Highly efficient chiral metal cluster systems derived from Ru3(CO)12 and chiral diiminodiphosphines for the asymmetric transfer hydrogenation of ketones
Hui
Zhang
a,
Chuan-Bo
Yang
a,
Yan-Yun
Li
a,
Zhen-Rong
Donga
a,
Jing-Xing
Gao
*a,
Hideaki
Nakamura
b,
Kunihiko
Murata
b and
Takao
Ikariya
*b
aDepartment of Chemistry, Institute of Physical Chemistry, State Key Laboratory for Physical Chemistry of Solid Surfaces, Xiamen University, Xiamen, 361005, Fujian, China
bGraduate School of Science and Engineering, Tokyo Institute of Technology & CREST, 2-12-1 O-okayama, Meguro-ku, Tokyo, 152-8552, Japan. E-mail: tikariya@o.cc.titech.ac.jp
First published on 2nd December 2002
Abstract
The chiral Ru cluster-based catalyst systems generated in situ from Ru3(CO)12 and chiral diiminodiphosphine tetradentate ligands effected asymmetric transfer hydrogenation of propiophenone in 2-propanol, leading to 1-phenyl-1-propanol in 94% yield and with 96% ee.
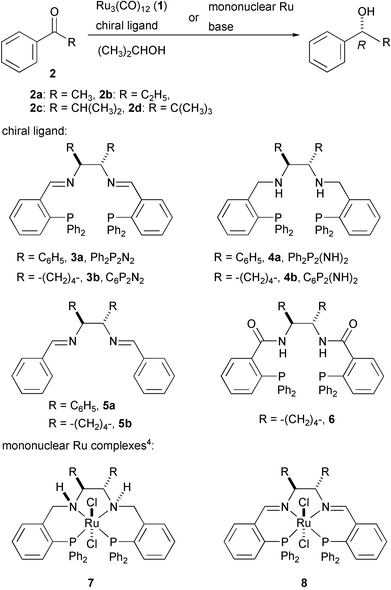
Enantioselective reduction of carbonyl compounds catalysed by well-defined transition metal complexes has been extensively studied as a practical synthetic tool to produce optically active alcohols during the last decade.1 In particular, asymmetric transfer hydrogenation with 2-propanol or formic acid catalysed by chiral molecular catalysts including lanthanide metal or group 8–10 metal complexes has been developed as a synthetic method complementary to asymmetric hydrogenation.2 Although an excellent catalyst performance in terms of reactivity and selectivity has been attained in the transfer hydrogenation of ketones by the mononuclear chiral metal complexes,2–4 neither efficient chiral metal clusters nor cluster-based chiral catalyst systems which give good results for the reaction have been reported.5 Herein we describe the first successful example of the enantioselective transfer hydrogenation of aromatic ketones catalysed by a cluster-based catalyst system generated in situ from commercially available Ru3(CO)12 (1) and chiral tetradentate diiminodiphosphine ligands.4
We first examined the effect of several kinds of chiral ligands combined with the trinuclear ruthenium carbonyl cluster 1 on the rate of reduction of aromatic ketones 2 with 2-propanol at 45 °C as shown in Scheme 1. In the absence of any added auxiliaries, the carbonyl cluster 1 was almost inert to the reduction of the ketones. However, the screening tests revealed that the asymmetric reduction of acetophenone 2a (substrate/Ru3 molar ratio of 200∶1) in 2-propanol containing a combined catalyst system generated from the reaction of 1 with the chiral tetradentate ligand N,N′-bis[o-(diphenylphosphino)benzylidene]-1,2-diphenylethylenediamine, Ph2P2N2 (3a), proceeded smoothly to give the corresponding optically active alcohol in 91% yield and with 81% ee. Addition of a strong base, K[OCH(CH3)2] (Ru3∶base = 1∶10) led to a slight improvement in the reactivity without any serious decrease in the enantioselectivity. Table 1 lists some experimental results. The Ph2P2N2-based complex provides better catalyst performance in terms of the reactivity and selectivity than those attained with the catalyst bearing N,N′-bis[o-(diphenylphosphino)benzylidene]cyclohexane-1,2-diamine, C6P2N2 (3b). Most notably, as the bulkiness of the alkyl substituents increases, the ee increases in the substituent order of CH3 < C2H5 < CH(CH3)2. The reaction of the more congested isobutyrophenone (2c) which is inert with mononuclear Ru catalyst systems,2–4 gave the corresponding alcohols in good to excellent yields and with up to 99% ee.
 | ||
| Scheme 1 | ||
| Ketone | Chiral catalyst | Base | Time/h | Yield (%) | Ee (%) |
|---|---|---|---|---|---|
| The reaction was carried out at 40 °C using a 0.1 mol dm–3 solution (5.0 mmol) in 2-propanol. Ketone∶Ru3 molar ratio of 200∶1. Added base: Ru3∶base = 1∶10. Yield was determined by GLC and NMR. The enantiomeric excess was determined by GLC using a chiral column (Chrompack CP-cyclodextrin-236-M-19 column). | |||||
| 2a | Ru3(CO)12/3a | No | 5 | 91 | 81 |
| 2a | Ru3(CO)12/3b | No | 5 | 11 | 83 |
| 2a | Ru3(CO)12/3a | Yes | 4 | 96 | 82 |
| 2b | Ru3(CO)12/3b | No | 5 | 94 | 95 |
| 2b | Ru3(CO)12/3a | Yes | 4 | 98 | 96 |
| 2b | Ru3(CO)12/3b | No | 5 | 72 | 92 |
| 2b | Ru3(CO)12/3b | Yes | 4 | 93 | 92 |
| 2c | Ru3(CO)12/3a | No | 5 | 48 | >99 |
| 2c | Ru3(CO)12/3a | Yes | 4 | 79 | >99 |
| 2c | Ru3(CO)12/3b | No | 5 | 30 | 94 |
| 2c | Ru3(CO)12/3b | Yes | 4 | 66 | 90 |
| 2d | Ru3(CO)12/3a | No | 5 | 6 | 79 |
| 2d | Ru3(CO)12/3b | No | 5 | 5 | 58 |
| 2b | Ru3(CO)12/4a | No | 5 | 0 | — |
| 2b | Ru3(CO)12/(S,S)-DPEN | No | 5 | 25 | 59 |
| 2b | Ru3(CO)12/(R,R)-CyDN | No | 5 | 16 | 40 |
| 2b | Ru3(CO)12/BINAP | No | 5 | 0 | — |
| 2b | Ru3(CO)12/DIOP | No | 5 | 0 | — |
| 2b | H4Ru4(CO)12/3a | No | 5 | 0 | — |
In sharp contrast to the diiminodiphosphine ligands, diaminodiphosphines such as N,N′-bis[o-(diphenylphosphino)benzyl]-1,2-diphenylethylenediamine (4a), Ph2P2(NH)2 or N,N′-bis[o-(diphenylphosphino)benzyl]cyclohexane-1,2-diamine (4b), C6P2(NH)24 did not exhibit any acceleration effect on the reaction under the conditions to those described in Table 1. Neither chiral diphosphine ligands, BINAP or DIOP nor chiral diimines, N,N′-bisbenzylidene-1,2-cyclohexanediamine (5b) gave any reduction product at all. The Trost ligand (6)6 combined with 1 did not produce an active catalyst for this transfer hydrogenation. The reaction of 2b with a binary system, 1 and (R,R)-1,2-cyclohexanediamine (CYDN) or (S,S)-1,2-diphenylethylenediamine (DPEN) proceeded very slowly and then stopped leading to the reduction product in low yield and low enantioselectivity. These results indicate that the structure of the PNNP tetradentate diiminodiphosphines is a crucial factor for ligand acceleration.
Our earlier results indicated that the diaminodiphosphine ligands, 4, combined with a mononuclear Ru complex (7) effects the asymmetric transfer hydrogenation of ketones leading to optically active alcohols with an excellent ee,4 while the mononuclear Ru complexes (8) bearing the diiminodiphosphines, 3, were completely inert to the same reaction. The NH moiety of the ligand in the mononuclear metal complex possibly participates in the formation of a six-membered cyclic transition state through hydrogen bonding to accelerate the reaction.2a,3a Contray to the mononuclear systems, in these Ru cluster systems, the diiminodiphosphines 3 were effective for the reduction of ketones with high enantioselectivity, suggesting that the reaction mode with the cluster system may be different from that proposed in the mononuclear Ru catalyst systems.7 Bhaduri and Sharma demonstrated that the transfer hydrogenation of carbonyl compounds with 2-propanol catalysed by a ruthenium carbonyl hydride cluster proceeded via a concerted Meerwein–Ponndorf–Verley (MPV)8a mechanism, not by way of the metal hydride species formed by β-hydride elimination in the metal alkoxide intermediate.2a,8b During the reaction the nuclearity of the catalyst does not change.8b A combined system of Ru4H4(CO)12 and 3a gave no reduction product under the reaction condition (Table 1). Based on the results obtained here as well as reported results,4,8b the hydrogen transfer between ketones and alcohols with these cluster-based catalyst systems possibly proceeds by way of the direct mode like an MPV mechanism. In fact, the more congested ketonic substrates provide better enantioselectivity although lower reactivity as shown in Table 1.
Although the nature of the active catalyst remains unknown, IR and 31P NMR spectroscopies of the catalytic systems provided us with further information about the real catalyst and the reaction mechanism as well as the nuclearity of the catalyst. The 31P NMR spectrum of the red solution obtained from the reaction of 1 and (S,S)-3b in a 1∶1 molar ratio in 2-propanol at 45 °C showed two major singlets around 31.5 and −13 ppm possibly due to the coordinated- and free-ligand, respectively. An addition of 10 equiv. of K[OCH(CH3)2] to the red solution caused an increase in the intensity of the singlet peak at 31.5 ppm and a decrease in the intensity of the free ligand peak, suggesting that the red complex generated in the solution phase could be related to the active species for the reaction. Although attempts to isolate the active catalysts from this reaction mixture failed because of their thermal instability, a relevant cluster complex bearing the chiral ligand, [(C2H5)4N][HRu3(CO)10{(S,S)-C6P2N2}],9 could be obtained as a red complex from the reaction of 1 and (S,S)-C6P2N2 (3b) in 2-propanol containing K[OCH(CH3)2] followed by an addition of [(C2H5)4N]I.† The isolable complex effected the transfer hydrogenation of acetophenone in 2-propanol containing no additional strong base to give (R)-1-phenylethanol in excellent yield albeit with lower enantioselectivity (96% yield, 47% ee, 5.5 h). These results suggest that the trinuclear cluster 1 combined with chiral ligand system most probably exists as the catalytically active species under the catalytic reaction conditions. This idea is possibly supported by the reaction rate, which shows a first order dependence on the concentration of 1 and by the fact that the mononuclear carbonyl complex, Ru(CO)3(PPh3)2 combined with 3b had no reactivity.
This study has demonstrated that chiral metal cluster complexes may offer new and unique opportunities in asymmetric catalysis and will stimulate further study on the development of efficient chiral cluster catalyst systems.
We are grateful to the National Natural Science Foundation of China (20073034), the Fujian Provincial Science and Technology Commission (2002F016), and Xiamen Science and Technology Commission (3502Z, 20021044) for financial support, and to Professor Ryoji Noyori (Nagoya University) and Professor Khirui Tsai (Xiamen University) for their valuable assistance.
Notes and references
- (a) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994 Search PubMed; (b) Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley, New York, 2nd edn., 2000 Search PubMed.
- (a) Recent reviews for asymmetric transfer hydrogenation: R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 Search PubMed; (b) M. J. Palmer and M. Wills, Tetrahedron: Asymmetry, 1999, 10, 2045 CrossRef CAS.
- (a) S. Hashiguchi, A. Fuji, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562 CrossRef CAS; (b) K. Murata, T. Ikariya and R. Noyori, J. Org. Chem., 1999, 64, 2186 CrossRef CAS; (c) M. Watanabe, K. Murata and T. Ikariya, J. Org. Chem., 2002, 67, 1712 CrossRef CAS.
- J.-X. Gao, T. Ikariya and R. Noyori, Organometallics, 1996, 15, 1087 CrossRef CAS.
- (a) G. Sœss-Fink and M. Jahncke, in Catalysis by Di- and Polynuclear Metal Cluster Complexes, ed. R. Adams and F. A. Cotton, Wiley-VCH, New York, 1998 Search PubMed, and references therein; (b) G. Gervasio, R. Gobetto, P. J. King, D. Marabello and E. Sappa, Polyhedron, 1998, 17, 2937 CrossRef CAS; (c) F. Prestopino, R. Persson, M. Monare, N. Focci and E. Nordlander, Inorg. Chem. Commun., 1998, 1, 302 CrossRef CAS; (d) S. Bhadouri, V. S. Darshane, K. Sharma and D. Mukesh, J. Chem. Soc., Chem. Commun., 1992, 23, 1738 RSC; (e) U. Mattedi, V. Beghetto and A. Scrivanti, J. Mol. Catal A: Chem., 1996, 109, 45 CrossRef.
- B. M. Trost, D. L. Van Vranken and C. Bingle, J. Am. Chem. Soc., 1992, 114, 9327 CrossRef CAS.
- During the preparation of catalyst from the reaction of 1 and diiminodiphosphine 3a in 2-propanol containing the base, the reduction of 3a to 4a did not occur.
- (a) J. D. Morrison and H. S. Mosher, in Asymmetric Organic Reactions, Prentice-Hall, New Jersey, 1971, ch. 5 Search PubMed; (b) S. Bhaduri and K. Sharma, J. Chem. Soc., Chem. Commun., 1988, 173 RSC.
- See Notes and References.
Footnote |
| † Isolation and identification of catalytically active intermediate complexes: a solution of Ru3(CO)12 (96 mg, 0.15 mmol) in 2-propanol (18 ml) was added a 0.4 M solution of (CH3)2CHOK in 2-propanol (2.25 ml, 0.9 mmol). Then the chiral ligand (S,S)-C6P2N2 (99 mg, 0.15 mmol) was added to the deep red solution. After stirring the reaction mixture at 45 °C for 45 min, a twofold excess of solid [(C2H5)4N]I (77 mg, 0.3 mmol) and water were added to effect precipitation of a brown-red solid, which was collected by filtration, and washed with water, and then with Et2O. Recrystallisation from CH2Cl2–Et2O gave brick-red crystals (117 mg, 57% yield). The crude compound was further purified by silica gel column chromatography with acetone as eluent to give an orange crystalline complex. IR (KBr) (νCO in the region 1900–2100 cm−1): 2077vw, 2015s, 1974vs, 1949s and 1911w cm−1; 31P NMR (CDCl3): δ +31.47. Anal. Calc. for [(C2H5)4N][HRu3(CO)9{(S,S)-C6P2N2}]·2H2O, C61H61N3O9P2Ru3·2H2O: C, 53.05; H, 4.75; N, 3.04. Found: C, 52.66; H, 4.15; N, 3.24%. |
| This journal is © The Royal Society of Chemistry 2003 |