Chirality transfer during alkylation of chiral amides
Takeo
Kawabata
*,
Orhan
Öztürk
,
Jianyong
Chen
and
Kaoru
Fuji
Institute for Chemical Research, Kyoto University, Uji, Kyoto-, 61-0011, Japan. E-mail: kawabata@scl.kyoto-u.ac.jp
First published on 6th December 2002
Abstract
Chiral amides derived from O-methyl mandelic acid and achiral amines underwent enantioselective α-methylation on treatment with LTMP followed by addition of methyl iodide; chirality transfer from an undeprotonated chiral amide into an achiral enolate in a mixed aggregate is supposed to be responsible for the asymmetric induction.
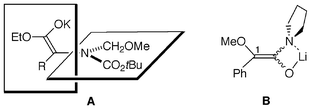
Asymmetric synthesis has been extremely developed during the last few decades and it is a mature area of science.1 Development of a conceptually novel method for asymmetric induction, however, is still of great importance. Seebach and Wasmuth have reported a pioneering work for enantioselective α-alkylation of an aspartic acid derivative, and proposed a mechanism involving a mixed aggregate of enolates.2 We have reported a novel method for enantioselective α-alkylation of α-amino acid derivatives which proceeds via chiral nonracemic enolates (A) with dynamic axial chirality.3–5 In the course of further study on asymmetric synthesis via enolate intermediates, we found an unprecedented asymmetric induction in alkylation of chiral amides derived from (S)-O-methyl mandelic acid and achiral amines (Scheme 1). We describe here the preliminary results and a possible mechanism for the asymmetric induction.
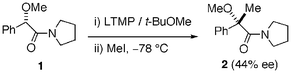 | ||
| Scheme 1 | ||
Amide 1 was readily prepared by condensation of (S)-O-methyl mandelic acid and pyrrolidine in the presence of 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide in 82% yield. Treatment of 1 with 1.1 equivalents of lithium 2,2,6,6-tetramethylpiperidide (LTMP) in tert-butyl methyl ether6 at −78 °C followed by addition of methyl iodide gave nonracemic product 2 (44% ee) in 34% yield with 54% recovery of 1. It was surprising for us that asymmetric induction appears to occur via enolate intermediate B that does not possess any elements of chirality.7
To investigate the structural requirements of substrates to cause asymmetric induction, several amides were prepared from (S)-O-methyl mandelic acid and achiral amines, and their α-methylation was examined (Table 1). Piperidine amide 3 underwent α-methylation in 41% ee by the same treatment as that for 1 (entry 1). N,N-Dimethylamide 5, N,N-diethylamide 7, and N,N-dibutylamide 11 gave α-methylated products in 44, 37 and 56% ee, respectively, by the similar treatment (entries 2, 3, and 5). N,N-Dibenzylamide 9 showed exceptionally low enantioselectivity (9% ee) on α-methylation (entry 4). The enantioselectivity of α-methylation was found to be highly solvent-dependent.6 While use of THF as a solvent resulted in the formation of racemic α-methylated product 12 (entry 7), use of cyclopentyl methyl ether8 (CPME) led to the highest asymmetric induction (64% ee, entry 8). Secondary amide 15 also underwent enantioselective α-methylation, albeit with low selectivity (14% ee, entry 12). Thus, any amides derived from (S)-O-methyl mandelic acid and achiral amines could undergo enantioselective α-methylation when treated with LTMP in tert-butyl methyl ether or CPME at −78 °C. N,N-Dibutylamide 11 and N-butyl-N-methylamide 13 showed the maximum asymmetric induction among the amides (entries 8 and 10).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Solvent | Product | Yieldb (%) | Eec (%) | Abs. confg. |
| a Typical procedure: n-BuLi (1.49 M in hexane, 0.37 mL, 0.55 mmol) was added to a solution of TMP (101 μL, 0.60 mmol) in 3.5 mL of dry t-butyl methyl ether at 0 °C, and the mixture was stirred for 10 min. After cooling to −78 °C, a solution of a substrate (0.5 mmol) in 1.5 mL of tert-butyl methyl ether was added dropwise. After stirring for 10 min, methyl iodide (0.31 mL, 5.0 mmol) was added and the resulting mixture was stirred at −78 °C for 20 h. b Numbers in parentheses indicate yields based on the recovered substrate. c Determined by HPLC analysis with chiral stationary phases: 2: Chiralpak AD, 2% i-PrOH–hexane; 4: Chiralcel OD, 1% i-PrOH–hexane; 6: Chiralpak AD, 1% i-PrOH–hexane; 8: Chiralpak AD, 1% i-PrOH–hexane; 10: Chiralpak AD, 5% i-PrOH–hexane; 12: Chiralcel OJ-R, 70% MeOH–H2O; 14: Chiralcel OJ-R, 70% MeOH–H2O; 16: Chiralpak AD, 2% i-PrOH–hexane. d Not determined. e 2.2 Mol equivalents of LTMP were used. f Cyclopentyl methyl ether. g Run in the presence of TMEDA (5.0 equiv.). h 2.2 Mol equivalents of LTMP were used. | ||||||||
| 1 | 3 | –(CH2)5– | t-BuOMe | 4 | 28 (82) | 41 | S | |
| 2 | 5 | Me | Me | t-BuOMe | 6 | 27 (68) | 44 | S |
| 3 | 7 | Et | Et | t-BuOMe | 8 | 31 (74) | 37 | d |
| 4 | 9 | CH2Ph | CH2Ph | t-BuOMe | 10 | 22 (50) | 9 | d |
| 5 | 11 | n-Bu | n-Bu | t-BuOMe | 12 | 25 (71) | 56 | S |
| 6e | 11 | n-Bu | n-Bu | t-BuOMe | 12 | 25 (44) | 49 | S |
| 7 | 11 | n-Bu | n-Bu | THF | 12 | 72 (95) | ∼0 | — |
| 8 | 11 | n-Bu | n-Bu | CPMEf | 12 | 30 (90) | 64 | S |
| 9g | 11 | n-Bu | n-Bu | CPMEf | 12 | 49 (79) | 33 | S |
| 10 | 13 | n-Bu | Me | CPMEf | 14 | 20 (34) | 62 | S |
| 11 | 13 | n-Bu | Me | THF | 14 | 20 (35) | ∼0 | — |
| 12h | 15 | t-Bu | H | t-BuOMe | 16 | 17 (39) | 14 | d |
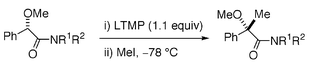
Chemical yields of the α-methylation were always low due to the unavoidable recovery (24–67%) of starting materials. Treatment of 11 with 1.1 equivalents of LTMP in tert-butyl methyl ether for 10 min followed by addition of CD3OD gave quantitative recovery of the substrate of 63% ee containing 35% deuterium. This suggests incomplete formation of the enolate under these conditions, provided that internal proton return9 is not significant. Because loss of the enantiomeric purity (37%) of recovered 11 almost corresponds to the degree of enolate formation (∼35% based on the deuterium contents), ee of recovered 11 may be a measure of the enolate formation. In entries 5, 8 and 9 of Table 1, ee’s of the recovered 11 were 86, 89 and 90%, respectively, which indicates insufficient enolate formation with 1.1 equivalents of LTMP. Use of 2.2 equivalents of LTMP resulted in the improvement of enolate formation, which is indicated by the ee (29%) of recovered 11, however, it did not improve the yield of α-methylation (entry 6).10
The absolute configuration of 2 was determined to be S by comparison of the optical rotation between 2 obtained by the present reaction and (S)-2 independently prepared from (S)-O-methyl atrolactic acid11 and pyrrolidine. The absolute configuration of 4, 6, 12, and 14 was also determined to be S by a similar manner. Thus, the stereochemical course of the α-methylation was retention in each case.
In order to investigate the mechanism of the present asymmetric induction, a crossover experiment between 3 and 11 was done. Treatment of a 1∶1 mixture of rac-3 and 11 (>99% ee) with LTMP (1.1 equivalents of the total amount of 3 and 11) in tert-butyl methyl ether at −78 °C followed by addition of methyl iodide afforded optically active4 (34% ee, 26% yield with 67% recovery) and 12 (49% ee, 25% yield with 59% recovery). Intermolecular chirality transfer was observed during their alkylation. These results strongly indicate that chirality transfer in a mixed aggregate consisting of an achiral enolate with a chiral undeprotonated starting material (C) is responsible for the asymmetric induction. Lower enantioselectivity observed in α-methylation in THF (Table 1, entries 7 and 11, ref. 6) or by addition of TMEDA (entries 8 vs. 9) is consistent with the proposed mechanism because formation of the mixed aggregate is unfavorable under these conditions. An enantiomerically enriched product yielded during the reaction (such as 12) was considered as another possible chiral ligand in the mixed aggregate. However, this seems unlikely because the enantioselectivity of alkylation of 11 did not depend on its conversion. Treatment of 11 with 1.1 equivalents of LTMP in CPME at −78 °C for 10 min followed by methyl iodide only for 30 min gave 12 of 67% ee in 7% yield (cf. 12 of 64% ee in 25% yield obtained by 20 h-treatment with methyl iodide, entry 8) and recovered starting material of 68% ee in 72% yield.
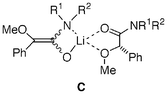
In conclusion, unprecedented asymmetric induction was found in α-methylation of chiral amides derived from optically active O-methyl mandelic acid and achiral cyclic, secondary, and primary amines. Chirality transfer by a mixed aggregate mechanism was assumed to be the origin of the asymmetric induction. Further investigation on the generality of the present asymmetric induction is currently underway.
We thank Zeon Corporation (Tokyo, Japan) for a generous gift of CPME.
Notes and references
- B. Stibbs, Can. Chem. News, 2002, 54, 26 Search PubMed; A. Pfaltz, Chimia, 2001, 55, 708 Search PubMed; H. Tye and P. J. Comina, J. Chem. Soc. Perkin Trans. 1, 2001, 1729 RSC.
- D. Seebach and D. Wasmuth, Angew. Chem., Int. Ed. Engl., 1981, 20, 971 CrossRef.
- Chiral properties of A are time- and temperature-dependent. Therefore, we denote this type of chirality ‘dynamic chirality’. The half-life to racemization of A is 22 h at −78 °C, see: T. Kawabata, H. Suzuki, Y. Nagae and K. Fuji, Angew. Chem., Int. Ed., 2000, 39, 2155 Search PubMed; T. Kawabata, J. Chen, H. Suzuki, Y. Nagae, T. Kinoshita, S. Chancharunee and K. Fuji, Org. Lett., 2000, 2, 3883 CrossRef CAS; T. Kawabata, S. Kawakami and K. Fuji, Tetrahedron Lett., 2002, 43, 1465 CrossRef CAS.
- For the related examples of asymmetric induction with enolates possessing axial chirality of a dynamic nature, see: B. Beagley, M. J. Betts, R. G. Pritchard, A. Schofield, R. J. Stoodley and S. Vohra, J. Chem. Soc., Chem. Commun., 1991, 924 Search PubMed; T. Gees, W. B. Schweizer and D. Seebach, Helv. Chim. Acta, 1993, 76, 2640 RSC; M. J. Betts, R. G. Pritchard, A. Schofield, R. J. Stoodley and S. Vohra, J. Chem. Soc., Perkin Trans. 1, 1999, 1067 CrossRef CAS.
- For asymmetric synthesis based on axial chirality of non-biaryl atropisomers, see: J. Clayden, Angew. Chem., Int. Ed. Engl., 1997, 36, 949 Search PubMed.
- Solvent effects on α-methylation of 1 are significant: use of THF or Et2O led to the production of 2 in 5% ee or 37% ee, respectively.
- A planar-chiral structure of enolate B is possible based on the restricted rotation of the C(1)–OMe bond. However, the barrier of the bond rotation is assumed to be too small to retain chirality even at −78 °C. For an example of the barrier of the related bond rotation, see: M. Nonella, C. Boullais, C. Mioskowski, E. Nabedryk and J. Breton, J. Phys. Chem. B, 1999, 103, 6363 Search PubMed.
- CPME is available from Zeon Corporation.
- E. Vedejs and L. Namkyu, J. Am. Chem. Soc., 1995, 117, 891 CrossRef CAS; C. Fehr, Angew. Chem., Int. Ed. Engl., 1996, 35, 2566 CrossRef.
- Several attempts at improving the yield of α-methylation were unsuccessful: e.g. prolonged enolate-formation (30–120 min), higher temperature (−60 °C) for enolate formation and/or methylation.
- E. L. Eliel, J. K. Koskimies and B. Lohri, J. Am. Chem. Soc., 1978, 100, 1614 CrossRef CAS; T. Kawabata, K. Yahiro and K. Fuji, J. Am. Chem. Soc., 1991, 113, 9694 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |