Electron-transporting materials for organic electroluminescent and electrophosphorescent devices
Gregory
Hughes
and
Martin R.
Bryce
*
Department of Chemistry and Centre for Molecular and Nanoscale Electronics, University of Durham, Durham, UK DH1 3LE. E-mail: m.r.bryce@durham.ac.uk
First published on 18th November 2004
Abstract
One of the requirements for efficient organic electroluminescent devices (OLEDs) is balanced charge injection from the two electrodes and efficient transport of both holes and electrons within the luminescent layer in the device structure. Many of the common luminescent conjugated polymers, e.g. derivatives of poly(phenylenevinylene) and poly(fluorene), are predominantly hole transporters (i.e. p-dopable). This article gives a brief overview of organic electroluminescence and electrophosphorescence and provides a more detailed consideration of ways in which electron transport in these systems has been enhanced by the incorporation of electron-deficient (i.e. n-dopable) small molecules and polymers into the devices, either as blends or by covalent attachment of sub-units to the luminophore or as an additional electron-transporting, hole-blocking (ETHB) layer adjacent to the cathode. The chemical structures of these systems are presented and their roles are assessed. Most of these ETHB molecules are electron-deficient aromatic nitrogen-containing heterocycles, e.g. derivatives of 1,3,4-oxadiazole, pyridine, pyrimidine, pyrazine, quinoline, etc. Non-aromatic thiophene-S,S-dioxide derivatives are also discussed. The article is written from an organic chemist's perspective.
 Gregory Hughes | Dr Gregory Hughes was born in Newcastle upon Tyne, UK. He obtained a BSc from Northumbria University. He has recently completed his PhD under Martin Bryce's supervision at the University of Durham on the synthesis of new electron transport materials for OLED applications. |
 Martin R. Bryce | Martin Bryce has been at the University of Durham since 1981 and became a full Professor in 1995. He was the Scientific Editor of J. Mater. Chem. from 1994–1999. His research interests embrace the synthesis of functional organic materials for optoelectronic device applications; nanoscale and supramolecular chemistry; and new synthetic methodology, especially applied to heterocycles. |
1. Introduction and background
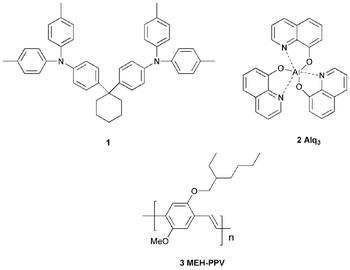
Electroluminescence (EL) from an organic material was first reported in 1963 by applying ca. 400 V across single crystals of anthracene.1 In 1982, vacuum-deposited thin films of anthracene were shown to exhibit blue EL when subjected to lower drive voltages, e.g. 30 V.2 However, the quantum efficiencies (defined as photons emitted/electrons injected) and the lifetimes of these devices were considerably lower than those based on inorganic systems which continued to be the focus of attention. It was the seminal work of Tang and VanSlyke with bilayers of low molecular weight organic molecules, reported in 1987, which initiated worldwide interest in the development of organic light-emitting devices (OLEDs).3 The device structure was conceptually simple, comprising evaporated thin layers of organic compounds, which were chosen for their ability to act as fluorophores and to transport charge carriers, sandwiched between two electrodes one of which was transparent. Green EL was observed with devices comprising the bis(triarylamine) 1 as the hole-transporting layer and tris(8-hydroxyquinoline)aluminium (Alq3) 2 as the emitter and electron-transporting layer in the configuration ITO/1/2/Mg:Ag. A brightness of >1000 cd m−2 at an operating voltage of <10 V was achieved.The next fundamental breakthrough was reported by Friend et al. in 1990, who demonstrated that the highly fluorescent conjugated polymer poly(p-phenylenevinylene) (PPV) could serve as the active material emitting yellow–green light in a single-layer OLED of configuration ITO/PPV/Al.4 The introduction of substituents into the PPV skeleton provides derivatives which are soluble in organic solvents, and in 1991 Braun and Heeger reported a red–orange emitting OLED based on poly[2-methoxy-5-(2-ethylhexyloxy)-p-phenylenevinylene] (MEH-PPV) 3 in the configuration ITO/MEH-PPV/Ca with an external quantum efficiency of 1%.5 These discoveries have led to intense worldwide interest in new materials for incorporation into OLEDs for practical applications, notably full-colour flat-panel displays. The world market for display devices using OLEDs is expected to grow to >$2500 m by 2007.6
The study of organic electroluminescence is a multi-disciplinary field which crosses the traditional boundaries of synthetic chemistry, applied and theoretical physics, materials science and device engineering. Many authoritative reviews are available.7 The present article will focus more specifically on electron-transporting materials and their roles in OLEDs, with particular emphasis on work published in the last five years. A review of this topic published in 1998 considered many examples from the earlier literature.8
The need for electron-transporting materials in OLED technology stems from the fact that many widely-used emitters, such as derivatives of poly(phenylenevinylene) and poly(fluorene), are predominantly hole-transporting (p-dopable) materials, i.e. they have low electron affinities. In a device structure this creates an imbalance of electron injection (from the low work-function cathode) and hole injection (from the high work-function anode). Hole injection predominates and charge recombination occurs near the polymer/cathode interface which results in a lowering of EL efficiency due to quenching of excitons by the metal electrode. One solution to this problem has been to use a low work function metal, such as calcium, as the cathode to lower the energy barrier to electron injection. However, an inherent drawback in this strategy is that such metals are highly sensitive to atmospheric degradation. A complementary approach involves synthetic chemistry to obtain electron-deficient (n-type) polymeric or low molecular weight materials suitable for incorporation into the devices. In this regard, aromatic nitrogen-containing heterocycles have proved to be particularly effective as electron-transporting, hole-blocking (ETHB) materials, in some cases also serving as the emitting component. The following strategies have met with success:
• Bilayer or multilayer structures can be assembled with one or more ETHB layers placed on top of the emissive polymer film (by spin-coating or thermal evaporation) before deposition of the cathode. This approach requires more complex fabrication procedures than for single-layer devices.
• Electron-deficient sub-units can be covalently bound to the emissive polymer, either by insertion into the main-chain, as end-capping groups, or as pendant side-groups. The synthesis of these polymers can be very demanding requiring multi-step routes and/or specific cross-coupling or copolymerisation reactions.
• ETHB materials can be blended (doped) into the emissive material prior to deposition. The benefit here is that the manufacture of these devices requires only a single spin-coating process. However, problems can be encountered with phase segregation and crystallisation of the organic components within the emissive layer during device operation, reducing efficiency and lifetimes.
A schematic drawing of a bilayer EL device and an energy level diagram are shown in Figs. 1 and 2. Balanced charge injection requires the anode and cathode work functions to match the HOMO and LUMO levels of the emissive compound and ETHB layers, respectively. Additional hole-transporting (HT) or electron-transporting (ET) layers may be added to provide multilayer devices. For example, copper phthalocyanine or PEDOT:PSS [poly(ethylenedioxythiophene) poly(styrene sulfonate)] are sometimes used as HT layers, (they also serve to planarise the anode) and a thin layer of LiF below the metal cathode can facilitate electron transport into the organic ETHB layer.7f In bilayer devices issues related to the morphology and carrier transport at the organic/organic interface are undoubtedly important, and can have a significant impact on device performance. Jenekhe et al. have discussed these aspects of polymer/polymer heterojunctions in some detail.9 Shirota et al.7d and Adachi et al.10 have also established that exciplex formation takes place at the interface of the ET and HT layers affecting the emission colour and efficiency. A low ionisation potential for the HT material favours exciplex formation leading to red-shifted emission bands.
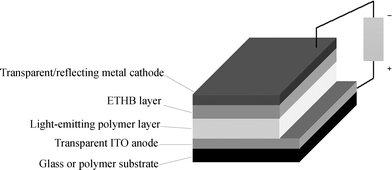 | ||
| Fig. 1 Schematic drawing of a bilayer OLED with an electron-transporting, hole-blocking (ETHB) layer adjacent to the cathode. | ||
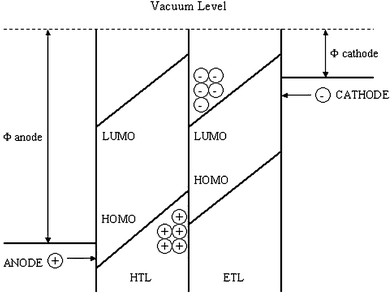 | ||
| Fig. 2 Schematic energy level diagram of a generalised bilayer OLED illustrating the accumulation of charge carriers at the interface of the hole-transport and electron-transport layers. (Redrawn from ref. 7h). | ||
Progress with ETHB materials in OLEDs will now be reviewed with the molecules being classified according to the molecular structure of the electron-deficient moiety. Electrophosphorescence will be considered separately in Section 13.†
2. 1,3,4-Oxadiazoles
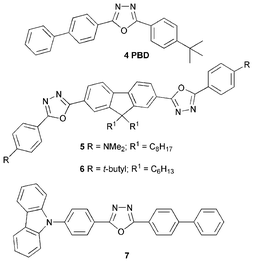
Since the initial studies using 5-(4-biphenyl)-2-(4-tert-butylphenyl)-1,3,4-oxadiazole (PBD) 4,11 2,5-diaryl-1,3,4-oxadiazoles have been widely exploited as ETHB materials in EL devices due to their electron-deficient nature, their high thermal stability and high photoluminescence quantum yield (PLQY). Brown et al.12 demonstrated that PBD was an effective ETHB material as a blend with a film-forming poly(methylmethacrylate) on top of a PPV layer. An 8–10 fold increase in the EL efficiency was achieved compared with a simple PPV device. Zhang et al. reported dramatic increases in quantum efficiences by dispersing PBD into the soluble green emitter poly(2-cholestanoxy-5-thexyldimethysilyl-1,4-phenylenevinylene).13Tang et al.14 reported compound 5 which is a 9,9′-dialkyfluorene derivative containing 1,3,4-oxadiazole and dialkylamine substituents as electron acceptor and electron donor units, respectively. Multilayer blue OLEDs (λmax 490 nm) were fabricated in the configuration ITO/HTL1/HTL2/5/Al2O3/Al (where HTL1 and HTL2 are different triarylamine derivatives). Chien et al.15 studied a related compound possessing a spirobifluorene unit to confer rigidity on the molecular structure. We have reported that OLEDs comprising blends of MEH-PPV and compound 6 in the configuration ITO/PEDOT/MEH-PPV-6/Al have external quantum efficiencies two orders of magnitude higher than that of a pure MEH-PPV device.16 Compound 6 is a blue-emitter. However, no emission from 6 was evident for the OLEDs containing blends of up to 90% of 6 in MEH-PPV, indicating that energy transfer from 6 to MEH-PPV is very efficient. Absorption and PL spectra of thin films of 6, MEH-PPV and a 90% blend are shown in Fig. 3.
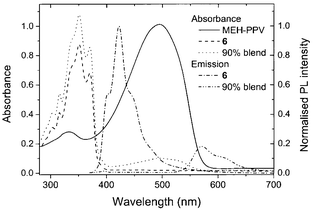 | ||
| Fig. 3 Absorption spectra of 6, MEH-PPV, and a 90 : 10 w/w blend of 6 and MEH-PPV; and PL spectra of 6 and a 90% blend excited at 350 nm. The PL spectra were normalised to the peak intensity of the spectrum of 6 (based on data in ref. 16). | ||
Electron-transporting 1,3,4-oxadiazole and hole-transporting carbazole moieties have been combined in compound 7. Guan et al.17 fabricated a multi-layered device with the configuration ITO/TPD/7/Alq3/Mg0.9Ag0.1/Ag (TPD = tetraphenyldiamine) which exhibited blue emission at λmax 470 nm, with a maximum luminance of 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 cd m−2 at a drive voltage of 15 V and a maximum luminous efficiency of 2.25 lm W−1.
200 cd m−2 at a drive voltage of 15 V and a maximum luminous efficiency of 2.25 lm W−1.
A series of 1,3,4-oxadiazole-pyridine and 1,3,4-oxadiazole-pyrimidine hybrids 8a–h have been studied in bilayer devices in our laboratory. For comparison, vinylene and phenylene analogues were investigated.18,19 In all cases EL emission for the bilayer devices originated from the MEH-PPV layer. Fig. 4 shows light output data for a single-layer MEH-PPV device and three bilayer LEDs with the structure ITO/MEH-PPV/ETHB/Al where the ETHB layer is vacuum-deposited 8a, 8b, or 8d. External quantum efficiency (EQE) values were 0.006%, 0.011%, 0.042% and 0.24%, respectively. The turn-on electric field for EL was lower for the bilayer structures than for the single-layer counterpart. The improved performance of 8d over the two analogues 8a and 8b was ascribed to increased electron deficiency due to the pyridine moiety.
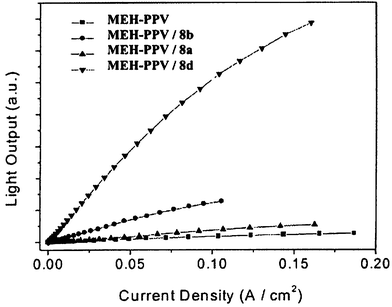 | ||
| Fig. 4 Light output versus current density for the single-layer MEH-PPV and bilayer LEDs of 1,3,4-oxadiazole hybrids 8; ITO anode, Al cathode (reproduced from ref. 18). | ||
For bilayer devices of the general configuration ITO/MEH-PPV(Ru)/8a–h/Al (where Ru is rubrene, blended 20% by weight into MEH-PPV) the addition of a layer of 8a–h increased electron injection, relative to a reference single-layer MEH-PPV(Ru) device,20 although the more electron-deficient pyrimidyl derivative 8h gave an EQE value of 0.12%, which is lower than the pyridyl systems, and comparable to that using 8b. Increases in EQEs were achieved by optimising the thickness of the layers, e.g. EQE 0.7% and brightness ca. 4000 cd m−2 at a current density of ca. 100 mA cm−2 was achieved for ITO/MEH-PPV(Ru)(90 nm)/8d(55 nm)/Al structures.19 Single-layer devices using 20% of the ETHB materials blended into the emissive MEH-PPV layer also showed improved EQEs (especially 8e).21
The 2,5-bis(2-pyridyl)-1,3,4-oxadiazole derivative 9 is also an effective ET material in single, blended-layer devices (EQE 0.26%)22 although higher turn-on voltages are required with 9 than for the pyridyl compounds 8d–g.
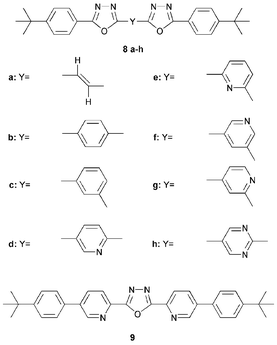
The amorphous films of low molecular weight 1,3,4-oxadiazoles can suffer from a lack of stability since crystallisation occurs with time or increased device operation temperature. More highly-branched, dendritic compounds have the advantage of forming stable amorphous glasses.23 Their non-planar structures should reduce intermolecular interactions in films and hence improve luminescence efficiency. Shirota and coworkers have successfully used tris(1,3,4-oxadiazole) derivative 10 as the ET layer in bilayer devices where the emitting layer is a p-dopable thiophene/oligothiophene system (n = 1–4) end-capped with triarylamine substituents. (Alq32 was also used instead of 10 as an ET layer).24 In this elegant study, the EL colour was tuned from light blue (n = 1) through to orange (n = 4) as the thiophene chain was extended.
Devices using 10 as the ETHB material in combination with various amorphous triphenylamine derivatives as HT materials in the configuration ITO/HTM/10/Mg : Ag emitted light resulting from the exciplex formed at the interface between the two organic layers. The emission colour was tuned from greenish-blue to orange depending on the ionisation potential of the HT material: a lower ionisation potential led to red-shifted emission.25
Cha and co-workers26 have synthesised the branched structures 11a and 11b with extended π-conjugation. Compound 11b has a high glass transition temperature (Tg 211 °C), is soluble in organic solvents, and forms good quality thin films by spin coating. In contrast, its tert-butyl counterpart 11a has poor solubility which led to thin films of inferior quality. An EL device ITO/PEDOT-PSS/11b/Li : Al emitted red light with an EQE of 0.02%. The (phenylethynyl)anthracene core is the only red emitting chromophore in the structure, which indicated that the outer diphenyloxadiazole moieties act only as electron transporters.
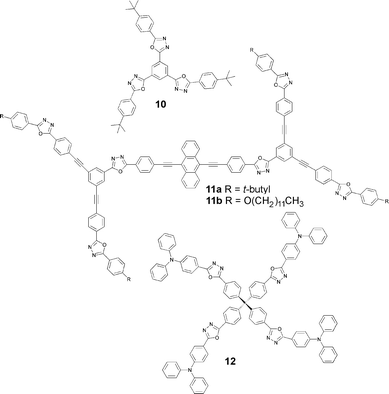
The thermal and EL properties of tetraphenylmethane-based compounds has remained largely unexplored. Yeh et al.27 produced a series of compounds with this core unit, e.g. the amorphous bipolar blue fluorescent compound 12, which has a high Tg of 150 °C. A single-layer device of the structure ITO/12/Ca/Ag showed blue EL at low applied voltage, turning to bluish-white at higher voltages. The device had an efficiency of ca. 0.7 cd A−1 at a current density of ca. 10 mA cm−2 and a maximum brightness of 1690 cd cm−2 under a driving voltage of ca. 14 V. Doping poly(vinylcarbazole) (PVK) with 12 in a 3 : 1 weight ratio for the device ITO/12 : PVK/Ca/Ag resulted in an efficiency of 0.8 cd A−1 with a current density of <8 mA cm−2.
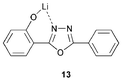
Liang et al.28 reported the blue emitting hydroxyphenyloxadiazole lithium complex 13 which served as the emitting layer (λmax 468 nm) in the device ITO/TPD/13/Al with a maximum luminance of 2900 cd m−2. A current efficiency of 3.9 cd A−1 and power efficiency of 1.1 lm W−1 was obtained. TPD was chosen for its hole transporting qualities. The efficiency of an ITO/NPB/Alq3/Al device increased considerably when 13 was inserted between the Alq32 layer and the Al electrode. The device specification which gave the best performance was: ITO/NPB(40 nm)/Alq3(60 nm)/13(2 nm)/Al with a maximum luminance of 18389 cd m−2 at a turn-on voltage of 3.0 V. Furthermore, this device exhibited high current and power efficiencies with values of 5.21 cd A−1 and 2.4 lm W−1, respectively. The impressive EL efficiency may be attributed to: (i) the oxadiazole segment in 13 has excellent ET ability and (ii) the lithium salt is favourable for electron injection.
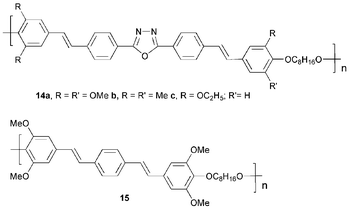
The synthesis of π-conjugated poly(oxadiazoles) has been achieved by direct analogy with their low molecular weight counterparts. Zheng and co-workers29 reported a series of alternating copolymers 14 containing 1,3,4-oxadiazole groups in the conjugated chain, together with flexible spacer units. Blue–green light was emitted by the copolymers as single-layer devices. The general device configuration was ITO/copolymer 14a–c/Ca/Al; EL maxima for devices based on 14a, 14b, 14c and the reference 15 were λmax 494, 509, 480 and 477 nm, respectively. The copolymers were also effective as ETHB layers in bilayer devices, e.g. for ITO/PPV/14c/Ca/Al devices PPV served as the emissive layer: no emission was observed from 14c. The brightness and efficiency of its single-layer counterpart ITO/PPV/Ca/Al was improved by more than a factor of 103 with the introduction of 14c. The bilayer device had a brightness of 2400 cd m−2 at 6.8 V with an EQE of 0.094%. This work demonstrated that the oxadiazole moiety in 14c moves the recombination zone away from the cathode interface and thereby increases the extent of hole/electron recombination in the emitting PPV layer.
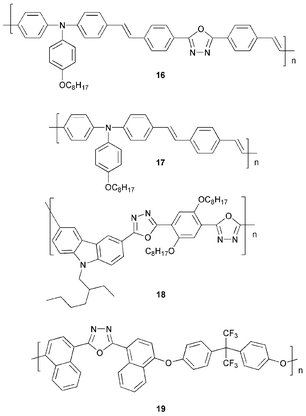
Bipolar polymers have been designed to harness both HT and ET properties. Zhang et al.30 have reported luminescent polymers containing hole-deficient triphenylamine and electron-deficient 1,3,4-oxadiazole units in the main chain. UV–Vis absorption spectra of thin films of 16 and 17 showed that the main absorption peak is red-shifted from λmax 470 in 17 to 495 nm in 16 due to the increased conjugation length which results from inclusion of the oxadiazole ring. A red-shift was also observed in the PL spectra, from λmax 545 to 555 nm. Devices in the configuration ITO/PEDOT/16 or 17/CsF/Al gave blue EL. The device using 16 had a maximum brightness of 3600 cd m−2 and a maximum EL efficiency of 0.65 cd A−1 equal to an EQE of 0.3%. In contrast, polymer 17 gave significantly reduced maximum brightness of 248 cd m−2 and lower EL efficiency of 0.042 cd A−1.
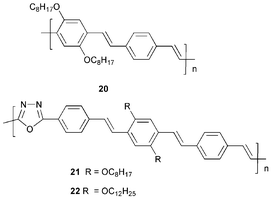
Another example of a bipolar material is 18.31 A device ITO/18/Al emitted blue light at 8 V with a current density of 1.14 cd m−2. Polymer 19 forms stable glasses with a Tg of 220 °C; it also has high thermal stability with a decomposition temperature of 415 °C.32 Optical data showed no absorption in the visible region of the spectrum [λmax (abs) 347 nm] and a fluorescence λmax at 433 nm. A bilayer device ITO/PPV/19/Al demonstrated that 19 possesses hole-blocking capabilities, showing increased operational performance in comparison to the reference single-layer device. A maximum brightness of 55 cd m−2 at a current density of 155 mA cm−2 was recorded at a onset voltage of 7.0 V, compared to the reference (2 cd m−2; 436 mA cm−2) although a lower onset voltage of 6.0 V was achieved.
Peng and Galvin33 studied the conjugated PPV-oxadiazole polymers 20 and 21. Their UV–vis absorption in THF solution (λmaxca. 450 nm) showed no observable effect of the oxadiazole ring. As solid films the PL of 21 was significantly red-shifted (by 35 nm) compared to 20 due to aggregation. As single-layer devices using ITO and Al as the contacts, 21 showed good LED characteristics with enhanced brightness compared to 20.
Polymer 22 has been reported recently by Mukroyannidis and Jenekhe et al.34 In a device ITO/PEDOT/22/Al, voltage-tunable EL colours (558 nm at 9 V; 527 nm at 16 V) were observed. The blue shift with increasing voltage is ascribed to conformational changes of the backbone with increasing temperature (possibly involving cis–trans isomerism). This coupling of thermochromism and EL is unusual.
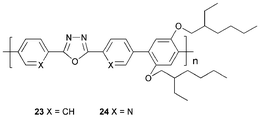
Wang et al.35 studied polymer 23 and its dipyridyl analogue 24. The former is the first polymeric material based on the PBD structure 4 as the repeating unit. TGA established that polymers 23 and 24 are highly stable up to 370 °C and 334 °C, respectively, and are amorphous with Tg values of 196 °C and 113 °C, respectively. They are blue emitters [λmax (PL; thin films) 444 (23) and 475 nm (24)]. For the device ITO/PEDOT/MEH-PPV/23/Al an EQE of 0.26% and brightness of 800 cd m−2 was readily achieved, with emission solely from the MEH-PPV layer.
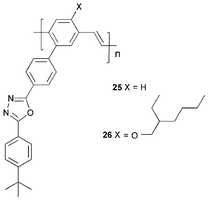
Lee et al. reported polymers 25 and 26 with PDB 4 attached as a pendant unit to the PPV backbone.36 EQEs for devices incorporating 25 and 26 were, respectively, 16 and 56 times the value of their PPV analogue. The ITO/PEDOT/26/Al : Li device has a maximum brightness of 1090 cd m−2 with an EQE of 0.045%. The 2-ethylhexyloxy substituent of 26 improves EL efficiency in comparison to 25, which is in line with earlier findings of Wudl et al. that long or bulky substituents enhance device efficiency by restricting the formation of polaron pairs.37 Further increases in EQE for PPV-related polymers have been achieved by incorporating additional oxadiazole units into both the main chain and pendant side chains.38
3. Oxazoles
There are very few examples of oxazole OLED materials. Compound 27 is a blue emitter and Jordan et al.39 reported a white-light emitting OLED ITO/TAD/27/Alq3/Al which has a maximum brightness of 4700 cd m−2 and a luminescent efficiency of 0.5 lm W−1, using TAD 29 as a hole-transporting layer. Substitution of 27 with 28, which does not emit light but possesses similar transport properties, gave EL emission typical of a TAD/Alq3 device. This indicates that 27 is responsible for the blue spectral components of the white emission, and not TAD. The proportion of red and green wavelengths was tailored by incorporating low percentages of the reddish dye DCM 1 into the Alq3 layer.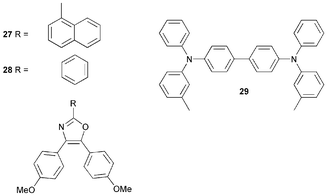
4. 1,2,4-Triazole
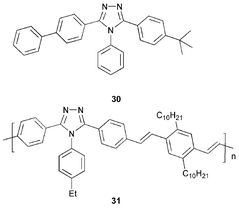
The literature contains fewer examples of low molecular weight triazole derivatives compared to their oxadiazole counterparts. The most commonly used is TAZ 30, which has been shown to be a superior hole-blocking material compared to PBD 4. The electron mobility in 30 is lower than Alq3.8 Kido et al.40 demonstrated that 30 can be efficiently applied as a hole-blocking layer in a triple-layer device ITO/PVK/30/Alq3/Mg : Ag; blue light with λmax 410 nm was emitted. In this device PVK functions as the emitting material and Alq3 acts as an additional ET layer. In the absence of 30 EL is red-shifted to λmax 530 nm with light emission originating from Alq3. White light was also realised for the same device configuration by doping PVK with fluorescent dyes.41 Jiang and co-workers42 showed that white light could also be produced from an ITO/CuPc/NPB/30 : rubrene/Alq3/Mg : Ag device in which 30 is a hole-blocking layer doped with 1.5% rubrene.Burn et al.43 reported a blue-emitting triazole-based conjugated polymer 31 which when used as an ETHB component led to significant improvement in device performance. For a bilayer device ITO/MEH-PPV/31/Al, light emission originated from the PPV layer and EQE reached 0.08% at a luminescence of 250 cd m−2 for a driving voltage of 14 V and a current density of 100 mA cm−2.
5. Pyridine
Compared to benzene, pyridine is π-electron-deficient, therefore the derived polymers have increased electron affinity, improved ET properties, and the symmetry of poly(phenylene) systems is broken. Poly(pyridine-2,5-diyl) (PPY) 32 has some attractive features: it has excellent resistance to photochemical and electrochemical oxidation; it has a PLQY of up to 37%; and is solution processible from formic acid giving solvent orthogonality with most commonly used materials, so enabling the simple fabrication of OLEDs. However, the efficiency of single-layer OLEDs using 32 with ITO and Al contacts is very low (EQE 0.001%).44 However, 32 is an excellent ET layer in devices.45 Monkman and co-workers46 showed that the inclusion of 32 as an ET layer in PPV-based devices gave an EQE of 0.25% when using Al contacts, i.e. ten times greater than similar devices without the layer of 32.Polymer 32 has also been used in conjunction with dendrimers containing a distyrylbenzene core and stilbene dendrons.44 Green light λmax 570 nm was observed which is characteristic of 32, indicating that charge recombination and light emission occurs within the layer of 32, with the dendrimer acting as an HT layer.
Ng et al.47 synthesised soluble blue-emissive alternating copolymers 33–35 comprising pyridine and dialkoxy-functionalised phenyl rings. These polymers exhibited high thermal stability with an onset of degradation at >300 °C. Single-layer devices were fabricated using ITO as the anode and magnesium–indium alloy as the cathode: blue light was emitted; λmax 444 (33), 432 (34) and 428 nm (35), respectively. Polymers 34 and 35 with meta-linkages, and hence reduced conjugation lengths, showed blue-shifted emission peaks in comparison to 33.
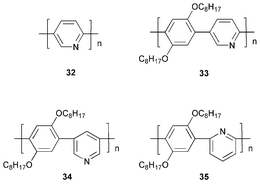
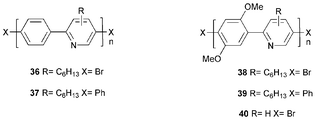
In our laboratory Wang et al.48 studied the novel pyridine-containing conjugated polymers 36–40. The PLQY values for films of 36 and 37 were ca. 15% and 13%, suggesting that non-radiative quenching of the excited polymers by terminal bromines of 37 was not a significant consideration. However, for films of 38 and 39, the corresponding PLQY values were ca. 5% and 17%, respectively. The effect of the methoxy groups was to blue shift PL emission for thin films from λmax 447 (36) to 438 nm (38). A comparison of the spectra of 38 and 40 revealed that the hexyl side chain led to a significant blue shift in both the absorption and emission profiles. This could be due to steric repulsion introduced by the hexyl group in 38 increasing the inter-ring torsion angles within the polymer chain, thus reducing conjugation. Alternatively, differences in the degree of intermolecular interaction could lead to the formation of excimers or aggregates, which would account for the substantial red shift in the spectra of 40.
Polymers 36 and 38 were used successfully as ET layers in devices with the architecture ITO/MEH-PPV/36 or 38/Al. Orange light was emitted entirely from the MEH-PPV layer, although efficiencies and brightness remained low.
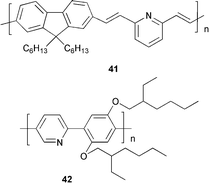
An alternating copolymer 41 composed of fluorenedivinylene as the light-emitting unit and pyridine was used by Kim and co-workers49 as an ET material in an OLED fabricated as a blend with PVK in a single-layer structure with ITO and Al electrodes. Blue EL emission with λmax 475 nm was observed with an EQE of 0.1%.
The copolymer 4250 has been synthesised in our laboratory and its PL and EL spectra (device configuration ITO/PEDOT/42/Ca/Al) are red-shifted upon protonation of the pyridine units. X-Ray crystallographic and spectroscopic studies of model oligomers show that protonation of the pyridine nitrogen and subsequent intramolecular hydrogen bonding to the adjacent alkoxy substituent planarises the backbone. This provides a novel proton-doping mechanism for driving heteroaromatic polymers into a near-planar configuration, thereby tuning the emission profiles.
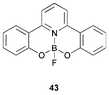
Low molecular weight pyridine-containing ET compounds 8d–g and 9 have been discussed in Section 2. The mixed pyridine-phenol boron complex 4351 exhibits strong blue emission at 445 nm in solvent and as thin films. In EL devices with TPD or PVK as HT layers, different coloured light was emitted (yellow and blue, respectively) suggesting that exciplex formation takes place at the HT/43 interface.
6. Pyrimidine
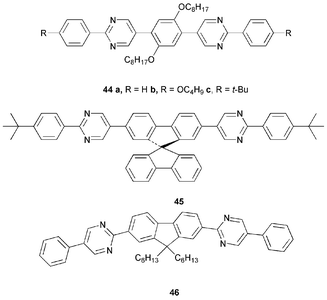
Pyrimidine has been less exploited than pyridine although it has a number of attractive features: (i) it is more electron-deficient than pyridine, and (ii) due to the lack of ortho–ortho interactions of C–H hydrogens, 2-arylpyrimidine derivatives are more planar and possess increased π-conjugation in comparison with 2-arylpyridine or biphenyl analogues.Pyrimidine-containing ET compound 8h was discussed in Section 2. Wong et al.52 reported that the penta(arylene) derivatives 44a–c with an alternating phenylene-pyrimidine structure exhibit high thermal stability and strong blue fluorescence at λmax 419 nm irrespective of the terminal substituents. PLQY values were 37, 54 and 37% for 44a–c, respectively. EL devices of the general configuration ITO/PEDOT-PSS/NCB/44b or 44c/Mg : Al(10 : 1)/Ag displayed EL spectra similar to the PL emission of 44b and 44c with EQEs of 1.3–1.8% and brightness > 2000 cd m−2. [NCB = 4-(N-carbazolyl)-4′-(N-phenylnaphthylamino)biphenyl].
The pyrimidine-containing spirobifluorene system 45 is also a blue emitter (PL λmax 430 nm) with exceptionally high PLQY in both solution and thin films: Φsol
(CHCl3) and Φfilm 100 and 80%, respectively. When 45 was used as a host material for perylene in a multilayer blue emitting device, a maximum brightness of ca. 80000 cd m−2 was achieved.53 A related bis(pyrimidyl)fluorene derivative 46 emits blue–green light (λmax 500 nm) in an OLED at high turn-on voltage, most likely emanating primarily from excimer states.54
7. Pyrazine
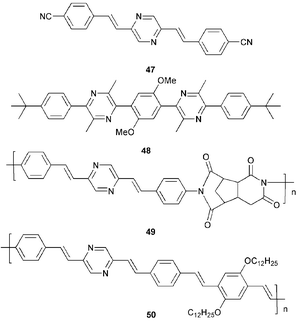
Grimsdale and co-workers55 demonstrated that the PL of distyrylpyrazines could be tuned from blue to green by varying the substituents on the phenyl rings. A di-N-oxide derivative displayed a significant red-shifted PL emission with a broad λmax at 540 nm. Liu et al.56 reported a series of distyrylpyrazine derivatives, e.g. compound 47. Multilayer OLEDs using 47 in conjunction with the HT material NPB and the host material TPBI gave bright blue emission, λmax 468 nm.We have reported bright blue OLEDs (λmax 444 nm) using 48 as the emissive material in the configuration ITO/PEDOT/48/Ca with no long-wavelength emission from π-aggregates or exciton states. The turn on voltage was 6.5 V and a maximum quantum efficiency of 0.09% was attainted.57
Only a few reports have concerned polymers containing pyrazine moieties. Wu et al.58 fabricated EL devices consisting of fluorescent polyimides with 2,5-distyrylpyrazine incorporated into the polymer backbone. A single-layer device of a Langmuir–Blodgett (LB) film of 49 using ITO as the anode and Mg : Al as the cathode emitted orange–red luminescence.
Peng and Galvin59 reported that polymer 50 is strongly fluorescent with a PL λmax 530 nm in THF solution and 550 nm as a solid film. Orange emission was observed for a single-layer device ITO/50/Al. The EQE was low: 0.012% at a current density of 1 mA mm−2. A PPV-based bilayer device including 50 as an additional transport layer showed only a slightly higher EQE of 0.015%.
8. 1,3,5-Triazine
1,3,5-Triazine derivatives have high thermal stability. Pang et al.60 reported that the star-shaped compounds 51 and 52 emit blue light in solution and in the solid state, with solution PLQY values of 43% and 78%, respectively. Multilayer EL devices were fabricated using the architecture ITO/CuPc/51 or 52/PDB/LiF/Al. Both devices displayed blue emission with EL λmax 396 and 444 nm for 51 and 52, respectively. Both devices had a high turn-on voltage of 15 V. Inclusion of the HT material TPD between the CuPc and the emitting layers lowered the turn-on voltage to 6 V. However, a second peak at λmax 532 nm corresponding to green emission was observed and ascribed to an exciplex state formed between TPD and 52. The corresponding device using 51 was not stable.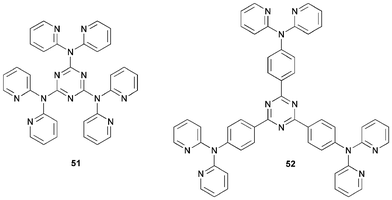
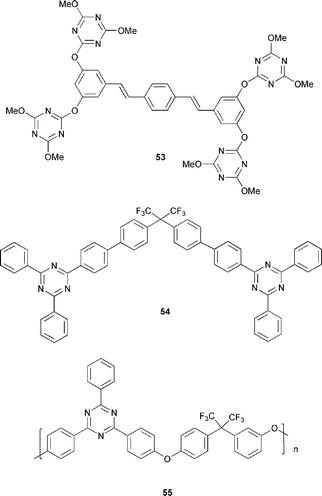
Burn et al.61 reported weak EL from the tetrakis(triazine) derivative 53. The PLQY of a thin film of 53 was 31%. The distyrylbenzene core is a known blue emitter and a single-layer OLED ITO/53/Ca emitted blue light with an EQE of 0.003%. The material was unstable during device operation.
Fink and co-authors62 studied triaryl-1,3,5-triazine derivatives with high Tg, e.g.54, which served as an ETHB layer in two different types of devices: (i) a polymer blend bilayer device, ITO/blend of PVK + TDAPB + Alq3 (4 : 1 : 1)/54/Mg : Ag (10 : 1) and (ii) a multilayer device, ITO/TDAPB/Alq3/54/Mg : Ag (10 : 1). Both devices exhibited green emission similar to the PL of Alq3. The influence of 54 in the multilayer device was evident by a threefold increase in the EQE accompanied by an increase in EL intensity (TDAPB is an HT layer).
Pösch and co-workers32 studied 55 (which is a polymeric analogue of 54) as an ETHB material in PPV-based OLEDs. Compound 55 has a high Tg of 241 °C, and is thermally stable with a decomposition temperature of 466 °C. The bilayer device ITO/PPV/55/Al gave a maximum brightness of 15 cd m−2 at a current density of 43 mA cm−2, in comparison to the reference PPV diode which had a maximum brightness of 2 cd m−2 at a current density of 436 mA cm−2. The bilayer device had a higher onset voltage of 8.5 V compared to 6.0 V for the reference.
9. Quinolines
As stated in Section 1, Alq32 was first used by Tang and VanSlyke3 and has remained the most widely used quinoline-based ET material. The hydroxyquinoline moieties are π-electron deficient, imparting the electron injection/transport property to this green light-emitting complex. Sometimes an additional ET layer e.g. 5,5′-bis(dimesitylboryl)2,2′-bithiophene is placed between the Alq3 and the cathode such that electron injection takes place more readily by a stepwise process, leading to improved emission from the Alq3 layer.63 In addition to its use in low molecular weight vapour-deposited OLEDs, Alq32 has been extensively used as an ET material in polymer OLED systems with considerable improvements in EQEs reported.64Polymeric quinoline derivatives possess high thermal and oxidative stability, and their films display outstanding mechanical properties. Kim et al.65 produced a series of polyquinolines 56a–d containing 9,9-di-n-hexylfluorene units in the main chain. Their optical absorption and luminescence properties vary with chain rigidity and conjugation length. Thermal stability is enhanced by the quinoline units as the Tg values of 56 (decomposition temperature > 380 °C) are much higher than those of polyfluorene (ca. 55 °C) or MEH-PPV (ca. 65 °C). The polymers show longer wavelength blue emission with increasing chain rigidity and conjugation length: for thin films λmax 414 nm (56a) to 494 nm (56d). To investigate their ETHB properties 56a and 56c were fabricated into multilayer OLEDs using the blue emissive statistical copolymer 57. The intensity of the emission and the EQE were enhanced by the presence of the additional layer of 56.
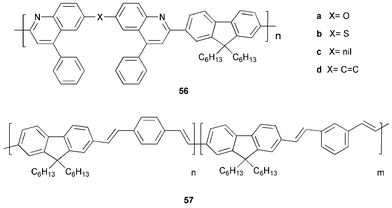
Jen and co-workers66 studied the thermally very stable copolymers containing biquinolines and 2,2-diphenylhexafluoropropane 58 or pyridine moieties 59. Yellow–green light with λmax 523 nm and a weak shoulder at ca. 440 nm was observed for ITO/CuPc/58/Al devices. For the single-layer ITO/59/Al device the turn-on voltages both for current and light were almost the same at 5.9 V, indicating balanced injection and transport of charge carriers, while for the bilayer device ITO/CuPc/59/Al the turn-on voltages for current and light were 9.7 and 8.1 V, respectively. An low EQE of 0.002% was recorded for the device ITO/59/Al which is comparable with single-layer MEH-PPV devices.
Jen et al.67 reported a copolymer 60 of alternating biquinoline and dialkoxyphenylenevinylene units in the main chain. EL spectra for the device configuration ITO/60/Al showed green–yellow emission at λmax 550 nm (EQE 0.06% and turn-on voltage 18 V) emanating from the extended π-system of 60 and not only from the MEH-PPV segment.
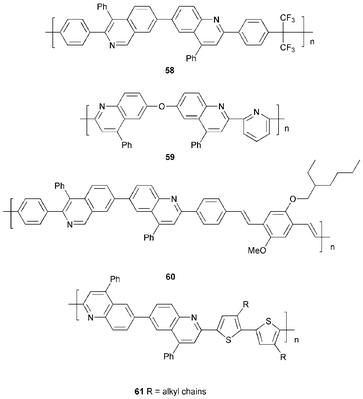
Jenekhe et al. have recently established that regioregular poly(4-alkylquinolines) are promising ET materials.68 Yellow EL (λmax 552 nm) with a turn-on voltage of 5 V, luminance of up to 86 cd m−2 at 10 V, and an EQE of up to 0.6% was observed when the polymers were used as the emissive layer in ITO/PEDOT/PVK/polymer/Al devices. The performance of MEH-PPV devices was enhanced by an additional poly(4-alkylquinoline) layer, with a luminance of up to 700 cd m−2 and an EQE of 3.0%. The orange–red EL was typical of MEH-PPV.
Copolymers 61 which incorporate di(phenylquinoline) and regioregular dialkylbithiophene moieties in the backbone have been studied recently.69 The low PL emission efficiency of 61 in thin films is in accord with their strong intramolecular charge-transfer character. The EL data for ITO/PEDOT/MEH-PPV/61/Al devices establish that polymers 61 function as ET materials, enhancing emission from the MEH-PPV layer. The best performance (1.4% EQE; 2170 cd m−2) was for the polymer with R = n-hexyl, possibly due to its higher molecular weight, which would reduce the number of potential trapping sites at the chain ends.
10. Quinoxaline
Wang and co-workers70 reported EL from bilayer OLEDs composed of the polycyclic quinoxaline dimer derivatives 62 and 63. Emission originated from exciplex formation at the interface between the two organic layers. PL measurements for 62 and 63 showed blue emission with λmax 450 nm and 500 nm, respectively. Yellow–orange emission (λmax 575 nm) was observed for a TPB/62 device, and red light (λmax 685 nm) for a TPB/63 device. The maximum luminances of the two devices were 600 cd m−2 and 70 cd m−2, with EQEs of 0.65% and 0.045%, respectively. Peak emissions of bilayer devices incorporating PVK as the hole-transporting layer were blue-shifted with respect to their TPB counterparts.Thomas et al.71,72 studied the bipolar compound 64 containing a quinoxaline acceptor and carbazole donors. Bright green/yellow emission was seen for the single-layer device ITO/64/Mg : Ag with λmax 548 nm, operating at a maximum brightness of 10437 cd m−2 and maximum luminance efficiency 0.6 lm W−1. Performance was enhanced by the inclusion of an Alq3 ET layer. The device ITO/64/Alq3/Mg : Ag emitted green/yellow light with λmax 544 nm; the maximum brightness and luminance efficiency were increased to 42942 cd m−2 and 2.5 lm W−1, respectively. Pyridopyrazine analogues of 64 were also studied in this work.
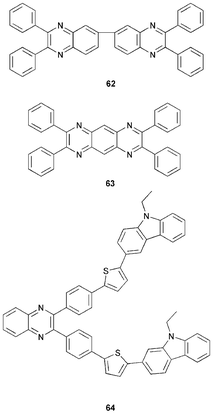
Quinoxaline polymers 6573 and 6632 function as ET layers in bilayer devices using MEH-PPV as the emitting layer.
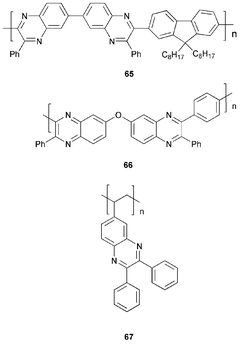
Feast et al.74 reported the homopolymer 67, and a related 2,3-diphenylquinoxaline copolymer with the HT material poly(4-vinyltriphenylamine) PVTPA. Polymer 67 has negligible emission properties, therefore, it was doped at a concentration of 0.5% (by weight) with the pyromethane laser dye PM580. OLEDs were made using PVTPA : 67 random copolymer and a 1 : 1 blend of the corresponding homopolymers; both devices produced green–yellow emission from the PM580 dopant at λmax 560 nm. For single-layer devices relatively high brightness was achieved: 2290 cd m−2 for the blend and 1557 cd m−2 for the random copolymer at the same current. Overall EQEs of 0.114 and 0.073% were recorded for the blend and copolymer, respectively.
11. Pyrazoloquinoline and dipyrazolopyridine
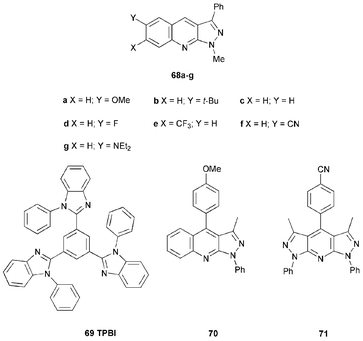
Tao et al.75 have studied a series of fluorescent 1-methyl-3-phenyl-1H-pyrazolo[3,4-b]quinolines 68a–g as dopants (2% w/w) in TPBI layers in the multilayer OLED structures ITO/NPB/CBP/TPBI : 68/TPBI 69/Mg : Ag. TPBI was chosen as the host material because it has a wide band gap and emits strongly at λmax 376 nm where most of the derivatives 68 have strong absorption. Two-layers of HT materials, NPB and CBP, were used with CBP providing an intermediate HOMO level by which holes can pass into the TPBI layer.76 EL spectra of the OLEDs are blue except for 68g which is green. The EQE for devices with 68a and 68b were 2.93 and 3.00%, respectively; somewhat lower values were found for 68c and analogues 68d–f which carry electron-withdrawing substituents. The device incorporating 68g has an EQE of 2.12% and a maximum brightness of 1169 cd m−2, compared to the maximum brightness of 584 cd m−2 for a blue device, obtained using 67a. Replacing 68 with 70 gave a bright blue device (λmax 454 nm) with luminance values as high as 13500 cd m−2 and an EQE of 3.4%.77
In the dipyrazolopyridine series Tao et al. reported that compound 71 is a very efficient dopant-emitter: devices had a brightness of 11200 cd m−2 at 14.2 V and an EQE of 3.2%.76,78
12. Thiophene-S,S-dioxide
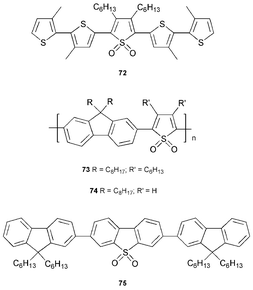
Thiophene, like the nitrogen heterocycles in Sections 2–11, displays considerable aromatic character. In contrast, thiophene-S,S-dioxide is not an aromatic system. Thiophene oligomers and polymers are among the most important optoelectronic materials, e.g. in fundamental studies on conducting polymers,79 as p-type semiconductors in thin-film field-effect transistors,80 electrochromic systems,81 molecular wires82 and as components of modified electrodes for chemosensor devices.83 This is due to a combination of their high chemical stability and ease of functionalisation with substituent groups which allows fine-tuning of the relevant properties. As mentioned in Section 1, PEDOT : PSS is often used as a non-emissive HT layer in OLEDs. However, oligo- and poly-thiophenes are generally not suitable as emissive layers due to their low PLQYs in the solid state arising from the formation of aggregates which quench PL, although they can have high PLQY values in solution.84Conjugated materials with thiophene-S,S-dioxide moieties in the backbone have been exploited, initially by Barbarella et al.,85 as n-type materials which are suitable as ET layers in OLEDs. An unusual feature of some oligomers, e.g.72, is that their PLQY values are remarkably higher in the solid state (37%; λmax 600 nm) than in solution (0.5% in CHCl3) due to specific self-assembly in the solid state (hydrogen bonding to the sulfone group) and their electronic structure.86 Blending compound 72 with PVK improves its film-forming properties, and devices ITO/PEDOT : PSS/72 : PVK/Ca/Al have a turn on voltage of only 2.1 V and a maximum luminance of 200 cd m−2 at 7 V.87 Emissive copolymers which incorporate thiophene-S,S-dioxide display promising EL characteristics. For example, devices based on poly(fluorene-co-thiophene-S,S-dioxide) 73 as the emissive layer (λmaxca. 550 nm ) reach a luminance of 340 lm m−1 at 22 V, and EQE of 0.14%. The relatively high turn on voltage (10 V) was reduced by inclusion of a LiF layer adjacent to the cathode to enhance electron injection.88
OLEDs based on blends of the guest copolymer 74 and the host poly(9,9-dioctylfluorene) show a marked increase in efficiency (150 fold) and luminance compared to those based on pure 74. There is efficient energy transfer from the higher energy gap poly(9,9-dioctylfluorene) to the lower energy gap polymer 74.89 For the blend, the contribution from poly(9,9-dioctylfluorene) to the EL emission is smaller than for the PL: direct electron-hole capture by polymer 74 is considered to contribute.
Perepichka et al. have recently synthesised 9,9-dialkylfluorene oligomers and polymers incorporating dibenzothiophene-S,S-dioxide units, e.g.75, which are highly emissive (PLQY 67% in CHCl3; 63% in thin film) and possess lower energy gaps than the corresponding pure dialkylfluorene analogues. Detailed photophysical and device studies using these materials are in progress.90
13. Electrophosphorescence
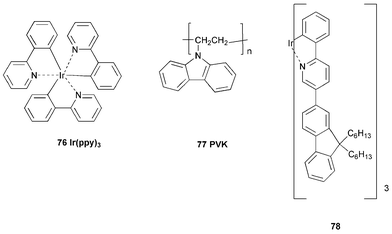
In OLEDs radiative decay (fluorescence) occurs from the spin-singlet excitons whereas spin-triplet states are non-emissive due to the requirements of spin conservation. Statistically electron-hole recombination should produce a triplet : singlet ratio of 3 : 1 which means the maximum efficiency will be limited to 25%,91 although there is evidence that for some polymers this ratio can be nearer 1 : 1.92 The energy of the non-emissive triplets can be harvested by energy-transfer to a phosphorescent dopant, e.g. transition metal complexes.93 Baldo et al. first demonstrated high-efficiency green OLEDs with the phosphorescent molecule fac-tris(2-phenylpyridine)iridium [Ir(ppy)3] 76.94 The commonly used concept is to blend a low molecular weight phosphorescent dye into a polymer matrix into which holes and electrons are injected. It is important that triplet excitons should not be transferred from the triplet emitter to the host, which means that the triplet energy of the host should be higher than that of the emitter. The use of PPV or poly(fluorene) derivatives (which have low triplet energies) is, therefore, limited to red or yellow triplet emitters. For higher energy triplet emitters (i.e., blue or green emitters) poly(9-vinylcarbazole) (PVK) is often used. However, PVK transports holes only, so an ET material needs to be added to get balanced electron and hole currents and thereby to enhance device performance.Vaeth and Tang95 reported emission from 76 doped into a poly(vinylcarbazole) (PVK) 77 host. To confine the emission zone to the Ir(ppy)3 : PVK layer a trilayered device was assembled. By replacing 10% of the PVK with PBD 4 and including the ET materials TPBI 69 and Alq32 a high external quantum efficiency of 8.5% was achieved. The complex device configuration was ITO/Ir(ppy)3 : PVK0.9PBD0.1/TPBI/Alq3/Mg0.9Ag0.1. Work on related cyclometallated complex 78 as the dopant (1% by weight) has given devices with an external quantum efficiency of 8%, luminance efficiency of 29 cd A−1 and brightness 3500 cd m−1 at 30 V (turn-on voltage ca. 10 V).96
Recently Yang and Neher have reported efficient single-layer polymer phosphorescent LEDs based on a green-emitting iridium complex and a PVK host co-doped with 4
(25% by weight) and hole-transporting molecules.97 Luminance efficiencies exceeding 30 cd A−1 were achieved, with brightness of ca. 20000 cd m−1 at 10 V.
Adachi et al. employed an ET layer of a 1,3,4-oxadiazole or a 1,2,4-triazole derivative as the host for [Ir(ppy)3] 76.98 More recently, Cao et al. have used similar ET materials in red-emitting electrophosphorescent devices (up to 12% external quantum efficiencies) with a different iridium complex and hosts PVK, poly(9,9-dioctylfluorene)-silsesquioxane and a poly(9,9-dioctylfluorene)-substituted triphenylamine derivative.99 1,3,5-Triazine ET materials have also been used as hosts in multilayer electrophosphorescent devices with [Ir(ppy)3] 76.100
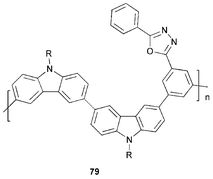
It remains a considerable challenge to design and synthesise host polymers which have a high triplet energy and at the same time suitable HOMO and LUMO levels for efficient and balanced charge injection. In this context new studies have reported dicarbazole–diaryloxadiazole copolymer 79 and analogues which may fulfill these criterea. The meta-linkage to the oxadiazole unit in 79 restricts the conjugation through the system.101 Using an iridium complex as a green triplet emitter dispersed in copolymer 79 as host, the device ITO/PEDOT : PSS/copolymer : emitter/Ba/Al shows a high efficiency of 23 cd A−1 which does not decrease at high current densities and high luminance levels.
14. Conclusions and future prospects
This article has reviewed a range of small molecules and polymers containing electron-deficient moieties which possess ETHB properties when they are incorporated into electroluminescent and electrophosphorescent devices. The need for ETHB materials is due to the fact that the common emitters have low electron affinities, e.g. MEH-PPV and poly(9,9-dialkyl)fluorene, and therefore hole injection and hole transport predominates. Most of the ETHB molecules are electron-deficient heteroaromatics, of which 2,5-diaryl-1,3,4-oxadiazole derivatives and Alq32 are typical examples. Non-aromatic thiophene-S,S-dioxides are also important in this context.As a result of a huge interdisciplinary research effort over the last 15 years, OLEDs are beginning to make significant inroads into the commercial market-place and the unabated interest in these products seems set to continue for the forseeable future with the promise of high-efficiency, low-cost, light-weight and even flexible thin displays.7j Small molecules and polymers continue to play pivotal roles in the development of this new technology. For some years to come they are likely to share broadly the same market as liquid crystal displays. While many charge-transporting organics have been synthesised there is still a need to find new materials which exhibit high fluorescence, controllable energy levels, pure red/green/blue emission colours and high stability under the conditions of device operation. Currently electrophosphorescent systems are setting the pace. Alongside the chemical aspects highlighted in this review article, the development of new device patterning and fabrication techniques, such as ink-jet printing,102 is of equal importance if the current ambitious goals for OLEDs are to be realised and fundamental changes in optoelectronic technology are to take place which will revolutionalise our everyday lives.103
Notes and references
- (a) M. Pope, H. P. Kallmann and P. Magante, J. Chem. Phys., 1963, 38, 2042 CrossRef CAS; (b) W. Helfrich and W. G. Schneider, Phys. Rev. Lett., 1965, 14, 229 CrossRef CAS.
- P. S. Vincett, W. A. Barlow, R. A. Hann and G. G. Roberts, Thin Solid Films, 1982, 94, 171 CrossRef CAS.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burn and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS.
- D. Braun and A. J. Heeger, Appl. Phys. Lett., 1991, 58, 1982 CrossRef CAS; 1991, 59, 878 (erratum).
- R. Stevenson, Chem. Ind., 2003, 33.
- Reviews: (a) A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed. Engl., 1998, 37, 402 CrossRef; (b) J. L. Segura, Acta Polym., 1998, 49, 319 CrossRef CAS; (c) R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Brédas, M. Lögdlund and W. R. Salanek, Nature, 1999, 397, 121 CrossRef CAS; (d) Y. Shirota, J. Mater. Chem., 2000, 10, 1 RSC; (e) U. Mitschke and P. Bäuerle, J. Mater. Chem., 2000, 10, 1471 RSC; (f) T. Fuhrmann and J. Salbeck, MRS Bull., May 2003, 354 Search PubMed; (g) For a review of the general area of organic electronic devices (including OLEDs) see: S. R. Forrest, Nature, 2004, 428, 911 Search PubMed; (h) For a monograph on organic materials for display technology (liquid crystal displays and OLEDs) see: S. M. Kelly, Flat Panel Displays, Royal Society of Chemistry, Cambridge, 2000 Search PubMed; (i) For a discussion of the different ways of measuring the efficiency of OLEDs see: S. R. Forrest, D. D. C. Bradley and M.. E. Thompson, Adv. Mater., 2003, 15, 1043 Search PubMed; (j) For an overview of the current status of OLED applications see: W. E. Howard, Sci. Am., February 2004, 64 Search PubMed.
- M. Thelakkat and H.-W. Schmidt, Polym. Adv. Technol., 1998, 9, 429 CrossRef CAS.
- X. Zhang and S. A. Jenekhe, Macromolecules, 2000, 33, 2069 CrossRef CAS.
- N. Tamoto, C. Adachi and K. Nagai, Chem. Mater., 1997, 9, 1077 CrossRef CAS.
- C. Adachi, T. Tsutsui and S. Saito, Appl. Phys. Lett., 1990, 56, 799 CrossRef CAS.
- A. R. Brown, D. D. C. Bradley, J. H. Burroughs, R. H. Friend, N. C. Greenham, P. L. Burn and A. Kraft, Appl. Phys. Lett., 1992, 61, 2793 CrossRef CAS.
- C. Zhang, S. Höger, K. Pakbaz, F. Wudl and A. J. Heeger, J. Electron. Mater., 1994, 23, 453 Search PubMed.
- H. Tang, F. Li and J. Shinar, Appl. Phys. Lett., 1997, 71, 2560 CrossRef CAS.
- Y. Chien, K. Wong, P. Chou and Y. Cheng, Chem. Commun., 2002, 2874 RSC.
- J. H. Ahn, C. Wang, C. Pearson, M. R. Bryce and M. C. Petty, Appl. Phys. Lett., 2004, 85, 1283 CrossRef CAS; S. Oyston, C. Wang, G. Hughes, A. S. Batsanov, I. F. Perepichka, M. R. Bryce, J. H. Ahn, C. Pearson and M. C. Petty, J. Mater. Chem., 2005 10.1039/b413066a.
- M. Guan, Z. Q. Bian, Y. F. Zhou, F. Y. Li, Z. J. Li and C. H. Huang, Chem. Commun., 2003, 2708 RSC.
- C. Wang, G.-Y. Jung, Y. Hua, C. Pearson, M. R Bryce, M. C. Petty, A. Batsanov, A. E. Goeta and J. A. K. Howard, Chem. Mater., 2001, 13, 1167 CrossRef CAS.
- C. Wang, G.-Y. Jung, A. S. Batsanov, M. R. Bryce and M. C. Petty, J. Mater. Chem., 2002, 12, 173 RSC.
- G.-Y. Jung, PhD Thesis, University of Durham, 2001.
- P. Cea, Y. Hua, C. Pearson, C. Wang, M. R. Bryce, F. M. Royo and M. C. Petty, Thin Solid Films, 2002, 408, 275 CrossRef CAS.
- P. Cea, Y. Hua, C. Pearson, C. Wang, M. R. Bryce, M. C. López and M. C. Petty, Mater. Sci. Eng., C, 2002, 22, 87 CrossRef.
- A. Kraft, Liebigs Ann., 1997, 7, 1463 Search PubMed.
- (a) T. Noda, H. Ogawa, N. Noma and Y. Shirota, Adv. Mater., 1997, 9, 239 CrossRef CAS; (b) T. Noda, H. Ogawa, N. Noma and Y. Shirota, J. Mater. Chem., 1999, 9, 2177 RSC.
- H. Ogawa, R. Okuda and Y. Shirota, Appl. Phys. A, 1998, 67, 599 CrossRef.
- S. W. Cha, S.-H. Choi, K. Kim and J.-I. Jin, J. Mater. Chem., 2003, 13, 1900 RSC.
- H.-C. Yeh, R.-H. Lee, L.-H. Chan, T.-Y. J. Lin, C.-T. Chen, E. Balasubramaniam and Y.-T. Tao, Chem. Mater., 2001, 13, 2788 CrossRef CAS.
- F. Liang, J. Chen, L. Wang, D. Ma, X. Jing and F. Wang, J. Mater. Chem., 2003, 13, 2922 RSC.
- M. Zheng, L. Ding, E. E. Gürel, P. M. Lahti and F. E. Karasz, Macromolecules, 2001, 34, 4124 CrossRef CAS.
- Z. Zhang, Y. Hu, H. Li, L. Wang, X. Jing, F. Wang and D. Ma, J. Mater. Chem., 2003, 13, 773 RSC.
- H. Meng, Z.-K. Chen, X.-L. Liu, Y.-H. Lai, S.-J. Chua and W. Huang, Phys. Chem. Chem. Phys., 1999, 1, 3123 RSC.
- P. Pösch, R. Fink, M. Thelakkat and H.-W. Schmidt, Acta Polym., 1998, 49, 487 CrossRef.
- Z. Peng, Z. Bao and M. E. Galvin, Chem. Mater., 1998, 10, 2086 CrossRef CAS.
- J. A. Mikroyannidis, I. K. Spiliopoulos, T. S. Kasimis, A. P. Kulkarni and S. A. Jenekhe, Macromolecules, 2003, 36, 9295 CrossRef CAS.
- C. Wang, M. Kilitziraki, L.-O. Pålsson, M. R. Bryce, A. P. Monkman and I. D. W. Samuel, Adv. Funct. Mater., 2001, 11, 47 CrossRef CAS.
- D. W. Lee, K.-Y. Kwon, J.-I. Jin, Y. Park, Y.-R. Kim and I.-W. Hwang, Chem. Mater., 2001, 13, 565 CrossRef CAS.
- F. Wudl, P. M. Allemand, G. Srdanov, Z. Ni and D. Mcbranch, ACS Symp. Ser., 1991, 455, 683 CAS.
- Z. Peng and J. Zhang, Synth. Met., 1999, 105, 73 CrossRef CAS.
- R. H. Jordan, A. Dodabalapur, M. Strukelj and T. M. Miller, Appl. Phys. Lett., 1996, 68, 1192 CrossRef CAS.
- J. Kido, K. Hongawa, K. Okuyama and K. Nagai, Appl. Phys. Lett., 1993, 63, 2627 CrossRef CAS.
- J. Kido, K. Hongawa, K. Okuyama and K. Nagai, Appl. Phys. Lett., 1994, 64, 815 CrossRef CAS.
- X. Jiang, Z. Zhang, W. Zhao, W. Zhu, B. Zhang and S. Xu, J. Phys. D: Appl. Phys., 2000, 33, 473 CrossRef CAS.
- A. W. Grice, A. Tajbakhsh, P. L. Burn and D. D. C. Bradley, Adv. Mater., 1997, 9, 1174 CrossRef CAS.
- M. Halim, I. D. W. Samuel, J. N. G. Pillow, A. P. Monkman and P. L. Burn, Synth. Met., 1999, 102, 1571 CrossRef CAS.
- M.-Y. Hwang, M.-Y. Hua and S.-A. Chen, Polymer, 1999, 40, 3233 CrossRef CAS.
- S. Dailey, M. Halim, E. Rebourt, L. E. Horsburgh, I. D. W. Samuel and A. P. Monkman, J. Phys.: Condens. Mater., 1998, 10, 5171 CrossRef CAS.
- S.-C. Ng, H.-F. Lu, H. S. O. Chan, A. Fujii, T. Laga and K. Yoshino, Adv. Mater., 2000, 12, 1122 CrossRef CAS.
- C. Wang, M. Kilitziraki, J. A. H. Macbride, M. R. Bryce, L. E. Horsburgh, A. K. Sheridan, A. P. Monkman and I. D. W. Samuel, Adv. Mater., 2000, 12, 217 CrossRef CAS.
- J. K. Kim, J. W. Yu, J. M. Hong, H. N. Cho, D. Y. Kim and C. Y. Kim, J. Mater. Chem., 1999, 9, 2171 RSC.
- A. P. Monkman, L.-O. Pålsson, R. W. T. Higgins, C. Wang, M. R. Bryce, A. S. Batsanov and J. A. K. Howard, J. Am. Chem. Soc., 2002, 124, 6049 CrossRef CAS.
- L. Yanqin, Y. Liu, W. Bu, J. Guo and Y. Wang, Chem. Commun., 2000, 1551 RSC.
- K.-T. Wong, T. S. Hung, Y. Lin, C.-C. Wu, G.-H. Lee, S. -M. Peng, C. H. Chou and Y. O. Su, Org. Lett., 2002, 4, 513 CrossRef CAS.
- C.-C. Wu, Y. Lin, H. H. Chiang, T. Y. Cho, C. W. Chen, K-T. Wong, Y. L. Liao, G.-H. Lee and S.-M. Peng, Appl. Phys. Lett., 2002, 81, 577 CrossRef CAS.
- G. Hughes, C. Wang, A. S. Batsanov, M. Fearn, S. Frank, M. R. Bryce, I. F. Perepichka, A. P. Monkman and B. P. Lyons, Org. Biomol. Chem., 2003, 1, 3069 RSC.
- A. C. Grimsdale, R. Cervini, R. H. Friend, A. B. Holmes, S. T. Kim and S. C. Moratti, Synth. Met., 1997, 85, 1257 CrossRef.
- M. W. Liu, X. H. Zhang, W. Y. Lai, X. Q. Lin, F. L. Wong, Z. Q. Gao, C. S. Lee, L. S. Hung, S. T. Lee and H. L. Kwong, Phys. Status Solidi A, 2001, 185, 203 CrossRef CAS.
- F. Türksoy, G. Hughes, A. S. Batsanov and M. R. Bryce, J. Mater. Chem., 2003, 13, 1554 RSC.
- A. Wu, T. Akagi, M. Jikei, M. Kakimoto, Y. Imai, S. Ukishima and Y. Takahashi, Thin Solid Films, 1996, 273, 214 CrossRef CAS.
- Z. Peng and M. E. Galvin, Chem. Mater., 1998, 10, 1785 CrossRef CAS.
- J. Pang, S. Freiberg, X.-P. Yang, M. D'Iorio and S. Wang, J. Mater. Chem., 2002, 12, 206 RSC.
- J. M. Lupton, L. R. Hemingway, I. D. W. Samuel and P. L. Burn, J. Mater. Chem., 2000, 10, 867 RSC.
- R. Fink, Y. Heischkel, M. Thelakkat and H.-W. Schmidt, Chem. Mater., 1998, 10, 3620 CrossRef CAS.
- T. Noda and Y. Shirota, J. Am. Chem. Soc., 1998, 120, 9714 CrossRef CAS.
- A. Bacher, I. Bleyl, C. H. Erdelen, D. Haarer, W. Paulus and H. W. Schmidt, Adv. Mater., 1997, 9, 1031 CrossRef CAS.
- J. L. Kim, J. K. Kim, H. N. Cho, D. Y. Kim, C. Y. Kim and S. I. Hong, Macromolecules, 2000, 33, 5880 CrossRef CAS.
- Y. Liu, H. Ma and A. K.-Y. Jen, J. Mater. Chem., 2001, 11, 1800 RSC.
- M. S. Liu, Y. Liu, C. Urian, H. Ma and A. K.-Y. Jen, J. Mater. Chem., 1999, 9, 2201 RSC.
- Y. Zhu, M. M. Alam and S. A. Jenekhe, Macromolecules, 2003, 36, 8958 CrossRef CAS.
- J. Tonzola, M. M. Alam, B. A. Bean and S. A. Jenekhe, Macromolecules, 2004, 37, 3554 CrossRef.
- J.-F. Wang, Y. Kawabe, S. E. Shaheen, M. M. Morrell, G. E. Jabbour, P. A. Lee, J. Anderson, N. R. Armstrong, B. Kippelen, E. A. Mash and N. Peyghambarian, Adv. Mater., 1998, 10, 230 CrossRef CAS.
- K. R. J. Thomas, J. T. Lin, Y.-T. Tao and C.-H. Chuen, Chem. Mater., 2002, 14, 2796 CrossRef.
- K. R. J. Thomas, J. T. Lin, Y.-T. Tao and C.-H. Chuen, J. Mater. Chem., 2002, 12, 3516 RSC.
- X. Zhan, Y. Liu, X. Wu, S. Wang and D. Zhu, Macromolecules, 2002, 35, 2529 CrossRef CAS.
- S. Dailey, W. J. Feast, R. J. Peace, I. C. Sage, S. Till and E. L. Wood, J. Mater. Chem., 2001, 11, 2238 RSC.
- Y.-T. Tao, E. Balasubramaniam, A. Danel, B. Jarosz and P. Tomasik, Chem. Mater., 2001, 13, 1207 CrossRef CAS.
- Y.-T. Tao, E. Balasubramaniam, A. Danel, B. Jarosz and P. Tomasik, Appl. Phys. Lett., 2000, 77, 933 CrossRef CAS.
- Y.-T. Tao, E. Balasubramaniam, A. Danel, A. Wisla and P. Tomasik, J. Mater. Chem., 2001, 11, 768 RSC.
- E. Balasubramaniam, Y.-T. Tao, A. Danel and P. Tomasik, Chem. Mater., 2000, 12, 2788 CrossRef.
- O. Patil, A. J. Heeger and F. Wudl, Chem. Rev., 1988, 88, 183 CrossRef CAS.
- Review: D. Fichou, J. Mater. Chem., 2000, 10, 571 Search PubMed.
- Review: L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481 Search PubMed.
- Review: T. Otsubo, Y. Aso and K. Takimiya, J. Mater. Chem., 2002, 12, 2565 Search PubMed.
- Reviews: (a) J. Roncali, J. Mater. Chem., 1999, 9, 1875 RSC; (b) L. M. Goldenberg, M. R. Bryce and M. C. Petty, J. Mater. Chem., 1999, 9, 1957 RSC.
- (a) C. Väterlein, H. Neureiter, W. Gebauer, B. Ziegler, M. Sokolowski, P. Bäuerle and E. Umbach, J. Appl. Phys., 1997, 82, 3003 CrossRef CAS; (b) P. Barta, F. Cacialli, R. H. Friend and M. Zagórska, J. Appl. Phys., 1998, 84, 6279 CrossRef CAS.
- G. Barbarella, L. Favaretto, M. Zambianchi, O. Pudova, C. Arbizzani, A. Bongini and M. Mastragostino, Adv. Mater., 1998, 10, 551 CrossRef CAS.
- (a) G. Gigli, G. Barbarella, L. Favaretto, F. Cacialli and R. Cingolani, Appl. Phys. Lett., 1999, 75, 439 CrossRef CAS; (b) L. Antlini, E. Tedesco, G. Barbarella, L. Favaretto, G. Sotgiu, M. Zambianchi, D. Casarini, G. Gigli and R. Cingolani, J. Am. Chem. Soc., 2000, 122, 9006 CrossRef CAS; (c) G. Barbarella, L. Favaretto, G. Sotgiu, M. Zambianchi, A. Bongini, C. Arbizzani, M. Mastragostino, M. Anni, G. Gigli and R. Cingolani, J. Am. Chem. Soc., 2000, 122, 11971 CrossRef CAS.
- G. Gigli, M. Ani, G. Barbarella, L. Favaretto, F. Cacialli and R. Cingolani, Physica E, 2000, 7, 612 CrossRef CAS.
- M. Pasini, S. Destri, W. Porzio, C. Botta and U. Giovanella, J. Mater. Chem., 2003, 13, 807 RSC.
- A. Charas, J. Morgado, J. M. G. Martinho, A. Fedorov, L. Alcácer and F. Cacialli, J. Mater. Chem., 2002, 12, 3523 RSC.
- I. F. Perepichka, I. I. Perepichka, L.-O. Pålsson and M. R. Bryce unpublished results.
- A. Köhler, J. S. Wilson and R. H. Friend, Adv. Mater., 2002, 14, 701 CrossRef CAS.
- J. S. Wilson, A. S. Dhoot, A. J. A. B. Seeley, M. S. Khan, A. Köhler and R. H. Friend, Nature, 2001, 413, 828 CrossRef CAS; M. Segal, M. A. Baldo, R. J. Holmes, S. R. Forrest and Z. G. Soos, Phys. Rev. B, 2003, 68, 75211 Search PubMed.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4 CrossRef CAS.
- K. M. Vaeth and C. Tang, J. Appl. Phys., 2002, 92, 3447 CrossRef CAS.
- X. Gong, S.-H. Lim, J. C. Ostrowski, D. Moses, C. J. Bardeen and G. C. Bazan, J. Appl. Phys., 2004, 95, 948 CrossRef CAS.
- X. H. Yang and D. Neher, Appl. Phys. Lett., 2004, 84, 2476 CrossRef CAS.
- C. Adachi, M. A. Baldo and S. R. Forrest, Appl. Phys. Lett., 2000, 77, 904 CrossRef CAS.
- C. Jiang, W. Yang, J. Peng, S. Xiao and Y. Cao, Adv. Mater., 2004, 16, 537 CrossRef CAS.
- H. Inomata, K. Goushi, T. Masuko, T. Konno, T. Imai, H. Sasabe, J. J. Brown and C. Adachi, Chem. Mater., 2004, 16, 1285 CrossRef CAS.
- A. van Dijken, J. J. A. M. Bastiaansen, N. M. M. Kiggen, B. M. W. Langeveld, C. Rothe, A. Monkman, I. Bach, P. Stössel and K. Brunner, J. Am. Chem. Soc., 2004, 126, 7718 CrossRef CAS.
- E. Tekin, B.-J. de Gans and U. S. Schubert, J. Mater. Chem., 2004, 14, 2627 RSC and references therein.
- For a detailed consideration of manufacturing and commercialisation issues in organic electronics see: J. R. Sheats, J. Mater. Res., 2004, 19, 1974 Search PubMed.
Footnote |
| † A feature of the development of OLED technology is a lack of consistency between different laboratories in terms of which techniques and parameters are used to assess device performance. This means it is often not possible to arrive at a meaningful comparison of data from different publications. For an authoritative discussion of these aspects, and formal definitions of the various terms used to measure OLED efficiences, see ref. 7i. |
| This journal is © The Royal Society of Chemistry 2005 |