A light-harvesting fluorinated fullerene donor-acceptor ensemble; long-lived charge separation†
Glenn A.
Burley
a,
Anthony G.
Avent
a,
Olga V.
Boltalina
b,
Ilya V.
Gol’dt
b,
Dirk M.
Guldi
c,
Massimo
Marcaccio
d,
Francesco
Paolucci
d,
Demis
Paolucci
d and
Roger
Taylor
a
aSchool of Chemistry, Physics and Environmental Sciences, University of Sussex, Brighton, UK BN1 9QJ. E-mail: G.Burley@sussex.ac.uk; R.Taylor@sussex.ac.uk
bChemistry Department, Moscow State University, Moscow, 119899, Russia. E-mail: ovb@thermo.chem.msu.ru
cRadiation Laboratory, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: guldi.1@nd.edu
dDipartimento di Chimica, Università di Bologna, 40126 Bologna, Italy. E-mail: paolucci@ciam.unibo.it
First published on 4th December 2002
Abstract
In a first example of a trannulene-based donor–acceptor dyad visible light photoexcitation generates a long-lived (870 ns) charge-separated state.
One of the most intensively investigated areas of fullerene chemistry concerns the photophysical properties of fullerene derivatives covalently tethered to one or more photoactive chromophores.1 The combination of the ability of [60]fullerene to accept up to six electrons2 with only small reorganisation energy upon reduction,1a renders it an attractive candidate for energy conversion and energy storage. A significant problem with the use of fullerenes as electron-acceptor units is the reduced electron affinity resulting from most derivatisations. However, fluorination of [60]fullerene enhances its electron affinity (e.g. values for C60F183 and C60F364 are ca. 3.1 and 3.48 eV compared with 2.67 eV for C605) thus overcoming the deficiencies of conventional derivatisation.
C60F18 is a unique member of the fluorofullerene family,6 possessing a flattened hemisphere comprising an aromatic face surrounded by a fluorinated crown; and a curved “normal” hemisphere akin to its all-carbon parent.7 Its high electron deficiency and strong visible light absorption (e.g. ε608 = 13,265 M−1 cm−1; ε667 = 20,580 M−1 cm−1 for 1) renders C60F18 a potential synthon for the construction of novel donor–acceptor arrays.
Recently, the first all-trans 18π annulenic fluorofullerene (named trannulene) via Bingel addition chemistry to C60F18 was reported.8 This simple one-step reaction has the potential for the attachment of a plethora of functionalities in three precise locations on the trannulene surface. As an initial investigation of the utility of these trannulenes for light-harvesting applications, we describe here the formation and photophysical properties of a novel multi-component donor–acceptor array 1 (Fig. 1).
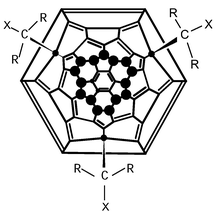 | ||
| Fig. 1 Structure of 1; R = CO2Et, X = CO2CH2AnthTTF, ● = F. | ||
Tetrathiafulvalene (TTF) moieties have been shown to be effective electron-transfer partners with [60]fullerene owing to their increase in aromaticity upon oxidation to form 1,3-dithiolium cations.9 However, toluene solutions of TTF react with C60F18 upon concentration of solvent in vacuo.10 The extended analogue of TTF (2) was used for this study to circumvent the reaction of the TTF nucleus with the fluorofullerene.
Acid deprotection of 3 (TBDMS = tert-butyldimethylsilyl) followed by malonylation of the alcohol 4 gave 5 as a yellow solid (Scheme 1). Due to the low acidity of the malonate protons of 5, it was converted to the corresponding methanetricarboxylate 2. Treatment of a toluene solution of C60F18 [from fluorination of [60]fullerene with MnF3–K2NiF6 (1∶5.5∶2.25 weight ratio) at 480 °C, 5.5%] and 2 with DBU afforded the emerald-green trannulene 1 (Scheme 2). The ethyl ester 6 was prepared as a reference.
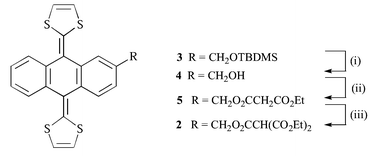 | ||
| Scheme 1 Reagents and conditions: (i) H+, THF, 94%; (ii) EtO2CCH2COCl, pyridine, DCM 0 °C, 71%; (iii) 1. NaH, 2. EtO2CCl, DMF, 0 °C, 42%. | ||
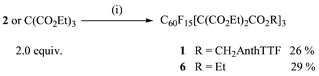 | ||
| Scheme 2 Reagents and conditions: (i) C60F18 (1.0 equiv.), DBU (1.9 equiv.), toluene. | ||
Electrochemical studies of 1 display a CV curve (Fig. 2) in which the redox processes of the models 2 and 6 are superimposed. This superimposition suggests no interaction between the electron donating and accepting moieties in the ground state. The redox couples at 0.62 and 0.15 V reveal slow heterogeneous electron-transfer kinetics. This corresponds to a three-electron oxidation attributed to the AnthTTF moiety, with approximate E1/2 = 0.53 V, and accords with previously reported studies on similar TTF compounds,11 confirmed by comparison with the CV of 2.
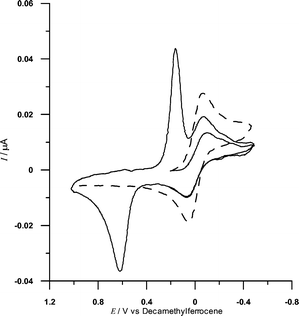 | ||
| Fig. 2 CV curves of 0.5 mM 1 (full line) and 0.5 mM 6 (dashed line) in 0.05 M NBu4+PF6−, tetrachloroethane solutions. Scan rate: 0.5 V s−1. T = 298 K. Working electrode: Pt. Potential measured with respect to a silver quasi-reference electrode and decamethylferrocene (internal standard). | ||
The reversible one-electron reduction peak (E1/2 = −0.005 V) in Fig. 2 is due to the reduction of the trannulene moiety, and is therefore also observed for 6 (Fig. 2). Note that these fullerene derivatives are much more easily reduced than C60. With respect to ferrocene, 6 is reduced at −0.54 V, while, under the same conditions, the first reduction of C60 is at −1.06 V, i.e.ca. 0.5 V more negative. Thus dyad 1 can be both easily oxidised and reduced, the approximate energy of the charge-separated state, AnthTTF˙+-C60F15˙−, being 0.54 eV.
Steady-state fluorescence experiments with 6 gave low quantum yields (Φ = 1.1 × 10−4). Matching the absorption of donor–acceptor system 1 at the 665 nm maximum (Fig. S2, see ESI†), where AnthTTF lacks any appreciable absorption, allowed us to probe the impact of the electron donor on the photoexcited fullerene. In fact, in dichloromethane the fluorescence quantum yield of 1 is reduced to 0.55 × 10−4 – Fig. S1, see ESI.† Despite their overall low quantum yields, fluorescence lifetimes were successfully determined for 6 and 1. In particular, a lifetime of 1.61 ns was derived for 6 from fitting the radiative decay around the 695 nm maximum to a mono-exponential fitting procedure. On the other hand, 1 gives rise to a lifetime of 0.68 ns, which corresponds to a moderate electron-transfer quenching efficiency of 43%, due to the semi-flexible nature of the linker.
From the difference between the 665 nm absorption and the 696 nm emission, we estimate a singlet excited state energy of 1.82 eV. This is sufficiently energetic to power in 1 a thermodynamically driven electron transfer to yield AnthTTF˙+-C60F15˙− (vide infra).
Transient absorption measurements shed light onto the electron transfer mechanism. Fig. 3 compares the differential absorption changes recorded with a 50 ns delay upon photoexciting solutions of 6 with those of 1 (2.0 × 10−5 M). In the case of 6, the features correspond to the long-lived (τ = 15 μs) and oxygen-sensitive (kq = 1.2 × 109 M−1 s−1) triplet excited state. The observed minima (∼400 and 665 nm) and shoulders (450 and 615 nm) match quite well the ground state absorption. Consequently, the bleaching relates to the depletion of the ground state. Also, new and particularly broad triplet–triplet transitions are noted in the near-IR region.
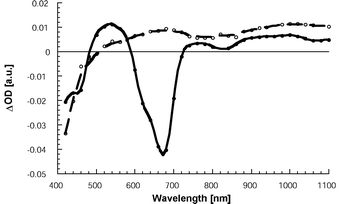 | ||
| Fig. 3 Transient absorption changes recorded 50 ns upon 337 nm laser excitation of 6 (full line) and 1 (dashed line) in oxygen-free dichloromethane (2.0 × 10−5 M). | ||
Quite different is the spectrum seen for 1, which bears no similarity to the triplet excited state of reference 6. Around 680 nm, instead of the strong ground-state bleaching, a transient maximum was recorded, which resembles the characteristic fingerprint of the one-electron oxidised AnthTTF˙+. The near-IR transition was ascribed, by analogy to C60 and several of its derivatives, to the one-electron reduced fullerene species, C60F15˙−. This confirms that the product of the intramolecular singlet excited state deactivation is AnthTTF˙+-C60F15˙−.
The charge-separated state is metastable and decays quantitatively to the singlet ground state. A mono-exponential rate law fits best the decay of both fingerprint absorptions, that is, C60F15˙− and AnthTTF˙+. From the corresponding fits we derived a radical pair lifetime of 870 ns.
In conclusion, a novel donor–acceptor ensemble was devised incorporating an all-trans 18π annulenic fluorofullerene (trannulene), and extended TTF. Relative to C60, one of the fundamental advantages of trannulenes lies in their improved chromophoric features. Strong visible light absorption, with a maximum at 665 nm (20,580 M−1 cm−1), renders them good light-harvesting building blocks. Once photoexcited with visible light, an energetically low lying (0.54 eV) and long-lived (870 ns) charge-separated state is generated via a rapid intramolecular electron transfer process.
EPSRC (UK), the University of Bologna, MIUR, C.N.R., Volkswagen Stiftung, and Basic Energy Sciences Office of the US Department of Energy (NDRL-4423) supported this work. We thank Prof. Nazario Martin for helpful discussion.
Notes and references
- (a) D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22 RSC; (b) D. M. Guldi and N. Martin, J. Mater. Chem., 2002, 12, 1978 RSC; (c) D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695 CrossRef CAS.
- L. Echegoyen and L. E. Echegoyen, Acc. Chem. Res., 1998, 31, 593 CrossRef CAS.
- K. Ohkubo, R. Taylor, O. V. Boltalina, S. Ogo and S. Fukuzumi, Chem. Commun., 2002, 1952 RSC.
- N. Liu, Y. Morio, F. Okino, H. Touhara, O. V. Boltalina and V. K. Pavlovich, Synth. Met., 1997, 86, 2289 CrossRef CAS.
- C. Brink, L. H. Andersen, P. Hvelplund, D. Mathur and J. D. Volstad, Chem. Phys. Lett., 1995, 233, 52 CrossRef CAS.
- R. Taylor, Chem. Eur. J., 2001, 7, 4074 CrossRef CAS.
- I. S. Neretin, K. A. Lyssenko, M. Y. Antipin, Y. L. Slovokhotov, O. V. Boltalina, P. A. Troshin, A. Y. Lukonin, L. N. Sidorov and R. Taylor, Angew. Chem., Int. Ed., 2000, 39, 3273 CrossRef CAS.
- (a) X. W. Wei, A. D. Darwish, O. V. Boltalina, P. B. Hitchcock, J. M. Street and R. Taylor, Angew. Chem., Int. Ed., 2001, 40, 2989 CrossRef CAS; (b) X. W. Wei, A. G. Avent, O. V. Boltalina, A. D. Darwish, P. W. Fowler, J. P. B. Sandall, J. M. Street and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 2002, 41 RSC; (c) A. D. Darwish, I. V. Kuvytchko, X. W. Wei, O. V. Boltalina, I. V. Gol′dt, J. M. Street and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 2002, 1118 RSC.
- J. L. Segura and N. Martin, Angew. Chem., Int. Ed., 2001, 40, 1372 CrossRef CAS.
- A. D. Darwish, A. G. Avent, O. V. Boltalina, I. Gol’dt, I. Kuvytchko, T. da Ros and R. Taylor, unpublished work.
- (a) S. G. Liu, I. Perez, N. Martin and L. Echegoyen, J. Org. Chem., 2000, 65, 9092 CrossRef CAS; (b) M. A. Herranz, S. Gonzalez, I. Perez and N. Martin, Tetrahedron, 2001, 57, 725 CrossRef CAS; (c) G. Kodis, P. A. Liddell, L. de la Garza, A. L. Moore, T. A. Moore and D. Gust, J. Mater. Chem., 2002, 12, 2100 RSC; (d) A. J. Moore and M. R. Bryce, J. Chem. Soc., Perkin Trans. 1, 1991, 157 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: fluorescence spectra of 1 and 6 and UV-vis absorption spectra of 1, 2 and 6. See http://www.rsc.org/suppdata/cc/b2/b209724a/ |
| This journal is © The Royal Society of Chemistry 2003 |