
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhenacylselenoesters allow facile selenium transfer and hydrogen selenide generation†
Utsav Dey
Sarkar‡
 ,
Mahima
Rana‡
and
Harinath
Chakrapani
*
,
Mahima
Rana‡
and
Harinath
Chakrapani
*
Department of Chemistry, Indian Institute of Science Education and Research Pune, Maharashtra, India. E-mail: harinath@iiserpune.ac.in
First published on 29th October 2024
Abstract
Hydrogen selenide (H2Se) is a precursor to several selenium-containing biomolecules and is emerging as an important redox-active species in biology, with yet to be completely characterized roles. Tools that reliably generate H2Se are key to achieving a better understanding of selenium biology. Here, we report the design, synthesis and evaluation of phenacylselenoesters as sources of H2Se. These compounds are prepared in two steps from commercial compounds, some are crystalline solids, and all are stable during storage. In the presence of esterase and a thiol in pH 7.4 buffer, these compounds produce H2Se, with half-lives of 5–20 min. We developed a colorimetric assay for the detection of gaseous H2Se by trapping it as zinc selenide (ZnSe), which is then converted to lead selenide (PbSe), which serves as a convenient visual indicator for this gas. The major organic products that are formed in nearly quantitative yields are relatively benign ketones and carboxylic acids. We provide evidence for these donors producing a thioselenide, a key intermediate in biological selenium metabolism. Finally, we compared sulfur and selenium transfer, both critical processes in cells. Phenacylthiol is relatively stable to cleavage by a thiol, and requires a sulfurtransferase enzyme to produce a persulfide and H2S. By contrast, the selenium analogue reacted with a thiol in the absence of this enzyme to produce H2Se. This result underscores the greater lability of the C–Se bond as compared with a C–S bond, and may have implications in biological selenium transfer. Together, phenacylselenoesters are easy to prepare, stable and generate H2Se under mild and biocompatible conditions. We anticipate that these will be valuable additions to the growing selenium redox toolbox.
Introduction
Selenium is an essential element, and selenocysteine is a vital component of proteins such as glutathione peroxidase (GPx), thioredoxin reductase (TrxR), and selenophosphate synthetase (SPS1 and SPS2), and is directly involved in their function.1–3 Selenoproteins also have a central role in stress response to reactive nitrogen species (RNS) and reactive oxygen species (ROS).4 Selenoenzymes, e.g., thioredoxin reductases, can reduce nucleotides during DNA synthesis and help regulate the intracellular redox state, and thyroid levels.5,6 Selenium exists in multiple oxidation states, and dietary selenium sources are converted to a key intermediate, hydrogen selenide (H2Se), for further conversion to selenocysteine, selenouridine, methylselenide, and other selenium-containing biomolecules (Fig. 1A).7–9 The reduction of environmental selenite (SeO32−) by glutathione (GSH) produces intermediate GS-Se-SG, which is reductively cleaved to generate H2Se through the thioselenide, GS-SeH as an intermediate (Fig. 1A).10 Thioselenides are also produced during the metabolism of selenocysteine into alanine and H2Se. Their sulfur counterparts, persulfides (RS-SH) have already been established as key biological intermediates, and the field of sulfur biology has benefitted from the large variety of tools at its disposal.11–13 Similarly, for selenium redox biology to grow, generating H2Se and related intermediates is crucial.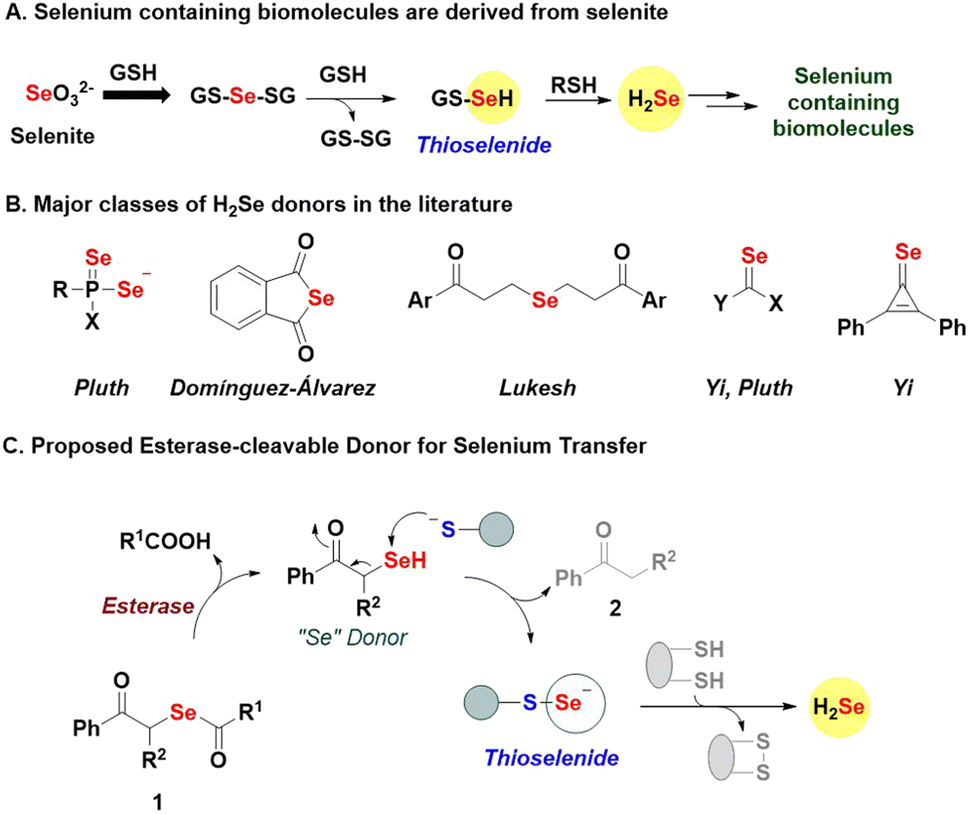 | ||
| Fig. 1 (A) Selenium containing biomolecules are derived from the reduction of inorganic SeO32− by glutathione (GSH) to generate the thioselenide, gluathioselenol GS-SeH. Cleavage of this intermediate by biothiols or reductases gives H2Se. (B) Major classes of hydrogen selenide donors reported in the literature. (C) Proposed esterase-activated thioselenide generation involves the cleavage of the selenoester bond to produce the donor, which can react with a thiol to produce a thioselenide and a ketone. Under reducing conditions, H2Se is formed from the thioselenide, or when the thioselenide is exposed to other cysteine-containing proteins, a protein thioselenide results. | ||
Several strategies to generate H2Se are known, and can be broadly classified based on the mechanism of activation, i.e., hydrolysis,14–17 pH,9 and thiol-activated donors,18,19 and combinations thereof (Fig. 1B).20,21 While these donors are useful, several release H2Se extremely slowly, some are relatively unstable and others generate relatively toxic byproducts. Here, we report phenacylselenoesters (1, Fig. 1C) as a new class of H2Se donors that are simple to prepare, stable, and capable of generating H2Se and transferring “Se” under mild and biocompatible conditions.
Phenacylselenoesters are expected to be cleaved by the enzyme esterase (ES) to produce a phenacylselenol (PhCOCH2SeH, “Se” donor, Fig. 1C).22,23 The key step is the reaction of the “Se” donor with a thiol to produce a thioselenide species and a ketone 2. Thioselenides can be reduced to generate H2Se or transfer selenium to a cysteine-containing protein to produce a protein thioselenide. The rate of selenium transfer can, in principle, be modulated by changing the ester (R1), which would alter the rate of hydrolysis by esterase, or by introducing sterics through the substituent R2. Together, this class of donors offers a robust and broadly applicable method for generating important selenium intermediates whose relevance in biology is being unravelled.
Results and discussion
Synthesis and characterization
The desired compounds 1a–1c were prepared in two steps – first, treatment of the acid chloride (R1COCl) with selenium powder and sodium borohydride produced R1COSe− as an intermediate, which was not isolated but directly reacted with the α-haloketone (R2COCH2Br) to give the desired phenacylselenoesters (Scheme 1).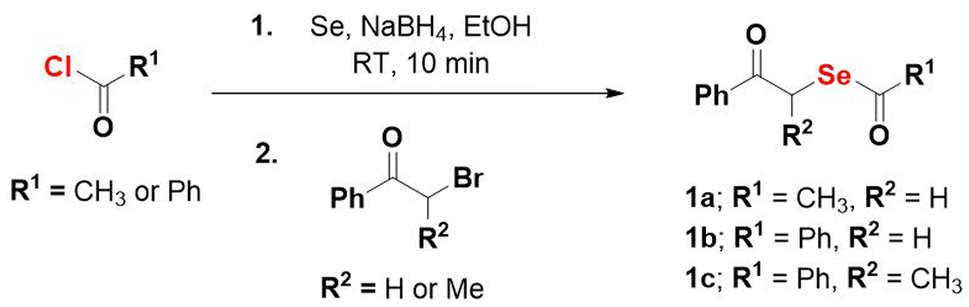 | ||
| Scheme 1 Synthesis of selenium donors 1a, 1b and 1c. Overall yields were 28–58%. | ||
Purification of the final compounds was performed using silica-gel column chromatography and yields ranged from 28 to 58%. The compound 1b was crystallised via a slow-cooling method and the structure of the molecule was confirmed through single-crystal X-ray diffraction measurements (Fig. 2).
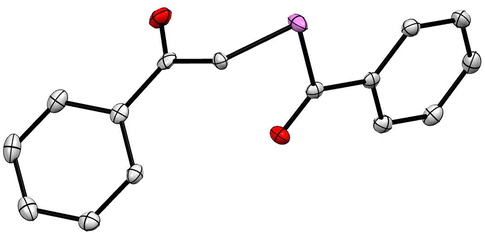 | ||
| Fig. 2 ORTEP diagram of 1b; H-atoms are not shown for clarity (CCDC number 2326967). Details of the crystallographic refinement parameters are listed in the ESI, Tables S1–S4.† Both the C–Se bond distances were found to be 1.94 Å, which is consistent with literature reports.45,46 | ||
Hydrogen selenide generation
In the presence of porcine liver esterase (ES), the donor 1 is expected to be cleaved to produce the phenacylselenol, which should then react with a thiol to produce a thioselenide (RS-Se−). This species in the presence of a bis-thiol such as dithiothreitol (DTT) should produce H2Se (Fig. 3A). Since DTT can play both roles, i.e., that of a selenium acceptor as well as an H2Se generator, we initially studied the reaction with ES and DTT. Previously, a lead acetate-based assay was used to estimate H2Se.24–27 Here, under acidic conditions, H2Se reacts with lead acetate to produce lead selenide (PbSe), which has a distinct absorbance at 400 nm.25–27 We modified this protocol by incorporating zinc acetate as an additive in pH 7.4, which traps the H2Se formed as ZnSe (Fig. 3A). An aliquot of the reaction mixture is then treated with lead acetate and HCl, and a brownish black PbSe confirms H2Se formation (Fig. 3A). Previously, the formation of H2Se has been shown through the use of sodium selenite in the presence of GSH.28 We confirmed the formation of PbSe under these conditions: Na2SeO3 (100 μM), GSH (400 μM), DTT (200 μM) and Zn(OAc)2 (400 μM) in pH 7.4 followed by treatment with lead acetate gave a brown coloration and significant absorbance at 400 nm (Fig. 3B, see the ESI, Fig. S1†).7,28 Using this revised protocol, the selenoesters (100 μM) were independently treated with ES (0.1 U mL−1) and DTT (200 μM) in pH 7.4 buffer and a significant increase in absorbance at 400 nm was seen for all three compounds in the presence of ES and DTT (Fig. 3B). In the presence of ES alone or DTT alone, only a small increase in PbSe was seen suggesting that the reaction to produce H2Se requires both ES and DTT (Fig. 3B). Furthermore, the PbSe yield depended on the concentration of DTT as well as ES (see the ESI, Fig. S2†). To unambiguously confirm the formation of lead (Pb) and selenium (Se), the precipitate formed in the reaction with 1b was analysed by energy-dispersive X-ray spectroscopy (EDXS) analysis. A significant component of both Pb and Se was observed in the mixture, supporting the generation of PbSe under these conditions (see the ESI, Fig. S3†).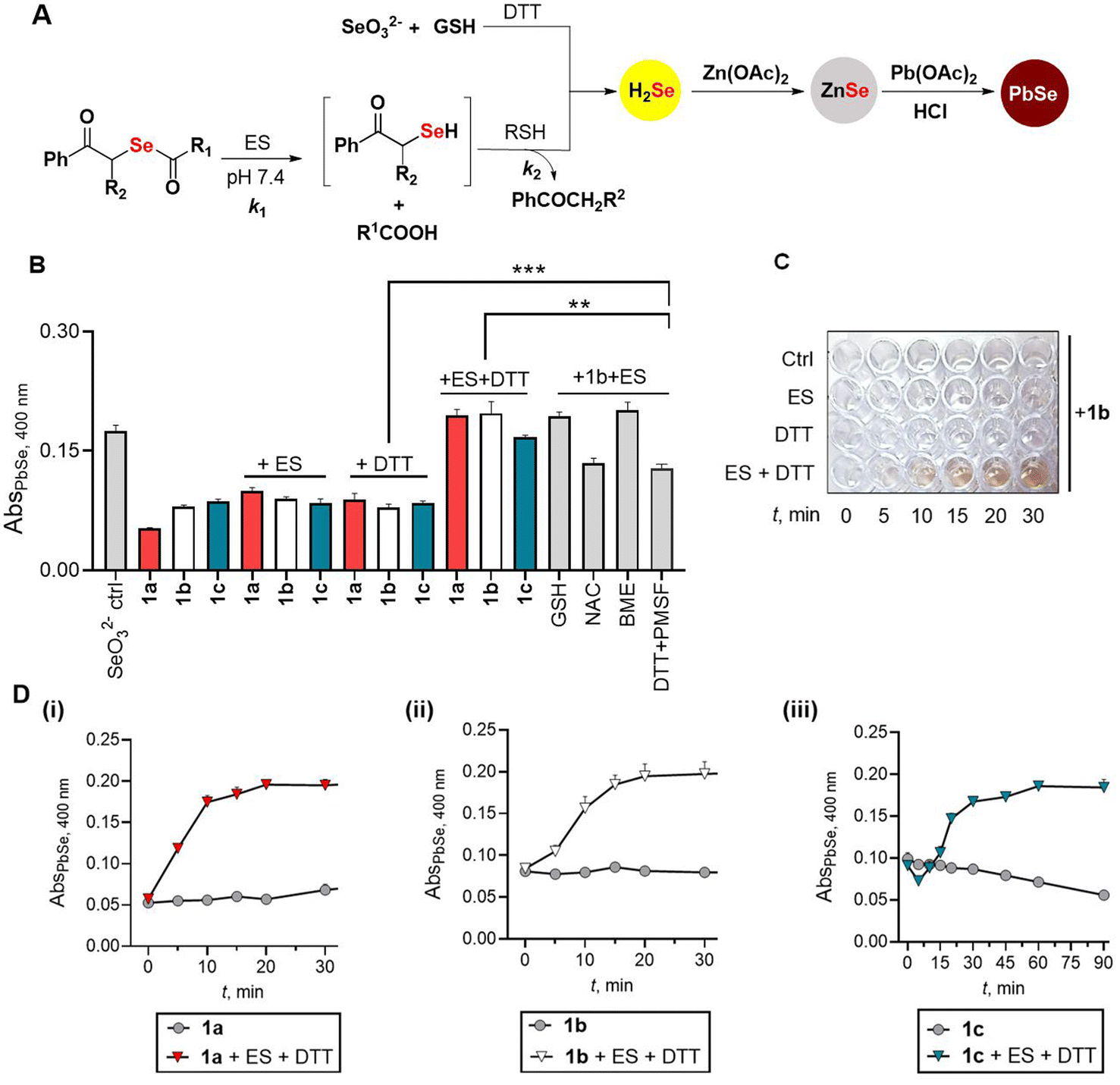 | ||
| Fig. 3 (A) Workflow for the detection of hydrogen selenide as PbSe. Absorbance at 400 nm was measured. The reaction of selenite and glutathione (GSH) is expected to produce GS-Se-SG, which reacts with DTT to generate H2Se; 1 is similarly expected to react with ES and DTT to generate H2Se. (B) PbSe assay of 1a, 1b and 1c (100 μM) with ES and different thiols (DTT = 200 μM, other thiols = 400 μM) in the presence of 400 μM of Zn(OAc)2 at 30 min. Addition of ES inhibitor PMSF (1 mM) decreases H2Se production. Student's two-tailed unpaired parametric t-test was used to determine significance: **p < 0.01, ***p < 0.001. All the results are represented as mean ± SD (n = 3 per group). SD stands for standard deviation. (C) A 96-well plate image during the reaction of 1b with ES and DTT, in the presence of zinc acetate: Ctrl is 1b (100 μM) alone, ES: ES (0.1 U mL−1), DTT indicates 1b + DTT (200 μM), and ES + DTT signifies the incubation of 1b, ES, and DTT together. (D) Time courses for the formation of PbSe during incubation of the compound alone or under the standard reaction conditions. All data are represented as mean ± SD (n = 3 per group). Data for: (i) 1a, (ii) 1b and (iii) 1c. | ||
Next, a time course study (snapshot of 1b is shown in Fig. 3C) showed a gradual increase in absorbance at 400 nm for all three compounds tested (Fig. 3D). Curve fitting of the formation of H2Se from 1a gave a rate constant kobs of 0.13 min−1 (see the ESI, Fig. S4†). Similarly, kinetic analysis of 1b gave kobs of 0.09 min−1 (see the ESI, Fig. S5†). In the case of 1c, a significant lag in H2Se generation was seen and overall, this compound was the slowest among the three tested (kobs = 0.03 min−1, see the ESI, Fig. S6†).
When the reaction with 1b and ES was conducted in the presence of other biothiols such as glutathione (GSH) or N-acetylcysteine (NAC), the liberation of H2Se was seen with the yield of H2Se being higher in the case of GSH (Fig. 3B). The reaction in the presence of β-mercaptoethanol gave a yield comparable to DTT. Hence, the yield of H2Se appeared to depend on the nature of the thiol acceptor. In the presence of GSH alone, 1b yielded H2Se, but at a slower rate when compared with 1b in the presence of GSH and ES (see ESI Scheme S1 and Fig. S7†). This is likely due to the cleavage of the selenoester in the presence of thiols. At higher concentrations of GSH (1 mM), the profile was similar to the standard reaction conditions (see ESI Fig. S7†).
We tested if H2Se was formed in the presence of amino acids without a sulfhydryl group, and in none of the cases, we see H2Se production (see ESI Fig. S8†) even in the presence of ES suggesting that a thiol was necessary to generate H2Se (see ESI Fig. S9†). To understand the role of ES, we used phenylmethylsulfonyl fluoride (PMSF), an inhibitor of ES.29 Under the standard reaction conditions, in the presence of PMSF, the yield of PbSe was diminished (Fig. 3B) suggesting the dependence of H2Se generation on the cleavage of the selenoester by esterase, although ES can be replaced with elevated concentrations of a thiol such as GSH. Next, we attempted to understand if the compound was able to generate H2Se in the presence of bovine serum albumin (BSA). However, in the presence of zinc acetate, we find significant coagulation of BSA, which renders estimation of PbSe difficult (see the ESI, Fig. S10†). In the absence of zinc acetate, an orange coloration was seen when 1b was treated with BSA; this result is consistent with elemental selenium formation through the generation of H2Se that was independently confirmed by sodium selenite and GSH (see ESI Fig. S11†).30
In recent years, colorimetric probes15,18 and fluorescence-based techniques31,32 have been employed for H2Se detection. Additionally, the released gaseous H2Se has also been captured by electrophiles and analyzed.9,14,15 The new modified protocol uses reagents that are inexpensive, and we anticipate that our protocol will be a convenient method for the estimation of H2Se.
Stability and decomposition profiles
To provide further insights into the reaction mechanism, the products of cleavage of 1b (Fig. 4A) were assessed using HPLC analysis. The selenoester 1b is expected to be hydrolysed to produce the intermediate phenacylselenol, which is in equilibrium with its corresponding conjugate base, the phenacylselenolate anion (pKa value of the selenol ∼ 5–6).33 Further reaction of this intermediate with a thiol produces H2Se and acetophenone. When 1b was incubated under the standard reaction conditions, disappearance of the selenoester, along with a nearly concomitant formation of benzoic acid, was observed (Fig. 4B and C).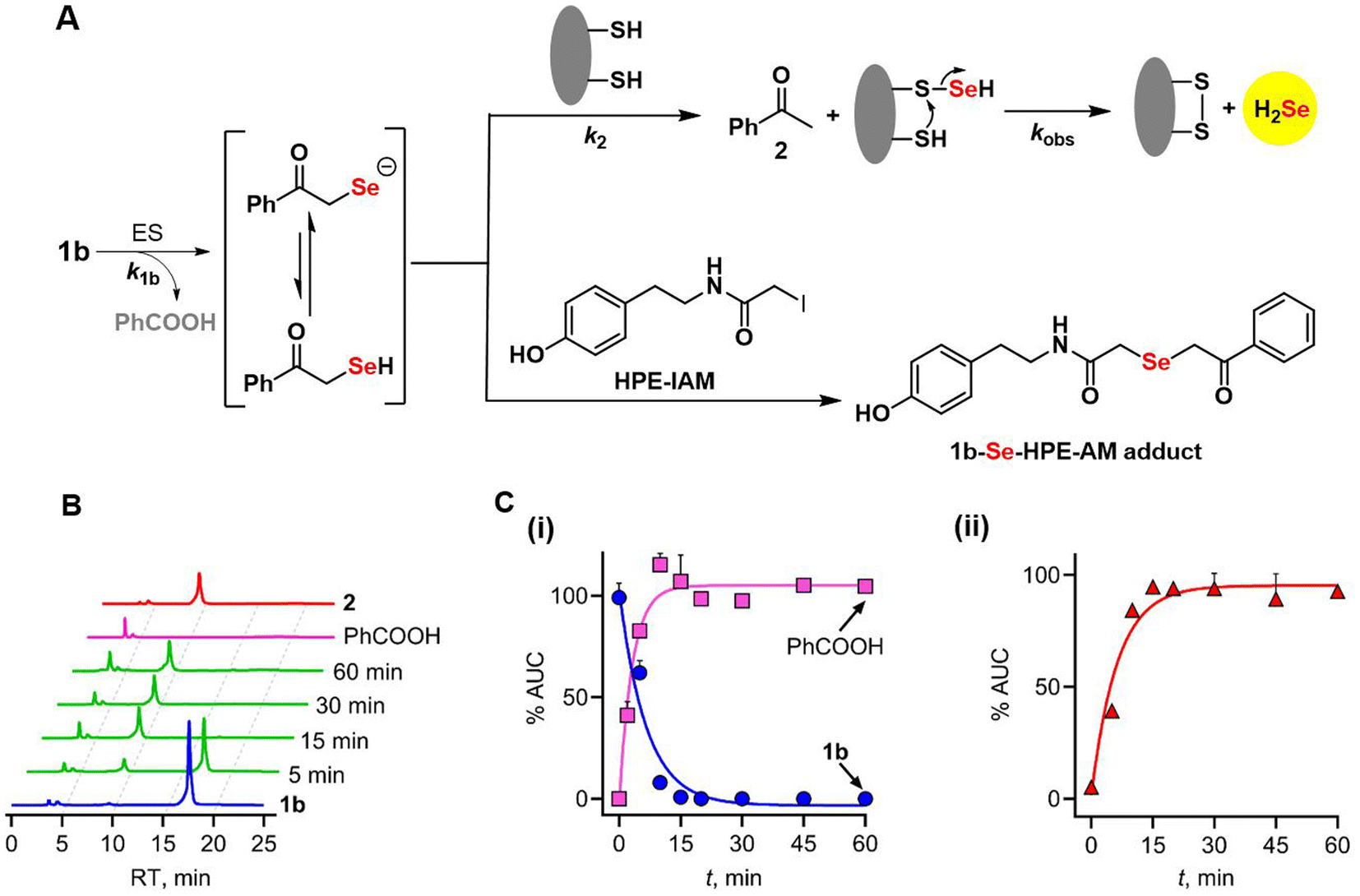 | ||
| Fig. 4 (A) Mechanism of cleavage of 1b in the presence of ES and DTT produces acetophenone 2 and H2Se. (B) Representative HPLC chromatograms of 1b (100 μM) in the presence of ES (0.1 U mL−1), DTT (200 μM) and zinc acetate (400 μM), showing the formation of PhCOOH and acetophenone 2 over 30 min. (C) Curve fitting gave (i) the apparent rate constant for decomposition of 1b, k1b of 0.15 min−1 and the formation of PhCOOH as 0.29 min−1 and (ii) apparent rate constant for the formation of 2 was found to be 0.16 min−1. | ||
The rate of disappearance (k1b) of 1b was estimated to be 0.15 min−1; the formation of benzoic acid was comparable (0.29 min−1) in magnitude while the yield of benzoic acid was >90% (see the ESI, Fig. S12†), supporting an efficient cleavage of the selenoester. The rate of acetophenone formation, k2, was determined to be 0.16 min−1, which again is nearly identical to k1b, and the yield of acetophenone was nearly quantitative. Based on these data, the yield of H2Se is estimated to be >90% as well.9 The kobs for H2Se generation from 1b was 0.09 min−1, which is comparable to k1b. The rate of formation of acetophenone being nearly identical to the rate of loss of 1b implies that phenacylselenol does not have a significant lifetime under these conditions. This short-lived intermediate was trapped in the presence of an electrophile (HPE-IAM) and detected by mass spectrometry (Fig. 4A, see the ESI, Scheme S2, Fig. S13†).
Selenoester 1b itself was stable in pH 7.4 buffer for 60 min (see the ESI, Fig. S14†), as well as pH 5.0 buffer (see ESI Fig. S15†). Due to aqueous solubility issues, these experiments were conducted in 50% MeCN. In pH 9 buffer (with 50% ACN), we found that the compound was stable for 30 min, but significant loss was observed in 60 min (see the ESI, Fig. S16†). However, we found no other major organic product being formed, and hence, the compound is likely precipitating out in alkaline buffer during extended durations. The selenoester was also found to be stable in the presence of H2O2 in MeCN (TLC analysis for 1a, 1b and 1c, see the ESI, Fig. S17†).34 Overall, the mechanistic picture is consistent with the cleavage of the selenoester by ES to produce an intermediate selenol, which reacts with DTT to produce H2Se. Previously, as a part of a structure–activity relationship study, selenoanhydride and selenoesters have been synthesized by Domínguez-Álvarez and co-workers and they demonstrated cytotoxicity and apoptosis induction in mouse T-lymphoma cells.35 However, it is unclear if these compounds generated H2Se. Based on our findings, we predict that these compounds will release H2Se under cell culture conditions used in their study, in the presence of esterases and biothiols.
Next, we evaluated the effect of introduction of a methyl group adjacent to the selenoester functional group on the decomposition profile. Under the standard reaction conditions, a gradual loss of 1c (k1c = 0.11 min−1) and the formation of benzoic acid as well as propiophenone over 60 min was observed (Fig. 5).
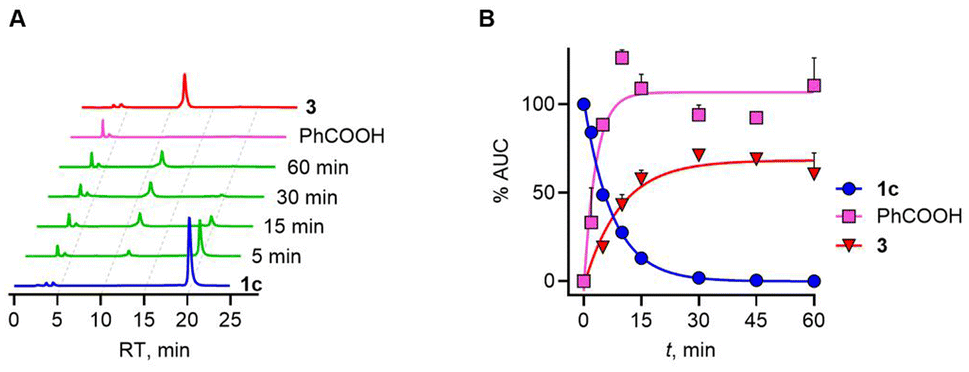 | ||
| Fig. 5 (A) Representative HPLC chromatograms of 1c (100 μM) in the presence of ES (0.1 U mL−1), DTT (200 μM) and zinc acetate (400 μM) over 60 min. (B) Kinetic representation of HPLC traces used for measuring the rate of (i) decomposition of 1c and formation of PhCOOH. (ii) Formation of propiophenone 3. Standard curve fitting using the equation gave apparent rate constants of loss of 1c = 0.11 min−1, formation of benzoic acid = 0.42 min—1, and formation of 3 = 0.11 min−1. | ||
The yield of benzoic acid was nearly quantitative under these conditions and this process is slightly slower. The rate of formation of propiophenone, which closely resembled the rate at which H2Se is generated, again supports no major accumulation of an intermediate. The presence of the methyl group seems to have no significant effect on the rate. Overall, the analysis shows that the rate-determining step is the cleavage of the selenoester to yield the selenol, which then reacts with a thiol to eventually generate H2Se through a thioselenide. This finding supports further analogue synthesis that can focus on varying rates of selenoester hydrolysis. Finally, 1a was found to be labile in pH 7.4 buffer and does not need esterase to be cleaved, and was not analysed further. Hence, the presence of a benzoate protective group provides better stability towards hydrolysis.
Sulfur and selenium transfer
Chalcogens have many similarities36 and hence discriminating between S and Se in biology is challenging.24,37 Incorporation of selenium by enzymes is a very specific process.38 However, in some cases, enzymes involved in sulfur metabolism do not distinguish between these elements resulting in a high Se-to-S ratio. The mechanisms of these processes are not well understood. Sulfur transfer by sulfur transferases has several important roles in biology, but little is known about selenium transfer. We investigated biochemical sulfur and selenium transfer using an enzyme 3-mercaptopyruvate sulfurtransferase (3-MST, see the ESI, Fig. S18†), which generates persulfide and hydrogen sulfide, and is involved in the persulfidation of several proteins in cells.39–41 This enzyme has a rhodanese domain and an active-site cysteine residue.42 We recently reported that the sulfur counterpart phenacylthioester 4, in the presence of ES, generated an artificial substrate 5 for 3-MST (Fig. 6A).43,44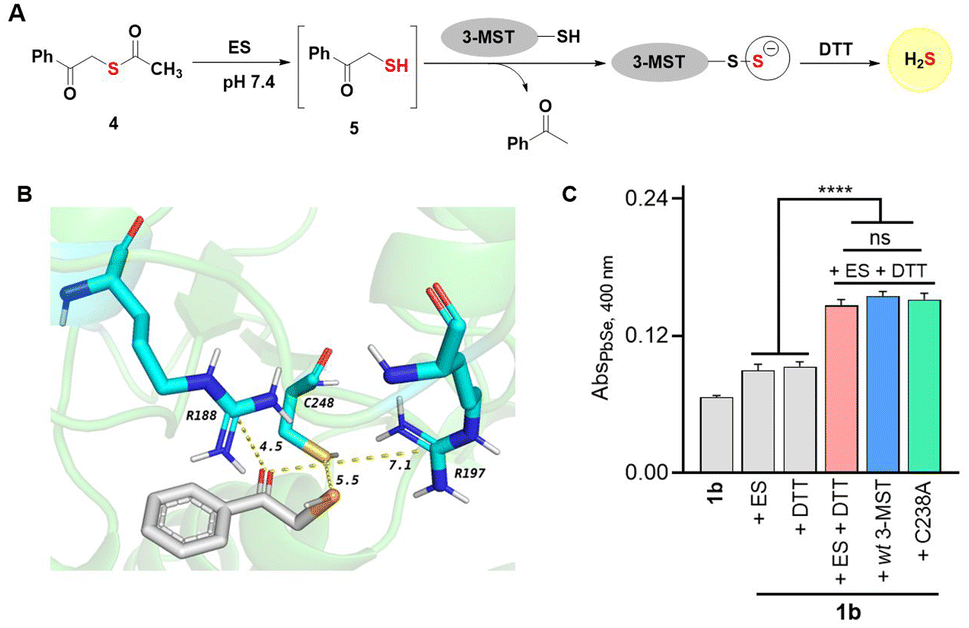 | ||
| Fig. 6 (A) Thioacetate 4 is cleaved by ES to produce the designed 3-MST substrate 5. The thiol is positioned to undergo a sulfur transfer reaction to produce persulfidated 3-MST (3-MST-SS−) along with acetophenone as a by-product. The 3-MST-SS− reacts with dithiothreitol (DTT) to generate H2S. (B) Molecular docking analysis of the active site of h3-MST with phenacylselenol intermediate derived from 1b. The distance between the sulfhydryl group of the active site cysteine residue and the Se is 5.5 Å. The R188 and R197 residues are also 4.5 Å and 7.1 Å from the carbonyl group, respectively. (C) Detection of H2Se using modified PbSe assay; no significant difference is observed between the background reaction of 1b with ES and DTT, and the reaction in the presence of wt 3-MST. The C238A single mutant also shows a similar response for H2Se generation. Analysis was carried out after 30 min. All data represented as mean ± SD (n = 3 per group). Student's two-tailed unpaired parametric t-test was carried out to determine significance. ****p < 0.0001 for the comparison between the control reactions vs. the reactions of 1b with ES and DTT alone, or with the addition of either E. coli wt 3-MST or C238A mutant. | ||
In the presence of 3-MST, ES and DTT, 5 produced H2S. The key step in this reaction is the cleavage of the C–S bond to produce the persulfidated 3-MST, i.e., (3-MST)-S-S− (Fig. 6A). The compound 5 in the presence of ES and DTT, but in the absence of 3-MST, did not generate H2S, suggesting that the C–S bond was not labile in the presence of nucleophiles.43 Since the electronegativities of S and Se are comparable, we evaluated if 3-MST could catalyse the cleavage of the C–Se bond under these conditions, presumably to produce a protein thioselenide as an intermediate. Molecular docking analysis of the phenacylselenol showed a favorable binding and proximity of the Se to the active site cysteine residue, much like the sulfur counterpart (Fig. 6B).39 Also, the active site arginine residues that are known to bind to the carbonyl group of 3-mercaptopyruvate are also at a favorable distance (see the ESI, Table S8†).41 If 1b was cleaved to produce a substrate for 3-MST, we would expect a higher yield of H2Se when treated with 3-MST. However, when 1b was reacted with 3-MST in the presence of ES and DTT, no significant difference between the background reaction and the reaction in the presence of 3-MST was seen (Fig. 6C and ESI, Fig. S20†).
We next studied the role of the active site cysteine, if any. The cysteine residue was replaced with alanine (C238A) and this mutant when tested under our standard reaction conditions also showed a similar time course for H2Se generation (see the ESI, Fig. S19†). These data support the lack of involvement of 3-MST in this process. Hence it appears that C–Se is significantly more labile than its C–S counterpart. This is likely due to the higher polarizability of Se and the longer C–Se bond length when compared with sulfur.45–47 The phenacylselenol is expected to be primarily in the anionic form (PhCOCH2Se−) in neutral pH, and hence resistant to attack by the cysteine of 3-MST. Alternatively, 3-MST may be specific in its reaction with sulfur transfer and may not have a propensity to react with a selenium center. These aspects will need to be further investigated and selenium transfer by sulfurtransferases will need to be examined in detail.
Finally, we evaluated if the donor 1b was capable of transferring selenium to glutathione to produce a thioselenide (Fig. 1A). This experiment with 1b was carried out in the presence of ES, GSH, and GS-SG (see the ESI, Scheme S3†), and we found evidence for the formation of GS-Se-SG (Fig. S20†), supporting the ability of 1b to produce this important intermediate in the biosynthesis of selenium-containing biomolecules.
Conclusions
Much like sulfur biology, the use of tools to study selenium biology is central to developing a better understanding of the redox chemical biology of this important element. Although numerous selenium donors are available, the compounds developed herein offer several advantages. They are easy to synthesize and undergo clean and efficient cleavage to produce H2Se and well-characterized products. In comparison, the majority of other donors mentioned in the literature are cumbersome to prepare, and some have extremely slow release rates from hours up to several days, while the rest produce potentially electrophilic and other reactive byproducts. Finally, the intermediate phenacylselenol can be protected with suitable groups and made responsive to stimuli other than ester cleavage, and these adaptations will not only aid with the study of fundamental selenium biology but also help exploit the therapeutic potential of H2Se.48–51Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written with inputs from all authors. All experiments and computational analyses were carried out by UDS and MR, supervised by HC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial assistance for this project was from IISER Pune and Science and Engineering Research Board (HC, CRG/2023/003892). The authors thank the laboratory of Dr Amrita B. Hazra, IISER Pune for their help with cloning and purification of 3-MST. UDS thanks the Council of Scientific and Industrial Research (CSIR), India, for a research fellowship.Notes and references
- L. H. Duntas and S. Benvenga, Endocrine, 2015, 48, 756–775 CrossRef CAS PubMed.
- Y. Saito, Redox Experimental Medicine, 2022, R149–R158 Search PubMed.
- C. M. Weekley and H. H. Harris, Chem. Soc. Rev., 2013, 42, 8870–8894 RSC.
- M. Benhar, Free Radical Biol. Med., 2018, 127, 160–164 CrossRef CAS PubMed.
- M. P. Rayman, Lancet, 2000, 356, 233–241 CrossRef CAS PubMed.
- K. H. Winther, M. P. Rayman, S. J. Bonnema and L. Hegedüs, Nat. Rev. Endocrinol., 2020, 16, 165–176 CrossRef CAS PubMed.
- K. A. Cupp-Sutton and M. T. Ashby, Antioxidants, 2016, 5, 42 CrossRef PubMed.
- R. F. Burk and K. E. Hill, Annu. Rev. Nutr., 2015, 35, 109–134 CrossRef CAS PubMed.
- R. A. Hankins, M. E. Carter, C. Zhu, C. Chen and J. C. Lukesh, Chem. Sci., 2022, 13, 13094–13099 RSC.
- H. Steve Hsieh and H. E. Ganther, Biochim. Biophys. Acta, Gen. Subj., 1977, 497, 205–217 CrossRef PubMed.
- K. Li, L. N. Zakharov and M. D. Pluth, J. Am. Chem. Soc., 2023, 145, 13435–13443 CrossRef CAS PubMed.
- Y. Ogasawara, G. Lacourciere and T. C. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9494–9498 CrossRef CAS PubMed.
- N. Lau and M. D. Pluth, Curr. Opin. Chem. Biol., 2019, 49, 1–8 CrossRef CAS PubMed.
- T. D. Newton and M. D. Pluth, Chem. Sci., 2019, 10, 10723–10727 RSC.
- T. D. Newton, S. G. Bolton, A. C. Garcia, J. E. Chouinard, S. L. Golledge, L. N. Zakharov and M. D. Pluth, J. Am. Chem. Soc., 2021, 143, 19542–19550 CrossRef CAS PubMed.
- A. Kharma, A. Misak, M. Grman, V. Brezova, L. Kurakova, P. Baráth, C. Jacob, M. Chovanec, K. Ondrias and E. Domínguez-Álvarez, New J. Chem., 2019, 43, 11771–11783 RSC.
- T. D. Newton, K. Li, J. Sharma, P. A. Champagne and M. D. Pluth, Chem. Sci., 2023, 14, 7581–7588 RSC.
- X. Kang, H. Huang, C. Jiang, L. Cheng, Y. Sang, X. Cai, Y. Dong, L. Sun, X. Wen, Z. Xi and L. Yi, J. Am. Chem. Soc., 2022, 144, 3957–3967 CrossRef CAS PubMed.
- Y. Long, W. Liang and L. Yi, J. Am. Chem. Soc., 2024, 146, 24776–24781 CrossRef PubMed.
- A. Krakowiak, L. Czernek, M. Pichlak and R. Kaczmarek, Int. J. Mol. Sci., 2022, 23, 607 CrossRef CAS PubMed.
- A. Krakowiak and S. Pietrasik, Biology, 2023, 12, 875 CrossRef CAS PubMed.
- H. Ishihara, S. Muto and S. Kato, Synthesis, 1986, 1986, 128–130 CrossRef.
- F. A. R. Barbosa, R. F. S. Canto, S. Saba, J. Rafique and A. L. Braga, Bioorg. Med. Chem., 2016, 24, 5762–5770 CrossRef CAS PubMed.
- M. A. Johnstone, S. J. Nelson, C. O'Leary and W. T. Self, Biochimie, 2021, 182, 166–176 CrossRef CAS PubMed.
- N. Esaki, T. Nakamura, H. Tanaka and K. Soda, J. Biol. Chem., 1982, 257, 4386–4391 CrossRef CAS PubMed.
- H. Mihara, M. Maeda, T. Fujii, T. Kurihara, Y. Hata and N. Esaki, J. Biol. Chem., 1999, 274, 14768–14772 CrossRef CAS PubMed.
- H. Mihara, T. Kurihara, T. Yoshimura, K. Soda and N. Esaki, J. Biol. Chem., 1997, 272, 22417–22424 CrossRef CAS PubMed.
- Y. Ogasawara, G. M. Lacourciere, K. Ishii and T. C. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1012–1016 CrossRef CAS PubMed.
- A. M. Gold and D. Fahrney, Biochemistry, 1964, 3, 783–791 CrossRef CAS PubMed.
- F. Shakeri, F. Zaboli, E. Fattahi and H. Babavalian, Adv. Mater. Sci. Eng., 2022, 25, 1–11 Search PubMed.
- Y. Tian, F. Xin, J. Jing and X. Zhang, J. Mater. Chem. B, 2019, 7, 2829–2834 RSC.
- F. Xin, Y. Tian and X. Zhang, Dyes Pigm., 2020, 177, 108274 CrossRef CAS.
- B. Thapa and H. B. Schlegel, J. Phys. Chem. A, 2016, 120, 8916–8922 CrossRef CAS PubMed.
- L. Wang, k. Zhu, W. Cao, C. Sun, C. Lu and H. Xu, Polym. Chem., 2019, 10, 2039–2046 RSC.
- E. Domínguez-Álvarez, M. Gajdács, G. Spengler, J. A. Palop, M. A. Marć, K. Kieć-Kononowicz, L. Amaral, J. Molnár, C. Jacob, J. Handzlik and C. Sanmartín, Bioorg. Med. Chem. Lett., 2016, 26, 2821–2824 CrossRef PubMed.
- M. J. Axley, A. Böck and T. C. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 8450–8454 CrossRef CAS PubMed.
- A. L. Johansson, R. Collins, E. S. J. Arnér, P. Brzezinski and M. Högbom, PLoS One, 2012, 7, e30528 CrossRef CAS PubMed.
- S. Müller, J. Heider and A. Böck, Arch. Microbiol., 1997, 168, 421–427 CrossRef PubMed.
- T. V. Mishanina, M. Libiad and R. Banerjee, Nat. Chem. Biol., 2015, 11, 457–464 CrossRef CAS PubMed.
- M. R. Filipovic, J. Zivanovic, B. Alvarez and R. Banerjee, Chem. Rev., 2018, 118, 1253–1337 CrossRef CAS PubMed.
- Y. Kimura, S. Koike, N. Shibuya, D. Lefer, Y. Ogasawara and H. Kimura, Sci. Rep., 2017, 7, 10459 CrossRef PubMed.
- P. K. Yadav, K. Yamada, T. Chiku, M. Koutmos and R. Banerjee, J. Biol. Chem., 2013, 288, 20002–20013 CrossRef CAS PubMed.
- P. Bora, S. Manna, M. A. Nair, R. R. M. Sathe, S. Singh, V. S. Sreyas Adury, K. Gupta, A. Mukherjee, D. K. Saini, S. S. Kamat, A. B. Hazra and H. Chakrapani, Chem. Sci., 2021, 12, 12939–12949 RSC.
- S. Manna, R. Agrawal, T. Yadav, T. A. Kumar, P. Kumari, A. Dalai, S. Kanade, N. Balasubramanian, A. Singh and H. Chakrapani, Orthogonal Persulfide Generation through Precision Tools Provides Insights into Mitochondrial Sulfane Sulfur, Angew. Chem., Int. Ed. Engl., 2024, 63, e202411133 CrossRef CAS PubMed.
- A. Krief and L. Hevesi, Organoselenium Chemistry I, Springer, Berlin, Heidelberg, 1st edn, 1988 Search PubMed.
- J. J. Determan and A. K. Wilson, Comput. Theor. Chem., 2013, 1017, 41–47 CrossRef CAS.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed.
- N. Lee, S. J. Park, M. Lange, T. Tseyang, M. B. Doshi, T. Y. Kim, Y. Song, D. I. Kim, P. L. Greer, J. A. Olzmann, J. B. Spinelli and D. Kim, bioRxiv, 2023, preprint, DOI:10.1101/2023.04.13.535674.
- S. Peng, H. Wang, Y. Xin, W. Zhao, M. Zhan, J. Li, R. Cai and L. Lu, Nano Today, 2021, 40, 101240 CrossRef CAS.
- A. K. Gilbert, T. D. Newton, M. H. Hettiaratchi and M. D. Pluth, Free Radical Biol. Med., 2022, 190, 148–157 CrossRef CAS PubMed.
- X. Pan, X. Song, C. Wang, T. Cheng, D. Luan, K. Xu and B. Tang, Theranostics, 2019, 9, 1794–1808 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2326967. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05788k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |