Layer-by-layer electrodeposition of redox polymers and enzymes on screen-printed carbon electrodes for the preparation of reagentless biosensors
Qiang
Gao
and
Xiurong
Yang
*
State Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin, 130022, China. E-mail: xryang@ns.ciac.jl.cn
First published on 18th November 2003
Abstract
Layer-by-layer electrodeposition of redox polymer/enzyme composition films on screen-printed carbon electrodes for fabrication of reagentless enzyme biosensors has been proposed and the resulting films were found to be very stable and rigid.
Enzyme immobilization on the sensing electrode surface is one of the most important points to consider in biosensor design. The immobilization method, which could control the amount and spatial distribution of the biocatalyst, was very promising.1 A layer-by-layer thin film fabrication technique has been introduced to produce complex multilayer thin films with molecular-level thickness control.2,3 Enzyme ‘wiring’ redox polymers, a fruitful strategy for the fabrication of biosensors, have been extensively employed in the preparation of biosensors for assaying substrates of the ‘wired’ enzyme.4,5 Recently, Gao et al.6 proposed a new method for the fabrication of biosensors by electrodeposition of redox polymers and co-electrodeposition of enzymes by coordinative crosslinking on a carbon electrode. The process involves the exchange of labile, inner-sphere chloride ligands of Os2+/3+ complexes of one chain with more strongly coordinated nitrogen ligands of a second chain. Amine-containing biomolecules were similarly incorporated into the electrodeposited redox polymer films as amines can also replace the inner sphere chlorides of the Os2+ complexes. As for most macromolecules, however, the formation and precipitation of the electrostatic adducts will occur when the positive charge of the redox polymer is about balanced by the negative charge of the biomolecule. It will adversely affect the electronic conductivity of the redox polymer films and limit the useful range of redox polymer/biomolecule ratio in the solution.7
Multilayer self-deposition, especially built up on electrostatic interactions, has offered an alternative approach for the immobilization of biomolecules.8 Further covalent stabilization of the resultant multilayer films, however, was desired not only to physically strengthen the film structure but also to eliminate potential desorption due to changes in pH or ionic strength.9 Here, we attempted to develop a glucose biosensor by layer-by-layer electrodeposition of redox polymer and glucose oxidase (GOD) using a screen-printed carbon electrode. The redox polymer, a water-soluble poly(vinylimidazole) complexed with osmium(4,4′-dimethyl-2,2′-bipyridine) chloride denoted PVI-Os, was prepared by a reported procedure.10Scheme 1 shows the structure of PVI-Os. The redox polymer films containing enzyme were made in two steps. First, the screen-printed carbon electrode was immersed in 10 mg ml−1 GOD in phosphate buffer (PB) (20 mM, pH 7.1) for 20 min to adsorb the first layer of enzyme. It was then removed and washed with PB. Then, the redox polymer film was electrodeposited from redox polymer solutions (0.65 mg mL−1) in PB (pH 7.1, 20 mM) by applying for 200 s a steady reducing potential (−1.4 V vs. Ag/AgCl). Such a two-step procedure was used to assemble the GOD/redox polymer films on screen-printed carbon electrodes with the desired number of layers. All resulting electrodes were washed with phosphate buffer (pH 7.1) and stored in a refrigerator (4 °C) when not in use.
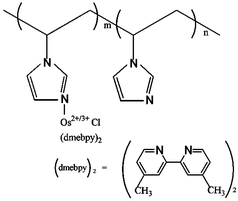 | ||
| Scheme 1 The structure of redox polymer PVI-Os. | ||
GOD has an isoelectric point of 4.2; thus, when pH > 4.2, the enzyme possesses a net negative charge, permitting their adsorption to the cationic redox polymer films. After each electrodeposition cycle creating a new GOD/redox polymer composition film, CVs were scanned in PB (pH 7.1, 20 mM) containing 0.15 M NaCl. Pairs of well-defined, chemically reversible CV peaks were observed centered about 130 mV (vs. Ag/AgCl), as shown in Fig. 1 and inset, characteristic of the Os2+/3+ redox couple; at 5 mV s−1 scan rate, the separation of the peaks was less than 20 mV for the enzyme electrodes as in the case of the pure redox polymer films, which means the redox polymer–enzyme films also have rapidly electron-exchanging redox couples. Reduction and oxidation peak currents grew with increasing number of electrodeposition composition films from one (a) to four (d) layers. For further electrochemical measurements, the electrode was then transferred into PB (pH 7.1, 20 mM, 0.15 M NaCl) containing 10 mM glucose. All the cyclic voltammograms, as shown in Fig. 1, registered from film modified electrodes were typical of enzyme-catalyzed and -mediated voltammograms, and the oxidation currents were significantly enhanced.
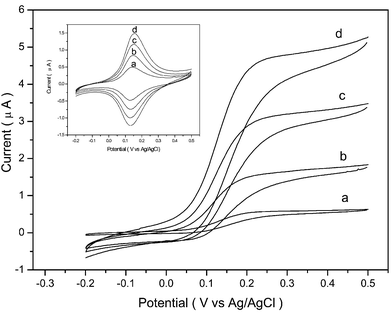 | ||
| Fig. 1 CVs of the film-modified screen-printed carbon electrodes as a function of the number of films: a) one; b) two; c) three; d) four layers in the presence or absence (inset) of 10 mM glucose. Electrolyte: 20 mM PB (pH 7.1) containing 0.15 M NaCl. Scan rate: 5 mV s−1. | ||
The effect of the number of GOD/redox polymer films on the electrocatalytic current was investigated by control experiments. The response of the biosensor with one to five layers of enzyme/redox polymer films to glucose was investigated. The anodic currents sampled at 200 mV (vs. Ag/AgCl) for 10 mM glucose with one to four layers of composite films were increased from 0.6 µA to 4.7 µA (Fig. 1a–d). This shows that the electrocatalytic current was enhanced with increasing number of layers, but four layers can make the electrocatalytic current reach a plateau value. So with five layers of enzyme, the electrocatalytic current to glucose was examined.
The relationship between peak current and scan rate was found to be linear with υ1/2 up to 100 mV s−1, which indicates that the charge-transport process within the film was diffusion-controlled. Further successive potential scanning at 100 mV s−1 for 1 h caused a decrease in current response of less than 5%, which means that the electroactive components were not free to diffuse away from the electrode surface. In terms of the separation of peak potential, comparison of the voltammogram with that of an electrode coating with pure redox polymer film showed no measurable difference at a slow scan rate. However, it increased with increasing scan rate, which indicated that incorporation of the enzyme polyanions in the redox polymer increased the density of the ionic bridges, reduced the mobility of the redox polymer's segments, reduced the segmental collisions leading to electron exchange between reduced and oxidized centers of the redox polymer, and as a result, reduced the conduction of the electrons in the films.11
Effect of dioxygen on the catalytic current was investigated by comparison of the glucose response under N2 and under air. Upon bubbling of nitrogen through the solution, the sensitive increased only by about 6%, showing that electron transport from the redox centers of the enzyme to those of the redox polymer was fast relative to electron transport to O2, the natural cosubstrate of GOD. The response of the present polymer/enzyme electrode, however, was still dependent on the oxygen concentration.
The flow-injection response of the multilayer PVI-Os/GOD modified screen-printed carbon electrode for glucose solutions of increasing concentration from 0.3 to 15 mM was investigated using a detection potential of 0.2 V (vs. Ag/AgCl). The present modified electrodes possessed a dynamic range of 0.3 to 10 mM at a flow rate of 0.8 ml min−1. The resulting calibration plot had a slope of 0.14 µA mM−1 and correlation coefficient, 0.997. A detection limit of 1 × 10−4 M could be estimated on the basis of the signal-to-noise (S/N = 3). In all cases the response was rapid and reproducible. The electrodes exhibited a very stable response during hours of continuous flow injection.
The effect of interference was determined by adding to a 10 mM glucose solution sequentially acetaminophen (to 0.2 mM), uric acid (to 0.4 mM), and then ascorbic acid (to 0.1 mM) and measuring at each step the change in current. There was little or no interference by acetaminophen and uric acid. In contrast, a large current response was observed for ascorbic acid. Thus, a preseparation or preoxidation of ascorbic acid12 was required for application of the biosensor to real sample analysis. The storage stability of the enzyme electrodes was tested over two weeks. When the enzyme electrodes were stored in the refrigerator at 4 °C, less than a 5% decrease in the response to 5 mM glucose was found over this period.
A layer-by-layer electrodeposition technique has been proposed to prepare glucose biosensors on screen-printed carbon electrodes. Redox hydrogen (PVI-Os) was introduced for dual purposes: the binding unit for the multilayered biocomposite film and the immobilizing matrix of the electron-transferring mediators for the reagentless biosensor. The resulting multilayer enzyme electrode responds rapidly to low glucose concentration, and increasing the number of GOD layers can improve the sensitivity of the biosensor. This method offers an alternative approach to immobilize biomolecules onto the surface of screen-printed carbon electrodes.
This work was supported by the National Natural Science Foundation of China (No. 20075027) and National Key Basic Research Development Project “Research on Human Major Disease Proteomics” (No. 2001CB5102).
Notes and references
- C. Bourdillon, C. Demaille, J. Moiroux and J. M. Saveant, Acc. Chem. Res., 1996, 29, 529–535 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232–1234 CrossRef CAS.
- J. Anzai, H. Takeshita, Y. Kobayashi, T. Osa and T. Hoshi, Anal. Chem., 1998, 70, 811–817 CrossRef CAS.
- Y. Degani and A. Heller, J. Am. Chem. Soc., 1998, 111, 2357–2358.
- C. N. Campbell, D. Gal, N. Cristler, C. Banditrat and A. Heller, Anal. Chem., 2002, 74, 158–162 CrossRef CAS.
- Z. Q. Gao, G. Binyamin, H. H. Kim, S. C. Barton, Y. C. Zhang and A. Heller, Angew. Chem., Int. Ed., 2002, 41, 810–813 CrossRef CAS.
- M. Dequaire and A. Heller, Anal. Chem., 2002, 74, 4370–4377 CrossRef CAS.
- K. Sirkar, A. Revzin and M. V. Pishko, Anal. Chem., 2000, 72, 2930–2936 CrossRef CAS.
- J. G. Franchina, W. M. Lackowski, D. L. Dermody, R. M. Crooks, D. E. Bergbreiter, K. Sirkar, R. J. Russell and M. V. Pishko, Anal. Chem., 1999, 71, 3133–3139 CrossRef CAS.
- T. J. Ohara, R. Rajagopalan and A. Heller, Anal. Chem., 1994, 66, 2451–2457 CrossRef CAS.
- T. de Lumley-woodyear, P. Rocca, J. Lindsay, Y. Dror, A. Freeman and A. Heller, Anal. Chem., 1995, 67, 1332–1338 CrossRef CAS.
- R. Maidan and A. Heller, J. Am. Chem. Soc., 1991, 113, 9003–9004 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |