Quadruple hydrogen bonded systems
Rint P. Sijbesma* and E. W. Meijer
Laboratory of Macromolecular and Organic Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: r.p.sijbesma@tue.nl; Fax: +40-2472655; Tel: +40-2472655
First published on 30th August 2002
Abstract
In this feature article, the development of linear quadruple hydrogen bonded systems is discussed, emphasizing applications in supramolecular chemistry and self-assembly.
Rint P. Sijbesma is associate professor in supramolecular polymer chemistry at the Eindhoven University of Technology, The Netherlands. He received his Ph.D. in Organic Chemistry at the University of Nijmegen in 1993, where he studied synthetic receptor molecules with professor Roeland Nolte. From 1992 to 1993, he worked as a postdoctoral research fellow with professor Fred Wudl (University of California, Santa Barbara) on the synthesis of water-soluble C60 derivatives. In 1993 he moved to Eindhoven as assistant professor, where he became associate professor in 2002 . His research interests are focused on the application of supramolecular concepts in polymer chemistry. |
E. W. ‘Bert’ Meijer is professor of Macromolecular and Organic Chemistry at the Eindhoven University of Technology, The Netherlands. He received his Ph.D. in Organic Chemistry at the University of Groningen with professor Hans Wijnberg. He was research scientist in the area of optoelectronic materials at Philips Research Laboratories from 1982 to 1989 and group leader of new polymeric materials at DSM Research from 1989 to 1992. Since 1992 he has been a full professor in Eindhoven and since 1995 he is also adjunct professor at the University of Nijmegen. His research interests are in dendrimers, supramolecular systems, and organic materials for electronics. |
Introduction
Hydrogen bonding is one of the most useful interactions in self-assembly,1 and is widely used because hydrogen bonds are directional and moderately strong.2 When stronger interactions are required than achievable with single hydrogen bonds, it is—at least conceptually—quite simple to increase the number of hydrogen bonds between components by using units that have arrays of hydrogen bonding sites. Like many concepts in supramolecular chemistry, the inspiration for this approach eventually comes from nature, where double and triple hydrogen bonds provide the means to read out and to replicate the information stored in DNA. Linear arrays of the kind found in nucleic acids—heterocyclic molecules with equidistant donor and acceptor sites—can be extended by adding rings or other functional units.3 The binary character of hydrogen bonding (a hydrogen bonding site is either a donor (D), or an acceptor (A)) leads to an information content of this type of linear arrays, which increases with the number of sites.4 While there are two different types of dimers for double hydrogen bond arrays, there are three triple hydrogen bonded dimers, and six different quadruple hydrogen bonded dimers, two of which contain self-complementary arrays [(DDAA)2 and (DADA)2] (Fig. 1). Obviously, when the spatial arrangement of sites in a hydrogen bonding unit is not restricted to be linear, the number of complementary pairs of arrays is unlimited. Multiple hydrogen bonding interactions between non-linear arrays of sites are very important in host–guest chemistry, e.g. for recognition of urea,5 barbiturates6 and other polar guests, and also plays a essential role in self-assembly of capsules7 and tubules.8 Self-assembly via double, triple and quadruple hydrogen bonds has been discussed recently,9 and the use of heteroaromatic modules for self-assembly using multiple hydrogen bonds has been reviewed.3 The present review will be restricted to linear quadruple hydrogen bonded arrays reported since 1988.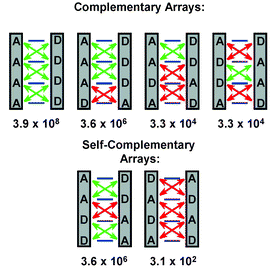 | ||
| Fig. 1 Dimers formed by linear arrays of four hydrogen bonding sites, and their stability constants (in M−1) in CDCl3 as predicted in ref 11. Attractive and repulsive secondary interactions are indicated by green and red double headed arrows, respectively. | ||
Several aspects contribute to the strength of quadruple hydrogen bonded complexes, and apart from the obvious effect of donor and acceptor strength of the individual sites, it has been suggested that there is a sizable contribution from secondary electrostatic interactions between adjacent sites in a complex, which are repulsive between like sites, while disparate sites attract each other.10 These effects have been used by Schneider to set up a linear free energy relationship for the prediction of the strength of multiple hydrogen bonded complexes in CHCl3.11 When increments are used of −8 kJ mol−1 for each primary interaction and −2.9 kJ mol−1 and +2.9 kJ mol−1 for each attractive and repulsive secondary interaction, respectively, it was found that stabilities of a set of 58 different complexes, was predicted with an average difference between calculated and observed values of just 1.7 kJ mol−1, corresponding to an average factor of 2 in Ka value. The six linear quadruple hydrogen bonded dimers (Fig. 1) have different numbers of attractive and repulsive secondary interactions, and their predicted complex stabilities are indicated. Apart from their strength, the possibility to achieve self-complementarity distinguishes linear quadruple hydrogen bonding units from units with three hydrogen bonding sites. While self-complementarity may limit the applicability in recognition, it offers distinct advantages in other fields of interest, such as the self assembly of supramolecular polymers,12–14 where the use of complementary units has the drawback that very precise control over stoichiometry is required to obtain high degrees of polymerization.
Considering the large body of literature on the use of double and triple hydrogen bonding units in self assembly, there are surprisingly few reports on linear arrays of more than three hydrogen bonds. Although it would be hard to prove, it seems likely that many scientists in the field of self assembly have been reluctant to design quadruple hydrogen bonding units because the heterocyclic chemistry involved in their synthesis was considered to be more troublesome than the gain in binding strength and recognition capabilities would justify. In the present feature article we will discuss quadruple hydrogen bonding units, both from our own work and that of others, and we hope to convince the reader that, at least for quadruple hydrogen bonding units, the fear to venture beyond the triple hydrogen bond is unwarranted, both in the sense of the limited synthetic effort required for their preparation, as well as for the enormous gain in binding strength achieved.
Bis-double hydrogen bonded arrays
A conceptually appealing way to arrive at linear quadruple hydrogen bonded arrays is to combine two double hydrogen bonded arrays in one molecule. One of the first examples of this approach emphasizes the informational content of a self complementary array consisting of two pyridone units. Ducharme and Wuest,15 have synthesized dipyridone 1, featuring a self complementary array of four hydrogen bonding sites and have compared its association properties with those of symmetric, non-self complementary dipyridone 2. The dimerization constant of 1 was determined to be higher than 6 × 104 M−1 in CHCl3, while the pyridone units of 2 are dimerized to the same extent as the model compound 2-pyridone, which has a dimerization constant of approximately 102 M−1.16 The crystal structures of 1 and 2 reflect the information encoded in the hydrogen bond arrays. Whereas dimers are found for 1, compound 2 forms planar polymeric chains of molecules. Related molecule 3 has been used by Wash et al. to probe the strength of acid–amide hydrogen bonds.17 It was shown that the stability constant of the dimer 32 exceeds 104 M−1 in CDCl3.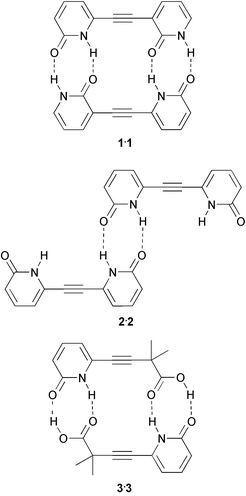
DADA arrays in pyridines, pyrimidines and triazines
Development of DADA arrays
Our involvement in the development of novel hydrogen bonding units stems from our interest in supramolecular polymers,12 reversible polymers in which the monomers are held together by non-covalent interactions. The construction of linear supramolecular polymers requires strong (but reversible) interactions that are highly directional. Multiple hydrogen bonding units are nearly ideal for this purpose, and we felt that development of building blocks which combine synthetic accessibility with strong complexation would be a prerequisite for success in the field. Our efforts had initially focused on synthetically highly accessible building blocks, e.g. triple hydrogen bonded complexes of melamine and imides,18 which have a Kdim value of approximately 102 M−1. Triple hydrogen bonding interactions between acylated diaminopyridines with thymines or uracil derivatives had been used in pioneering work by the group of Jean-Marie Lehn.19 However, the association constant (Ka) of 800 M−1 in CHCl3 of this combination is moderate at best, and only leads to polymers with a high degree of polymerization (DP) when the molecules are in the liquid crystalline state.Remarkably, acylation of the diaminopyridine is a highly effective method to strengthen the interaction in the complexes with ADA arrays, as it increases the dimerization constant approximately tenfold from Ka = 84 M−1 for the complex of diaminopyridine 5 with N-propylthymine 4, to Ka = 800 M−1 for the complex of 4 with diacetylaminopyridine 6.20 In an effort to increase the complexation strength of the triazine–thymine couple, we prepared diacylated triazine derivative 7, expecting to find a binding constant in the order of 104 M−1, as compared to a value of 890 M−1 for the complex of a non-acylated triazine with 4.20
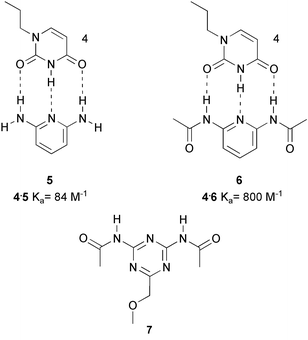
Much to our surprise, the complex of 7 with N-propylthymine turned out to have a Ka value not higher than 6 M−1. Thus, instead of raising the complex stability, a dramatic drop in stability resulted from acylation of the triazine amino groups! However, the experiment turned out to be an instructive failure. Because we had decided to routinely check for dimerization of the separate components in our study of DAD–ADA hydrogen bonded complexes, the weak, but significant dimerization of 7 (Kdim = 37 M−1) had not escaped our notice. But why would dimerization of 7 be stronger than in diacylaminopyridine? It occurred to us that the key to understanding the dimerization might lie in the different conformational properties of amidopyridines and amidotriazines. Although trans amides are generally more stable than cis amides,21 a trans conformation of the amide groups in 7, would result in considerable electrostatic repulsion between the carbonyl groups and the nitrogen atoms in the triazine ring. When the amides are cis, however, an ADADA array of five hydrogen bonding sites is formed, of which four sites can be involved in dimerization via a DADA array (Scheme 1).
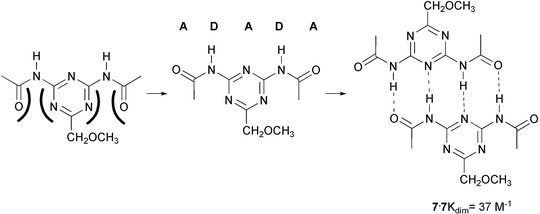 | ||
| Scheme 1 | ||
The initial attempts to find stronger triple hydrogen bonded complexes had thus led to the accidental discovery of quadruple hydrogen bonds, but stronger complexes had not been found. Instead, the dimer of 7 had a disappointingly low stability, considering the number of hydrogen bonds involved. We were aware of the fact that the DADA dimer had been predicted to be the least stable of all quadruple hydrogen bonded complexes, based on Jorgensen's10 concept of secondary interactions. Inspection of the structure of the dimer shows that in a ADADA dimer, two additional repulsive interactions are present, which we coined ‘spectator repulsions’, because the carbonyl groups causing the repulsion are not involved in hydrogen bonding. These spectator repulsions are readily eliminated, by removing one of the acyl groups, and we found that the Kdim value is indeed higher by a factor of 4 (Kdim = 530 M−1) in monoacylated diaminotriazine 8a.22
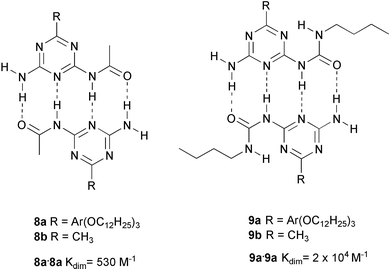
The crystal structure of the methyl derivative 8b (Fig. 2(a)) confirmed the hypothesis that the dimers are held together by quadruple hydrogen bonds.
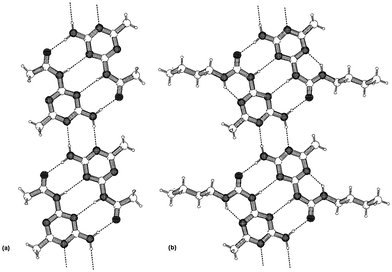 | ||
| Fig. 2 PLUTON representation of the hydrogen bonding patterns in the crystal structures of: (a) 2-(acetylamino)-4-amino-6-methyl-s-triazine 8b, (b) 2-butylureido-4-amino-6-methyl-s-triazine 9b. | ||
Incidentally, a search for similar structures in the crystallographic database revealed that we were not the first to find dimerization via linear arrays of four hydrogen bonding sites. Dimerization via such an array had been found in the crystal structure of acylated pyrimidine derivative 10 in 1992,23 although the stability of the dimers in solution was not investigated.
A further increase in complex stability was observed in preorganized ureidotriazine 9. Here, an intramolecular hydrogen bond fixes the DADA array in a planar conformation as can be seen in the crystal structure of 9b (Fig. 2(b)), resulting in a dimerization constant of 2 × 104 M−1. Yet, this turned out not to be the limit of stability for DADA arrays in solution. A further dimerization study, which included amido and ureido pyrimidines 11–13, showed dimerization constants ranging from 170 M−1 (for the dimer of 11), to larger than 2 × 105 (for the dimer of 13), corresponding to a 17.5 kJ mol−1 difference in stability of dimers that are held together by identical arrays, and consist of atoms with comparable donor and acceptor strength.
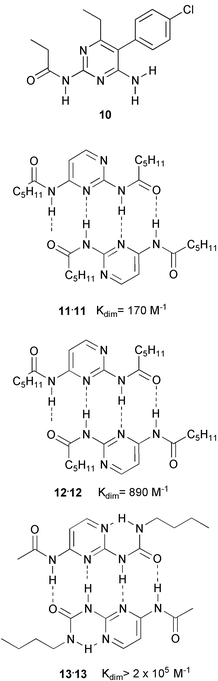
These large differences indicate the limitations of the additivity rules based on the concepts of secondary interactions.11 In particular the stabilities of the dimers of 9 and 13 are in excess of the predicted values for DADA arrays by 10.3 and 16.0 kJ mol−1, respectively. We have ascribed part of the difference to a high degree of preorganization in these compounds, but other factors, such as overall charge distribution may play a role as well.
The ease of synthesis of diaminotriazines, and the sizable dimerization constant of the mono-ureido derivative encouraged us to employ this unit as a building block for self-assembly of highly organized supramolecular structures. In a number of investigations, ureidotriazine unit 9a, provided with a trialkoxyphenyl group, has been used as an element that induces a high degree of order due to the strong anisotropy of its disk shaped dimer.24
Compound 9a is liquid crystalline over a large temperature range, and X-ray diffraction has shown that the molecules are organized in dimers, which stack in columns with hexagonal order. IR measurements indicate that the strength of the quadruple hydrogen bonds is such that upon isotropization at 183 °C, the molecules remain dimerized to a large extent.
DADA arrays in supramolecular polymers
The relatively strong dimerization, and the simple preparation of ureidotriazines are attractive features for the use of this unit in bifunctional compounds which can assemble into linear polymeric aggregates, termed ‘supramolecular polymers’. We have recently shown that combining hydrogen bonding with solvophobic interactions in bifunctional trialkoxyphenyl derivatives of ureidotriazines results in a hierarchical aggregation process, which is solvent dependent.25 While bifunctional molecules 14a are present as monomers in DMSO, they self assemble into random coil polymers in CDCl3.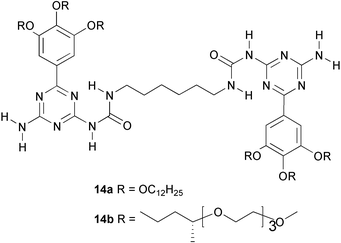
In dodecane, solvophobic interactions induce stacking of dimerized ureidotriazine units in a similar fashion as was observed in the mesophase of the monofunctional analog. This process results in highly ordered polymers with a helical columnar architecture (Fig. 3).
 | ||
| Fig. 3 Proposed mode of aggregation of molecules 14 in helical columns. | ||
Analogs 14b, with chiral oligoethylene side chains instead of alkyl side chains were also studied.25,26 The side chains render this compound water soluble. Amazingly, quadruple hydrogen bonding of this compound persists in this solvent because a hydrophobic microenvironment is created by stacking of the planar trialkoxyphenyltriazine moieties, in which disruption of the hydrogen bonds by water molecules is prevented.
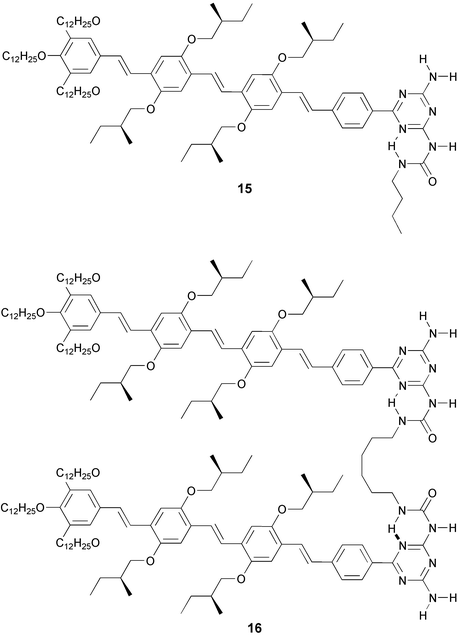
Dimerization of oligo(p-phenylenevinylene)-functionalized ureidotriazine 15, (Kdim = 2.1 ± 0.3 × 104 M−1 in CDCl3) is the first step in a hierarchical self assembly process that leads to chiral π-conjugated stacks of dimers in dodecane. These supramolecular architectures are our building blocks for supramolecular electronics. Bifunctional compound 16 forms random coil polymers in chloroform, while in dodecane, aggregates are less organized than those of 15, presumably because the hexyl spacers frustrate the formation of well-ordered helical stacks.
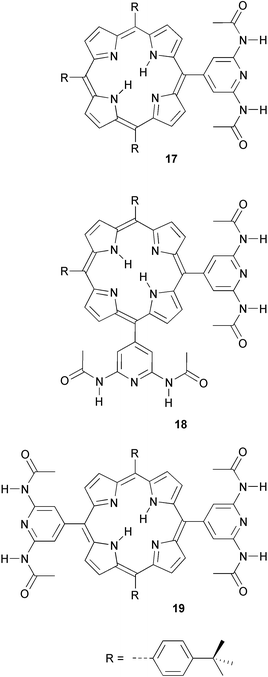
Self-complementary DADA hydrogen bonding in diacetamidopyridine has been used to prepare supramolecular tapes as well as cyclic tetramers in solution.27 The association constant of porphyrin 17 with one diacetamidopyridyl group was determined to be 160 M−1 in CDCl3 using NMR dilution experiments. Bifunctional compound 18 is preorganized to form tetrameric cycles, while by combining monofunctional compound 17 with bifunctional derivative 19, oligomers with different average degrees of polymerization were obtained. The proposed structure of the dimer of 17, with a DADA quadruple hydrogen bonding array was later questioned by Ercolani,28 based on the known preference of amides for an trans conformation.29 With the acetamido groups in the cis conformation, the most probable mode of association would be via double hydrogen bonds. It was also pointed out by Ercolani, that although the average degree of polymerization can be adjusted by adding monofunctional 17 to 19, there is no independent control over the polydispersity, because a statistical mixture of oligomers is still formed. In fact, the effects of adding 17 on both the degree of polymerization and the polydispersity are indistinguishable from the effects of diluting a solution of pure 19.
DDAA arrays in ureidopyrimidinones
Development of ureidopyrimidinone hydrogen bonding units
With the DADA arrays based on ureidotriazines, our group had developed easily accessible and versatile hydrogen bonding units, but for the purpose of obtaining linear supramolecular polymers with a higher degree of polymerization, a unit with a substantially higher association constant would be attractive. The limits of stability of DADA dimers had probably been reached with pyrimidine 13 (Ka = 2 × 105 M−1), and its preparation requires chromatographic separation and a relatively expensive starting material, diaminopyrimidine. We therefore set out to find molecules which would dimerize via a self complementary DDAA array and which would be simple to prepare. Ureidopyrimidinones (UPy’s) 20 seemed to be good candidates, because they are easily synthesized in two steps by reaction of β-keto esters with guanidine30 followed by acylation of the resulting isocytosine with an isocyanate.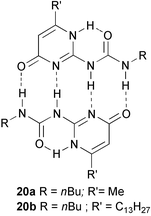
We noted that the synthesis of several ureidopyrimidinones had been published,31 but dimerization via quadruple hydrogen bonds had not been reported, possibly because dimerization in this fashion requires the molecules to be present in the non-standard [1H] tautomeric form. Fortunately, synthesis of 20a, from commercially available compounds methyl isocytosine and butylisocyanate led to a highly soluble product, which could be crystallized to afford crystals for X-ray diffraction.32 The results of crystal structure determination (Fig. 4) justified the assumptions of the design, as it shows dimers of 20a, which are held together by quadruple hydrogen bonds via a DDAA array.
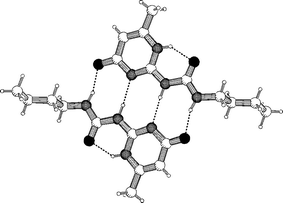 | ||
| Fig. 4 Crystal structure of 20a, in which the molecules are dimerized via DDAA arrays of hydrogen bonding sites. | ||
The molecules are preorganized by an intramolecular hydrogen bond from the urea NH group to one of the nitrogen atoms in the pyrimidine ring. The NMR spectrum of 20a in CDCl3 is relatively simple, and the position of the NH signals indicates extensive hydrogen bonding. The most encouraging feature of the spectrum was the complete lack of chemical shift changes upon dilution down to 10−4 M, indicating that dimerization of 20a persists at low concentrations, and that the Kdim value should be very high. Because the dimerization constant in chloroform was beyond the range that could be determined by 1H NMR dilution experiments, measurements were performed in more polar solvent mixtures (CDCl3–DMSO). Extrapolation of these values to pure chloroform gave a lower limit for Kdim of 106 M−1. In later work,33 thermodynamic parameters of dimerization were studied by using the excimer fluorescence in dimers of pyrene labeled derivative 21, and the Kdim value was determined to be approximately 2 × 107 M−1 in chloroform, and 108 M−1 in toluene at 298 K. Statistical mixtures of homo and heterodimers of UPy derivatives 22 and 23 have been used to determine the exchange rate, and hence the pre-exchange lifetime of the dimers by NMR. In CDCl3 the lifetime of the dimer is approximately 0.1 s. at 298 K, while in toluene, the kinetic stability of the dimer is increased to give a lifetime of 1.7 s at room temperature. Ikegami and Arai showed that anthracenyl labeled UPy derivative 24 and N,N-dimethylaniline labeled UPy derivative 25, give exciplex emission at concentrations as low as 10−5 M in chloroform, completely in line with the Kdim value determined with compound 21.
The thermal stability of the ureidopyrimidinone group is important for applications, because chemical degradation of this group at high temperatures may limit its use in supramolecular polymers. Armstrong and Buggy34 studied the stability of 20a as a model compound. Above 225 °C, the melting point of this compound, it underwent thermal degradation in three stages. Degradation was proposed to start with cleavage of the butane isocyanate.
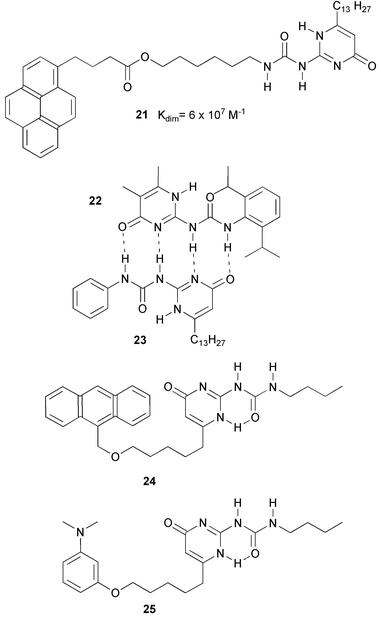
An important aspect of the ureidopyrimidinone unit as a building block in self-assembly is the presence of different tautomeric forms (Scheme 2). Three different forms have been observed in solution. In DMSO, a solvent which strongly competes for hydrogen bonding, the molecules are in the monomeric [3H] tautomeric form. In CDCl3, the molecules are present as a mixture of [1H] and pyrimidinol tautomers. Both tautomers are dimeric.
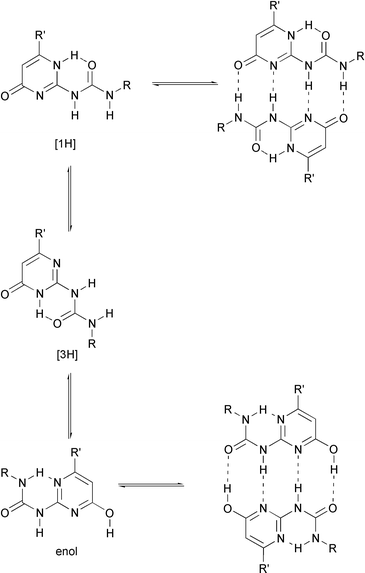 | ||
| Scheme 2 Equilibria between monomers and dimers of the three tautomeric forms of ureidopyrimidinones (UPy’s). | ||
Substituents on the 6-position of the heterocycle were found to have a strong effect on the position of the equilibrium between these tautomers; electron withdrawing substituents such as CF3 and p-nitrophenyl groups strongly favor the pyrimidinol form. The pyrimidinol tautomer dimerizes via a DADA array, and is expected to form weaker complexes due to a higher number of secondary repulsions in DADA dimers. Although the presence of the pyrimidinol tautomer does not compromise the ability of the unit to form very strongly associated dimers, as determined by excimer fluorescence studies (see above), the stability of the ureidopyrimidinone dimers might be even higher when the molecules could be fixed in their [1H] tautomeric form.
In an inventive approach to heterocyclic quadruple hydrogen bonding units that do not suffer from weakening effects of tautomerism, Corbin and Zimmerman have prepared 26, a molecule that is able to dimerize regardless of the tautomeric form it is in35 (Scheme 3) Moreover, the relative spatial arrangement of the substituents does not change upon tautomerization, an important feature for the reliability of the self-assembly processes. Recently, the same authors36 reported the dimerization of 27, with a Ka value of 259 M−1. This low value is due to a number of factors. The molecules need to unfold for dimerization via four hydrogen bonds, and repulsive spectator interactions further reduce the stability of the complex.
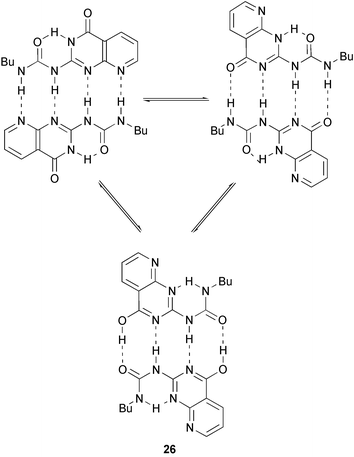 | ||
| Scheme 3 Equilibria of 26, which forms dimers regardless of tautomeric form. | ||
Ureidopyrimidinones in self-assembly and in supramolecular polymers
Oligo(p-phenylene vinylene) derivatives 28, provided with a UPy group were shown to form strong dimers. These compounds open new possibilities for the design of electronic materials.37 Strong dimerization of UPy groups of 29 in combination with threading of protonated amines in the crown ether ring of this compound has been used recently to self assemble four molecules into an organized complex.38The UPy group has also been used to organize fullerenes in supramolecular dimers.39,40 Two independent studies report the functionalization of C60 with UPy. Photophysical measurements on dimers of 30 showed strong electronic coupling through the hydrogen bond edge. However, cyclic voltammetry of 30 in toluene–acetonitrile showed that there is very little interaction between C60 units in the dimers. The C60 reduction waves in 31 were also affected very little compared to a model compound without the UPy unit attached. It was shown that a statistical mixture of dimers was formed when equimolar amounts of 31 and 20a were mixed in CDCl3.
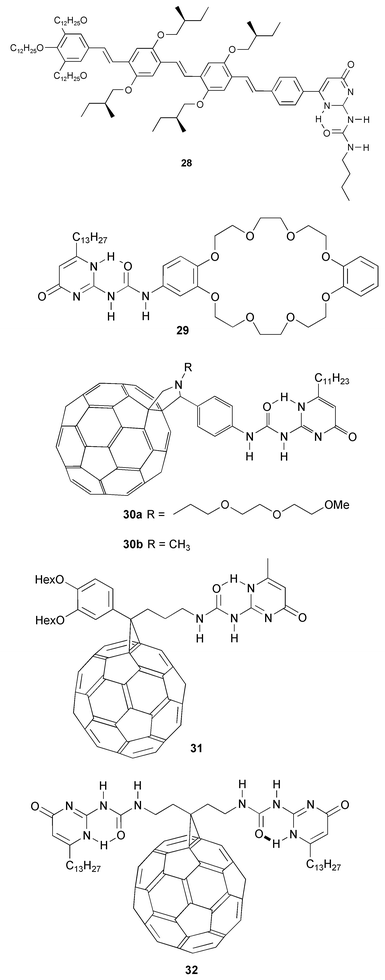
Hummelen’s group has also reported the synthesis of C60 derivative 32 with two UPy groups, which was shown to form supramolecular polymers at 100 mM in CDCl3, while complex 1H NMR spectra were formed at low concentration, due to the formation of cyclic species. The polymers hold promise as well-organized materials for organic photovoltaic devices with increased efficiency.41
Combining two hydrogen bonding units in highly preorganized scaffolds is a simple and effective way to obtain dimers with very high stability. This approach, when applied to double or triple hydrogen bonding arrays42,43 leads to dimers that can be considered as artificial dinucleotides, and which are stable in DMSO–CDCl3 mixtures. We have shown that dimers of 33, in which two UPy units are connected by a tetramethyl xylylene spacer, also have a high kinetic stability.44
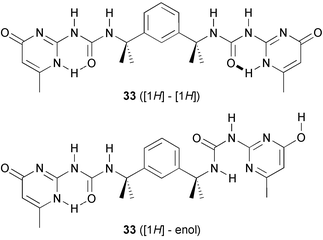
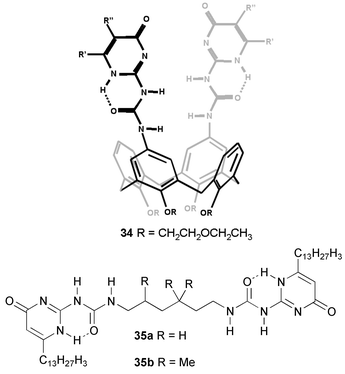
Different isomers of this dimer exist in the solid state (Fig. 5), differing in the relative arrangement of the dimerized UPy groups, or in the tautomeric form in which these groups exist. In solution, the symmetric dimers are found, in which all UPy groups are the [1H] tautomer, as well as an asymmetric isomer in which half of the UPy groups is the enol tautomer (Scheme 4) It was shown that in CDCl3, isomerization of the dimer, a process which is expected to proceed as least as fast as complete dissociation, proceeds with a time constant of approximately 44 min. Thus, an exceedingly stable 8-H bonding unit is formed by the simple combination of the UPy unit with a preorganized spacer. Similar preorganization is responsible for the strong dimerization of calixarene 34 reported by de Mendoza and co-workers.45 The stability of this dimer in CDCl3 is too high to allow observation of dissociation using NMR. Therefore, the extent of dissociation in DMSO–CDCl3 solvent mixtures was investigated, and Ka values of 2500 M−1 and 572 M−1 were calculated in solutions containing 64% and 73% of DMSO, respectively. It was also demonstrated that addition of a triflate salt leads to dissociation of the dimer, presumably due to complexation of the triflate anion to the urea part of the UPy units.
![Anti-[1H]–[1H] (a), and syn-[1H]–[1H] (b) forms of dimers of 33 found in different crystal modifications.](/image/article/2003/CC/b205873c/b205873c-f5.gif) | ||
| Fig. 5 Anti-[1H]–[1H] (a), and syn-[1H]–[1H] (b) forms of dimers of 33 found in different crystal modifications. | ||
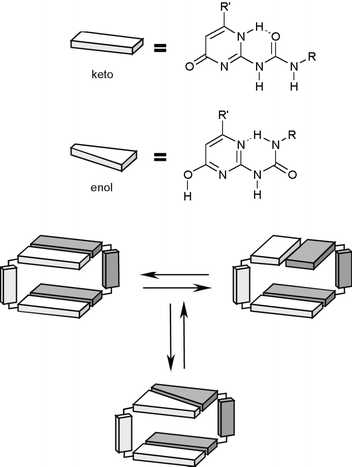 | ||
| Scheme 4 Equilibrium between isomers of dimeric 33. | ||
It can be calculated that with a dimerization constant of 6 × 107 of the ureidopyrimidinone group, bifunctional derivatives that are not preorganized to form cycles, may reach high degrees of polymerization. We decided to investigate the possibilities of obtaining supramolecular polymers by connecting two UPy units by a C6-spacer (compound 35a). This bifunctional compound forms viscous solutions in chloroform, and the pure material is a rubber like solid, which crystallizes upon standing.46
Addition of monofunctional UPy derivative 20b to solutions of 35a has a dramatic effect on the viscosity (Fig. 6). Like in conventional condensation polymerization, monofunctional molecules act as ‘chain stoppers’, and reduce the average degree of polymerization. In solutions of 35, the reduction of viscosity takes place immediately, showing that the polymer chains are continuously breaking and recombining, and quickly establish a new equilibrium degree of polymerization. Analysis of the effect of varying amounts of chain stoppers on solution viscosity allowed us to estimate the degree of polymerization in 40 mM solutions of pure 35a to be in the order of 700 monomer units.
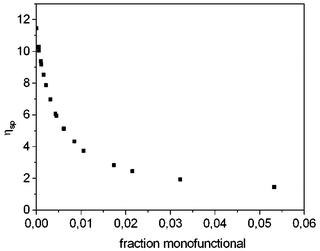 | ||
| Fig. 6 Specific viscosity of a 40 mM chloroform solution of bifunctional UPy derivative 35a as a function of the fraction of added monofunctional molecules 20b. | ||
A protected monofunctional UPy derivative with a photocleavable o-nitrobenzyl group has been used as a stopper molecule that can be activated by light. When a viscous solution of 35a, containing small amounts of the o-nitrobenzyl protected derivative 36 was irradiated with UV light, the stopper molecules were activated, and the DP of the polymer dropped, resulting in a large decrease in viscosity47 (Fig. 7)
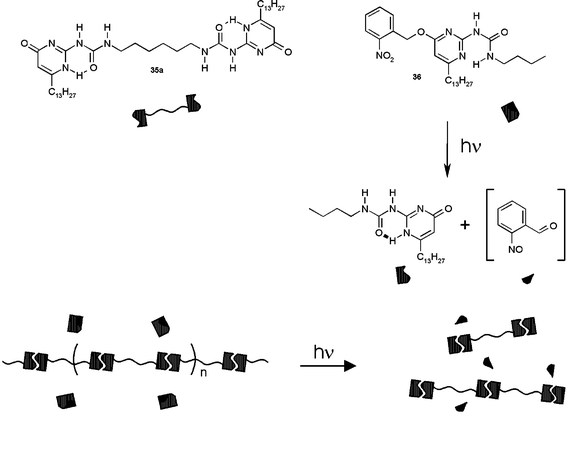 | ||
| Fig. 7 Photodeproctection of 36, leading to a decrease in the average DP of a solution of 35a. | ||
Bifunctional UPy derivatives have been prepared with a range of oligomeric or polymeric spacers, such as polysiloxanes,48 poly(ethylene/butylenes), polyethers, polyesters and polycarbonates.49 Introduction of UPy end groups results in dramatic changes of the mechanical properties of the polymers, and materials were obtained that combine many of the mechanical properties of conventional macromolecules with the low melt viscosity of organic compounds.
Further control over the topology of supramolecular polymers is attained when trifunctional monomers are used, which lead to the formation of reversible networks.50
Recently, Coates and co-workers have incorporated vinyl-substituted unit 37 in poly(1-hexene), using a nickel-based polymerization catalyst which is tolerant to functional Lewis basic groups. With small amounts of UPy-units incorporated, the polyolefins show thermoplastic elastomeric properties.51
Supramolecular polymers based on UPy, are in dynamic equilibrium with cyclic species. In equimolar mixtures of compounds 35a and 38 in chloroform, the preferential formation of cyclic heterodimer 35a·38 can be observed due to the distinct signals of this medium-sized ring in the 1H NMR spectrum. When the concentration of this species is plotted as a function of the total solute concentration, the presence of a critical concentration is evident, below which little polymer is present, and above which the concentration of cycles remains constant. This phenomenon, which is predicted by theory, has been observed for polycondensations under equilibrating conditions, but it is generally difficult to observe in truly reversible systems.52 Entropy driven ring-opening polymerization is another remarkable phenomenon that has been observed in UPy-based supramolecular polymers. In solutions of 35b, the equilibrium between cycles and polymers shifts to favor polymerization when the temperature is increased. As a result, the viscosity of the solutions increases upon increasing the temperature.53
Squaramide dimers
Recently, N-carbamoyl squaramides 39 were introduced as readily available units which dimerize via four hydrogen bonds.54 In contrast to the units based on triazine, isocytosine, or Zimmerman’s pyrimidopyrimidinone unit, the dimers are held together by two bifurcated instead of four separate hydrogen bonds, as is evident from the crystal structure of this compound.. Nevertheless, no signs of dissociation of the dimers is observed by NMR spectroscopy in CDCl3 down to concentrations of 0.5 mM, suggesting strong dimerization. Dilution studies in a 95∶5 solvent mixture CDCl3–CD3CN allowed determination of Kdim, which has values of up to 9800 M−1 for 39b in this solvent.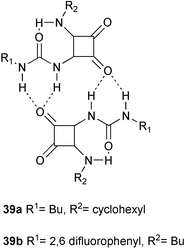
Complementary arrays
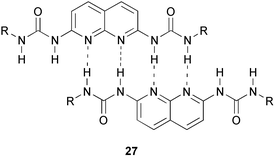
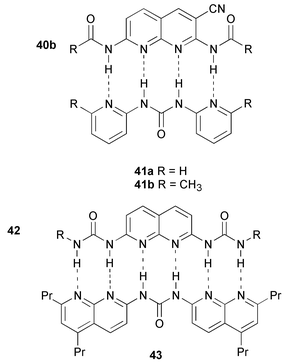
The possibilities of complementary quadruple hydrogen bond arrays remains largely unexplored. Of the four different types of quadruple hydrogen bonded complexes that are possible, only the binding properties of the DAAD–ADDA couple have been reported.35,55 Two of the tautomeric forms of 26 contain a ADDA array, and its complexation with the complementary DAAD array of 2,7-bis(acylamino)-1,8-naphthyridine 40 has been studied in solution.35 Addition of a small excess of 40a led to the dissociation of dimers of 26, and the formation of a complex with 40a. The complex 26·40 is consequently even more stable than the dimer 26·26 (Ka = >3 × 107). In a related approach to DAAD–ADDA dimers, Lüning and Kuhl chose to study the combination of a 2,7-bis(acylamino)-1,8-naphthyridine with dipyridyl ureas.55The formation of heterocomplexes was studied with 1H NMR titrations. In contrast to the complex reported by Corbin and Zimmerman, binding constants are relatively low (2000 and 160 M−1, for the complexes of 40b with 41a and 41b, respectively), possibly due to steric interactions between R substituents on both components. An additional aspect that may negatively influence the complex stability of dimers involving pyridyl ureas is the presence of an intramolecular hydrogen bond in the monomer which must be disrupted in order to form a ADDA array. Similar effects destabilize the complex of 42 with 43 which is held together by six hydrogen bonds.36 Here the intramolecular hydrogen bond in monomeric 42 is observed not only in solution but also in the solid state. As a result, the Ka value of this complex is 5 × 105 M−1, at least two orders of magnitude lower than predicted for a six-fold hydrogen bonded complex.
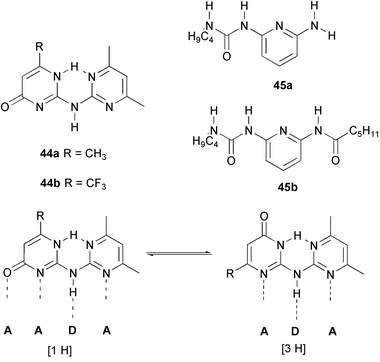
In an attempt to create a pair of hydrogen bonding units which dimerize via complementary ADAA and DADD arrays, our group prepared dipyrimidinylamines 44 with the aim to form stable complexes with pyridylureas 45.56 However, X-ray crystal structure determination of 44a showed that the compound exists in the [3H] tautomeric form, which displays a ADA, instead of a ADAA array of hydrogen bonding sites. This tautomeric form persists in solution, as was shown with NOE experiments. As a result of the tautomerization, no binding of pyridylureas was observed.
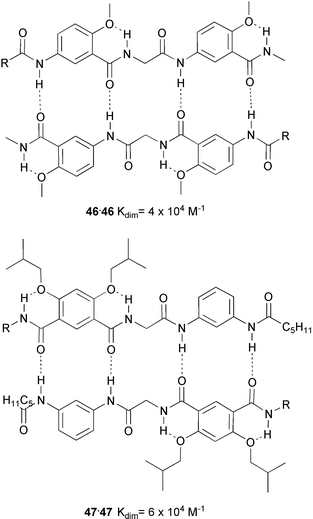
Recently, Bing Gong and coworkers have introduced a new type of recognition unit featuring linear arrays of hydrogen bonding sites.57 These units are based on combining residues from 3-aminobenzoic acid, isophthalic acid and 1,3-diaminobenzene in oligoamides which are held in a planar arrangement by intramolecular hydrogen bonding of amide N–H groups to ether oxygen atoms.
The validity of the concept was demonstrated by the stability of complexes 46·46 and 47·47, in CHCl3 solution (Ka values are 4 × 104 M−1 and 6.5 × 104 M−1, respectively). The proposed mode of complexation was confirmed by the crystal structure determination of the dimers.
In later work,58 Bing Gong has demonstrated that this concept can be extended to six-hydrogen bonded molecular duplexes with Ka values of the order of 109 M−1. Comparison of two closely related duplexes, with single mismatched binding sites indicate that the mismatch leads to a significant (approximately 40-fold) reduction in binding constant, but that the remaining five hydrogen bonds are sufficiently strong to keep the components together in solution.59
Outlook
The development of a range of linear quadruple hydrogen bonding units in the past five years, has shown that these units may combine synthetic accessibility with high association constants. These properties make the hydrogen bonding units ideal building blocks for self assembly, or ‘non-covalent synthesis’1 of functional materials and supramolecular polymers. Although even stronger binding is desirable in some cases, the slower kinetics of the strongest complexes may limit their reversibility. Recent development of molecules with complementary quadruple hydrogen bonding arrays fills a distinct need for such recognition units. These arrays, as well as compounds with non-linear arrays, which have been developed as hydrogen bonding receptors,60 allow further control in self-assembly and recognition processes, such as the directed formation of switchable supramolecular block-copolymers,61 or the selective sequestration of tagged species (catalysts or synthetic targets) on a solid phase.62 At present, supramolecular polymers based on quadruple hydrogen bonds are used in thermoplastic elastomers and π-conjugated materials, and technological applications of supramolecular polymers processed out of solution are foreseen in the near future.Notes and references
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS.
- G. A. Jeffrey, An introduction to hydrogen bonding, Oxford University Press, Oxford, 1997 Search PubMed.
- S. C. Zimmerman and P. S. Corbin, Struct. Bonding (Berlin), 2000, 96, 63–94 Search PubMed.
- The number of different arrays with n sites is ½(2n + 21/2n) (n = even) or ½(2n + 21/2(n − 1)) (n = odd).
- T. W. Bell and Z. Hou, Angew. Chem., Int. Ed., 1997, 36, 1536–1538 CrossRef CAS.
- S. K. Chang, D. Van Engen, E. Fan and A. D. Hamilton, J. Am. Chem. Soc., 1991, 113, 7640–7645 CrossRef CAS.
- J. Rebek, Chem. Soc. Rev., 1996, 25, 255–264 RSC.
- D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988–1011 CrossRef CAS.
- D. C. Sherrington and K. A. Taskinen, Chem. Soc. Rev., 2001, 30, 83–93 RSC.
- W. L. Jorgensen and J. Pranata, J. Am. Chem. Soc., 1990, 112, 2008 CrossRef; J. Pranata, S. G. Wierschke and W. L. Jorgensen, J. Am. Chem. Soc., 1991, 113, 2810 CrossRef CAS.
- J. Sartorius and H.-J. Schneider, Chem. Eur. J., 1996, 2, 1446–1453 CrossRef CAS.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS.
- Supramolecular Polymers, ed. A. Ciferri, Marcel Dekker inc., New York, 2000 Search PubMed.
- N. Zimmerman, J. S. Moore and S. C. Zimmerman, Chem. Ind. (London), 1998, 604–610 Search PubMed.
- Y. Ducharme and J. D. Wuest, J. Org. Chem., 1988, 53, 5787–5789 CrossRef CAS.
- M. H. Krackov and C. M. Lee, J. Am. Chem. Soc., 1965, 87, 892–896 CrossRef CAS.
- P. L. Wash, E. Maverick, E. Chiefari and D. A. Lightner, J. Am. Chem. Soc., 1997, 119, 3806 CrossRef.
- R. F. M. Lange and E. W. Meijer, Macromolecules, 1995, 28, 782–783 CrossRef CAS; R. F. M. Lange and E. W. Meijer, Macromol. Symp., 1996, 102, 301–308 Search PubMed.
- C. Fouquey, J.-M. Lehn and A.-M. Levelut, Adv. Mater., 1990, 2, 254–257 CrossRef CAS; J.-M. Lehn, Macromol. Chem., Macromol. Symp., 1993, 69, 1–17 Search PubMed; T. Gulik–Krzywicki, C. Fouquey and J.-M. Lehn, Proc. Natl. Acad. Sci.USA, 1993, 90, 167; M. Kotera, J.-M. Lehn and J.-P. Vigneron, J. Chem. Soc., Chem. Commun., 1994, 197–200 RSC; M. Kotera, J.-M. Lehn and J.-P. Vigneron, Tetrahedron, 1995, 51, 1953–72 CrossRef CAS.
- F. H. Beijer, R. P. Sijbesma, J. A. J. M. Vekemans, E. W. Meijer, H. Kooijman and A. L. Spek, J. Org. Chem., 1996, 61, 6371–6380 CrossRef CAS.
- A. R. Katritzky and I. Ghiviriga, J. Chem. Soc., Perkin Trans. 2, 1995, 1653 Search PubMed.
- F. H. Beijer, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, Angew. Chem., Int. Ed., 1998, 37, 75–78 CrossRef CAS.
- R. J. Griffin and P. R. Lowe, J. Chem. Soc., Perkin Trans. 1, 1992, 1811–1819 RSC.
- F. H. Beijer, PhD Thesis, Technische Universiteit Eindhoven, 1998.
- J. H. K. K. Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167–170 CrossRef CAS.
- L. Brunsveld, J. A. J. M. Vekemans, J. H. K. K. Hirschberg, R. P. Sijbesma and E. W. Meijer, Proc. Natl. Acad. Sci., 2002, 99, 4977–4982 Search PubMed.
- C. M. Drain, X. Shi, T. Milic and F. Nifiatis, Chem. Commun., 2001, 287–288 RSC.
- G. Ercolani, Chem. Commun., 2001, 1416–1417 RSC.
- F. H. Beijer, R. P. Sijbesma, J. A. J. M. Vekemans, E. W. Meijer, H. Kooijman and A. L. Spek, J. Org. Chem., 1996, 61, 6371–6380 CrossRef CAS.
- D. J. Brown, R. F. Evans, W. B. Cowden and M. D. Fenn, The pyrimidines, Wiley, New York, 1994, p. 193–196 Search PubMed.
- A. Kreutzberger and H. Schimmelpfennig, Arch. Pharm., 1981, 314, 34–41 Search PubMed; J. Krepelka; A. Cerny, R. Kotva, J. Vachek and M. Melka, Collect. Czech. Chem. Commun., 1986, 51, 1140–1149; G. Vasilev, Dokl. Bolg. Akad. Nauk., 1990, 43, 57–60 Search PubMed (Chem. Abstr., 115, 49603); J. Fan and C. C. Cheng, J.Heterocycl. Chem., 1993, 30, 1273–1276 Search PubMed.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- G. Armstrong and M. Buggy, Mater. Sci. Eng., C: Biomimetic Supramol. Syst., 2001, 18, 45–49 Search PubMed.
- P. S. Corbin and S. C. Zimmerman, J. Am. Chem. Soc., 1998, 120, 9710–9711 CrossRef CAS.
- P. S. Corbin and S. C. Zimmerman, J. Am. Chem. Soc., 2000, 122, 3779–3780 CrossRef CAS.
- A. El–ghayoury, E. Peeters, A. P. H. J. Schenning and E. W. Meijer, Chem. Commun., 2000, 1969–1970 RSC.
- Y. Tokunaga and T. Seo, Chem. Commun., 2002, 970–971 RSC.
- J. J. Gonzalez, J. de Mendoza, S. Gonzalez, E. M. Priego, N. Martin, C. Luo and D. M. Guldi, Chem. Commun., 2001, 163–164 RSC.
- M. T. Rispens, L. Sanchez, J. Knol and J. C. Hummelen, Chem. Commun., 2001, 161–162 RSC.
- L. Sanchez, M. T. Rispens and J. C. Hummelen, Angew. Chem., Int. Ed., 2002, 41, 838–840 CrossRef CAS.
- J. L. Sessler and R. Wang, J. Org. Chem., 1998, 63, 4079–4091 CrossRef CAS.
- H. Kagechika, I. Azamuya, A. Tanati, K. Yamaguchi and K. Shudo, Tetrahedron Lett., 1999, 40, 3423–3426 CrossRef CAS.
- B. J. B. Folmer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1999, 121, 2001–2007 CrossRef CAS.
- J. J. Gonzalez, P. Prados and J. de Mendoza, Angew. Chem., Int. Ed., 1999, 38, 525–528 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS.
- B. J. B. Folmer, E. Cavini, R. P. Sijbesma and E. W. Meijer, Chem. Commun., 1998, 1847–1848 RSC.
- J. H. K. K. Hirschberg, F. H. Beijer, H. A. van Aert, P. C. M. M. Magusin, R. P. Sijbesma and E. W. Meijer, Macromolecules, 1999, 32, 2696–2705 CrossRef.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- R. F. M. Lange, M. van Gurp and E. W. Meijer, J. Pol. Sci. A, Pol. Chem., 1999, 37, 3657–3670 Search PubMed.
- L. R. Rieth, R. F. Eaton and G. W. Coates, Angew. Chem., Int. Ed., 2001, 40, 2153–2156 CrossRef CAS.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, Macromolecules, 2001, 34, 3815–3818 CrossRef.
- B. J. B. Folmer, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2001, 123, 2093–2094 CrossRef CAS.
- A. P. Davis, S. M. Draper, G. Dunne and P. Ashton, Chem. Commun., 1999, 2266, 2265–2266 RSC.
- U. Luning and C. Kuhl, Tetrahedron Lett., 1998, 39, 5735–5738 CrossRef CAS.
- S. H. M. Söntjens, J. T. Meijer, H. Kooijman, A. L. Spek, M. H. P. van Genderen, R. P. Sijbesma and E. W. Meijer, Org. Lett., 2001, 3, 3887–3889 CrossRef CAS.
- B. Gong, Y. Yan, H. Zeng, E. Skrzypczak–Jankunn, Y. W. Kim, J. Zhu and H. Ickes, J. Am. Chem. Soc., 1999, 121, 5607–5609 CrossRef CAS.
- H. Zeng, R. S. Miller, R. A. Flowers and B. Gong, J. Am. Chem. Soc., 2000, 122, 2635–2644 CrossRef CAS.
- H. Zeng, H. Ickes, R. A. F. Ii and B. Gong, J. Org. Chem., 2001, 66, 3574–3583 CrossRef CAS.
- J. H. Hartley, T. D. James and C. J. Ward, J. Chem. Soc., Perkin Trans. 1, 2000, 3155–3184 RSC.
- J. Ruokolainen, R. Makinen, M. Torkkeli, T. Makela, R. Serimaa, G. ten Brinke and O. Ikkala, Science, 1998, 280, 557–560 CrossRef CAS.
- S.-Q. Zhang, K. Fukase, M. Izumi, Y. Fukase and S. Kusumoto, Synlett, 2001, 590–596 CAS.
| This journal is © The Royal Society of Chemistry 2003 |