A new solid acid catalyst: the first phosphonate and phosphonic acid functionalised microporous polysilsesquioxanes†
Magdalena
Jurado-Gonzalez
,
Duan
Li Ou
,
Bradley
Ormsby
,
Alice C.
Sullivan
and
John R. H.
Wilson
Department of Chemistry, Queen Mary and Westfield College, Mile End Road, London, UK E1 4NS. E-mail: a.c.sullivan@qmw.ac.uk
First published on 14th December 2000
Abstract
A novel microporous solid acid, the phosphonic acid modified polysilsesquioxane [(HO)SiCH2CHPO(OH)2- CH2CH2SiO2(OH)]n P2, from the new single precursor compound [(EtO)3SiCH2CHPO(OCH2CH3) 2CH2CH2- Si(OEt)3] 1, is reported, along with the catalytic activity of P2 for the pinacol–pinacolone rearrangement.
The development of functionalised porous silicas1a,b and polysilsesquioxane materials1c,d for various applications is proceeding at great pace as evidenced by recent reviews and reports on synthesis and applications in this area. Functional groups or their precursors may be attached to silica via compounds (RO)3Si(CH2)nX where X may be further modified if required. Such materials offer a wide spectrum of potential applications for example as acid, base, or oxidation catalysts or asymmetric carbon–carbon coupling catalysts, or as biocompatible surface groups.1a,b,2 In the context of the present report efforts are also being directed towards the synthesis of functionalised porous polysilsesquioxanes, where the functional group is an integral part of a single framework precursor. Examples include polysilsesquioxanes with built-in NLO and luminescence chromophores,3a,b built-in crown-ether4 or cyclam5 groups for ion sensing applications or ferrocenyl groups for electrochemical applications.6
It occurred to us that phosphonic acid functionalised silicas and polysilsesquioxanes might offer a new class of solid acid materials with wide-ranging potential to act as catalysts, ion exchange and separation materials. There were no examples of such materials previously reported in the literature.
We report here on unique examples of the uniformly modified phosphonate and phosphonic acid polysilsesquioxanes [(HO)SiCH2CHPO(OCH2CH3)2CH 2CH2SiO2(OH)]nP1 and [(HO)SiCH2CHPO(OH)2CH2CH2SiO 2(OH)] P2 prepared from the new precursor compound [(EtO)3SiCH2CHPO(OCH2CH3) 2CH2CH2Si(OEt)3], 1, 1,4-bis(triethoxysilyl)-2-(diethylphosphonato)butane. Compound 1 was formed‡ as shown in eqn. (1) by radical addition of HPO(OEt)2 to the ene fragment in 1,4-bis(triethoxysilyl)but-2-ene7 and was fully characterised by solution phase NMR, mass spectrometry and elemental analysis.
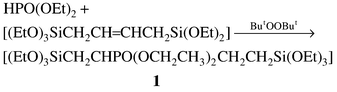 | (1) |
P1 was obtained as a transparent monolith after acid-catalysed sol–gel processing of 1 in THF.§ The solid state 13C CP MAS NMR spectrum of P1 had peaks close to those observed for compound 1; the 29Si spectrum showed one broad resonance at −63 ppm consistent with predominantly T2 type silicon sites while the 31P NMR showed a single peak at 35 ppm similar to that of 1. Treatment of P1 with concentrated hydrochloric acid gave the corresponding phosphonic acid modified material P2.¶ The absence of any Qn type resonances in the 29Si CP MAS NMR of P2 indicates that the transformation from ester to acid occurred without any Si–C cleavage. The decrease in C∶P ratio and changes in the NMR spectra from P1 to P2 (in particular the disappearance of the signals due to the phosphonate ester group −PO(OCH2CH3)2 at 63 ppm, see Fig. 1), is consistent with the formation of the phosphonic acid from the ester. Average formulae for these materials derived from C∶P ratios and based on predominantly T2 environments (66% condensation) are [(HO)SiCH2CHPO(OEt)2CH2CH2- SiO2(OH)]nP1 and [(HO)SiCH2CHPO(OH)2CH2CH2- SiO2(OH)]nP2.
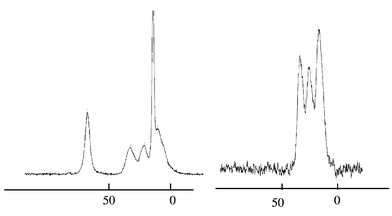 | ||
| Fig. 1 | ||
The material P2 was found to be microporous by nitrogen sorption porosimetry with very narrow pore size distribution.
It is our intention initially to utilise the phosphonic acid functionality in these and related materials we have prepared for various catalytic transformations. Preliminary experiments based on catalytic pinacol–pinacolone rearrangement and dehydration of cyclohexanol were used to test the reactivity of the phosphonic acid functionality in P2. A range of solid acid catalysts have been reported to catalyse pinacol–pinacolone rearrangements; examples include metal substituted aluminophosphates,8 lanthanide substituted zeolites,9 heteropolytungstates,10 various SAPOs (silicoaluminophosphates)11 and polyphosphoric acid.12
A reaction between P2 (0.2 g) and pinacol (6.0 g) at 140 °C (no solvent) resulted in 80% conversion to pinacolone after 12 h. Control experiments|| using mesoporous or microporous silicas under the same conditions afforded no rearranged product. Attempted dehydration reactions using P2 (0.25 g) and cyclohexanol (25 cm3) (C6H11OH∶PO(OH)2 = 60∶1) at 120 °C resulted in about 15% conversion to cyclohexene (1H NMR of the cooled reaction mixture) after 12 h. In control experiments using mesoporous or microporous silica under the same conditions no cyclohexene was produced. Industrial processes for production of cyclohexene require high temperature and pressure whereby cyclohexanol is fed to activated catalysts such as silica, alumina or zinc aluminate at 380–450 °C (see ref. 13 for example). Neat phosphoric acid used in large excess is also active between 160–170 °C. 14
A detailed systematic study of both reaction types will be carried out to optimise the catalyst performance.
Our material P2 and related materials we have prepared15 are also subjects for other studies including rearrangements, condensations, ion exchange and binding of biological molecules.
Acknowledgements
We thank EPSRC for a quota studentship to Bradley Ormsby, and Grant GR/M78281; Greg Coumbarides (QMW), Peter Haycock and Harold Toms of the ULIRS High Field NMR Service at QMW, Abil Aliev ULIRS solid state NMR service at University College London, for NMR. We also thank the University of London Central Research Fund and the Faculty Research Support Fund of Queen Mary and Westfield College for support.Notes and references
- (a) P. M. Price, J. H. Clark and D. J. Macquarrie, J. Chem. Soc., Dalton Trans., 2000, 101 RSC; (b) J. H. Clark and D. Macquarrie, Chem. Commun., 1998, 853 RSC; (c) D. A. Loy and K. J. Shea, Chem. Rev., 1995, 95, 1431 CrossRef CAS; (d) R. J. P. Corriu and D. Leclercq, Angew. Chem., Int. Ed. Engl., 1996, 35, 1420 CrossRef.
- I. Rodriguez, S. Iborra, A. Corma, F. Rey and J. L. Jorda, J. Chem. Soc., Chem. Commun., 1999, 593 RSC; J. A. Elings, R. Ait-Meddour, J. H. Clark and D. J. Macquarrie, J. Chem. Soc., Chem. Commun., 1998, 2707 RSC; R. Ciriminna, J. Blum, D. Anvir and M. Pagliaro, J. Chem. Soc., Chem. Commun., 2000, 1441 RSC; Y. Wang, T. Ju Su, R. Green, Y. Tangt, D. Styrkas, T. N. Danks, R. Bolton and J. R. Lu, J. Chem. Soc., Chem. Commun., 2000, 587 RSC; S. J. Bae, S. -Wook Kim, T. Hyeon and B. Moon Kim, J. Chem. Soc., Chem. Commun., 2000, 31 RSC.
- H. W. Oviatt, K. J. Shea, S. Kalluri, Y. Shi, W. H. Steier and L. R. Dalton, Chem. Mater., 1995, 7, 493 CrossRef CAS; A.-C. Franville, D. Zambon and R. Mahiou, Chem. Mater., 2000, 12, 428 CrossRef CAS.
- S. Brandes, R. J. P. Corriu, F. Denat, G. Dubois, R. Guilard and C. Reye, J. Chem. Soc., Chem. Commun., 1999, 2283 RSC.
- C. Chuit, R. J. P. Corriu, G. Dubois and C. Reye, J. Chem. Soc., Chem. Commun., 1999, 723 RSC.
- P. Audebert, P. Calas, G. Cerveau, R. J. P. Corriu and N. Costa, J. Electroanal. Chem., 1994, 372, 275 CrossRef CAS.
- R. J. P. Corriu, J. J. E. Moreau, P. Thepot and M. Wong Chi Man, Chem. Mater., 1992, 4, 1217 CrossRef CAS.
- B. Y. Hsu and S. F. Cheng, Microporous Mesoporous Mater., 1998, 21, 505 CrossRef CAS.
- C. P. Bezouhanova and F. A. Jabur, J. Mol. Catal., 1994, 87, 39 CrossRef CAS.
- Y. Toyoshi, T. Nakato and T. Okuhara, Bull. Chem. Soc. Jpn., 1998, 71, 2817 CAS; Y. Toyoshi, T. Nakato, R. Tamura, H. Takahashi, H. Tsue, K. Hirao and T. Okuhara, Chem. Lett., 1998, 135 CrossRef CAS.
- F. A. Jabur, V. J. Penchev and C. P. Bezoukhanova, J. Chem. Soc., Chem. Commun., 1994, 1591 RSC.
- M. A. Kakimoto, T. Seri and Y. Imai, Bull. Chem., Soc. Jpn., 1988, 61, 2643 CAS.
- T. K. Shioyama and O. K. Bartlesville, Phillips Petroleum Company, US1980000113948 April 7, 1981..
- W. M. Dehn and K. E. Jackson, J. Am. Chem., Soc., 1933, 55, 4285.
- A. Aliev, D. Li Ou, B. Ormsby and A. C. Sullivan, J. Mat. Chem., 2000, 10, 2758 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. 2 nitrogen sorption isotherm of P2 and Fig. 3 BJH pore size distribution of P2. See http://www.rsc.org/suppdata/cc/b0/b008192m/ |
| ‡ 1,4-Bis(triethoxysilyl)-2-(diethylphosphonato)butane 1: 1,4-bis(triethoxysilyl)but-2-ene (30.0 g, 78.8 mmol) and diethyl phosphite (21.8 g, 158 mmol, 20.3 ml) and di-tert-butyl peroxide (0.58 g, 3.9 mmol, 0.72 ml) were heated at 140 °C for 18 h under nitrogen. The resultant mixture was distilled under reduced pressure to yield 1 (22.5 g, 55%) bp 147 °C, 0.4 mm Hg; Calcd C 46.3, H 9.1. Found C 46.1, H 9.0; m/z 541.5 (M + Na)+ 519.5 (M+), 473.4 (M − OCH2CH3)+; Calcd for C20H48Si2P 519.2574; Found 519.2569; NMR: [(6CH35CH2O)3Si 1CH22CH{PO(O9CH2 10CH3)2}- 3CH24CH2Si(O7CH 28CH3)3] superscripts indicate proton or carbon environments as appropriate 1H(CDCl3) δ 0.51–1.03 (complex m, 4H, 1CH2, 4CH2), 1.05 (t, 9 H, 8CH3,3JHH 8 Hz), 1.06 (t, 9 H, 6CH3,3JHH 8 Hz), 1.27 (t, 6 H, 10CH3,3JHH 8 Hz), 1.56–1.91 (complex m, 3H, 2CH, 3CH2), 3.66 (q, 7CH2, 3JHH 8 Hz), 3.69 (q, 5CH2, 3JHH 8 Hz), 3.95 (m, 9CH2, 3JPH 10 Hz); 13C (CDCl3) δ 8.17 (d, 4CH2, 3JPC 4 Hz ), 8.92, 9.01 (d, 1CH2, 2JPC 6.5 Hz), 16.30, 16.39 (d, 10CH3, 2JPC 6.5 Hz ), 18.10 (s, 6CH3,8CH3), 23.56 (s, 3CH2, 2JPC 4 Hz), 32.16, 34.35 (d, 2CH, JPC 138 Hz), 58.09 (s, 5CH2), 58.29 (s, 7CH2), 61.16, 61.25 (d, 9CH2, 2JPC 6.5 Hz); 29Si (CDCl3) δ −45.65 (s, 4C-Si), −47.66, −47.94 (d, 1C-Si, 3JPSi 40 Hz); 31P (CDCl3) δ 36.13 (s). |
| § P1: Compound 1 (4.45 g, 8.58 mmol), THF (37 ml) and 1 M HCl (0.8 ml) were stirred under dynamic nitrogen for 1 h and stored in a polythene bottle. Gelation occurred after 11 d. The transparent monolithic gel obtained was air dried for 1 week and then dried at 60 °C in an oven for 24 h. A transparent monophasic crack-free glass was produced. This was powdered, washed with water, ethanol and ether consecutively, and then dried under vacuum at 120 °C for 24 h; 13C CP MAS δ 14.3, 16.9, 23.2, 33.2, 63.1; 29Si CP MAS δ −63.5; 31P CP MAS δ 34.5; Calculated average formula based on T2 environments [(HO)SiCH2CHPO(OEt)2CH2CH2SiO 2(OH)]n C∶P = 3.1, Found C/P = 2.8; Calculated for P1·3H2O C, 26.2; H, 6.3, P 8.5. Found C 26.6, H 6.0, P 9.6%. |
| ¶ P2: Powdered P1 (1.00 g) and concentrated HCl (100 ml) were refluxed for 24 h. The mixture was filtered through a fritted funnel and washed with excess H2O to remove all traces of HCl, followed by ethanol and ether. The residue was dried under vacuum at 120 °C for 24 h; 13C CP MAS δ 16.5, 23.1, 33.6; 29Si CP MAS NMR δ −66.2; 31P CP MAS NMR δ 33.5; Average formula based on T2 environments [(HO)SiCH2CHPO(OH)2CH2CH2SiO 2(OH)]n C∶P = 1.55, Found C∶P = 1.35; Calculated P2·2H2O C 16.3, H 5.1, P 10.5. Found C 14.2, H 4.3, P 10.5%; BET surface area 354 m2 g−1; micropore surface area 345 m2 g−1; micropore volume 0.192 cm3 g−1; total pore volume 0.219 cm3 g−1. |
| || Control experiments were carried out with microporous and mesoporous silicas prepared by standard methods from TEOS and with samples of these treated with concentrated HCl and washed in the manner described above for P2. |
| This journal is © The Royal Society of Chemistry 2001 |