Development of a microfabricated cytometry platform for characterization and sorting of individual leukocytes
Alexander
Revzin†
a,
Kazuhiko
Sekine
a,
Aaron
Sin
a,
Ronald G.
Tompkins
a and
Mehmet
Toner
*b
aCenter for Engineering in Medicine and Surgical Services, Massachusetts General Hospital, Harvard Medical School, and Shriners Hospital for Children, Boston, MA 02114, USA
bShriners Hospital for Children, 51 Blossom St., Boston, MA 02114, USA. E-mail: mtoner@hms.harvard.edu; Fax: (617) 371-4950; Tel: (617) 371-4883
First published on 12th October 2004
Abstract
Organizing leukocytes into high-density arrays makes these cells amenable to rapid optical characterization and subsequent sorting, pointing to clinical and basic science applications. The present paper describes development of a cytometry platform for creating high-density leukocyte arrays and demonstrates retrieval of single cells from the array. Poly(ethylene glycol) (PEG) photolithography was employed to fabricate arrays of microwells composed of PEG hydrogel walls and glass attachment pads 20 µm × 20 µm and 15 µm × 15 µm in size. PEG micropatterned glass surfaces were further modified with cell-adhesive ligands, poly-L-lysine, anti-CD5 and anti-CD19 antibodies, in order to engineer specific cell–surface interactions within the individual wells. Localization of the fluorescently-labeled proteins in the glass attachment pads of PEG microwells was visualized by fluorescence microscopy. Glass slides micropatterned with PEG and cell-adhesive ligands were exposed to T-lymphocytes for 30 min. These anchorage-independent cells became selectively captured in the ligand-modified microwells forming high-density cell arrays. Cell occupancy in the microwells was found to be antibody-dependent, reaching 94.6 ± 2.3% for microwells decorated with T-cell specific anti-CD5 antibodies. Laser capture microdissection (LCM) was investigated as a method for sorting cells from the array and retrieval of single selected cells was demonstrated.
Introduction
White blood cells or leukocytes are blood constituents offering significant insight into human physiology and pathophysiology of infections, allergic conditions, and malignancies.1 Leukocytes are a heterogeneous mixture of several cellular subsets (granulocytes, lymphocytes and monocytes) defined by morphological and immunostaining characteristics. Leukocyte subsets provide important clinical information regarding patient care. For example, enumeration of CD4+ T-lymphocytes is a key step in detection and evaluation of infections such as human immunodeficiency virus (HIV) or malaria.2–4 Similarly, in patients treated for bacterial infections levels of neutrophils (granulocytes) are reflective of treatment efficacy.5 Leukemia is often assessed by interpretation of patterns and intensity of cell surface antigen expression of several leukocyte types.6,7Flow cytometry (FC) is a standard tool used to identify leukocyte subsets based on cell morphology and antigenicity.2,8 While extremely useful for rapid, multiparametric analysis of leukocyte populations, FC has several shortcomings. It permits a given cell to be observed only once, requires large numbers of cells to conduct analysis and provides only limited morphological information. Laser-scanning cytometry (LSC), a technique based on observation of cells affixed to glass slides, was proposed to complement FC as a multiparametric cell analysis method.9 Because the cells are fixed on the surface, LSC allows for direct visualization of cell morphology using light microscopy, permits kinetic analysis of cells and requires a small cell sample.10–14 Ability to carefully cross-correlate cell morphology and immunostaining has made LSC attractive for immunophenotyping of leukocyte subsets.13–15
In the emerging field of multiparametric, microscopy-based cell analysis the emphasis has been correctly placed on the development of sensitive instrumentation. However, it will also be necessary to design “smart” glass slides for specific cell–surface interactions. For example, LSC instruments are equipped with motorized precision stages in order to allow the same cell to be observed multiple times. Even then, returning to a given cell is very difficult if the cells are randomly distributed on the slide. This and other problems associated with cell capture and distribution on the surface may be resolved through the development of glass substrates enabling cells to be positioned in a controlled and well-defined fashion.
Several microfabrication and surface engineering strategies have been developed for the very purpose of designing cell–surface interactions on the micrometer scale.16–20 The majority of these strategies micropattern protein- and cell-resistant material, poly(ethylene glycol) (PEG),19,21 to define non-adhesive domains on the substrate. Microcontact printing has been employed extensively and successfully to pattern microdomains of PEG-thiols on gold in order to define attachment of cells.16,22,23 Other PEG surface modification strategies have been used to pattern mammalian cells on glass substrates.24,25 Glass is one of the most convenient substrates for cellular observation, therefore, the ability to micropattern cells on this optically transparent material is important. Recently, a PEG micropatterning method closely resembling negative-tone lithography was proposed26 and employed to create high-density arrays of mammalian cells on protein-modified glass substrates.27,28
Optical characterization of arrayed cells is important, however, the information it provides is limited to cellular phenotype. A more insightful analysis of the genome or the proteome would require removal of the selected cells from the array. While microfabrication29 and surface engineering30 cell-release strategies are still in the early stages of development, laser-mediated cell retrieval technologies are already commercially available.31,32 For example, laser capture microdissection (LCM), designed as a tool for procurement of pure cell populations from heterogeneous tissue slices, is being used extensively for genomic and proteomic analysis of isolated cells.33–36 LCM allows for single cells to be selected and retrieved during direct microscopic visualization under brightfield or fluorescence illumination, hence, it is an excellent method for selective removal of specific cells from the high-density array.
The present paper demonstrates formation of high-density arrays of model leukocytes on glass and proposes retrieval of selected individual cells using laser microdissection technology. To organize cells into arrays, we fabricated microwells composed of PEG hydrogel walls and 400 µm2 and 225 µm2 square glass attachment pads. In order to capture anchorage-independent T-lymphocytes, glass attachment pads were selectively modified with lymphocyte-specific antibodies and other cell-adhesive ligands. Capture of the T-lymphocytes on the glass substrate was found to be ligand-dependent. While PEG microwells decorated with T-cell specific anti-CD5 antibody had the cell occupancy of 94.6 ± 2.3%, microwells modified with non-specific ligands, avidin and anti-CD19 antibody, had the cell occupancy of 4.8 ± 4% and 13.3 ± 3 .1% respectively. Retrieval of individual cells from the cell array was accomplished with laser capture microdissection (LCM).
Experimental
Materials
Poly(ethylene glycol) diacrylate (PEG-DA) (MW 575), 2,2′-dimethoxy-2-phenyl acetophenone (DMPA), anhydrous toluene, sodium azide, bovine serum albumin (BSA), avidin, poly-L-lysine, ethanol, dessicator and glovebag were purchased from Aldrich Chemical Co. (Milwaukee, WI, USA). Silane adhesion promoter, 3-acryloxypropyl trichlorosilane, was purchased from Gelest, Inc. (Morrisville, PA, USA). Sulfuric acid and hydrogen peroxide were obtained from J.T. Baker (Phillipsburg, NJ, USA). Glass slides (75 × 25 mm), tissue culture flasks and serological pipettes were obtained from Fisher Scientific (Fair Lawn, NJ, USA). Phosphate buffered saline (PBS) 1×, Roswell Park Memorial Institute (RPMI-1640) cell culture media, fetal bovine serum (FBS) and penicillin/streptomycin were purchased from Gibco (Gaithersburg, MD, USA). Cell Tracker™ Orange [5-(and 6-)-(((4-chloromethyl)-benzoyl)amino)tetramethyl rodamine, CMTMR] and Green [5-chloromethylfluorescein diacetate, CMFDA] were purchased from Molecular Probes (Eugene, OR, USA). Biotinylated mouse anti-human anti-CD5 and anti-CD19 antibodies were purchased from BD Pharmingen (San Diego, CA, USA). Avidin-fluorescein (FITC) was purchased from Pierce Biotechnology (Rockford, IL, USA). Human acute lymphoblastic leukemia T-cells (MOLT 3) and Burkitt's lymphoma B-cells (Raji) were purchased from American Type Culture Collection (Manssas, VA, USA). Cyanine (Cy)-3 and -5 fluorescent dyes were bought from Amersham Biosciences (Piscataway, NJ, USA).Photolithographic patterning of PEG hydrogel on glass
Prior to PEG micropatterning, glass substrates were modified with silane adhesion promoter according to the procedure described previously.27 Briefly, 75 × 25 mm glass slides were immersed for 30 min in “piranha” solution consisting of a 3∶1 ratio of aqueous solutions of 50% v/v of sulfuric acid and 30% w/v of hydrogen peroxide. After removal from the “piranha” bath, glass slides were copiously rinsed with DI water and dried under nitrogen. Modification of glass substrates with silane adhesion promoter was achieved by immersing glass slides in 2 mM solution of 3-acryloxypropyl trichlorosilane in anhydrous toluene for 15 min. After removal from the silane solution, slides were rinsed in toluene and dried with nitrogen. Silanized glass slides were stored at 4 °C prior to further use.PEG hydrogel patterns were fabricated from the precursor solution of PEG-DA (MW 575) with 1% w/v photoinitiator, DMPA. This solution was spun at 700 to 1000 rpm for 6 s onto the acrylated silane-treated 75 × 25 mm glass slides using a spin-coater (Machine World Inc., Redding, CA, USA). The uniform layer of PEG-DA precursor solution on glass was then exposed through a chrome/sodalime photomask (Advance Reproductions, North Andover, MA, USA) to 365 nm, 15 mW cm−2 UV light from Q 2001 mask aligner (Quintel Co., San Jose, CA, USA). Exposure times ranged from 1 to 2 s. Regions of PEG-DA exposed to UV underwent free-radical polymerization and became cross-linked, while unexposed regions were dissolved in DI water after 5 min of development. The height of the resultant hydrogel microstructures varied from 2 to 10 µm as measured with Dektak3 surface profiler (Veeco Instruments, Santa Barbara, CA, USA). PEG hydrogel microwells of two types were fabricated. In the first case, a 100 × 100 element array contained wells with individual dimensions of 20 µm thick PEG walls and 400 µm2 glass attachment pads. The second microwell format contained 150 × 150 array elements with individual wells comprised of 10 µm wide PEG walls and 225 µm2 glass attachment pads. High-resolution images of the hydrogel microstructures were obtained using a JSM 5600LV scanning electron microscope (SEM) (JEOL Inc., Peabody, MA, USA) operating at 5 to 10 mV accelerating voltage. In order to avoid charging effects, substrates were sputter-coated with gold–palladium to a thickness of 6 nm prior to SEM experiments. The same protocol was followed for preparation and imaging of samples containing fixed lymphocytes, and PEG microstructures without the cells.
Modifying surfaces with cell-adhesive ligands
To modify surfaces with poly-L-lysine (PLL), PEG micropatterned surfaces were placed into 100 µg mL−1 PLL solution in DI water for 30 min. The glass substrates were removed and rinsed thoroughly with DI water. PLL modified surfaces were stored at 4 °C prior to cell seeding experiments. Glass attachment sites of PEG microwells were coated with antibodies through avidin–biotin binding as follows: 25 mm × 25 mm glass pieces containing PEG patterns were placed into 10 µg mL−1 solution of avidin in 1× PBS (pH 7.4) for 1 h. Avidin-modified glass substrates were thoroughly washed with DI water and placed into 3 µg mL−1 solution of biotinylated antibody in 1× PBS. After 2 h incubation at room temperature antibody-coated glass surfaces were removed, rinsed with DI water and gently dried under nitrogen. The same procedure was followed for anti-CD5 and anti-CD19 antibodies. Antibody-coated surfaces were used in cell seeding experiments within 2 h of surface modification to avoid antibody denaturation. Fluorescence was used to visualize confinement of avidin-FITC, anti-CD5-Cy3, anti-CD19-Cy3 and PLL-Cy3 in the glass attachment sites of the microwells. Labeling of biotinylated anti-CD5 and anti-CD19 antibodies, and PLL with Cy3 was done according to instructions from the fluorochrome manufacturer.37 Immobilization of fluorescently-labeled ligands in the microwells was routinely imaged using a Nikon Eclipse TE2000 inverted microscope (Nikon, Japan) with an attached SPOT digital camera (Diagnostic Instruments Inc., Burlingame, CA, USA). Zeiss LSM 5 Pascal confocal microscope (Carl Zeiss Inc., Thornwood, NY, USA) was employed to collect composite, brightfield and fluorescence images of fluorochrome-labeled ligands decorating attachment sites of PEG microwells.Forming high-density arrays of leukocytes
MOLT 3 and Raji lymphocytes were cultured in suspension in 175 cm2 tissue culture flasks at 37 °C in humidified atmosphere with 5% CO2/90% air. Cells were incubated in RPMI-1640 supplemented with 110 mg L−1 sodium pyruvate, 2mM L-glutamine, 10% fetal bovine serum, 200 U mL−1 penicillin and 200 µg mL−1 streptomycin. These cells are not anchorage dependent, thus, the cells suspended in RPMI-1640 were collected from the tissue culture flask without trypsinization. Cells were then centrifuged at 800 rpm for 5 min and resuspended in 1× PBS at concentrations of 0.5, 1 and 2 × 106 cells mL−1 as determined by hemocytometer counting. Viability of cells was assessed by trypan blue exclusion. Raji and MOLT 3 cells were labeled with 5 µM CellTracker™ Orange and Green in accordance with manufacturer's instructions.38 For the experiments involving fluorescent staining, cells were cultured in phenol red-free RPMI-1640 cell culture media to avoid autofluorescence. Stained cells were observed through fluorescein (480 ± 30 nm/535 ± 40 nm) and rodamine (540 ± 25 nm/605 ± 50 nm) excitation/emission filters of the Nikon Eclipse inverted microscope. The Zeiss LSM 5 Pascal confocal microscope was employed to collect composite, fluorescence and brightfield, images of stained cells.Pictorial overview of the cell seeding process is presented in Fig. 1A. For cell seeding experiments, 25 mm × 25 mm glass pieces, micropatterned with PEG and modified with cell-adhesive ligands, were placed into 35 mm diameter petri dishes. 2 mL of cell suspension in 1× PBS was introduced into the petri dishes containing micropatterned glass templates and incubated for 30 min at room temperature. Micropatterned templates were then removed, transferred into a clean petri dish and rinsed twice with 2 mL of 1× PBS. PBS was introduced into the dish from the side to avoid shearing the cells. The same procedure was followed for several cell concentrations and ligand-immobilized templates. Upon completion of the experiments cells were fixed in 1% glutaraldehyde v/v in 1× PBS for 30 min. Cell occupancy in the microwells was enumerated according to the formula:
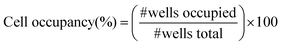
 | ||
| Fig. 1 (A) Diagram of the cell seeding process. Step 1: Cell suspension is introduced into a petri dish containing a PEG micropatterned and protein-modified glass substrates. Step 2: Cells sediment, cover the surface conformally and interact with both PEG and glass regions of the surface. Step 3: After 30 min of incubation cells exposed to protein-modified regions of glass become attached, cells interacting with PEG regions do not attach and are aspirated. (B) Modifying glass attachment sites of PEG microwells with antibodies through avidin-biotin conjugation. Step 1: Fabricate array of microwells. Step 2: Place micropatterned template in avidin (10 µg mL−1 in PBS) for 1 h. Rinse surfaces with DI water. Step 3: Place glass substrate in solution of biotinylated antibody (3 µg mL−1 in PBS) for 2 h. Wash away unattached antibody molecules with DI water. Step 4: Add leukocytes and incubate for 30 min. | ||
Typically, cell occupancy in a randomly selected region of the array comprised of ∼400 wells was manually counted at 100× and 200× magnifications. Number of cells per well was assessed using the same counting protocol. For all experiments, the number of samples was n ≥ 4.
Capture efficiency was defined as surface density of captured cells/surface density of total cells. For 2 × 106 total cells introduced into a 35 mm diameter petri dish, surface density of sedimented cells was 2079 cells mm−2. In the case of 20 µm × 20 µm attachment site dimensions, microwell density was estimated to be 625 wells mm−2. The number of cells captured in the microwells covering area equivalent to 1 mm2 was assessed using cell occupancy and cell per well estimates.
Cell retrieval by laser capture microdissection (LCM)
In the LCM experiments, cell-seeding procedure was the same as described in the previous section. However, alternative cell fixation protocol was followed. Glass slides containing cell arrays were placed for 5 min in ethanol solutions of increasing concentrations, 75%, 95% and 100%, followed by 5 min incubation in xylene. This procedure was necessary to dehydrate the samples and to ensure reproducible cell capture on the transfer film. Cell capture was accomplished using PixCell® IIe Laser Capture Microdissection System (Arcturus, Mountain View, CA, USA). Cells, selected under brightfield illumination, were brought into contact with a CapSure® LCM cap containing a thin transfer film of ethylene vinyl acetate (EVA). A focused laser beam coaxial with microscope optics was pulsed to locally melt EVA, fusing the transfer film to selected cells. When the LCM cap was removed from the micropatterned surface, cells remained adhered to the EVA transfer film. Presence of cells on the transfer film was verified by light microscopy and SEM.Results and discussion
In the present paper, PEG photolithography was combined with selective immobilization of leukocyte-adhesive ligands to enable formation of high-density leukocyte arrays on glass. Laser-mediated cell retrieval technology was employed to remove individual cells of interest from the cell array. The proposed strategies are key components of the cytometry platform for rapid identification of leukocyte subsets and retrieval of pure cell populations for subsequent genomic or proteomic analysis. Such a platform would be a valuable tool with applications in clinical hematology, leukocyte biology and rare cell detection.Modifying PEG microwells with antibodies
In our prior studies, PEG photolithography proved to be an excellent method for patterning anchorage-dependent epithelial cells, fibroblasts and hepatocytes, on glass.27,28 While leukocytes are not anchorage dependent, adhesion of these cells can be induced by decorating surfaces with polycationic molecules,39 ECM proteins40 or cell-specific antibodies.41 The latter option is particularly attractive because of the specificity of antibodies to cell surface antigens and the potential for antibody-mediated capture of selected leukocyte subsets.Schematic presented in Fig. 1B demonstrates avidin-biotin mediated coupling of antibody molecules to glass attachment sites of PEG microwells. Adsorption of avidin on micropatterned glass substrates was localized to the glass attachment pads as verified by fluorescence microscopy (see Fig. 2A). Importantly, no attachment of avidin to PEG hydrogel regions was seen. This observation held true for other cell-adhesive ligands investigated in the present study. The avidin attachment step provided biotin binding sites and enabled modification of microwells with biotinylated antibodies. Avidin-modified glass surfaces were immersed in biotin–antibody solution in PBS. Antibody molecules bound selectively onto avidin-modified attachment sites of PEG wells as seen from Fig. 2B. Assembly of biotinylated anti-CD5 and anti-CD19 antibodies in the PEG microwells followed the same protocol. The concentration of 3 µg mL−1 of antibody in PBS used in our studies was similar to antibody concentrations reported in previous lymphocyte capture studies.41 As demonstrated in the next section, antibody molecules provided a sufficient number of antibody–antigen bonds to enable effective capture of lymphocytes.
 | ||
| Fig. 2 (A) Composite, brightfield and fluorescence, image of avidin–FITC immobilized in the attachment sites of PEG microwells (×400). (B) Composite picture of biotinylated anti-CD-Cy3 antibody bound to avidin-modified PEG microwells (×400). (C) SEM image of high-density arrays of MOLT 3 T-cells captured in 20 µm × 20 µm wells. Inset shows higher magnification view of cells captured in antibody-coated attachment sites (×1100). | ||
Forming high-density arrays of T-lymphocytes
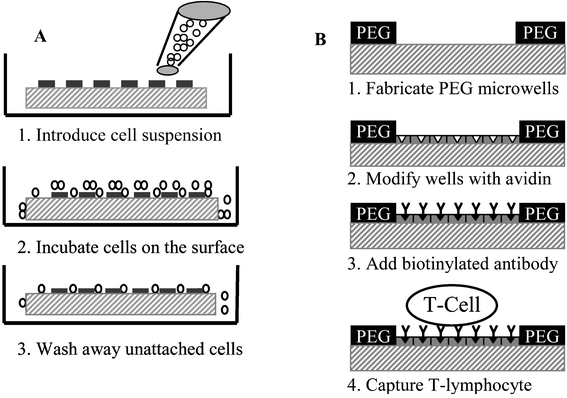
Glass substrates, micropatterned with PEG and modified with cell-adhesive ligands, were seeded with cells as shown in Fig. 1A. Micropatterned templates were covered with cell suspension and incubated for 30 min. During this time, cells settled down and covered the surface entirely, interacting with PEG- and ligand-modified regions alike. However, leukocyte attachment occurred only in the microdomains modified with cell-adhesive molecules, PLL and antibodies. Upon aspiration of cell suspension, unattached cells were removed, revealing arrays of bound leukocytes. Fig. 2C shows a representative result of the leukocyte patterning experiment where MOLT 3 T-cells were captured on anti-CD5-decorated 400 µm2 attachment sites. This image, presenting an array of 240 wells almost entirely occupied by T-cells, is indicative of the large-scale organization of cells in a 100 × 100 array of microwells. Presently, the photomask is designed to cover only small regions of 75 × 25 mm glass slide with the microwell pattern. By simply redesigning the photomask, microwell array may be expanded to completely cover a glass slide. Such an array would contain ∼1.1 × 106 microwells with individual dimensions of 20 µm × 20 µm attachment pads and 20 µm wide PEG walls. Therefore, it is possible to form cell arrays containing over a million cells.Given the cell seeding method, it is intuitive that cell occupancy in the microwells should depend on the cell concentration in solution. To study this interdependence, MOLT 3 T-cells were seeded at different concentrations onto arrays of anti-CD5-coated microwells with 20 µm × 20 µm individual dimensions. Cell occupancy was assessed by manually counting presence of cells in a randomly selected region of ∼400 microwells. Results of these experiments, presented in Fig. 3A, clearly point to a cell concentration dependence in cell array formation. At lower cell concentrations of 0.5 and 1 × 106 cells mL−1, cell occupancy was 43 ± 3.3% and 84 ± 6.8% respectively, whereas in the case of 2 × 106 cells mL−1, concentration cell occupancy reached 94.6 ± 2.3%.
 | ||
| Fig. 3 Quantification of T-cell patterning in antibody-modified 400 µm2 PEG wells. Cell occupancy is based on manual counts of cells in ∼400 wells; for all data points, n ≥ 4. (A) Effects of the T-lymphocyte concentration in solution on the occupancy of PEG microwells. (B) Cell concentration dependency of MOLT 3 T-cells occupying individual wells. (C) Specificity of T-cell interactions with ligand-modified microwells. Anti-CD5 is a T-lymphocyte specific antibody whereas avidin and anti-CD19 are non-specific adhesive ligands. | ||
The number of cells occupying individual 20 µm × 20 µm wells was analyzed using the same concentration range. As seen from Fig. 3B, microwells seeded at lower concentration (0.5 × 106 cells mL−1) were predominantly (65 ± 1.6%) occupied by single cells. As the cell concentration increased to 1 and 2 × 106 cells mL−1, single cell per well occurrence declined to 27.4 ± 4% and 12.3 ± 4.3% respectively. Fabricating smaller wells will lead to improved single cell occupancy at higher cell concentrations.
In addition to cell occupancy, cell capture efficiency was estimated. Capture efficiency, defined as the number of cells captured on the surface vs. the total number of cells placed on the surface, was assessed based on the results presented in Fig. 3(A,B) and petri dish dimensions. The capture efficiency for 0.5 and 2 × 106 cells mL−1 was found to be 41% and 43% respectively, while the estimated value for 1 × 106 cells mL−1 was 62%. Therefore, while providing best cell occupancy results, the highest concentration was sub-optimal from the standpoint of cell capture efficiency. These results underscore the importance of optimizing seeding conditions to satisfy the requirements for both quality of the formed cell array and cell capture yield.
The capture of T-cells in anti-CD5-modified wells, demonstrated in Fig. 2C, was expected given the specificity of this antibody to the CD5 surface antigen expressed by T-lymphocytes. To compare and quantify interactions of MOLT 3 T-cells with specific and non-specific ligands, micropatterned glass substrates were coated with avidin, anti-CD19 and anti-CD5 antibodies. Avidin is a biotin binding protein employed in the antibody-surface coupling scheme, while anti-CD19 is specific to antigen (CD19) expressed exclusively by B-cells. Both molecules are not specific to the T-cells investigated. Micropatterned templates decorated with individual ligands were exposed to MOLT 3 T-cells suspended in PBS at 2 × 106 cells mL−1. Results presented in Fig. 3C point toward specificity of T-cell–surface interactions. Avidin and anti-CD19 decorated microwells had cell occupancy of 5% and 13%, while 95% of anti-CD5-modified wells were occupied.
Surface modification with antibodies is important for applications requiring enrichment of a selected leukocyte subset. However, other applications (e.g. leukocyte differential counts) calling for unbiased surface presentation of multiple leukocyte types would require microwells to be modified with generically adhesive ligands. In order to create generically cell-adhesive surfaces, PEG microwells were coated with poly-L-lysine (PLL). This polycation imparts positive charge on the surface and promotes electrostatic attachment of negatively charged cells.39 When physisorbed from solution, PLL exclusively coated glass attachment sites and did not deposit onto PEG hydrogel regions of the micropatterned substrate (see Fig. 4A). MOLT 3 T-cells did interact with PLL modified surfaces as seen from Fig. 4B showing T-cells captured in PLL-coated 15 µm × 15 µm PEG wells. This figure also shows that 225 µm2 wells are largely occupied by individual lymphocytes. Attachment of cells in the corner, seen in the inset of Fig. 4B, was only observed in high aspect ratio PEG wells. PLL-containing solution may be difficult to remove from the corners of the wells, resulting in higher PLL concentration and offering a possible explanation for cell adhesion behavior.
 | ||
| Fig. 4 (A) Fluorescence imaging demonstrating selective immobilization of PLL-Cy3 in 20 µm × 20 µm PEG microwells (×100). (B) SEM of MOLT 3 T-lymphocytes residing in 15 µm × 15 µm PEG wells. Glass attachment pads of the microwells were coated with PLL. (C) Mixed population of two cell types, T-lymphocytes labeled with CellTracker™ Green and Raji B-lymphocytes labeled with CellTracker™ Orange, in the 20 µm × 20 µm PEG microwells (×800). | ||
To demonstrate potential for placing a mixed leukocyte population in the microwell array, two pure leukocyte subsets, MOLT 3 T-cells and Raji B-cells, were labeled with CellTracker™ Green and Orange respectively, reconstituted in a 1∶1 cell mixture and incubated with micropatterned templates. Fig. 4C shows composite images of differentially labeled T- and B-cells occupying the same array. While for practical applications leukocytes will have to be immunostained with fluorescent antibodies, Fig. 4C is symbolic of the desired outcome: formation of the arrays of cells with phenotype-specific fluorescent signatures. Leukocyte populations in the array could be rapidly identified and quantified using optical cytometry instruments.
Cell retrieval by laser capture microdissection (LCM)
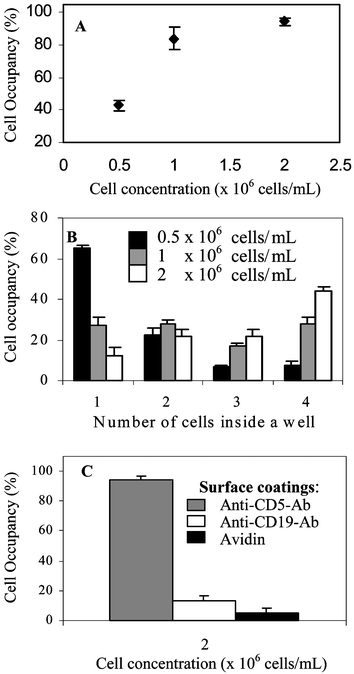
Optical analysis is attractive as a means of rapid characterization of arrayed cells, however, this analysis is limited to cellular phenotype. Powerful tools available for genotype analysis, from single-cell PCR42 to DNA microarrays,43,44 would require that cells be physically removed from the array for subsequent lysing and downstream processing. In this paper, LCM was investigated as an enabling tool for removal/sorting of cells from the array. LCM-mediated T-cell removal from the PEG microwells is presented in Fig. 5. Prior to LCM experiments, cells were fixed according to the protocol described in the Experimental section. Because the LCM system includes a light microscope, cells can be chosen for removal under brightfield or fluorescence illumination. Fluorescence is particularly convenient for leukocyte characterization where immunofluorescent labeling is commonly employed. The region of the micropatterned substrate containing chosen cells was brought into contact with an optically transparent LCM cap, containing thermoplastic EVA transfer film (Step 2, Fig. 5A). A pulse from a semiconductor IR laser melted the EVA film, enveloping selected cells. A focused laser beam (∼10 µm diameter) allowed melting of the transfer film around single cells (Step 3, Fig. 5A). Cells remained on the transfer film after removal of the cap from the surface. Fig. 5B shows nine MOLT 3 T-cells immobilized in PEG microwells prior to the LCM experiment. After completion of the experiment the same nine cells were transferred from the microwells (Fig. 5C) onto the LCM cap (Fig. 5D), making cells available for downstream processing and molecular analysis. In general, we found LCM to be an exciting prospective technology for retrieval of single cells from the array. In the context of blood analysis, LCM may enable identification of malignant, infected, or fetal cells from the high-density array of peripheral blood leukocytes.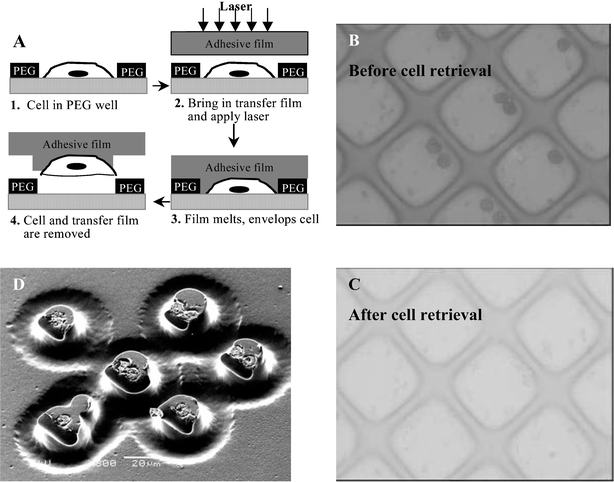 | ||
| Fig. 5 Retrieval of individual leukocytes from the cell array using LCM system. (A) Overview of cell removal. Step 1: Cells of interest are identified using light microscopy. Step 2: LCM cap containing a transfer film is brought into contact with the cell array after which focused laser beam is pulsed. Step 3: Transfer film melts and fuses with cells lying underneath. When the LCM cap is removed, cells remain preferentially attached to the transfer film. The cap is placed into an Eppendorf tube containing DNA, mRNA, or protein preparation buffers. (B) Nine cells are identified for retrieval and covered with a cap containing the transfer film (×400). (C) The same region after laser activation and removal of the transfer film (×400). (D) SEM image of the same nine lymphocytes imbedded in the LCM cap (×800). | ||
Conclusions
This paper investigated surface engineering strategies leading to formation of arrays of model leukocytes, MOLT 3 T-cells, on glass. PEG photolithography was used to fabricate arrays of microwells with PEG walls and glass attachment sites, defining microdomains for adhesion of proteins or cells. Capture of cells on microdomains modified with T-cell specific and non-specific adhesive ligands was investigated. Best results were obtained when PEG microwell arrays were modified with T-cell specific anti-CD5 antibody. In this case, 95% of microwells in a 100 × 100 array were occupied with T-lymphocytes. In addition, laser-mediated sorting of cells was investigated and retrieval of single selected T-lymphocytes from the cell array was demonstrated using LCM technology. We envision the surface micropatterning strategies presented here as key components of a microfabricated cytometry platform for analysis of peripheral blood leukocytes. Such platform will enable presentation of leukocytes in a high-density array format for rapid optical characterization. Employed in concert with optical cytometry and cell-retrieval technologies, this microfabricated platform will be a valuable tool for clinical diagnostics, detection and analysis of rare blood cells, and leukocyte biology.Acknowledgements
Confocal microscopy was performed at CNY-6 Confocal Microscopy Core at Massachusetts General Hospital with the assistance from Mr Igor Bagayev. SEM was performed at the W. M. Keck Microscopy Facility at the Whitehead Institute for Biomedical Engineering. LCM was conducted at Harvard Center for Neurodegeneration and Repair, Advanced Tissue Resources Center. We thank Dr Charles Vanderburg for assistance with operation of LCM instrument. AS acknowledges postdoctoral fellowship from the Croucher Foundation. Financial support for this work was provided by the National Institutes of Health (P41 EB002503 and T32GM07035). The microfabrication work was performed at the BioMEMS Resource Centre.References
- M. L. Turgeon, Clinical Hematology, Lippincott Williams & Wilkins, 3rd edn., 1999 Search PubMed.
- B. Brando, D. Barnett, G. Janossy, F. Mandy, B. Autran, G. Rothe and B. Scarpati, Cytometry, 2000, 42, 327–346 CrossRef CAS.
- T. Hanscheid, Clin. Lab. Haematol., 1999, 21, 235–245 Search PubMed.
- P. J. Norris and E. S. Rosenberg, J. Mol. Med., 2002, 80, 397–405 CrossRef.
- B. Houwen, Lab. Hematol., 2001, 7, 89–100 Search PubMed.
- D. Campana and F. G. Behm, J. Immunol. Methods, 2000, 243, 59–75 CrossRef CAS.
- C. D. Jennings and K. A. Foon, Blood, 1997, 90, 2863–2892 CAS.
- J. P. McCoy and W. R. Overton, Am. J. Clin. Pathol., 1996, 106, 82–86 Search PubMed.
- Z. Darzynkiewicz, E. Bedner, X. Li, W. Gorczyca and M. R. Melamed, Exp. Cell Res., 1999, 249, 1–12 CrossRef CAS.
- E. Bedner, P. Burfeind, W. Gorczyca, M. R. Melamed and Z. Darzynkiewicz, Cytometry, 1997, 29, 191–196 CrossRef CAS.
- E. Bedner, M. R. Melamed and Z. Darzynkiewicz, Cytometry, 1998, 33, 1–9 CrossRef CAS.
- D. A. Rew, L. R. Reeve and G. D. Wilson, Cytometry, 1998, 33, 355–361 CrossRef CAS.
- R. J. Clatch and J. R. Foreman, Cytometry, 1998, 34, 36–38 CrossRef CAS.
- R. J. Clatch, J. R. Foreman and J. L. Walloch, Cytometry, 1998, 34, 3–16 CrossRef CAS.
- A. Gerstner, W. Laffers, F. Bootz and A. Tarnok, J. Immunol. Methods, 2000, 246, 175–185 CrossRef CAS.
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335–373 Search PubMed.
- A. Folch and M. Toner, Annu. Rev. Biomed. Eng., 2000, 2, 227 Search PubMed.
- R. Singhvi, A. Kumar, G. P. Lopez, G. N. Stephanopoulos, D. I. C. Wang, G. M. Whitesides and D. E. Ingber, Science, 1994, 264, 696–698 CrossRef CAS.
- C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Science, 1997, 276, 1425–1428 CrossRef CAS.
- D. Lehnert, B. Wehrle-Hall, C. David, U. Weiland, C. Ballestrem, B. A. Imhof and M. Bastermeyer, J. Cell Sci., 2004, 117, 41–52 Search PubMed.
- K. Prime and G. Whitesides, Science, 1991, 1164–1167 CAS.
- M. Mrksich and G. M. Whitesides, Annu. Rev. Biophys. Biomol. Struct., 1996, 25, 55–78 CrossRef CAS.
- Y. Xia and G. M. Whitesides, Angew. Chem. Int. Ed., 1998, 37, 550–575 CrossRef CAS.
- A. Tourovskaia, T. Barber, B. T. Wickes, D. Hirdes, B. Grin, D. G. Castner, K. E. Healy and A. Folch, Langmuir, 2003, 19, 4754–4764 CrossRef CAS.
- D. Irimia and J. O. M. Karlsson, Biomed. Microdevices, 2003, 5, 185–194 CrossRef CAS.
- A. Revzin, R. J. Russell, V. K. Yadavalli, W.-G. Koh, C. Deister, D. D. Hile, M. B. Mellott and M. V. Pishko, Langmuir, 2001, 17, 5440–5447 CrossRef CAS.
- A. Revzin, R. G. Tompkins and M. Toner, Langmuir, 2003, 19, 9855–9862 CrossRef CAS.
- A. Revzin, P. Rajagopalan, A. W. Tilles, F. Berthiaume, M. L. Yarmush and M. Toner, Langmuir, 2004, 20, 2999–3005 CrossRef CAS.
- R. B. Maxwell, A. L. Gerhardt, M. Toner, M. L. Gray and M. A. Schmidt, J. Microelectromech. Syst., 2003, 12, 630–640 CrossRef.
- W. S. Yeo, M. N. Yousaf and M. Mrksich, J. Am. Chem. Soc., 2003, 125, 14994–14995 CrossRef CAS.
- M. R. Emmert-Buck, R. F. Bonner, P. D. Smith, R. F. Chuaqui, Z. Zhuang, S. R. Goldstein, R. A. Weiss and L. A. Liotta, Science, 1996, 274, 998–1001 CrossRef CAS.
- K. Schutze, H. Posl and G. Lahr, Cell. Mol. Biol., 1998, 44, 735–746 Search PubMed.
- F. Fend, M. R. Emmert-Buck, R. Chuaqui, K. Cole, J. Lee, L. A. Liotta and M. Raffeld, Am. J. Pathol., 1999, 154, 61–66 Search PubMed.
- V. Knezevic, C. Leethanakul, V. E. Bichsel, J. M. Worth, V. V. Prabhu, J. S. Gutkind, L. A. Liotta, P. J. Munson, E. F. Petricoin and D. B. Krizman, Proteomics, 2001, 1, 1271–1278 CrossRef CAS.
- K. Miura, E. D. Bowman, R. Simon, A. C. Peng, A. I. Robles, R. T. Jones, T. Katagiri, P. He, H. Mizukami, L. Charboneau, T. Kikuchi, L. A. Liotta, Y. Nakamura and C. C. Harris, Cancer Res., 2002, 62, 3244–3250 CAS.
- C. M. Michener, A. M. Ardekani, E. F. Petricoin, L. A. Liotta and E. C. Kohn, Cancer Detect. Prev., 2002, 26, 249–255 Search PubMed.
- Product catalogue: fluorescent labeling and detection reagents, Amersham Biosciences, 2002 Search PubMed.
- Handbook of Fluorescent Probes and Research Chemicals, Molecular Probes, Eugene, OR, 6th edn., 1996 Search PubMed.
- A. Macieira-Coelho and S. Avrameas, Proc. Natl. Acad. Sci. USA, 1972, 69, 2469–2473 CAS.
- D. Hauzenberger, N. Martin, S. Johansson and K.-G. Sundquist, Exp. Cell Res., 1996, 222, 312–318 CrossRef CAS.
- L. J. Wysocki and V. L. Sato, Proc. Natl. Acad. Sci. USA, 1978, 75, 2844–2848 CAS.
- S. Hahn, X. Y. Zhong, C. Troeger, R. Burgemeister, K. Gloning and W. Holzgreve, Cell. Mol. Life Sci., 2000, 57, 96–105 CrossRef CAS.
- T. R. Golub, D. K. Slonim, P. Tamayo, C. Huard, M. Gaasenbeek, J. P. Mesirov, H. Coller, M. L. Loh, J. R. Downing, M. A. Caligiuri, C. D. Bloomfield and E. S. Lander, Science, 1999, 286, 531–537 CrossRef CAS.
- D. J. Lockhart and E. A. Winzeler, Nature, 2000, 405, 827–836 CrossRef CAS.
Footnote |
| † Present address: Biomedical Engineering Department, UC Davis, Davis, CA, USA. |
| This journal is © The Royal Society of Chemistry 2005 |