Solvothermal synthesis of [Cr10(μ-O2CMe)10(μ-OR)20] ‘chromic wheels’ with antiferromagnetic (R = Et) and ferromagnetic (R = Me) Cr(III)···Cr(III) interactions
Eric J. L.
McInnes†
*a,
Christopher
Anson‡
a,
Annie K.
Powell‡
a,
Andrew J.
Thomson
a,
Sandrine
Poussereau
b and
Roberta
Sessoli
b
aSchool of Chemical Sciences, University of East Anglia, Norwich, UK NR4 7TJ
bUniversita degli Studi di Firenze, Dipartimento di Chimica, Via Maragliano 75/77, 50144, Firenze, Italy
First published on 19th December 2000
Abstract
Two decanuclear cyclic Cr(III) complexes have been synthesised in high yield by solvothermal techniques: magnetic susceptibility studies reveal ferromagnetic Cr⋯Cr exchange in one, and antiferromagnetic Cr⋯Cr exchange in the other.
The synthesis and magnetic characterisation of molecules with large numbers of unpaired electrons have attracted intense study since the discovery that molecular aggregates stabilising high ground spin states can display the phenomenon of single-molecule magnetism.1 Cyclic complexes are of particular interest as models of infinite one-dimensional chain compounds and in the study of quantum-size effects,2,3 and are known for several first row d-transition ions.4 Among these, Fe(III) rings have received most attention5 and can generally be made under ambient conditions. By contrast, few cyclic chromium complexes have been reported,6 and require much harsher synthetic conditions owing to the kinetic inertness of Cr(III).7
Solvothermal techniques allow high reaction temperatures in low boiling solvents, whilst maintaining the advantages of solution chemistry (e.g. crystallisation of products). However, there are few reports of solvothermal syntheses of molecular clusters.8 We report here the solvothermal synthesis of [Cr10(μ-O2CMe)10(μ-OR)20] (R = Me 1, Et 2) in high yield. These are the highest nuclearity cyclic clusters yet reported for Cr(III), and 1 displays the rarely observed phenomenon of ferromagnetic Cr(III)···Cr(III) exchange.
Heating trinuclear basic chromium acetate, [Cr3(μ3-O)(μ-O2CMe)6(H 2O)3]Cl·6H2O (100 mg), in MeOH or EtOH (10 ml) in a Teflon-lined autoclave at 200 °C for 1 d followed by slow (0.1 °C min−1) cooling to room temperature over ca. 2 d yields large, dark-green crystals of 1 or 2, respectively (60–70%). Single-crystal X-ray diffraction analyses9 reveal cyclic decanuclear structures (Fig. 1), similar to the ‘molecular ferric wheels’ reported by the groups of Lippard5a and Winpenny.5b The Cr10 rings are close to planar, with each pair of neighbouring Cr(III) ions bridged by one μ-acetate and two μ-alkoxide groups. In both compounds the rings are closely aligned perpendicular to the crystal {1 0 −1} plane. In 1, the Cr10 rings in any given layer adopt a staggered configuration relative to those in adjacent layers (Fig. 2), while in 2 they are more closely ‘eclipsed’.
![Molecular structure (50% thermal ellipsoids) of
[Cr10(O2CMe)10(OMe)20]
1. Bond length ranges: Cr–O(Me) 1.935(7)–1.985(8)
Å, Cr–O(acetate) 1.969(8)–2.005(7) Å.
Cr···Cr distances: 2.985(3)–2.994(3) Å.
Bond angle ranges: Cr–O(Me)–Cr 98.0(3)–100.3(4)°. The
analogous ranges for 2 are: Cr–O(Et) 1.951(6)–1.980(6)
Å, Cr–O(acetate) 1.981(7)–1.998(7) Å,
Cr···Cr 2.992(2)–2.997(2) Å,
Cr–O(Et)-Cr 98.5(3)–100.1(3)°.](/image/article/2001/CC/b007773i/b007773i-f1.gif) | ||
| Fig. 1 Molecular structure (50% thermal ellipsoids) of [Cr10(O2CMe)10(OMe)20] 1. Bond length ranges: Cr–O(Me) 1.935(7)–1.985(8) Å, Cr–O(acetate) 1.969(8)–2.005(7) Å. Cr···Cr distances: 2.985(3)–2.994(3) Å. Bond angle ranges: Cr–O(Me)–Cr 98.0(3)–100.3(4)°. The analogous ranges for 2 are: Cr–O(Et) 1.951(6)–1.980(6) Å, Cr–O(acetate) 1.981(7)–1.998(7) Å, Cr···Cr 2.992(2)–2.997(2) Å, Cr–O(Et)-Cr 98.5(3)–100.1(3)°. | ||
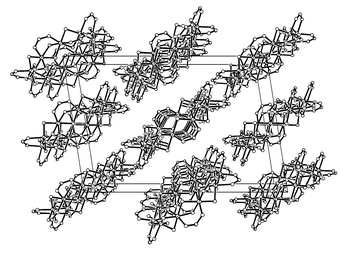 | ||
| Fig. 2 Packing diagram of 1 viewed down the crystallographic b axis. All Me groups have been omitted for clarity. | ||
The magnetic behaviour of 2 is relatively straightforward: χTvs. T decreases slowly from 300 to 120 K and then more quickly to 2 K (Fig. 3). This is indicative of weak, intra-ring antiferromagnetic exchange between Cr(III) ions. The best simulation10 is obtained with J = +0.9 cm−1 (Fig. 3). Introduction of inter-cluster or zero-field splitting (ZFS) effects do not improve the simulation. Corroboration of this number comes from fitting 1/χvs. T (linear to low temperature, data not shown) to the Curie–Weiss law, giving a Curie constant of C = 18.8 cm3 mol−1 K and a small Weiss constant of Θ = −3.4 K. The Cr···Cr exchange can then be calculated from Θ,6b giving J = +0.94 cm−1.
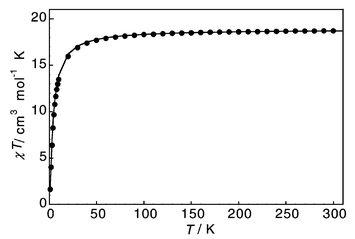 | ||
| Fig. 3 χ T vs. T for 2 (●) and best fit with J = +0.9 cm−1 (––––). | ||
The magnetic behaviour of 1 is more complicated. χTvs. Tincreases from 18.3 to 20.0 cm3 mol−1 K between 300 and 22 K, indicating the presence of ferromagnetic interactions, before decreasing down to 2 K (Fig. 4). The high-temperature data (above 50 K) can be fitted to the Curie–Weiss law with C = 18.2 cm3 mol−1 K and Θ = +4.1 K. The decrease of χT below 20 K is indicative of an antiferromagnetic contribution to χT. The χTvs. T curve can be modelled with a ferromagnetic intra-molecular exchange between neighbouring Cr(III) ions of J = −4.5 cm−1 and a weak antiferromagnetic inter-molecular exchange of Jinter = +0.26 cm−1 (see Fig. 4). It is worth noting that it is the ferromagnetic nature of the intra-ring coupling in 2 that allows detection of an antiferromagnetic inter-ring interaction at temperatures below 20 K: a similar inter-ring coupling in 1 would be barely detectable. The ferromagnetic interaction is predominant as evidenced by the initial increase in χT down to 20 K. The decrease of χT below 20 K could not be modelled by inclusion of ZFS effects, or by having ferromagnetic exchange in one of the two independent molecules of 1 and antiferromagnetic exchange in the other.
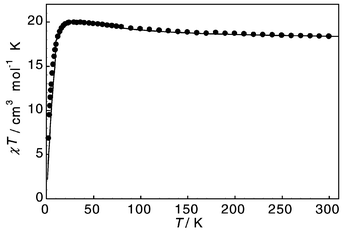 | ||
| Fig. 4 χ T vs. T for 1 (●) and best fit with J = −4.5 cm−1 and Jinter = +0.26 cm−1 (––––). | ||
The magnetic behaviour of 2 (antiferromagnetic intra-ring exchange, resulting in a non-magnetic S = 0 ground state) is common for cyclic complexes. However, J = +0.9 cm−1 is very weak compared to other cyclic Cr(III) complexes (typically +6 to +10 cm−1).6 The ferromagnetic interactions observed in 1 are highly unusual: there are very few previous examples of ferromagnetic Cr(III)···Cr(III) exchange in molecular systems11 and, furthermore, only two previous examples of ferromagnetically coupled cyclic complexes of any metal ion.12
The so-called GHP model13 correlates the exchange constant in ‘planar’ {Cr2(μ-OR)2} dimers with (i) Cr–O(R)–Cr bridging angle (phis), (ii) Cr–O(R) bond length (r) and (iii) dihedral angle between the bridging plane and the O–R vector (θ). Our systems are complicated by the non-planarity of the {Cr2(μ-OR)2} bridges imposed by the μ-O2CMe groups: these bridges are also likely involved in the exchange pathways. However, we note that both 1 and 2 have an average phis ≈ 99° and in the GHP model are in the ‘crossover’ region between (weak) antiferromagnetic and ferromagnetic coupling regimes. This is consistent with the observed small values of |J| ≈ 0.9–5 cm−1. Furthermore, the smaller average θ values observed in 2 than in 1 are consistent with the more antiferromagnetic exchange in the former: this effect has been observed previously in ethoxide vs. methoxide bridged Cr(III) dimers.14 Thus, we attribute the differing magnetic behaviour of 1 and 2 to a combination of the above effects.
Acknowledgements
We thank the EPSRC and Italian MURST for financial support. This work was also supported by 3MD EU network contact no. ERB 4061 PL 97-0197.Notes and references
- J. Yoo, E. K. Brechin, A. Yamaguchi, M. Nakano, J. C. Huffman, A. L. Maniero, L. C. Brunel, K. Awaga, H. Ishimoto and G. Christou and D. N. Hendrickson, Inorg. Chem., 2000, 39, 3615 CrossRef CAS.
- A. Caneschi, D. Gatteschi, C. Sangregorio, R. Sessoli, L. Sorace, A. Cornia, M. A. Novak, C. Paulsen and W. Wernsdorfer, J. Magn. Magn. Mater., 1999, 200, 182 CrossRef CAS.
- D. Gatteschi, R. Sessoli and A. Cornia, Chem. Commun., 2000, 725 RSC.
- R. E. P. Winpenny, Comments Inorg. Chem., 1999, 20 A, 233 Search PubMed.
- (a) K. L. Taft, C. D. Delfs, G. C. Papaefthymiou, S. Foner, D. Gatteschi and S. J. Lippard, J. Am. Chem. Soc., 1994, 116, 823 CrossRef CAS; (b) C. Benelli, S. Parsons, G. A. Solan and R. E. P. Winpenny, Angew. Chem., Int. Ed. Engl., 1996, 35, 1825 CrossRef CAS.
- (a) I. M. Atkinson, C. Benelli, M. Murrie, S. Parsons and R. E. P. Winpenny, Chem. Commun., 1999, 285 RSC; (b) M. Eshel, A. Bino, I. Felner, D. C. Johnston, M. Luban and L. L. Miller, Inorg. Chem., 2000, 39, 1376 CrossRef CAS; (c) N. V. Gerbeleu, Y. T. Struchkov, G. A. Timco, A. S. Batsanov, K. M. Indrichan and G. A. Popovich, Dokl. Akad. Nauk. SSSR, 1990, 313, 1459 Search PubMed.
- F. E. Mabbs, E. J. L. McInnes, M. Murrie, S. Parsons and R. E. P. Winpenny, Chem. Commun., 1999, 643 RSC; S. Parsons, A. A. Smith and R. E. P. Winpenny, Chem. Commun., 2000, 579 RSC.
- S. O. H. Gutschke, D. J. Price, A. K. Powell and P. T. Wood, Angew. Chem., Int. Ed. Engl., 1999, 38, 1088 CrossRef CAS.
- Crystal data: for 1: C40H90O40Cr10, M = 1731.12, monoclinic, space group P21/n, a = 17.663(5), b = 15.968(4), c = 26.142(4) Å, β = 98.89(3)°, U = 7285(3) Å3, T = 293 K, Z = 4 (two half molecules in the asymmetric unit), μ(Mo-Kα) = 1.52 mm−1, 12837 reflections collected, 10960 unique (Rint = 0.0712), final wR2 (all data) = 0.1918, R1 [I > 2σ(I)] = 0.0887. For 2: C60H130O40Cr10, M = 2011.64, monoclinic, space group P21/n, a = 9.446(2), b = 16.052(3), c = 28.659(4) Å, β = 91.037(14)°, U = 4344.8(14) Å3, T = 293 K, Z = 2, μ(Mo-Kα) = 1.28 mm−1, 9844 reflections collected, 7645 unique (Rint = 0.0552), wR2 (all data) = 0.2634, R1 [I > 2σ(I)] = 0.0820. Data collected on a Siemens P4 diffractometer to 2θ = 50° and corrected for Lorentz, polarisation and absorption effects. Structures were solved by direct methods and refined by full-matrix least squares on F2 (all data) (SHELXTL). Eight of the 20 (O)Me groups in 1 showed two-fold disorder, while many of the (O)Et groups in 2 were highly disordered. Non-hydrogen atoms were refined anisotropically, except for carbon atoms of minor disorder components; hydrogen atoms were placed in calculated positions. CCDC 182/1858. See http://www.rsc.org/suppdata/cc/b0/b007773i/ for crystallographic files in .cif format..
- Magnetic susceptibilities for 1 and 2 were measured on a SQUID magnetometer in the temperature ranges 2–60 and 2–300 K in applied fields of 0.1 and 1 T, respectively, and diamagnetic corrections applied. Corrected χT values for 1 and 2 at 300 K were 18.3 and 18.7 cm3 mol−1 K, respectively. The expected value for 10 Cr(III), S = 3/2 ions is 18.4 cm3 mol−1 K. In order to calculate χT for 10 interacting S = 3/2 ions we must diagonalise the Heisenberg spin-Hamiltonian matrices (H = Σ JijSiSj) for the 116304 spin states of the cluster: the largest matrix (19425 × 19425) is beyond our present computational capabilities. Therefore, we have extrapolated the results from calculations on 6 and 8 interacting S = 3/2 ions. An average g-value of 1.98 was assumed for all simulations. Here, a positiveJ value implies an antiferromagnetic interaction..
- For example, R. P. Scaringe, W. E. Hatfield and D. J. Hodgson, Inorg. Chem., 1977, 16, 1600 CrossRef CAS; A. Bino, D. C. Johnston, D. P. Goshorn, T. R. Halbert and E. I. Stiefel, Science, 1988, 241, 1479 CrossRef CAS.
- A. J. Blake, C. M. Grant, S. Parsons, J.M. Rawson and R. E. P. Winpenny, J. Chem. Soc., Chem. Commun., 1994, 2363 RSC; E. Rentschler, D. Gatteschi, A. Cornia, A. C. Fabretti, A-L. Barra, O. I. Schegolikhina and A. A. Zhdanov, Inorg. Chem., 1996, 35, 4427 CrossRef CAS.
- J. Glerup, D. J. Hodgson and E. Pederson, Acta. Chem. Scand. Ser. A, 1983, 37, 161 Search PubMed.
- H. R. Fischer, D. J. Hodgson and E. Pedersen, Inorg. Chem., 1984, 23, 4755 CrossRef CAS.
Footnotes |
| † Present address: Chemistry Department, The University of Manchester, Oxford Road, Manchester, UK M13 9PL. E-mail: eric.mcinnes@man.ac.uk |
| ‡ Present address: Institüt fur Anorganische Chemie, Universitat Karlsruhe, Engesserstrasse Geb 30.45, 76128 Karlsruhe, Germany. |
| This journal is © The Royal Society of Chemistry 2001 |