Kinetics of formation of the novel peroxide FC(O)OO(O2)SF
María E.
Tucceri
,
María P.
Badenes
,
Adela E.
Croce
and
Carlos J.
Cobos
*
Instituto de Investigaciones Fisicoquímicas Teóricas
y Aplicadas (INIFTA), Departamento de Química, Facultad de Ciencias Exactas, Universidad Nacional de La Plata, CONICET, CICBA, Casilla de Correo 16, Sucursal 4, (1900) La Plata, Argentina.. E-mail: cobos@inifta.unlp.edu.ar
First published on 14th December 2000
Abstract
The high-pressure rate coefficient for the formation of the new peroxide FC(O)OO(O2)SF from recombination of FC(O)O and FS(O2)O radicals has been determined by laser flash photolysis at 296 K; density functional theory calculations indicate peroxide stabilization and allow estimation of an O–O bond dissociation energy of 20.6 ± 3 kcal mol−1.
Early syntheses of fluorinated peroxides involved the coupling of oxy-radicals to form the peroxide bond.1 In fact, the co-photolysis of the mixtures of peroxides SF5OOSF5/CF3OOCF3 and SF5OOSF5/FS(O2)OO(O2)SF have been employed by Cady and coworkers to prepare SF5OOCF3 and SF5OO(O2)SF.1 The fluorinated peroxides allow study of the role that the electron-withdrawing effect of electronegative groups plays on their reactivity, energetics and conformation. Here we report the determination of the rate coefficient for the recombination of FC(O)O and FS(O2)O radicals to form the new peroxide FC(O)OO(O2)SF at 296 K. In addition, the rate coefficient for the reaction of FS(O2)O with CO has been determined for the first time.
A laser flash photolysis–absorption spectroscopy configuration described in detail elsewhere,2–7 was employed in the present experiments. Typically, samples of 30–40 mbar of FS(O2)OF in the presence of 130–300 mbar of CO, 15 mbar of O2 and up to 900 mbar of SF6 were irradiated with the emission of an excimer laser operating on the 193 nm ArF transition. No more than 30 single shot experiments from fresh samples were averaged and analysed up to 5 ms for each set of conditions. In the photolysis, electronically excited FS(O2)O radicals in the B 2E state are initially formed and afterwards collisionally deactivated to the X 2A2 ground state via a manifold of low-lying vibrationally excited states.4,5 After ca. 250 μs the excited radicals are thermalized.5 On the other hand, photolytically generated F atoms are rapidly consumed by recombination with CO to form FCO radicals, which subsequently recombine with O2 and yield FC(O)O2 radicals. Finally, these radicals form FC(O)O radicals by self-reaction. The whole reaction mechanism is detailed in refs. 6–9. FCO and FC(O)O2 are almost quantitatively consumed at 200 μs. Thus, the well established mechanisms involved in the FS(O2)O2–5 and FC(O)Ox (x = 0, 1, 2)6–9 radical chemistries lead to the conclusion that over 300 μs only FS(O2)O and FC(O)O survive and the mechanism reduces to eqns. (1)–(5)
| FC(O)O + FC(O)O → FC(O)OO(O)CF | (1) |
| FS(O2)O + FS(O2)O → FS(O2)OO(O2)SF | (2) |
| FC(O)O + FS(O2)O → FC(O)OO(O2)SF | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) FS(O2)O + CO →
FSO2 + CO2 FS(O2)O + CO →
FSO2 + CO2 | (4) |
| FS(O2)O +
FSO2
→
FS(O2)O(O2)SF | (5) |
Under the present conditions, reactions (1) and (2) are pressure independent with high-pressure rate coefficients of k∞,1 = 5.5 × 10−138 and k∞,2 = 4.6 × 10−14 cm3 molecule−1 s−1, respectively.3 Second-order plots of the absorbance monitored at 450 nm after photolysis of FS(O2)OO(O2)SF/CF4 and FS(O)2OF/CO/O2/SF6 mixtures are depicted in Fig. 1. In the first case, the generated FS(O)2O radicals [absorption cross-sections σ(FS(O2)O) = 3.64 × 10−18 cm2 molecule−13] react exclusively according to reaction (2). In the latter case, a fast component due to the above mentioned thermalization of excited FS(O2)O radicals is followed by a second component with a slope much higher than the observed for the first mixture. Absorbance vs. time profiles were numerically fitted employing the mechanism described by reactions (1)–(5). The modelling leads to radical concentrations such that the absorbance may be mostly attributed to FS(O)2O absorption with a small contribution due to FC(O)O radicals [σ(FC(O)O) = 6.7 × 10−19 cm2 molecule−18]. Moreover, the calculations show that the higher slope observed in signal (B) of Fig. 1 is predominantly due to FS(O2)O consumption by reaction (3). At longer times and higher CO pressures these radicals are also consumed in reaction (4). However, the low FSO2 concentration precludes the determination of k5, for which reasonable values ranging from 2 × 10−12 to 7 × 10−11 cm3 molecule−1 s−1 do not affect the modelling results. Between ca. 175 and 1060 mbar the rate coefficients determined for reaction (3) remain independent on total pressure such that they can be certainly ascribed to the limiting high pressure value. All experimental results are very well reproduced using the k∞,1, k∞,2 and k5 values given above as well as k∞,3 = (1.2 ± 0.3) × 10−12 cm3 molecule−1 s−1 and k4 = (1.8 ± 0.7) × 10−17 cm3 molecule−1 s−1 for reactions (3) and (4). The errors quoted are 2σ. The value of k∞,3 is normal for this type of reaction10 while the low value of k4 is quite consistent with the measured activation energy of 7 kcal mol−1.11 The experimental study is supplemented by density functional theory thermochemical computations to determine the bond dissociation energies of O–O, C–O and O–S bonds in FC(O)OO(O2)SF. For this, standard enthalpies of formation of the peroxide and the relevant radicals were calculated. The value for FC(O)OO(O2)SF was estimated using the isodesmic reaction: FOOF + FC(O)OH + HSO3F → FC(O)OO(O2)SF + 2FOH. Energy calculations were carried out on optimized geometries and harmonic frequencies evaluated employing the hybrid B3LYP density functional with the 6-311++G(d,p) basis set.12 Unless otherwise indicated, the experimental enthalpies of formation of the species involved in this and other isodesmic reactions given here are from ref. 10. According to the experimental uncertainties, an error level for all derived thermochemical properties of this work of ±3 kcal mol−1 is estimated. From the calculated enthalpy of the isodesmic reaction of ΔH0r = 44.7 kcal mol−1, the value ΔH0f,298(FC(O)OO(O2)SF) = −229.4 kcal mol−1 was derived. The enthalpy of formation for FS(O2)O was estimated from the experimental bond dissociation energy, De(FS(O2)O–F) = 33.1 kcal mol−1,2 and ΔH0f,298(FS(O2)OF) = −136.9 kcal mol−1 obtained from the isodesmic reaction FO2 + HSO3F → FS(O2)OF + HO2 (ΔH0r = 40.0 kcal mol−1). In this way, ΔH0f,298(FS(O2)O) = −122.8 kcal mol−1 results. Using the above enthalpies of formation for FC(O)OO(O2)SF and FS(O2)O together with ΔH0f,298(FC(O)O) = − 86.0 kcal mol−1,13 we obtain the enthalpy change ΔH0298(FC(O)O–O(O2)S F) = 20.6 kcal mol−1 which is similar to the value measured for the FS(O2)O–O(O2)SF bond of 22.1 kcal mol−1.3
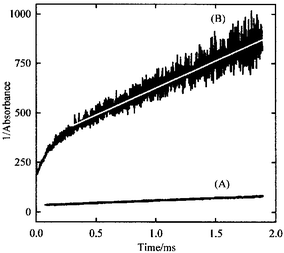 | ||
| Fig. 1 Plot of the inverse of the absorbance vs. time. (A) 8.1 mbar of FS(O2)OO(O2)SF and 16.4 mbar of CF4; (B) 29.3 mbar of FS(O2)OF, 132.4 mbar of CO, 14.3 mbar of O2 and 890.0 mbar of SF6. The solid line is the result of the modelling described in the text. | ||
Finally, using ΔHf,298(FS(O2)OO) = −110.2 kcal mol−1, estimated using the isodesmic reaction FO2 + HSO3F → FS(O2)OO + FOH (ΔH0r = 40.4 kcal mol−1), and ΔH0f,298(FCO) = 44.6 kcal mol−1,7 ΔH0f,298(FC(O2)OO) = −76.1 kcal mol−1,6 and ΔH0f,298 (FSO2) = −96.2 kcal mol−1,14 dissociation energies for other bonds in FC(O)OO(O2)SF were evaluated. The resulting values are: ΔH0298(FC(O)OO–(O2)S F) = 57.1 and ΔH0298(FC(O)–OO(O2)S F) = 74.6 kcal mol−1. These results indicate that no energetically feasible exit channels for the peroxide decomposition exist. The minimum-energy pathways for the recombination reaction (3) and for FC(O)–OO(O2)SF and FC(O)OO–(O2)SF dissociations show a smooth energy profile without a maximum: no transition state was found on the B3LYP/6-311 + G(d) surface. Thus the enthalpy changes can be assimilated to the respective bond dissociation energies. Scarcely probable is the competition between the reaction FC(O)O + FS(O2)O → CO2 + FS(O2)OF and reaction (3). This assumption is supported by the fact that most fluorine abstraction reactions by either FS(O2)O or other radicals exhibit relatively large activation energy values (ca. 10–30 kcal mol−1) and consequently very small room temperature rate coefficients. In particular, for the similar reaction FNO2 + FS(O2)O → NO2 + FS(O2)OF an activation energy of ca. 30 kcal mol−1 can be estimated from the measured value of 10 kcal mol−1 for the reverse reaction,15 the enthalpies of formation of FNO2 and NO2 molecules10 and the above values for FS(O2)O and FS(O2)OF. The present results indicate that after formation by reaction (3), the new peroxide is mostly collisionally stabilized at room temperature. However, the low O–O bond dissociation energy leads to significant decomposition as temperatures rises, such that a gaseous sample of FC(O)OO(O2)SF finally degrades to the more stable peroxide FC(O)OO(O)CF.6–9
Further experimental and theoretical work on FC(O)OO(O2)SF is underway.
Acknowledgements
This work was supported by the Universidad Nacional de La Plata, the CONICET and the CICBA.Notes and references
- C. I. Merrill, S. M. Williamson, G. H. Cady and D. F. Eggers, Inorg. Chem., 1962, 1, 215 CrossRef CAS.
- A. E. Croce de Cobos, C. J. Cobos and E. Castellano, J. Phys. Chem., 1989, 93, 274 CrossRef CAS.
- C. J. Cobos, A. E. Croce de Cobos, H. Hippler and E. Castellano, J. Phys. Chem., 1989, 93, 3089 CrossRef CAS.
- C. J. Cobos, A. E. Croce and E. Castellano, J. Photochem. Photobiol. A: Chem., 1991, 59, 143 Search PubMed.
- A. E. Croce, C. J. Cobos and E. Castellano, J. Photochem. Photobiol. A: Chem., 1990, 55, 135 Search PubMed.
- M. P. Badenes, E. Castellano, C. J. Cobos, A. E. Croce and M. E. Tucceri, Chem. Phys. Lett., 1999, 303, 482 CrossRef CAS.
- M. P. Badenes, E. Castellano, C. J. Cobos, A. E. Croce and M. E. Tucceri, Chem. Phys., 2000, 253, 205 CrossRef CAS.
- M. M. Maricq, J. J. Szente, T. S. Dibble and J. S. Francisco, J. Phys. Chem., 1994, 98, 12294 CrossRef CAS.
- T. J. Wallington, T. Ellermann, O. J. Nielsen and J. Sehested, J. Phys. Chem., 1994, 98, 2346 CrossRef CAS.
- W. B. DeMore, S. P. Sander, D. M. Golden, R. F. Hampson, M. J. Kurylo, C. J. Howard, A. R. Ravishankara, C. E. Kolb and M. J. Molina, Chemical Kinetics and Photochemical Data for Use in Stratospheric Modeling. Evaluation Number 12, JPL Publication97-4, California Institute of Technology, Pasedena, CA, 1997. http://jpldataeval.jpl.nasa.gov/ Search PubMed.
- E. Vasini and H. J. Schumacher, Z. Phys. Chem. Neue Folge, 1975, 94, 39 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98, Revision A.7, Gaussian, Inc., Pittsburgh PA, 1998. Search PubMed.
- T. S. Dibble and J. S. Francisco, J. Phys. Chem., 1994, 98, 11694 CrossRef CAS.
- M. P. Badenes, M. E. Tucceri and C. J. Cobos, Z. Phys. Chem., 2000, 214, 1193 Search PubMed.
- R. Gatti and H. J. Schumacher, Z. Phys. Chem. Neue Folge, 159, 62, 1968 Search PubMed.
| This journal is © The Royal Society of Chemistry 2001 |