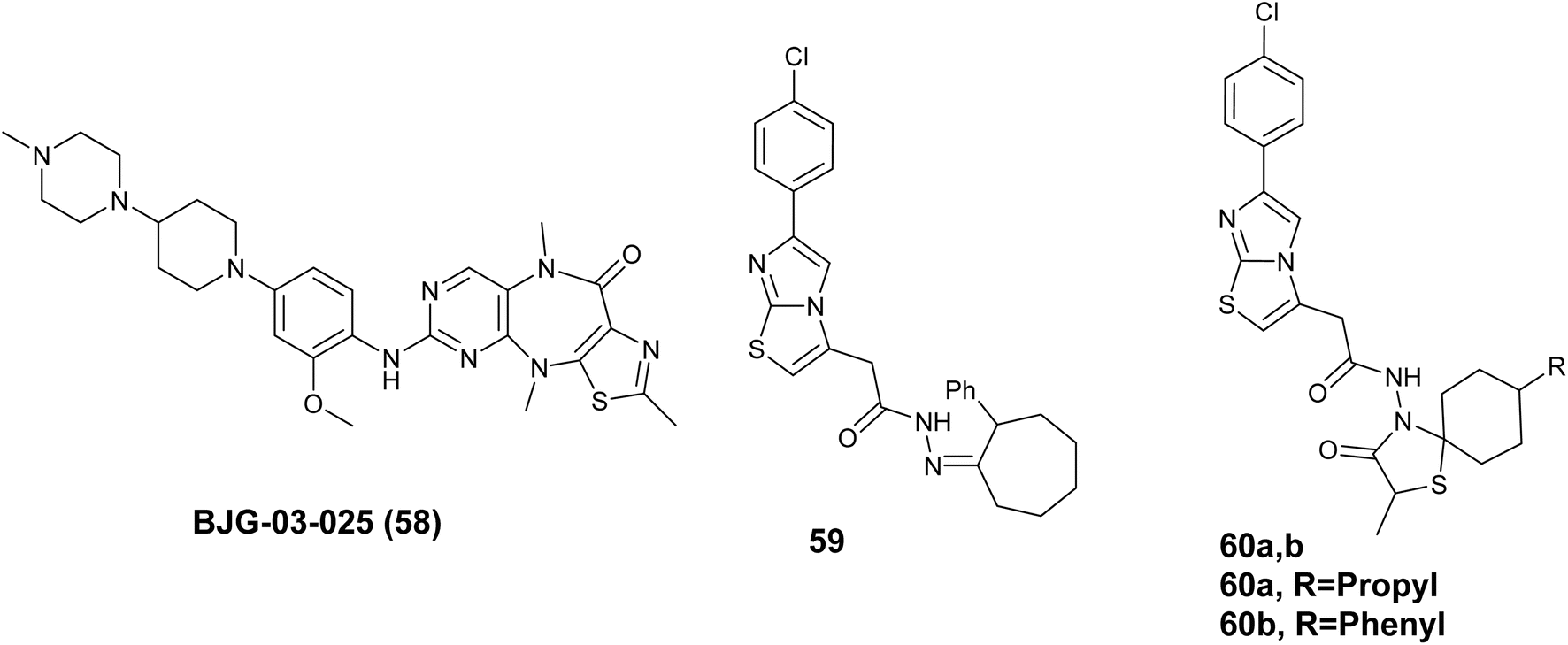 | ||
| Fig. 22 Thiazole derivatives as focal adhesion kinase (FAK) inhibitors (58–60a–b). | ||
A new group of imidazo[2,1-b]thiazole compounds was created and tested for their ability to cause cell death and apoptosis in glioma C6 cancer cells. The findings indicated that the investigated compounds unveiled significant activity toward the tested cells, particularly derivative 59, Fig. 22 which demonstrated an IC50 value of 4.83 ± 0.28 μM with minimal toxicity towards normal NIH/3T3 cells. Additionally, compound 60a, Fig. 22 exhibited higher apoptotic activity against the C6 cancer cell line compared to cisplatin 5.7 and 2.2%, in the order given. Furthermore, the most promising compounds were further examined as focal adhesion kinase (FAK) inhibitors. The findings showed that the compounds tested had significant inhibitory effects on FAK, with compound 60b, Fig. 22 being identified as the most potent inhibitor of FAK on the C6 cancer cell line 72.54 ± 3.6%.101
Hubbard et al. have documented advancements in this field through the utilization of the single-drug multi-target strategy, focusing on a group of IGF-1R inhibitors derived from imidazo[2,1-b] thiazole. The imidazo[2,1-b]thiazole compound series, exemplified by compound 61, Fig. 23 demonstrated notable efficacy against IGF-1R in A431 cells, exhibiting an IC50 value of 41 nM, along with moderate potencies towards EGFR and ErbB2 at 350 and 310 nM, respectively.103
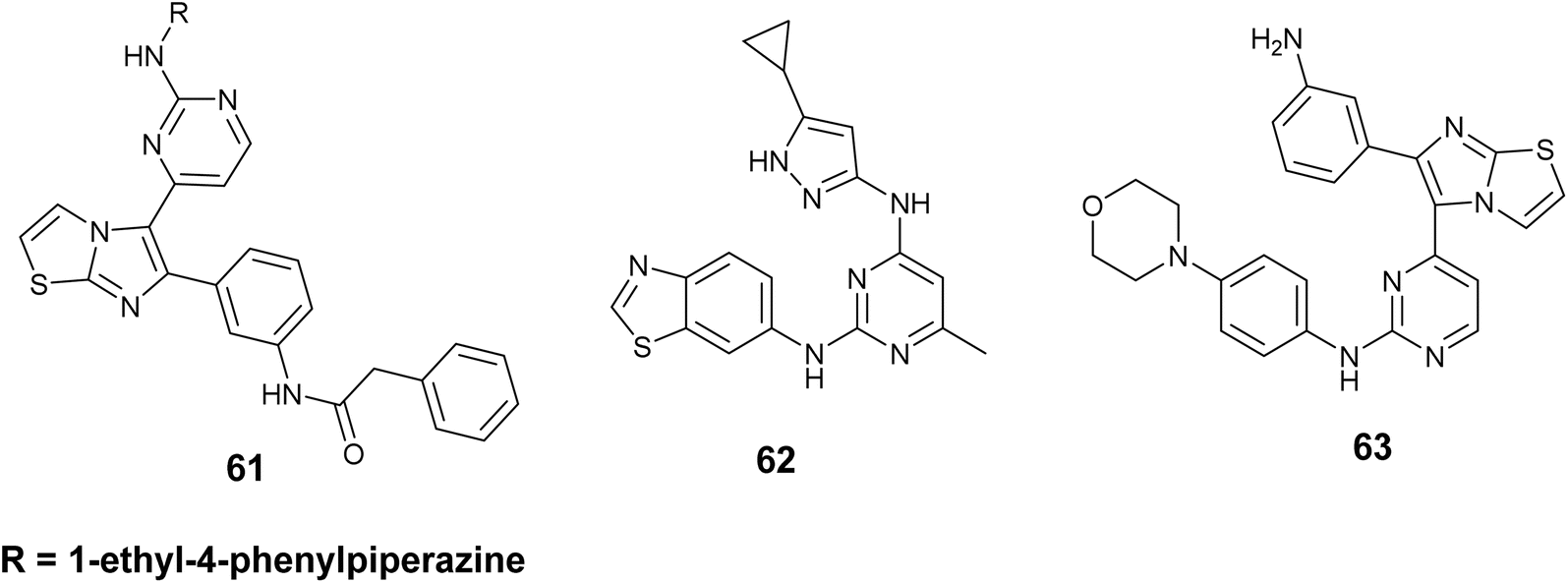 | ||
| Fig. 23 Thiazole derivatives as insulin-like growth factor type 1 receptor (IGF-1R) (61–63). | ||
Additionally, Tandon and colleagues developed and synthesized compound 62, Fig. 23 as a dual inhibitor targeting both IGF-1R and EGFR, which was derived from a bis-arylamino-pyrimidine scaffold. This compound exhibited potent activities against IGF-1R and EGFR, with the value of IC50; 74 and 45 nM, respectively. Furthermore, derivative 62 demonstrated notable anti-proliferative effects across various cell lines, including A431, HT29, and A549 (IC50 = 170, 274, and 111 nM, respectively). Notably, derivative 62 exhibited sensitivities in inhibiting proliferation in H1975 cells, with a GI50 of 1.3 M. It is important to highlight that H1975 cells carry an EGFR mutation (T790M) and are known to be resistant to erlotinib, as evidenced by a GI50 of 47 M.104
Gadekar and colleagues outlined the development, creation, evaluation of a group of compounds derived from a pyrimidine-containing imidazothiazole structure as dual kinase inhibitors that target IGF1R and EGFR for potential use in cancer treatment. The findings indicated that numerous compounds exhibited significant activity at the nanomolar level. Notably, compound 63, Fig. 23 demonstrated the most potent and encouraging effect on IGF1R, IC50 = 52 nM and EGFR IC50 = 35.5 nM, in addition to possessing a favorable pharmacokinetic profile.105
Sanachai and colleagues employed both experimental and theoretical approaches to investigate new substances that could be effective in fighting cancer targeting JAK2 and EGFR. They prepared derivatives of aromatic alkyl-amino analogs based on a thiazole scaffold and evaluated their inhibitory effects on JAK2 and EGFR proteins, Additionally, their ability to combat cancer in human cancer cell lines. In vitro cytotoxicity screening results indicated significant activity of the tested compounds, notably compound 64, Fig. 24 exhibited notable efficacies against HEL cell lines IC50 = 6.89 ± 0.38 μM, while derivative 65 demonstrated strong effectiveness against tested cell A431, IC50 = 8.04 ± 0.90 μM. The most promising compounds were further assessed as inhibitors of JAK2 and EGFR proteins, revealing that compound 66, Fig. 24 displayed significant activity against JAK2 (IC50 = 17.64 ± 1.68 nM), and derivative 65, Fig. 24 displayed the most significant activity targeting EGFR (IC50 = 28.46 ± 3.09 nM). Based on molecular docking analysis, these three compounds suit the active site well and form hydrogen bonds to Lys857, Leu932, and Glu930 Situated in the area where the enzyme bends and JAK2's catalytic site, it hydrophobically contacts Leu983.107
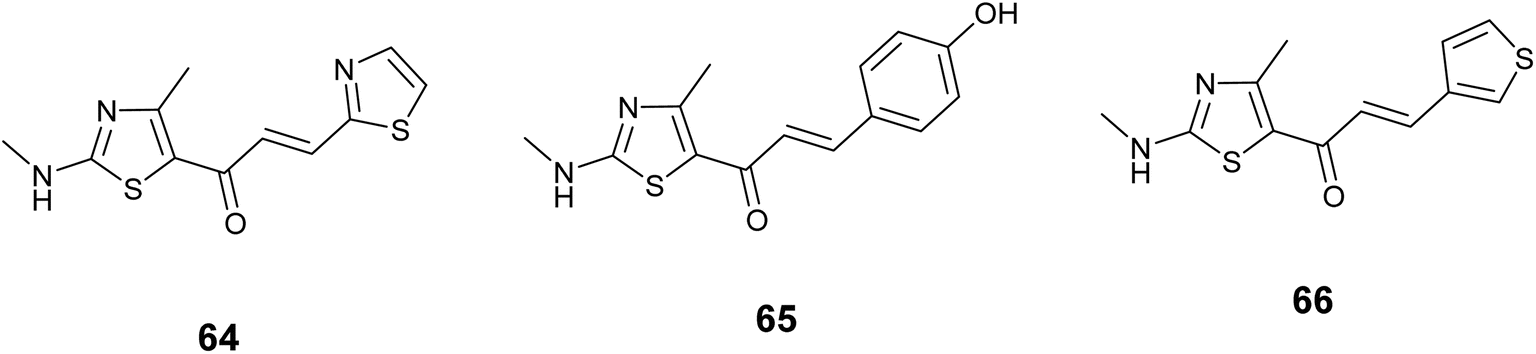 | ||
| Fig. 24 Thiazole derivatives as Janus kinase (JAK) inhibitors (64–66). | ||
Pan and colleagues synthesized two thiazole amine-based compounds and assessed the effect of these compounds on human mast cells that mutated the KIT gene. Their findings revealed that compound 67, Fig. 25 displayed the highest activity, effectively blocking the activation of KIT and the subsequent signaling molecules Stat3 and Stat5. Moreover, compound 67 showed notable suppression of cell survival in KBM5 cells with wild-type Bcr-Abl and BaF3 cells that have FIP1L1-PDGFRa expression, with IC50 values of 56.0 nM and 20.0 nM, respectively. Moreover, Compound 67 inhibited the growth of cells expressing the D816V KIT mutation, triggered cell death, and reduced the levels of Mcl-1 and survivin in cancerous cells with the mutant KIT.109
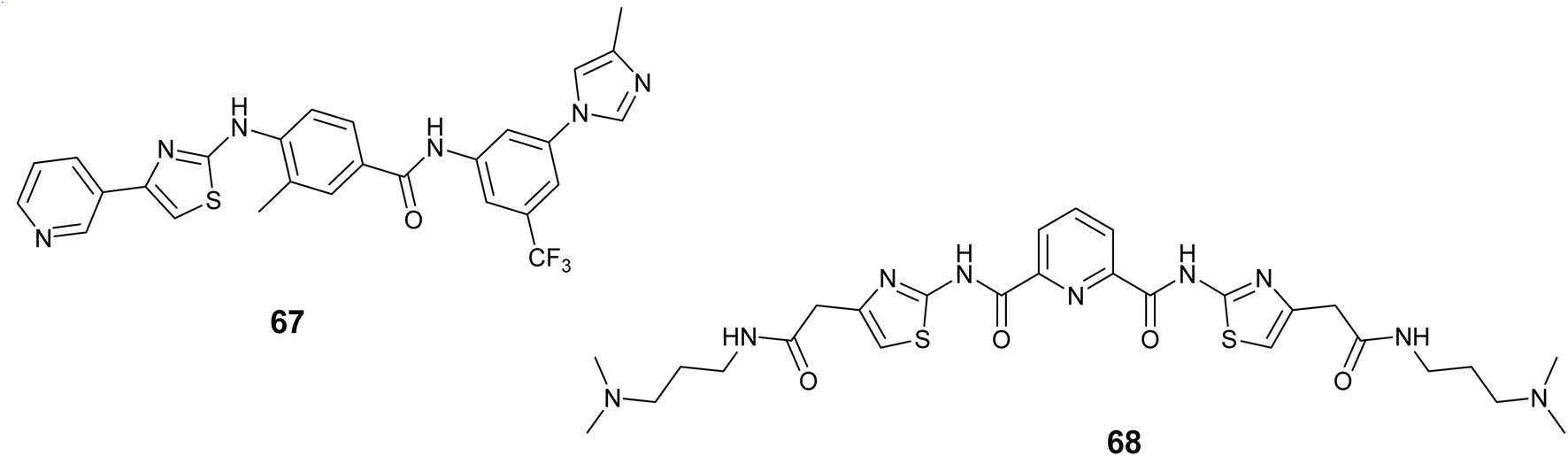 | ||
| Fig. 25 Thiazole derivatives as KIT inhibitors (67 and 68). | ||
In 2021, Paul and colleagues introduced a novel thiazole polyamide (68), Fig. 25 that exhibits high affinity binding to the c-KIT1 G-quadruplex (G4) at sub-micromolar levels, leading to the suppression of c-KIT proto-oncogene expression. Compound 68 demonstrated notable efficacy against the c-KIT1 G4, as evidenced by a fluorescence increase of up to tenfold upon interaction with the c-KIT1 G4, DC50 = 0.88 μM and a Kd = 0.69 μM. Furthermore, derivative 68 was found to enhance the presence of G4-specific antibody (BG4) foci and identify G4 structures within cancer cells.110
Lv et al. conducted an investigation on thiazolyl-pyrazoline derivatives to assess their efficacy as inhibitors of EGFR kinase activity. Among the compounds studied, compound 69, Fig. 26 showed the strongest inhibitory effect on EGFR TK, with IC50 = 0.06 μM. Furthermore, Compound 69 showed notable inhibitory effects on the growth of MCF-7 cells in laboratory tests, with an IC50 value of 0.07 μM, which is similar to the standard medication erlotinib (IC50; 0.02 μM). Molecular docking analysis revealed that Compound 69 formed three hydrogen bonds when bound to the EGFR kinase.112
 | ||
| Fig. 26 Thiazole derivatives as epidermal growth factor receptor (EGFR) inhibitors (69–73). | ||
A new series of novels containing a thiazole core and/or fused structure was created, produced, and tested for their ability to fight cancer in human cell lines, as well as their ability to inhibit EGFR in laboratory settings. Although all the created compounds showed limited effectiveness in fighting EGFR-related tumors, compound 70, Fig. 26 demonstrated the highest potency toward the human cell line HeLa, with IC50 = 0.42 μM, and also displayed significant activity toward EGFR with IC50 = 55 nM. Additionally, these compounds showed negligible cellular toxicity towards human normal cell lines HL7702.113
In 2020, Srour and colleagues created a new set of compounds based on a combination of benzoimiazole and thiazole to act as inhibitors of the epidermal growth factor receptor (EGFR). These newly synthesized derivatives were evaluated in a laboratory setting to determine their ability to inhibit the activity of EGFR tyrosine kinase. The findings indicated that Compound 71a, Fig. 26 showed the most activity, with an IC50 value of 71.67 nanomolar. Additionally, the findings of the MTT assay indicated that compound 71b, Fig. 26 exhibited notable cytotoxic effects on the MCF-7 human breast cancer cell line, with an IC50 value of 5.96 μM, in comparison to the IC50 value of the reference drug erlotinib, which was 4.15 μM. Compound 71c also exhibited notable efficacy as an agent that inhibits EGFR tyrosine kinase and acts as an anti-breast cancer treatment, with IC50 values of 109.71 ± 3.55 nM and 6.30 ± 0.37 μM, respectively. Additionally, compound 71c, Fig. 26 elicited apoptotic responses and halted the cell cycle at the G2/M checkpoint, consequently interfering with the mitotic progression in MCF-7 cells. Furthermore, compound 71c promoted the expression of oncogenic indicators including caspase-3, p53, and Bax/Bcl-2, while concurrently suppressing the functionality of the PARP-1 enzyme.114
Al-Warhi and colleagues conducted a study in which they synthesized various thiazolyl-pyrazoline derivatives to assess their efficacy as inhibitors of cell proliferation in the lung (A549) and breast (T-47D) cell line derived from cancer cells using the MTT assay. The findings showed that the thiazolyl-pyrazoline derivatives displayed greater anti-proliferative effects on the T-47D breast cancer cells compared to the A549 lung cancer cells. Specifically, compounds 72a and 72b, Fig. 26 demonstrated notable efficacy against the two tested cell lines A549 with the value of IC50; 3.92 and 6.53 μM, while T-47D cells the value of IC50; 0.88 and 0.75 μM, respectively. The most favorable compounds were subsequently evaluated for their ability to inhibit EGFR. with compound 72c, Fig. 26 displays the highest activity (IC50 = 83 ± 40 nM). Moreover, compounds 72a and 72b were found to cause sub-G1 phase arrest and cellular apoptosis, aligning with the expected outcomes of inhibiting EGFR.115
In 2023, Raghu et al. conducted a study where they synthesized and evaluated a novel series of quinazoline-based thiazole scaffold for their potential anti-proliferative properties toward MCF-7, HepG2, and A549 cancer cell lines in vitro. The results indicated that all compounds exhibited varying degrees of cytotoxicity, Compound 73 Fig. 26 exhibited the highest level of efficacy among the cell lines that have been examined (MCF-7, HepG2, and A549), displaying IC50 values of 2.86 ± 0.31, 5.91 ± 0.45, and 14.79 ± 1.03 μM, respectively. Furthermore, derivative 71, identified as the most active compound, was also evaluated as an EGFR inhibitor. Notably, compound 73 showed strong inhibitory effects against wild-type, L858R/T790 M, and L858R/T790 M/C797S mutant EGFR kinases, with IC50 values of 2.17, 2.81, and 3.62 nM, respectively (Table 2).116
| Compounds | Activity values of IC50 | |
|---|---|---|
| Serine/threonine thiazole-based kinase inhibitors | ||
| PI3K/AKT/mTOR signaling pathway inhibitors | 18 | 0.50–4.75 μM |
| 19 | 0.30–0.45 μM | |
| 20 | 9.0 nM | |
| 21 | 290 nM | |
| 22 | 0.02 μM | |
| 23 | 0.719 & 0.131 μM | |
| 24 | 2.33 nM | |
| Cyclin-dependent kinases (CDKs) inhibitors | 25 | 0.64 to 2.01 μM |
| 26 | 54.8 nM | |
| 27 | ND | |
| 28 | 0.35 μM | |
| Aurora kinase inhibitors | 29 | 79 nM |
| 30 | 140 nM | |
| 31 | Ki = 8.0 & 9.2 nM | |
| 32 | ND | |
| Inhibitors of the enzyme casein kinases II (CK2) | 33 | 0.4 μM |
| 34 | 1.9 μM | |
| 35 | 3.4 μM | |
| B-RAF inhibitors | 36 | 0.05 μM |
| 37 | 19 nM | |
| 38a | 9.30 nM | |
| 38b | 1.20 nM | |
| 39 | 8.2 & 0.978 nM | |
| 40 | 23.1 nM | |
| Inhibitors of glycogen synthase kinase 3β | 41 | 100 nM |
| 42 | 0.29 nM | |
| 43 | 1.1 nM | |
| 44 | 2.55 μM | |
| 45 | 7.35 μM | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Inhibitors of kinases that are based on tyrosine thiazole | ||
| Inhibitors of the c-MET protein | 46 | 17.6 nM |
| 47 | 0.06 nM | |
| 48 | 2.54 nM | |
| Inhibitors of the receptor for vascular endothelial growth factor (VEGFR) | 49 | 0.5 μM |
| 50 | % Inhibition = 85.72% | |
| 51 | 91 nM | |
| 52 | ND | |
| Anaplastic lymphoma kinase (ALK) inhibitors | 53 | 24.1 nM |
| 54 | 11–24 nM | |
| 55 | 0.8 nM | |
| FMS-like tyrosine kinase 3 (FLT3) inhibitors | 56 | 22 nM |
| 57 | 0.022 μM | |
| Inhibitors of focal adhesion kinase (FAK) | 58 | 20 nM |
| 59 | Inhibition% = 68.71% | |
| 60a | Inhibition% = 47.78% | |
| 60b | Inhibition% = 72.54% | |
| Insulin-like growth factor type 1 receptor (IGF-1R) | 61 | 41 nM |
| 62 | 74 nM | |
| 63 | 52 nM | |
| Janus kinase (JAK) inhibitors | 64 | 6.89 μM |
| 65 | 8.04 μM | |
| 66 | 17.64 nM | |
| KIT inhibitors | 67 | 56.0 nM & 20.0 nM |
| 68 | DC50 = 0.88 μM & a Kd = 0.69 μM | |
| Epidermal growth factor receptor (EGFR) inhibitors | 69 | 0.06 μM |
| 70 | 55 nM | |
| 71a | 71.67 nM | |
| 71b | 109.71 nM | |
| 71c | 6.30 μM | |
| 72a | ND | |
| 72b | ND | |
| 72c | 83 nM | |
| 73 | 2.17, 2.81 &3.62 nM | |
6. Molecular modeling studies
Molecular docking techniques were employed to elucidate the binding interactions between the active compounds and the binding pocket of the target protein. Therefore, compound 18 was selected for molecular docking studies against PI3Kc (PDB code 3QKO) due to its superior efficacy as an anticancer agent. It exhibited a notable fit by establishing five hydrogen bonds with the residues Val882, Tyr867, Lys833, Asp964, and Asp841, as illustrated in Fig. 27.51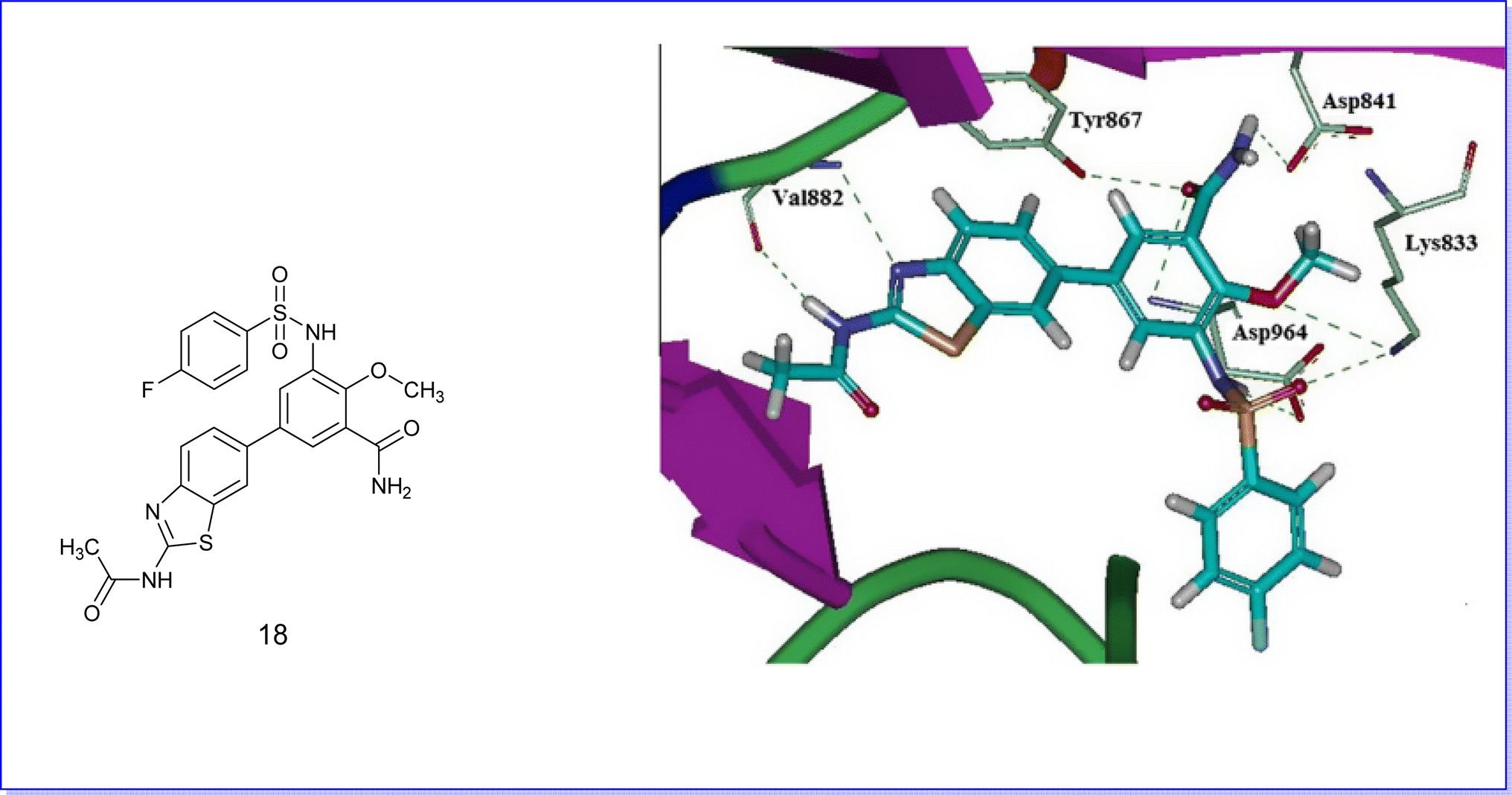 | ||
| Fig. 27 Docking mode of compound 18 with PI3Kc. | ||
Moreover, the compound 34 demonstrated favorable docking within the active site of the CK2 protein (PDB code CX-5279), as illustrated in Fig. 28. The results indicated effective binding at the active site, characterized by the formation of three hydrogen bonds involving the hydroxyl group, carboxyl group, and thiazole nitrogen with the amino acids SER51, LYS77, and LYS68. Additionally, a pi–pi interaction was observed.70
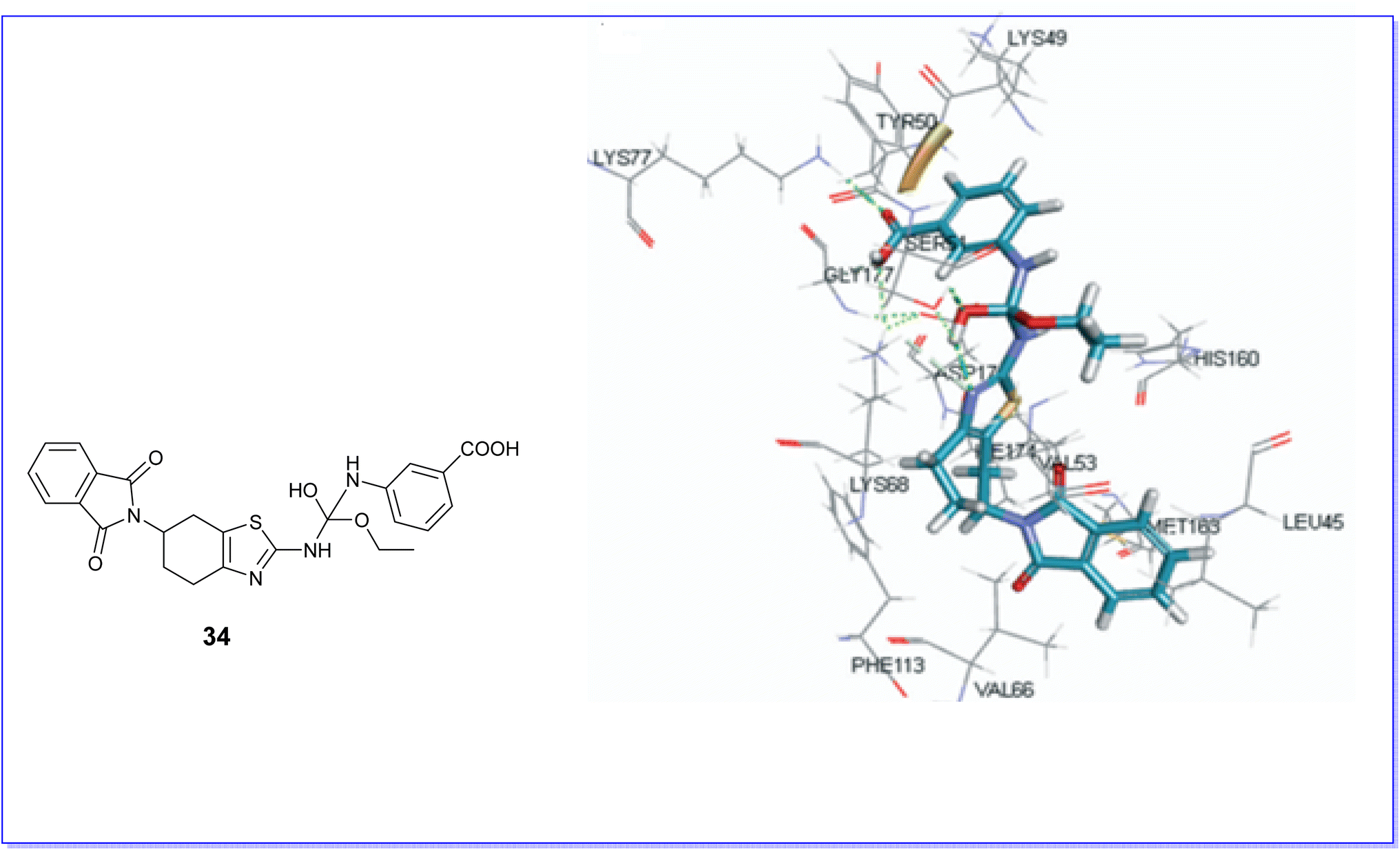 | ||
| Fig. 28 Molecular docking of compound 34 with CK2 protein. | ||
Furthermore, the two compounds, cefixime (44) and ceftriaxone (45), underwent molecular docking analysis against GSK3β (PDB code 6Y9S). The findings revealed that both compounds exhibited the most advantageous docking scores of −7.36 and −7.06, respectively, Fig. 29. These favorable scores are attributed to the formation of hydrogen bonds between the aminothiazole group of the compounds and the hinge residues of GSK3b.83
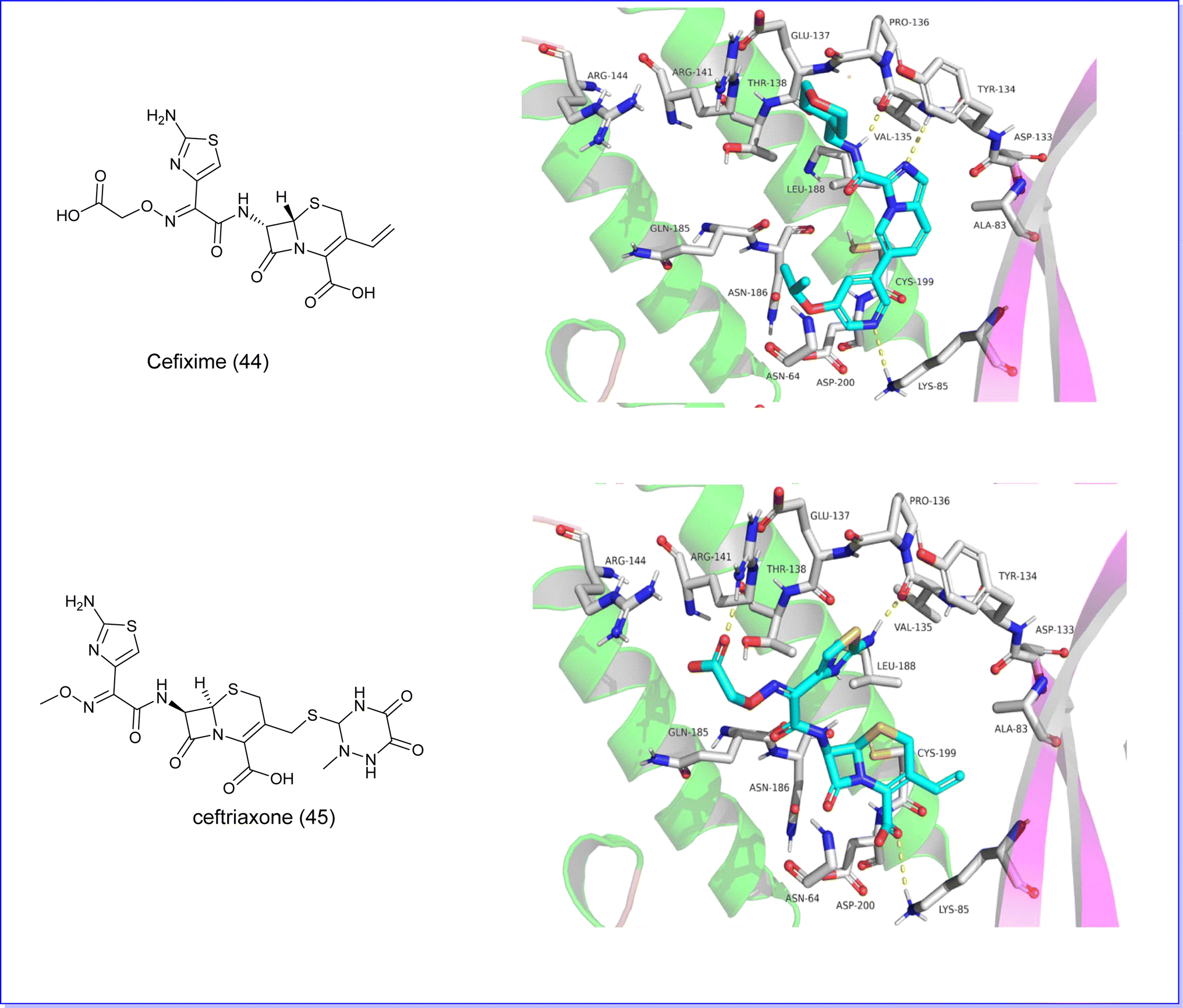 | ||
| Fig. 29 (A) Molecular docking of cefixime (44) GSK3 protein. (B) Molecular docking of ceftriaxone (45) with GSKS protein. | ||
The study of the molecular docking aimed to elucidate the interaction between compound 46 and c-Met protein (PDB code 3LQ8). As illustrated in Fig. 30, the results demonstrated the establishment of two hydrogen bonds: the first bond formed between the nitrogen atom of the quinoline and the Met1160 residue, while the second bond was formed through the interaction of the carbonyl oxygen atom of the urea moiety with Asp1222. Furthermore, a π–π interaction was noted between the phenyl ring and Phe1223. Both the π–π and hydrogen bond interactions played a significant role in stabilizing the conformation of the ligand–protein complex.85
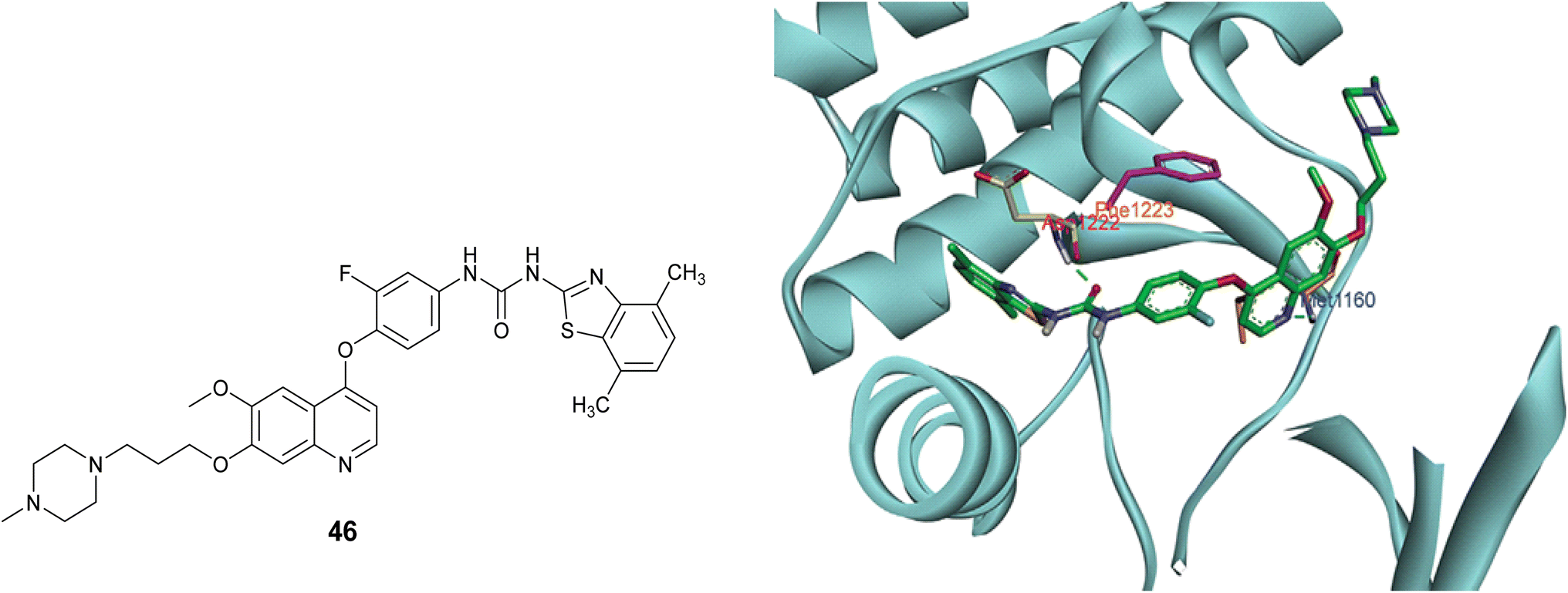 | ||
| Fig. 30 The active site of c-Met interacting with compound 46. | ||
Moreover, compound 60a underwent molecular docking analysis against Focal Adhesion Kinase (FAK), utilizing the PDB code 2ETM, Fig. 31. The findings revealed that Compound 60a exhibited a more favorable docking score in comparison to the other tested compounds. Furthermore, it demonstrated effective interaction with the target by establishing hydrogen bonds with Cysteine 502 of FAK.101
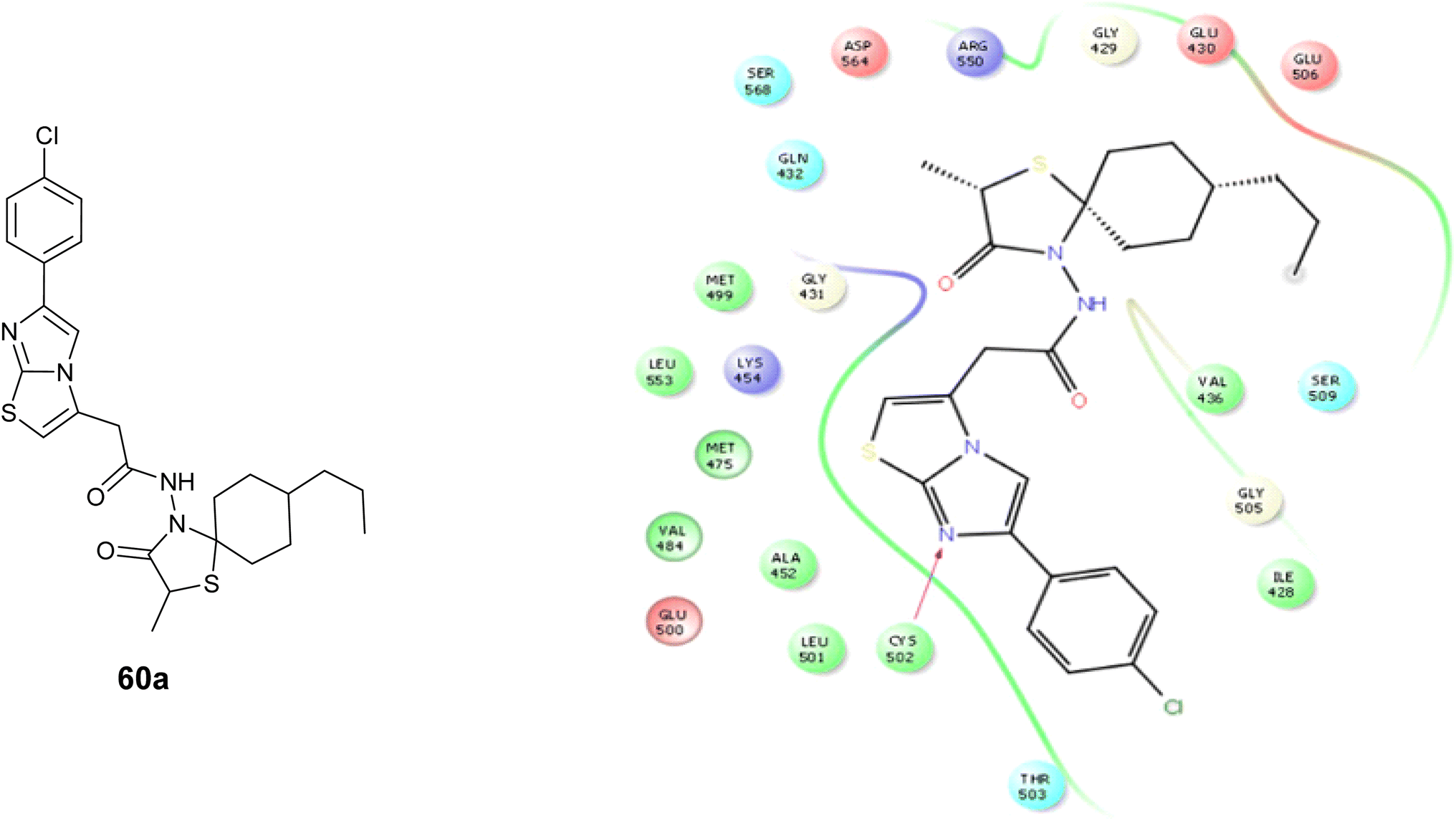 | ||
| Fig. 31 Molecular modeling for compound 60a with focal adhesion mont (FAK). | ||
In addition, Compound 69 was analyzed for its molecular docking properties (Fig. 32), revealing a favorable binding affinity to the EGFR kinase (PDB code 1fgk). The results indicated the formation of three hydrogen bonds with the residues Leu768, Gln767, and Cys751. Additionally, the modeling suggested the presence of a π–cation interaction between the thiazole ring of compound 69 and the residue Lys828.112
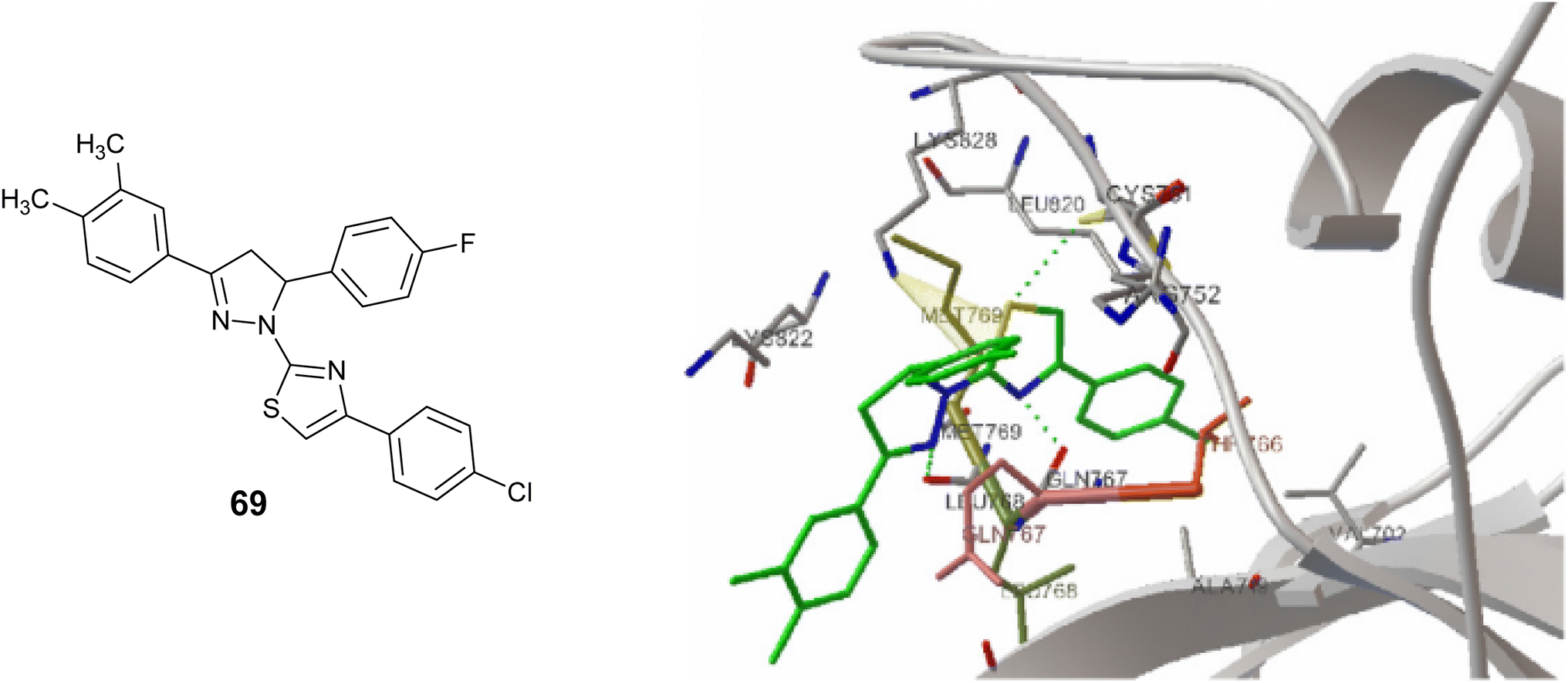 | ||
| Fig. 32 Molecular docking modeling of compound 69 with EGFR Masse. | ||
7. Conclusion and future aspects
This review highlights the emerging opportunities in utilizing thiazole-based heterocyclic derivatives for their potential anticancer properties. The review underscores the diverse applications of thiazole scaffold compounds in the development of drugs for combating various types of cancer. Despite the significant progress made in the field of thiazole-containing compounds, further focused research efforts are necessary. Such as, the advancement of appropriate synthesis methodologies utilizing green chemistry principles is crucial in propelling the future development of thiazole-containing compounds. Also, an in-depth investigation into the pharmacodynamic and pharmacokinetic characteristics of identified potent compounds with potential anticancer properties. Another appealing feature involves conducting research on the identification and isolation of thiazole-containing compounds that are naturally found in plants and marine organism. Finally, the findings of this analysis suggest that thiazole-derived compounds have potential as inhibitors of various enzyme targets and metabolic pathways. Further exploration into the potential adverse effects of thiazole-containing compounds represents a promising avenue for future scientific inquiry. The creation of enhanced analogues with improved efficacy, pharmacokinetics, and reduced side effects would represent a significant advancement in the field with potential benefits for human health and well-being.Data availability
All the data supporting this review article have been included in the manuscript.Author contributions
Conceptualization, M. S. E., A. S.; data curation, M. S. E., H. A. A. I., A. F. K.; formal analysis, A. S., H. A. A. I., A. F. K.; funding acquisition, M. S. E.; investigation, M. S. E., H. A. A. I., A. F. K.; methodology, A. S., resources, M. S. E., H. A. A. I., A. F. K., A. S.; validation, A. S.; visualization, A. S.; writing – original draft preparation, A. S.; writing – review and editing, M. S. E., H. A. A. I., A. F. K. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
Authors declare no conflict of interest. The authors alone are responsible for the content and writing of the paper.Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at Northern Border University, Arar, KSA for funding this research work through the project number “NBU-FFR-2024-80-02”.References
- R. J. Obaid, E. U. Mughal, N. Naeem, M. M. Al-Rooqi, A. Sadiq, R. S. Jassas, Z. Moussa and S. A. Ahmed, Pharmacological significance of nitrogen-containing five and six- membered heterocyclic scaffolds as potent cholinesterase inhibitors for drug discovery, Process Biochem., 2022, 120, 250–259 CrossRef CAS.
- E. Kabir and M. Uzzaman, A review on biological and medicinal impact of heterocyclic compounds, Results Chem., 2022, 4, 100606 CrossRef CAS.
- S. Kumari, K. Maddeboina, R. D. Bachu, S. H. Boddu, P. C. Trippier and A. K. Tiwari, Pivotal role of nitrogen heterocycles in Alzheimer's disease drug discovery, Drug Discovery Today, 2022, 27(10), 103322 CrossRef CAS.
- M. Sani and M. Zanda, Recent advances in the synthesis and medicinal chemistry of SF5 and SF4Cl compounds, Synthesis, 2022, 21, 4184–4209 CrossRef.
- C. Wentrup, Origins of organic chemistry and organic synthesis, Eur. J. Org Chem., 2022, 7(25), e202101492 CrossRef.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, A review on recent advances in nitrogen-containing molecules and their biological applications, Molecules, 2020, 25(8), 1909 CrossRef CAS.
- R. Mishra, P. K. Sharma, P. K. Verma, I. Tomer, G. Mathur and P. K. Dhakad, Biological potential of thiazole derivatives of synthetic origin, J. Heterocycl. Chem., 2017, 54(4), 2103–2116 CrossRef CAS.
- M. Issac, M. Aknin, A. Gauvin-Bialecki, N. De Voogd, A. Ledoux, M. Frédérich, Y. Kashman and S. Carmeli, Cyclotheonellazoles A–C, potent protease inhibitors from the marine sponge Theonella aff. swinhoei, J. Nat. Prod., 2017, 80(4), 1110–1116 CrossRef CAS PubMed.
- D. R. Abou-Hussein and D. T. Youssef, Mirabolides A and B; new cytotoxic glycerides from the Red Sea sponge Theonella mirabilis, Mar. Drugs, 2016, 14(8), 155 CrossRef PubMed.
- K. C. Huang, Z. Chen, Y. Jiang, S. Akare, D. Kolber-Simonds, K. Condon, S. Agoulnik, K. Tendyke, Y. Shen, K. M. Wu and S. Mathieu, Apratoxin A shows novel pancreas- targeting activity through the binding of Sec 61, Mol. Cancer Ther., 2016, 15(6), 1208–1216 CrossRef CAS.
- Y. Onda, Y. Masuda, M. Yoshida and T. Doi, Conformation-based design and synthesis of apratoxin A mimetics modified at the α, β-unsaturated thiazoline moiety, J. Med. Chem., 2017, 60(15), 6751–6765 CrossRef CAS PubMed.
- Z. Xia and C. D. Smith, Total synthesis of dendroamide A, a novel cyclic peptide that reverses multiple drug resistance, J. Org. Chem., 2001, 66(10), 3459–3466 CrossRef CAS.
- I. Nickeleit, S. Zender, F. Sasse, R. Geffers, G. Brandes, I. Sörensen, H. Steinmetz, S. Kubicka, T. Carlomagno, D. Menche, I. Gütgemann and A. Argyrin, reveals a critical role for the tumor suppressor protein p27kip1 in mediating antitumor activities in response to proteasome inhibition, Cancer cell, 2008, 14(1), 23–35 CrossRef PubMed.
- R. Dahiya, S. Dahiya, N. K. Fuloria, S. Kumar, R. Mourya, S. V. Chennupati, S. Jankie, H. Gautam, S. Singh, S. K. Karan and S. Maharaj, Natural bioactive thiazole-based peptides from marine resources: structural and pharmacological aspects, Mar. Drugs, 2020, 18(6), 329 CrossRef CAS.
- A. Singh, D. Malhotra, K. Singh, R. Chadha and P. M. Bedi, Thiazole derivatives in medicinal chemistry: Recent advancements in synthetic strategies, structure activity relationship and pharmacological outcomes, J. Mol. Struct., 2022, 1266, 133479 CrossRef CAS.
- S. H. Ali and A. R. Sayed, Review of the synthesis and biological activity of thiazoles, Synth. Commun., 2021, 51(5), 670–700 CrossRef CAS.
- M. F. Abo-Ashour, W. M. Eldehna, A. Nocentini, H. S. Ibrahim, S. Bua, H. A. Abdel-Aziz, S. M. Abou-Seri and C. T. Supuran, Novel synthesized SLC-0111 thiazole and thiadiazole analogues: Determination of their carbonic anhydrase inhibitory activity and molecular modeling studies, Bioorg. Chem., 2019, 87, 794–802 CrossRef CAS PubMed.
- A. Yadav, P. Verma, B. Chauhan and A. P. Mishra, Insilco Study, Synthesis of Thiazole Molecules As Possible Dihydrofolate Reductase Inhibitors Against Malaria, J. Pharm. Negat. Results, 2023, 1909–1916 CAS.
- N. D. Santos, N. D. Junior, J. F. de Oliveira, D. M. Duarte, J. C. dos Santos Soares, D. S. Marques, A. C. da Silva Santos, F. Nogueira, V. R. Pereira, M. C. de Lima and I. J. da Cruz Filho, Synthesis, characterization, antioxidant and antiparasitic activities new naphthyl- thiazole derivatives, Exp. Parasitol., 2023, 248, 108498 CrossRef CAS PubMed.
- A. A. Al-Karmalawy, D. S. El-Gamil, R. El-Shesheny, M. Sharaky, R. Alnajjar, O. Kutkat, Y. Moatasim, M. Elagawany, S. T. Al-Rashood, F. A. Binjubair and W. M. Eldehna, Design and statistical optimisation of emulsomal nanoparticles for improved anti-SARS-CoV-2 activity of N-(5-nitrothiazol-2-yl)-carboxamido candidates: in vitro and in silico studies, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 2202357 CrossRef PubMed.
- T. Al-Warhi, D. M. Elimam, Z. M. Elsayed, M. M. Abdel-Aziz, R. M. Maklad, A. A. Al-Karmalawy, K. Afarinkia, M. A. Abourehab, H. A. Abdel-Aziz and W. M. Eldehna, Development of novel isatin thiazolyl-pyrazoline hybrids as promising antimicrobials in MDR pathogens, RSC Adv., 2022, 12(48), 31466–31477 RSC.
- G. Kumar and N. P. Singh, Synthesis, anti-inflammatory and analgesic evaluation of thiazole/oxazole substituted benzothiazole derivatives, Bioorg. Chem., 2021, 107, 104608 CrossRef CAS.
- P. C. Sharma, K. K. Bansal, A. Sharma, D. Sharma and A. Deep, Thiazole-containing compounds as therapeutic targets for cancer therapy, Eur. J. Med. Chem., 2020, 188, 112016 CrossRef CAS.
- T. Al-Warhi, M. M. Elbadawi, A. Bonardi, A. Nocentini, A. A. Al-Karmalawy, N. Aljaeed, O. J. Alotaibi, H. A. Abdel-Aziz, C. T. Supuran and W. M. Eldehna, Design and synthesis of benzothiazole-based SLC-0111 analogues as new inhibitors for the cancer-associated carbonic anhydrase isoforms IX and XII, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 2635–2643 CrossRef CAS.
- A. Leoni, A. Locatelli, R. Morigi and M. Rambaldi, Novel thiazole derivatives: a patent review (2008–2012, Part 1), Expert Opin. Ther. Pat., 2014, 24, 201 CrossRef CAS PubMed.
- M. Gümüs, M. Yakan and I. Koca, Recent advances of thiazole hybrids in biological applications, Future Med. Chem., 2019, 11, 1979 CrossRef PubMed.
- S. Hu-Lieskovan, S. Mok, B. H. Moreno, J. Tsoi, L. Robert, L. Goedert, E. M Pinheiro, R. C. Koya, T. G Graeber, B. Comin-Anduix and A. Ribas, Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF (V600E) melanoma, Sci. Transl. Med., 2015, 7, 279 Search PubMed.
- L. Chen, J. Hu, J. Xu, C. Shao and G. Wang, Biological and Chemical Diversity of Ascidian-Associated Microorganism, Mar. Drugs, 2018, 16, 362 CrossRef CAS PubMed.
- M. T Chhabria, S. Patel, P. Modi and P. S. Brahmkshatriya, Thiazole: A Review on Chemistry, Synthesis and Therapeutic Importance of its Derivatives, Curr. Top. Med. Chem., 2016, 16, 2841 CrossRef.
- S. Sharma, M. Devgun, R. Narang, S. Lal and A. C. Rana, Thiazoles: A retrospective study on synthesis, structure-activity relationship and therapeutic significance, Indian J. Pharm. Educ. Res., 2022, 56, 646–666 CrossRef CAS.
- A. M. Borcea, I. Ionut, O. Crisan and O. Oniga, An overview of the synthesis and antimicrobial, antiprotozoal, and antitumor activity of thiazole and bisthiazole derivatives, Molecules, 2021, 26(3), 624 CrossRef CAS PubMed.
- T. Eicher and S. Hauptmann, The Chemistry of Heterocycles. Structure, Reactions, Syntheses, and Applications, John Wiley & Sons, 2013 Search PubMed.
- S. H. Ali and A. R. Sayed, Review of the synthesis and biological activity of thiazoles, Synth. Commun., 2021, 51(5), 670–700 CrossRef CAS.
- I. G. Shahin, K. O. Mohamed, A. T. Taher, A. S. Mayhoub and A. E. Kassab, Recent Advances in the Synthesis of Thiazole Ring Mini Review, Mini-Rev. Org. Chem., 2023, 20(3), 270–284 CrossRef CAS.
- D. X. Duc and N. T. Chung, Recent Development in the Synthesis of Thiazoles, Curr. Org. Synth., 2022, 19(6), 702–730 CrossRef CAS PubMed.
- U. Fathy, Thiazole derivatives: prospectives and biological applications, J. Sulfur Chem., 2024, 45, 1–43 CrossRef.
- M. Kalhor, Z. Vahedi and H. Gharoubi, Design of a new method for one-pot synthesis of 2-amino thiazoles using trichloroisocyanuric acid in the presence of a novel multi-functional and magnetically catalytic nanosystem: Ca/4-MePy-IL@ ZY-Fe 3 O 4, RSC Adv., 2023, 13(14), 9208–9221 RSC.
- J. Y. Al-Humaidi, S. M. Gomha, N. A. A. El-Ghany, B. Farag, M. E. Zaki, T. Z. Abolibda and N. A. Mohamed, Green Synthesis and Molecular Docking Study of Some New Thiazoles Using Terephthalohydrazide Chitosan Hydrogel as Ecofriendly Biopolymeric Catalyst, Catalysts, 2023, 13(9), 1311 CrossRef CAS.
- M. S. Shafik, Copper (II) complex immobilized on magnetic nanoparticles catalyzed synthesis of oxazole and thiazole derivatives, J. Synth. Chem., 2023, 1(3), 163–170 Search PubMed.
- T. B. Nguyen and P. Retailleau, Base-catalyzed three-component reaction between chalcones, isothiocyanates, and sulfur: Access to thiazole-2-thiones, Org. Lett., 2021, 23(14), 5344–5348 CrossRef CAS PubMed.
- B. Madhav, S. N. Murthy, B. S. P. A. Kumar, K. Ramesh and Y. V. D. Nageswa, A tandem one-pot aqueous phase synthesis of thiazoles/selenazoles, Tetrahedron Lett., 2012, 53, 3835–3838 CrossRef CAS.
- D. Castagnolo, M. Pagano, M. Bernardini and M. Botta, Domino Alkylation-Cyclization Reaction of Propargyl Bromides with Thioureas/Thiopyrimidinones: A New Facile Synthesis of 2-Aminothiazoles and 5H-Thiazolo[3,2-a]pyrimidin-5-ones, Synlett, 2009, 13, 2093–2096 Search PubMed.
- H. Bouherrou, A. Saidoun, A. Abderrahmani, L. Abdellaziz, Y. Rachedi, F. Dumas and A. Demenceau, Synthesis and biological evaluation of new substituted hantzsch thiazole derivatives from environmentally benign one-pot synthesis Using silica supported.tungstosilisic acid as reusable catalyst, Molecules, 2017, 22(5), 757 CrossRef PubMed.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA, 2018, 68(6), 394–424 Search PubMed.
- H. Ding, X. Yu, C. Hang, K. Gao, X. Lao, Y. Jia and Z. Yan, Ailanthone: A novel potential drug for treating human cancer, Oncol. Lett., 2020, 20(2), 1489–1503 CrossRef CAS PubMed.
- F. Ardito, M. Giuliani, D. Perrone, G. Troiano and L. Lo Muzio, The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy, Int. J. Mol. Med., 2017, 40(2), 271–280 CrossRef CAS PubMed.
- M. A. Arslan, O. Kutuk and H. Basaga, Protein kinases as drug targets in cancer, Curr, Cancer Drug Targets, 2006, 6, 623–634 CrossRef CAS PubMed.
- K. K. Bhanumathy, A. Balagopal, F. S. Vizeacoumar, F. J. Vizeacoumar, A. Freywald and V. Giambra, Protein tyrosine kinases: their roles and their targeting in leukemia, Cancers, 2021, 13(2), 184 CrossRef CAS.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, The protein kinase complement of the human genome, Science, 2002, 298(5600), 1912–1934 CrossRef CAS PubMed.
- Q. Zhou, V. W. Lui and W. Yeo, Targeting the PI3K/Akt/mTOR pathway in hepatocellular carcinoma, Future Oncol., 2011, 7(10), 1149–1167 CrossRef CAS PubMed.
- H. Li, X. M. Wang, J. Wang, T. Shao, Y. P. Li, Q. B. Mei, S. M. Lu and S. Q. Zhang, Combination of 2-methoxy-3-phenylsulfonylaminobenzamide and 2-aminobenzothiazole to discover novel anticancer agents, Bioorg. Med. Chem., 2014, 22(14), 3739–3748 CrossRef CAS PubMed.
- X. Xie, H. Li, J. Wang, S. Mao, M. Xin, S. Lu, Q. Mei and S. Zhang, Synthesis and anticancer effects evaluation of 1-alkyl-3-(6-(2-methoxy-3- sulfonylaminopyridin- 5-yl)benzo[d]thiazol-2-yl)urea as anticancer agents with low toxicity, Bioorg. Med. Chem., 2015, 23, 6477–6485 CrossRef CAS PubMed.
- R. A. Fairhurst, M. Gerspacher, P. Imbach-Weese, R. Mah, G. Caravatti, P. Furet, C. Fritsch, C. Schnell, J. Blanz, F. Blasco, S. Desrayaud, D. A. Guthy, M. Knapp, D. Arz, J. Wirth, E. Roehn-Carnemolla and V. H Luu, Identification and optimisation of 4,5-dihydrobenzo [1,2-d:3,4-d]bisthiazole and 4,5-dihydrothiazolo [4,5- h]quinazoline series of selective phosphatidylinositol-3 kinase alpha inhibitors, Bioorg. Med. Chem. Lett., 2015, 25, 3575–3581 CrossRef CAS PubMed.
- S. Cao, R. Cao, X. Liu, X. Luo and W. Zhong, Design, synthesis and biological evaluation of novel benzothiazole derivatives as selective PI3Kβ inhibitors, Molecules, 2016, 21(7), 876 CrossRef PubMed.
- R. Z. Batran, E. Y. Ahmed, H. M. Awad, K. A. Ali and N. A. Latif, EGFR and PI3K/m-TOR inhibitors: design, microwave assisted synthesis and anticancer activity of thiazole–coumarin hybrids, RSC Adv., 2023, 13(42), 29070–29085 RSC.
- M. E. Salem, M. Samir, A. H. Elwahy, A. M. Farag, A. M. Selim, A. A. Alsaegh, M. Sharaky, N. Bagato and I. T. Radwan, Design, synthesis, docking study, cytotoxicity evaluation, and PI3K inhibitory activity of Novel di-thiazoles, and bis (di-thiazoles), J. Mol. Struct., 2024, 1301, 137379 CrossRef CAS.
- S. Ghafouri-Fard, T. Khoshbakht, B. M. Hussen, P. Dong, N. Gassler, M. Taheri, A. Baniahmad and N. A. Dilmaghani, A review on the role of cyclin dependent kinases in cancers, Cancer Cell Int., 2022, 22(1), 325 CrossRef CAS.
- M. A. Assiri, T. E. Ali, A. K. Alsolimani, A. A. Shati, M. Y. Alfaifi and S. E. Elbehairi, Ultrasound-assisted synthesis of novel Schiff bases from 3-(2-oxo-2 H-chromen- 3-yl)-1-(4-phenylthiazol-2-yl)-1 H-pyrazole-4-carboxaldehyde and their cytotoxicity, apoptosis, cell cycle, molecular docking, and ADMET profiling, Synth. Commun., 2024, 10, 1–28 Search PubMed.
- H. Shao, S. Shi, D. W. Foley, F. A. Y. Abbas, X. Liu, F. Lam, S. Huang, X. Jiang, N. Baharin, P. M. Fischer and S. Wang, Synthesis, structure-activity relationship, and biological evaluation of 2,4,5-trisubstituted pyrimidine CDK inhibitors as potential anti-tumor agents, Eur. J. Med. Chem., 2013, 70, 447 CrossRef CAS PubMed.
- A. A. Aly, S. Bräse, A. A. Hassan, N. K. Mohamed, L. E. El-Haleem, M. Nieger, N. M. Morsy, M. B. Alshammari, M. A. Ibrahim and E. M. Abdelhafez, Design, synthesis, and molecular docking of paracyclophanyl-thiazole hybrids as novel CDK1 inhibitors and apoptosis inducing anti-melanoma agents, Molecules, 2020, 25(23), 5569 CrossRef CAS PubMed.
- A. M. El-Naggar, M. A. El-Hashash and E. B. Elkaeed, Eco-friendly sequential one-pot synthesis, molecular docking, and anticancer evaluation of arylidene-hydrazinyl- thiazole derivatives as CDK2 inhibitors, Bioorg. Chem., 2021, 108, 104615 CrossRef CAS.
- S. M. Gomha, M. E. Zaki, D. Maliwal, R. R. Pissurlenkar, M. S. Ibrahim, M. Fathalla and A. M. Hussein, Synthesis, in-silico studies, and biological evaluation of some novel 3-thiazolyl-indoles as CDK2–inhibitors, Results Chem., 2023, 6, 101209 CrossRef CAS.
- R. Du, C. Huang, K. Liu, X. Li and Z. Dong, Targeting AURKA in Cancer: molecular mechanisms and opportunities for Cancer therapy, Mol. Cancer, 2021, 20, 1–27 CrossRef.
- T. Pradhan, O. Gupta, G. Singh and V. Monga, Aurora kinase inhibitors as potential anticancer agents: Recent advances, Eur. J. Med. Chem., 2021, 221, 113495 CrossRef CAS PubMed.
- C. B. Andersen, Y. Wan, J. W. Chang, B. Riggs, C. Lee, Y. Liu, F. Sessa, F. Villa, N. Kwiatkowski, M. Suzuki and L. Nallan, Discovery of selective aminothiazole aurora kinase inhibitors, ACS Chem. Biol., 2008, 3(3), 180–192 CrossRef CAS PubMed.
- S. Wang, C. A. Midgley, F. Scaërou, J. B. Grabarek, G. Griffiths, W. Jackson, G. Kontopidis, S. J. McClue, C. McInnes, C. Meades and M. Mezna, Discovery of N-phenyl-4-(thiazol- 5-yl) pyrimidin-2-amine aurora kinase inhibitors, J. Med. Chem., 2010, 53(11), 4367–4378 CrossRef CAS PubMed.
- S. Bathula, M. Sankaranarayanan, B. Malgija, I. Kaliappan, R. R. Bhandare and A. B. Shaik, 2-Amino Thiazole Derivatives as Prospective Aurora Kinase Inhibitors against Breast Cancer: QSAR, ADMET Prediction, Molecular Docking, and Molecular Dynamic Simulation Studies, ACS Omega, 2023, 8(46), 44287–44311 CrossRef CAS.
- Y. Chen, Y. Wang, J. Wang, Z. Zhou, S. Cao and J. Zhang, Strategies of Targeting CK2 in Drug Discovery: Challenges, Opportunities, and Emerging Prospects, J. Med. Chem., 2023, 66, 2257–2281 CrossRef CAS.
- M. V. Protopopov, G. P. Volynets, S. A. Starosyla, V. S. Vdovin, S. S. Lukashov, Y. V. Bilokin, V. G. Bdzhola and S. M. Yarmoluk, Identification of 1, 3-thiazole-5- carboxylic Acid Derivatives as Inhibitors of Protein Kinase CK2, Curr. Enzyme Inhib., 2018, 14(2), 152–159 CrossRef CAS.
- T. R. Pardhi, M. S. Patel, V. Sudarsanam and K. K. Vasu, Design, synthesis, and evaluation of 4, 5, 6, 7-tetrahydrobenzo [d] thiazole-based novel dual kinase inhibitors of CK2 and GSK3β, MedChemComm, 2018, 9(9), 1472–1490 RSC.
- B. Bestgen, I. Krimm, I. Kufareva, A. A. Kamal, W. G. Seetoh, C. Abell, R. W. Hartmann, R. Abagyan, C. Cochet, M. Le Borgne and M. Engel, 2-Aminothiazole derivatives as selective allosteric modulators of the protein kinase CK2. 1. identification of an allosteric binding site, J. Med. Chem., 2019, 62(4), 1803–1816 CrossRef CAS PubMed.
- L. J. Andrews, Z. A. Thornton, S. S. Saincher, I. Y. Yao, S. Dawson, L. A. McGuinness, H. E. Jones, S. Jefferies, S. C. Short, H.-Y. Cheng, A. McAleenan, J. P. T. Higgins and K. M. Kurian, Prevalence of BRAF V600 in glioma and use of BRAF Inhibitors in patients with BRAF V600 mutation-positive glioma: systematic review, Neuro-Oncology, 2022, 24(4), 528–540 CrossRef CAS PubMed.
- M. Y. Zhao, Y. Yin, X. Yu, C. B. Sangani, S. Wanga, A. Lu, L. Yang, P. Lv, M. Jiang and H. Zhu, Synthesis, biological evaluation and 3D-QSAR study of novel 4,5- dihydro-1H-pyrazole thiazole derivatives as B-RAF(V600E) inhibitors, Bioorg. Med. Chem., 2015, 23, 46–54 CrossRef CAS PubMed.
- M. S. Abdel-Maksoud, M. R. Kim, M. I. El-Gamal, M. M. Gamal El-Din, J. Tae, H. S. Choi, K. Lee, K. H. Yoo and C. Oh, Design, synthesis, in vitro antiproliferative evaluation, and kinase inhibitory effects of a new series of imidazo[2,1-b] thiazole derivatives, Eur. J. Med. Chem., 2015, 95, 453–463 CrossRef CAS PubMed.
- M. S. Abdel-Maksoud, U. M. Ammar and C. H. Oh, Anticancer profile of newly synthesized BRAF inhibitors possess 5-(pyrimidin-4-yl) imidazo [2, 1-b] thiazole scaffold, Bioorg. Med. Chem., 2019, 27(10), 2041–2051 CrossRef CAS.
- U. M. Ammar, M. S. Abdel-Maksoud, K. I. Mersal, E. M. Ali, K. H. Yoo, H. S. Choi, J. K. Lee, S. Y. Cha and C. H. Oh, Modification of imidazothiazole derivatives gives promising activity in B-Raf kinase enzyme inhibition; synthesis, in vitro studies and molecular docking, Bioorg. Med. Chem. Lett., 2020, 30(20), 127478 CrossRef CAS PubMed.
- H. S. Anbar, M. I. El-Gamal, H. Tarazi, B. S. Lee, H. R. Jeon, D. Kwon and C. H. Oh, Imidazothiazole-based potent inhibitors of V600E-B-RAF kinase with promising anti-melanoma activity: Biological and computational studies, J. Enzyme Inhib. Med. Chem., 2020, 35(1), 1712–1726 CrossRef CAS.
- A. Y. Khormi, T. A. Farghaly, A. Bayazeed, Y. O. Al-Ghamdi, H. G. Abdulwahab and M. R. Shaaban, Novel thiazole derivatives incorporating phenyl sulphonyl moiety as potent BRAFV600E kinase inhibitors targeting melanoma, RSC Adv., 2022, 12(42), 27355–27369 RSC.
- D. Karati, K. K. Shaoo, K. R. Mahadik and D. Kumr, Glycogen synthase kinase-3β inhibitors as a novel promising target in the treatment of cancer: Medicinal Chemistry Perspective, Result. Chem., 2022, 4, 100532 CrossRef CAS.
- D. Sharma, A. Malhotra and R. Bansal, An overview of discovery of thiazole containing heterocycles as potent GSK-3β inhibitors, Curr. Drug Discovery Technol., 2018, 15(3), 229–235 CrossRef CAS.
- T. D. Gould, H. Einat, R. Bhat and H. K. Manji, AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test, Int. J. Neuropsychopharmacol., 2004, 7(4), 387–390 CrossRef CAS PubMed.
- P. Sivaprakasam, X. Han, R. L. Civiello, S. Jacutin-Porte, K. Kish, M. Pokross, H. A. Lewis, N. Ahmed, N. Szapiel, J. A. Newitt and E. T. Baldwin, Discovery of new acylaminopyridines as GSK-3 inhibitors by a structure guided in-depth exploration of chemical space around a pyrrolopyridinone core, Bioorg. Med. Chem. Lett., 2015, 25(9), 1856–1863 CrossRef CAS PubMed.
- H. Nassar, W. Sippl, R. A. Dahab and M. Taha, Molecular docking, molecular dynamics simulations and in vitro screening reveal cefixime and ceftriaxone as GSK3β covalent inhibitors, RSC Adv., 2023, 13(17), 11278–11290 RSC.
- Y. Dong, J. Xu, B. Sun, J. Wang and Z. Wang, MET-targeted therapies and clinical outcomes: A systematic literature review, Mol. Diagn. Ther., 2022, 26, 203–227 CrossRef.
- H. Lei, G. Hu, Y. Wang, P. Han, Z. Liu, Y. Zhao and P. Gong, Design, Synthesis, and Biological Evaluation of 4-Phenoxyquinoline Derivatives Containing Benzo [d] thiazole-2-yl Urea as c-Met Kinase Inhibitors, Arch. Pharm., 2016, 349(8), 651–661 CrossRef CAS.
- A. E. Abdallah, R. M. Mohareb and E. A. Ahmed, Novel Pyrano [2, 3-d] thiazole and Thiazolo [4, 5-b] pyridine Derivatives: One-pot Three-component Synthesis and Biological Evaluation as Anticancer Agents, c-Met, and Pim-1 Kinase Inhibitors, J. Heterocycl. Chem., 2019, 56(11), 3017–3029 CrossRef CAS.
- X. Nan, Q. X. Wang, S. J. Xing and Z. G. Liang, Design, synthesis, and biological evaluation of thiazole/thiadiazole carboxamide scaffold-based derivatives as potential c-Met kinase inhibitors for cancer treatment, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 2247183 CrossRef PubMed.
- X.-J. Liu, H.-C. Zhao, S.-J. Hou, H.-J. Zhang, L. Cheng, S. Yuan, L.-R. Zhang, J. Song, S.-Y. Zhang and S.-W. Chen, Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy, Bioorg. Chem., 2023, 13, 3106425 Search PubMed.
- U. Bhanushali, S. Rajendran, K. Sarma, P. Kulkarni, K. Chatti, S. Chatterjee and C. S. Ramaa, 5-Benzylidene-2,4-thiazolidenedione derivatives: design, synthesis and evaluation as inhibitors of angiogenesis targeting VEGR-2, Bioorg. Chem., 2016, 67, 139–147 CrossRef CAS PubMed.
- T. Al-Warhi, M. Abualnaja, O. A. Abu Ali, N. M. Alyamani, F. G. Elsaid, A. A. Shati, S. Albogami, E. Fayad, A. H. Abu Almaaty, K. O. Mohamed and W. M. Alamoudi, Design, Synthesis and Cytotoxicity Screening of New Thiazole Derivatives as Potential Anticancer Agents through VEGFR-2 Inhibition, Symmetry, 2022, 14(9), 1814 CrossRef CAS.
- M. M. Al-Sanea, A. Hamdi, A. A. Mohamed, H. W. El-Shafey, M. Moustafa, A. A. Elgazar, W. M. Eldehna, H. Ur Rahman, D. G. Parambi, R. M. Elbargisy and S. Selim, New benzothiazole hybrids as potential VEGFR-2 inhibitors: design, synthesis, anticancer evaluation, and in silico study, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 2166036 CrossRef.
- A. Sandor, I. Fizesan, I. Ionut, G. Marc, C. Moldovan, I. Oniga, A. Pîrnău, L. Vlase, A. E. Petru, I. Macasoi and O. Oniga, Discovery of A Novel Series of Quinazoline– Thiazole Hybrids as Potential Antiproliferative and Anti-Angiogenic Agents, Biomolecules, 2024, 14(2), 218 CrossRef CAS.
- Y. Liu, C. Chen, C. Rong, X. He and L. Chen, Anaplastic lymphoma kinase tyrosine kinase inhibitor-associated cardiotoxicity: a recent five-year pharmacovigilance study, Front. Pharmacol., 2022, 13, 858279 CrossRef CAS PubMed.
- Z. Liu, X. Yue, Z. Song, X. Peng, J. Guo, Y. Ji, Z. Cheng, J. Ding, J. Ai, M. Geng and A. Zhang, Design, synthesis and pharmacological evaluation of 2-(thiazol-2-amino)-4- arylaminopyrimidines as potent anaplastic lymphoma kinase (ALK) inhibitors, Eur. J. Med. Chem., 2014, 86, 438–448 CrossRef CAS PubMed.
- Q. Huang, T. W. Johnson, S. Bailey, A. Brooun, K. D. Bunker, B. J. Burke, M. R. Collins, A. S. Cook, J. J. Cui, K. N. Dack and J. G. Deal, Design of potent and selective inhibitors to overcome clinical anaplastic lymphoma kinase mutations resistant to crizotinib, J. Med. Chem., 2014, 57(4), 1170–1187 CrossRef CAS.
- B. Acharya, D. Saha, D. Armstrong, N. R. Lakkaniga and B. Frett, FLT3 Inhibitors for acute myeloid leukemia: successes, defeats, and emerging paradigms, RSC Med. Chem., 2022, 13, 798–816 RSC.
- C. Novartis, Thiazole and pyrazole derivatives as Flt-3 kinase inhibitors, WO Pat. 2005047273, 2005 Search PubMed.
- X. D. Lin, H. W. Yang, S. Ma, W. W. Li, C. H. Zhang, W. J. Wang, R. Xiang, L. L. Li and S. Y. Yang, Discovery of 6-phenylimidazo [2, 1-b] thiazole derivatives as a new type of FLT3 inhibitors, Bioorg. Med. Chem. Lett., 2015, 25(20), 4534–4538 CrossRef CAS PubMed.
- Z. Zhang, J. Li, S. Jiao, G. Han, J. Zhu and T. Liu, Functional and clinical characteristics of focal adhesion kinases in cancer progression, Front. Cell Dev. Biol., 2022, 10, 1040311 CrossRef PubMed.
- B. J. Groendyke, B. Nabet, M. L. Mohardt, H. Zhang, K. Peng, E. Koide, C. R. Coffey, J. Che, D. A. Scott, A. J. Bass and N. S. Gray, Discovery of a pyrimidothiazolodiazepinone as a potent and selective focal adhesion kinase (FAK) inhibitor, ACS Med. Chem. Lett., 2020, 12(1), 30–38 CrossRef.
- F. Basoğlu, N. Ulusoy-Güzeldemirci, G. Akalın-Çiftçi, S. Çetinkaya and A. Ece, Novel imidazo [2, 1-b] thiazole-based anticancer agents as potential focal adhesion kinase inhibitors: Synthesis, in silico and in vitro evaluation, Chem. Biol. Drug Des., 2021, 98(2), 270–282 CrossRef.
- S. L. Lin, C. Y. Lin, W. Lee, C. F. Teng, W. C. Shyu and L. B. Jeng, Mini review: molecular interpretation of the IGF/IGF-1R Axis in cancer treatment and stem cells-based therapy in regenerative medicine, Int. J. Mol. Sci., 2022, 23(19), 11781 CrossRef CAS.
- S. D. Fidanze, S. A. Erickson and G. T. Wang, et al, Imidazo[2,1-b]thiazoles: Multi-targeted inhibitors of both the insulin-like growth factor receptor and members of the epidermal growth factor family of receptor kinases, Bioorg. Med. Chem. Lett., 2010, 20, 2452–2455 CrossRef CAS PubMed.
- R. Tandon, S. Kapoor and S. Vali, et al, Dual epidermal growth factor receptor (EGFR)/insulin-like growth factor-1 receptor (IGF-1R) inhibitor: a novel approach for overcoming resistance in anticancer treatment, Eur. J. Pharmacol., 2011, 67, 56–65 CrossRef.
- P. K. Gadekar, G. Urunkar, A. Roychowdhury, R. Sharma, J. Bose, S. Khanna, A. Damre and S. Sarveswari, Design, synthesis and biological evaluation of 2, 3- dihydroimidazo [2, 1-b] thiazoles as dual EGFR and IGF1R inhibitors, Bioorg. Chem., 2021, 115, 105151 CrossRef CAS PubMed.
- B. Rah, R. A. Rather, G. R. Bhat, A. B. Baba, I. Mushtaq, M. Farooq, T. Yousuf, S. B. Dar, S. Parveen, R. Hassan and F. Mohammad, JAK/STAT signaling: molecular targets, therapeutic opportunities, and limitations of targeted inhibitions in solid malignancies, Front. Pharmacol., 2022, 13, 821344 CrossRef CAS PubMed.
- K. Sanachai, T. Aiebchun, P. Mahalapbutr, S. Seetaha, L. Tabtimmai, P. Maitarad, I. Xenikakis, A. Geronikaki, K. Choowongkomon and T. Rungrotmongkol, Discovery of novel JAK2 and EGFR inhibitors from a series of thiazole-based chalcone derivatives, RSC Med. Chem., 2021, 12(3), 430–438 RSC.
- S. Godesi, J. Lee, H. Nada, G. Quan, A. Elkamhawy, Y. Choi and K. Lee, Small Molecule c-KIT Inhibitors for the Treatment of Gastrointestinal Stromal Tumors: A Review on Synthesis, Design Strategies, and Structure–Activity Relationship (SAR), Int. J. Mol. Sci., 2023, 24(11), 9450 CrossRef CAS PubMed.
- Y. Jin, K. Ding, D. Wang, M. Shen and J. Pan, Novel thiazole amine class tyrosine kinase inhibitors induce apoptosis in human mast cells expressing D816V KIT mutation, Cancer Lett., 2014, 353(1), 115–123 CrossRef CAS PubMed.
- R. Paul, D. Dutta, T. Das, M. Debnath and J. Dash, G4 Sensing Pyridyl-Thiazole Polyamide Represses c-KIT Expression in Leukemia Cells, Chem.–Eur. J., 2021, 27(33), 8590–8599 CrossRef CAS PubMed.
- A. Sebastian, V. Pandey, C. D. Mohan, Y. T. Chia, S. Rangappa, J. Mathai, C. P. Baburajeev, S. Paricharak, L. H. Mervin, K. C. Bulusu, J. E. Fuchs, A. Bender, S. Yamada, Basappa, P. E. Lobie and K. S. Rangappa, Novel adamantanyl-based thiadiazolyl pyrazoles targeting EGFR in triple-negative breast cancer, ACS Omega, 2016, 1412–1424 CrossRef CAS PubMed.
- P. C. Lv, D. D. Li, Q. S. Li, X. Lu, Z. P. Xiao and H. L. Zhu, Synthesis, molecular docking and evaluation of thiazolyl-pyrazoline derivatives as EGFR TK inhibitors and potential anticancer agents, Bioorg. Med. Chem. Lett., 2011, 21(18), 5374–5377 CrossRef CAS.
- X. Deng, T. Tan, Q. An, Z. Ma, C. Jin, Q. Wang and C. Meng, Hum, Synthesis, characterization, and biological activity of a novel series of benzo [4, 5] imidazo [2, 1-b] thiazole derivatives as potential epidermal growth factor receptor inhibitors, Molecules, 2019, 24(4), 682 CrossRef CAS PubMed.
- A. M. Srour, N. S. Ahmed, S. S. Abd El-Karim, M. M. Anwar and S. M. El-Hallouty, Design, synthesis, biological evaluation, QSAR analysis and molecular modelling of new thiazol-benzimidazoles as EGFR inhibitors, Bioorg. Med. Chem., 2020, 28(18), 115657 CrossRef CAS PubMed.
- T. Al-Warhi, A. M. El Kerdawy, M. A. Said, A. Albohy, Z. M. Elsayed, N. Aljaeed, E. B. Elkaeed, W. M. Eldehna, H. A. Abdel-Aziz and M. A. Abdelmoaz, Novel 2-(5-Aryl-4, 5-dihydropyrazol-1-yl) thiazol-4-one as EGFR inhibitors: synthesis, biological assessment and molecular docking insights, Drug Des., Dev. Ther., 2023, 1457–1471 Search PubMed.
- M. S. Raghu, H. A. Swarup, T. Shamala, B. S. Prathibha, K. Y. Kumar, F. Alharethy, M. K. Prashanth and B. H. Jeon, Design, synthesis, anticancer activity and docking studies of novel quinazoline-based thiazole derivatives as EGFR kinase inhibitors, Heliyon, 2023, 9(9), e20300 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |