
The chemistry of oleates and related compounds in the 2020s
Pavel V.
Ivchenko
 *ab and
Ilya E.
Nifant'ev
ab
*ab and
Ilya E.
Nifant'ev
ab
aA. V. Topchiev Institute of Petrochemical Synthesis, RAS, Leninsky av., 29, 119991 Moscow, Russian Federation. E-mail: inpv@org.chem.msu.ru; phpasha1@yandex.ru
bDepartment of Chemistry, M. V. Lomonosov Moscow University, Leninskie Gory, 1-3, 119991 Moscow, Russian Federation
First published on 8th November 2024
Abstract
The use of renewable resources as an alternative to fossil raw materials is one of the topical directions of modern science. Consistent efforts have been devoted to finding the most prospective feedstocks and developing efficient methods for their processing. Among them, plant oils represent a promising source for the fuel, chemical, polymer, pharmaceutical and cosmetic industries. Unsaturated fatty acids (FAs) and their derivatives, particularly oleates and ricinoleates, are candidate starting materials for both organochemical modifications to obtain functional materials and catalytic transformations to α-olefins, unsaturated carboxylic acids and their derivatives, branched hydrocarbons and other valuable compounds required in the fine chemistry and polymer industry. In the 2020s, significant progress has been made in the field of oleochemistry, and in the present work, we attempt to provide the broadest overview of the chemistry of oleates and related compounds, i.e., derivatives of unsaturated FAs. Our review addresses the issues of raw material processing and purification as well as the catalytic and organochemical transformation of unsaturated FAs and FA esters with and without the cleavage of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. The most promising actual directions of research, their compliance with the principles of green chemistry and the prospects for further development are also discussed in this work.
C bonds. The most promising actual directions of research, their compliance with the principles of green chemistry and the prospects for further development are also discussed in this work.
Introduction
The modern manufacturing of fine chemicals is focused on the use of renewable feedstocks. In this case, advanced oleochemistry is aimed at the development of efficient methods for the synthesis and purification of raw materials from natural oils and fats and the use of these materials for the production of value-added products. Among them, methyl oleate (MO) and similar starting compounds have great potential for the synthesis of a wide range of desirable substances. Some of the fundamental issues associated with oleochemistry were addressed in recent broader reviews on biomass conversion.1,2 Also, numerous recent reviews describe and discuss narrower fields of the preparation;3–10 catalytic transformations,2,3,11–18 including metathesis;13,19–21 organochemistry;2,13,22–24 polymer chemistry25–29 and applications2,15,24,30 of oleates and related compounds.However, known (and continuously optimized) processes and materials as well as newly developed reactions and their products require comparative assessment in terms of organic chemistry and science of catalysis and their compliance with the 12 principles of green chemistry (GRC),31 which is becoming increasingly important. Generally, the principles of GRC are not different from the requirements of modern chemical technology. Compliance with these principles deepen the importance of GRC in multidisciplinary studies and makes it possible to introduce clear markers to identify prospective and populist scientific findings. The aim of this review is to present and critically examine, considering the requirements for greener processes and products,32 the main issues of the chemistry of oleates and related compounds reported in scientific periodicals from 2020 and propose the most promising avenues for further research in this field. Considering the defined objective, this manuscript is subdivided into different sections devoted to the preparation and purification of oleates (in a broad sense) and the chemistry of oleates with breaking, functionalization, and retention of the CC bond. Due to the high importance and relevance of catalytic metathesis and biobased polymers, these issues are discussed in separate sections.
Preparation and purification of oleates and related compounds
Vegetable oils and animal fats, triglycerides (TGs) of fatty acids (FAs, Fig. 1), are the main natural sources of oleates and related compounds; however, it should be noted that lipids can be obtained from raw materials distinct from plants and animals, e.g. glycerol and carbohydrates metabolized by some germs.33 The content of oleate depends on the type of the raw material.34 Some vegetable oils have a high oleic acid (OA) content, in particular, peanut (36%–72%), olive (65%–85%), high-oleic rapeseed (canola, 50%–66%), palm (38%–44%), high-oleic sunflower (82%–86%) and super-high-oleic safflower (up to 92%) oils;35–37 however, according to other data, the OA content in canola oil can reach 75%–84%.38 The choice of the oleate source evidently depends on the price of the feedstock, amounting to USD 896, 1069, 1079 and 1118 per ton of palm, sunflower, soja and rapeseed oil, respectively (July 2024),39 which are substantially higher than the price of crude oil (∼600 USD per ton).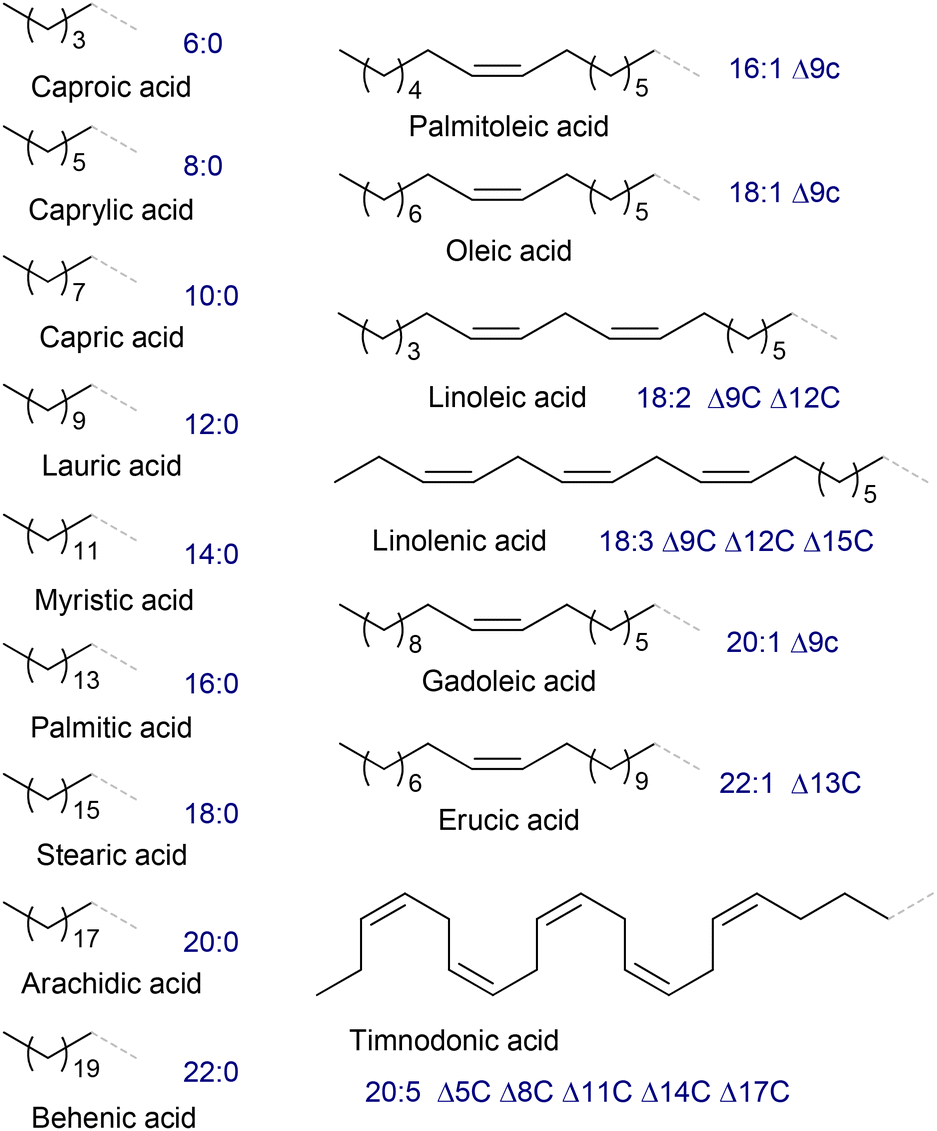 | ||
| Fig. 1 Structures of R and lipid codes of the main fatty acids (FAs) RCOOH in oils and fats. | ||
Natural oils can be subjected to hydrolysis and subsequent esterification or direct transesterification with the formation of fatty acid esters. Fatty acid methyl esters (FAMEs, Scheme 1a) are the most common intermediates for the production of fine oleochemicals. High yields of pure oleate-enriched FAMEs can be achieved only if feedstocks of high purity have been used (for example, virgin edible oils, which brings us back to the problem of fuel vs. food/feed competition). Mostly, FAMEs are considered fuel (biodiesel),40,41 with the worldwide biodiesel production estimated to reach 50 billion liters in 2025.42 However, most of the modern oleochemical processes, as discussed in the present review, are catalytic, and the purity of the raw materials and high MO content are critically important for the catalysts to achieve high selectivity and activity.
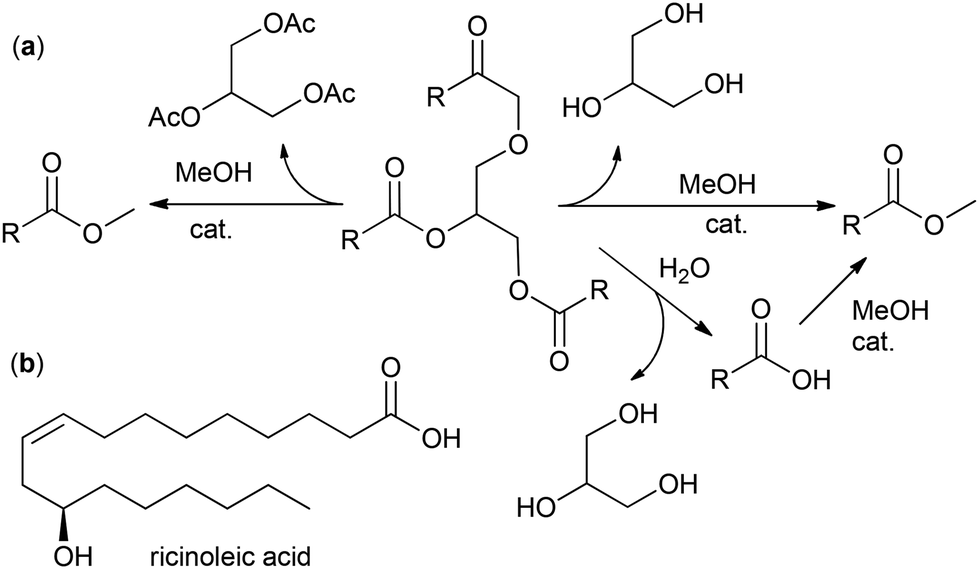 | ||
| Scheme 1 (a) Chemical transformation of a triglyceride to the fatty acid methyl ester (FAMEs) RCOOMe. (b) Structural formula of ricinoleic acid. | ||
Therefore, the methods for biodiesel production43–47 do not necessarily apply to the preparation of MA and its analogs intended for the use in fine oleochemistry. In a recent technoeconomic analysis of using waste and virgin oils for biodiesel production,48 Al-Sakkari et al. convincingly demonstrated that the homogeneous methanolysis of waste oil in the presence of KOH is economically reasonable. The conversion of virgin oils to FAMEs using advanced heterogeneous catalytic approaches is feasible when the aim is to use FAMEs in fine oleochemistry.
An alternative approach to FAMEs is the use of alkyl acetate (e.g. MeOAc) instead of MeOH; this “interesterification” reaction results in formation of glycerol ester (e.g. triacetin, a by-product of great added value) instead of glycerol (Scheme 1a).49 The direct transformation of plant materials into FAMEs, bypassing the oil separation stage, seems attractive50 but will hardly be able to give pure product.
In this section, we present and discuss the actual benefits and drawbacks of hydrolysis/esterification, direct transesterification and acyl exchange (interesterification) approaches for oil and fat splitting, mainly focusing on the preparation of MO. The problems and prospects of OA enrichment via the reduction of polyunsaturated acids and solutions concerning the purification of MO are also discussed. Some of the actual approaches described and discussed in this section do not relate directly to the synthesis of MO but can potentially be used for this purpose. Castor oil holds a special place among plant oils due to its unique composition of 85%–96% glyceride of ricinoleic acid (RA, Scheme 1b),36 and the preparation and purification of RA are also discussed in this section.
Hydrolysis/esterification of triglycerides
In search for milder and more selective methods for the hydrolysis of TGs, enzymatic catalysis has long attracted attention from researchers, and in the 2020s, numerous significant works were published. The use of enzymatic catalysis for the hydrolysis of TGs encounters the following challenges: (i) the formation of mono- and diglycerides during the hydrolysis of TGs, (ii) limited chemical stability of enzymes, and (iii) desirability of reusing the catalyst. The study by Mateos et al.53 exemplifies the first problem, where Araujia sericifera latex lipase (0.05%) demonstrated specific enzymatic activity of ∼720 μmol mg−1 h−1 (60% conversion of triglycerides towards free FAs) after 0.5 h at 25 °C; the subsequent esterification of FAs was efficient under these mild conditions, but the transesterification of monoglycerides only showed 20% efficiency.53 Similar selectivity can be suppressed based on the selection of appropriate mixtures of lipases of various genesis.54 To increase the enzyme activity, R2NH/CO2-based ionic liquids (ILs), which do not cause enzyme denaturation, were proposed as an additives. For example, bis(2-ethylhexyl)-ammonium bis(2-ethylhexyl)carbamate (8 wt%) provided ∼94% yield of FAs after 8 h hydrolysis of refined palm oil when using 0.2 wt% of lipase.55
The problem of enzyme stability was successfully solved by immobilization. To cite a few recent examples, Candida rugosa lipase, supported on methacrylate resin, and surface functionalized with octadecyl groups, showed moderate (60%) conversions to FAs but high thermal stability (60 °C).56 The immobilization of Aspergillus oryzae lipase on a metal–organic framework (MOF) prepared by the reaction of ZrO(NO3)2 with 2-aminoterephthalic acid in the presence of OA increased the thermal stability and activity of the catalyst, and the yield of FAs in the hydrolysis of soybean oil (55 °C, 12 h, 0.15 wt% catalyst) reached 98.3% and exceeded 82% after seven times of recycling.57 The use of immobilized enzymes seems highly attractive, ensuring compliance with the principles of green chemistry.
Conventional acid-catalyzed esterification is still employed in the production of esters from FAs, and thus the optimization of the reaction conditions, as before, has attracted attention from researchers. In particular, Supeno et al. showed that the esterification of oleic acid, catalyzed by H2SO4, proceeds in high yields using a homogenizer device at ambient temperature (entry 1, Table 1).59
| Entry | Raw material | Catalyst (wt%) | Conditions and comments | FAME yield, % | Ref. |
|---|---|---|---|---|---|
| 1 | OA | H2SO4 (0.7 M) | [MeOH]/[OA] = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 20 °C, 30 min :1, 20 °C, 30 min |
96.1% | 59 |
| 2 | OA (99.9%) | γ-Al2O3 catalyst bed (l = 10 cm) in tubular reactor | [MeOH]/[OA] = 20:1, 20 MPa, 275 °C, 1 min |
>91% | 60 |
| 3 | OA | ZnO/TiO2 supported on glass beads | Continuous flow rotating reactor under recycle mode, near-IR and microwave irradiation | 93.6% | 61 |
| 4 | OA (99%) | MOF from HfCl4, furan-2,5-dicarboxylic and stearic acids (4.1) | [MeOH]/[OA] = 19.5:1, 49 °C, 9.5 h; recyclable (6×) |
98.6% | 72 |
| 5 | OA (99%) | H3PW12O40 doped [(–O–)3Si(CH2)2Si(–O–)3]n nanotubes (6) | 120 °C, 1 mL OA, 3.8 mL MeOH, 10 h; recyclable | 94.7% | 69 |
| 6 | OA (99%) | H3PMo12O40-doped COF (7) | [MeOH]/[OA] = 8:1, 70 °C, 8 h; recyclable |
95% | 70 |
| 7 | OA | Sulfated biochar (4) | [MeOH]/[FA] = 40:1, 76 °C, 4 h; recyclable |
98.8% | 62 |
| 8 | Sunflower oil FAs | Lewatit MonoPlus SP 112 cation exchange resin (10) | [MeOH]/[FA] = 10:1, 60 °C, 24 h; recyclable |
60% | 64 |
| 9 | OA | Amorphous carbon-based solid acid (8) | [MeOH]/[OA] = 15:1, 60 °C, light irradiation |
∼90% | 73 |
| 10 | OA | Sulfated ZrO2 powder (8) | [MeOH]/[OA] = 15:1, 60 °C, light irradiation |
∼40% | 73 |
| 11 | OA | Sulfamic acid-modified UiO-66 MOF (7.6) | [MeOH]/[OA] = 21.9:1, 85 °C, 1.8 h; recyclable 5× (85%) |
94.4% | 63 |
| 12 | OA | Sulfated lignin-based biomass (9) | [MeOH]/[OA] = 16:1, 85 °C, 1 h; recyclable |
99% | 65 |
| 13 | OA | Sulfamic acid-functionalized PVC (1.5) | [MeOH]/[OA] = 12:1, 65 °C, 2 h; recyclable |
95.5% | 66 |
| 14 | OA | Sulfated poly(styrene-co-n-acylglycerol) (1) | [MeOH]/[OA] = 60:1, 90 °C, 12 h |
96% | 67 |
| 15 | OA | [N,N,N-Tris(propanesulfonic)aniline][EtOH]3 (2.1) | [EtOH]/[OA] = 14:1, 60 °C, 2.5 h; liquid catalyst |
97.7% | 74 |
| 16 | OA | C. Antarctica lipase B on acrylic resin (0.25 wt%) | [MeOH]/[OA] = 1.5:1, 55 °C, 45 h |
99% | 75 |
| 17 | OA | TiO2 (20) | [MeOH]/[OA] = 55:1, 55 °C |
98% | 76 |
In heterogeneous esterification, simple inorganic phases, e.g. γ-Al2O360 and ZnO/TiO2,61 were studied. It should be noted that the relatively high MO yields reported in these works were achieved through the use of a specific reactor design and/or microwave and IR irradiation. It should also be noted that in the study by Tavlarides et al.,60 supercritical methanol was used.
Sulfated supports and polymers represent another group of transesterification catalysts. In particular, sulfated biochar (4 wt%) demonstrated up to 98.8% yield in OA methanolysis when using MeOH vapor.62 A sulfamic acid-modified UiO-66 metal–organic framework (MOF) showed up to 94.4% productivity when 7.6 wt% of the catalyst was used at an [MeOH]/[OA] ratio of 21.9:1.63 Otopkova et al.64 studied the activity and stability of different types of cation-exchange resins in the methanolysis of FAs prepared from sunflower oil. Up to 10× catalyst reusability was achieved for Lewatit MonoPlus SP 112 resin with a gel structure, but the conversion of FAs was only 60%. Sulfated lignin-based biomass showed substantially higher catalytic activity (up to 99% conversion). The reusability of this catalyst was confirmed by consecutive reaction cycles with a conversion of 98.0%, 96.4%, and 88.2%.65 The use of sulfamic acid-functionalized PVC (1.5 wt%) provided the 98.1% yield of MO after 2 h of reaction at 65 °C.66 Sulfated poly(styrene-co-n-acylglycerol) was also studied, and yields of MO of up to 96% were achieved.67
Heteropolyacids are well-known and efficient catalysts for the esterification of FAs,68 and several works on the use of heteropolyacids have been published in recent years. Wang et al. proposed the use of H3PW12O40-functionalized organosilicon nanotubes as a recyclable catalyst for the methanolysis of OA.69 Zhao and Li70 introduced H3PMo12O40 in a covalent organic framework (COF), which was prepared from 1,3,5-triformylphloroglucinol and ethidium bromide, and this catalytic system demonstrated high activity at an [MeOH]/[OA] ratio of 8:1 and 70 °C. For the ethanolysis of OA in the presence of H4SiW12O40·H2O,71 the suitability of the pseudo-homogeneous kinetic model with average reaction order n = 1.9 and activation energy Ea = 50.1 kJ mol−1 was demonstrated.
A commercial immobilized enzyme (C. Antarctica lipase B immobilized on acrylic resin) demonstrated high selectivity in the formation of MO, but with a relatively low reaction rate (∼99% conversion was achieved after 30 h at 55 °C).75
Liquid–liquid two-phase esterification can provide deeper homogenization of the catalyst. In 2023, Zhou et al. reported the use of a liquid acidic catalyst, [PhN((CH2)3SO3H)3][EtOH]3, in the ethanolysis of OA, and the yield of ethyl oleate reached up to 97.7%.74
When using the electrochemical method for esterification, the yield of FAMEs was 42%.77 Photocatalytic esterification, catalyzed by TiO2, was studied by Welter et al.; however, the high yield of FAMEs was achieved at an unacceptably high catalyst loading (20%) and [MeOH]/[OA] ratio of 55:1.76
It should also be mentioned that MeOH can be replaced by MeOAc in the esterification of FAs, e.g., calcined and sulphated spent bleaching earth was used as a catalyst (12 wt%) for the conversion of palm FA distillate, and 89.9% yield of FAMEs was obtained at the [MeOAc]/[FA] ratio of 12:1 and 100 °C after 3 h.78
When comparing the results of the above-mentioned recent works considering green chemistry principles, unacceptably high MeOH/OA ratios in many developed methods are immediately apparent.60,62,63,65,67,69,72–74,76 Relatively high OA conversions in an acceptable time were achieved in a few cases,62,65,72,76 and considering the catalyst loading and reusability, only the use of sulfated lignin-based biomass (Table 1, entry 12)65 generally complies with the actual requirements of green catalytic processes. However, heterogeneous catalysts do not show indisputable advantages over conventional homogeneous acid catalysis except higher environmental friendliness.
Transesterification of triglycerides
The transesterification of TGs, the most widely used method for converting oils and fats into FAMEs,3 was the subject of hundreds of works. In recent years, these studies were positioned as catalytic, with the catalysts belonging to basic, acidic and coordination types. We think that a similar classification is redundant; for practical purposes, the homogeneity or heterogeneity of the catalysts and the nature of reactive sites (acidic or basic) are far more important. It should be noted that the high content of FAs in the raw material complicates the use of basic catalysts that are more efficient in the transesterification of purified TGs in comparison with acidic catalysts. Consequently, the development of esterification catalysts is still ongoing, and two-stage processes (esterification of the FAs remnants, followed by transesterification using the same alcohol) are economically reasonable.79,80 In this section, we describe and discuss the results of recent studies on the use of homogeneous and heterogeneous catalysts in the transesterification (mainly, methanolysis) of TGs. Finally, the brief discussion at the end of this section is devoted to the interesterification process, which is still being explored.:1, 0.50 wt% of catalyst, 65 °C and 105 min. The combination of ultrasound and IR radiation facilitated the methanolysis of rice bran oil, and optimization of the reaction conditions (60 wt% MeOH, 1 wt% of KOH, 7.8 min) led to a FAME yield of 97.7%.82 Further optimization of the conventional methods for the synthesis of FAMEs remains actual, for example, in 2024 Photaworn et al. reported the results of their study aimed at improving FAME production using a pilot-scale continuous multiple baffle reactor with increased turbulence and mixing intensity.83 The yield of refined palm oil-based FAMEs of 97.7% was obtained with a residence time of 10 min (60 °C, [MeOH]/[oil] = 5.5:1, 1.0 wt% MeOK as a catalyst).
The problem of saponification during the MeONa (or NaOH) catalyzed transesterification of TGs was studied in more detail by Chanakaewsomboon et al. from the standpoint of colloid chemistry,84 and MeOK (or KOH)-catalyzed processes were also studied by this group.85 They showed that the soap layer, as a barrier resisting the transfer of alcohol and catalyst solution, is formed beyond the interface of the MeO−/MeOH solution droplets in oil medium. The glycerol forms a thicker layer between the MeO−/MeOH droplet and soap layer. A high content of FFA, water and MeO− contribute to the formation of soap. The content of 0.3 wt% soap in crude FAMEs was found to be the maximum limit, given that excess soap led to a decrease in the yield of FAMEs. As shown by Hayyan et al.,86 the excess soap can be removed by choline chloride based deep eutectic solvents; however, a threefold excess of extractant is needed for almost complete removal of the soap, which is completely unacceptable in view of GRC principles.
To avoid saponification at the stage of methanolysis of TGs with a high FA content, Bai et al. once again confirmed the efficiency of preliminary acid-catalyzed methanolysis;87 the yield of FAME in the second NaOH-catalyzed transesterification stage reached 98.5%.
The mass transfer between phases can be intensified by the use of a rotor–stator spinning disc reactor (RS-SDR); the NaOH-catalyzed conversion of refined sunflower oil to FAMEs was 82% with a productivity of 7780 mol mR−3 min−1.88 An alternative approach to improve the efficiency of the transesterification of TGs was demonstrated recently by Thakkar et al. using the example of KOH-catalyzed methanolysis of castor oil using a hydrodynamic cavitation reactor,89 at 60 °C, [MeOH]/[oil] ∼ 10:1 and using 1.06 wt% of catalyst, and after 1 h the yield of FAMEs was 92.3%.
The problem of saponification can be solved by avoiding the use of alkali catalysts. For example, the Fe(III) complexes [(2-O-C6H4CHNCH2)2]FeX (X = Cl, OAc) demonstrated up to 98% conversion of waste cooking oil to FAMEs (180 °C, 2 h, [MeOH]/[oil] = 20:1), with the catalyst loading of 0.1 mol%.90
In this way, the use of alkali catalysts in liquid-phase transesterification is highly efficient; however, the disadvantages of this method include the need to use refined oils as the raw materials, utilization of alkaline wastewater, and the separation of glycerol, which is energy-consuming. Because of this, many attempts have been devoted to developing catalysts and processes that confirm to the GRC principles in terms of waste prevention, safer chemicals and solvents, as well as atom economy and energy efficiency.
| Entry | Raw material | Catalyst (wt%) | Conditions and comments | FAME yield, % | Ref. |
|---|---|---|---|---|---|
| 1 | Cottonseed, soybean, jatropha, castor, waste cooking oils | K2CO3-doped CaO (5) | 65 °C, [MeOH]/[oil] = 12:1, 30 min recyclable (10×) |
∼99% | 92 |
| 2 | Soybean oil | Na2CO3-doped blast furnace dust (9.8) | 75 °C, [MeOH]/[oil] = 13.7:1, 1.62 h; recyclable (15×) |
99% | 7 |
| 3 | Soybean oil | Li2ZrO3 (6) | 65 °C, [MeOH]/[oil] = 8:1, 2 h; recyclable (7×) |
98%–99% | 98 |
| 4 | Triolein | CaO (3) | 60 °C, [MeOH]/[oil] = 9:1, 5 h |
96% | 116 |
| 5 | Sunflower oil | MgO/MgAl0.4Fe1.6O4 (20) | 117 °C, [MeOH]/[oil] = 14.6:1, 5 h magnetically separable; recyclable (6×) |
98.8% | 117 |
| 6 | Chicken fat waste | CaO (1.5) | 65 °C, [MeOH]/[oil] = 10:1, 2 h |
93% | 118 |
| 7 | Palm oil | Porous K2O@CaO (3) | 80 °C, [MeOH]/[oil] = 15:1, 2 h; not recyclable |
88% | 96 |
| 8 | Catharanthus roseus seed oil | Ba2+ doped CaO | 58 °C, [MeOH]/[oil] = 15:1, 70 min recyclable, ⅔ of activity at run 5 |
91.8% | 97 |
| 9 | Soybean oil | K2O and K2CO3 loaded zeolite-geopolymer hybrid material (2) | 70 °C, [MeOH]/[oil] = 16:1, 3 h recyclable (5×) |
97.6% | 93 |
| 10 | Waste cooking oil | 20 wt% MeONa/zeolite Y (2.1) | 60 °C, [MeOH]/[oil] = 16:1, 30 min |
99% | 94 |
| 11 | Refined sunflower oil | Na2SiO3 (20) | RS-SDR, 60 °C, [MeOH]/[oil] = 9:1, 8 min |
72% | 88 |
| 12 | Palm oil | M2SiO3 (M = Li, Na, K) (3) | 60 °C, [MeOH]/[oil] = 12:1, 60 min |
87.5% | 95 |
| 13 | Palm oil | H2NCH2CH2NH2 loaded chitosan cross-linked with epichlorohydrin (8) | 25 °C, MeOH/oil v/v ratio 1:1; recyclable (3×) |
nd | 119 |
| 14 | Mustard oil | NiO–CdO–Nd2O3 nanocomposite (0.8) | 80 °C, [MeOH]/[oil] = 15:1, 5 h; recyclable (3×) |
80% | 120 |
| 15 | Bischofia javanica seed oil | Bischofia javanica leaf extract on ZrO2 (2.5) | 70 °C, [MeOH]/[oil] = 6:1, 2 h; recyclable (4×) |
95.8% | 121 |
| 16 | Waste cooking oil | Guanidine/bacterial cellulose (7.5) | 70 °C | 99% | 91 |
| 17 | Pyrus glabra seed oil | Hydroxyapatite (0.5) | 80 °C, [MeOH]/[oil] = 6:1, 30 min recyclable, ⅔ of activity at run 6 |
89.2% | 99 |
| 18 | Jatropha oil | Mg2+ doped CoFe2O4 (3) | [MeOH]/[oil] = 11.5:1, 35 min; ultrasonic activation |
95.5% | 100 |
| 19 | Jatropha oil | Zn/Fe DMCC (5) | 160 °C, [MeOH]/[oil] = 10:1, 3 h, 12 bar; recyclable (85%, 5×) |
95% | 108 |
| 20 | Canola oil | H3PW12O40/mesoporous aluminosilicate (3) | 200 °C, [MeOH]/[oil] = 20:1, 7 h, 40 bar; recyclable (5×) |
82.3% | 102 |
| 21 | Canola oil | H3PW12O40/mesoporous aluminosilicate (5.9) | 200 °C, [MeOH]/[oil] = 27:1, 8 h; recyclable |
89% | 103 |
| 22 | Oleic acid + castor oil | Ce3+ and F− doped H3PW12O40 (4) | 110 °C, [MeOH]/[oil] = 18:1, 1.5 h; recyclable (76%, 5×) |
93% | 104 |
| 23 | Waste cooking oil | Sulfated TiO2 nanowires (5) | 65 °C, [MeOH]/[oil] = 26:1, 3 h |
99.5% | 105 |
| 24 | Waste cooking oil | MoO3 impregnated in Nb2O5 (6) | 145 °C, [MeOH]/[oil] = 20:1, 2.5 h recyclable (>80%, 7×) |
94.2% | 106 |
| 25 | Jatropha curcas oil | Sulfated Zn2+ and Fe3+-doped carbon (7.7) | 96 °C, [MeOH]/[oil] = 23:1, 50 min recyclable (>82%, 7×) |
97.1% | 107 |
| 26 | Jatropha curcas oil | Sn4+ doped sulfated MMT clay (7) | 150 °C, [MeOH]/[oil] = 20:1, 7.5 h |
93.1% | 122 |
| 27 | Rapeseed, waste corn oils | Peganum harmala spice seed extract modified graphene oxide-CuFe2O4 (8) | 65 °C, [MeOH]/[oil] = 13:1, 7 h magnetic, recyclable |
81% | 101 |
| 28 | Waste cooking oil | B. subtilis lipase immobilized on Fe3O4/PVA (10.1) | 34.7 °C, [MeOH]/[oil] = 4:1, reactant flow rate = 16.95 mL min−1; magnetic field intensity 132.28 Oe |
88.5% | 113 |
| 29 | Waste Periplaneta Americana oil | Eversa® Transform lipase immobilized on Fe3O4/biochar (14) | 30 °C, [MeOH]/[oil] = 7:1, 14 h |
95.7% | 115 |
In recent years, alkali metal carbonates have been found to be sufficiently basic for transesterification. In particular, among K2CO3-doped oxides of Mg(II), Ca(II) and Zn(II), CaO-based catalysts showed the best characteristics. The transesterification of different oils and animal fats in the presence of 5 wt% of this type of catalyst ([MeOH]/[oil] = 12:1, 65 °C) was completed after 30 min.92 Impregnating blast furnace dust with Na2CO3 solution, followed by calcination at 500 °C, led to the formation of the active catalyst;7 99% yield of FAME at a 9.8 wt% catalyst loading was achieved, and this catalyst seems prospective in view of its reusability (up to 15 cycles). A K2O and/or K2CO3-loaded X-type zeolite-geopolymer hybrid material (2 wt%) efficiently catalyzed the methanolysis of soybean oil with the yield of FAME reaching up to 97.6%.93 MeONa/zeolite Y demonstrated high catalytic activity (up to 99% yield of FAMEs based on waste cooking oil).94 For this catalytic system, the surface formation of Na2SiO3 was proposed, but stoichiometric Na2SiO3 was moderately active.88 In a more recent study, freshly prepared alkali metal silicates showed higher productivity.95
The doping of K2O on porous CaO was achieved via the treatment of calcined waste chicken eggshell by an aq. KI solution and subsequent calcination;96 however, the results were mediocre (Table 2, entry 7). Ba2+ doping on CaO resulted in a more efficient catalyst.97
Solid-state catalysts prepared via the reaction of ZrO2 with alkali metals and subsequent oxidative calcination at 800 °C showed high activities, and the Li-based catalyst Li2ZrO3 demonstrated excellent productivity (99% conversion after 2 h at 6:1 MeOH to oil ratio) and reusability.98 Of certain interest is the use of hydroxyapatite, prepared by the chemical precipitation method (d = 28–51 nm and specific surface area of 21.9 m2 g−1) in the methanolysis of Pyrus glabra seed oil under microwave irradiation.99 At [MeOH]/[oil] = 6:1, the yield of FAMEs reached up to 89.2%. An easy-to-separate ferromagnetic catalyst, Mg2+-doped CoFe2O4, at 3 wt% loading provided 95.5% yield of FAMEs from Jatropha oil.100 A natural organobase (Peganum harmala spice seed extract)-immobilized heterogeneous catalyst was also proposed for the production of biodiesel.101
Acidic catalysts, studied in recent years, demonstrated different productivity and selectivity. In particular, H3PW12O40 supported on mesoporous aluminosilicate showed 82%–89% conversion in the methanolysis of canola oil.102,103 The introduction of Ce3+ and F− in H3PW12O40 increased the yield of FAMEs to 93% when a mixture of oleic acid and castor oil was used.104 Sulfated TiO2 nanowires, prepared from anatase TiO2 by molten salt method (5 wt%), provided up to 99.5% conversion of waste cooking oil to FAMEs.105 MoO3-impregnated Nb2O5 showed high efficiency in the methanolysis of waste cooking oil.106 The complex catalyst, prepared from Zn2+ and Fe3+-doped carbon with subsequent treatment by 4-diazoniobenzene sulfonate, demonstrated relatively high activity and reusability (Table 2, entry 25).107 A double metal cyanide catalyst, prepared from ZnCl2 and K3[Fe(CN)6] in the presence of tBuOH and Pluronic P-123, also showed high productivity (Table 2, entry 19).108
Heterogenized enzymes were also studied in methanolysis. For example, lipase from Pseudomonas fluorescens supported on mesoporous CaCO3 microparticles showed higher productivity in comparison with free lipase;109 however, the data provided do not allow the productivity to be assessed. The immobilization of lipase from Rhizopus oryzae on Fe3O4-doped mesoporous silica also resulted in an increase in the transesterification selectivity and activity.110Rhizopus oryzae lipase on functionalized poly(methyl methacrylate) demonstrated high stability;111 the productivity of the catalyst increased in the presence of acids in the starting TGs, and the similar effect of acidity was observed when using commercial immobilized Rhizomucor miehei lipase.112B. subtilis lipase immobilized on Fe3O4/poly(vinyl acetate) (PVA) under optimized conditions showed conversions of up to 88.5%.113 The use of “whole-cell” enzymatic catalysis114 is of theoretical interest but seems hardly applicable in practice. Very interesting and somehow breakthrough results were achieved when using Fe3O4/biochar-immobilized lipase, providing 95.7% yield of the FAMEs.115
The cost of eliminating water from waste oils before transesterification may be up to 40% of the price of FAMEs; however, this problem can be solved via the integration of a pervaporation unit with a modified reactive distillation column.123 In this work (2020), the price of WCO-based FAMEs was estimated to be 1.04 USD per kg. In the study by Oke et al.,124Azadirachta indica FAMEs were priced at 1.06 USD per kg, and in a later work,116 MO was priced at 1.12 USD per kg. In this study, the CaO-catalyzed methanolysis of triolein was compared with sulfonated carbon-catalyzed methanolysis using an economic analysis. In the case of the CaO-catalyzed process, the computed return on investment (ROI) was 105.36%.
It should be noted that for catalytic systems studied in recent years, e.g. chitosan modified with ethylenediamine and cross-linked with epichlorohydrin119 and guanidine/bacterial cellulose,91 high yields of FAMEs were not achieved, or detailed experimental data were not given. The mention of frankly weird catalytic components such as CaO2125 also raises questions.
In this way, numerous studies to develop transesterification catalysts have been performed recently. A great part of these works focused on the development of catalysts that can provide high yields of FAMEs when using feedstocks with a high FA content. Conventional homogeneous catalysts (MeONa, NaOH, etc.) are not efficient towards similar starting materials due to saponification, which complicates the separation of the reaction products and decreases the yield of FAMEs. The use of acid heterogeneous catalysts has its own advantages, given that the residual FA is subjected to esterification under the reaction conditions.
It should also be noted that the use of supercritical MeOH and EtOH in the transesterification of TGs represents a non-catalytic alternative to conventional methods for the preparation of FAMEs.126–128 However, the main drawbacks of this method are the high required [ROH]/[oil] ratios (>20:1), reaction temperatures (>300 °C) and pressures (>200 bar). In recent years, research in this field has continued; however, the optimal conditions found are still not suitable for the production of FAMEs. For example, in the methanolysis of waste beef tallow, the optimized [MeOH]/[oil] ratio was 45:1 at 335–365 °C;129 for the methanolysis of candlenut oil, the maximum FAME yield of 96.4% was obtained after 22 min at 285 °C, 115 bar, and [MeOH]/[oil] = 30:1;130 the methanolysis of Jojoba oil with 95.7% FAME yield was conducted within 23 min at 287 °C and 123 bar at [MeOH]/[Oil] = 30:1;131 the optimized conditions for the methanolysis of Aegle marmelos oil were 325.5 °C, [MeOH]/[oil] = 41:1, and 23 min.132 In 2023, Show et al. reported the results of their study on the catalyst-free methanolysis of waste cooking oil, where 94.2% yield of FAMEs was obtained at 275 °C and 7 bar at [MeOH]/[oil] = 12.4:1.133 Very recently, Demir and Akgün proposed the use of a complex heterogeneous catalyst for the methanolysis of Jatropha curcas oil,134 and ∼97.5% yield of FAMEs was achieved with 10 wt% catalyst loading at 300 °C, 90 bar, [MeOH]/[oil] = 40:1 after 3 min; hence, the function of the catalyst was shortening the reaction time, thus is the juice worth the squeeze?
When the data presented in Table 2 is critically compared to the GRC principles, some of the works deserve particular attention. Relatively low MeOH/TG ratios could be successfully applied when using Li2ZrO3 (Table 2, entry 3),98 CaO (Table 2, entry 4),116 a biobased organocatalyst (Table 2, entry 15),121 hydroxyapatite (Table 2, entry 17),99 Zn/Fe DMCC (Table 2, entry 19)108 and immobilized enzyme (Table 2, entries 27 and 29).101,115 However, high conversions of the substrate were achieved only in a few cases, and among the works published in the 2020s, the studies by Dai et al.98 and Guo et al.115 deserve particular attention, providing the highest yields of FAMEs. In conclusion, the great diversity catalysts under study did not show compliance with GRC principles, and further investigations in this field have a high probability of the development of efficient catalysts for the heterogeneous transesterification of TGs that can compete with conventional homogeneous catalysts.
Acyl exchange (interesterification of triglycerides)
Despite the fact that catalytic methanolysis of TGs remains the main industrial method for the production of FAMEs, studies on the interesterification of TGs are continuing, partly because of the economic attractiveness of the side reaction product, triacetin, whose synthesis from industrially derived glycerol is an energy- and time-consuming process.135 In an early study,136 the MeOK-catalyzed interesterification of TGs by MeOAc was found to be slower than methanolysis, the conversion of diacetinmonoglycerides to triacetin was the rate-limiting step of interesterification, and the process was complicated by its high degree of reversibility. Most of the works in this field were performed in 2000–2010, as reflected in the review by Marx,137 and thus here we will only discuss several actual examples.In 2020, Akkarawatkhoosith et al. reported the use of an ion-exchange resin as a catalyst for the reaction between palm oil and EtOAc, and 97% yield of ethyl esters was achieved at 113 °C using an [EtOAc]/[oil] ratio of 16.7:1 after 13 h.138 Nunes and Castilhos studied interesterification between soybean oil and MeOAc, in the presence of nanocrystalline CaO (10 wt%) at 325 °C and [MeOAc]/[oil] ratio of 40:1, and yield of FAMEs was 62.3%.139 In the same year, interesterification under supercritical conditions in the absence of a catalyst was studied, and at a 2:1 w/w MeOAc/oil ratio, ∼57% yield of FAMEs was achieved based on grease trap waste lipids.49 Under supercritical conditions (325 °C, 150 bar) EtOAc converted spent coffee grounds oil to FAMEs with 91.8% yield, but the yield of triacetin was only 4.2%.140 The catalytic activity of NbOPO4 in the reaction of soybean oil with MeOAc was studied by Albarello et al.141 High conversions were observed at 345 °C, and the yield of FAMEs was ∼64% due to the thermal decomposition of the reaction products. The use of ultrasonic-induced cavitation allowed a decrease in the [MeOAc]/[oil] ratio to 8:1, and after 50 min at 50 °C, the yield of mahua oil-based FAMEs was 96%.142 SnO/γ-Al2O3 at 210 °C demonstrated moderate productivity (33.5% FAME, 1.5% triacetin).143
In all cases, the content of triacetin in the reaction products did not match the stoichiometry of the process. In addition, the separation of triacetin and diacetinmonoglycerides from the resulting mixture has not been elaborated. The products of interesterification can be used without separation as biodiesel, but their applicability as raw materials for catalytic oleochemistry is questionable. In summary, interesterification is inferior to methanolysis-based processes in parameters of atom economy and energy efficiency, and therefore does not quite match GRC principles. The relatively low number of publications on this topic confirms the dubious prospects of the actual use and industrial implementation of interesterification.
C18:1 enrichment of FAMEs via hydrogenation or hydrogen transfer
The high content of poly(unsaturated) fatty acids (PUFAs) in TGs complicates the further use of FAMEs in fine oleochemistry because catalytic processes require high purity and compositional uniformity of the raw materials. Given that most valuable processes start from monounsaturated acid derivatives (mainly oleates), an additional step of partial and selective hydrogenation of PUFAs and their derivatives to (C18:1) products appears to be attractive. The partial/selective hydrogenation of PUFAs was discussed in recent reviews,9,144 and the target reaction and main side processes are presented in Scheme 2. Promising data obtained during the studies in this field were published in 2020s, and in this section, we attempt to summarize these results and evaluate the prospects of (C18:1) enrichment. | ||
| Scheme 2 Hydrogenation or hydrogen transfer reduction of FAMEs to increase the content of C18:1 esters, target compound and side products using the example of C18 methyl esters. | ||
C bond migration during this process (Scheme 2) are the obvious tasks in the development of new catalytic systems. In the 2020s, attempts were made to solve these problems, with varying degrees of success. Before discussing recent results, it should be mentioned here that Ni catalysts were first applied to oil hydrotreatment at the beginning of the 20th century, Cu (Cu/Cr) systems were intensively studied in the 1960s, and then platinum group metals emerged as promising catalysts.144
The hydrogenation of canola and sunflower oils using the commercial Lindlar catalyst (Pd/CaCO3, poisoned with lead) was studied by Gallucci et al. in 2020.145 Under the optimized reaction conditions (4 bar of H2, 180 °C, 4 mgcat mLoil−1), the conversions of linoleic (C18:2) and linolenic (C18:3) acids were 84.6% and 90.1%, respectively, with the low formation of C18:0 acid (<10%). However, the content of (E)-C18:1 in the experiments with sunflower oils reached up to 51%.
In 2020, Quaranta and Cornacchia reported that C18:3-enriched FAMEs can be hydrogenated under mild conditions (50 °C, 1 bar H2, 1 h) over 0.1% of Pd(5%)/C with C18:1 selectively of up to 97.8%;146 however, the C18:2 content was reduced moderately (from 21.5% to 16.7%), and the content of (E)-C18:1 reached up to 39%. In a later study by this group, the partial hydrogenation of C18:2-rich FAMEs (66.9 and 75.1 mol% C18:2, prepared from technical linoleic acid and tobacco seed oil, respectively) was performed in n-heptane under mild conditions (15 °C, 1 bar of H2) within 45–60 min in the presence of 2–6 wt% of Pd(5%)/C; the yield of C18:1 products was 83%–92% with the selectivity of 93%–95%.147 (E)-C18:1 formation and CC scattering were 20.3%–34.8% and 15%–24%, respectively. When using nanoporous carbon as a support for Pd nanoparticles (0.5 wt% of catalyst, 80 °C, 5 bar of H2), palm oil-based FAMEs (8.5% C18:2, 36.1% (Z)-C18:1, 4.8% C18:0) were converted to the product with 0.9% content of C18:2 ester, but up to 11.6% (E)-C18:1 ester was formed, and the content of C18:0 ester increased to 8–9%.148
Superficial Pd nanoparticles supported on carbonaceous SBA-15 were studied for the hydrogenation of palm oil oil-derived FAMEs (80–100 °C, 5–8 bar of H2, Pd/oil weight ratio of 6.7–8.3 × 10−5), more than 95% conversion of C18:2 was achieved, but the selectivity for the formation of (Z)-C18:1 ester was low, and partial isomerization of the starting oleate to (E)-C18:1 ester and excess amount of C18:0 were detected.149 The more complex Pd–Pt/MCM-41 catalyst (1 wt%) at 100 °C and 4 bar of H2 provided >90% conversion of C18:2 and C18:3 esters in sunflower oil-based FAME to (Z)-C18:1 and (E)-C18:1 esters in an ∼2:1 ratio, respectively.150 The hydrogenation of soybean oil-derived FAMEs over Pd/SiO2 at 100 °C and 4 bar of H2 proceeded a higher rate, but lower selectivity in comparison with the Pd/MCM-41 system.151 Further tuning of the porosity of the Pd–Pt/MCM-41 catalyst in the hydrogenation of soybean oil-based FAMEs allowed a 79.1 wt% content of C18:1 to be to achieved (57.0 wt% (Z)-C18:1); the initial content of (Z)-C18:1 was 28.9 wt%, and the content of methyl stearate increased from 3.7 to 4.9 wt%.152
The complexity of the catalytic problem and limitations of Pd-based catalysts are illustrated by the data presented in Table 3. In this table, the results from comparative experiments with Pd catalysts supported on carbon (Sibunit 159k, T-900) and different metal oxides are presented.153 The catalytic activity decreased in the order of Cr2O3 > T-900 > V2O5 ≈ ZrO2 ≫ Ga2O3 ≈ Ta2O5 > TiO2 ≫ FW-70. At the same time, the nature of the support had minimal effect on the product distribution.
| Support | IV | C14:0 | C16:0 | C18:0 | (Z)-C18:1 | (E)-C18:1 | (Z)-C18:2 | (E)-C18:2 |
|---|---|---|---|---|---|---|---|---|
| a 180 °C, 5 bar, 50 g of oil, 10 mg of catalyst; terminated after the absorption of the calculated amount of H2 required for the formation of a product with an iodine value (IV) of 71–75. b Data for feed oil. | ||||||||
| —b | 202.1 | 0.08 | 7.34 | 3.71 | 25.60 | 0 | 61.33 | 0.93 |
| Sibunit 159k | 74.2 | 0.07 | 7.57 | 9.13 | 37.82 | 41.16 | 1.35 | 1.88 |
| T-900 | 75.8 | 0.08 | 7.59 | 8.73 | 39.65 | 39.71 | 1.35 | 2.65 |
| α-Al2O3 | 74.6 | 0.08 | 7.53 | 9.22 | 36.41 | 42.02 | 1.37 | 2.39 |
| ZrO2 (ds = 1.7 nm) | 74.1 | 0.08 | 7.60 | 11.20 | 36.36 | 38.53 | 2.03 | 3.21 |
| ZrO2 (ds = 3.2 nm) | 73.8 | 0.07 | 7.48 | 9.57 | 36.51 | 42.26 | 0.96 | 2.17 |
| V2O5 | 76.7 | 0.07 | 7.50 | 7.45 | 43.16 | 36.63 | 1.34 | 2.95 |
| Ga2O3 | 75.5 | 0.08 | 7.52 | 8.57 | 38.42 | 40.40 | 1.43 | 2.62 |
| TiO2 | 76.5 | 0.07 | 7.49 | 9.41 | 39.89 | 36.16 | 2.26 | 3.75 |
| Ta2O5 | 75.8 | 0.09 | 7.64 | 8.03 | 39.61 | 39.64 | 1.22 | 2.82 |
| Cr2O3 | 74.8 | 0.08 | 7.55 | 8.35 | 40.25 | 39.59 | 1.16 | 2.05 |
The formation of active Pd0–O2−–Ce3+ sites on the surface of CeO2 nanoparticles was proposed as a prospective way for the development of heterogeneous catalysts for the hydrogenation of FAMEs.154 The MO selectivity of 52.6% at 93.8% conversion for sunflower oil-based feedstock was achieved. DFT calculations revealed a qualitative difference between the (111) and (100) crystal planes of CeO2 in the absorption of Pd, starting with C18:2 ester and isomeric C18:1 esters.
Ni-based catalysts represent less active alternatives to Pd-based systems; however, the problems of (Z)-/(E)-isomerization and C18:0 formation are still not completely resolved. For example, a Ce-doped Ni/Al catalyst showed high (Z)-C18:1 selectivity in the hydrogenation of canola oil, but upon reaching almost complete C18:3/C18:2 hydrogenation, the content of (Z)-C18:1 became less than the initial value.155 Hinchiranan et al.156 studied an Ni catalyst supported on electrospun silica fibers, at 1 bar H2 and 140 °C, and this catalyst showed 71% conversion of C18:2 ester with high selectivity for the formation of (Z)-C18:1 product (the content of (E)-C18:1 ester was usually less than 3%), but the TOF of this catalyst was only ∼140 h−1.
An Ni/bentonite catalyst demonstrated up to 75% conversion of C18:2 ester in the hydrogenation of Jatropha oil FAMEs, and the contents of C18:2, (Z)-C18:1, (E)-C18:1 and C18:0 were 7.64, 42.7, 24.06 and 14.67 wt%, respectively (3.3 wt% of catalyst, 200 °C, 3 bar, 1 h).157 Dell'Anna et al.158 showed that the Ni-containing product from the copolymerization of Ni-2-(acetoacetoxy)ethyl methacrylate and CH2CHCONMe2 in the presence of N,N′-methylene bis-acrylamide cross-linker demonstrated high C18:1 selectivity in the hydrogenation of soybean oil (Fig. 2) and waste cooking oil, although (Z)-/(E)-isomerization could not be avoided. This catalyst demonstrated good recyclability for at least five subsequent runs. Pd/Ni on nanoporous carbon showed low activity and poor recyclability.159
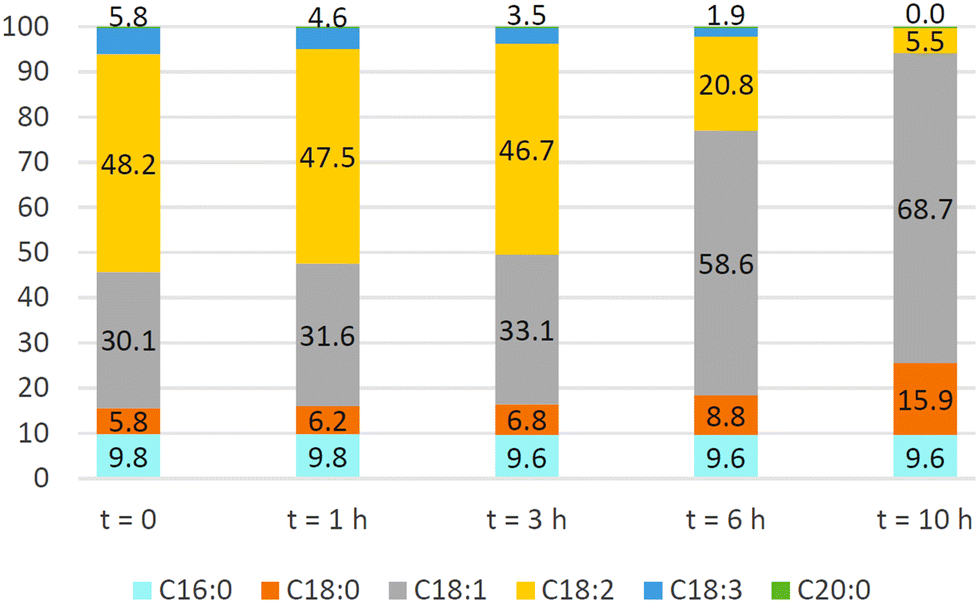 | ||
| Fig. 2 Soybean oil FAME composition at different reaction times during hydrogenation with Ni-containing polymer (0.059 mmol of Ni), FAME (0.5 mmol, 125 mg), and MeOH (5 mL), at 100 °C under H2 (10 bar). In all the histograms, the percent of C20:0 methyl ester is equal to 0.3%.158 Reprinted with permission, Copyright (2022) MDPI. | ||
The use of Cu-based catalysts sometimes allows the problem of C18:0 formation to be reduced but (Z)-/(E) isomerization also proceeds on these catalysts.144 The Cu/SiO2 catalyst prepared by the hydrolysis precipitation method provided conversions of up to 93.3% for C18:3 and 90.4% for C18:2 (8 mgCat mLoil−1, 180–200 °C, 4 bar of H2), and the maximum amount of total (E)-isomers at 200 °C and 12 bar of H2 was ∼23%; however, the formation of C18:0 ester did not exceeded 4%.160
Concluding this section, the studies on the use of novel techniques for the production of partially hydrogenated FAMEs using non-thermal parallel-plate dielectric barrier discharge (DBD) plasma should be mentioned. Without any catalyst, soybean oil FAMEs (130 mL) under an H2/He atmosphere after 20 h at 25 °C were converted to partially hydrogenated FAMEs, and the 18:2 ester content declined by half, whereas the 18:1 ester content increased from 23.6% to 34.1% (2% of (E)-isomer), but side by side, the stearate content increased from 4.2% to 16.6% in the latter process.161 A change in the design of the laboratory setup did not result in any measurable improvements in the 18:1 ester content.162 The use of the DBD reaction system in the RANEY®-Ni-catalyzed hydrogenation of soybean oil FAMEs resulted in a decrease in PUFA content by 57 mol%, and the selectivity for C18:1 formation was ∼78%.163
When analyzing the processes for partial selective C18:2/C18:3 hydrogenation from the point of view of GRC principles, Ni-based catalytic systems appear to be the most promising for further study and practical use. Pd-based systems are more active, but (Z)-/(E)-isomerization is accompanied by CC bond migration to some extent. Also, if this is the case, the idea of C18:1 enrichment for further use in fine catalytic oleochemistry becomes meaningless.
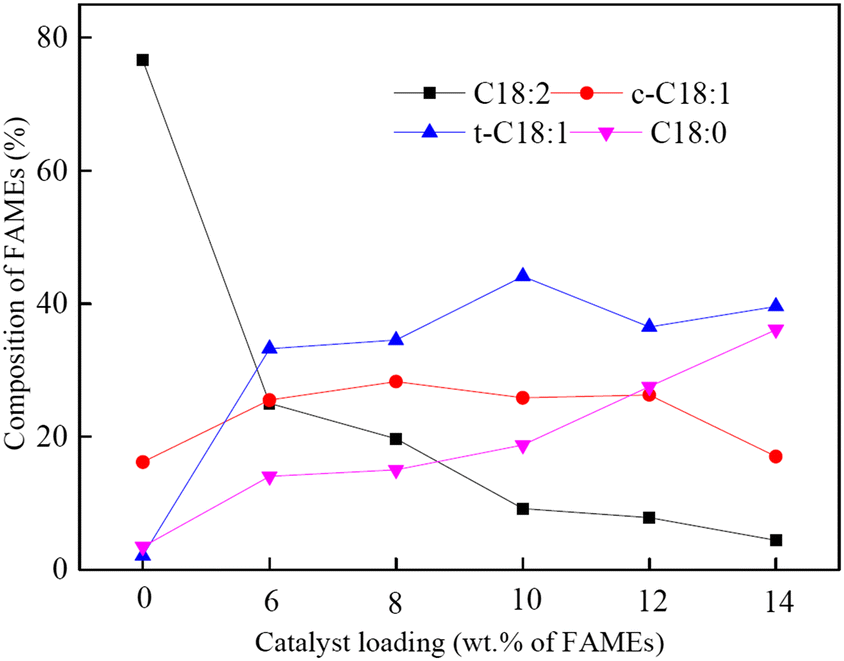 | ||
| Fig. 3 Effect of catalyst loading on the composition of FAMEs (7 g of FAMEs, 1.14 g of NaBH4, 40 mL of water, 85 °C, 150 min, pH 12).165 Reprinted with permission, Copyright (2021), Elsevier B. V. | ||
Bentonite modified by cetyltrimethylammonium bromide was used as a support for a Pd catalyst for hydrogen transfer using HCOONH4.166 The results of this study were modest, given that 8 wt% of catalyst and tenfold excess of reducing agent provided ∼70% conversion of C18:2 ester, and (Z)-C18:1 and (E)-C18:1 products were formed in an ∼1:1 ratio.
A comparative study on commercial Pd/Al2O3, Cu and Ni catalysts in a one-pot reaction combining the transesterification of soybean oil and partial hydrogenation of FAMEs obtained in supercritical methanol was carried out by Lee et al. in 2020.167 Relatively high conversions of TGs to FAMEs and C18:2 to C18:1 and C18:0 esters were observed when using Pd/Al2O3, and for all the catalysts, the decomposition of glycerol was observed; in this way, in the one-pot process, glycerol also serves as a hydrogen donor. Lee et al.168 proposed the use of Pd/ZSM-5 as a catalyst for the partial hydrogenation of FAMEs in supercritical MeOH. At 300 °C, 100 bar, 45:1 MeOH:oil ratio and 0.5 mg Pd per gOil loading, the FAME yield was 97.1%. In these experiments, ZSM-5 with an Si/Al2 ratio of 30, 70 and 270 with a 0.5 wt% of Pd loading was studied in comparison with the Pd/AlO3 system. In all cases, (Z)-/(E)-isomerization and C18:0 ester formation were substantial, and CC bond isomerization was also observed. RANEY® Ni as the catalyst and iPrOH as the hydrogen donor turned out to be active in the partial hydrogenation of Jatropha oil FAMEs (especially under the influence of microwave irradiation),169,170 and other alcohols were less efficient; however, (E)-C18:1 ester mainly contributed to the formation of the C18:1 products (Fig. 4).169
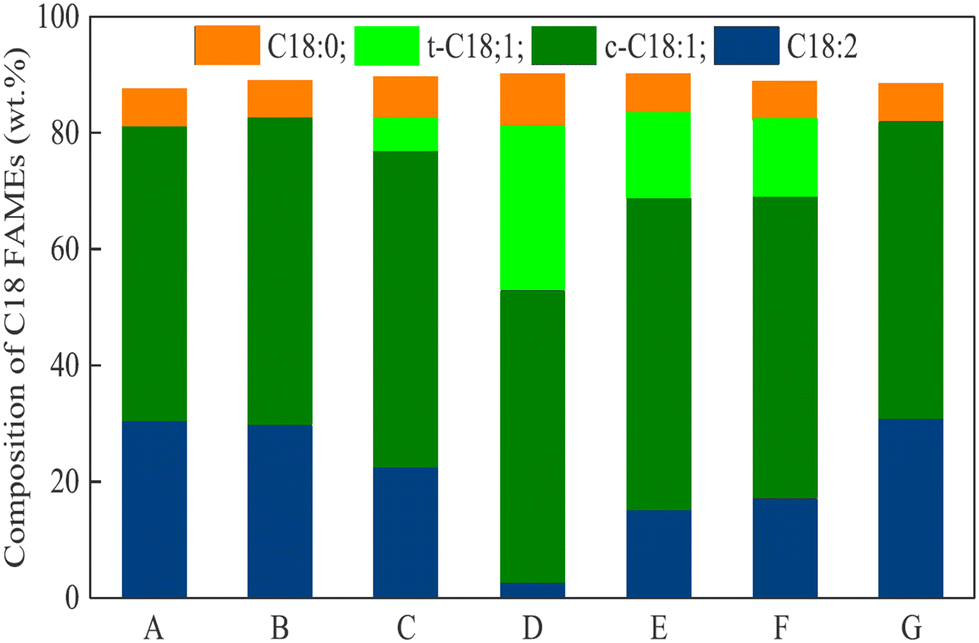 | ||
| Fig. 4 Changes in C18 FAME composition for different hydrogen donors under microwave-assisted hydrogen transfer reaction: (A) feed biodiesel, (B) MeOH, (C) EtOH, (D) iPrOH, (E) sec-butanol, (F) sec-pentanol, and (G) tBuOH.169 Adopted with permission, Copyright (2020), Elsevier B. V. | ||
In summary, the hydrogenation or hydrogen-transfer reduction of FAMEs increases the content of C18:1 esters; however, the isomerization and migration of CC bonds may hinder the further use of the reaction products in fine oleochemistry. At the current stage of investigations, it is worth talking only about the improvement of the quality of oils to the requirements of the food industry and upgrading of the characteristics of biodiesel to meet ASTM standards, and there are no catalytic systems providing complete elimination of C18:2 and C18:3 components with a marked increase in (Z)-C18:1 content: (E)-C18:1 and C18:0 products are also formed. The idea of oleate enrichment via selective hydrogenation seems promising, but known catalytic systems are inconsistent with GRC principles, and the further design of (Z)-C18:1-selective hydrogenation catalysts is still in demand.
Purification of oleates
In a recent study by Kangsadan et al., electrically driven separation (EDS) technology with a high voltage alternating current source was used for removing glycerol and other impurities from FAMEs prepared via the NaOH-catalyzed methanolysis of refined palm oil.173 The high efficiency of the EDS method in comparison with the commonly used gravitational settling was best illustrated by the value of remaining concentration of NaOH in the FAMEs obtained, where the separation efficiency reached up to 99.8% after 5 min vs. several days when using gravitational settling. In 2024, the EDS method was efficiently applied for the purification of non-edible oil-based FAMEs.174 EDS technology for the pre-treatment of FAMEs represents a vivid example of a method developed in compliance with GRC principles to avoid wastewater formation.
:1 [OA]/[sodium oleate] (“acid soaps”) was used for >99% purification of olive oil-based OA, and the yields were 39%–43%. The co-crystallization of FAs with urea alone was not particularly effective for mixtures with a high content of linoleic acid and relatively low (<50 wt%) content of OA,177 and prior low-temperature crystallization from MeOH178 was needed. However, complexation with urea can be used for reducing the saturated FA content in FA mixtures,179 which was used previously in the first stage of the separation of OA by low-temperature crystallization from petroleum ether.180 Another example of the use of urea complexation with FAs is reflected in an older study illustrating the actual “waste to treasures” concept by an example of the separation of OA with 95.5% purity from olive mill wastewater.181
OA with a purity of 98.7–98.9% was prepared from a mixture of FAs with 82% OA content via a rather complex five-step process including two distillation and three low-temperature crystallization steps.182
The purification of MO can also be performed via low-temperature crystallization, e.g. the combination of fractional distillation at 1 torr and double fractional crystallization (acetone, −60 °C) allowed to MO with 99.7 mol% purity to be obtained,183 but the composition of the starting FAMEs as well as the isolated yield of MO were not specified in this work.
In the laboratory, the use of high-pressure liquid chromatography (HPLC), proposed by Gunstone et al. in 1984,184 remains the most rapid method that allows MO (>99% purity) and other FA esters to be obtained on ∼10 g scale using 5% Et2O in petroleum ether as the eluent.
The development of new methods for the separation of FAMEs remains challenging. Recently, Bowden et al. developed a method for the separation of FAs or FAMEs based on their degrees of unsaturation using mixed matrix membranes containing nanometer-sized covalent organic frameworks (COF) having narrow pore size distributions (1.3, 1.8, 2.3, and 3.4 nm).185 As the degree of unsaturation increased, the FAs or FAMEs had a slower flux, in particular, when using COF with a pore size of 1.8 nm, methyl stearate (18:0) had a 5.9× faster flux than methyl linolenate (18:3).
Another prospective chemical technique based on the formation of Ag+ π-complexes with unsaturated compounds was proposed in the study by Liu et al.186 The use of the deep eutectic solvent [CF3SO3]Ag/acetamide provided more efficient binding of 18:2 and 18:3 esters, while 18:1 ester remained in the organic phase (n-hexane). As a result, the separation of MO of >98% purity was achieved.
In summary, conventional “old” methods for obtaining high-purity OA and MO are still employed and there are no competitive new methods for the deep purification of FAs and FAMEs, especially considering GRC principles (Table 4). Besides HPLC, which is useful in the laboratory but unlikely to be applied on a pilot and industrial level, the methods for the deep purification of oleates are labor-intensive and often require large amounts of toxic solvents (e.g. MeOH) and additional reagents (e.g. NaOH and HCl). Therefore, it is not strange that researchers investigating MO-based catalytic processes such as cross-metathesis (see below) choose to use FAMEs with a high oleate content, for example, obtained from high-oleic sunflower oil,187 without deep purification.
| Entry | Starting oleate content, wt% | Description of the separation method | Achieved oleate content, wt% | Yield, % | Ref. |
|---|---|---|---|---|---|
| a On an accrual basis. b No data. c Total yield. | |||||
| Purification of OA | |||||
| 1 | 80.2 | (i) Formation and separation of the urea complex, 1 kg FAs, 3.6 kg urea, 9 L MeOH, 83.9% FAs after hydrolysis; (ii) fractional distillation (10-plate column); (iii) crystallization from acetone (12 mL g−1), −50 °C | (i) 84; (ii) 90.4; (iii) 97.7 | (i) 88%; (ii) 43%a; (iii) 26%a | 175 |
| 2 | n. d.b | (i) Formation and separation of the urea complex, 5 kg FAs, 5 kg urea, 15 L MeOH, removal of the solvents; (ii) repeating of the stage I; (iii) acid soap crystallization, 2.8 kg FAs, 5.7 L MeOH, 0.5 eq. NaOH. Crystallization: 1st – 1.5 mL MeOH per g FAs, 2nd and 3rd – 2.5 mL MeOH per g FAs; (iv) distillation, 195–197 °C per 2 Torr | (i) n. d.; (ii) 87.1; (iii) n. d.; (iv) >99 (no impurities by GC) | (i) 77%; (ii) 74%; (iii) 43.4%; (iv) 89% | 176 |
| 3 | 42.2 | (i) Crystallization from MeOH, FA/MeOH 1:15 (w/v), −15 °C, 24 h; (ii) formation and separation of the urea complex |
88 | 86%c | 177 |
| 4 | 82 | (i) Cooling to −4 °C (separation of C18:0); (ii) distillation (182–184 °C/0.8 Torr); (iii) crystallization from acetone (−25 °C, 4:1 v/w); (iv) crystallization from aq. MeOH (−10 °C, 3:1 v/w); (v) distillation (0.5 Torr) |
98.7–98.9 | n. d. | 182 |
| 5 | 75 | (i) Formation and separation of urea complex with saturated acids; (ii) four crystallizations from petroleum ether (−60 °C) | 99.0 | ∼25% | 180 |
| Purification of MO | |||||
| 6 | 81.5 | (i) Preparation from olive oil (MeONa catalyst) and fractional distillation (164–179 °C per 4 Torr); (ii) formation and separation of urea complex, crystallization from acetone at −35 °C (separation of saturated esters) and at −60 °C; (iii) fractional distillation (184 °C per 4.2 Torr) | (i) 89; (ii) 97.2; (iii) 98.9 | (i) 61%; (ii) 70%; (iii) 94% | 175 |
| 7 | n. d. | (i) Distillation (3 Torr); (ii) crystallization from acetone (−37 °C, 10:1 v/w), separation of the filtrate; (iii) crystallization from acetone (−60 °C, 20:1 v/w) |
99.7 | n. d. | 183 |
| 8 | 77 | HPLC (petroleum ether/Et2O 95:5 v/v) |
>99 | >99% | 184 |
The separation of MO (OA) from other FAMEs (FAs) is just one facet of a more general problem in the preparation of oleate feedstocks for precision catalytic experiments. Besides substrate homogeneity (e.g. purity of oleate affecting the composition of the products of catalytic processes), the chemistry of oleates and related compounds is complicated by autoxidation with the formation of isomeric hydroperoxides.188 These hydroperoxides are unstable,189 and their decomposition during the treatment of the oil feedstocks may result in great variety of oxygenated organic compounds.190 Many of these oxygenates can be catalytic poisons. Furthermore, their analysis and selective elimination are difficult to perform in practice, and before precision catalytic experiments non-selective methods for deep purification, such as multiple-week storage over active absorbents (Magnesol, active Al2O3) are used.191 When creating a production, the feedstock separation, purification and storage before use should be developed considering the need to avoid autoxidation, but how to avoid the formation of undesirable autoxidation products in the growing plants? There is no definitive answer to this question and probably never will be.
Preparation and purification of ricinoleic acid
The preparation of RA from castor oil was the subject of a recent review,8 and the methods for the separation of castor oil were also discussed very recently.192 Castor oil is comprised of 89–92 mol% of RA, and its other main components are linoleic (∼4%), oleic (∼3%), stearic (∼1%), and linolenic (<1%) acids. The high content of RA in castor oil significantly simplifies its treatment. The industrially implemented Twitchell hydrolysis is based on the use of lipophilic sulfonic acids as a catalysts and provides up to 98% conversion of castor oil, while the Colgate-Emery process gives only 92% conversion in the case of RA. The deep purification of RA via distillation is complicated due to the lower thermal stability of this acid in comparison with OA and other FAs. Base-catalyzed hydrolysis is used in the laboratory despite the obvious flaws caused by the need for additional acidification and extraction stages.8 However, due to the high content of the target compound in castor oil, its purification via the separation of metal ricinoleates was efficient in the case of RA.193 The formation of estolides is an evident and inevitable side-process in the hydrolysis of castor oil, and higher yields and purity of RA can be achieved using a two-stage process that includes methanolysis of the oil with subsequent hydrolysis of more reactive methyl ricinoleate194 under milder conditions.8 Consequently, the conventional industrial and laboratory methods for the synthesis of RA are energy- and material-consuming.Therefore, only natural enzymatic approaches have attracted the attention from researchers. In the 2020s, several scientific papers were published. Bose and Goswami proposed the use of 1-butyl-3-methylimidazolium hexafluorophosphate IL to facilitate the hydrolysis of castor oil catalyzed by Porcine pancreas lipase, and the positive impact of IL on the yield of RA was detected, but only 52% yield of RA was obtained.195 The higher efficiency of immobilized lipase demonstrated by similar catalytic systems in the hydrolysis of oleate-enriched TGs with regard to castor oil revealed an increase in the substrate conversion.196 At the same time, only partial conversion (26.3%) was achieved, and thus the practical application of this method seems doubtful. Very recently, porcine pancreas lipase was immobilized on a composite carrier, consisting of Fe3O4 and MOF, surface modified by an N-(3-aminopropyl)imidazole-based IL.197 Under the optimized conditions, the conversion was 45.7%, and the catalytic system retained 89.4% of its initial activity after 10 cycles.
Microbial conversion is a cleaner and greener approach for the production of ricinoleic acid from castor oil. In 2023, Sarma et al. separated and identified Aspergillus flavus BU22S, which is capable of producing RA.198 The conversion of castor oil to RA by Aspergillus flavus BU22S was optimized using the response surface methodology (RSM), but the real yield of RA was only 47 g kgoil−1, whereas the oil loss was 127 g kgoil−1.199 With the use of the JHYL-R146 strain, FAs with an RA and linoleic acid content of 74% and 11% were obtained, respectively.200 Obviously, the microbial approach is unlikely to have any practical use at present, given that further studies are needed.
In this way, the recently studied “greener” approaches for the separation of RA do not comply with GRC principles in the parameter of atom economy. It is quite possible that highly active and selective catalysts for the hydrolysis or transesterification of castor oil will be found in the near future. However, existing technologies are quite effective and there are no demands for complex and expensive studies in this field.
Metathesis of oleates and related compounds
Fundamentals of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C bond metathesis and its use in oleochemistry
C bond metathesis and its use in oleochemistry
The catalytic metathesis of unsaturated compounds entails the redistribution of olefin fragments by the scission and formation of CC bonds. The active catalytic species represent carbene complexes, and the reaction proceeds via four-membered metallacycles, resulting in the formation of new olefins and new carbene complexes (Scheme 3a).201 Metathesis is attractive due to its high atom-efficiency, and a range of metathesis-based processes has been successfully applied in the chemical industry, where the production of α-olefins from ethylene and other large-scale processes typically use heterogeneous Re, Mo, W or Ni-containing catalysts.202 Metathesis has also found wide application in fine chemistry and pharmaceuticals,201 and the search for efficient homogeneous and single-site supported catalysts has consistently attracted attention from researchers.201,203
 | ||
| Scheme 3 (a) Main idea of catalytic metathesis. (b) Metathesis in oleochemistry: self-metathesis (SM) and cross-metathesis (CM) using the example of MO; acyclic diene metathesis (ADMET) polymerization and ring-closing metathesis (RCM) using the example of dec-9-enoates. | ||
Therefore, it is natural that the metathesis of oleates, their derivatives and related compounds has been intensively studied in recent years.19–21,27,204–206 The presence of an internal CC bond in the non-symmetrical oleate molecule allows it to undergo self-metathesis (SM) and cross-metathesis (CM), when using esters of unsaturated alcohols and OA (or ω-unsaturated dec-9-enoic or undec-10-enoic acid, obtained from OA and RA, respectively), acyclic diene metathesis polymerization (ADMET) and ring-closing metathesis (RCM) can be performed (Scheme 3b).
In the 1990s and early 2000s, the metathesis of oleates and other derivatives of unsaturated fatty acids was studied using the conventional homogeneous Ru-based Grubbs I, II catalysts, Hoveyda–Grubbs I, II catalysts, Mo-based Schrock catalysts (Fig. 5) and their analogs. Similar catalysts were efficient in SM and ADMET processes, but remarkable progress in the CM of oleates and RCM of alkenyl oleates and related compounds was achieved in recent years with the use of new catalytic systems (see below).
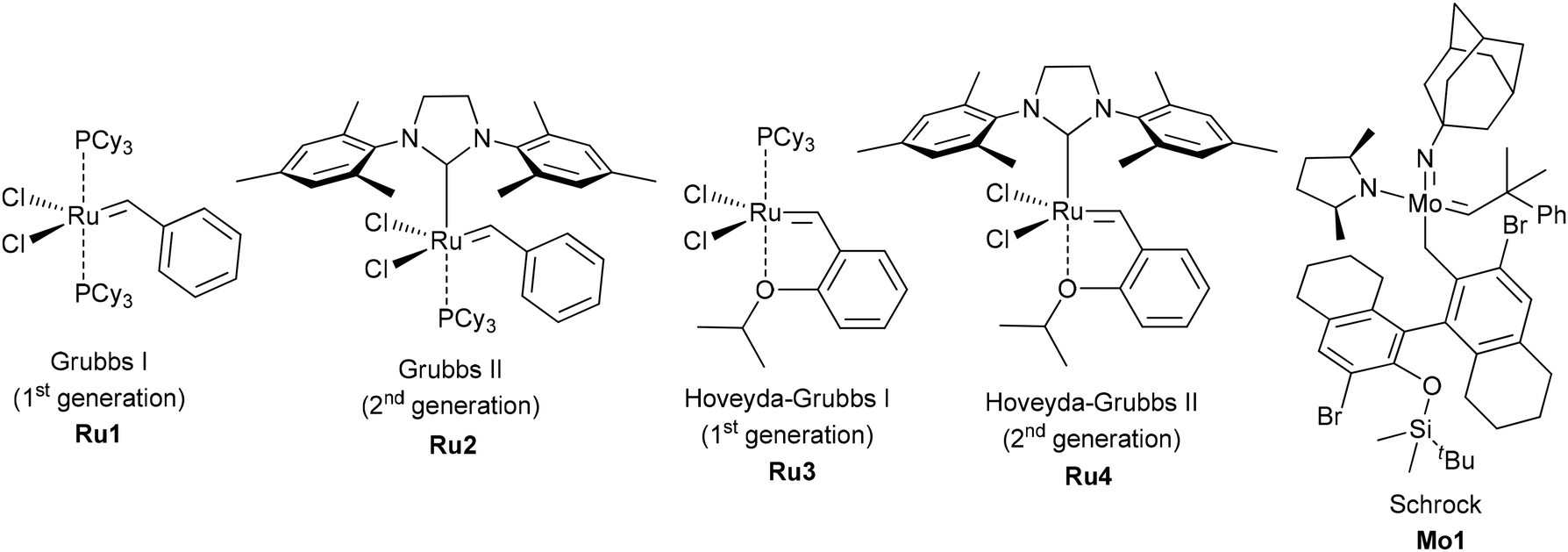 | ||
| Fig. 5 Conventional catalysts studied in the metathesis of oleates in 1990s–2000s. | ||
Self-metathesis of oleates
The self-metathesis of MO yields dimethyl octadec-9-enedioate and octadec-9-ene (Scheme 3a). This reaction is reversible, and equilibrium can be reached rapidly even with the use of the supported Hoveyda–Grubbs II catalyst Ru4.207 The reaction products can be formed in (E)- and (Z)-configurations, and the reverse process may lead to the (E)-isomer of MO, methyl elaidate. In 2020, Carrasco et al. showed that Ru4 and its analogs, bearing 2,3-iPr2C6H3 groups in the N-heterocyclic carbene (NHC) ligand and various trans-ligands with respect to the NHC unit, demonstrate similar catalytic behavior, and the composition of the reaction mixture was thermodynamically controlled.208 In this way, the SM of oleates is of dubious practical value, but octadec-9-enedioic acid may still find application in the synthesis of polyesters.27In 2023, Krzesiński et al. studied SM of technical grade FAMEs (MO and ∼20 wt% of methyl linoleate), catalyzed by different Ru complexes.209 To minimize the yield of the undesired product Me(CH2)7CHCHCH2CHCH(CH2)7COOMe, which complicates the separation of dimethyl octadec-9-enedioate, the screening of available catalysts was conducted, and Ru4 (100 ppm) was selected given that this complex provided the formation of the target diester and side product in a ratio of more than 95:5. Diester, prepared from 300 g of pretreated technical grade MO, was isolated by fractional distillation, purified by crystallization from MeOH at −30 °C (37% yield, (E)-/(Z)- ratio of 97:3), and then used in the synthesis of copolymers. In other works (for example ref. 210), SM of MO was studied as a model process, demonstrating the catalytic characteristics of the complexes.
A much more interesting catalytic process, including the self-metathesis of MO, was proposed by Mecking et al.211 Under the action of [1,2-C6H4(CH2PtBu2)2]Pd(OTf)2 (Pd1), dimethyl octadec-9-enedioate isomerized to α,β-unsaturated diester (the driving force for this process is the lower solubility of the product in MeOH), which under the action of Ru4, formed a longer-chain diester (Scheme 4). The CHCH bond conjugated with the COOMe fragment turned out to be inert in self-metathesis, and the addition of but-2-ene or hex-3-ene was needed in the final stage.
 | ||
| Scheme 4 “Chain doubling” strategy for the preparation of ultra-long-chain α,ω-diester.211 | ||
Cross-metathesis of oleates
The cross-metathesis of oleates is more attractive in terms of obtaining value-added products. From the points of view of value and ease of separation of the reaction products, the ethenolysis of oleates (Scheme 3) is the most promising and economically beneficial due to the use of the inexpensive olefin (ethylene) and increased interest in dec-1-ene and dec-9-enoic acid derivatives.19 The added attraction of the ethenolysis of oleates is driven by the possibility of producing ethylene from bioethanol212,213 and recent progress in the development of nonenzymatic methods for ethanol production.214 In this way, the ethenolysis of oleates may be considered a green process completely based on renewable raw materials.Cross-metathesis using higher α-olefins was successfully commercialized,205e.g. but-1-ene-based technology for natural oil conversion215,216 was implemented by Elevance in a plant oil refinery with a capacity of 180 kMT in Gresik (Indonesia).217 An ethylene-based process was scaled-up relatively recently, in 2016, by Elevance Renewable Sciences Inc. and Versalis,218 but no additional reports about its full-scale industrial implementation were found in recent scientific periodicals. The delayed development of technology for the ethenolysis of oleates is primarily attributed to the problems of catalytic activity and deactivation of the catalyst (see below), and ways to solve these problems have been found over the last few years.
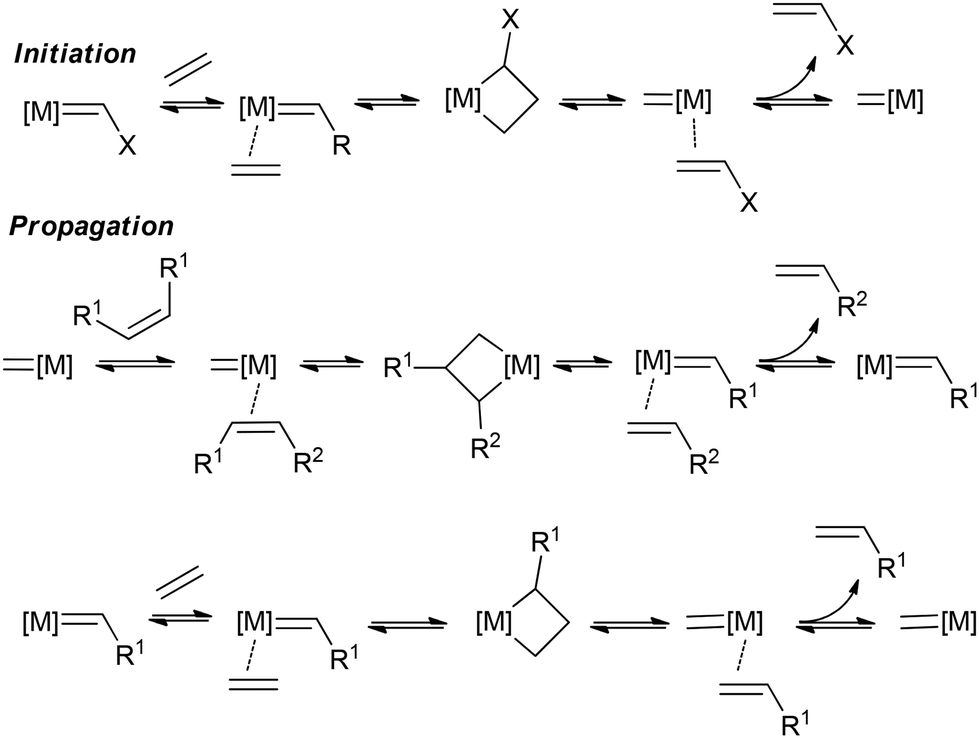 | ||
| Scheme 5 Common mechanism for the cross-metathesis in the example of ethenolysis.219 | ||
Ru-based catalysts are usually highly active in metathesis reactions in the absence of ethylene, but loss of activity was observed when ethylene was used as a reagent.220 As was shown in the study by Nascimento et al.,221 the decomposition of Ru-based catalysts during ethenolysis can occur via two pathways, i.e., bimolecular decomposition involving RuCH2 species and β-hydride elimination (Scheme 6). Also, the rate of decomposition directly depends on the type of ligand L, NHC or cyclic alkyl amino carbene (CAAC), the substituents in these ligands, and the halogen atoms at the Ru center (Fig. 6). DFT modeling of bimolecular coupling revealed a complex overall mechanism with the formation of inactive “LRuX2” species (Scheme 6c). Combined experimental and theoretical studies showed the lower stability of CAAC complexes in comparison with NHC derivatives in bimolecular decomposition, which makes it necessary to search for catalytic systems that are quite active at ppm concentrations. Alternatively, the experimental finding that CAAC-Ru complexes are more resistant to β-H elimination in comparison with Ru1–Ru4 and other NHC-Ru systems222 is of great importance.
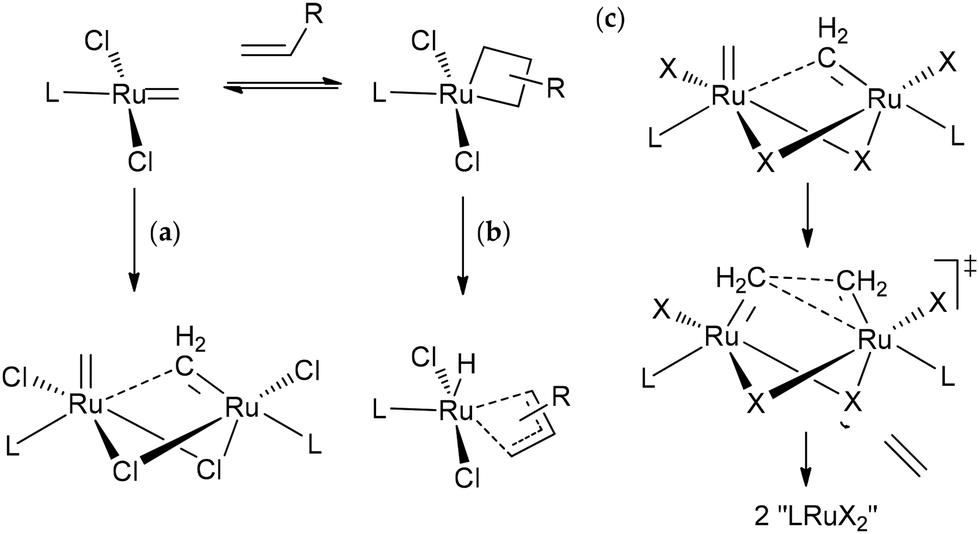 | ||
| Scheme 6 Intrinsic decomposition pathways in Ru-catalyzed ethenolysis: (a) bimolecular decomposition and (b) β-hydride elimination. Key steps in the bimolecular decomposition of identified by DFT calculations (c).221 | ||
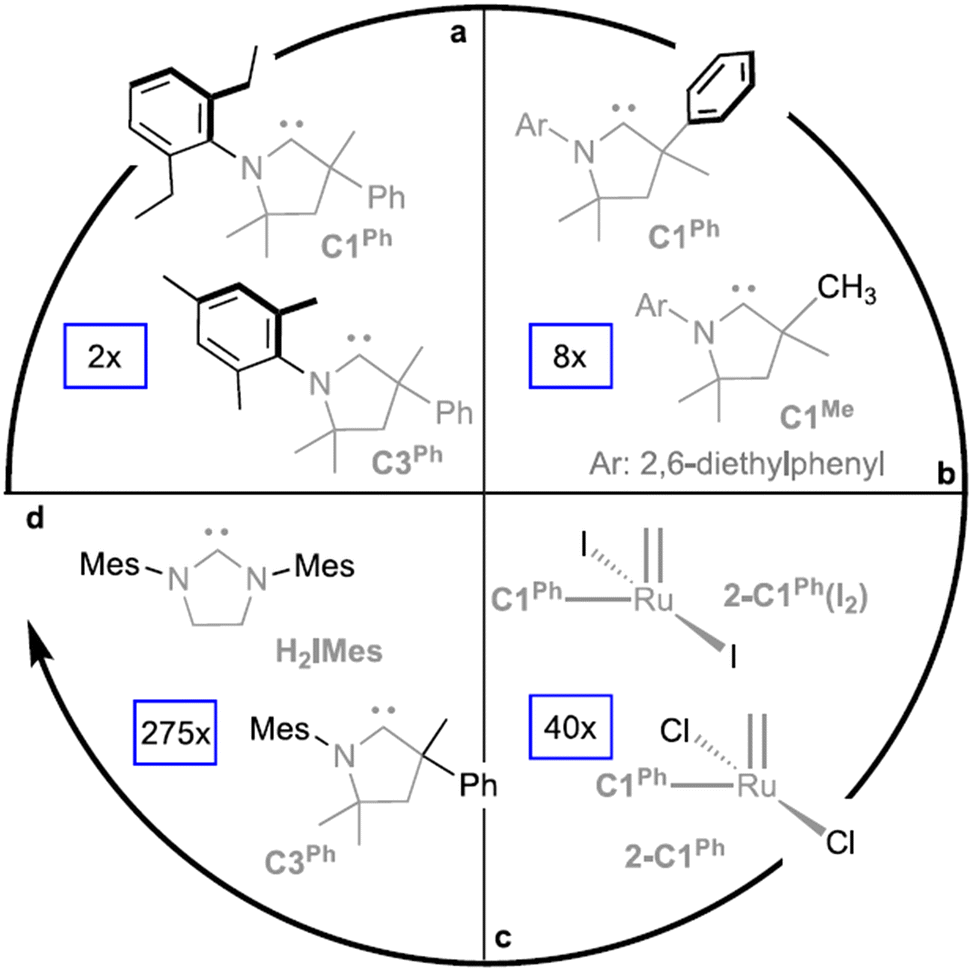 | ||
| Fig. 6 Relative rates (text in blue boxes) of bimolecular decomposition as a function of the structural changes shown in black: (a) NAr substituents. (b) Substitution at Cα (the quaternary center α to the carbene carbon). (c) Anionic ligand: chloride vs. iodide. (d) NHC vs. CAAC: H2IMes vs. its closest analog, C3Ph.221 Reprinted with permission, Copyright (2021), the American Chemical Society. | ||
A year later, Młodzikowska-Pieńko and Trzaskowski conducted a comparative theoretical study on the decomposition of different Ru complexes and showed that bimolecular coupling is the preferable deactivation pathway.223 In this study, CAAC complexes were shown to be less susceptible to decomposition via β-hydride elimination in comparison with NHC systems, thus confirming the experimental results reported by Nascimento and Fogg.222 In a later study,224 the higher stability of CAAC-Ru complexes against β-hydride elimination was attributed to the trans-effect of the ligand. In this study, it was also shown that the trans-effect hinders the unproductive catalytic cycle with the involvement of RuCH2 species and the C2H4 molecule.
It should also be considered that oleochemicals represent a specific type of raw materials with distinctive impurities, including water. The hydrolytic degradation of Grubbs I and II catalysts Ru1 and Ru2 during ethenolysis was attributed to the formation of [Cy3PMe]+ species, respectively.225 The role of water in the deactivation of more advanced NHC and CAAC Ru complexes not containing the PCy3 ligand was clarified recently by Blanco and Fogg.226 They showed that the coordination of H2O to the Ru atom accelerates both β-elimination in the metallacyclobutane intermediate and bimolecular decomposition of the catalysts, even though in the absence of alkene, the NHC and CAAC Ru complexes were stable in the presence of traces of water.
Another important aspect of the action of Ru-based catalysts is the prospect of their immobilization or support. An attempt to prepare a supported Ru catalyst using Hoveyda–Grubbs II complex Ru4 was reported by Nieres et al. in 2023.227 They proposed that Ru4 interacts with the silica support via hydrogen bonds through the chloride ligands, and possibly by coordination of the Ru center to the ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OH surface groups. The experimental studies of the stability of this catalyst showed that the main identified organic product from the decomposition of the supported catalyst in wet air is 2-isopropoxybenzaldehyde, and the proposed mechanism of this process was confirmed by DFT modeling (Fig. 7). We think that the computation results are of general importance for NHC-Ru catalysts used under insufficiently inert conditions, where the participation of O2 in the presence of H2O in the deactivation of the catalysts in real catalytic processes cannot be underestimated.
Si–OH surface groups. The experimental studies of the stability of this catalyst showed that the main identified organic product from the decomposition of the supported catalyst in wet air is 2-isopropoxybenzaldehyde, and the proposed mechanism of this process was confirmed by DFT modeling (Fig. 7). We think that the computation results are of general importance for NHC-Ru catalysts used under insufficiently inert conditions, where the participation of O2 in the presence of H2O in the deactivation of the catalysts in real catalytic processes cannot be underestimated.
 | ||
| Fig. 7 Energy profile of insertion of O2 to the RuC bond, with subsequent release of 2-isopropoxybenzaldehyde. All energies are given in kJ mol−1, and relevant distances in Å are included in each structure.227 Reprinted with permission, Copyright (2023), Wiley-VCH. | ||
The conventional Ru-based catalysts (Grubbs I, Grubbs II, and Hoveyda–Grubbs I) turned out to be ineffective in cross-metathesis oleochemistry.21,228,229 In 2018, Grisi et al. proposed the replacement of the mesitylene fragments in the Hoveyda–Grubbs II catalyst with alkyl or cycloalkyl fragments without a positive outcome as applied to MO ethenolysis.230 In 2020, Ullah et al. reported that Hoveyda–Grubbs II catalyst Ru4 showed a TON of 92000 in the ethenolysis of corn oil FAMEs at 50 ppm loading of the catalyst (50 °C, 4 bar),231 but these data are not quite consistent with the results in the other studies. In 2022, Öztürk et al. reported that Ru4 can be encapsulated in hollow mesoporous silica, which led to the formation of a more stable and productive catalyst for the ethenolysis of MO and FAMEs.232 However, the problem of low catalytic activity of conventional metathesis catalysts was resolved by the design of new Ru complexes.
CAAC-based Ru complexes (Fig. 8) were proposed and intensively studied in the ethenolysis of MO by Grubbs et al. in the mid-2010s.191 When used in experiments with ethylene of 99.95% purity (10 Bar, 40 °C) and high-purity MO (degassed and stored over Al2O3 for over 1 month), these complexes showed different productivities depending on the structure of the CAAC ligand (Table 5). Some catalysts demonstrated TONs of more than 105, but the values of conversion (<42%) and yield (<39%) are insufficient for the industrial implementation of these catalysts.
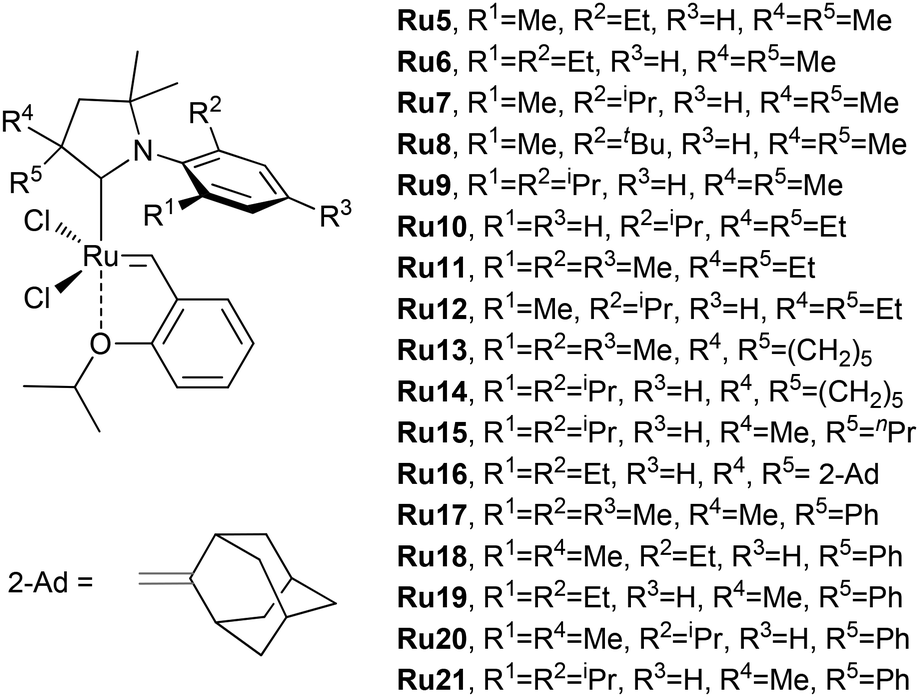 | ||
| Fig. 8 Cyclic alkyl amino carbene (CAAC) complexes studied in the ethenolysis of MO by Grubbs et al.191 | ||
| Entry | Catalyst | Conv.b,c, % | Selectivityd,e, % | Yieldf, % | TONg |
|---|---|---|---|---|---|
| a Reaction conditions: catalyst (3 ppm), C2H4 (10 bar, 99.95% purity), 40 °C, 3 h. b Determined by GC using dodecane as the internal standard. c Conversion = 100 − [(final mol of MA) × 100]/[initial mol of MA]. d Selectivity for ethenolysis products over self-metathesis products. e Selectivity = 100 × (mol of products)/[(mol of products) + (2 × mol of self-metathesis products)]. f Yield = conversion × selectivity/100. g TON = yield × (initial mol of MA/mol of catalyst)/100. | |||||
| 1 | Ru5 | 37% | 86% | 32% | 110000 |
| 2 | Ru6 | 42% | 88% | 37% | 120000 |
| 3 | Ru7 | 59% | 92% | 54% | 180000 |
| 4 | Ru8 | 18% | 94% | 17% | 57900 |
| 5 | Ru9 | 19% | 97% | 18% | 60000 |
| 6 | Ru10 | 22% | 63% | 14% | 47000 |
| 7 | Ru11 | 26% | 86% | 22% | 73000 |
| 8 | Ru12 | 42% | 92% | 39% | 130000 |
| 9 | Ru13 | 19% | 78% | 14% | 47000 |
| 10 | Ru14 | 13% | 97% | 13% | 43000 |
| 11 | Ru15 | 16% | 97% | 15% | 50000 |
| 12 | Ru16 | <5% | — | — | — |
| 13 | Ru17 | 41% | 83% | 34% | 110000 |
| 14 | Ru18 | 46% | 85% | 39% | 130000 |
| 15 | Ru19 | 48% | 88% | 43% | 140000 |
| 16 | Ru20 | 57% | 94% | 54% | 180000 |
| 19 | Ru21 | 47% | 98% | 46% | 150000 |
| 1 | Ru5 | 37% | 86% | 32% | 110000 |
| 2 | Ru6 | 42% | 88% | 37% | 120000 |
To increase catalytic activity in the ethenolysis of MA and the stability of CAAC-based Ru complexes, Skowerski proposed the replacement of the CH(2-iPrOC6H4) fragment in the catalysts presented in Fig. 5 with CAAC and the synthetically available 3-phenylindenylidene fragment (Fig. 9).233 At 5 ppm concentrations, complex Ru26 showed comparable activities with Ru21, whereas complexes Ru22 and Ru24 showed negligible activities. The reaction of Ru26 with CuCl resulted in dimer [(CAAC)(3-phenylindenylidene) RuCl(μ-Cl)]2Ru29 with increased productivity and higher initiation rate in comparison with Ru26.234
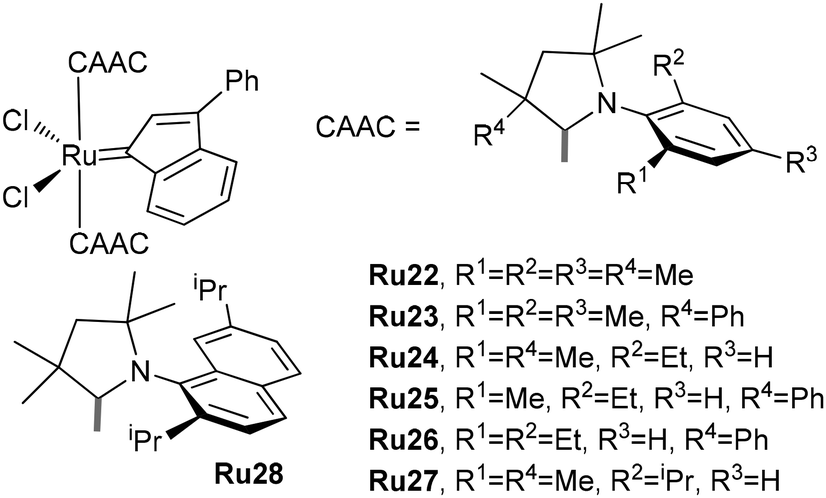 | ||
| Fig. 9 Bis-CAAC Ru complexes studied by Skowerski et al.233 | ||
In 2020–2023, numerous works on the further development of Ru-based catalysts based on CAAC ligands and their structural analogs for the ethenolysis of MO were published, with varying success. For example, in 2020, Grubbs et al. synthesized Ru complexes based on six-membered CAAC ligands; at 40 °C, 10 bar of C2H4 pressure, and 20 ppm [Ru] loading, these new complexes showed moderate productivity (TON = 116–1720), and in the comparative experiment, complex Ru21 had TON = 24180.235 The design of photo-switchable CAAC complexes containing a PhNNC6H4 substituent236 is only of theoretical interest (37% MO conversion at 2 mol% catalyst loading).
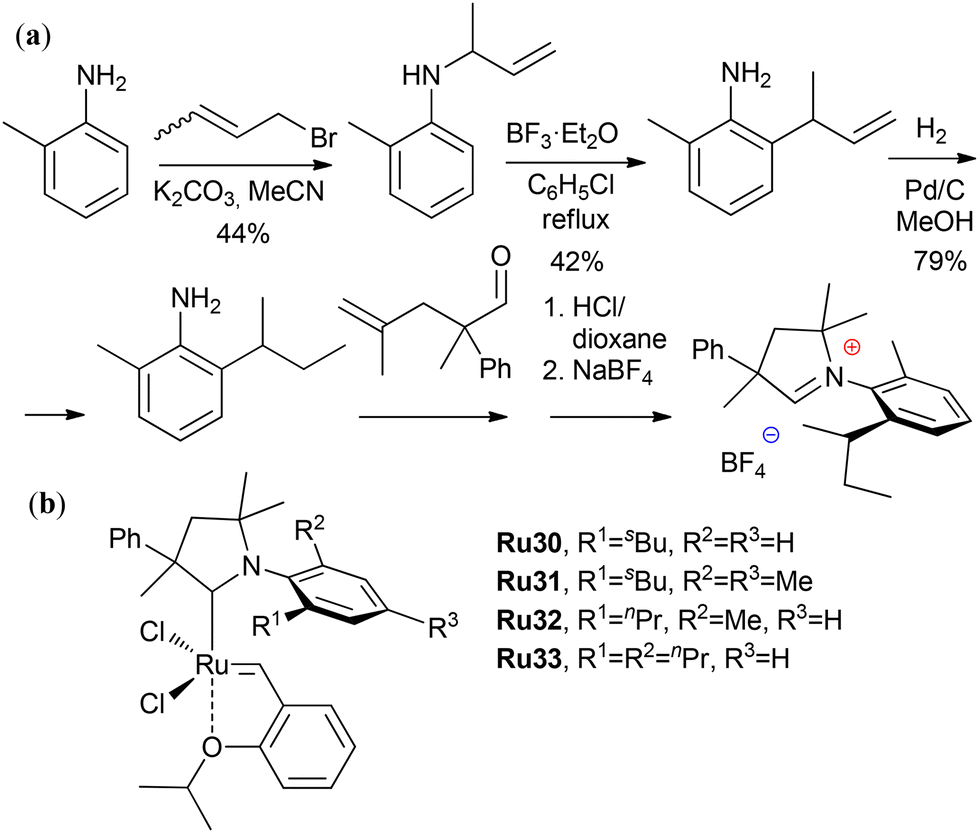
The synthetic availability of the CAAC-Ru catalyst is a crucial factor for assessing the efficiency of the process as a whole. For example, complexes Ru7 and Ru20 had high activities (Table 5) but limited availability.191 In 2023, Grela et al. reported research on finding simple, efficient and scalable approaches for the preparation of CAAC ligands and their corresponding complexes. In particular, the employment of aza-Claisen rearrangement and subsequent reduction of the resulting anilines proved to be an efficient method for the synthesis of inexpensive analogs of Ru7 and Ru20 (Scheme 7).237 The sec-butyl-substituted complex Ru30 at a 3 ppm loading was superior to Ru7 in the ethenolysis of MO, achieving a TON of up to 192000.
 | ||
| Scheme 7 (a) Synthesis of CAAC ligand precursor using aza-Claisen rearrangement (using the example of 2-sBu-6-Me-aniline). (b) Synthetically available CAAC-Ru complexes.237 | ||
Another way to increase the synthetic availability of CAAC-Ru complexes is based on the use of ibuprofen-derived aldehyde, resulting in ligand precursors and catalysts (Fig. 10) with increased lipophilicity and solubility in n-hexane (an order of magnitude higher than that of Ru20).210 The results of the MO ethenolysis experiments with the ibuprofen-derived CAAC Ru catalysts are presented in Table 6.
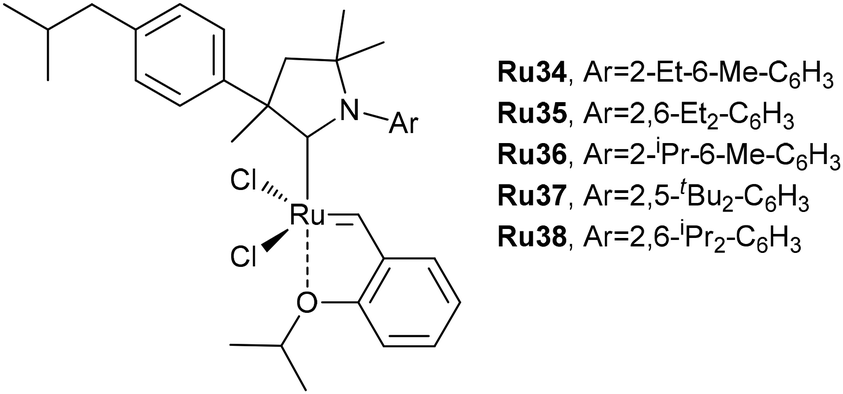 | ||
| Fig. 10 Ibuprofen-derived CAAC Ru catalysts.210 | ||
210
| Catalyst | Loading, [ppm] | C2H4 grade | Conv., % | Select., % | Yield, % | TON |
|---|---|---|---|---|---|---|
| a 99.95% purity. b 99.995% purity. | ||||||
| Ru20 | 3 | 3.5a | 42% | 96% | 43% | 144000 |
| 1 | 4.5b | 28% | 97% | 27% | 274000 |
|
| Ru34 | 3 | 3.5 | 36% | 90% | 32% | 107000 |
| 1 | 4.5 | 18% | 89% | 16% | 160000 |
|
| Ru35 | 3 | 3.5 | 54% | 91% | 49% | 164000 |
| 1 | 4.5 | 24% | 92% | 22% | 221000 |
|
| Ru36 | 3 | 3.5 | 57% | 95% | 54% | 181000 |
| 1 | 4.5 | 38% | 96% | 36% | 362000 |
|
| Ru37 | 3 | 3.5 | 68% | 87% | 59% | 197000 |
| 1 | 4.5 | 30% | 90% | 27% | 274000 |
|
| Ru38 | 3 | 3.5 | 55% | 97% | 54% | 179000 |
| 1 | 4.5 | 27% | 99% | 27% | 270000 |
|
In most CAAC-Ru complexes, the N–Ar fragment occupies the space above the benzylidene ligand. Polish researchers proposed that the structure of CAAC provides “inverted” coordination of the carbene ligand,238 and synthesized several new complexes with a similar inverted coordination (Fig. 11) and high catalytic activity in ethenolysis (Table 7).
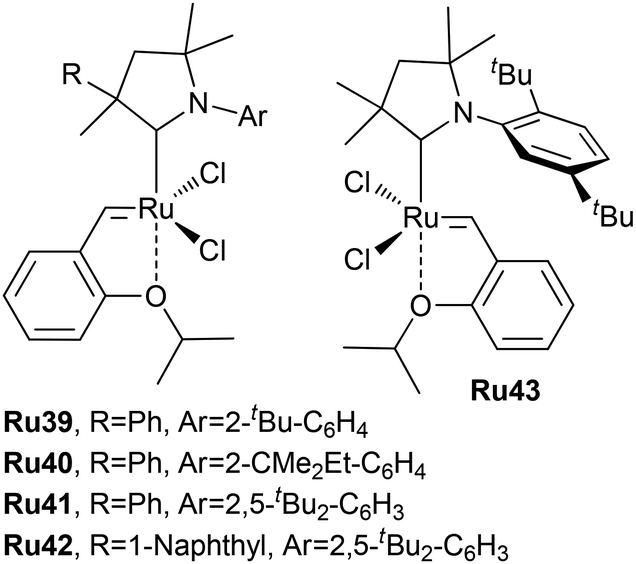 | ||
| Fig. 11 CAAC Ru complexes with “inverted” coordination Ru39–Ru42 and complex Ru43, analog of Ru41 with “normal” coordination.238 | ||
238
| Catalyst | Loading, [ppm] | Conv., % | Select., % | TON |
|---|---|---|---|---|
| a For 3 ppm of catalyst, ethylene purity of 99.95%, and for 1 and 0.5 ppm, ethylene purity of 99.995%. | ||||
| Ru7 | 3 | 59% | 92% | 180000 |
| 1 | 34% | n.r. | 340000 |
|
| Ru20 | 3 | 54% | 96% | 173000 |
| 0.5 | 19% | 97% | 360000 |
|
| Ru39 | 3 | 83% | 88% | 244000 |
| 0.5 | 40% | 93% | 744000 |
|
| Ru40 | 3 | 81% | 89% | 242000 |
| 0.5 | 29% | 93% | 536000 |
|
| Ru41 | 3 | 80% | 90% | 240000 |
| 0.5 | 23% | 90% | 409000 |
|
| Ru42 | 3 | 80% | 89% | 237000 |
| 0.5 | 38% | 95% | 723000 |
|
| Ru43 | 3 | 56% | 35% | 65000 |
| 0.5 | 13% | 8% | 20000 |
|
Additional experiments with 0.5 ppm catalyst loading revealed the fundamental preference of catalyst Ru40 in comparison with benchmark complexes Ru7 and Ru20, where complex Ru40 with an “inverted” geometry turned out to be insensitive to the quality of ethylene, demonstrating the same activity (TON ∼ 740000) when using ethylene of 99.995% and 99.95% purity. Besides, the new complexes showed higher thermal stability in comparison with Ru20. The authors proposed that the inversion of the CAAC ligand creates an open site around the RuCH fragment, which probably allows more facile coordination of the substrate.
In research on the design of efficient CAAC-Ru catalysts, the recent study by Gawin et al.187 deserves special attention. To increase the stability and productivity of the catalysts, a new spirocyclic alkyl amino carbene (SCAAC) type of ligands was proposed. Starting from the corresponding cyclic ketones, the SCAAC ligand precursors (Scheme 8) were obtained in 15%–55% yield. The SCAAC Ru complexes with a “normal” geometry (XRD data, Fig. 12) were studied in the ethenolysis of MO and FAMEs in comparison with benchmark CAAC complexes (Table 8).
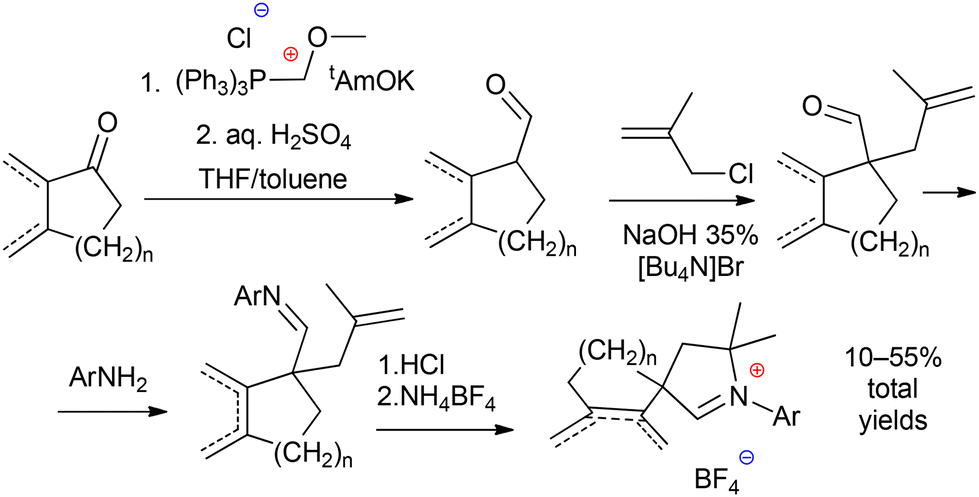 | ||
| Scheme 8 Synthesis of SCAAC ligand precursors.187 | ||
 | ||
| Fig. 12 New spirocyclic CAAC Ru complexes.187 | ||
| Catalyst | Loading, [ppm] | Conv.a, % | Yielda, % | Select.a, % | TON |
|---|---|---|---|---|---|
| a Per methyl dec-9-enoate. | |||||
| Ru7 | 1 | 18.9% | 17.1% | 90.7% | 171000 |
| 0.5 | 14.5% | 13.7% | 94.3% | 274000 |
|
| Ru20 | 1 | 26.2% | 25.4% | 91.7% | 254000 |
| 0.5 | 16.4% | 16.3% | 99.6% | 326000 |
|
| Ru21 | 1 | 26.2% | 25.4% | 96.8% | 254000 |
| 0.5 | 17.7% | 17.4% | 98.3% | 348000 |
|
| Ru44 | 1 | 20.2% | 14.5% | 71.5% | 145000 |
| Ru45 | 1 | 37.0% | 31.9% | 86.3% | 319000 |
| Ru46 | 1 | 52.4% | 50.2% | 95.8% | 502000 |
| 0.5 | 31.9% | 30.9% | 96.9% | 618000 |
|
| Ru47 | 1 | 56.2% | 53.8% | 95.7% | 538000 |
| Ru48 | 1 | 49.2% | 47.1% | 95.7% | 471000 |
| Ru49 | 1 | 56.4% | 53.5% | 94.9% | 535000 |
| Ru50 | 1 | 66.1% | 62.3% | 94.2% | 623000 |
| 0.5 | 50.4% | 48.2% | 95.6% | 964000 |
|
| Ru51 | 1 | 53.4% | 51.3% | 96.0% | 513000 |
| Ru52 | 1 | 68.2% | 64.3% | 94.3% | 643000 |
| Ru53 | 1 | 53.2% | 50.8% | 95.5% | 508000 |
| Ru54 | 1 | 55.5% | 52.8% | 95.1% | 528000 |
| Ru55 | 1 | 49.5% | 48.0% | 96.8% | 480000 |
| 0.5 | 30.5% | 29.6% | 97.2% | 592000 |
|
| Ru56 | 1 | 4.0% | 3.8% | 96.0% | 38000 |
| Ru57 | 1 | 22.9% | 22.3% | 97.4% | 223000 |
In the experiments with purified MO, the catalyst loading of 0.1 ppm was used, and the complex Ru50 showed unprecedented catalytic activity, with the TON of 2610000. The studies on the geometry of the complexes failed to determine the relationship between structure and catalytic activity, causing researchers to pay attention to catalyst stability issues. Firstly, the relationship between initiation rate and catalytic productivity was established. It was found that the Ru complexes with a too high or too low initiation rate demonstrated low activities, whereas the complexes with intermediate kinit (Ru46 and Ru50) were active. Another important aspect was deactivation. In decomposition experiments, complexes Ru7 and Ru20 decomposed rapidly, Ru21 was more stable, and Ru46 and Ru50 showed remarkable thermal stability. Further experimental and theoretical studies revealed the highest stability of Ru50 against both the β-hydride elimination and bimolecular deactivation pathways. In this way, SCAAC Ru complexes represent the most promising candidates for industrial use at present.
However, most of the catalytic studies on the ethenolysis of MO and FAMEs were limited by the measurement of the substrate conversion, product yields and selectivity using gas chromatography (GC), and thus descriptions of the preparative examples with distillation of the reaction products are rare. For example, when using 25 ppm of Ru6, the ethenolysis of rapeseed oil FAME (0.87 kg) resulted in the separation of 132 g of dec-1-ene (94% purity) and 267 g of methyl dec-9-enoate (89% purity).216
Besides CAAC-based Ru catalysts, complexes of Ru with other types of ligands were studied in recent years. Byun et al. synthesized a series of imidazo[1,5-a]pyridin-3-ylidene carbene derivatives (Fig. 13a).239 The ethenolysis of MO required 500–100 ppm loading of these catalysts, which demonstrated up to 50% conversion at 60 °C after 3 h. The highest TON was 6700 (20 ppm of Ru63, conversion of 19%, and selectivity of 71%). In 2020, the same group of researchers described the synthesis of “abnormal” fluorinated imidazo[1,5-a]pyridine NHC Ru complexes (Fig. 13b) and their use in the ethenolysis of MO, also using Cy3P–CuCl cocatalyst.240
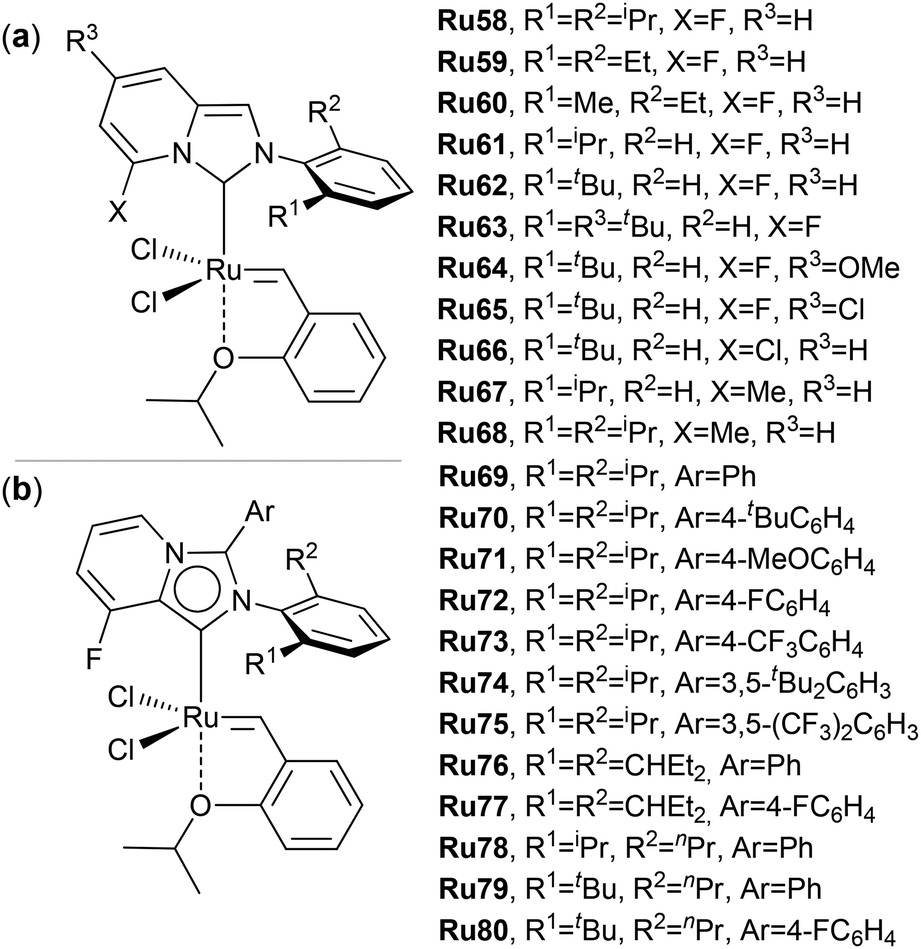 | ||
| Fig. 13 (a) Imidazo[1,5-a]pyridin-3-ylidene carbene complexes.239 (b) “Abnormal” imidazo[1,5-a]pyridin-3-ylidene carbene complexes,240 studied in the ethenolysis of MO. | ||
The highest TON of 110000 was demonstrated by complex Ru80, with moderate conversion (33%) and high selectivity (95%). Fig. 14 illustrates the limited potential of similar catalytic systems for industrial application.
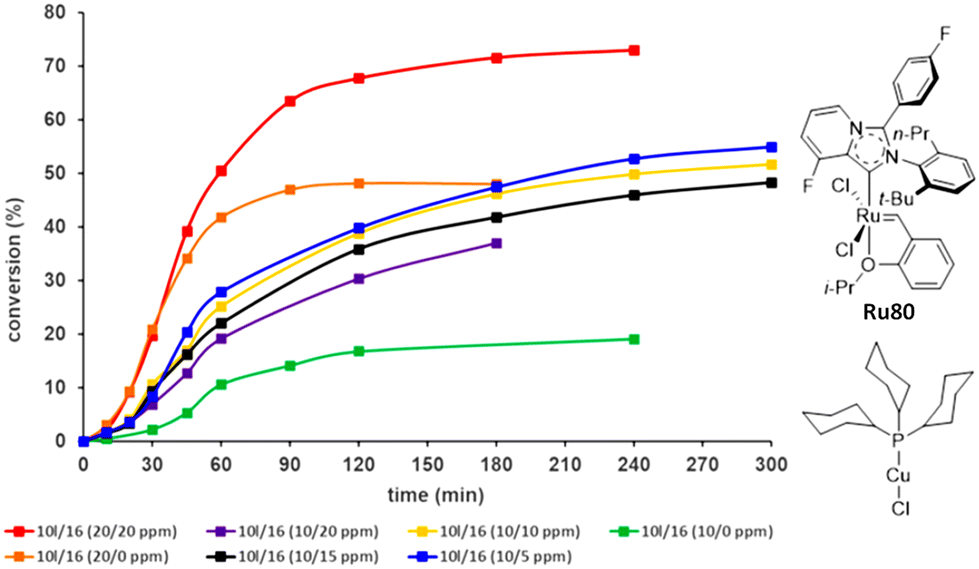 | ||
| Fig. 14 Reaction profiles for the ethenolysis of MO with catalysts Ru80 and Cy3P–CuCl at [Ru/Cu] loadings of 20/20 ppm (red), 20/0 ppm (orange), 10/20 ppm (purple), 10/15 ppm (black), 10/10 (yellow), 10/5 ppm (blue), and 10/0 ppm (green); reaction conditions: 40 °C, 10 bar C2H4.240 Reprinted with permission, Copyright (2020), the American Chemical Society. | ||
The results of the DFT modeling failed to explain the effect of the Cu(I) co-catalyst in terms of the decrease in the activation energy.240 However, the fact that the process was accelerated in the presence of Cu(I) should not be ignored in the further development of catalysts for MO → dec-1-ene and methyl dec-9-eneoate transformation.
A series of acyclic aminooxycarbene (AAOC)-ligated Ru complexes, Ru81–Ru84 (Fig. 15), was synthesized and studied in the ethenolysis of MO by Hong et al.241 At a loading of 50 ppm, these complexes were comparable to CAAC-based catalysts in terms of activity and selectivity (Table 9), and when the concentration of Ru84 decreased to 5 ppm, a yield of 50% and TON of 100000 were achieved.
 | ||
| Fig. 15 Acyclic aminooxycarbene Ru complexes.241 | ||
| Catalyst | Conv., % | Selectivitya, % | Yield, % | TON |
|---|---|---|---|---|
| a As a ratio of the sum of the target products (mol) to the total amount of target products and doubled sum of the side products, internal olefin and the ester of dicarboxylic acid (mol). | ||||
| Ru81 | <1% | 92% | <1% | 230 |
| Ru82 | 82% | 89% | 73% | 14500 |
| Ru83 | 80% | 90% | 72% | 14000 |
| Ru84 | 86% | 91% | 78% | 16000 |
| Ru6 | 80% | 87% | 70% | 14000 |
| Ru9 | 61% | 93% | 57% | 11400 |
A family of indol-2-ylidene Ru complexes, Ru85–Ru90 (Fig. 16), was synthesized and studied in the ethenolysis of MO by Kim et al.,242 and the least sterically hindered complex Ru89 showed TON = 61200 and selectivity of 95%. Complexes Ru85–Ru90 also demonstrated high thermal stability.
 | ||
| Fig. 16 Indol-2-ylidene Ru complexes.242 | ||
Dimerization of the RuCH2 species is one of the deactivation pathways. Thus, to avoid this and solve the problem of elimination of the catalyst from the reaction products, heterogenization of the catalyst can be considered as a prospective approach. Well-defined supported or immobilized metathesis catalysts were the subject of the early review by Copéret and Basset,243 and several attempts have been made in recent years to prepare efficient heterogeneous metathesis catalysts.
An attempt to prepare active CAAC Ru catalysts included supporting complexes containing an N,N-dimethylaniline substituent (Fig. 17) on the ion-exchange resin Amberlyst-15.244 However, experiments on the ethenolysis of MO produced discouraging results, where the maximum TON of only 360 was achieved after 24 h at 40 °C.
 | ||
| Fig. 17 CAAC Ru complexes bearing N,N-dimethylaniline functionalities.244 | ||
In 2020, Hwang et al. prepared and studied CM catalysts by grafting MeReO3 on a spinel-type mesoporous ZnAl2O4 support.245 However, these catalysts demonstrated low activities (TON 3.0–5.6 at 45 °C and 10 bar C2H4), making further studies meaningless. The same year, the scientific groups of Gauvin and Taoufik synthesized and studied W- and Mo-based catalysts supported on mesoporous silica, and the heterogeneous catalyst prepared from OMo(CH2tBu)3Cl showed a TON of up to 5000.246 Recently, Tangyen et al. reported the results of their study on the immobilization of inherently inactive MoOCl4 on the surface of mesoporous SiO2.247 The best results were achieved at 7 mol% Mo loading (80 °C, 2 bar of ethylene, 1 h), 82% conversion of MO and >99% selectivity. The high catalyst loading is not compatible with GRC principles, but the use of the highly toxic SnMe4 as an activator for the catalyst puts an end to the prospects of the practical use of similar catalytic systems. Obviously, the use of heterogeneous catalysts allows the problem of metal removal to be avoided, and thus important in homogeneous catalysis, but the invented systems must not go beyond actual requirements of modern technologies (synonymous with the 12 GRC principles) and common sense.
Concluding this section devoted to ethenolysis of MO, the studies on tandem catalytic systems should be mentioned. In the study by M. Chen and C. Chen,248 the ethenolysis of MO in the presence of CM catalyst Ru19 was followed by copolymerization, catalyzed by complexes Pd2 or Pd3 (Fig. 18 and Table 10). This tandem process resulted in a terpolymer that represents LLDPE with polar functionalities.
 | ||
| Fig. 18 Pd copolymerization catalysts.248 | ||
The ethenolysis of MO in combination with CC bond migration processes in supercritical CO2 (scCO2) was studied by Mecking et al. in 2023.249 Using Ru26 and [1,2-C6H4(CH2PtBu2)2]Pd(OTf)2 (Pd1) as the catalysts, statistical mixtures of α-olefins and methyl ω-alkenylcarboxylates were obtained from FAMEs. As can be seen in Fig. 19, the composition and ratio of the reaction products were dependent on the composition of the raw material, FAMEs prepared from high-oleic sunflower oil (3% 16:0, 3% 18:0, 93% 18:1, 1% 18:2) and soybean oil (11% 16:0, 4% 18:0, 24% 18:1, 49% 18:2, 12% 18:3).
 | ||
| Fig. 19 Sequential catalysis of plant oils. (a) FAME composition of high-oleic sunflower and soybean oil. (b) GC of the obtained product mixture (HC3:1 to HC10:1 corresponds to alkenes and E3:1 to E10:1 to esters) starting from FAME, EVE – ethyl vinyl ether, quenching agent. (c) and (d) Product distribution of alkenes (blue) and esters (yellow) starting from high-oleic sunflower and soybean oil, respectively. Dots represent the theoretical product distributions from complete ethenolysis and isomerization. (e) Biorefining concept comprising sequential ethenolysis, isomerization and ethenolysis catalysis for the polyunsaturated FAME 18:2 and FAME 18:3. Reaction conditions: (1) ethenolysis 0.06 mol% Ru26, 55 °C, 3 bar ethylene, p (total) = 250 bar, catalyst added as solution in 6 mL CH2Cl2, 24 h; (2) isomerization 0.4 mol% Pd3 per double bond, 85 °C, p (total) = 500 bar, catalyst added as solution in 5.9 mL CH2Cl2 and 0.1 mL MeOH, 48 h; (3) ethenolysis 0.4 mol% Ru26 per double bond, 55 °C, 15 bar ethylene, p (total) = 450 bar, catalyst added as a solution in 6 mL CH2Cl2, 24 h.249 Reprinted with permission, Copyright (2023), Wiley-VCH. | ||
The preference for higher α-olefins in comparison with ethylene was clearly demonstrated on the example of propenolysis of MO (Scheme 9), catalyzed by “abnormal” NHC complexes,240 resulting in multicomponent reaction mixtures (Table 11). The reaction equilibria could be shifted to cross-metathesis products by increasing the α-olefin/MO ratio; however, an increase in the α-olefin concentration and α-olefin/MO ratio facilitated deactivation of the catalyst.252
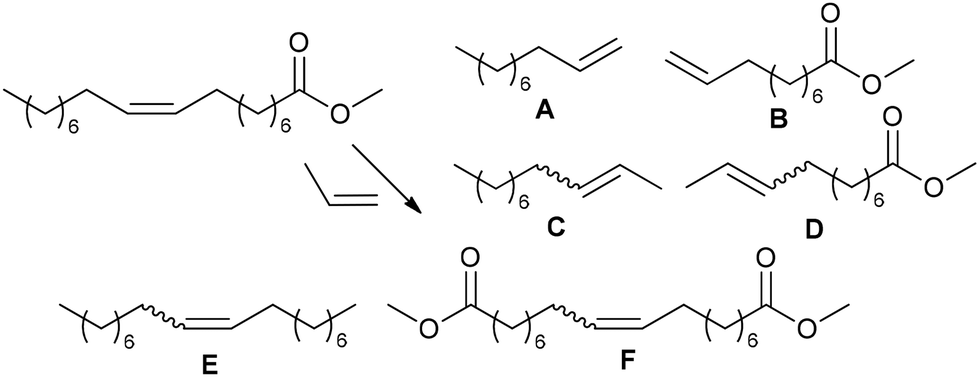 | ||
| Scheme 9 Propenolysis of MO.240 | ||
| Entry | Catalyst, ppm | Cy3CuCl, ppm | Conv., % | A/B/C/D yield, % | A/B TON |
|---|---|---|---|---|---|
| a 5.5 bar of C3H6. | |||||
| 1 | Ru79, 5 | 5 | 47% | 23/26/23/21% | 47000/53000 |
| 2 | Ru79, 3 | 3 | 12% | 23/27/22/23% | 77000/90000 |
| 3 | Ru79, 1 | 2 | 17% | 11/12/8/6% | 110000/120000 |
| 4 | Ru80, 3 | 3 | 50% | 28/27/24/20% | 93000/91000 |
| 5 | Ru80, 1 | 2 | 21% | 13/13/9/7% | 130000/130000 |
| 6a | Ru80, 1 | 2 | 33% | 21/18/16/11% | 210000/180000 |
However, the idea of using functionalized olefins in the cross-metathesis of oleates is still feasible. In 2020, Grisi et al. reported the results of their study on the catalytic activity of Ru4 and NHC Ru complexes Ru93–Ru96 (Fig. 20) in the CM of ethyl oleate with (Z)-but-2-ene-1,4-diyl diacetate.253 At high catalyst loadings (1 mol%), complex anti-Ru95 containing cyclohexyl and 2-isopropylphenyl substituents surpassed the activity and selectivity of CM using the benchmark catalyst Ru4.
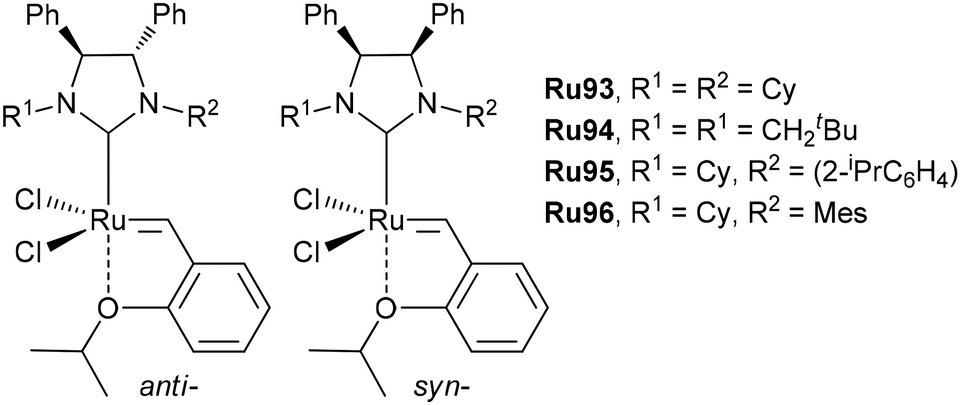 | ||
| Fig. 20 NHC Ru complexes studied in CM with (Z)-AcOCH2CHCHCH2OAc.253 | ||
A valuable result was obtained by Kajetanowicz et al.210 during the study of the cross-metathesis of methyl dodec-10-enoate with acrylonitrile (2 eq.), where CAAC complexes Ru20, Ru34 and Ru35 (300 ppm) at 70 °C provided at least 96% yield of nitrile 22, a precursor of a valuable monomer used in the production of Arkema's Nylon-11 (Rilsan® Polyamide 11) (Scheme 10).
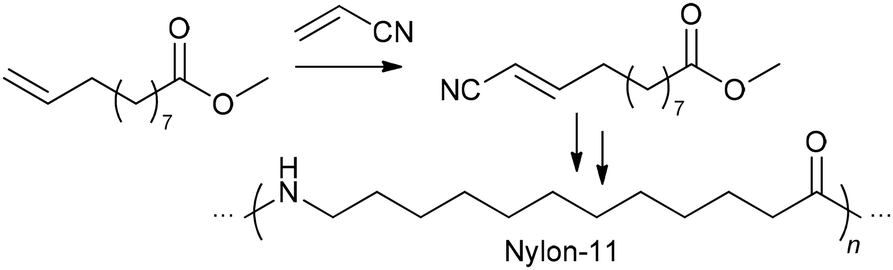 | ||
| Scheme 10 CM of methyl dodec-10-enoate to produce nitrile 22,210 a precursor for Nylon-11 production. | ||
From the GRC point of view, the cross-metathesis of oleates represents, in theory, an extremely prospective approach to obtain valuable products in terms of atom economy, reagent and solvent safety and energy efficiency. However, not all scientists believe this. The main scientific problem in the industrial implementation of the most prospective and cost-effective ethenolysis of MO lies in the separation of the reaction products. Elimination of the active catalyst species represents a great challenge, especially when using highly active catalysts (<1 ppm loading). This problem is evidently underestimated in pursuit of the highest activity rates, but eventually this problem needs to be resolved. The recent studies by Grela et al.238 and Gawin et al.187 provided an excellent basis for the creation of brilliant oleochemical technology fully compliant with GRC principles. The goal remains to develop efficient methods for the separation of the reaction products with elimination of the catalyst remnants. However, a solution to this problem may not be easy.
Ring-closing metathesis of oleates and related compounds
The idea of RCM (Scheme 3) implies the presence of CC structural fragments quite distant from each other in the substrate molecule. As applied to oleochemistry, RCM results in the formation of macrocyclic unsaturated lactones. Evidently, unsaturated esters of both OA and derivatives of OA and RA, ω-unsaturated dec-9-enoic and undec-10-enoic acids, can be regarded as substrates of this reaction. Among the ω-unsaturated acids, undec-10-enoic is the most synthetically available, and several works have been devoted to studying the RCM of alkenyl undec-10-enoates. In an early study,254 RCM of dec-9-enyl undec-10-enoate and undec-10-enyl undec-10-enoate, catalyzed by Cl2Ru(PCy3)CHCHCPh2 (Ru97, ∼1 mol%, benzene, 60 °C) resulted in the formation of 20-membered (Z/E 57:43) and 21-membered (Z/E 60:40) macrolactones in the yields of 83 and 82%, respectively. ω-Alkenyl oleates gave 19- and 20-membered (Z/E 71:29) lactones in yields of 65% and 63%, respectively. The substrate concentration was in the range of 5–7 mM. These data are given here as a reference point for a comparative analysis with the results in more recent works, as described and discussed below.
A series of Ru complexes was studied in the cyclization of CH2CH(CH2)8COO(CH2)4CHCH2 (10 mM), and the best yield (83%) was obtained for Grubbs II catalyst Ru2.255 Cyclization of the same substrate (5 mM solution in toluene) in the presence of Ru26 (Scheme 11) at 60 °C resulted in the formation of cyclic lactone (E/Z = 65:35) in 91% yield (GC data).233 Dimeric derivative Ru29 was found to be even more active in this reaction at a concentration of 100 ppm.234
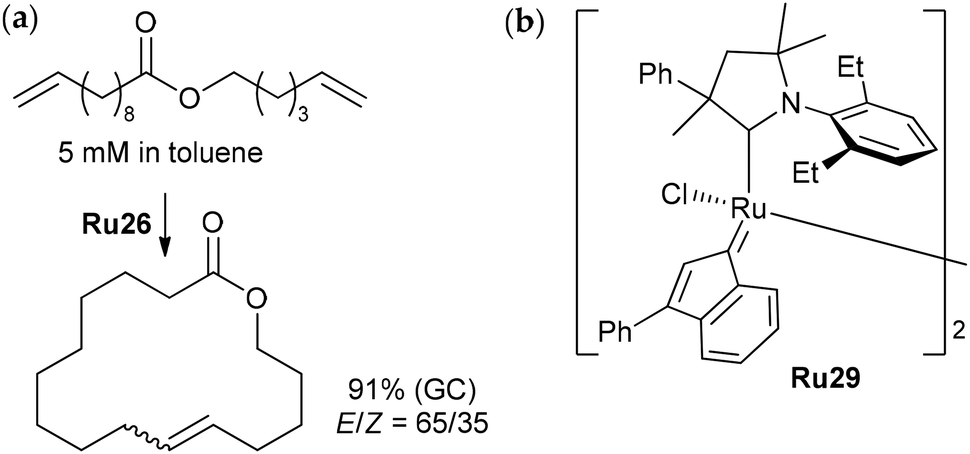
One of the main problems in the synthesis of macrolactones is the need for low concentrations of the substrates (Ziegler-Ruggli dilution principle). In compliance with the dilution principle, even Grubbs and Hoveyda–Grubbs catalysts were proven to be quite efficient in the RCM of but-3-en-1-yl oleate and related compounds at a concentration of 1 mM.256 71%–84% yields of macrolactones were reported for RCM reactions in CH2Cl2 at a substrate concentration of 50 mM using 1 mol% of Grubbs II catalyst,257 which is contrary to the results in previous and recent investigations.
The fundamental study by Grela et al.258 aimed to use the inherent reversibility of olefin metathesis to produce macrolactones at concentrations much higher than commonly used for RCM (1–5 mM). They proposed that under carefully selected conditions, backbiting of ADMET oligomers can yield the RCM macrocyclic product, which can be removed from the reaction mixture in vacuo, considering the possibility of the migration of the CC bond. ADMET of 5-hexenyl undec-10-enoate and subsequent depolymerization in the presence of 1 mol% of catalyst II (Fig. 21) resulted in the formation of (E/Z) mixtures of 11–21-membered macrocycles with a maximum yield of 24% (Fig. 22).
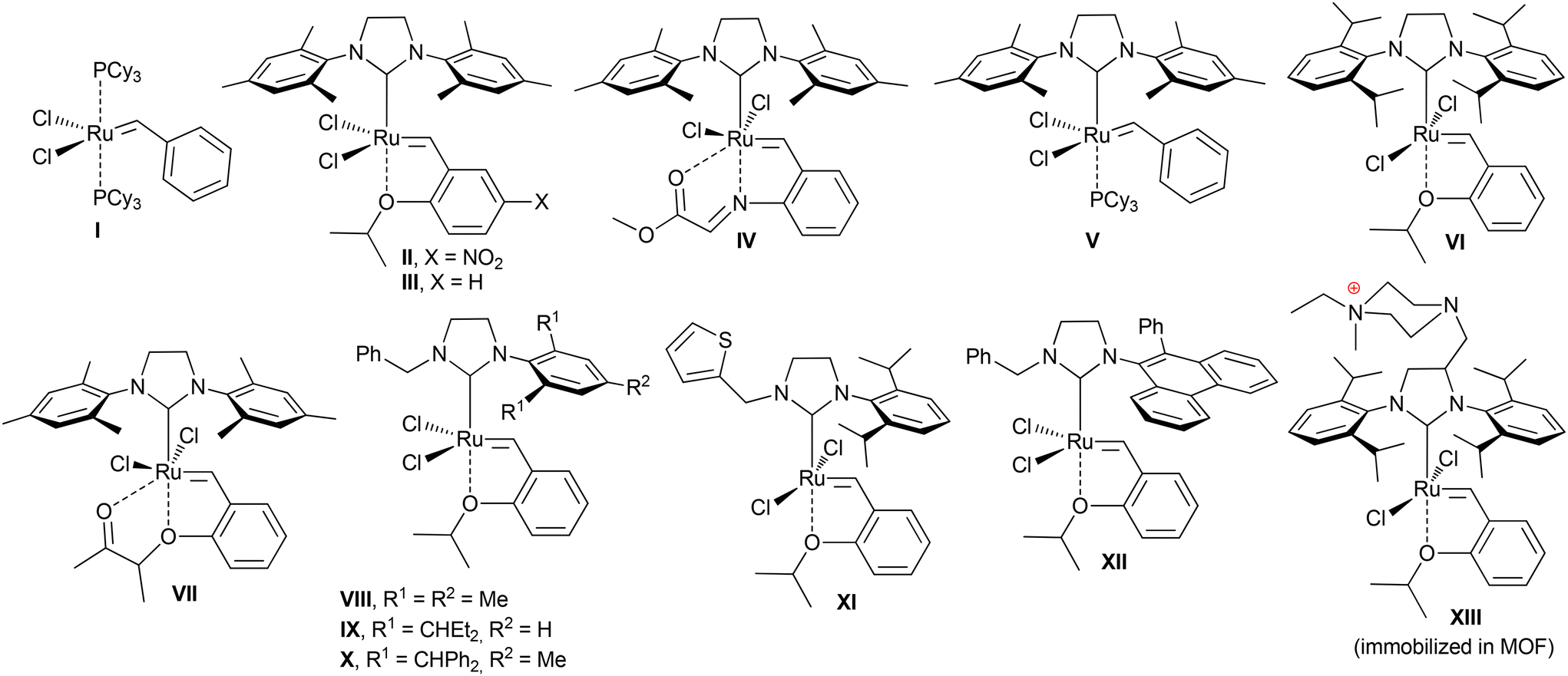 | ||
| Fig. 21 Ru complexes studied in RCM of alkenyl oleates.258 | ||
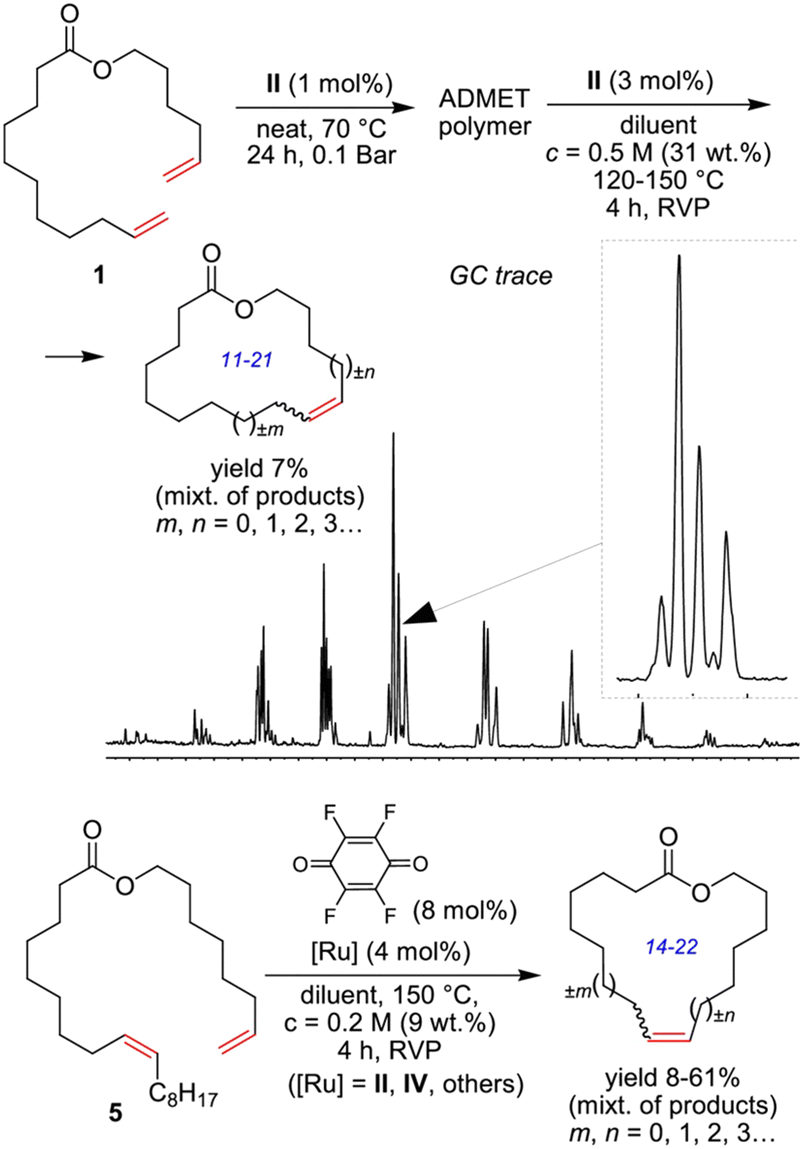 | ||
| Fig. 22 ADMET, and then RCM depolymerization approach to macrolactones and GC trace of the product.258 Reprinted with permission, Copyright (2018), the American Chemical Society. | ||
More promising results were obtained when ADMET and RCM were conducted in the presence of an additive known to cease CC bond isomerization, 2,3,5,6-tetrafluorobenzoquinone; however, CC bond migration and the formation of macrocyclic lactones of different ring sizes was observed. A comparative study on Ru complexes was conducted in experiments in paraffin oil at 110 °C under high vacuum (10−6 mbar), with the substrate concentration of 0.2 M. The results of the experiments were qualitatively dependent on the type of the catalyst used (Fig. 23).
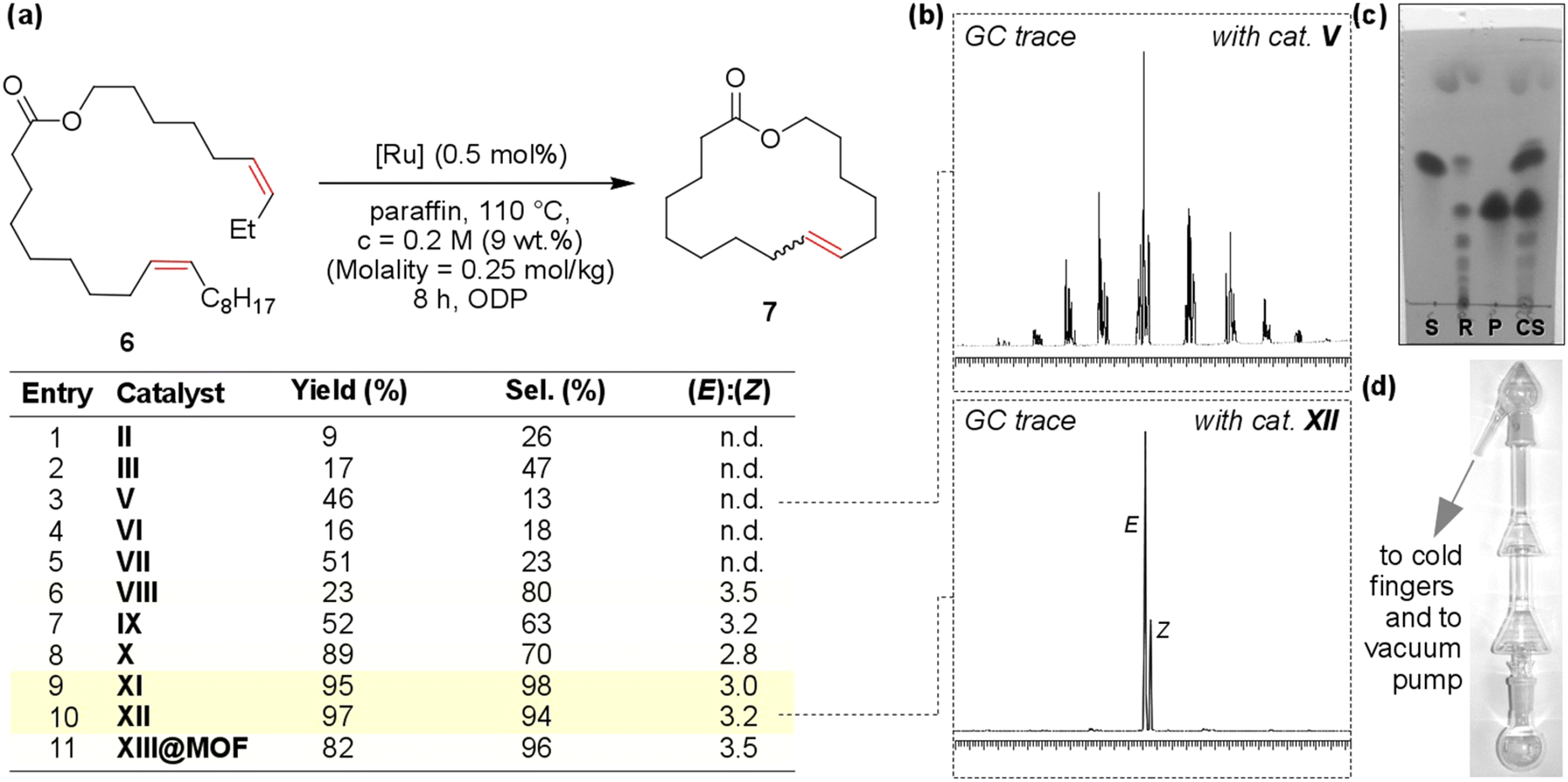 | ||
| Fig. 23 RCM experiments. (a) Yield and selectivity for the formation of macrolactone (selectivity = (Int. of E + Z)/(Int. of all products)). (b) GC traces of products obtained using complexes V and XI. (c) TLC analyses of reaction mixture from experiment ceased after 20 min. S – standard of the substrate, R – sample of reaction mixture, P – standard of the product, and CS – all three together. (d) Example of a reaction/distillation Hickman glass apparatus.258 Reprinted with permission, Copyright (2018), the American Chemical Society. | ||
Under the optimized conditions, several alkenyl oleates were transformed to 13-, 16-, 17- and 19-membered unsaturated lactones, and the yields were 60%–93%, and (E)/(Z) ratio was 2.1–4.9 depending on the cycle size. The most important finding in the study by Grela et al.258 was the need for appropriate catalyst design, where endowing Ru catalysts with high thermal stability is necessary for RCM selectivity when the separation of the RCM product is performed by vacuum distillation.
Also, in 2019, Buchmeiser et al. proposed an alternative route to solve the problem of the substrate concentration in RCM.259 They utilized the factor of spatial confinement by preparing functionalized Ru-based catalyst Ru98 (Scheme 12a), selectively immobilized inside ordered mesoporous silica SBA-15 with pore diameters, d, of 50 and 62 Å. It was shown that the selectivity of macrocyclization substantially increased with an increase in the hydrodynamic diameter of the substrate (Scheme 12b) when using the SBA-1550-based catalyst, e.g. dodecane-1,12-diol-based diester was formed in up to 60% of the RCM product at 25 mM concentration.
 | ||
| Scheme 12 (a) Synthesis of Ru catalyst capable of immobilization on silica. (b) Structures and hydrodynamic diameters (DOSY-NMR data) of the RCM substrates.259 | ||
Besides the macrolactone yield and selectivity of RCM (the absence of CC bond migration), the (E)/(Z) ratio is also important. One intriguing result was obtained by Grela et al. when studying the catalytic activities of Ru complexes containing a dithiocatechol fragment. Consequently, complex Ru99 showed very low activity but unprecedented (Z)-selectivity (Scheme 13) in the formation of unsaturated lactone at high substrate concentrations (200 mM instead of the typically used 5 mM).260
 | ||
| Scheme 13 Structures of the complexes Ru99260 and Ru100262 (a) and their unprecedented (Z)-selectivity in RCM (b). | ||
Generally, complex Ru99 demonstrates unusual chemistry; for example, it decomposes in solution under O2 with a formation of OCHC6H4-2-OiPr and corresponding RuO species.261 Recently, Grela et al. reported the synthesis and study of complex Ru100 with a similar structure (Scheme 13) but higher thermal stability.262 Under the reactive distillation conditions (110 °C, 8 h), 0.5 mol% of Ru100 led to 16-membered lactone with 78% yield, and the (E)/(Z) ratio was 98:2. Starting from methallyl oleate, 13-membered lactone was obtained in 66% yield (1 mol% of Ru100).
Derivatives of dec-9-enoic acid can be rightfully referred to as oleochemical products, and their alkenyl esters were studied in RCM. For example, sulfur-chelated complexes Ru101 and Ru102 were inactive in RCM of undec-9-en-1-yl dec-9-enoate at 20 °C but showed moderate activity at 100 °C; complex Ru101 with a CF3 substituent was more active and selective (89% conversion after 1 h, and using 2 mol% of catalyst and 20 mM substrate concentration), the (E)/(Z) ratio was 91:9. The scope of the Ru101-catalyzed reaction as applied to OA derivatives is presented in Scheme 14.263
 | ||
| Scheme 14 Structures of Ru100 and Ru101 (a) and reaction scope for macrocyclization of the unsaturated esters of dec-9-enoic acid, catalyzed by Ru100 (b).263 | ||
As a result of further studies,264 complexes Ru103–Ru105 in the CAAC family (Fig. 24a) were found to be efficient RCM catalysts at ∼100 ppm loading, and the cyclization of CH2CH(CH2)8COO(CH2)4CHCH2 (10 mM) yielded 55% of 16-membered lactone (80 °C, 6 h, 100 ppm of Ru103), but CH2CH(CH2)7COO(CH2)2CHCH2 was inert towards these catalysts.
 | ||
| Fig. 24 Ru-based catalysts for RCM of ω-alkenyl dec-9-enoates (a)264 and complex Mo2, as an efficient catalyst for the RCM of ω-alkenyl oleates (b).265 | ||
Mo-based catalysts are usually inferior to Ru-based systems in activity, and only two articles devoted to Mo catalysts of RCM were published recently. The idea of the use of the ordered mesoporous support SBA-15 was implemented for Mo complexes of N-heterocyclic carbenes,266 and up to 98% RCM selectivity was achieved at 1 mol% loading of the catalyst and up to 100 mM concentration of the substrates. In the recent work by Grela et al.,265 the high thermal stability of Mo-based metathesis catalyst Mo2 (Fig. 24a) was successfully used in the vacuum distillation method. Given that molybdenum alkylidenes did not cause CC migration even at 100–120 °C, the title reaction proceeded with very high selectivity, leading to well-defined unsaturated macrocyclic lactones. For example, non-6-yl oleate was converted to 16-membered lactones ((E)/(Z) = 4.7) with 92% yield after 8 h at 110 °C using 2.6 mol% of catalyst.
Acyclic diene metathesis polymerization and related processes
ADMET polymerization of alkenyl oleates and related compounds seems to be an obvious pathway to unsaturated polyesters. ROMP of unsaturated lactones represents an alternative to ADMET, but in recent years this direction has not been explored.In terms of synthetic availability, ricinoleic acid (RA)-based unsaturated lactones represent promising candidates for ROMP. These lactones could be easily prepared via the condensation of RA, and the product of this reaction represents a mixture of unsaturated cyclic mono-, di-, and tri-esters and higher cyclic oligomers. It should be noted that the first attempts to prepare the corresponding high-MW RA homopolymers by ring-opening transesterification polymerization (ROTEP) failed,267 and high-MW RA polymers were only recently obtained by transesterification in 2024.268 However, three years earlier, Ogawa and Hillmyer synthesized homopolymers with Mw > 400 kDa via ROMP of mono-, di- and mixed RA macrolactones, catalyzed by 0.01 mol% of commercial Grubbs II catalyst Ru2.269 The fundamental difference between the RA polymers obtained by ROMP and polycondensation methods was (Z)-/(E)-isomerization in the former case, as illustrated in Fig. 25.
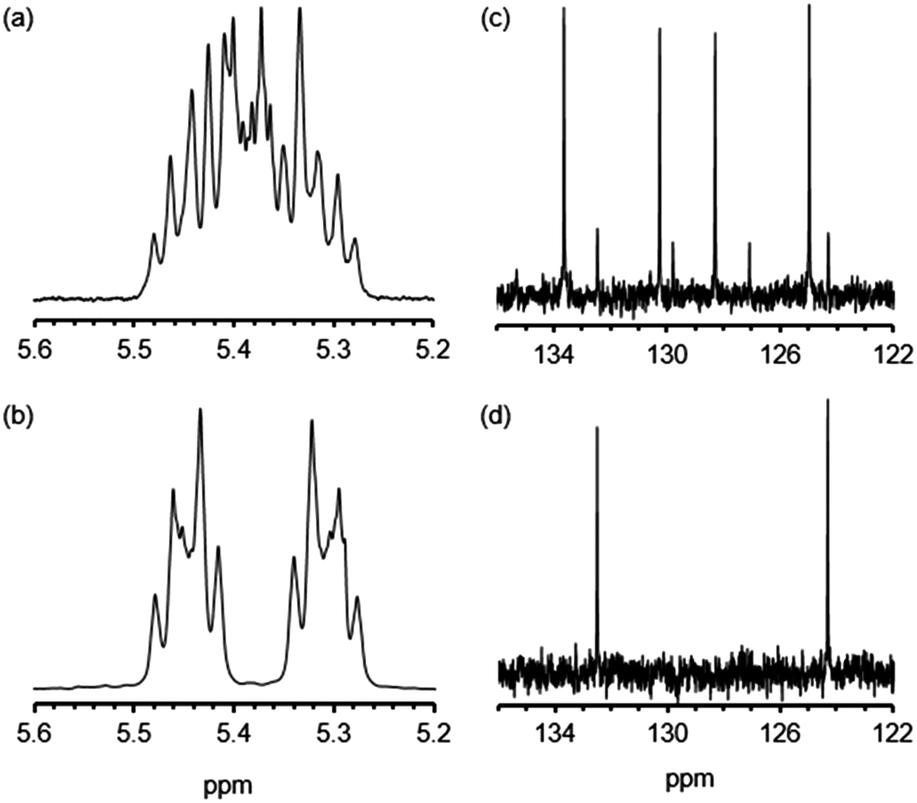 | ||
| Fig. 25 1H (400 MHz) and 13C (101 MHz) NMR spectra in CDCl3 of the olefin region of poly(RA) synthesized via ROMP ((a) and (c)) and via self-polycondensation ((b) and (d)).269 Reprinted with permission, Copyright (2021), The Royal Society of Chemistry. | ||
As a result, the higher-MW homopolymer obtained via ROMP had a glass transition temperature, Tg, in the range of −65 to −67 °C (for comparison, the Tg of poly(RA) not containing (E)-isomeric units was −73 °C), and represented a gel-like material in contrast with the previously reported physical state for poly(RA) containing only (Z)-isomeric units (viscous liquid).
To conclude this section, we note that catalytic metathesis reactions have the possibility to be the most atom-efficient and greener processes of modern oleochemistry. Among the variety of processes, the ethenolysis of MO with the formation of dec-1-ene and methyl dec-9-enoate is of particular practical interest; however, other cross-metathesis reactions should not be overlooked. In the last few years, a quantum leap has occurred, and scientists have reported ways to overcome the hurdle of 0.1 ppm catalyst loading.187 However, when evaluating these outstanding results, the necessity of the deep purification of MO also needs to be considered. The cost of similar purification may turn out to be the major contribution to the price of the products. Thus, to solve this problem effectively and sustainably, unique full-cycle production technologies should be developed, including cultivation and processing of oilseed with tight quality control, and employing the most advanced methods for the transesterification of TGs.
Other transformations with complete breaking of the CC bond
Complete oxidative breaking of CC bond in oleates
On the laboratory scale, the multistep transformation of OA to azelaic acid was proposed by Brenna et al., and the reaction sequence included the enzymatic epoxidation of OA, followed by hydrolysis, oxidation and oxidative coupling (Scheme 15a).274 Starting from 91% commercial OA, 77% of dihydroxystearic acid was obtained by crystallization; the overall yield at the latter stages was ∼93%, and the isolated yields were 73% (azelaic acid) and 77% (pelargonic acid).
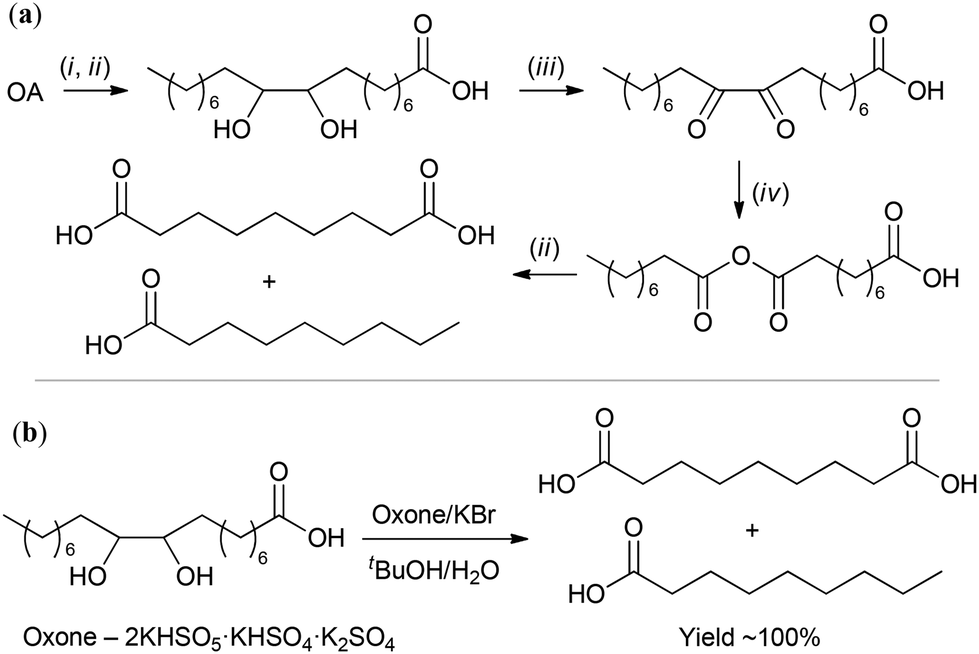 | ||
| Scheme 15 (a) Synthesis of azelaic acid from OA. Reagents and conditions: (i) H2O2 35%, Novozyme 435, acetonitrile, 5 h, 50 °C; (ii) NaHSO3-saturated solution, H2SO4 2 M, 3 h, r.t.; (iii) atmospheric O2, cat. Fe(NO3)3/TEMPO/NaCl, toluene, 5 h, 100 °C; and (iv) 35% H2O2, toluene, 3 h, 30 °C.274 (b) Synthesis of azelaic acid from 9,10-dihydroxyoctadecanoic acid.275 | ||
A very simple and efficient method for the oxidative cleavage of diols based on the use of Oxone/KBr in MeCN/H2O or tBuOH/H2O medium was successfully applied to 9,10-dihydroxyoctadecanoic acid, and in the presence of 20 mol% KBr, the conversion was 98%; when using 9,10-dihydroxystearic acid, the yields of pelargonic and azelaic acids were almost quantitative (Scheme 15b).275 However, these approaches have obvious drawbacks from a GRC standpoint, and thus and scientists have turned their attention to the development of more efficient catalytic solutions with the use of waste-free oxidants.
Catalytic approaches for the oxidative cleavage of OA and oleates can be based on the use of both H2O2 and molecular oxygen, given that the use of other oxidants does not seem economically and environmentally viable and are relatively poorly represented in the current literature. Despite the industrial implementation of H2O2-based process, research in this direction continued in 2020s.
A catalytic system based on H2WO4/H2O2 was applied in the direct oxidative cleavage of olive, rapeseed and sunflower oils, and the reaction conditions were optimized to minimize the formation of 9,10-dihydroxystearic glycerides; and the derivatives of azelaic acid were obtained in 71%, 48% and 60% yields, (at [W]/[Oil] 1:800, 1:200 and 1:400, 100 °C, 24 h 7.5 g of 60% H2O2 per 5 g oil), respectively.276 The H3PW12O40-catalyzed oxidative coupling of MO by H2O2 resulted in a moderate yield of azelaic acid, but when using methyl 9,10-dihydroxystearate, 80% selectivity was achieved.277 The direct oxidation of high-oleic sunflower oil to glyceryl triazelate using 35% H2O2 was conducted using Ru(acac)3 (2 mol%) and pyridine-2,6-dicarboxylic acid catalytic system, and the product bearing 2.83 –COOH fragments per molecule was obtained.278
An alternative catalytic approach is based on the use of O2 as an oxidant. This reaction is complicated by the formation of hydroperoxides. In recent years, several attempts have been made to search for catalysts for this process. In particular, Dibenedetto et al. proposed the use of mixed oxide catalysts (containing oxides of Ce, Nd, La, K and Bi) optionally doped with TiO2 in the oxidation of high-oleic sunflower oil (6–8 bar O2, 140–160 °C), and yields in the range of 40–56% of azelaic acid were obtained.279
Another alternative is based on a two-stage process, where firstly, TG, oleate or OA is transformed to 9,10-dihydroxy derivatives, which are further oxidized. In the case of dihydroxylated high-oleic TG (83.8% OA), the yield of azelaic acid after 5 h (80 °C, 25 bar O2) was up to 62% when using the Cu/Al2O3 catalyst (1 wt%).280
Among the oxidants besides H2O2 and O2, N-methylmorpholine-N-oxide, considered a “green” reagent (?), was used in combination with NaIO4 (1:1 mol/mol).281 In the presence of an Ru-based supported catalyst (polydopamine-functionalized carbon microspheres as a support), 95% yield of azelaic acid was achieved at a 4:4:1 [N-methylmorpholine-N-oxide]/[NaIO4]/[OA] ratio. There will be no additional comment concerning the feasibility of this method; however, in 2023, related studies were continued.282
When analyzing the catalytic efficiency, by-product formation and the need to use additional reagents and solvents, recent attempts to find catalytic approaches for the oxidative coupling of MO do not meet the GRC criteria. A two-stage process based on the use of H2O2/H2WO4 at the dihydroxylation stage and O2/Co(OAc)2 at the oxidative cleavage stage was subjected to detailed analysis,273 which confirmed its applicability. From an environmental perspective, this method is not without flaws, considering the proven toxicity of W283 and Co.284 However, the development of new catalytic approaches should consider the results of this assessment, given that some newly developed approaches do not seem to be any better.
 | ||
| Scheme 16 Common scheme for the oxidative cleavage of MO with the formation of aldehydes.274 | ||
An attempt to perform a similar transformation was performed by Rossi et al. in 2023,285 where they studied the oxidation of MO by O2, catalyzed by supported AgAu nanoparticles. The reaction was complicated by the formation of intermediate oxirane, dihydroxylated MO, methyl octadec-8,10-dienoate and isomeric aldehydes and ω-oxoesters. Consequently, the reaction rates were low, as well as the selectivity (Fig. 26). The idea of the formation of aldehydes is attractive considering their high reactivity in further possible transformations, but the complexity of the product composition brings into doubt the idea of this approach.
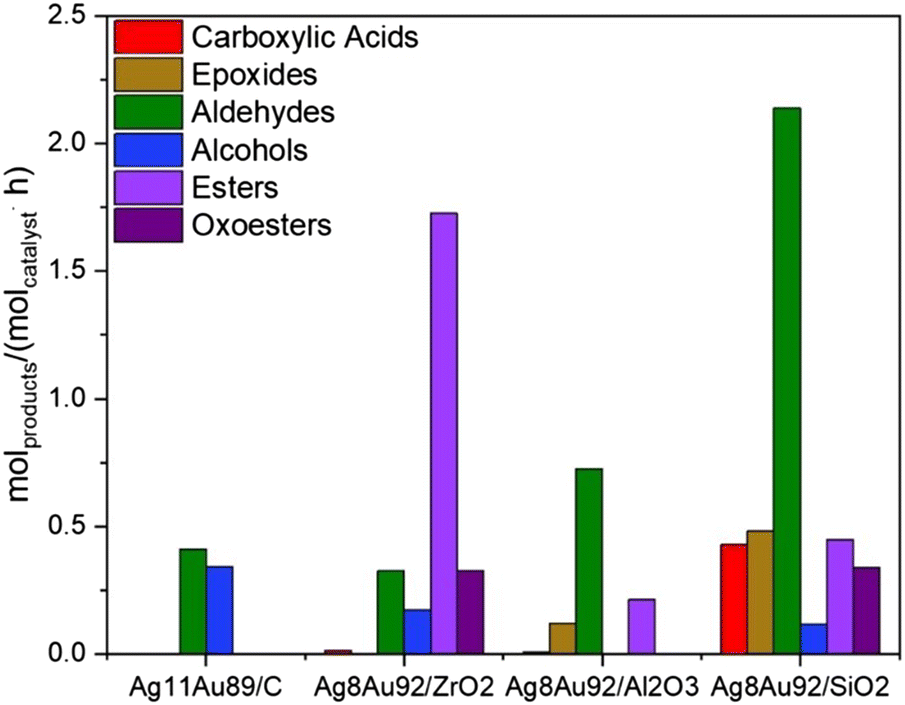 | ||
| Fig. 26 Productivity of oxidative cleavage of MO according to the variation of the support in bimetallic catalytic systems. Reaction conditions: in bulk, 1 h, 10 bar O2, 80 °C, ∼0.2 mol% of catalyst (Ag + Au).285 Reprinted with permission, Copyright (2023), Wiley-VCH. | ||
Base-induced cleavage of ricinoleic acid
One of the most important applications of RA is the production of sebacic acid via base-catalyzed scission.271,286 At high temperatures and in the presence of alkali, sebacic acid and octan-2-ol are formed, while at lower temperatures, the formation of 10-hydroxydecanoic acid and octan-2-one was observed, but in low yields (Scheme 17).287 | ||
| Scheme 17 Alkali scission of ricinoleic acid.287 | ||
Despite the successful industrial implementation of the production of sebacic acid from RA, the studies on this process continued in 2020s. In particular, castor oil and its derivatives were used as the raw material for alkali fusion in the presence of Pb3O4 (1%). At 280 °C, the optimized oleochemical/NaOH ratios (w/w) were 15:14, 15:14, 15:12 and 15:14, and the maximum yields of sebacic acid were 68.8%, 77.7%, 80.1% and 78.6% for castor oil, methyl ricinoleate, sodium ricinoleate and RA, respectively.288
In recent work,289 the subsequent oxidation of methyl ricinoleate to the corresponding 12-oxo derivative was followed by isomerization to the trans-10,11 isomer, dihydroxylation of CC bond, oxidative cleavage to methyl 10-oxodecanoate, oxidation and hydrolysis. The total yield of sebacic acid amounted to 16.7%. The comparison of these two approaches with the cleavage of RA clearly demonstrates the preference for the catalytic approach, which allows to multistage reactions to be conduct in one step. However, this process288 should not be regarded as ‘green’ due to the use of a toxic290 Pb-containing catalyst and formation of large amounts of alkaline effluents. The method proposed very recently by Zhang et al.291 is based on the use of Fe2O3 on activated carbon. Supporting the catalyst decreased the accumulation of iron in the product, and 83.5% yield of sebacic acid was achieved when using RA as a starting compound. However, a high excess of 40% alkali was still required (1:1 w/w).
Pyrolysis of ricinoleic acid
Undec-10-enoic acid (or its methyl ester) can be obtained via the pyrolysis of castor oil or methyl ricinoleate with heptanal as a by-product.286 This reaction is industrially implemented, despite the fact that the yields of the target products do not exceed 50%.292 An increase in the yield of these products was the obvious goal of recent studies in this field.With the use of an inductively heated reactor coupled with atomization feeding, 56.1% yield of methyl undec-10-enoate was achieved at the optimized reaction temperature (520 °C).293 Under lower temperatures, dehydration becomes substantial, which was confirmed recently by DFT modeling of the process.294 It should be noted that this modeling was based on calculations of different bond dissociation energies, which implies the free-radical mechanism of the process, but at the same time, McLafferty-type rearrangement (Scheme 18) cannot be ruled out.295
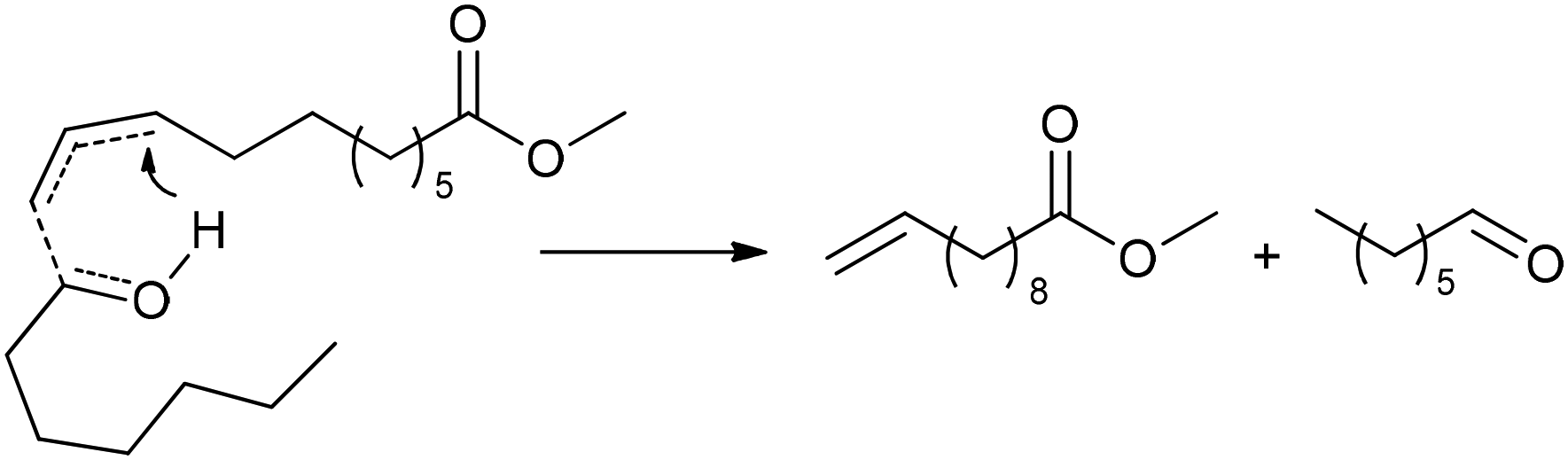 | ||
| Scheme 18 Pyrolysis of methyl ricinoleate via McLafferty rearrangement (one of the proposed reaction mechanisms).295 | ||
During a recent study on the pyrolysis of methyl ricinoleate in a micro fixed bed reactor, the optimal preheated and reaction temperatures were found to be 350 °C and 550 °C, and the yields of methyl undec-10-enoate and heptanal were 46.7 and 27.3 wt%, respectively.296 In this study, the free-radical mechanism was proposed, and the activation barrier was estimated to be 37.8, 43.1 or 45.2 kcal mol−1 depending on the functional used in the DFT calculations (B3LYP, RPBE or PBE, respectively).
Microwave-assisted pyrolysis using SiC as a microwave absorbent bed was conducted on lab-scale (1 kW, 126 mm diameter, 0.9 kg h−1) and scaled-up (7 kW, 580 mm, 7.0 kg h−1) reactors, and the yield of methyl undec-10-enoate amounted to 63.1% at 500 °C.297 When using a standard tubular reactor and dilution of methyl ricinoleate by water vapor, the optimized yield of methyl undec-10-enoate was only 23.8%.298
Thermal cracking of ricinoleates is still the main synthetic approach for the preparation of undec-10-enoic acid. However, high energy consumption and low atom efficiency with the formation of large amounts of potentially toxic side products are the obvious shortcomings of this method. In summary, both alkali and thermal scission of RA represent environmentally dirty productions, but there is still no alternative to date.
Reactions with partial breaking of CC bond
Catalytic hydrogenation of CC bond
As can be seen in Scheme 19, the catalytic hydrogenation of oleates and related compounds may result in qualitatively different products, including saturated FAs and esters, saturated alcohols, and unsaturated alcohols. The hydrogenation of carboxylic acids, esters, and related compounds over heterogeneous catalysts was the subject of a recent large overview by Qu, Junge and Beller.299 The partial catalytic hydrogenation of C18:2 and C18:3 esters was discussed previously.
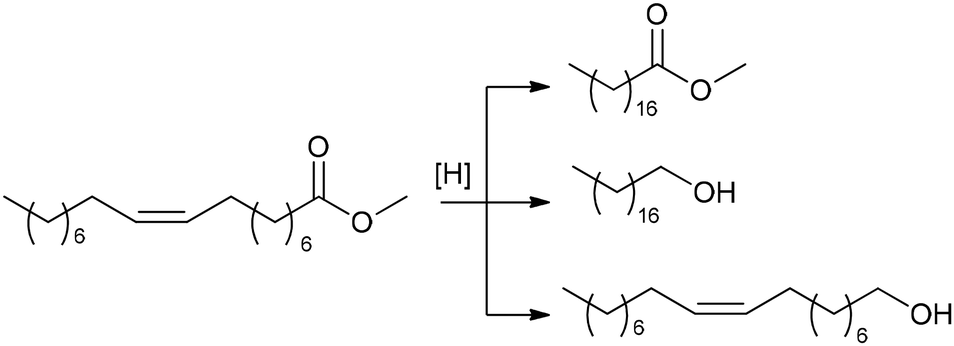 | ||
| Scheme 19 Hydrogenation of FAMEs (using the example of MO). | ||
Currently, the hydrogenation of OA with the formation of stearic acid is unlikely to present practical interest, but new catalytic solutions should be mentioned here, given that they can be applied in other hydrogenation processes. For example, the induction heating of Pd- or Pt-decorated Fe3O4 prevents coke formation,300 a common problem in high-temperature catalytic processes.
The hydrogenation of FAs to fatty alcohols represents higher practical interest, which is presently the subject of research. Hydrogen transfer with the use of iPrOH and CoOx catalysts turned out to be unproductive (∼0.4 mmol gcat−1 h−1 when using plant oils as a starting materials).301 Hydrogen transfer with the use of MeOH and Cu/SiO2 provided higher yields of the alcohols (82%) based on FAMEs.302 However, it is unlikely to expect a breakthrough in the development of the synthesis of fatty alcohols based on hydrogen transfer. In comparison with industrially implemented catalytic hydrogenation, these methods are more energy intensive with low atom economy.
Epoxidation and chemistry of oxiranes
Oxiranes derived from FAMEs, including the product of MO epoxidation methyl 9,10-epoxy stearate, can be potentially used as PVC plasticizers, diluents for paints, corrosion inhibitors, additives for lubricants and coatings, and raw materials for the production of surfactants and polyurethanes.3 Very recently, the anticancer activity of palm oil FAME epoxides against WiDr, T47D and HeLa cells was detected.303 To date, the epoxidation of FAMEs and TGs is conducted using peracids (HCO3H and MeCO3H) as oxidants (Prilezhaev reaction, Scheme 20), and the latter process is industrially implemented.304 The epoxidation of TGs, FAs and fatty acid esters was the subject of a recent mini-review by Lewandowski et al.305 | ||
| Scheme 20 Epoxidation of oleates using the example of MO: (a) generation of peracid and (b) Prilezhaev reaction. | ||
Peracids are formed in situ from H2O2 and HCOOC or glacial AcOH. Among the oxidants, H2O2 is attractive due to its high active oxygen content and formation of water as the only “green” byproduct. However, the activation of H2O2 requires the use of catalysts and the catalytic approaches for the epoxidation of vegetable oils were recently reviewed.306 Obviously, RCOOH can also be viewed as a peculiar catalyst (OOH transfer agent) in the reaction of FAMEs with H2O2, but we decided to discuss the recent studies based on Prilezhaev reaction and catalytic processes separately.
CHCH2– fragment. A detailed study of H2O2/AcOH in the epoxidation of linseed oil (C18:1, C18:2 and C18:3 content 19.8, 16.8 and 54.8 mol%, respectively) in the presence of Amberlite IR120-H ion exchange resin resulted in the development of a kinetic model providing a relative epoxy yield of >85%.308 Also, the kinetic study showed that one CC bond per linolenic acid chain has higher reactivity in comparison with other CC bonds in linseed oil, and the difference increased from 6.5× to 41× with an increase in temperature from 60 °C to 85 °C.308 Studies on the oxidation of OA, FAMEs or TGs by H2O2/HCOOH continue to be performed,309–312 with an unclear level of practical significance. From a GRC position, the recent work by Azmi et al.313 deserves separate mention, where they found that one half of HCOOH can be replaced by AcOH during the preparation of an oxidant without the loss of the product yield (∼82%), which is certainly desirable in terms of safety. The safety requirements also necessitate the use of aqueous solutions of H2O2, and thus water is also a reaction product. Consequently, the yield of the oxiranes decreased over time due to hydrolysis, and optimization of the reaction time complicated the scaling of the process. The typical yields of oxirane in the Prilezhaev reaction usually do not exceed 85%.
C bond (Scheme 21).316
 | ||
| Scheme 21 Mechanism for the epoxidation of MO catalyzed by tungsten peroxo complexes.316 | ||
Heterogenization of the catalyst in the epoxidation process is attractive in terms of technological effectiveness, and there have been multiple attempts to develop active and selective heterogeneous catalysts for the epoxidation of FAMEs. The heterogeneous catalysts obtained via the isomorphic substitution of Ti(IV) in different zeolites (ZSM-35, ZSM-5, and BEA) showed a low catalytic performance in MO epoxidation in the bulk, and the performance per number of Ti sites reached up to 17.1 ± 1.8 mol mol−1.317 A Ti silicate-based catalyst was almost inactive.318 The mechanism for the epoxidation of MO on the surface of TiO2 was studied in 2020,319 and formation of oxygen vacancies was proposed as a cause for the deactivation of titania-based catalysts.
To enhance the mass transfer in MO epoxidation, 2D channel catalysts were developed by Yang et al. in 2023.320 For this purpose, (EtO)4Si was hydrolyzed in the presence of [(C16H33)NMe3]Br, and the resulting material was grafted with (EtO)3Si(CH2)3NH2 and supported on H3PMo12O40, and a similar MCM-41-based catalyst was also prepared for comparison. The first catalyst (∼17 wt%) at [H2O2]/[MO] = 4 after 4 h at 90 °C showed 91.7% conversion and 90.0% selectivity; the reusability test revealed negligible loss of activity after 5 runs. The MOF-based catalyst H3PMo12O40/Cr(1,3,5-C6H3(COO)3) showed moderate efficiency (up to 73%) in FAME conversion to oxiranes.321
The catalytic behavior of MOx/Al2O3 catalysts was compared recently in MO oxidation with the use of H2O2, and even more interesting, molecular oxygen.322 Oxidation by H2O2 was conducted using an [H2O2]/[MO] ratio of 8:1 at 80 °C, and the results are presented in Fig. 27. When using H2O2 as the oxidant, the Mo and Zn catalysts exhibited a higher catalytic performance, and the MO conversion was below 10% for the Cu and Mn catalysts due to the decomposition of H2O2. The comparison of MoOx/Al2O3 and CuOx/Al2O3 catalysts in the epoxidation of MO by O2 demonstrated the preference for the Cu-based system, where in the presence of 15 wt% of CuOx/Al2O3 (3 h, 80 °C, 10 bar O2), the conversion was 67.5%. This difference was attributed to the higher content of the active O2− species on the surface of CuOx/Al2O3 revealed during the studies of O2 absorption.
 | ||
| Fig. 27 Conversion of MO over transition metal-based Al2O3 catalysts.322 Reprinted with permission, Copyright (2023), the Elsevier B. V. | ||
Several recent works have been devoted to enzymatic catalysis in the epoxidation of oleates. In the case of Novozym 435 under the optimized conditions (5 h at 50 °C, 0.15 M oleic acid, 0.27 M H2O2 and 5 mg mL−1 Novozym 435 in MeCN), the conversion of OA and isolated yield of the oxirane were 98% and 83%, respectively.274 Novozym 435 catalyzed the epoxidation of tall oil FAs with 81% conversion.323 Further development of the enzymatic approach for the epoxidation of FAs is unthinkable without an objective evaluation of the reaction mechanism, and in 2023 the relevant research was conducted.324 The detailed study of the reaction kinetics allowed the choice between chemo-enzymatic epoxidations via the percarboxylic acid route (Scheme 22) and direct epoxidation, in favor of the first reaction pathway. The kinetic studies indicated that the perhydrolysis step is the rate-determining step, with the epoxidation step being significantly faster.
 | ||
| Scheme 22 Chemo-enzymatic epoxidation route.324 | ||
In summary, catalytic alternatives to Prilezhaev epoxidation using H2O2 have not been able to provide higher yields of the oxiranes to date. High loadings of heterogeneous catalysts are needed, and in the case of homogeneous catalysts, there is the additional problem of metal separation. In general, greener methods for the epoxidation of oleates are waiting to be researched and developed.
To increase the yield of the oxiranes, there is some merit to avoiding the use of the water in epoxidation. In similar studies, tBuOOH was viewed as a more soluble alternative in organic solvents to H2O2. Mo-based catalysts have been proven to be effective when using this oxidant, e.g. heterogeneous [MoO3(2,2′-biimidazole)]·H2O (1 mol%) after 24 h at 70 °C demonstrated 97% conversion and 94% selectivity in MO epoxidation;325 the complex of [MoO3 with (5-(2-pyridyl-1-oxide)tetrazole)] (1 mol%)326 under the same conditions showed ∼100% conversion and 96% selectivity, and the complex of MoO2Cl2 with 5-(2-pyridyl)-2H-tetrazole had similar characteristics.327 The Mo2O6(pyrazine) hybrid material with a structure consisting of perovskite-like MoO3 layers pillared by pyrazine molecules displayed excellent selectivity in the formation of diepoxide from methyl linoleate.328 Complex bimetallic catalysts, prepared by the reaction of MoO2Cl2 and other metal chlorides (TaCl5, NbCl5 or WCl6) with acetophenone, led to 92%–96% epoxide selectivity at 84%–95% MO conversion at 70 °C and loading of 5.6 gcat molMO−1.329 The attempt to use host–guest complexes of (η5-C5H5)Mo(CO)3Me with cucurbit[n]urils in MO epoxidation failed.330 All the above-mentioned experiments with tBuOOH were conducted in C6H5CF3 medium. The use of additional solvent, more expensive oxidant and high catalyst loading do not allow tBuOOH-based methods to be considered as greener in comparison with conventional H2O2/HCOOH-based process.
The hydrolysis of oxiranes is a unique process; however, the reaction can be complicated by the formation of side products containing keto groups. Acid-catalyzed hydrolysis in the presence of a large excess of water usually proceeds with high yields,331 but the use of additional solvents is often needed. The idea of using heterogeneous catalysts with the minimization of the water loading seems attractive, but as was shown in the study by Dorado et al.,332 in the presence of sulfated perfluorinated polymers (Nafion and Aquivion), the content of keto esters in some experiments exceeded 50%. Under the optimized conditions, the yield of diol (Scheme 23a) was 92%.
 | ||
| Scheme 23 Functionalization of MO-based oxirane: hydrolysis (a);331 synthesis of β-hydroxy hydroperoxides (b);333 hydroxyalkylation, including the formation of polyols335 or PEG nonionic surfactants (c);337 preparation of zwitterionic surfactants (d);338 acrylate monomers (e);339 phosphonitrogenated lubricant additives (f);340 and cyclic carbonates (g). Only one of two possible structural isomers is shown. | ||
The reaction with H2O2 with the formation of β-hydroxy hydroperoxides (Scheme 23b) proved to be more interesting in comparison with hydrolysis, but in early studies, neat or ethereal H2O2 was used. Recently,333 Duguet et al. successfully employed 50% aq. H2O2 in combination with H3PW12O40, and the yield of 92% was achieved at 10 °C after 16 h for 10% solution in tert-amyl alcohol. Satisfactory yields were obtained for other unsaturated esters (73%–80%) and methyl linoleate (55%). The thermal decomposition of methyl 9(10)-hydroperoxy-10(9)-hydroxyoctadecanoate resulted in the formation of nonanal and methyl 9-oxononanoate (EtCN, 100 °C, 2.5 h), which can be used in further transformations, and the main side product was Me(CH2)7CH(OH)C(O)(CH2)7COOMe.
Ring-opening of oxiranes by alcohols (hydroxyalkoxylation, Scheme 23c) generates precursors for lubricants, polyurethanes and other materials. This process is usually catalyzed by acids; in 2023, numerous ionic liquids were screened, and the high efficiency of [(C12H25)NMe2(CH2)3SO3H][MeSO3] was demonstrated when applied to linear and branched primary alcohols.334 Aliphatic polyols are demanded as a raw material for the production of polyurethanes. For the synthesis of polyols, the ring-opening of MO-based oxiranes by glycerol was proposed, and under the optimized conditions (RSM, [glycerol]/[ester] 10:1, 120 °C, 0.18 wt% BF3·Et2O catalyst), almost full conversion to glycerol monoethers (Scheme 23c) were achieved,335 and the products were successfully used in the synthesis of polyurethanes.336 Ring-opening by poly(ethylene glycol) resulted in nonionic surfactants (Scheme 23c).337 A zwitterionic surfactant was obtained in three steps including acetal formation (reaction with PhCH2CH2CHO), amidation and quaternization (Scheme 23d).338
Acylation with acrylic acid (100 °C, hydroquinone) resulted in the acrylic monomer (Scheme 23e), which is successfully used in copolymers for potential use as archaeological consolidants.339 Subsequent treatment of methyl epoxyoleate by H2NCH2CH2NH2, P2O5 and triethanolamine resulted in a complex dimeric product (Scheme 23f), which was proven to be effective as a lubricating additive for paraffin oils and biolubricants.340
It is well-known that oxiranes can be transformed to cyclic carbonates by reaction with CO2 (Scheme 23g), and this “green” process continues to attract attention from researcher,341 with the carbonation of TGs and their derivatives the subject of recent reviews.342,343 In the 2020s, the focus has been on the development of heterogeneous catalysts. For example, a catalyst based on 1-hydroxypropyl-3-n-butylimidazolium chloride, NbCl5 and protonated carboxymethyl cellulose was prepared and studied by Leveneur et al.;344,345 68.5% conversion was achieved after 6 h (170 °C, 30 bar CO2) with a catalyst loading of 0.042 g mL−1. The heterogeneous organocatalysts developed and studied by the Leveneur and Salmi groups represented SiO2 or Zn/SBA-15-supported derivatives of tertiary amines, pyridine or imidazole (Scheme 24).346 Carbonation experiments (140 °C, 30 bar CO2, 7.4 wt% of the catalyst) showed the preference for 4-(pyrrolidin-1-yl)pyridine (4-PP)-based catalysts, where the SiO2-based system provided higher selectivity, whereas the Zn/SBA-15-based catalyst was more active. However, the conversion of oxirane did not exceed 76% after 23 h. The detailed kinetic study of 4-PP@SBA-15 revealed the causes for the formation of side products, keto esters, via Meinwald rearrangement.347
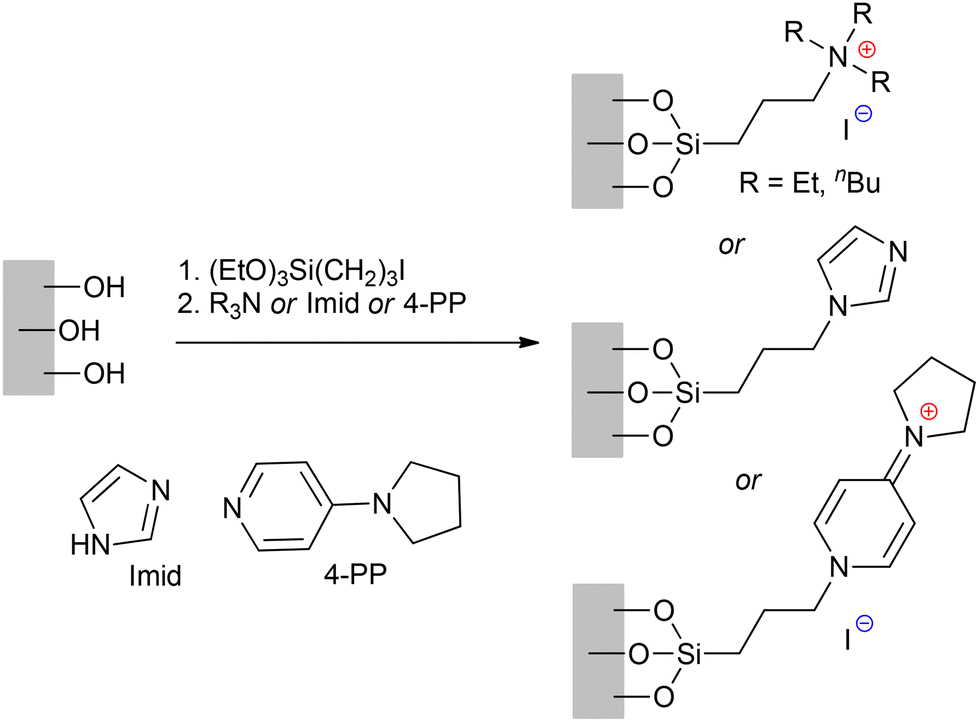 | ||
| Scheme 24 Preparation of supported catalysts for the carbonation of MO epoxide.346 | ||
In the presence of nano-sized SnO2 (100 mg) and [Bu4N]I (0.5 mol%), the conversion of MO (1.5 mmol) was only 42% (80 °C, 20 bar, 48 h).348
Organocatalytic (including homogeneous) approaches for the carbonation of epoxides have also been examined in the 2020s. For example, ascorbic acid was proposed as a hydrogen bond donor for this process, and in the presence of ascorbic acid (1.5 mol%) in combination with [Bu4N]Cl (5 mol%), >99% conversion and selectivity were achieved in the epoxidation of MO (48 h, 10 bar CO2, 100 °C), and the isolated yield of cyclic carbonate was 90%.349 The formation of keto esters was observed in a recent comparative study on the use of [Bu4N]Br and [Bu4N]I in the carbonation of epoxidized linseed oil, which was more pronounced when using [Bu4N]I.350 Phosphonium ILs showed high selectivity in the carbonation of epoxidized soybean oil, and after 5 h at 160 °C and 40 bar CO2, 2 mol% of [Ph3P(CH2)11Me]Br provided >99% conversion and 84% selectivity.351
To avoid the separation of the oxiranes, the tandem catalytic approach for the preparation of carbonates starting from MO was proposed by Perosa et al.304 (Oct3NMe)2WO4 ionic liquid (5 mol%) catalyzed the epoxidation of MO by 30% H2O2 (2 eq., 75 °C, 4 h) with almost quantitative yield. After separation of the epoxidized MO, the same catalyst was used for carboxylation. Also, more importantly, a single-reactor process for the synthesis of carbonated MO was successfully carried out, where the combination of (Oct3NMe)2WO4 (5 mol%) and KBr (5 mol%) allowed a yield of 99% of cyclic carbonate (cis-/trans- ratio 98:2) to be achieved.
One of the promising applications of carbonated oils and FA derivatives is the preparation of non-isocyanate polyurethanes via interaction with diamines. In the study by Leveneur et al.,352 the steric effect of amine (nBuNH2, nBuMeNH, nBuEtNH, and nBu2MeNH) on the aminolysis of carbonated MO was explored. For the reaction with nBuNH2, Ea of aminolysis and side amidation reactions was estimated to be 42.1 and 45.4 J mol−1 in the case of nBuEtNH, respectively, and nBu2MeNH amidation was negligible.
Epoxidized OA can polymerize at elevated temperatures with the formation of viscous liquid or solid materials (Tg ∼ −19 °C) depending on the degree of polymerization;311 however, the prospects for practical use of these polymers are unclear.
Dihydroxylated FAs are widely used in further transformations to valuable products, which is amply reflected in many previous works. In the 2020s, the oxidative cleavage of diols was described in several works, as mentioned above.274,275
Based on methyl 9,10-dihydroxyoctadecanoate, the corresponding 9,10-dioxo ester was obtained in two stages (O2/Pd(OAc)2/neocuproine and O2/VOCl2 sequentially, 72% yield) and transformed to substituted imidazoles.354
When comparing the epoxidation and dihydroxylation of MO and related compounds, it should be noted that almost quantitative yield of the reaction product can be achieved in the latter case. The combination of catalytic dihydroxylation353 and catalytic oxidative cleavage275 may form the basis for the further development of green technology for the production of azelaic and pelargonic acids that meets the most of the GRC criteria.
Estolides
Estolides are well-known oligomeric products obtained via acid-catalyzed reactions between FAs (Scheme 25) or between TGs and FAs.355 Estolides are biobased lubricant base stock, which have many advantages such as biodegradability and improved low-temperature characteristics. A topical review on this was published in 2020.356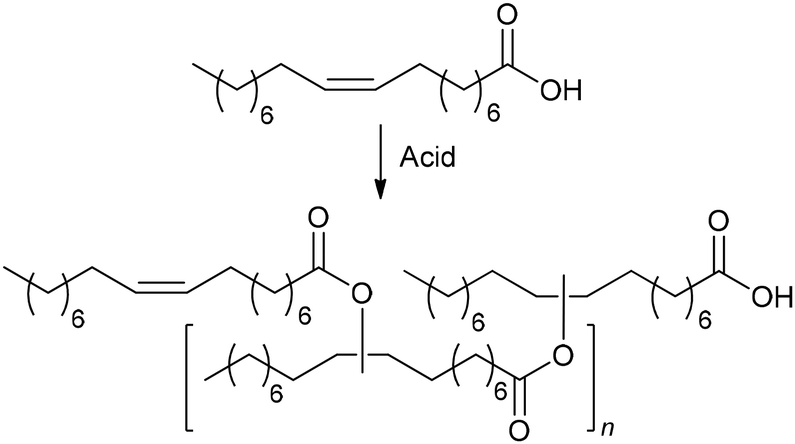 | ||
| Scheme 25 Synthesis of FA-based estolides.356 | ||
In recent years, several works have been devoted to the further optimization of the synthesis of estolides, including ultrasound-assisted preparation from OA357 and optimization of the reaction conditions to achieve the required viscosity and low-temperature characteristics.358 The studies of estolide esters as a Group 5 oil base stock are still ongoing.359 It should be noted that 12-hydroxystearic acid-based estolides were synthesized and comprehensively studied recently.360
The use of branched FAs (see below) turned out to be prospective for the further improvement of the properties of estolides. The comparison of the properties of estolides obtained by the reaction of OA or iso-oleic acid containing one methyl branch with saturated FAs showed no particular advantages for acidic forms, but 2-ethylhexyl esters of branched estolides demonstrated excellent low-temperature characteristics, e.g. pour point (PP) of −60 °C.361
Catalytic hydroformylation and related processes
In 2020s, only a few studies were performed. The reaction of ethyl ricinoleate or castor oil with CO and H2 in a mixture of toluene with different nitrogen bases (80 °C, 80 bar CO/H2 1:1, 24 h), catalyzed by Rh(acac)(CO)2 (0.4 mol%), resulted in the formation of complex mixtures of products; the highest yield of the primary alcohols was ∼80%.363 The idea of using biphasic systems to enable recycling of water-soluble homogeneous catalysts was successfully realized by Seidensticker et al. in 2019.364 Hydroformylation in continuous biphasic mode using a jet-loop reactor was studied in detail very recently by the same group,365 and MO and methyl undec-10-enoate were used as starting materials, achieving 35% and >80% steady-state yields, respectively.
 | ||
| Scheme 26 Methoxycarbonylation of methyl undec-10-enoate.366 | ||
Transformations with retention of the CC bond
Synthesis of oleyl alcohol and related compounds
Fatty acid alcohols are widely used in the production of detergents, lubricants, cosmetics, plasticizers and other widely sought-after goods;11 in 2022, the global fatty alcohol market was estimated to be 3.9 million tons.367 Among the unsaturated fatty alcohols, oleyl alcohol is the most produced and demanded product, and the technologies for its production have long been developed and widely used. These technologies are based on the catalytic hydrogenation of MO and C18:1-enriched FAMEs with the use of Cu/Cr-based catalysts under severe reaction conditions (250–350 °C, 100–200 bar).368 Alternative synthetic approaches to fatty alcohols are discussed in reviews,11,369 covering the articles published before 2020, a recent review on the deoxygenation of FAs and esters15 includes one actual reference on the subject, and below we discuss the recent works in this field.The results of the study on the Rh–Sn–B/Al2O3 catalyst in the hydrogenation of OA revealed the optimal reaction conditions (290 °C, 20 bar), providing 82%–83% yield of oleyl alcohol.370 It is essential that the side product, oleyl oleate, does not contribute as an intermediate. The Rh–Sn–B/TiO2 catalyst provided 88.3% yield of oleyl alcohol, and it was shown that the Rh/Sn ratio makes the Rh hydrogenating activity more selective for the hydrogenation of the carbonyl group.371 During further optimization of Rh–Sn–B/γ-Al2O3 systems,372 the key role of NaBH4 at the catalyst preparation stage was clearly demonstrated. It should be noted that experiments with Rh–Sn–B catalysts were conducted in n-dodecane (12 g gOA−1), making it difficult to scale-up the process. When the hydrogenation of FAMEs over Rh–Sn–B/γ-Al2O3 was conducted in the bulk, only 39% yield of oleyl alcohol was achieved.373
To mitigate the reaction conditions and increase the catalyst productivity, extra attention has been paid to the development of homogeneous catalytic systems. In view of the high efficiency of NHC complexes of Mn in the hydrogenation of the –COOR group, which was discovered in 2023,374 in 2024, Beller et al. conducted a study on the hydrogenation of ethyl oleate catalyzed by complexes Mo3–Mo8 (Scheme 27).375 Complexes Mo6 and Mo7 showed similarly high catalytic activities with 90–91% conversion and 73–76% yield of oleyl alcohol. The difference between the conversion and yield was mainly due to the formation of oleyl oleate. In all cases, no CC bond hydrogenation was observed, which underlines the crucial difference between these complexes and noble metal-based catalysts. Under the optimized conditions (0.5 mol% Mo6, 5 mol% tBuOK, 80 °C, 50 bar H2, 24 h), the conversion of the ester and the yield of oleyl alcohol were 98% and 95%, respectively.
 | ||
| Scheme 27 Structures and catalytic properties (conversion/selectivity) of complexes Mo3–Mo8 in the hydrogenolysis of ethyl oleate (1 mol% [Mo], 5 mol% tBuOK, 60 °C, 50 bar H2, 16 h in dioxane).375 | ||
To elucidate the mechanism of the catalytic process, NMR spectroscopic studies, control experiments, and DFT computations (M06L-SCRF/TZVP level of theory) were performed. Mo–CH2CN species and μ-H dimeric intermediate were detected, and the common reaction mechanism was proposed (Fig. 28).
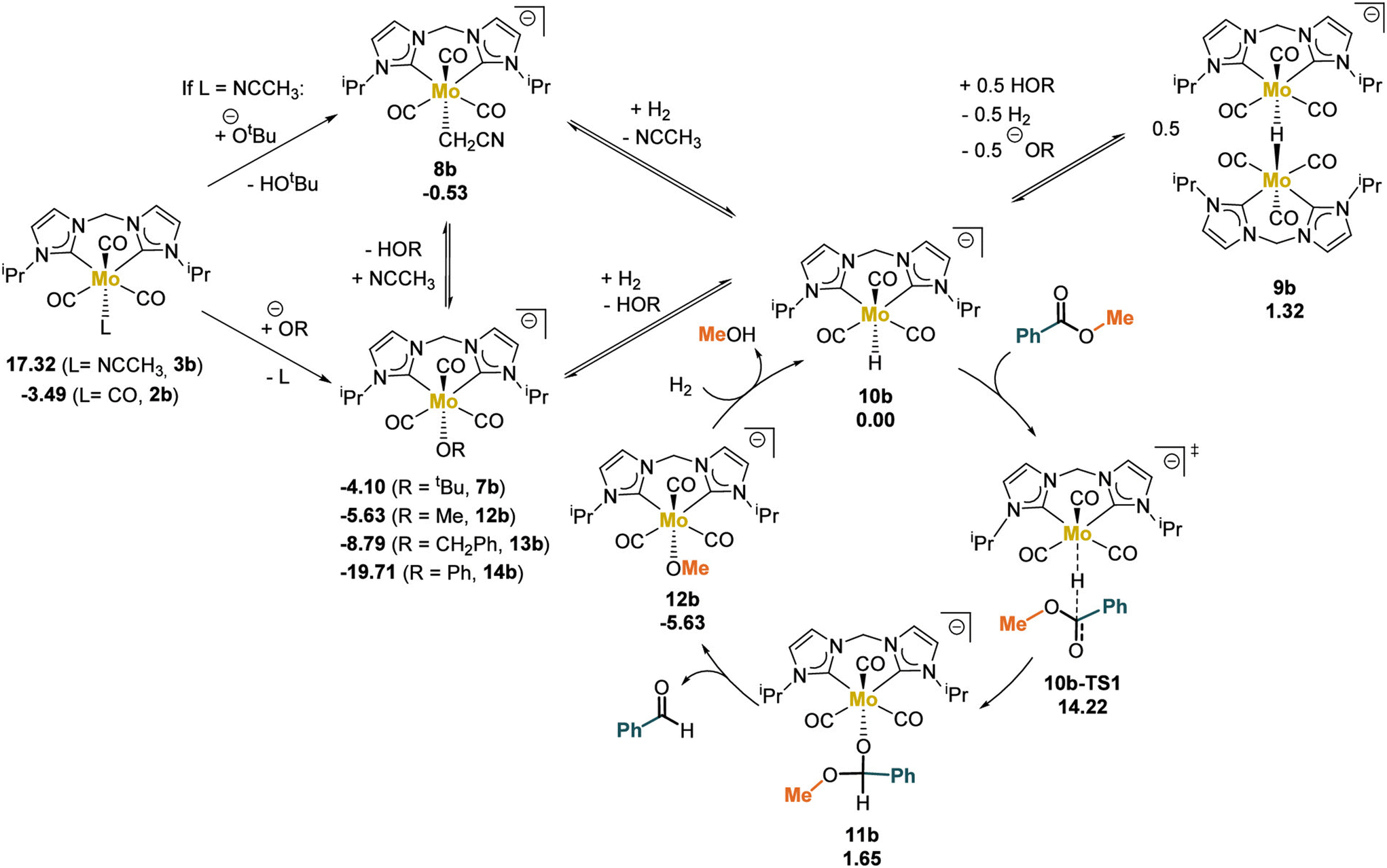 | ||
| Fig. 28 DFT-supported mechanistic rationale for the Mo-catalyzed hydrogenation of esters (using the example of PhCOOMe).375 Reprinted with permission, Copyright (2024), the American Chemical Society. | ||
The reduction of MO using complex hydrides is an evident method for the synthesis of oleyl alcohol in the laboratory. In 2020,376 the substantially lower reactivity of MO in comparison with methyl laurate was revealed, and MOx/Al2O3 (M = Mo, Fe, Ce) were proposed as reaction promoters.377 In both studies, the [MeOH]/[NaBH4] molar ratio of 6:1 was used, which is hardly justified in view of comprehensive studies of the reactivity of NaBH4 in MeOH.378
Very recently, Ph2SiH2 was used for the catalytic reduction of TGs in the presence of (2-tBuS-C6H4SiMe2)Rh(Ph3P)3H (0.5 mol%) with the formation of unsaturated fatty alcohols and glycerol after hydrolysis in the final stage.379 Besides the target products, 13%–28% of aldehydes was formed depending on the type of oil.
Oleyl alcohol is a highly demanded oleochemical product, and the further development of its production from FA esters is quite crucial today. Industrially implemented methods are energy consuming and unsafe (200–220 °C, 50–300 bar of H2),11 and the recent study by Beller et al.375 is of great importance because of the prospects of hydrogenation under milder conditions due to the use of an advanced catalyst.
Catalytic deoxygenation via decarboxylation
An alternative approach for the valorization of TGs to biofuels is to convert them into the corresponding Cn or Cn−1 hydrocarbons via deoxygenation. Deoxygenation combines different reactions (Scheme 28), resulting in the formation of both “green” hydrocarbon fuels (decarboxylation or hydrodeoxygenation) and more valuable α-olefins or carbonyl compounds. Some of the processes, as presented in Scheme 28, do not enable retention of the CC bond and are less interesting in the context of our review; thus, in this section, we focus on the decarboxylation of oleates with retention of the CC bond.
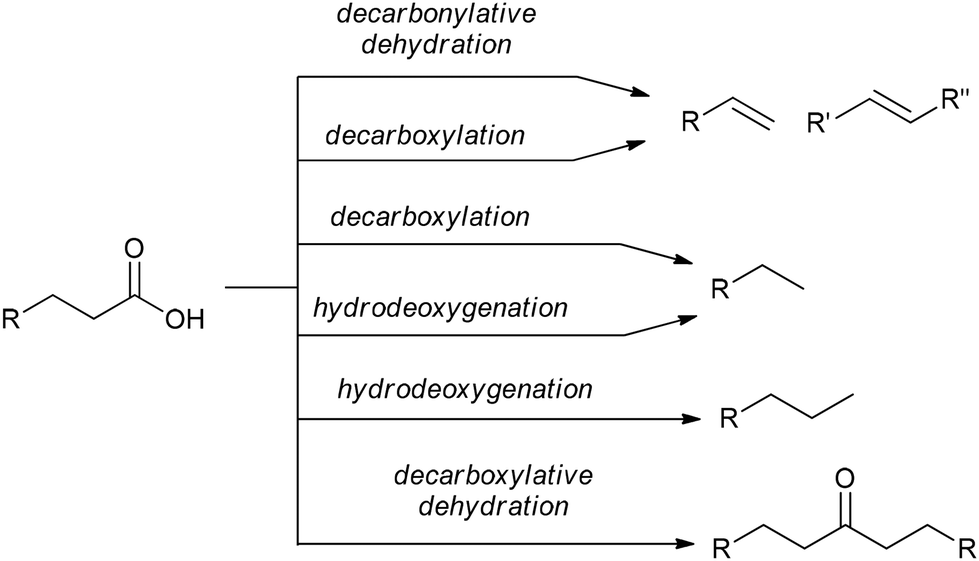 | ||
| Scheme 28 Variety of deoxygenation processes based on the example of carboxylic acids. | ||
Thermal decarboxylation of aliphatic carboxylic acids requires harsh reaction conditions, is complicated by side reactions, and has no potential for practical implementation. However, decarboxylation of FAs with the formation of hydrocarbons is receiving attention from researchers with regard to green diesel production. The relatively recent (2022) review by Chen et al.16 covers the results published up to 2021. The conventional catalytic approaches are based on the hydrodecarboxylation of FAs to generate C1-shortened alkanes that are neither products nor raw materials of fine oleochemistry.
The retention of CC bonds during decarboxylation can be facilitated by enzymatic catalysis, and the further progress in FA decarboxylation is associated with the discovery of light-activated fatty acid photodecarboxylase in 2017.380 The results and prospects of the studies in this field were discussed in recent thematic reviews.17,381,382 In 2020,383 it was shown that a two-step cascade reaction involving lipase and Chlorella variabilis NC64A photodecarboxylase (CvFAP) can be used to generate hydrocarbons from different commercially available oils in amounts of up to 24 g L−1. In the model reactions with palmitic acid, the apparent quantum yield of 40% was achieved;384 no significant difference between CvFAP containing E. coli and broken cells was obtained.385 Further optimization of CvFAP-catalyzed decarboxylation was performed again for palmitic acid, and the use of a microfluidic photobioreactor resulted in a conversion of 96.7% (flow rate of 10 μL min−1, blue light intensity of 200 μmol m−2 s−1, 30 °C, [CvFAP] = 2.4 mg mL−1, [FA] = 12 mM); the TOF of CvFAP was estimated to be 19.2 h−1.386 In 2023, FAs from waste oils were studied as real substrates;387 the preference for unsaturated FAs in comparison with palmitic acid was established, and the hydrocarbon production rate of 18.4 mM h−1 was obtained at a flow rate of 10 μL min−1.
The reaction mechanism for decarboxylation on photodecarboxylase is presented in Scheme 29, and the key role of the flavin cofactor was proven experimentally in real time.388 In the absence of a substrate, the photoexcited, high redox-potential flavin is assumed to oxidise the nearby active site amino acid, leading to irreversible inactivation.389 This deactivation is the main problem hampering the practical use of photodecarboxylases. In 2021,390 Hollmann et al. showed that the photostability of CvFAP can be improved by employing medium-chain length carboxylic acids (e.g. caprylic acid) “to keep the enzyme busy”.
 | ||
| Scheme 29 Catalytic cycle of the photoenzymatic decarboxylation of carboxylic acids catalyzed by Chlorella variabilis NC64A photodecarboxylase.390 | ||
The enzymatic photodecarboxylation of FAs is viewed as a clean approach for sustainable hydrocarbon fuel production in environmental chemical engineering.387 However, significant progress towards developing the catalytic conversion of TGs to saturated hydrocarbons (in particular by using Ni,391–394 Fe,395 and Fe/Ni396 catalysts) has been made in recent years. This conventional approach is capable of competing with the photoenzymatic method. An undeniable advantage of the use of CvFAP, and in prospect, new genetic engineering biocatalysts is the retention of CC bonds, and one can hope for advances in R&D in this field to meet the actual standards of green chemistry.
Allyl substitution
Ally substitution-type reactions have not attracted substantial attention from the researchers in the 2020s. The products of the nitration of FAMEs were proposed as a cetane number enhancer in 2007,397 and complex mixtures of compounds were obtained depending on the nature of the nitration agent. In 2024,398 further studies on the nitration of MO were conducted using a mixture comprised of 65 wt% HNO3 and Ac2O in a 28:72 w/w ratio and the reaction was conducted at 15 °C, 25 °C and 40 °C. The allyl substitution by the NO2 group was the main reaction pathway (80%–87% selectivity), and the product proved to be a promising candidate for the replacement of 2-ethylhexyl nitrate in diesel formulations as a cetane improver.
It is also worth paying attention to recent studies on C–H allylic oxygenation. The reaction of MO with H2O2 (20 eq.) was conducted in MeCN/H2O in the presence of aq. NH3 with the use of Mo2C as a catalyst; the yield of isomeric hydroperoxides (Scheme 30) was 66%.399 In this process, singlet oxygen served as the oxidant.
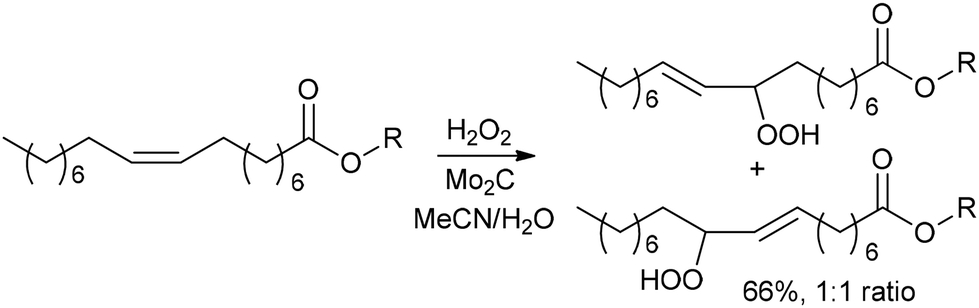 | ||
| Scheme 30 C–H allylic oxygenation of MO.399 | ||
It is relevant to note here that the allylic autoxidation of MO and related compounds proceeds in more complex reaction pathways with the formation of mixtures of positional and (Z)/(E) isomers.189 The chemical picture is even more complicated in the case of polyunsaturated FAs.400,401 Given the critical importance of the processes of autoxidation of FAs in the food industry and fine catalytic oleochemistry, a comprehensive review focusing on the analysis of lipid hydroperoxides was recently reported by Kontogianni and Gerothanassis.402
Functionalization of –COOH (–COOR) group
Simple functionalization of the COOH or COOR group in oleates was presented in only a few recent publications (which is not surprising given that at present, this approach has been sufficiently elaborated). The expansion of the range of oleate-based surfactants, lubricants and other demanded products often requires the preparation of FA esters other than FAMEs (Fig. 29a). Transesterification of FAMEs is the simplest route to obtain these compounds, and optimization of the yields of the products is important from a practical point of view. A comparative study of (iPrO)4Ti, KF/Al2O3 and MgO showed that the latter catalyst provides the highest yields of esters when using C8 + aliphatic alcohols.337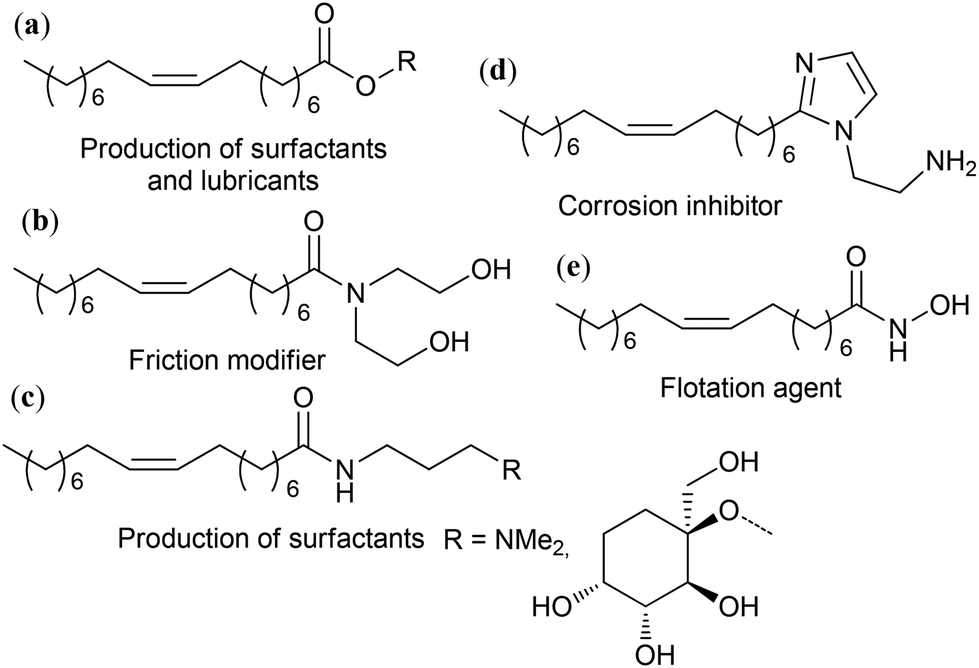 | ||
| Fig. 29 Structures and applications of the functional derivatives of OA. | ||
The amidation of FAMEs with N,N-diethanolamine in the presence of 5 wt% of 2.5-Li@CaO–Ca(OH)2-450 nanocatalyst (Li-doped CaO nanoparticles, calcined at 450 °C) was completed after 30 min at 110 °C.403 This catalyst demonstrated good recyclability (up to 8 runs with 99% FAME conversion), and was also used for the conversion of triglycerides (waste cooking, Karanja and Jatropha oils) into amides of FAs (Fig. 29b). The prepared amide derivatives acted as cetane number and lubricity improvers for diesel fuels. The product of the reaction of MO with N1,N1-dimethylpropane-1,3-diamine (Fig. 29c, R = NMe2) is in demand for the production of surfactants, where high efficiency due to the replacement of the conventional base catalysts with a Cr-based MOF in amidation was demonstrated recently.404 Another prospective surfactant was a fructose derivative of OA (Fig. 29c).405 The OA imidazoline derivative (Fig. 29d) and L-cysteine showed a synergistic effect as a carbon steel corrosion inhibitor.406 Hydroxamic acid (Fig. 29e) showed high efficiency in the flotation separation of bastnaesite and barite, an important process during the separation of Ce and rare-earth metals,407 and spodumene/albite separation.408 Epoxidized hydroxamic OA turned out to be efficient in fluorite/calcite separation.409
Functionalization of the –COOH(R) group in OA and related compounds is carried out in each specific case using methods depending on the structure of the target molecule. The conventional organochemical approaches are still used, and further optimization of one or other method in light of the GRC principles will become feasible only when the corresponding product is in demand.
Other transformations
In this section, we summarize the reactions resulting in the functionalization of the starting compounds via intermediate migration of the CC bond, and the processes accompanied by more deep destruction and/or skeletal isomerization of the hydrocarbon fragment.
Isomerizing processes via CC bond shift
:60 [Pd]/[MO] ratio, dimethyl nonadec-1,19-dioate was formed, and ethyl erucate behaved similarly. In their further study of oleates and erucates,411 the corresponding diols HO(CH2)xOH and diamines H2N(CH2)xNH2 (x = 19, 23) were prepared in high yields. Very similar results were obtained independently by Köckritz et al.412 Although MO in the presence of Pd-diphosphine catalyst forms a statistical mixture of isomers with a 94% content of internal CC bonds, the high reactivity of the terminal –CHCH2 fragment413 determines the structure of the final reaction product. The preparation of dimethyl nonadec-1,19-dioate from technical grade MO using Pd2(dba)3/1,2-C6H4(CH2PtBu2)2/MsOH was optimized recently by Herrmann et al.,414 where the homogeneous Pd catalyst remained in the MeOH reaction medium and was recycled for up to eight runs. The latter work represents a marked example of the development of an atom-efficient catalytic process not using the standard method based on the limited solubility of the reaction product in the reaction medium containing the dissolved catalyst. The prospects of the use of dimethyl nonadec-1,19-dioate in the synthesis of polyester plastics bring added value to this work in terms of GRC.
Skeletal isomerization processes
Due to the absence of branched FAs in plant oils and fats, the catalytic transformation and functionalization of oleates and related compounds usually result in organic products with a linear structure. However, derivatives of branched hydrocarbons are highly in demand in the chemical industry, which has attracted increasing attention to the skeletal isomerization of natural oils, FAs and FA esters. A corresponding thematic review was published in 2021,18 and thus in this section, we discuss the resent studies in this field.In 2021, Sels et al. reported the results of their study aimed at replacing the expensive ferrierite isomerization catalysts with available ZSM-5 zeolites.416 In the presence of 2.5 wt% of catalyst, 90% conversion of FA was achieved after 6 h at 250 °C, and the yield of branched acids amounted to 70%. The accessibility of the active Brønsted acid sites was found to be a key factor determining catalytic activity, and the post-treatment of ZSM-5 was successfully used to obtain a highly active “hierarchical” ZSM-5 catalyst. It should be noted that a thorough analysis of the reaction products with the determination of the structure of each component was not performed.
Ferrierite-based catalysts are still relevant, and in 2024, synthetic ferrierite-like zeolites were studied in MO isomerization.417 In batch reactor experiments in the presence of 5 wt% of catalyst, 95%–98% conversions were achieved after 8 h at 260 °C, and the yield of branched FAMEs was 56%–60%, but the spent catalyst contained 12–20 wt% of coke. In the continuous flow process, the weight hourly space velocity was 3.5 h−1 for 50% conversion at 285 °C. The positive impact of the formation of a hierarchical micro/mesoporous structure was also detected in this study.
It should also be noted here that the results of the recent study by Gooßen et al.418 suggest ambiguous interpretation of the results from previous studies. Using H-mordenite as a catalyst at 290 °C, near equilibrium isomerization of rapeseed oil FAMEs was achieved with up to 49% recovery of the linear monomeric products; the main side products were oligomers (3%–15%) and skeletal isomerization products (10%–23%). At higher temperatures, the content of isomerization products increased to make significant contributions to the product composition (Fig. 30).
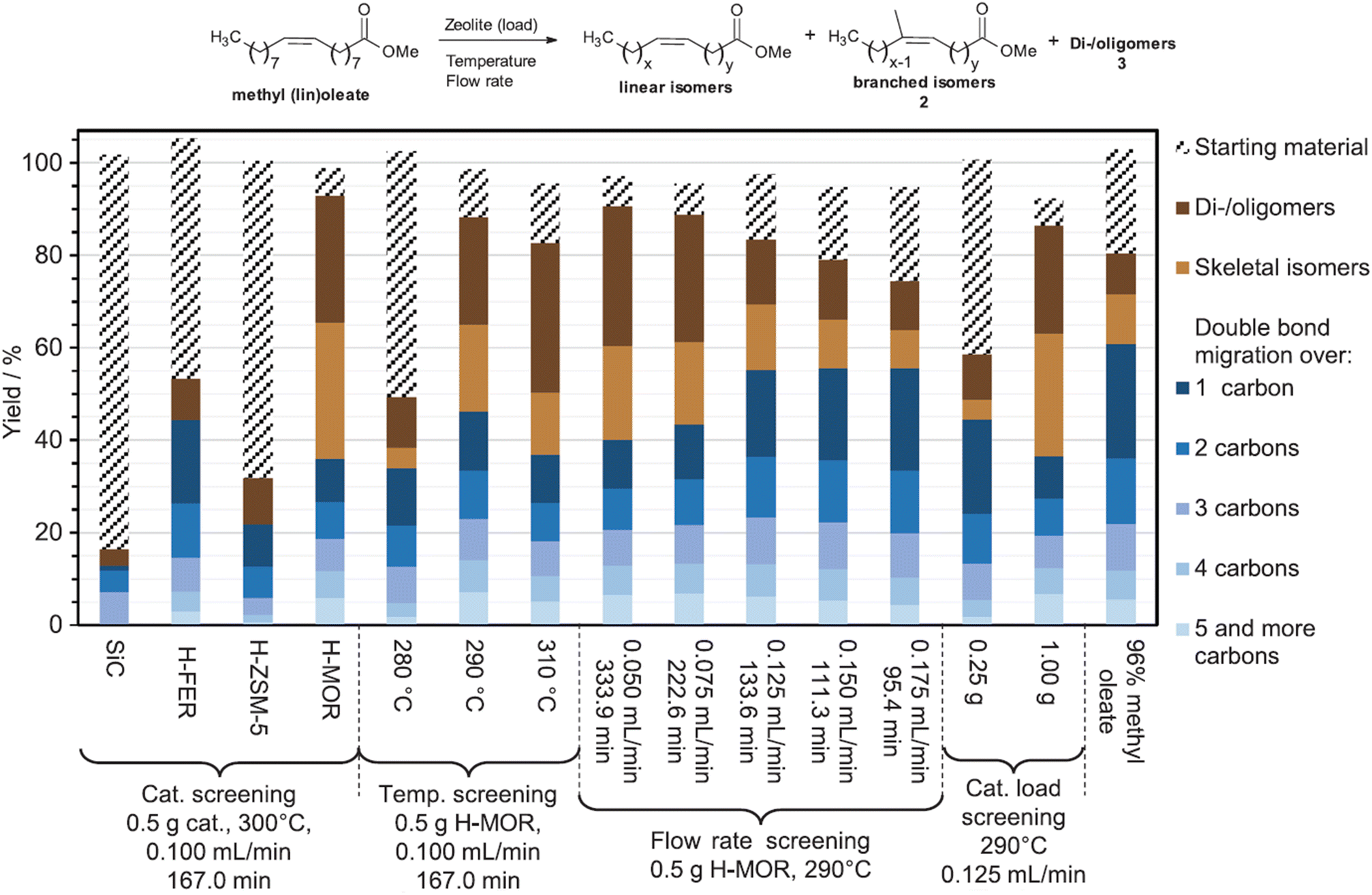 | ||
| Fig. 30 Distribution of double bond positions in product mixtures from the condition screening in a flow reactor. The values in mL min−1 denote the flow rate, and the values in min denote the mean residence time of the substrate. Deviations from 100% mass balance are due to the error of the analytic methods. Catalyst abbreviations: SiC – carborundum, H-FER – ferrierite, H-ZSM-5 – zeolite ZSM-5, and H-MOR – H-mordenite.418 Reprinted with permission, Copyright (2023), Wiley-VCH. | ||
Isomerized OA derivatives (or, after hydrogenation, isomerized stearates) can find application in the synthesis of various value-added products. For example, trimethylolpropane triisostearate and triisooleate exhibited higher oxidative stability, viscosity indexes (VI) and better cold flow properties compared to poly-α-olefin base stocks.419 At first glance, the isomerization of FAs and FAMEs, conceptually similar to catalytic cracking in the petroleum industry, provides good opportunities for the production of sought-after branched compounds. However, the isomerization of oleates in contrast to petrochemical processes is accompanied to a much greater extent with oligomerization and coke formation, and it is the greener nature of the raw material that determines the pollution-intensive character of the isomerization process.
Complex catalytic processes with destruction of the main chain
In contrast to catalytic deoxygenation, resulting in the formation of paraffins, the combination of the active catalyst with high reaction temperatures (300 °C and higher) may result in deeper transformations of oleates and related compounds. Besides the decarboxylation or reduction of –COOH(R), depending on the reaction medium (the presence of H2 or O2), the processes of dehydrogenation, oxidation, cracking and isomerization can proceed, leading to the formation of complex product mixtures. These products are primarily considered fuels or raw materials for the production of ethylene and short-chain α-olefins. In this section, we briefly report some of the recent results of the studies in this field.In 2023, Le et al. studied the use of V catalysts supported on mesoporous silica with a three-dimensional pore architecture in the oxidative dehydrogenation of OA.420 At 450 °C, very complex mixtures of products were formed. It should be noted that in addition to liquid products, 24%–28% of gaseous products were formed. With an increase in the temperature, the content of olefins decreased, and the content of oxygenates increased.
The hydrocracking of MO using an Fe/SiO2–Al2O3 solid acid catalyst (380 °C, 5 bar) also resulted in the formation of complex mixtures of the products.421 Remarkably, full conversion of the substrate with high C16–C18 selectivity was followed by the formation of at least 25% of high-MW reaction products. The high selectivity in the hydrodeoxygenation (>90%) of FAMEs for diesel-range alkanes (C15–C18) with partial hydrocracking was substantially increased when using the Ru/Fe catalyst even at 250 °C, whereas obvious cracking was observed from 210 °C over the monometallic Ru catalyst.422
In OA/MeOH cracking (500 °C) using Ni/Ce, supported on LaY molecular sieves, the hydrocarbon selectivity of ∼93% with 75% C8–C17 content was achieved;423 however, the reaction products contained high amounts (up to 60%) of aromatic hydrocarbons.
In 2024, the use of the “green” solvent scCO2 was proposed for the decarboxylation of OA in the presence of MoO3/γ-Al2O3 catalyst.424 Up to 98% decarboxylation and 49% liquid fuel yield were achieved. It should be noted that in the absence of H2, up to 10% of C18 + hydrocarbons were formed.
The aforementioned processes aimed to find green alternatives for hydrocarbon production. However, with the one exception (Principle 7 of GRC, renewable feedstocks), at the current stage, the conversion of FAs into hydrocarbons does not correspond to economic and environmental criteria for advanced processing technologies. Thus, further studies are needed, at least to provide diversification of the feedstocks, but economic viability is not likely to be achieved in the near future.
Complex enzymatic cascades
Complex sequences of enzymatic processes may result in valuable products, but their assignment to one or another type of reaction is problematic. A one-pot process using a combination of Micrococcus luteus alcohol dehydrogenase (ADH), an engineered Vibrio fluvialis amine transaminase, and Chlorella variabilis NC64A photodecarboxylase was applied in the synthesis of 9-aminoheptadecane and (S,Z)-heptadec-9-en-7-amine from oleic and ricinoleic acids, respectively. The combination of ADH, Pseudomonas putida KT2440 Baeyer–Villiger monooxygenase and photodecarboxylase resulted in the formation of n-octylnonanoate and n-hexyl undec-3-enoate. The conversion reached up to 90% with a rate of up to 37 U g−1.425 Undoubtedly similar studies are very promising from a GRC point of view, but there is still much work to do in this area.Complex organochemical processes
The products of simple but multistage organochemical modifications of oleates are (or positioned to be) in-demand products. In 2020s, several works in this direction were published. In particular, an OA-based surfactant was prepared via a three-stage method (Scheme 31a) and showed a higher oil washing efficiency in comparison with the commercial cocamidopropyl betaine and petroleum sulfonate.426 The same surfactant was also proposed for enhanced oil recovery.427 Subsequent thiol–ene click reaction with thioglycolic acid and amidation by N-phenyl-p-phenylenediamine resulted in the formation of amidoester (Scheme 31b) with promising antiwear and antioxidant properties when applied to polyol base oils.428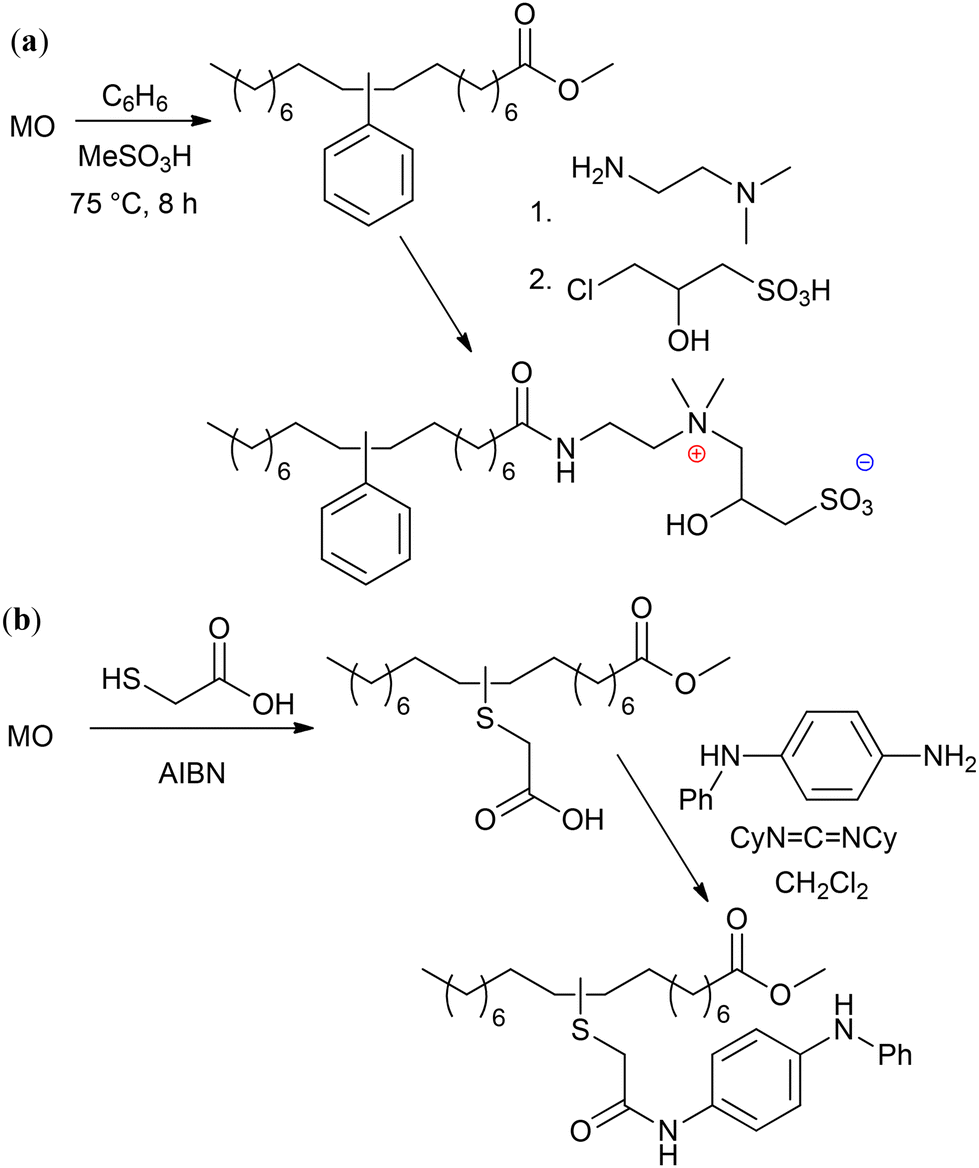 | ||
| Scheme 31 Examples of multi-stage organochemical transformations of MO. | ||
If we omit applicability and relevance, several studies deserve attention, at least as an exercise of the imagination in oleochemistry. For example, in the study by Zhao et al.,302 biobased fatty alcohols were transformed to 1-bromoalkanes, and subsequent Grignard reaction with esters and deoxygenation over Pd/C resulted in the formation of single-component three-arm long chain hydrocarbons. A similar structure is close to the “golden standard” for Group 4 poly-α-olefin engine oils,429 and the properties of similar compounds are of interest for tribology. The kinematic viscosity (KV), pour point (PP) and viscosity index (VI) of the hydrocarbons were not studied in detail in this work, and the only comparison of the hydrocarbon containing C11, C10 and C10 chains (KV100 = 3.2, VI = 153, PP = −42 °C) with Mobil PAO4 oil (KV100 = 3.8, VI = 143, PP = −66 °C) is not in favor of the former, except for its high VI value. The aforementioned processes are based on conventional toxic and expensive organic reagents, and their evaluation in terms of GRC, to put it mildly, is difficult.
Triglyceride-based polymers
(Co)polymers of ricinoleic acid and its derivatives
In early studies, ROTEP of unsaturated cyclic oligomeric lactones, products of the self-condensation of RA, was initiated by standard catalytic systems such as Sn(Oct)2 and Y(OTf)3, but the Mn of poly(RA) did not exceeded 4.4 kDa.267 In 2020, the polycondensation method was proposed for the synthesis of the copolymer of RA and 4-hydroxycinnamic acid, and the corresponding hetero-dimeric monomers were prepared and introduced in polycondensation, achieving the highest Mn of 24.3 kDa.430 Recently, high-MW poly(RA) was synthesized via the solution polycondensation of methyl ricinoleate in 1-alkyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ILs, while retaining the (Z)-configuration of the monomer units, and homopolymers with Mn up to 64.7 kDa were obtained.268 Poly(RA) had a Tg = −72.8 °C and Td = 319 °C, enabling it to be considered as a biobased degradable elastomer.Given that undec-10-enoic acid can be seen as an RA derivative, it would be pertinent to mention here the synthesis of diesters and diols, containing SiMe2OSiMe2 inserts (Scheme 32), and copolymers therefrom.431 With the use of these “building blocks”, polyurethane, polycarbonate, polyesters and polyamides were prepared. It is noteworthy that the hydrolytic degradation of the copolymers proceeds through the scission of the Si–O bond.
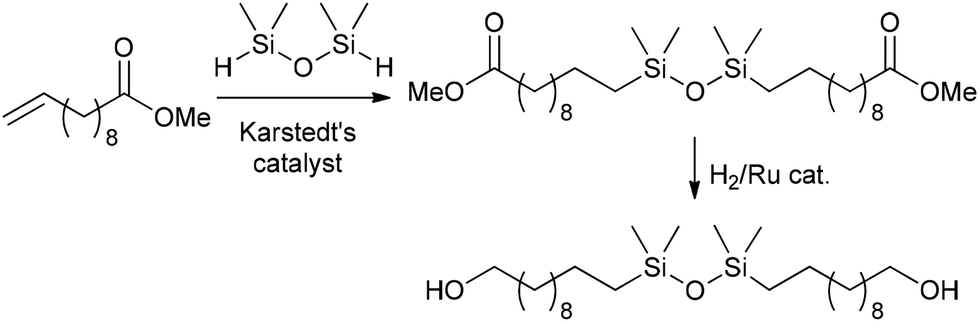 | ||
| Scheme 32 Synthesis of diester and diol from methyl undec-10-enoate.431 | ||
Oleate-based copolymers
(E)-Dimethyl octadec-9-enedioate, prepared by the self-metathesis of technical grade MO, was introduced in (nBuO)4Ti-catalyzed polycondensation with HO(CH2)nOH (n = 2, 3, 5), cyclohexane-1,4-diol and octane-1,8-diamine.209 Besides polyamide (Tm = 150 °C), the obtained copolymers had relatively low Tm (9–42 °C); all copolymers by 20 h heating under vacuum (200 °C, ∼20 h) were insoluble in common organic solvents. The polycondensation of (E)-dimethyl octadec-9-enedioate with 1,4-C6H4(CH2NH2)2 and NH(CH2CH2NH2)2 was catalyzed by 1,5,7-triazabicyclo[4,4,0]dec-5-ene (TBD) and conducted at 160 °C without and with microwave irradiation,432 and the thermal and mechanical properties of the polyamides depended on the method used.Saturated long-chain dicarboxylic acids seem more promising comonomers for the preparation of polyesters. In the 2010s,410,411 Mecking et al. synthesized polyesters based on the products of the isomerizing alkoxycarbonylation of FAMEs. For example, polyester –[O(CH2)19OC(O)(CH2)17C(O)]n– had Tm = 103 °C. Copolymers of other diols were also prepared, and the dependence of Tm of the polyesters on the lengths of the (CH2)x fragments in the diacids and diols is presented in Fig. 31. Obviously, the melt temperatures of the novel polyesters were remarkably enhanced in comparison with shorter chain diacids.
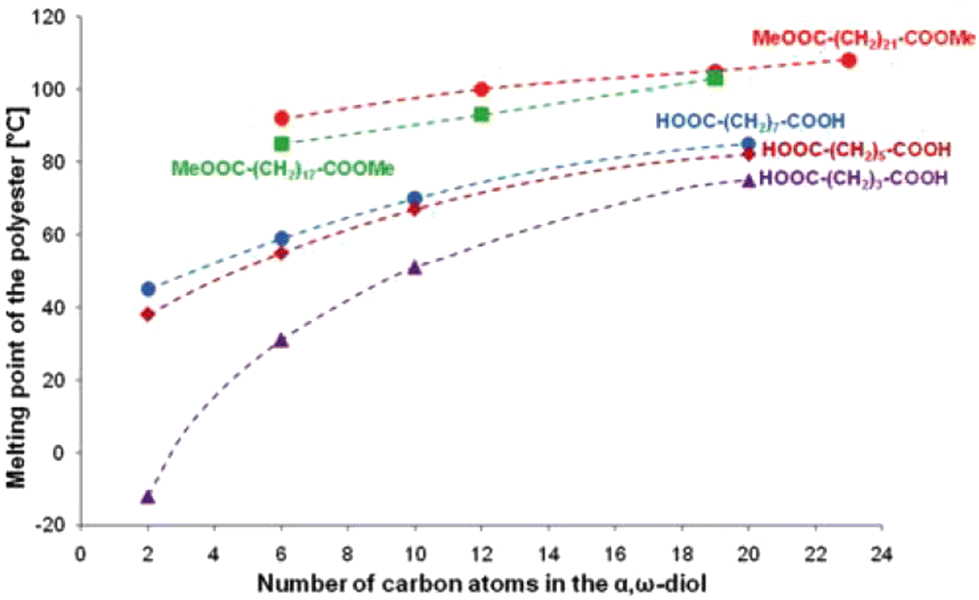 | ||
| Fig. 31 Comparison of the melting points of different polyesters obtained by the polycondensation of dimethyl-1,19-nonadecanedioate and diethyl-1,23-tricosanedioate and of azelaic, pimelic, and glutaric acid with α,ω-diols of different chain lengths.411 Reprinted with permission, Copyright (2011), the American Chemical Society. | ||
An alternative approach to the use of long-chain dicarboxylic acids is based on the self-metathesis of oleates (C18 diacid) or other unsaturated esters with subsequent hydrogenation of the CC bond. The corresponding copolymers were synthesized in the 2010s,205 and in 2021 Mecking et al. performed a separate study focusing on the mechanical properties and processibility of similar polyesters (based on the example of –[O(CH2)18OC(O)(CH2)16C(O)]n–) in comparison with polycarbonate –[O(CH2)18OC(O)]n– and commercial high-density polyethylene (HDPE).433 It was found that all the copolymers crystallized similarly with the formation of aligned hydrocarbon segments with a high degree of crystallinity. Polyester and polycarbonate showed better 3D printability in comparison with HDPE. The additional advantage of biobased polyesters and polycarbonate is the ease of their chemical recycling via transesterification under mild conditions (120 °C in EtOH or MeOH), even in the presence of poly(ethylene terephthalate), not to mention polyolefin plastics.
In 2017,211 polyesters of the formula –[O(CH2)xOC(O)(CH2)x−2C(O)]n– with Tm = 113 and 120 °C for x = 32 and 48, respectively, and copolymers with commercial diols –[O(CH2)yOC(O)(CH2)x−2C(O)]n– were obtained starting from long-chain α,ω-unsaturated diesters (obtained by self-metathesis of MO). Polyester –[O(CH2)48OC(O)(CH2)46C(O)]n– demonstrated discontinuous lamellar thickening characterized by a discrete increase in Tm with an increase in the crystallization time.434 A double melting peak (Tm = 112.7 °C and 117.8 °C for the second heat, 10 °C min−1) appeared due to the melting of the crystals with the same crystallographic packing but different thicknesses. In 2023,435 polyesters –[O(CH2)xOC(O)(CH2)x−2C(O)]n– (x = 12, 18, 32, 48) were studied, and their base characteristics are presented in Table 12. For x = 18, 32 and 48, the formation of crystals with a non-integer or integer number of ester-ester layers in the same crystallographic form was detected. It should be noted that rapid quenching of the polyesters resulted in the formation of unlayered crystals, which was complete for x = 48 and partial for polyesters with a shorter (CH2)x spacer.436 This complex behavior of biobased PE-like materials is obviously essential for their further processing.
435
| x | Ester groups per 100 CH2 | M w, kDa | Đ M, kDa | T c, °C | T m, °C | ΔHm, J g−1 |
|---|---|---|---|---|---|---|
| 12 | 9.1 | 80 | 2.5 | 71.2 °C | 83.9 °C | 126 |
| 18 | 5.9 | 90 | 1.9 | 85.6 °C | 97.8 °C | 143 |
| 32 | 3.2 | 77 | 2.5 | 99.8 °C | 110.9 °C | 160 |
| 48 | 2.1 | 26 | 1.9 | 108.8 °C | 117.8 °C | 180 |
In 2024, Eck and Mecking summarized the interim results of the studies of PE-like biobased polyesters,437 and FA-derived analogs of PE also received attention in a similar review devoted to PE-like materials.438 In these works, the similarity in the mechanical properties between –[O(CH2)18OC(O)(CH2)16C(O)]n– (Young's modulus E ≈ 900 MPa, stress at yield σy ≈ 22 MPa, elongation at break ε ≈ 500%) and HDPE (E ≈ 900 MPa, σy ≈ 27 MPa, ε ≈ 900%), as well as excellent water vapor permeability for a biobased polyester, were noted. For comparison,437 –[O(CH2)18OC(O)(CH2)2C(O)]n– had E = 730 MPa, σy = 19 MPa, ε = 330% and Tm = 96 °C, and this material attracted particular attention from Mecking's group due to its high potential for processing, inter alia, melt spinning to yield synthetic fibers (Fig. 32).
 | ||
| Fig. 32 Upscaling and processing of PE-2,18. (a) PE-2,18 pellets obtained from a 10 kg-scale polycondensation reaction employing commercially available, technical grade monomers. (b) Fabric made from melt-spun PE-2,18 fibers.437 Reprinted with permission, Copyright (2024), the American Chemical Society. | ||
The retention of the CC bond in copolymers is not conducive to achieving good thermal properties. According to one recent result, copolymers based on biobased 9-decen-1-yl diesters of adipic or azelaic acid were synthesized by Moser et al. via two methods, i.e., ADMET polymerization and photoinitiated thiol–ene reaction with HS(CH2)2SH.439 The obtained copolymers had Tm of less than 70 °C.
Oleate-based polyesters are undoubtedly promising alternatives to conventional polyolefin plastics. Besides their promising mechanical characteristics, these copolymers have undoubted advantages, as manifested in their ease of chemical recycling in the presence of conventional plastics. The prospects for the gradual replacement of polyolefins and PET with these new materials are justified in terms of GRC, but the current estimated cost of similar materials is too high for immediate industrial implementation.
Ring-opening transesterification polymerization of macrolactones
ROTEP of macrolactones, obtained from renewable feedstocks, is an alternative approach to polyesters, containing long aliphatic fragments. Although the ROTEP of conventional smaller cyclic esters (ε-caprolactone, lactides, six-membered cyclic carbonates, etc.) is dictated by the monomer ring strain (enthalpy-driven process), in the case of macrolactones, the ROTEP is an entropy-driven process, favored by a decrease in temperature. As previously mentioned, ROTEP of RA-based macrolactones is not effective for the synthesis of high-MW polymers.267 Alternatively, different unsaturated macrolactones are accessible via RCM of alkenyl oleates and related compounds. These macrolactones usually represent mixtures of (E)- and (Z)-isomers with different stability and reactivity, which complicates their polymerization. In recent years, a few works on ROTEP of unsaturated macrolactones were published. The results of these studies are of general importance for ROTEP of the entire family of unsaturated macrolactones.In 2020, Naddeo et al. used pyridylamido zinc(II) complex Zn1 as a catalyst for the BnOH-initiated homo- and copolymerization of ω-6-hexadecenlactone (Scheme 33).440 The homopolymer of ω-6-hexadecenlactone had Tm = 55 °C, which is close to Tm of poly(εCL). Poly(ω-6-hexadecenlactone-co-εCL) containing 73 mol% of εCL units had Tm = 42 °C. The melting point of poly(ω-6-hexadecenlactone-co-ω-pentadecenlactone) gradually decreased from 90 °C to 55 °C with an increase in the content of unsaturated lactone in the copolymer. This copolymer was functionalized via radical thiol–ene reaction with eugenol-derived 4-(3-mercaptopropyl)-2-methoxyphenol to obtain a polyethylene-like polyester material with antimicrobial properties (antimicrobial E. coli test) resulting from the presence of 2-methoxyphenol fragments.441
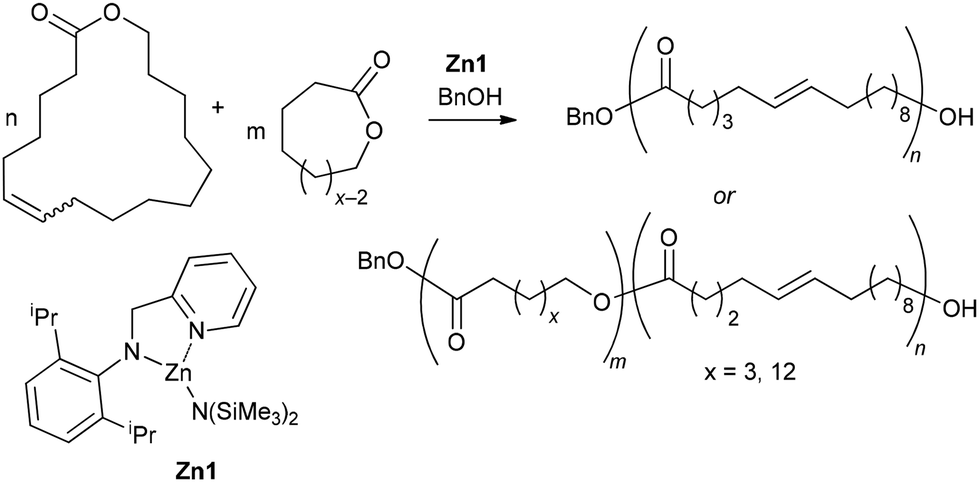 | ||
| Scheme 33 (Co)polymerization of ω-6-hexadecenlactone.440 | ||
Globalide, a 16-membered unsaturated macrolactone (Scheme 34), is potentially available from natural sources and can also be viewed as an appropriate monomer for the study of ROTEP and functionalization of unsaturated polyesters. To date, enzymatic catalysis is the main method for the ROTEP of globalide, and the Novozym 435-catalyzed process was optimized recently with the use of supercritical CO2/propane reaction medium.442
 | ||
| Scheme 34 Globalide, its homopolymer, and functionalization via thiol–ene click chemistry.444 | ||
Candida Antarctica recombinant Lipase B catalyzed the polymerization of globalide with the formation of a polymer (Mn = 30.7 kDa, ĐM = 1.38) that was simultaneously functionalized by HS-containing antimicrobial dyes and cross-linked with (CH2OCH2CH2SH)2via thiol–ene reaction.443 The resulting organogels showed excellent antimicrobial activity against S. aureus and E. coli. To increase the biocompatibility of poly(globalide) and vary its physical properties, the polymer was functionalized by HS(CH2)6OP(O)(OR)2 (R = Et, Ph).444 With an increase in the degree of functionalization (DF), Tm gradually decreased from 40 °C to 35 °C, and the material became amorphous when DF reached 36 mol%.
Another study on the functionalization of poly(globalide) aimed at introducing the RGD peptide to improve the cell adhesive and proliferative characteristics of the material.445 As shown in Fig. 33, electrospun mats were cross-linked by HS(CH2)5SH, functionalized by 2-(Boc-amino) ethanethiol, and after deprotection, coupled with RGD peptide via EDC/NHS chemistry. The functionalized ES material showed similar tensile strength as non-functionalized cross-linked fibers, but higher elasticity. The in vitro tests revealed an increase in the adhesion and proliferation of human mesenchymal stem cells.
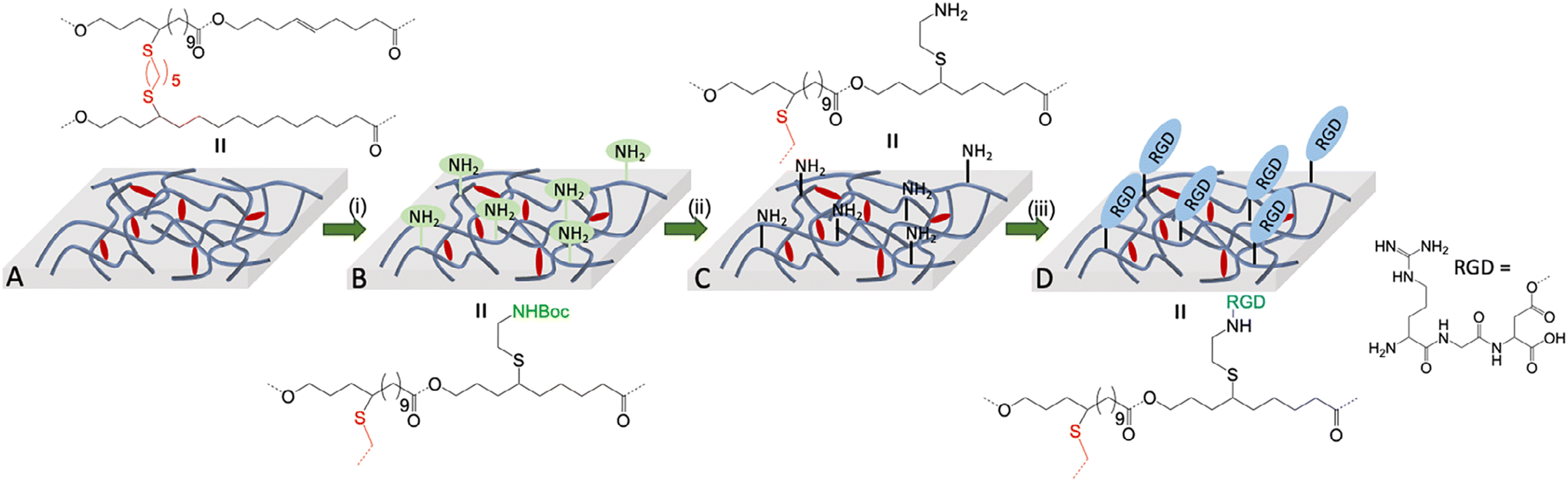 | ||
| Fig. 33 Reaction steps to produce RGD-functional electrospun fibers from cross-linked poly(globalide).445 Reprinted with permission, Copyright (2022), Wiley-VCH. | ||
Poly(globalide) (synthesized by Novozym 435-catalyzed ROP, Mn = 51 kDa, Tm = 44 °C) was used in the preparation of bilayered films with regenerated cellulose nanofibers via layer-by-layer casting.446 The bilayered films showed superior keratinocyte growth in comparison with pristine poly(globalide), demonstrating the high potential of these materials in skin tissue engineering.
Unsaturated macrolactones, derivatives of ω-alkenyl oleate, undec-10-enoate of dec-9-enoate, were not studied in ROTEP. Saturated macrolactones from HO(CH2)xCOOH (x = 10, 12, 14), prepared by the ozonolysis/reduction of Malania oleifera Chum oil FAs and subsequent intramolecular cyclization,447 can also be potentially used in ROP. These monomers are waiting to be explored. Also, it should be noted that unsaturated macrolactones can be introduced in metathesis copolymerization, as was shown recently in the example of ω-6-hexadecenlactone and norbornene,448 which opens up additional opportunities to create new polymer materials.
In conclusion of this section, particular attention should be paid to the common method of the improvement of the mechanical characteristics of biobased aliphatic polyesters via the use of branching agents, which was demonstrated very recently using the example of ADMET polymerization of dianhydro-D-glucityl bis(undec-10-enoate) in the presence of glycerol tris(undec-10-enoate).449 A similar approach gave excellent results when applied to petroleum-based polyesters,450 and thus why not apply this approach more widely for the improvement of the characteristics of biobased saturated polyesters obtained by transesterification?
Summary and outlook
In the media, green chemistry is usually associated with the prevention of environmental pollution and the use of renewable feedstocks. Unfortunately, many researchers share this view and are positioning their studies as “green” without considering the principles of GRC. These principles are abundantly clear in terms of modernist science. In fact, GRC principles represent a set of requirements for modern and efficient processes and technologies that deserve further study, development and industrial implementation. The GRC principles serve as a type of filter for the selection of prospective directions in modern oleochemistry from huge amounts of data reported in hundreds of publications.The use of catalysts is one of the principles of GRC. Among the trends in modern chemistry, the search for “greener” organocatalytic and enzymatic alternatives to the commonly used metal-containing catalysts is quite applicable to oleochemistry. In our review, the content was divided into thematic sections based on the reaction types, but it seems reasonable here to compare metal-based catalysts and organocatalysts in several processes in which both types of catalysts were studied. The best results for each type of catalyst are presented in Table 13. As can be seen, if organocatalysis or enzymatic catalysis is applicable, it is quite competitive with metal-based catalysts.
| Process | Metal-based catalytic approach | Organocatalytic/enzymatic approach | ||
|---|---|---|---|---|
| Description and result | Ref. | Description and result | Ref. | |
|
a Best results for MeOK-catalyzed methanolysis: 10 min, 60 °C, [MeOH]/[oil] = 5.5:1, 1.0 wt% MeOK, yield 97.7%.
|
||||
| Esterification of FAs | Hf-containing MOF (4.1 wt%); [MeOH]/[OA] = 19.5:1, 49 °C, 9.5 h. Yield 98.6% |
72 |
C. Antarctica lipase B on acrylic resin (0.25 wt%); [MeOH]/[OA] = 1.5:1, 55 °C, 45 h. Yield 99% |
75 |
| Methanolysis of TGsa | Li2ZrO3 (6 wt%); [MeOH]/[oil] = 8:1, 65 °C, 2 h; recyclable (7×). Yield 98%–99% |
98 | Eversa® Transform lipase immobilized on Fe3O4/biochar (14 wt%); [MeOH]/[oil] = 7:1, 30 °C, 14 h. Yield 95.7% |
115 |
| Epoxidation of OA or MO | H3PMo12O40 supported on 2D channel silica (17 wt%); [H2O2]/[MO] = 4, 90 °C, 4 h. Yield 82.5% | 320 | [H2O2]/[HCOOH]/[OA] = 20:2:1, 80 °C, 2 h. Yield 85.3% |
311 |
| Novozym 435, 5 mg mL−1; 0.27 M H2O2, 0.15 M OA in MeCN, 50 °C, 5 h. Yield 83% | 274 | |||
| Carboxylation of epoxidized MO | 1-Hydroxypropyl-3-n-butylimidazolium chloride, NbCl5, carboxymethyl cellulose (∼4 wt%); 170 °C, 30 bar CO 2, 6 h. Yield <68.5% | 344 | Ascorbic acid (1.5 mol%) [Bu4N]Cl (5 mol%), 10 bar CO2, 100 °C 48 h. Yield 90% | 349 |
FAs and FAMEs, with all their merits and shortcomings, represent the main starting materials for the production of fine oleochemicals. Recent studies on the synthesis of FAs and FAMEs from TGs were mainly focused on biobased fuels. However, actual catalytic solutions, facilitating the separation and purification of FAMEs, can be applied for the preparation of the starting materials for subsequent catalytic reactions. The production of FAMEs as biodiesel and FAs from triglycerides is industrially adopted (block A in Fig. 34) as a large-scale oleochemical process. Several other processes have also been developed and implemented in the oleochemical industry, including the production of azelaic acid from oleates,271,272,273 sebacic and undec-10-enoic acid from castor oil,286 production of oleyl alcohol,11,369 and butenolysis of MO250,251 (block A in Fig. 34). This block also includes pilot trials of the ethenolysis of MO.218 Among these processes, the cross-metathesis of oleates seems to be the most efficient and atom-consuming in terms of GRC principles.
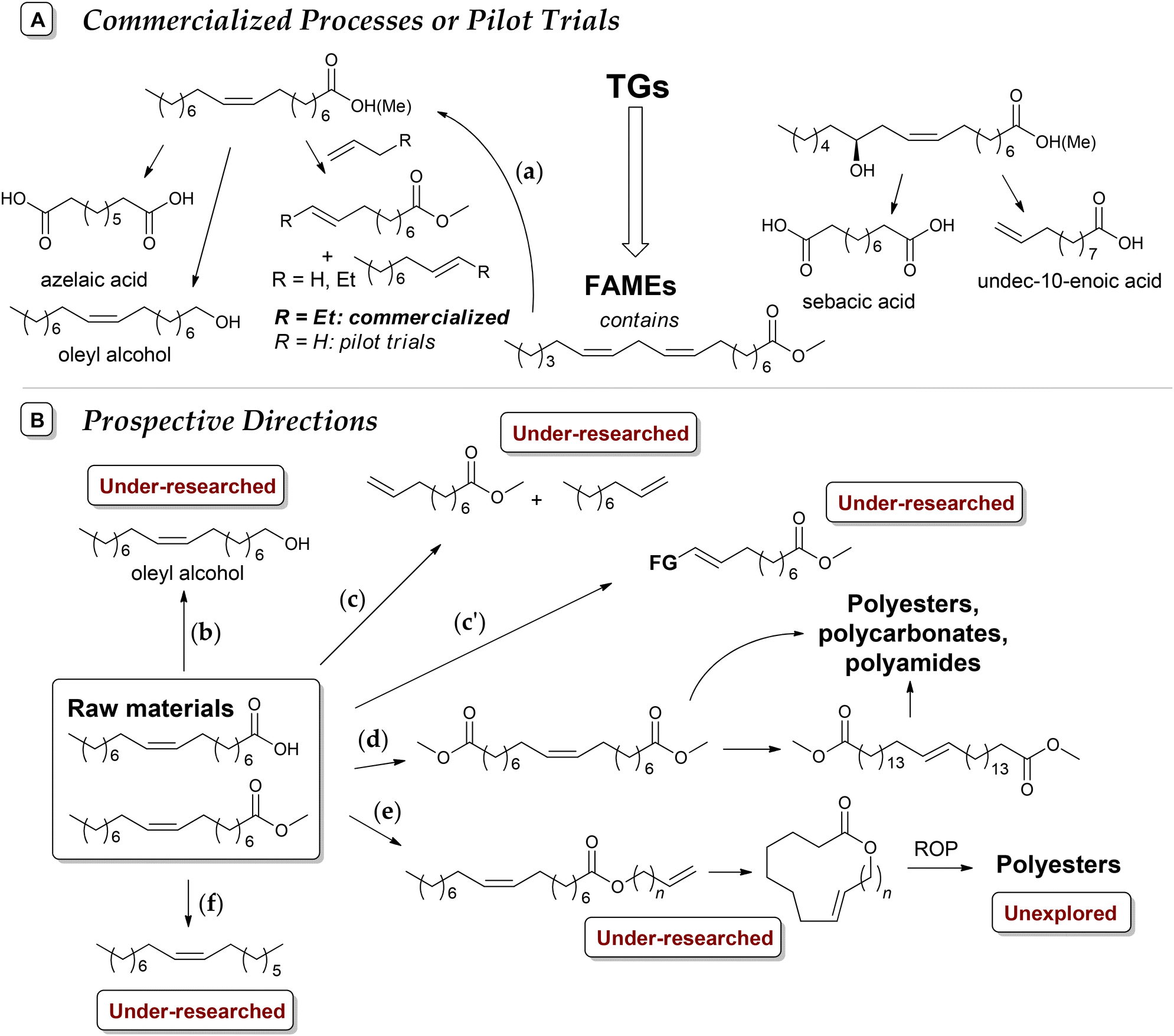 | ||
| Fig. 34 Industrially implemented processes (A) and prospective directions (B) of advanced oleochemistry (based on the example of MO). | ||
Fig. 34 also summarizes the promising but still under-researched or unexplored (B) processes of actual oleochemistry, in particular pathway (a) highlights the important aspect of the preparation of oleates, and the opportunity for the selective hydrogenation of polyunsaturated FAMEs to increase oleate content. However, this catalytic task remains unresolved to date.
The most feasible and prospective directions in oleate-based chemistry include industrially implemented processes that need further improvement. The synthesis of in-demand oleic alcohol (b) is still based on the use of low-active heterogeneous catalysts under harsh reaction conditions, and the search for more active and selective catalysts remains relevant; modest progress in this field was only achieved last year.375 A catalytic breakthrough in the ethenolysis of oleates (c) was literally achieved in recent years using SCAAC Ru complexes (TON > 106),187 but the problems of the purity of the raw material, elimination of the catalyst and real separation of the reaction products with acceptable isolated yields have not been entirely resolved. Industrially implemented approaches for cross-metathesis with the use of unsaturated counterparts other than ethylene are limited by α-olefins, and thus the use of “polar” monomers in the cross-metathesis of oleates (c′) deserves attention.
Oleate self-metathesis (d) opens ways to obtain biobased plastics; however, there is much that can be done in this area to improve the mechanical properties, rheology and processibility of similar materials. RCM of ω-alkenyl oleates and related compounds (e) has also been long studied, and in recent years efficient methods and catalysts that can overcome the limitations of the dilution principle have been developed. However, the products of this process, a wide variety of unsaturated lactones, are still totally unexplored in ROTEP and poorly studied in ROMP. Progress in catalytic decarboxylation with retention of the CC bond (f) was achieved through the use of photoenzymatic catalysis with all its inherent drawbacks. Thus, is there no alternative to the more traditional catalytic solution?
In this way, modern oleochemistry still has room to grow. However, it should be understood that the renewability of the feedstock does not make any process with the use of TGs, oleates and their analogs a suitable basis for “green” technology. All the principles of green chemistry should be considered when developing new catalytic and organochemical solutions; otherwise, the study becomes meaningless at least within the green economy framework.
Abbreviations
| acac | Acetylacetonate, MeCOCHCOMe |
| AcOH | Acetic acid |
| ADMET | Acyclic diene metathesis polymerization |
| CAAC | Cyclic alkyl amino carbene |
| CM | Cross-metathesis |
| COF(s) | Covalent organic framework(s) |
| DF | Degree of functionalization |
| DMCC | Double metal cyanide catalyst |
| E a | Activation energy |
| Et2O | Diethyl ether |
| EtOH | Ethanol |
| FA(s) | Fatty acid(s) |
| FAMEs | Fatty acid methyl esters |
| GC | Gas chromatography |
| GRC | Green chemistry |
| HPLC | High pressure liquid chromatography |
| IL(s) | Ionic liquid(s) |
| iPrOH | Isopropanol |
| MeCN | Acetonitrile |
| MeOAc | Methyl acetate |
| MeOH | Methanol |
| MO | Methyl oleate |
| MOF(s) | Metal–organic framework(s) |
| MsOH | Methanesulfonic acid |
| NHC | N-Heterocyclic carbene |
| OA | Oleic acid |
| OAc | Acetate |
| PEG | Poly(ethylene glycol) |
| Pluronic P-123 | (CH2CH2O)20-b-(CH2CHMeO)70-b-(CH2CH2O)20 |
| PUFA(s) | Poly(unsaturated) fatty acid(s) |
| RA | Ricinoleic acid |
| RCM | Ring-closing metathesis |
| ROH | Aliphatic alcohol |
| ROTEP | Ring-opening transesterification polymerization |
| SCAAC | Spirocyclic alkyl amino carbene |
| scCO2 | Supercritical CO2 |
| SM | Self-metathesis |
| t BuOH | tert-Butanol |
| TG(s) | Triglyceride(s) |
| T g | Glass transition temperature |
| TON | Turnover number |
| U | The enzyme unit, the amount of enzyme that catalyses the reaction of 1 μmol of substrate per minute |
Author contributions
Conceptualization: P. V. I. and I. E. N.; funding acquisition: P. V. I.; methodology: P. V. I. and I. E. N.; writing – original draft: P. V. I. and I. E. N.; writing – review & editing: P. V. I. and I. E. N.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed with a financial support of the Russian Science Foundation (grant No. 24-43-20016). Analysis of the greener alternative to common plastics was made in the frameworks of TIPS RAS State Plan.References
- Y. Nakagawa, M. Yabushita and K. Tomishige, RSC Sustainability, 2023, 1, 814 RSC.
- S. Pandey, B. S. Rajput and S. H. Chikkali, Green Chem., 2021, 23, 4255 RSC.
- A. S. Belousov, A. L. Esipovich, E. A. Kanakov and K. V. Otopkova, Sustainable Energy Fuels, 2021, 5, 4512 RSC.
- C. Len, V. Duhan, W. Ouyang, R. Nguyen and B. Lochab, Front. Chem., 2023, 11, 1306182 CrossRef CAS PubMed.
- I. M. Rizwanul Fattah, H. C. Ong, T. M. I. Mahlia, M. Mofijur, A. S. Silitonga, S. M. Ashrafur Rahman and A. Ahmad, Front. Energy Res., 2020, 8, 101 CrossRef.
- M. U. Qadeer, M. Ayoub, M. Komiyama, M. U. K. Daulatzai, A. Mukhtar, S. Saqib, S. Ullah, M. A. Qyyum, S. Asif and A. Bokhari, J. Cleaner Prod., 2021, 309, 127388 CrossRef CAS.
- X.-M. Wang, Y.-N. Zeng and Y.-R. Wang, Renewable Energy, 2023, 207, 385 CrossRef CAS.
- F. O. Nitbani, P. J. P. Tjitda, H. E. Wogo and A. I. R. Detha, J. Oleo Sci., 2022, 71, 781 CrossRef CAS PubMed.
- A. Psalidas, E. Emmanouilidou and N. C. Kokkinos, Energies, 2024, 17, 561 CrossRef CAS.
- F. Long, W. Liu, X. Jiang, Q. Zhai, X. Cao, J. Jiang and J. Xu, Renewable Sustainable Energy Rev., 2021, 148, 111269 CrossRef CAS.
- M. A. Sánchez, G. C. Torres, V. A. Mazzieri and C. L. Pieck, J. Chem. Technol. Biotechnol., 2017, 92, 27 CrossRef.
- N. Barteczko, M. Grymel and A. Chrobok, Catal. Commun., 2023, 177, 106662 CrossRef CAS.
- U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. R. Meier and J. O. Metzger, Angew. Chem., Int. Ed., 2021, 60, 20144 CrossRef CAS.
- A. Chatterjee, S. H. Hopen Eliasson and V. R. Jensen, Catal. Sci. Technol., 2018, 8, 1487 RSC.
- Y. Zhou, J. Remón, Z. Jiang, A. S. Matharu and C. Hu, Green Energy Environ., 2023, 8, 722 CrossRef CAS.
- B.-S. Chen, Y.-Y. Zeng, L. Liu, L. Chen, P. Duan, R. Luque, R. Ge and W. Zhang, Renewable Sustainable Energy Rev., 2022, 158, 112178 CrossRef CAS.
- X. Guo, A. Xia, W. Zhang, Y. Huang, X. Zhu, X. Zhu and Q. Liao, Bioresour. Technol., 2023, 367, 128232 CrossRef CAS PubMed.
- R. Maghrebi, M. Buffi, P. Bondioli and D. Chiaramonti, Renewable Sustainable Energy Rev., 2021, 149, 111264 CrossRef CAS.
- J. Spekreijse, J. P. M. Sanders, J. H. Bitter and E. L. Scott, ChemSusChem, 2017, 10, 470 CrossRef CAS PubMed.
- V. Yelchuri, K. Srikanth, R. B. N. Prasad and M. S. L. Karuna, J. Chem. Sci., 2019, 131, 39 CrossRef.
- W. Yan, Z. You, K. Meng, F. Du, S. Zhang and X. Jin, Chin. J. Chem. Eng., 2022, 48, 44 CrossRef CAS.
- T. A. Majrashi, A. Sabt, H. A. Abd El Salam, G. H. Al-Ansary, M. F. Hamissa and W. M. Eldehna, RSC Adv., 2023, 13, 13655 RSC.
- B. R. Moser, S. C. Cermak, K. M. Doll, J. A. Kenar and B. K. Sharma, J. Am. Oil Chem. Soc., 2022, 99, 801 CAS.
- A. D. Smith, A. G. Tennyson and R. C. Smith, Sustainable Chem., 2020, 1, 209 Search PubMed.
- F. Stempfle, P. Ortmann and S. Mecking, Chem. Rev., 2016, 116, 4597 CrossRef CAS.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012 CrossRef CAS.
- K. Nomura and N. W. B. Awang, ACS Sustainable Chem. Eng., 2021, 9, 5486 CrossRef CAS.
- J. A. C. Silva, L. M. Grilo, A. Gandini and T. M. Lacerda, Polymers, 2021, 13, 1722 CrossRef CAS PubMed.
- G. Hayes, M. Laurel, D. MacKinnon, T. Zhao, H. A. Houck and C. R. Becer, Chem. Rev., 2023, 123, 2609 CrossRef CAS PubMed.
- C. K. Ho, K. B. McAuley and B. A. Peppley, Renewable Sustainable Energy Rev., 2019, 113, 109261 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC.
- P. Jessop, Green Chem., 2020, 22, 13 RSC.
- A. Pérez-Rodríguez, C. M. Flores-Ortiz, G. Ma. Chávez-Camarillo, E. Cristiani-Urbina and L. Morales-Barrera, Fermentation, 2023, 9, 443 CrossRef.
- V. Dubois, S. Breton, M. Linder, J. Fanni and M. Parmentier, Eur. J. Lipid Sci. Technol., 2007, 109, 710 CrossRef CAS.
- D. J. Anneken, S. Both, R. Christoph, G. Fieg, U. Steinberner and A. Westfechtel, in Ullmann's Encyclopedia of Industrial Chemistry, VCH, Weinheim, 2006, vol. 14, pp. 73–116 Search PubMed.
- P. K. Ojha, D. K. Poudel, A. Rokaya, S. Maharjan, S. Timsina, A. Poudel, R. Satyal, P. Satyal and W. N. Setzer, Compounds, 2024, 4, 37 CrossRef CAS.
- SHO safflower, can be found under https://www.csiro.au/en/research/plants/crops/Oil-crops/SHO-safflower (accessed: 15 September 2024).
- T. Chang, J. Wu, X. Wu, M. Yao, D. Zhao, C. Guan and M. Guan, PLoS One, 2022, 17, e0272798 CrossRef CAS PubMed.
- Durchschnittlicher Preis der wichtigsten pflanzlichen Öle im Welthandel von Juli 2021 bis Juli 2024, can be found under https://de.statista.com/statistik/daten/studie/1312207/umfrage/preis-von-pflanzlichen-oelen-im-welthandel-art-monate/, (accessed: 15 September 2024).
- S. Shylesh, A. A. Gokhale, C. R. Ho and A. T. Bell, Acc. Chem. Res., 2017, 50, 2589 CrossRef CAS PubMed.
- R. Singh, A. Arora and V. Singh, Bioresour. Technol., 2021, 326, 124772 CrossRef CAS.
- OECD Library. OECD-FAO Agricultural Outlook 2021–2030, can be found under https://www.oecd-ilibrary.org/sites/89d2ac54-en/index.html?itemId=/content/component/89d2ac54-en, (accessed: 15 September 2024).
- E. F. Aransiola, T. V. Ojumu, O. O. Oyekola, T. F. Madzimbamuto and D. I. O. Ikhu-Omoregbe, Biomass Bioenergy, 2014, 61, 276 CrossRef CAS.
- S. N. Gebremariam and J. M. Marchetti, Energy Convers. Manage., 2018, 168, 74 CrossRef CAS.
- H. H. Mardhiah, H. C. Ong, H. H. Masjuki, S. Lim and H. V. Lee, Renewable Sustainable Energy Rev., 2017, 67, 1225 CrossRef CAS.
- P. Azadi, R. Malina, S. R. H. Barrett and M. Kraft, Renewable Sustainable Energy Rev., 2017, 76, 1479 CrossRef CAS.
- G. Knothe and L. F. Razon, Prog. Energy Combust. Sci., 2017, 58, 36 CrossRef.
- E. G. Al-Sakkari, M. G. Mohammed, A. A. Elozeiri, O. M. Abdeldayem, M. M. Habashy, E. S. Ong, E. R. Rene, I. Ismail and I. Ashour, Front. Energy Res., 2020, 8, 583357 CrossRef.
- C. P. Trentini, B. T. Ferreira de Mello, N. Postaue, N. Stevanato, L. Cardozo-Filho and C. da Silva, J. Supercrit. Fluids, 2020, 164, 104896 CrossRef CAS.
- Y. Chen, F. Long, Q. Huang, K. Wang, J. Jiang, J. Chen, J. Xu and X. Nie, Bioresour. Technol., 2022, 364, 128038 CrossRef CAS PubMed.
- F. O. Nitbani, P. J. P. Tjitda, B. A. Nurohmah and H. E. Wogo, J. Oleo Sci., 2020, 69, 277 CrossRef CAS PubMed.
- M. A. Peters, C. T. Alves, J. Wang and J. A. Onwudili, ACS Omega, 2022, 7, 46870 CrossRef CAS PubMed.
- P. S. Mateos, M. B. Navas, S. R. Morcelle, C. Ruscitti, S. R. Matkovic and L. E. Briand, Catal. Today, 2021, 372, 211 CrossRef CAS.
- G. P. R. Souza, T. B. A. Correia, W. S. M. Reis, E. H. Bredda, P. C. M. Da Rós and E. B. Pereira, Catal. Lett., 2022, 153, 689 CrossRef.
- W. Z. Ng, E.-S. Chan, Y. S. Tan, M. Y. Liow, C. W. Ooi, B. T. Tey and C. P. Song, ACS Sustainable Chem. Eng., 2024, 12, 7566 CrossRef CAS.
- A. Anand, P. Gnanasekaran, A. M. Allgeier and L. R. Weatherley, Food Bioprod. Process., 2020, 123, 164 CrossRef CAS.
- G. Kuang, Z. Wang, X. Luo, Z. Geng, J. Cui, M. Bilal, Z. Wang and S. Jia, Int. J. Biol. Macromol., 2023, 242, 124807 CrossRef CAS.
- M. Toikka, P. Kuzmenko, A. Samarov and M. Trofimova, Fuel, 2022, 319, 123730 CrossRef CAS.
- M. Supeno, J. P. Sihotang, Y. V. Panjaitan, D. S. Y. Damanik, J. Br. Tarigan and E. K. Sitepu, RSC Adv., 2023, 13, 33107 RSC.
- J. Zhang, J. Liu, X. Huang, S. Choi, R. Zhu, S. Tang, J. Q. Bond and L. L. Tavlarides, Fuel, 2020, 268, 117359 CrossRef CAS.
- P. Karan and R. Chakraborty, Chem. Eng. J., 2023, 455, 140032 CrossRef CAS.
- B. Zhang, M. Gao, W. Tang, X. Wang, C. Wu and Q. Wang, Energy Convers. Manage., 2023, 278, 116708 CrossRef CAS.
- S. P. Gouda, D. Shi, S. Basumatary, H. Li, R. Piloto-Rodríguez and S. L. Rokhum, Chem. Eng. J., 2024, 480, 148154 CrossRef CAS.
- K. V. Otopkova, A. L. Esipovich, E. A. Kanakov, T. A. Charykova, V. E. Baydachenko and T. A. Ryabova, Kinet. Catal., 2022, 63, 666 CrossRef CAS.
- N. Yadav, G. Yadav and Md. Ahmaruzzaman, Sci. Rep., 2023, 13, 9074 CrossRef CAS PubMed.
- Z. Wang, R. Zhang, D. Liu, D. Jiang, T. You, P. Niu, Q. Chen, K. He, L. Ren and L. Xu, Res. Chem. Intermed., 2023, 49, 5407 CrossRef CAS.
- A. T. Jensen, R. Gambetta and F. Machado, Macromol. React. Eng., 2023, 17, 2200055 CrossRef CAS.
- M. A. Hanif, S. Nisar and U. Rashid, Catal. Rev. Sci. Eng., 2017, 59, 165 CrossRef CAS.
- W. Wang, J. Zhougu, C. Liu, N. Wang, S. Shi, Z. Liu, P. Shen, F. Shi and J. Hu, Fuel, 2024, 361, 130693 CrossRef CAS.
- Y. Zhao and G. Li, J. Inorg. Organomet. Polym, 2023 DOI:10.1007/s10904-023-02932-1.
- P. Prasertpong, S. Shimpalee and N. Tippayawong, Energy Rep., 2020, 6, 66 CrossRef.
- Y. Li, K. Zhu, Y. Jiang, L. Chen, H. Zhang, H. Li and S. Yang, Fuel Process. Technol., 2023, 239, 107558 CrossRef CAS.
- T. Furusawa, T. Ebisawa, A. Toyoshima, Y. Mori and Y. Taniguchi, Chem. Eng. J., 2022, 429, 132524 CrossRef CAS.
- Z. Zhou, X. Chen, J. Worth, C. Ye, J. Chen and T. Qiu, AIChE J., 2023, 69, e18098 CrossRef CAS.
- M. Martinez-Garcia, W. Van Hecke, H. Peeters, D. Gabriels, P. Van der Weeën, W. Dejonghe and Y. Satyawali, J. Biotechnol., 2024, 379, 78 CrossRef CAS PubMed.
- R. A. Welter, H. Santana, L. G. de la Torre, M. Robertson, O. P. Taranto and M. Oelgemöller, ChemPhotoChem, 2022, 6, e202200007 CrossRef CAS.
- P. Moredi, M. Saidi and A. T. Najafabadi, Process Saf. Environ. Prot., 2021, 147, 684 CrossRef.
- A. O. Esan, S. M. Smith and S. Ganesan, Process Saf. Environ. Prot., 2022, 166, 402 CrossRef CAS.
- I. Sarantopoulos, E. Chatzisymeon, S. Foteinis and T. Tsoutsos, Energy Sustainable Dev., 2014, 23, 110 CrossRef CAS.
- C. He, Y. Mei, Y. Zhang, L. Liu, P. Li, Z. Zhang, Y. Jing, G. Li and Y. Jiao, Bioresour. Technol., 2020, 304, 123017 CrossRef CAS PubMed.
- A. Ahmad, A. K. Yadav and A. Singh, Korean J. Chem. Eng., 2023, 40, 2941 CrossRef CAS.
- A. H. Sebayang, F. Kusumo, J. Milano, A. H. Shamsuddin, A. S. Silitonga, F. Ideris, J. Siswantoro, I. Veza, M. Mofijur and S. R. Chia, Fuel, 2023, 346, 128404 CrossRef CAS.
- A. Thipdech, K. Prasertsit and S. Photaworn, Chem. Eng. Process., 2024, 195, 109614 CrossRef CAS.
- I. Chanakaewsomboon, C. Tongurai, S. Photaworn, S. Kungsanant and R. Nikhom, J. Environ. Chem. Eng., 2020, 8, 103538 CrossRef CAS.
- I. Chanakaewsomboon, K. Phoungthong, A. Palamanit, V. Seechamnanturakit and C. K. Cheng, Biomass Convers. Biorefin., 2023, 13, 9237 CrossRef CAS.
- A. Hayyan, Y.-S. Ng, M. K. Hadj-Kali, M. U. M. Junaidi, E. Ali, A. K. Aldeehani, K. H. Alkandari, F. D. H. Alajmi, A. T. H. Yeow, M. Y. Zulkifli, L. Z. Kai and M. A. Hashim, Biomass Convers. Biorefin., 2022, 12, 113 CrossRef CAS.
- H. Bai, J. Tian, D. Talifu, K. Okitsu and A. Abulizi, Fuel, 2022, 318, 123697 CrossRef CAS.
- A. Chaudhuri, E. B. Temelli, C. J. W. Hop, V. P. Sureshkumar and J. van der Schaaf, Ind. Eng. Chem. Res., 2022, 61, 6831 CrossRef CAS.
- K. Thakkar, S. S. Kachhwaha and P. Kodgire, Fuel, 2022, 308, 121907 CrossRef CAS.
- M. Melchiorre, A. Amoresano, P. H. M. Budzelaar, M. E. Cucciolito, F. Mocerino, G. Pinto, F. Ruffo, A. Tuzi and R. Esposito, Catal. Lett., 2022, 152, 3785 CrossRef CAS.
- C. I. Gogoaşă, C. E. Răducanu, L. E. Petraş, D. R. C. Tîrpan, G. Vasilievici, A. L. Mîrţ, T. Dobre and O. C. Pârvulescu, Catalysts, 2023, 13, 1431 CrossRef.
- D. Kumar, C. H. Park and C. S. Kim, Int. J. Energy Res., 2020, 44, 4568 CrossRef CAS.
- N. Supamathanon, K. Boonserm, N. Osakoo, J. Wittayakun, S. Prayoonpokarach, N. Chanlek and W. Dungkaew, Renewable Energy, 2023, 202, 1460 CrossRef CAS.
- K. A. Shiferaw, J. M. Mathews, E. Yu, E.-Y. Choi and N. H. Tarte, Inorganics, 2023, 11, 163 CrossRef.
- R. Manurung, R. Hasibuan and A. G. A. Siregar, Case Stud. Chem. Environ. Eng., 2024, 9, 100543 CrossRef CAS.
- P. Maneechakr and S. Karnjanakom, J. Environ. Chem. Eng., 2021, 9, 106542 CrossRef CAS.
- M. Karthikeyan, S. Sundararaman, A. Kumar, J. P. Deivasigamani and M. Rajasimman, Environ. Res., 2023, 222, 115336 CrossRef PubMed.
- Y.-M. Dai, Y.-Y. Li, J.-H. Lin, I.-H. Kao, Y.-J. Lin and C.-C. Chen, Fuel, 2021, 286, 119392 CrossRef CAS.
- N. Mohammadi, N. Ostovar, R. Niromand and F. Absalan, Sustainable Chem. Pharm., 2023, 36, 101272 CrossRef CAS.
- X. Yang, W. Liu, R. Zhao and A. Raise, Ind. Crops Prod., 2023, 204B, 117319 CrossRef.
- T. Tamoradi, A. R. Kiasat, H. Veisi, V. Nobakht and B. Karmakar, Sci. Rep., 2022, 12, 19652 CrossRef CAS PubMed.
- F. Esmi, S. Masoumi and A. K. Dalai, Catalysts, 2022, 12, 658 CrossRef CAS.
- F. Esmi, A. K. Dalai and Y. Hu, Fuel, 2023, 348, 128594 CrossRef CAS.
- Q. Wu, Q. Shu, W. Guo and X. Xing, Fuel, 2024, 361, 130668 CrossRef CAS.
- C. Fadilah, C. Kurniawan, M. Ridwan, M. Al Muttaqii, E. Agustian, A. S. Andreani, A. A. Dwiatmoko and I. Yati, React. Kinet., Mech. Catal., 2023, 136, 1529 CrossRef CAS.
- V. L. de Brito, M. A. Gonçalves, H. C. L. dos Santos, G. N. da Rocha Filho and L. R. V. da Conceição, Renewable Energy, 2023, 215, 118947 CrossRef CAS.
- S. Ao, S. P. Gouda, M. Selvaraj, R. Boddula, N. Al-Qahtani, S. Mohan and S. L. Rokhum, Energy Convers. Manage., 2024, 300, 117956 CrossRef CAS.
- P. Kumar, L. Matoh, V. C. Srivastava and U. L. Štangar, Renewable Energy, 2020, 148, 946 CrossRef CAS.
- Y. S. Choi, H. W. Jeon and E. T. Hwang, Bioresour. Technol., 2024, 412, 131394 CrossRef PubMed.
- M. Zhang, S.-H. Jun, Y. Wee, H. S. Kim, E. T. Hwang, J. Shim, S. Y. Hwang, J. Lee and J. Kim, Int. J. Biol. Macromol., 2022, 222, 2368 CrossRef CAS PubMed.
- J. López-Fernández, M. D. Benaiges and F. Valero, Bioresour. Technol., 2021, 334, 125233 CrossRef PubMed.
- G. C. Heinzl, D. A. Mota, V. Martinis, A. S. Martins, C. M. F. Soares, N. Osório, J. Gominho, K. M. Nampoothiri, R. K. Sukumaran, H. Pereira and S. Ferreira-Dias, Bioresour. Technol., 2022, 346, 126646 CrossRef CAS PubMed.
- J. Liu, G. Chen, B. Yan, W. Yi and J. Yao, Bioresour. Technol., 2022, 355, 127253 CrossRef CAS.
- J. Iyyappan, J. Jayamuthunagai, B. Bharathiraja, A. Saravanaraj, R. P. Kumar and S. Balraj, Bioresour. Technol., 2022, 363, 127893 CrossRef CAS.
- J. Guo, Y. Wang and Z. Fang, Bioresour. Technol., 2024, 394, 130237 CrossRef CAS.
- L. K. Qian and S. J. Huey, Asia-Pac. J. Chem. Eng., 2023, 18, e2994 CrossRef CAS.
- H. Nayebzadeh, F. Naderi and B. Rahmanivahid, Fuel, 2020, 271, 117595 CrossRef CAS.
- M. Rahmayanti, I. Fatimah, A. Y. Ikhsani and D. N. Azizah, Inorg. Chem. Commun., 2024, 165, 112604 CrossRef CAS.
- I. P. Cantika, M. A. Zulfikar and H. Rusli, J. Kim. Sains Apl., 2023, 26, 230 CrossRef CAS.
- M. Zeeshan, S. Ghazanfar, M. Tariq, H. M. Asif, A. Hussain, M. Usman, M. A. Khan, K. Mahmood, M. Sirajuddin and M. Imran, Renewable Energy, 2023, 210, 800 CrossRef CAS.
- M. Ameen, M. Zafar, M. F. Ramadan, M. Ahmad, T. Makhkamov, A. Bokhari, M. Mubashir, L. F. Chuah and P. L. Show, Environ. Technol. Innovation, 2023, 30, 103101 CrossRef CAS.
- L. Chen, L. He, B. Zheng, G. Wei, H. Li, H. Zhang and S. Yang, Fuel Process. Technol., 2023, 250, 107903 CrossRef CAS.
- N. Petchsoongsakul, K. Ngaosuwan, W. Kiatkittipong, D. Wongsawaeng and S. Assabumrungrat, Renewable Energy, 2020, 162, 1906 CrossRef CAS.
- E. O. Oke, O. Adeyi, B. I. Okolo, C. J. Ude, J. A. Adeyi, K. K. Salam, U. Nwokie and I. Nzeribe, Bioresour. Technol., 2021, 332, 125141 CrossRef CAS.
- J. Prameswari, W. Widayat, L. Buchori and H. Hadiyanto, Environ. Sci. Pollut. Res., 2023, 30, 98832 CrossRef CAS.
- S. Saka and D. Kusdiana, Fuel, 2001, 80, 225 CrossRef CAS.
- S. Karki, N. Sanjel, J. Poudel, J. H. Choi and S. C. Oh, Appl. Sci., 2017, 7, 632 CrossRef.
- K. de Boer and P. A. Bahri, Biomass Bioenergy, 2011, 35, 983 CrossRef CAS.
- R. García-Morales, A. Zúñiga-Moreno, F. J. Verónico-Sánchez, J. Domenzain-González, H. I. Pérez-López, C. Bouchot and O. Elizalde-Solis, Fuel, 2022, 313, 122706 CrossRef.
- M. A. Shaah, M. S. Hossain, F. Allafi, M. O. Ab Kadir and M. I. Ahmad, RSC Adv., 2022, 12, 9845 RSC.
- N. K. Singh, Y. Singh and A. Sharma, Biomass Bioenergy, 2022, 156, 106332 CrossRef CAS.
- S. S. Selvan, P. S. Pandian, A. Subathira and S. Saravanan, Biofuels, 2021, 12, 797 CrossRef.
- A. Ahmed, A. Ali, M. Mubashir, H. R. Lim, K. S. Khoo and P. L. Show, J. Phys. Energy, 2023, 5, 024003 Search PubMed.
- V. Demir and M. Akgün, ChemistrySelect, 2024, 9, e202304678 CrossRef CAS.
- A. Sandid, T. Attarbachi, R. Navarro-Tovar, M. Pérez-Page, V. Spallina and J. Esteban, Chem. Eng. J., 2024, 496, 153905 CrossRef CAS.
- A. Casas, M. J. Ramos and Á. Pérez, Chem. Eng. J., 2011, 171, 1324 CrossRef CAS.
- S. Marx, Fuel Process. Technol., 2016, 151, 139 CrossRef CAS.
- N. Akkarawatkhoosith, A. Kaewchada, C. Ngamcharussrivichai and A. Jaree, Bioenergy Res., 2020, 13, 542 CrossRef CAS.
- A. L. B. Nunes and F. Castilhos, Fuel, 2020, 267, 117264 CrossRef CAS.
- W. Supang, S. Ngamprasertsith, W. Sakdasri and R. Sawangkeaw, J. Supercrit. Fluids, 2022, 186, 105586 CrossRef CAS.
- M. Albarello, A. L. Barrachini Nunes, L. J. Vernier and F. de Castilhos, Bioenergy Res., 2024, 17, 518 CrossRef CAS.
- S. S. Gandhi, P. R. Gogate and V. D. Pakhale, Int. J. Green Energy, 2022, 20, 1514 CrossRef.
- C. Prestigiacomo, M. Biondo, A. Galia, E. Monflier, A. Ponchel, D. Prevost, O. Scialdone, S. Tilloy and R. Bleta, Fuel, 2022, 321, 124026 CrossRef CAS.
- M. L. Spiekermann and T. Seidensticker, Catal. Sci. Technol., 2024, 14, 4390 RSC.
- U. P. Laverdura, L. Rossi, F. Ferella, C. Courson, A. Zarli, R. Alhajyoussef and K. Gallucci, ACS Omega, 2020, 5, 22901 CrossRef CAS PubMed.
- E. Quaranta and D. Cornacchia, Renewable Energy, 2020, 157, 33 CrossRef CAS.
- E. Quaranta, A. Dibenedetto, A. Colucci and D. Cornacchia, Fuel, 2022, 326, 125030 CrossRef CAS.
- P. Udomsap, S. Meesiri, N. Chollacoop and A. Eiad-Ua, Nanomaterials, 2021, 11, 1431 CrossRef CAS PubMed.
- S.-Y. Chen, A. Chang, A. N. Rungsi, L. Attanatho, C.-L. Chang, J.-H. Pan, A. Suemanotham, T. Mochizuki, H. Takagi, C.-M. Yang, A. Luengnaruemitchai and H.-H. Chou, Appl. Catal., A, 2020, 602, 117707 CrossRef CAS.
- A. Na Rungsi, A. Luengnaruemitchai, N. Chollacoop, S.-Y. Chen, T. Mochizuki, H. Takagi and Y. Yoshimura, Appl. Catal., A, 2020, 590, 117351 CrossRef.
- N. Numwong, P. Prabnasak, P. Prayoonpunratn, P. Triphatthanaphong, C. Thunyaratchatanon, T. Mochizuki, S.-Y. Chen, A. Luengnaruemitchai and T. Sooknoi, Fuel Process. Technol., 2020, 203, 106393 CrossRef CAS.
- A. Na Rungsi, T. H. Truong, C. Thunyaratchatanon, A. Luengnaruemitchai, N. Chollacoop, S.-Y. Chen, T. Mochizuki, H. Takagi and Y. Yoshimura, Fuel, 2021, 298, 120658 CrossRef CAS.
- A. V. Romanenko, P. A. Simonov, M. A. Kulagina, S. I. Udalova, I. N. Voropaev and G. A. Bukhtiyarova, Catal. Ind., 2023, 15, 374 CrossRef.
- M. Zha, H. Yan, R. Li, Y. Sun, R. Wang, Y. Liu, X. Chen, X. Zhou, X. Feng and C. Yang, Mol. Catal., 2023, 538, 112996 CrossRef CAS.
- M. Konkol, R. Bicki, M. Kondracka, K. Antoniak-Jurak, P. Wiercioch and W. Próchniak, React. Kinet., Mech. Catal., 2016, 119, 595 CrossRef CAS.
- S. Phumpradit, P. Reubroycharoen, P. Kuchonthara, C. Ngamcharussrivichai and N. Hinchiranan, Catalysts, 2020, 10, 993 CrossRef CAS.
- T. Zhu, L. Zhang, Z. Li, G. Wei, Z. Xin, D. Xiong and Z. Ou, Waste Biomass Valorization, 2021, 12, 465 CrossRef CAS.
- A. M. Fiore, G. Romanazzi, C. Leonelli, P. Mastrorilli and M. M. Dell'Anna, Catalysts, 2022, 12, 506 CrossRef CAS.
- T. Longprang, N. Kaewtrakulchai, W. Kiatkittipong, A. Srifa, N. Chollacoop, A. Eiad-Ua and S. Assabumrungrat, Arabian J. Chem., 2024, 17, 105800 CrossRef CAS.
- U. P. Laverdura, L. Rossi, C. Courson, A. Zarli and K. Gallucci, Energies, 2023, 16, 7201 CrossRef CAS.
- G. Kongprawes, D. Wongsawaeng, P. Hosemann, K. Ngaosuwan, W. Kiatkittipong and S. Assabumrungrat, Int. J. Energy Res., 2021, 45, 4519 CrossRef CAS.
- K. Puprasit, D. Wongsawaeng, K. Ngaosuwan, W. Kiatkittipong and S. Assabumrungrat, J. Food Eng., 2022, 334, 111167 CrossRef CAS.
- W. Zhao, C. Hua, X. Zhang, X. Qi, K. Tanongkiat and J. Wang, Plasma Sci. Technol., 2021, 23, 095506 CrossRef CAS.
- L. Zhang, Z. Xin, L. Gao, Z. Li, G. Wei and R. Mo, J. Chem. Sci., 2021, 133, 58 CrossRef CAS.
- Z. Xin, G. Wei, L. Zhang, L. Gao, Z. Li and W. Zhao, Fuel, 2021, 299, 120877 CrossRef CAS.
- L. Gao, L. Zhang, B. Gu, L. Liang, Y. Zhou, G. Wei, J. Liu and R. Pei, J. Mater. Sci., 2022, 57, 5964 CrossRef CAS.
- H.-S. Lee, H. Seo, D. Kim and Y.-W. Lee, J. Supercrit. Fluids, 2020, 156, 104683 CrossRef CAS.
- H.-S. Lee, J. Lee, H. Seo, H. Kang, D. H. Kim and Y.-W. Lee, Fuel Process. Technol., 2021, 218, 106870 CrossRef CAS.
- L. Zhang, Z. Xin, Z. Liu, G. Wei, Z. Li and Y. Ou, Renewable Energy, 2020, 147, 695 CrossRef CAS.
- L. Zhang, Z. Xin, Z. Liu, Y. Ou, Z. Ye, Z. Li and G. Wei, Fuel, 2020, 270, 117510 CrossRef CAS.
- G. Mendow and C. A. Querini, Chem. Eng. J., 2013, 228, 93 CrossRef CAS.
- H. Yu, P. Yu and Y. Luo, J. Electr. Eng. Technol., 2017, 12, 820 CrossRef.
- R. Ampairojanawong, A. Boripun, S. Ruankon, T. Suwanasri, K. Cheenkachorn and T. Kangsadan, Electrochem, 2023, 4, 123 CrossRef CAS.
- S. S. Almady, A. I. Moussa, M. M. Deef, M. F. Zayed, S. M. Al-Sager and A. M. Aboukarima, Sustainability, 2024, 16, 2896 CrossRef CAS.
- D. Swern and W. E. Parker, J. Am. Oil Chem. Soc., 1952, 29, 614 CrossRef CAS.
- L. J. Rubin and W. Paisley, J. Am. Oil Chem. Soc., 1960, 37, 300 CrossRef CAS.
- M. Bahadi, N. Salih and J. Salimon, Appl. Sci. Eng. Prog., 2021, 14, 1 Search PubMed.
- A. A.-W. Japir, J. Salimon, D. Derawi, B. H. Yahaya, M. Bahadi, S. Al-Shuja'a and M. R. Yusop, Oilseeds Crops Oils Lipids, 2018, 25, A203 Search PubMed.
- D. G. Hayes, Y. C. Bengtsson, J. M. Van Alstine and F. Setterwall, J. Am. Oil Chem. Soc., 1998, 75, 1403 CrossRef CAS.
- F. D. Gunstone, J. McLaughlan, C. M. Scrimgeour and A. P. Watson, J. Sci. Food Agric., 1976, 27, 675 CrossRef CAS PubMed.
- R. Elkacmi, N. Kamil, M. Bennajah and S. Kitane, BioMed Res. Int., 2016, 1397852 Search PubMed.
- E. Mularczyk and J. Drzymala, Sep. Sci. Technol., 1989, 24, 151 CrossRef CAS.
- S. M. Hyde and D. Verdin, Trans. Faraday Soc., 1968, 64, 144 RSC.
- E. Bascetta, F. D. Gunstone and C. M. Scrimgeour, Lipid, 1984, 19, 801 CrossRef CAS.
- N. P. R. Ranasinghe Arachchige, N. W. Xiong and N. B. Bowden, ACS Appl. Nano Mater., 2023, 6, 6715 CrossRef CAS PubMed.
- X. Fu, Y. Liu, C. Sun and W. Liu, Fuel, 2024, 362, 130880 CrossRef CAS.
- R. Gawin, A. Tracz, P. Krajczy, A. Kozakiewicz-Piekarz, J. P. Martínez and B. Trzaskowski, J. Am. Chem. Soc., 2023, 145, 25010 CAS.
- N. A. Porter, K. A. Mills and R. L. Carter, J. Am. Chem. Soc., 1994, 116, 6690 CrossRef CAS.
- C. Ding, L. Wang, Y. P. Yao and C. Li, Food Chem., 2022, 392, 133298 CrossRef CAS PubMed.
- Z. Zhou, Y.-L. Li, F. Zhao, R. Xin, X.-H. Huang, Y.-Y. Zhang, D. Zhou and L. Qin, J. Agric. Food Chem., 2022, 70, 16410 CrossRef CAS PubMed.
- V. M. Marx, A. H. Sullivan, M. Melaimi, S. C. Virgil, B. K. Keitz, D. S. Weinberger, G. Bertrand and R. H. Grubbs, Angew. Chem., Int. Ed., 2015, 54, 1919 CrossRef CAS PubMed.
- J. M. Avramovic, A. M. Marjanović Jeromela, M. S. Krstić, B. M. Kiprovski, A. V. Veličković, D. D. Rajković and V. B. Veljković, Sep. Purif. Rev., 2024, 1–23 CrossRef.
- B. Vaisman, A. Shikanov and A. J. Domb, J. Am. Oil Chem. Soc., 2008, 85, 169 CrossRef CAS.
- K. K. Adama and O. A. Anani, Heliyon, 2023, 9, e16536 CrossRef CAS PubMed.
- A. L. Bose and D. Goswami, Chem. Eng. Commun., 2020, 207, 972 CrossRef CAS.
- D. Bhattacharjee and D. Goswami, J. Dispersion Sci. Technol., 2021, 42, 947 CrossRef CAS.
- L. Xu, Q. Yang, H. Liu, Q. Li, M. Li, X. Liu, S. Chen, X. Wang and H. Suo, Mol. Catal., 2024, 568, 114521 CrossRef CAS.
- S. Singh, S. Sharma, S. J. Sarma and S. K. Brar, Environ. Prog. Sustainable Energy, 2023, 42, e14172 CrossRef CAS.
- S. Singh, S. Sharma, S. J. Sharma and S. K. Brar, Fermentation, 2023, 9, 318 CrossRef CAS.
- K. Park and J.-S. Hahn, Metab. Eng., 2024, 81, 197 CrossRef CAS PubMed.
- A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243 CrossRef CAS PubMed.
- M. Ghashghaee, Rev. Chem. Eng., 2017, 34, 595 CrossRef.
- C. Copéret, Z. J. Berkson, K. W. Chan, J. de Jesus Silva, C. P. Gordon, M. Pucino and P. A. Zhizhko, Chem. Sci., 2021, 12, 3092 RSC.
- J. C. Mol, Green Chem., 2002, 4, 5 RSC.
- S. Chikkali and S. Mecking, Angew. Chem., Int. Ed., 2012, 51, 5802 CrossRef CAS PubMed.
- J. Morvan, M. Mauduit, G. Bertrand and R. Jazzar, ACS Catal., 2021, 11, 1714 CrossRef CAS.
- J. Zelin, A. F. Trasarti and C. R. Apesteguía, Catal. Commun., 2013, 42, 84 CrossRef CAS.
- M. R. Carrasco, C. Nikitine, M. Hamou, C. de Bellefon, C. Thieuleux and V. Meille, Catalysts, 2020, 10, 435 CrossRef CAS.
- P. Krzesiński, V. César, K. Grela and P. Ortiz, RSC Sustainability, 2023, 1, 2033 RSC.
- A. Sytniczuk, F. Struzik, K. Grela and A. Kajetanowicz, Chem. Sci., 2023, 14, 10744 RSC.
- T. Witt, M. Häußler, S. Kulpa and S. Mecking, Angew. Chem., Int. Ed., 2017, 56, 7589 CrossRef CAS PubMed.
- I. Rossetti, A. Tripodi and G. Ramis, Int. J. Hydrogen Energy, 2020, 45, 10292 CrossRef CAS.
- R. E. Cardozo, N. M. Clauser, F. E. Felissia, M. C. Area and M. E. Vallejos, Green Chem., 2024, 26, 4092 RSC.
- L. Feng, J. Guo, J. Pang, M. Yin, Y. Zhao, P. Wu and M. Zheng, Green Chem., 2024, 26, 8564 RSC.
- J. A. Brekan and G. E. Gerhardt, Lube-Tech, 2016, 103, 34 Search PubMed.
- A. Kajetanowicz, M. Chwalba, A. Gawin, A. Tracz and K. Grela, Eur. J. Lipid Sci. Technol., 2020, 122, 1900263 CrossRef CAS.
- Elevance starts shipment from Indonesia biorefifinery. Green Chemicals Blog, can be found under https://greenchemicalsblog.com/elevance-starts-shipment-from-indonesia-biorefinery/, 2012 (accessed: 15 September 2024).
- Partner collaborations advanced Elevance technology, can be found under https://biomassmagazine.com/articles/partner-collaborations-advanced-elevance-technology-12816 (accessed: 15 September 2024).
- P. Tobón, S. Gómez, A. Restrepo and F. Núñez-Zarur, Organometallics, 2021, 40, 119 CrossRef.
- A. H. Hoveyda, Z. Liu, C. Qin, T. Koengeter and Y. Mu, Angew. Chem., Int. Ed., 2020, 59, 22324 CrossRef CAS PubMed.
- D. L. Nascimento, M. Foscato, G. Occhipinti, V. R. Jensen and D. E. Fogg, J. Am. Chem. Soc., 2021, 143, 11072 CrossRef CAS.
- D. L. Nascimento and D. E. Fogg, J. Am. Chem. Soc., 2019, 141, 19236 CrossRef CAS.
- K. Młodzikowska-Pieńko and B. Trzaskowski, Organometallics, 2022, 41, 3627 CrossRef.
- G. Occhipinti, D. L. Nascimento, M. Foscato, D. E. Fogg and V. R. Jensen, Chem. Sci., 2022, 13, 5107 RSC.
- W. L. McClennan, S. A. Rufh, J. A. M. Lummiss and D. E. Fogg, J. Am. Chem. Soc., 2016, 138, 14668 CrossRef CAS PubMed.
- C. O. Blanco and D. E. Fogg, ACS Catal., 2023, 13, 1097 CrossRef CAS PubMed.
- P. D. Nieres, V. A. Vaillard, J. Zelín, N. I. Neuman, A. F. Trasarti, C. R. Apesteguía and S. E. Vaillard, ChemCatChem, 2023, 15, e202300010 CrossRef CAS.
- P. D. Nieres, J. Zelin, A. F. Trasarti and C. R. Apesteguia, Catal. Sci. Technol., 2016, 6, 6561 RSC.
- G. L. P. Aydos, B. C. Leal, O. W. Perez-Lopez and J. Dupont, Catal. Commun., 2014, 53, 57 CrossRef CAS.
- C. Ambrosio, V. Paradiso, C. Costabile, V. Bertolasi, T. Caruso and F. Grisi, Dalton Trans., 2018, 47, 6615 RSC.
- R. A. Pradhan, M. Arshad and A. Ullah, J. Ind. Eng. Chem., 2020, 84, 42 CrossRef CAS.
- M. Aşkun, K. Sagdic, F. Inci and B. Ö. Öztürk, Catal. Sci. Technol., 2022, 12, 6174 RSC.
- R. Gawin, A. Kozakiewicz, P. A. Guńka, P. Dąbrowski and K. Skowerski, Angew. Chem., Int. Ed., 2017, 56, 981 CrossRef CAS PubMed.
- D. L. Nascimento, A. Gawin, R. Gawin, P. A. Guńka, J. Zachara, K. Skowerski and D. E. Fogg, J. Am. Chem. Soc., 2019, 141, 10626 CrossRef CAS PubMed.
- A. E. Samkian, Y. Xu, S. C. Virgil, K.-Y. Yoon and R. H. Grubbs, Organometallics, 2020, 39, 495 CrossRef CAS.
- S. Park, S. Byun, H. Ryu, H. Hahm, J. Lee and S. Hong, ACS Catal., 2021, 11, 13860 CrossRef CAS.
- A. Sytniczuk, F. Struzik, V. Purohit, K. Grela and A. Kajetanowicz, Catal. Sci. Technol., 2023, 13, 3682 RSC.
- A. Sytniczuk, A. Kajetanowicz and K. Grela, Chem. Catal., 2023, 3, 100713 CrossRef CAS.
- S. Byun, H. Seo, J.-H. Choi, J. Y. Ryu, J. Lee, W. Chung and S. Hong, Organometallics, 2019, 38, 4121 CrossRef CAS.
- S. Byun, S. Park, Y. Choi, J. Y. Ryu, J. Lee, J.-H. Choi and S. Hong, ACS Catal., 2020, 10, 10592 CrossRef CAS.
- S. Byun, D.-A. Park, S. Kim, S. Kim, J. Y. Ryu, J. Lee and S. Hong, Inorg. Chem. Front., 2022, 9, 323 RSC.
- M. Kim, H. Kim, S. Kim, S. Hong and E. Lee, Organometallics, 2022, 41, 1905 CrossRef CAS.
- C. Copéret and J.-M. Basset, Adv. Synth. Catal., 2007, 349, 78 CrossRef.
- J. Deme, M. Nagyházi, Z. May, J. Hancsók, J. Valyon, S. Kéki, R. Tuba and G. Turczel, React. Kinet., Mech. Catal., 2022, 135, 2519 CrossRef CAS.
- M. Lee, Y. H. Han and D. W. Hwang, Catal. Commun., 2020, 144, 106088 CrossRef CAS.
- P. Rouge, K. C. Szeto, Y. Bouhoute, N. Merle, A. De Mallmann, L. Delevoye, R. M. Gauvin and M. Taoufik, Organometallics, 2020, 39, 1105 CrossRef CAS.
- N. Tangyen, W. Natongchai, S. Del Gobbo and V. D'Elia, ACS Omega, 2024, 9, 19712 CrossRef CAS PubMed.
- M. Chen and C. Chen, Angew. Chem., Int. Ed., 2020, 59, 1206 CrossRef CAS PubMed.
- S. de Roo, F. Einsiedler and S. Mecking, Angew. Chem., Int. Ed., 2023, 62, e202219222 CrossRef CAS PubMed.
- C. S. Higman, J. A. M. Lummiss and D. E. Fogg, Angew. Chem., Int. Ed., 2016, 55, 3552 CrossRef CAS PubMed.
- C. Bruneau and C. Fischmeister, in Organometallics for Green Catalysis. Topics in Organometallic Chemistry, ed. P. Dixneuf and J. F. Soulé, Springer, New York, 2018, vol. 63, pp. 77–102 Search PubMed.
- J. Zelin, P. D. Nieres, A. F. Trasarti and C. R. Apesteguía, Appl. Catal., A, 2015, 502, 410 CrossRef CAS.
- V. Paradiso, R. Contino and F. Grisi, Catalysts, 2020, 10, 904 CrossRef CAS.
- K. E. Litinas and B. E. Salteris, J. Chem. Soc., Perkin Trans. 1, 1997, 2869 RSC.
- A. Dumas, S. Colombel-Rouen, I. Curbet, G. Forcher, F. Tripoteau, F. Caijo, P. Queval, M. Rouen, O. Baslé and M. Mauduit, Catal. Sci. Technol., 2019, 9, 436 RSC.
- R. Rana, Shreya, R. Upadhyay and S. K. Maurya, ACS Omega, 2021, 6, 25381 CrossRef CAS PubMed.
- V. Yelchuri, T. Azmeera and M. S. L. Karuna, Eur. J. Chem., 2023, 14, 273 CrossRef CAS.
- A. Sytniczuk, M. Dąbrowski, Ł. Banach, M. Urban, S. Czarnocka-Śniadała, M. Milewski, A. Kajetanowicz and K. Grela, J. Am. Chem. Soc., 2018, 140, 8895 CrossRef CAS PubMed.
- F. Ziegler, J. Teske, I. Elser, M. Dyballa, W. Frey, H. Kraus, N. Hansen, J. Rybka, U. Tallarek and M. R. Buchmeiser, J. Am. Chem. Soc., 2019, 141, 19014 CrossRef CAS PubMed.
- Ł. Grzesiński, M. Milewski, M. Nadirova, A. Kajetanowicz and K. Grela, Organometallics, 2023, 42, 2453 CrossRef PubMed.
- E.-J. Y. Boisvert, H. C. Max and D. E. Fogg, ACS Catal., 2023, 13, 2885 CrossRef CAS.
- Ł. Grzesiński, M. Nadirova, J. Guschlbauer, A. Brotons-Rufes, A. Poater, A. Kajetanowicz and K. Grela, Nat. Commun., 2024, 15, 8981 CrossRef PubMed.
- R. S. Phatake, N. B. Nechmad, O. Reany and N. G. Lemcoff, Adv. Synth. Catal., 2022, 364, 1465 CrossRef CAS.
- A. Del Vecchio, J. Talcik, S. Colombel-Rouen, J. Lorkowski, M. R. Serrato, T. Roisnel, N. Vanthuyne, G. Bertrand, R. Jazzar and M. Mauduit, ACS Catal., 2023, 13, 6195 CrossRef CAS.
- A. Sytniczuk, M. Milewski, M. Dąbrowski, K. Grela and A. Kajetanowicz, Green Chem., 2023, 25, 2299 RSC.
- F. Ziegler, H. Kraus, M. J. Benedikter, D. Wang, J. R. Bruckner, M. Nowakowski, K. Weißer, H. Solodenko, G. Schmitz, M. Bauer, N. Hansen and M. R. Buchmeiser, ACS Catal., 2021, 11, 11570 CrossRef CAS.
- R. Slivniak and A. J. Domb, Biomacromolecules, 2005, 6, 1679 CrossRef CAS PubMed.
- L. Wang, H. Yuan, X. Ma, Z. Li, H. Sun, X. Zhang, X. Huang, Q. Peng and Y. Tan, RSC Sustainability, 2024, 2, 2541 RSC.
- R. Ogawa and M. A. Hillmyer, Polym. Chem., 2021, 12, 2253 RSC.
- A. Soutelo-Maria, J.-L. Dubois, J.-L. Couturier and G. Cravotto, Catalysts, 2018, 8, 464 CrossRef.
- B. Cornils and P. Lappe, in Ullmann's Encyclopedia of Industrial Chemistry, VCH, Weinheim, 2005, pp. 1–19 Search PubMed.
- A. Todea, C. Deganutti, M. Spennato, F. Asaro, G. Zingone, T. Milizia and L. Gardossi, Polymers, 2021, 13, 4091 CrossRef CAS PubMed.
- G. A. De Leon Izeppi, J.-L. Dubois, A. Balle and A. Soutelo-Maria, Ind. Crops Prod., 2020, 150, 112411 CrossRef CAS.
- E. Brenna, D. Colombo, G. Di Lacce, F. G. Gatti, M. C. Ghezzi, F. Tentori, D. Tessaro and M. Viola, Molecules, 2020, 25, 1882 CrossRef CAS PubMed.
- V. Dorado, C. I. Herrerías and J. M. Fraile, Tetrahedron, 2023, 139, 133450 CrossRef CAS.
- M. Melchiorre, V. Benessere, M. E. Cucciolito, C. Melchiorre, F. Ruffo and R. Esposito, ChemistrySelect, 2020, 5, 1396 CrossRef CAS.
- J. Vondran, M. Peters, A. Schnettger, C. Sichelschmidt and T. Seidensticker, Catal. Sci. Technol., 2022, 12, 3622 RSC.
- L. S. Correa and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2023, 125, 2200171 CrossRef.
- F. Nocito, I. Orlando, F. Digioia, M. Aresta and A. Dibenedetto, ACS Sustainable Chem. Eng., 2021, 9, 6459 CrossRef CAS.
- A. Vassoi, T. Tabanelli, A. Sacchetti, F. Di Gioia, L. Capuzzi and F. Cavani, ChemSusChem, 2021, 14, 2375 CrossRef CAS PubMed.
- S. Gámez, E. de la Torre and E. M. Gaigneaux, ChemCatChem, 2022, 14, e202201134 CrossRef.
- S. Gámez, E. de la Torre and E. M. Gaigneaux, Ind. Eng. Chem. Res., 2023, 62, 4928 CrossRef.
- A. M. Bolt, Adv. Pharmacol., 2023, 96, 119 CAS.
- L. Leyssens, B. Vinck, C. Van Der Straeten, F. Wuyts and L. Maes, Toxicology, 2017, 387, 43 CrossRef CAS PubMed.
- R. T. P. da Silva, D. O. Silva, P. F. M. de Oliveira, R. Bellabarba, P. Johnston, J. Smit, J. Holt, M. Betham and L. M. Rossi, ChemPlusChem, 2023, 88, e202300268 CrossRef CAS PubMed.
- S. Singh, S. Sharma, S. J. Sarma and S. K. Brar, Environ. Prog. Sustainable Energy, 2023, 42, e14008 CrossRef CAS.
- M. J. Diamond, R. G. Binder and T. H. Applewhite, J. Am. Oil Chem. Soc., 1965, 42, 882 CrossRef CAS.
- S. Yu, J. Cui, X. Wang, C. Zhong, Y. Li and J. Yao, J. Am. Oil Chem. Soc., 2020, 97, 663 CrossRef CAS.
- G. Bhukya and S. S. Kaki, Eur. J. Lipid Sci. Technol., 2022, 124, 2100244 CrossRef CAS.
- A. Kumar, A. Kumar, M. M. S. Cabral-Pinto, A. K. Chaturvedi, A. A. Shabnam, G. Subrahmanyam, R. Mondal, D. K. Gupta, S. K. Malyan, S. S. Kumar, S. A. Khan and K. K. Yadav, Int. J. Environ. Res. Public Health, 2020, 17, 2179 CrossRef CAS PubMed.
- Q. Zhang, Z. Wang, Z. Qin, B. Li and Z. Guo, Molecules, 2024, 29, 4504 CrossRef CAS PubMed.
- H. Guobin, L. Zuyu, Y. Suling and Y. Rufeng, J. Am. Oil Chem. Soc., 1996, 73, 1109 CrossRef CAS.
- X. Mao, Q. Xie, X. Yi, Y. Duan, S. Yu, Z. Wu, X. Liang and Y. Nie, Appl. Therm. Eng., 2021, 194, 117093 CrossRef CAS.
- X. Mao, Q. Xie, Y. Duan, S. Yu and Y. Nie, Materials, 2022, 15, 1565 CrossRef CAS.
- G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Polymers, 2010, 2, 440 CrossRef CAS.
- S. Y. Dhanuskar, A. Modak, D. Mhatre, S. N. Naik and K. K. Pant, ChemistrySelect, 2023, 8, e202204680 CrossRef.
- Y. Duan, P. Yuan, S. Huang, L. Wang, J. Deng, S. Yu, Q. Xie and Y. Nie, Chem. Eng. Process., 2023, 184, 109293 CrossRef CAS.
- C. V. Rodrigues, C. L. Marcele, B. Vanderleia, S. E. Luiz, W. V. Rodolfo, M. H. França and E. Laércio, Chem. Ind. Chem. Eng. Q., 2023, 29, 263 CrossRef.
- R. Qu, K. Junge and M. Beller, Chem. Rev., 2023, 123, 1103 CrossRef CAS.
- C. L. Roman, N. da Silva Moura, S. Wicker, K. M. Dooley and J. A. Dorman, ACS Appl. Nano Mater., 2022, 5, 3676 CrossRef CAS PubMed.
- Y. Xiao, Y. Liu, X. Zhang, J. Hou, X. Liu, Y. Yuan and X. Liao, Catal. Commun., 2022, 165, 106448 CrossRef CAS.
- S. Chen, T. Wu, Y. Fang and C. Zhao, Renewable Energy, 2022, 186, 280 CrossRef CAS.
- B. R. Fatmayanti, Jumina, B. Purwono, Y. S. Kurniawan, H. D. Pranowo and E. N. Sholikhah, ChemistrySelect, 2024, 9, e202400752 CrossRef CAS.
- R. Calmanti, N. Sargentoni, M. Selva and A. Perosa, Catalysts, 2021, 11, 1477 CrossRef CAS.
- G. Lewandowski, M. Musik, K. Malarczyk-Matusiak, Ł. Sałaciński and E. Milchert, Mini-Rev. Org. Chem., 2020, 17, 412 CrossRef CAS.
- P. T. Wai, P. Jiang, Y. Shen, P. Zhang, Q. Gu and Y. Leng, RSC Adv., 2019, 9, 38119 RSC.
- Y.-B. Huang, M.-Y. Yao, P.-P. Xin, M.-C. Zhou, T. Yang and H. Pan, RSC Adv., 2015, 5, 74783 RSC.
- M. R. Janković, O. M. Govedarica and S. V. Sinadinović-Fišer, Ind. Crops Prod., 2020, 143, 111881 CrossRef.
- D. R. Sawitri, P. Mulyono, Rochmadi and A. Budiman, J. Teknol., 2021, 83, 157 CrossRef.
- M. B. Mahadi, I. S. Azmi, M. Z. Ab Kadir, N. Mohamed, M. A. Rahman and M. J. Jalil, Biomass Convers. Biorefin., 2024, 14, 17395 CrossRef CAS.
- M. Bahadi, N. Salih and J. Salimon, Biointerface Res. Appl. Chem., 2021, 11, 14359 CAS.
- I. S. Azmi, M. J. Jalil and A. Hadi, Biomass Convers. Biorefin., 2024, 14, 7847 CrossRef CAS.
- I. S. Azmi, T. A. Z. T. Ozir, I. Md. Rasib, S. D. Nurherdiana and M. J. Jalil, Biomass Convers. Biorefin., 2024, 14, 13303 CrossRef CAS.
- J. Vondran, J. Pela, D. Palczewski, M. Skiborowski and T. Seidensticker, ACS Sustainable Chem. Eng., 2021, 9, 11469 CrossRef CAS.
- D. Yu. Yushchenko, Z. P. Pai, Yu. V. Uchenova and T. B. Khlebnikova, Kinet. Catal., 2023, 64, 270 CrossRef.
- M. Jabbour, I. Ben Talouba, L. Balland and N. Mouhab, Ind. Eng. Chem. Res., 2024, 63, 1773 CrossRef CAS.
- S. Klinyod, K. Yomthong, P. Iadrat, P. Kidkhunthod, K. Choojun, T. Sooknoi and C. Wattanakit, Chem. – Asian J., 2024, e202400669 CrossRef CAS PubMed.
- X. Gao, B. Luo, Y. Hong, P. He, Z. Zhang and G. Wu, Front. Chem. Sci. Eng., 2023, 17, 772 CrossRef CAS.
- S. Praserthdam, M. Rittiruam, K. Maungthong, T. Saelee, S. Somdee and P. Praserthdam, Sci. Rep., 2020, 10, 18952 CrossRef CAS.
- C. Zhou, F. Li, C. Wang, R. Cao, Y. Liu, Y. Yin, H. Chen, Z. Wan, Y. Zhu and W. Yang, Mol. Catal., 2023, 547, 113406 CrossRef CAS.
- A. L. Esipovich, E. A. Kanakov, T. A. Charykova and K. V. Otopkova, Catalysts, 2023, 13, 138 CrossRef CAS.
- E. Jeon, J.-Y. Park, M.-C. Kim, S.-J. Lee and D.-K. Kim, Catal. Today, 2023, 411–412, 113901 CrossRef.
- M. Kirpluks, R. Pomilovskis, E. Vanags, A. Abolins, I. Mierina and A. Fridrihsone, Process Biochem., 2022, 122, 38 CrossRef CAS.
- W. Wikström, A. F. Aguilera, P. Tolvanen, R. Lassfolk, A. Medina, K. Eränen and T. Salmi, Ind. Eng. Chem. Res., 2023, 62, 9169 CrossRef.
- T. R. Amarante, P. Neves, F. A. Almeida Paz, A. C. Gomes, M. Pillinger, A. A. Valente and I. S. Gonçalves, Catal. Sci. Technol., 2021, 11, 2214 RSC.
- M. S. Nunes, D. M. Gomes, A. C. Gomes, P. Neves, R. F. Mendes, F. A. Almeida Paz, A. D. Lopes, M. Pillinger, A. A. Valente and I. S. Gonçalves, Catalysts, 2023, 13, 565 CrossRef CAS.
- M. S. Nunes, A. C. Gomes, P. Neves, R. F. Mendes, F. A. Almeida Paz, A. D. Lopes, M. Pillinger, I. S. Gonçalves and A. A. Valente, Catal. Today, 2023, 423, 114273 CrossRef CAS.
- D. M. Gomes, A. F. Silva, A. C. Gomes, P. Neves, A. A. Valente, I. S. Gonçalves and M. Pillinger, Catal. Today, 2023, 418, 114050 CrossRef CAS.
- D. M. Gomes, X. Yao, P. Neves, N. Pinna, P. A. Russo and A. A. Valente, Catal. Sci. Technol., 2024, 14, 646 RSC.
- P. Neves, A. C. Gomes, R. P. Monteiro, M. J. Santos, A. A. Valente, A. D. Lopes, I. S. Gonçalves and M. Pillinger, Appl. Organomet. Chem., 2024, 38, e7412 CrossRef CAS.
- B. Testud, D. Pintori, E. Grau, D. Taton and H. Cramail, Green Chem., 2017, 19, 259 RSC.
- V. Dorado, C. I. Herrerías and J. M. Fraile, Mol. Catal., 2023, 547, 113282 CrossRef CAS.
- T. De Dios Miguel, N. D. Vu, M. Lemaire and N. Duguet, ChemSusChem, 2021, 14, 379 CrossRef CAS.
- D. D. Winfield and B. R. Moser, J. Am. Oil Chem. Soc., 2023, 100, 237 CrossRef CAS.
- N. Kamairudin, S. S. Hoong, L. C. Abdullah, H. Ariffin and D. R. A. Biak, Molecules, 2021, 26, 648 CrossRef CAS PubMed.
- N. Kamairudin, L. C. Abdullah, S. S. Hoong, D. R. A. Biak and H. Ariffin, Polymers, 2023, 15, 3028 CrossRef CAS PubMed.
- J. K. Ogunjobi, T. J. Farmer, J. H. Clark and C. R. McElroy, ACS Sustainable Chem. Eng., 2023, 11, 1857 CrossRef CAS.
- F.-H. Liu, C.-L. Gao, C.-C. Zhang, H.-Z. Gang, B.-Z. Mu and S.-Z. Yang, J. Surfactants Deterg., 2023, 26, 135 CrossRef CAS.
- M. Cutajar, F. Machado, V. C. Crucitti, S. Braovac, R. A. Stockman, S. M. Howdle and S. E. Harding, Sci. Rep., 2022, 12, 18411 CrossRef PubMed.
- F. Jie, M. Liao, S. Jiang, C. Song, C. Tang and B. Chen, J. Mech. Sci. Technol., 2023, 37, 6639 CrossRef.
- P. Tyagi, D. Singh, N. Malik, S. Kumar and R. S. Malik, Mater. Today, 2023, 65, 133 CrossRef CAS.
- A. Centeno-Pedrazo, J. Perez-Arce, Z. Freixa, P. Ortiz and E. J. Garcia-Suarez, Ind. Eng. Chem. Res., 2023, 62, 3428 CrossRef CAS.
- V. Aomchad, À. Cristòfol, F. D. Monica, B. Limburg, V. D'Elia and A. W. Kleij, Green Chem., 2021, 23, 1077 RSC.
- X. Cai, P. Tolvanen, P. Virtanen, K. Eränen, J. Rahkila, S. Leveneur and T. Salmi, Int. J. Chem. Kinet., 2021, 53, 1203 CrossRef CAS.
- X.-S. Cai, H.-X. Guo, X.-J. Zhao, J.-J. Zhao, H.-M. Liu, S. Leveneur and X.-D. Wang, Chem. Eng. Sci., 2024, 291, 119964 CrossRef CAS.
- W. Y. Perez-Sena, K. Eränen, N. Kumar, L. Estel, S. Leveneur and T. Salmi, J. CO2 Utiliz., 2022, 57, 101879 CrossRef CAS.
- T. Salmi, W. Y. Perez-Sena, F. Ciccarelli, K. Eränen, A. Medina, T. Cogliano, M. Di Serio, J. Wärnå, S. Leveneur and V. Russo, Chem. Eng. Sci., 2024, 285, 119578 CrossRef CAS.
- S. Saengsaen, S. Del Gobbo and V. D'Elia, Chem. Eng. Res. Des., 2023, 191, 630 CrossRef CAS.
- W. Natongchai, S. Pornpraprom and V. D′ Elia, Asian J. Org. Chem., 2020, 9, 801 CrossRef CAS.
- J. F. Nieto-Alarcón, D. A. González, E. Vigueras-Santiago and S. Hernández-López, J. Chem., 2023, 2023, 3169663 Search PubMed.
- A. Centeno-Pedrazo, J. Perez-Arce, S. Prieto-Fernandez, Z. Freixa and E. J. Garcia-Suarez, Mol. Catal., 2021, 515, 111889 CrossRef CAS.
- A. F. G. Agudelo, W. Y. Pérez-Sena, N. Kebir, T. Salmi, L. A. Ríos and S. Leveneur, Chem. Eng. Sci., 2020, 228, 115954 CrossRef.
- N. Araji, G. Chatel, A. Moores, F. Jérôme and K. De Oliveira Vigier, New J. Chem., 2020, 44, 11507 RSC.
- M. Bouchakour, M. Daaou and N. Duguet, Eur. J. Org. Chem., 2021, 1647 CrossRef CAS.
- S. C. Cermak, T. A. Isbell, J. W. Bredsguard and T. D. Thompson, in Fatty Acids. Chemistry, Synthesis, and Applications, ed. M. U. Ahmad, AOCS Press, Urbana, IL, 2017, pp. 431–475 Search PubMed.
- Y. Chen, G. Biresaw, S. C. Cermak, T. A. Isbell, H. L. Ngo, L. Chen and A. L. Durham, J. Am. Oil Chem. Soc., 2020, 97, 231 CrossRef CAS.
- R. Thakur, P. Sanap, P. Gogate and A. Pratap, Chem. Eng. Process., 2023, 193, 109533 CrossRef CAS.
- P. Sanap, D. Sonawane, S. Patil and A. Pratap, Ind. Crops Prod., 2022, 188, 115711 CrossRef CAS.
- G. B. Bantchev and S. C. Cermak, Fuel, 2022, 309, 122190 CrossRef CAS.
- R. Thakur, P. Sanap, S. Patil and A. Pratap, Ind. Crops Prod., 2023, 205, 117435 CrossRef CAS.
- G. Biresaw, Y. Chen, L. Chen, H. Ngo, K. Wagner, K. E. Vermillion and S. C. Cermak, Ind. Crops Prod., 2022, 182, 114857 CrossRef CAS.
- T. Vanbésien, E. Monflier and F. Hapiot, Eur. J. Lipid Sci. Technol., 2016, 118, 26 CrossRef.
- C. Becquet, F. Berche, H. Bricout, E. Monflier and S. Tilloy, ACS Sustainable Chem. Eng., 2021, 9, 9444 CrossRef CAS.
- N. Herrmann, J. Bianga, T. Gaide, M. Drewing, D. Vogt and T. Seidensticker, Green Chem., 2019, 21, 6738 RSC.
- T. F. H. Roth, M. Häusler, D. Vogt and T. Seidensticker, Catal. Today, 2024, 439, 114803 CrossRef CAS.
- J. Vondran, R. Moeschke, T. Deysenroth and T. Seidensticker, Eur. J. Lipid Sci. Technol., 2023, 125, 2200126 CrossRef CAS.
- Fatty Alcohol Market Analysis, can be found under https://www.chemanalyst.com/industry-report/fatty-alcohol-market-634, 2023 (accessed: 15 September 2024).
- R. D. Rieke, D. S. Thakur, B. D. Roberts and G. T. White, J. Am. Oil Chem. Soc., 1997, 74, 341 CrossRef CAS.
- P. Munkajohnpong, C. Kesornpun, S. Buttranon, J. Jaroensuk, N. Weeranoppanant and P. Chaiyen, Biofuels, Bioprod. Biorefin., 2020, 14, 986 CrossRef CAS.
- C. A. Fonseca Benítez, V. A. Mazzieri, C. R. Vera, V. M. Benitez and C. L. Pieck, React. Chem. Eng., 2021, 6, 726 RSC.
- C. A. Fonseca Benítez, V. A. Mazzieri, M. A. Sánchez, M. A. Vicerich, V. M. Benitez and C. L. Pieck, Can. J. Chem. Eng., 2023, 101, 2800 CrossRef.
- M. A. Vicerich, M. A. Sánchez, C. F. Benítez and V. A. Mazzieri, Catal. Lett., 2024, 154, 5806 CrossRef CAS.
- M. A. Sánchez, M. A. Vicerich, C. F. Benítez, F. Nardi, F. J. Passamonti and V. A. Mazzieri, React. Kinet., Mech. Catal., 2024, 137, 2173 CrossRef.
- N. F. Both, A. Spannenberg, H. Jiao, K. Junge and M. Beller, Angew. Chem., 2023, 135, e202307987 CrossRef.
- N. F. Both, J. Thaens, A. Spannenberg, H. Jiao, K. Junge and M. Beller, ACS Catal., 2024, 14, 4082 CrossRef CAS.
- A. Vallejo Orrego, C. A. Ferretti and V. K. Díez, J. Am. Oil Chem. Soc., 2020, 97, 1029 CrossRef CAS.
- A. Vallejo Orrego, C. A. Ferretti and V. K. Díez, J. Am. Oil Chem. Soc., 2023, 100, 477 CrossRef CAS.
- C. P. Prasanth, J. Ebbin, A. Abhijith, D. S. Nair, I. Ibnusaud, J. Raskatov and B. Singaram, J. Org. Chem., 2018, 83, 1431 CrossRef CAS PubMed.
- U. Prieto-Pascual, I. Bustos, Z. Freixa, A. Kumar and M. A. Huertos, RSC Sustainability, 2024, 2, 1052 RSC.
- D. Sorigué, B. Légeret, S. Cuiné, S. Blangy, S. Moulin, E. Billon, P. Richaud, S. Brugière, Y. Couté, D. Nurizzo, P. Müller, K. Brettel, D. Pignol, P. Arnoux, Y. Li-Beisson, G. Peltier and F. Beisson, Science, 2017, 357, 903 CrossRef PubMed.
- T. M. Hedison, D. J. Heyes and N. S. Scrutton, Curr. Res. Chem. Biol., 2022, 2, 100017 CrossRef CAS.
- Y. Sui, X. Guo, R. Zhou, Z. Fu, Y. Chai, A. Xia and W. Zhao, Mol. Biotechnol., 2023 DOI:10.1007/s12033-023-00775-2.
- Y. Ma, X. Zhang, W. Zhang, P. Li, Y. Li, F. Hollmann and Y. Wang, ChemPhotoChem, 2020, 4, 39 CrossRef CAS.
- H. T. Duong, Y. Wu, A. Sutor, B. O. Burek, F. Hollmann and J. Z. Bloh, ChemSusChem, 2021, 14, 1053 CrossRef CAS PubMed.
- X. Guo, A. Xia, F. Li, Y. Huang, X. Zhu, W. Zhang, X. Zhu and Q. Liao, Energy Convers. Manage., 2022, 255, 115311 CrossRef CAS.
- F. Li, A. Xia, X. Guo, Y. Huang, X. Zhu, W. Zhang, R. Chen and Q. Liao, Renewable Sustainable Energy Rev., 2023, 183, 113507 CrossRef CAS.
- F. Li, A. Xia, X. Guo, W. Zhang, Y. Huang, X. Zhu, X. Zhu and Q. Liao, J. Environ. Chem. Eng., 2023, 11, 110748 CrossRef CAS.
- R. Wu, X. Li, L. Wang and D. Zhong, Angew. Chem., Int. Ed., 2022, 61, e202209180 CrossRef CAS PubMed.
- B. Lakavath, T. M. Hedison, D. J. Heyes, M. Shanmugam, M. Sakuma, R. Hoeven, V. Tilakaratna and N. S. Scrutton, Anal. Biochem., 2020, 600, 113749 CrossRef CAS PubMed.
- Y. Wu, C. E. Paul and F. Hollmann, ChemBioChem, 2021, 22, 2420 CrossRef CAS PubMed.
- K. N. Papageridis, N. D. Charisiou, S. Douvartzides, V. Sebastian, S. J. Hinder, M. A. Baker, S. AlKhoori, K. Polychronopoulou and M. A. Goula, Renewable Energy, 2020, 162, 1793 CrossRef CAS.
- M. Gousi, E. Kordouli, K. Bourikas, E. Simianakis, S. Ladas, G. D. Panagiotou, C. Kordulis and A. Lycourghiotis, Catal. Today, 2020, 355, 903 CrossRef CAS.
- M. Ameen, M. T. Azizan, A. Ramli, S. Yusup and B. Abdullah, Catal. Today, 2020, 355, 51 CrossRef CAS.
- Y. Wu, J. Duan, X. Li, K. W. Wu, J. Wang, J. Zheng, S. Li, D. Wang and Z. Zheng, Renewable Energy, 2023, 218, 119372 CrossRef CAS.
- S. Zulkepli, H. V. Lee, N. Abd. Rahman, L. T. Chuan, P. L. Show, W.-H. Chen and J. C. Juan, Fuel, 2022, 308, 121860 CrossRef CAS.
- U. Gupta, M. Yadav, B. Saini, R. Krishnapriya and R. K. Sharma, Fuel, 2024, 360, 130588 CrossRef CAS.
- L. Canoira, R. Alcántara, S. Torcal, N. Tsiouvaras, E. Lois and D. M. Korres, Fuel, 2007, 86, 965 CrossRef CAS.
- I. B. Talouba, A. Diop, K. Neveu, L. Balland, N. Brodu and N. Mouhab, Termochim. Acta, 2024, 731, 179647 CrossRef.
- M. G. Kallitsakis, D. K. Gioftsidou, M. A. Tzani, P. A. Angaridis, M. A. Terzidis and I. N. Lykakis, Organics, 2022, 3, 173 CrossRef CAS.
- R. Ahmed, P. C. Varras, M. G. Siskos, H. Siddiqui, M. I. Choudhary and I. P. Gerothanassis, Molecules, 2020, 25, 4902 CrossRef CAS PubMed.
- Y. Yao, T. Wang, Z. Qiang, W. Du and C. Li, J. Agric. Food Chem., 2024, 72, 704 CrossRef CAS PubMed.
- V. G. Kontogianni and I. P. Gerothanassis, Molecules, 2022, 27, 2139 CrossRef CAS PubMed.
- D. Kumar, C. H. Park and C. S. Kim, Ind. Crops Prod., 2020, 158, 113001 CrossRef CAS.
- K. K. Shirshin, A. L. Esipovich, V. I. Strakhova and Y. V. Sak, React. Kinet., Mech. Catal., 2023, 136, 741 CrossRef CAS.
- H.-C. Lin, M. Kidonakis, J. P. Kaniraj, I. Kholomieiev, B. Fridrich, M. C. A. Stuart and A. J. Minnaard, Green Chem., 2024, 26, 4715 RSC.
- Y. Zhu, Q. Sun, Y. Wang, J. Tang, Y. Wang and H. Wang, Corros. Sci., 2021, 185, 109414 CrossRef CAS.
- Y. Wang, D. Wang, L. Xu, K. Xue, X. Zhang, X. Shi, C. Liu and J. Meng, Miner. Eng., 2023, 204, 108405 CrossRef CAS.
- W. Zhang, Z. Guo, F. Lyu, W. Sun, Z. Gao and M. Tian, Miner. Eng., 2023, 199, 108117 CrossRef CAS.
- Z. Guo, M. Tian, Z. Gao and W. Sun, J. Mol. Liq., 2023, 387, 122563 CrossRef CAS.
- D. Quinzler and S. Mecking, Angew. Chem., Int. Ed., 2010, 49, 4306 CrossRef CAS PubMed.
- F. Stempfle, D. Quinzler, I. Heckler and S. Mecking, Macromolecules, 2011, 44, 4159 CrossRef CAS.
- G. Walther, J. Deutsch, A. Martin, F.-E. Baumann, D. Fridag, R. Franke and A. Köckritz, ChemSusChem, 2011, 4, 1052 CrossRef CAS PubMed.
- P. Roesle, C. J. Dürr, H. M. Möller, L. Cavallo, L. Caporaso and S. Mecking, J. Am. Chem. Soc., 2012, 134, 17696 CrossRef CAS.
- N. Herrmann, K. Köhnke and T. Seidensticker, ACS Sustainable Chem. Eng., 2020, 8, 10633 CAS.
- R. F. Herzog, T. Huber and H. M. Riepl, J. Organomet. Chem., 2021, 956, 122112 CrossRef CAS.
- D. Kerstens, H. De Peuter, I. Khalil, S. Van Praet, J. Van Aelst and B. F. Sels, ACS Sustainable Chem. Eng., 2021, 9, 4357 CrossRef CAS.
- J. F. Sierra-Cantor, O. Gimello, C.-A. Guerrero-Fajardo, F. Di Renzo, H. Petitjean, M. Riviere, C. Gérardin and N. Tanchoux, Appl. Catal., B, 2024, 344, 123602 CrossRef CAS.
- M. Kondratiuk, D. Gopinath, A. Elrrays and L. J. Gooßen, Eur. J. Lipid Sci. Technol., 2023, 125, 2200163 CrossRef CAS.
- M. I. Sarker, H. Ngo, B. K. Sharma, K. M. Wagner, K. C. Jones and M. J. Powell, Tribol. Int., 2023, 186, 108649 CrossRef CAS.
- D. Le, N. Chaidherasuwet, A. Rueangthaweep, C. Kulsing and N. Hinchiranan, Catal. Today, 2023, 407, 260 CrossRef CAS.
- B. Saini, M. Yadav, S. K. Jha, R. Krishnapriya, P. Kang, V. Kant, R. Singhal and R. K. Sharma, Sustainable Energy Fuels, 2023, 7, 2568 RSC.
- X. Cao, J. Zhao, S. Jia, F. Long, Y. Chen, X. Zhang, J. Xu and J. Jiang, Chem. Eng. J., 2024, 481, 148345 CrossRef CAS.
- K. Zhang, X. Liu, J. Bi, A. BaQais, B. B. Xu, M. A. Amin, Y. Hou, X. Liu, H. Li, H. Algadi, J. Xu and Z. Guo, New J. Chem., 2023, 47, 18272 RSC.
- A. S. Morgan, Md Z. Hossain, M. B. I. Chowdhury and P. Charpentier, J. Supercrit. Fluids, 2024, 205, 106120 CrossRef CAS.
- H.-J. Cha, S.-Y. Hwang, D.-S. Lee, A. R. Kumar, Y.-U. Kwon, M. Voß, E. Schuiten, U. T. Bornscheuer, F. Hollmann, D.-K. Oh and J.-B. Park, Angew. Chem., 2020, 132, 7090 CrossRef.
- W. Wang, M.-Y. Liang, J.-Q. Lang, H. I. Mtui, S.-Z. Yang and B.-Z. Mu, Biomass Convers. Biorefin., 2023 DOI:10.1007/s13399-023-04803-8.
- W. Wang, M.-Y. Liang, J.-Q. Lang, H. I. Mtui, S.-Z. Yang and B.-Z. Mu, Green Mater., 2023, 12, 50 Search PubMed.
- P. Gupta, S. Akhtar, Nisha, R. S. Negi, S. K. Porwal and R. K. Singh, J. Am. Oil Chem. Soc., 2024, 101, 501 CrossRef CAS.
- I. E. Nifant'ev, A. A. Vinogradov, A. A. Vinogradov, I. V. Sedov, V. G. Dorokhov, A. S. Lyadov and P. V. Ivchenko, Appl. Catal., A, 2018, 549, 40 CrossRef.
- A. Yamamoto, K. Nemoto, M. Yoshida, Y. Tominaga, Y. Imai, S. Ata, Y. Takenaka, H. Abe and K. Sato, RSC Adv., 2020, 10, 36562 RSC.
- C. Cheng, J. X. Shi, E.-H. Kang, T. F. Nelson, M. Sander, K. McNeill and J. F. Hartwig, J. Am. Chem. Soc., 2024, 146, 12645 CrossRef CAS PubMed.
- R. Ahmadi and A. Ullah, ACS Sustainable Chem. Eng., 2020, 8, 8049 CrossRef CAS.
- M. Häußler, M. Eck, D. Rothauer and S. Mecking, Nature, 2021, 590, 423 CrossRef PubMed.
- S. F. Marxsen, M. Häuβler, S. Mecking and R. G. Alamo, Polymer, 2020, 191, 122282 CrossRef CAS.
- S. F. Marxsen, M. Häuβler, M. Eck, S. Mecking and R. G. Alamo, Polymer, 2023, 282, 126181 CrossRef CAS.
- S. F. Marxsen, M. Häuβler, S. Mecking and R. G. Alamo, ACS Appl. Polym. Mater., 2021, 3, 5243 CrossRef CAS.
- M. Eck and S. Mecking, Acc. Chem. Res., 2024, 57, 971 CrossRef CAS PubMed.
- S. T. Schwab, M. Baur, T. F. Nelson and S. Mecking, Chem. Rev., 2024, 124, 2327 CrossRef CAS PubMed.
- B. R. Moser, K. M. Doll and N. P. J. Price, J. Am. Oil Chem. Soc., 2023, 100, 149 CrossRef CAS.
- M. Naddeo, I. D'Auria, G. Viscusi, G. Gorrasi, C. Pellecchia and D. Pappalardo, J. Polym. Sci., 2020, 58, 528 CrossRef CAS.
- M. Naddeo, G. Vigliotta, C. Pellecchia and D. Pappalardo, React. Funct. Polym., 2020, 155, 104714 CrossRef CAS.
- C. Guindani, W. A. G. Jaramillo, G. Candiotto, E. A. Rebelatto, F. W. Tavares, J. C. Pinto, P. M. Ndiaye and M. Nele, J. Supercrit. Fluids, 2022, 186, 105588 CrossRef CAS.
- J. V. Rowley, P. Wall, H. Yu, G. Tronci, D. A. Devine, J. J. Vernon and P. D. Thornton, ACS Appl. Polym. Mater., 2020, 2, 2927 CrossRef CAS.
- A. E. Polloni, V. Chiaradia, R. J. F. C. do Amaral, C. Kearney, B. Gorey, D. de Oliveira, J. V. de Oliveira, P. H. H. de Araújo, C. Sayer and A. Heise, Polym. Chem., 2020, 11, 2157 RSC.
- F. C. Sales de Oliveira, R. J. F. Correa do Amaral, L. E. Cardoso dos Santos, C. Cummins, M. M. Morris, C. J. Kearney and A. Heise, J. Biomed. Mater Res., Part A, 2022, 110, 257 CrossRef PubMed.
- H. R. Amaral, J. A. Wilson, R. J. F. C. do Amaral, I. Pasçu, F. C. S. de Oliveira, C. J. Kearney, J. C. C. Freitas and A. Heise, Carbohydr. Polym., 2021, 252, 117201 CrossRef CAS PubMed.
- P. Liu, W. Li and X. Liu, BMC Chem., 2022, 16, 46 CrossRef CAS PubMed.
- A. Martínez, D. Zárate-Saldaña, J. Vargas and A. A. Santiago, Int. J. Mol. Sci., 2022, 23, 4521 CrossRef PubMed.
- L. O'Hari, P. Go, M. M. Abdellatif, R. Makino, D. Shimoyama, S. Higashi, H. Hirano and K. Nomura, Polymers, 2024, 16, 468 CrossRef PubMed.
- I. E. Nifant'ev, V. V. Bagrov, P. D. Komarov, S. O. Ilyin and P. V. Ivchenko, Polymers, 2022, 14, 1720 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |