DOI:
10.1039/B405719H
(Communication)
Lab Chip, 2005,
5, 14-19
Differentiation-on-a-chip: A microfluidic platform for long-term cell culture studies
Received
16th April 2004
, Accepted 17th June 2004
First published on 26th July 2004
Abstract
Here we demonstrate a microfluidic perfusion system suitable for a long-term (>2 week) culture of muscle cells spanning the whole process of differentiation from myoblasts to myotubes. Cell-adhesive surface microdomains alternating with a robust cell-repellent coating mimic in vivo spatial cues for muscle cell assembly and allow for confining the fusion of myoblasts into aligned, isolated multinucleated myotubes. The microfluidic system provides accurate control of the perfusion rates and biochemical composition of the environment surrounding the cells. Comparing muscle cell-specific differentiation markers and the timing of fusion, we observed no differences in differentiation between microfluidic and traditional cultures. All differentiation assays were fully microfluidic, i.e. they were performed by sequentially changing the fluids in the microchannels. By delivering fluorescent markers using heterogeneous laminar flows, it was possible to confine a membrane receptor labeling assay to a region smaller than a myotube. Our method can serve as an improved in vitro model for studying muscle cell differentiation and for characterizing extracellular molecules and mechanisms involved in neuromuscular differentiation.
Introduction
Conventional cell culture techniques have changed little over the last several decades and essentially consist of growing cells on a homogeneous large surface (polystyrene or glass dishes or wells) immersed in a homogeneous bath. However, in vivo cells respond to spatially and temporally organized signals in the surrounding microenvironment. Microfluidic systems have been developed by several groups to attempt to create microenvironments of greater physiological relevance and high-throughput platforms for cell behaviour analysis.1,2 Shuler and co-workers have developed a microscale model of multi-organ interactions consisting of microfluidically connected chambers that contain mammalian cell cultures and represent “key” human organs, to study and predict toxicity of chemicals at the organism level.3,4 Beebe and colleagues have manipulated and grown mouse embryos inside microfluidic elastomeric channels and observed improved and more in vivo-like development rates compared to traditional methods.5–8 Walker et al. have successfully cultured insect cells in microchannels for up to 7 days and showed slower growth rates than in conventional microscale cultures.9 Takayama et al. have pioneered the use of multiple laminar streams in microfluidic channels to position10 and sort cells,11 to deliver biologically-active molecules to selected cell domains12,13 and, most importantly, to stimulate signalling pathways in single cells in a physiologically-relevant way.14 Matsue and colleagues have prepared a pattern of cardiac myocytes inside a microfluidic channel and exposed it to heterogeneous flow to demonstrate the capability of this method for high-throughput drug screening and cell toxicity studies.15 With notable exceptions (e.g. ref. 9), most work to date has reported short-term (<1 day) cell cultures, which have less stringent cell maintenance requirements than long-term cultures (e.g. pH and osmotic pressure stability, constant gas and nutrient concentration). While traditional non-microfluidic (static) cultures can be straightforwardly maintained by occasional medium replacements, microfluidic cultures cannot be treated as static because the average volume of medium per cell is typically one order of magnitude smaller than in macroscopic cultures; constant flow poses a challenge because the medium must be pre-warmed (for mammalian cells) and pre-equilibrated with specific gas compositions. Reliable fluidic control is particularly paramount for perfusion-based cell differentiation studies because differentiation processes are biochemically delicate and they take several days to complete. To the best of our knowledge, no long-term microfluidic cultures of mammalian cells have been previously demonstrated. Here we report a microfluidic system that allows for maintaining cell cultures for more than 2 weeks using a gravitational flow. As a test, we demonstrate that the microfluidic culture supports C2C12 cell growth and differentiation. C2C12 cells are myogenic cells that in vitro can be induced to fuse into multinucleated myotubes, providing a test bed for differentiation and synaptogenesis. Furthermore, the system can be combined with cell-adhesive and cell-repellent micropatterns to produce cellular microstructures within the microfluidic environment. Over one week, the tracks of C2C12 cells transform into precisely aligned myotubes. Thus, the cellular micropatterns allow for controlling the width, spacing, and orientation of the myotubes with respect to the flow. Our long-term perfusion setup is simple and can be transferred to any biological laboratory equipped with a cell culture incubator. Long-term viability is of great interest not only for differentiation studies but also for a variety of applications encompassing cell-based biosensors, high-throughput assays, and basic cell biology research.
Experimental
Surface modification for selective cell attachment
Micropatterns of cell adhesive films were prepared using oxygen plasma etching and elastomeric masks as reported previously16 except for modifications noted below. An interpenetrating network (IPN) of P(AAm-co-EG) was homogeneously grafted on borosilicate 2-inch glass discs (Erie Scientific, Portsmouth, NH). The synthesis of IPN consists of a three-step derivatization and has been described in detail elsewhere.17,18 To selectively remove IPN, polydimethylsiloxane (PDMS) microchannels open on both ends were placed onto the IPN surface and exposed to an oxygen plasma (Branson/IPC 2000 barrel etcher, 150 W, 0.75 Torr) for 3 cycles of 8 min to allow for substrate cooling between cycles; excessive heating can cause detachment of the PDMS structures (PDMS expands when heated) and, potentially, IPN degradation. The IPN areas exposed to the plasma were etched away, exposing glass substrate amenable to protein adsorption; the IPN areas covered by PDMS were protected from the plasma by virtue of the conformal contact that PDMS forms with dry, smooth surfaces. The microchannels were typically ∼7 mm long, 50 µm deep and 15–40 µm wide and were separated by 200 µm. Without removing the microchannels from the IPN/glass surface, they were next used to selectively deliver cell attachment-promoting molecules to the etched glass areas. First, a buffer layer of poly-D-lysine solution (PDL, 100 µg mL−1) was suctioned into the microchannels and left at room temperature for 1 h; PDL serves to improve protein adsorption and preserve protein biological activity. The microchannels were then flushed with sterile de-ionized (dI) water and re-filled with diluted (∼1:6 in Dulbecco's Modified Eagle's Medium (DMEM)) growth factor-reduced Matrigel Matrix (BD Biosciences, Bedford, MA), which was allowed to adsorb overnight at 4 °C. Finally, the microchannels were flushed with sterile dI water and dried. Finally, the PDMS mask was removed. The microfluidic perfusion device was sealed atop the Matrigel/IPN pattern, and cells were seeded through the inlet. Cells were seeded on these substrates immediately after surface patterning and attaching the microfluidic device. Sometimes, etch-patterned poly-D-lysine-coated substrates were stored at 4 °C for up to 2 weeks.
Microfluidic device fabrication
The microfluidic device was designed to have two orthogonal fluidic networks (μFN), one for short-term perfusion (dubbed “ST-μFN”) and the other one for long-term perfusion (dubbed “LT-μFN”). The ST-μFN consists of three inlet channels (Fig. 1C, inlets 3–5) converging into a long main channel (width 900–1500 µm, height 250 µm) for creating heterogeneous laminar flows. The LT-μFN consists of 16 parallel microchannels (width 53 µm, height 50 µm, separation 1100µm) that “cross” the main channel (Fig. 1C, inlet 1). All devices were fabricated using PDMS replica molding.19 The master molds were fabricated by two-layer photolithography using the photoresist SU-8 2035–2100 (MicroChem Corp, Newton, MA) according to the manufacturer's datasheets. Layer I defined the three converging channels and LT-μFN, whereas Layer II defined the main channel with adjacent portions (1/4) of the inlet channels (Fig. 1A–B). Layer I was spun to a thickness of 50 µm, prebaked to remove the SU-8 solvent, exposed to the UV light through a mask (with the ST-μFN design and alignment marks), and developed in SU-8 developer. Next, Layer II was spun to a thickness of 200 µm, prebaked, aligned with the Layer I pattern, postbaked, and developed. PDMS prepolymer (Sylgard 184 kit, Dow Corning) was prepared by mixing PDMS base with a curing agent in a 10:1 ratio by weight and degassing the mixture under house vacuum. Finally, the PDMS prepolymer was cast against the master mold. The inlets and the outlets were made from silicone tubing embedded in (i.e. chemically bonded to) PDMS. The PDMS was cured overnight at 65 °C and autoclaved the next day (autoclaving provides sterility, and presumably completes the PDMS cross-linking process). To ensure water-tight seal with the substrates, the devices were O2 plasma-activated (30 s at 150 W and 0.75 Torr) and bonded to the patterned substrate immediately after exposure. Some PDMS was also “hand-painted” around the edges of the device to reinforce the attachment.
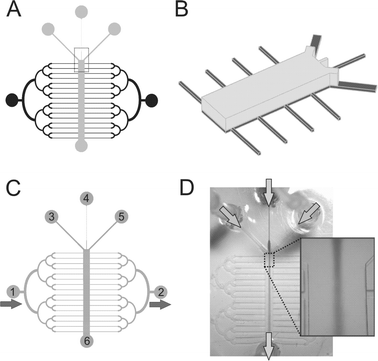 |
| | Fig. 1 Schematics and operation of the microfluidic device. (A) The short-term microfluidic perfusion network (ST-μFN) is shown in gray and the long-term microfluidic perfusion network (LT-μFN) is shown in black. (B) Schematics of the fragment (boxed in (A)) of the master mold showing two layers: Layer I is shown in dark gray and Layer II is shown in light gray. (C–D) Two modes of operation of the device: (C) medium is gravity-fed through the LT-μFN from inlet “1” to “2” to maintain a viable cell culture; “3–5” are inlets and “6” is the outlet for the ST-μFN; (D) hydrodynamic focusing in the device: a central stream carrying a soluble factor gets “squeezed” by two side streams; Arrows indicate the direction of the flows. | |
Cell culture
The murine skeletal muscle cell line C2C12 was purchased from American Type Cell Culture (Manassas, VA). C2C12 myoblast cultures were maintained for 2–3 weeks in DMEM (Life Technologies) supplemented with 20% fetal bovine serum (FBS, HyClone Inc, Logan, UT) and 1% penicillin-streptomycin-fungizone (antibiotic-antimycotic, Life Technologies). Myoblast fusion (a major milestone in muscle cell differentiation) was induced by switching the cell culture medium to DMEM containing 2% horse serum (“differentiating medium”, DM). Prior to starting the experiment, C2C12 cultures were maintained in T75 flasks (Corning) inside an incubator at 37 °C in a humidified atmosphere containing 5% CO2. Cells were dissociated from the culture flasks at 50–75% confluence with 0.25% trypsin and 1 mM EDTA in Hanks' Balanced Salt Solution (Life Technologies), resuspended in DMEM containing 20% serum, counted in a hemocytometer, injected into the microfluidic devices at 2,000,000 cells mL−1, and allowed to attach and spread for 15–30 min before continuous perfusion was established. When myoblasts reached confluency within lines (typically within 1 day), the growth medium was switched to DM.
Operation of the microfluidic device
We used gravity to drive the medium flow from a source reservoir (p35 dish) connected to the inlet(s) of the LT-μFN via a barbed fitting elbow connector and silicone tubing (average flow rate ∼0.5 mL day−1). At this flow rate, no cell death was observed; at slower flow rates, the occurrence of cell death was increasingly high. The LT-μFN source reservoir was replenished daily with fresh pre-warmed growth medium. The waste reservoir was cleared daily. To replace the growth medium with DM, the connector with the tubing was unplugged from the device, the source reservoir and the tubing were emptied and re-filled with DM, and then the tubing was connected again to the device inlet. The device, along with the source and waste reservoir, was placed inside of a p150 dish, covered, and kept in a conventional incubator at 37 °C, 5% CO2, and 85% humidity. Following myotube formation (∼1 week after seeding), the perfusion was stopped by blocking both the inlet and the outlet of the LT-μFN; next, all three inlets of the ST-μFN were gravity-fed from a set of elevated “constant-flow syringes”
(CFS) based on the design of a similar device (Warner Instruments, Hamden, CT). The hydrostatic head pressure generated by the elevation was ∼8 cms H2O. Each CFS consists of two nested containers (fabricated with traditional syringes, hence their name) that are static and provide nearly constant flow by a clever mechanism of dripping from one container to the other. The two-layer design of the ST-μFN (featuring a constriction segment with high flow resistance) ensures that the fluctuations in gravity head caused by the dripping have a minimal effect on the flow rates.
α-Bungarotoxin (BTX) conjugated to a green fluorescent Alexa Fluor 488 (BTX*) was purchased from Molecular Probes (Eugene, OR). In order to visualize acetylcholine receptors (AChRs), the cells (either fixed or alive) were incubated in DM or phosphate-buffered saline (PBS) containing 500 ng mL−1 of BTX* and 1%
(w/vol) bovine serum albumin (BSA), rinsed in PBS and imaged with a fluorescence microscope (see below). For experiments where cells were fixed, 10 min exposures to 2% paraformaldehyde were used. Allura food dye (red, MW = 496) was used (2.5 µg mL−1) to visualize BTX*-carrying streams; we have not noticed any toxicity effects derived from the exposure of the myotubes to the food dye. The live cytoplasmic dye CellTracker Blue CMAC, MW = 209, was purchased from Molecular Probes and used according to the manufacturer's protocol.
Fluorescence microscopy
Fluorescence and phase-contrast images were acquired on an inverted microscope (Diaphot TE 300, Nikon) with a CCD camera (SPOT, Diagnostic Instruments, Sterling Heights, MI) using commercial imaging software (MetaMorph, Universal Imaging Corporation, Downingtown, PA). Fluorescence images were obtained in grayscale (12-bit).
Results and discussion
Microfluidic system
The microfluidic cell culture device was made by molding in PDMS using standard soft lithography techniques as described in the Methods. Schematics and a corresponding image of the microfluidic device are presented in Fig. 1. Fig. 1B illustrates that the device is made with channels of two different heights (Layer I and Layer II). The device contains two microchannel networks perpendicular to each other: (a) a “short-term microfluidic perfusion network”
(ST-μFN), comprising the “main channel” that contains the myotube micropatterns (parallel lines separated by 200 µm) and starts at the point of convergence of three inlet channels; the three inlets are used to switch liquids required for the biochemical assays and also serve the purpose of generating heterogeneous laminar flow streams for relatively short time durations (∼0–10 h); and (b) a “long-term microfluidic perfusion network”
(LT-μFN) comprising 16 “perfusion microchannels” orthogonal to the main channel that are used to perfuse the cells for days in order to maintain a viable culture. The LT-μFN and the ST-μFN intersect at the main channel (where cells are cultured), but are never in operation simultaneously; when fluid is run through the LT-μFN, the ST-μFN is capped, and vice versa. The main channel was 900, 1000, or 1500 µm wide (typical length range of myotubes in a random culture), and about 2 cm long (can potentially accommodate ∼80 micropatterned lines of cells orthogonal to the main channel). The LT-μFN's perfusion microchannels were 50 µm wide. Note that the LT-μFN is designed as a symmetric arbor with binary splitting so as to provide equal flow to all the perfusion channels. The LT-μFN's perfusion channels were designed to provide much higher flow resistance (and therefore, carry much lower flow rates for a given head pressure) than the ST-μFN's main channel, thus consuming less reagents and subjecting cells to less shear stress during the long time periods (days) over which cells proliferate and differentiate. Fig. 1 shows how this microfluidic device is operated during long-term cell perfusion (Fig. 1C) or during short-term exposure to heterogeneous flows from up to three inlets (Fig. 1D).
Flow analysis
Average fluid velocities (v) were estimated from the volume discharged from the device (CFS placed 8 cm above the plane of the device) through the outlet (of either LT-μFN or the ST-μFN) averaged over the period of 1 h (ST-μFN) or 1 day (LT-μFN): ∼17 µL min−1
(v
∼ 1133 µm s−1) for the ST-μFN's main channel (1000 µm-wide) and ∼0.02 µL min−1
(v
∼ 126 µm s−1) for each of the 16 LT-μFN channels. Given the dimensions of the main microchannel (width 900–1500 µm, height 250 µm) and small flow velocities (v
∼ 1 mm s−1), the Reynolds numbers are very small (i.e. < 1), typical of laminar flow in microfluidic channels.20 The fluid shear stresses at the microchannel walls can be calculated using classical fluid dynamics formulas for a parabolic flow profile.21 Shear stress for the ST-μFN's main channel flows, to which cells were subjected for short time periods (up to 4 h), was estimated to be 1.16 10−2 N m−2, while shear stress due to perfusion flow in LT-μFN was 1.29 10−3 N m−2. These short-term and long-term shear stresses are much lower than the shear stresses shown to compromise surface attachment of adherent cells (0.5 to 10.0 N m−2,22), affect cell viability, phenotype, metabolism or protein expression.23,24 In agreement with past work on other cell types and functions, our estimated shear stresses were not observed to noticeably affect C2C12 cell viability, morphology, growth, or differentiation.
Long-term micropatterned microfluidic cultures
The formation of skeletal muscle tissue in vivo involves the determination of precursor cells to the myoblastic pathway, fusion of myoblasts into multinucleated myotubes and maturation into muscle fibers. While in intact tissues muscle fibers are densely packed in a parallel orientation, in traditional culture systems myogenic cells form randomly-oriented, highly branched syncitia.25 Thus, by dictating the spatial organization of the myotubes using surface chemistry, we gain the ability to (1) mimic the highly organized in vivo morphology more accurately than in randomly-oriented cultures, (2) grow isolated myotubes, and (3) design the position/orientation of the cells with respect to the flow.
Device assembly.
A schematic illustration of the process of cellular micropatterning within the microfluidic channels is shown in Fig. 2. As described in detail in the Experimental section, a cell-adhesive micropattern is fabricated on a glass substrate prior to assembling the microfluidic device; this glass substrate is used later as the floor of the device (Fig. 2A). We first prepare micropatterned lines of cell-adhesive Matrigel (adsorbed onto poly-D-lysine, which is adsorbed on glass) separated by a cell-repellent IPN background. The Matrigel micropattern is quickly dried with HEPA-filtered air and covered with the dry autoclaved PDMS microchannels (Fig. 2A). Subsequently, the device (first the LT-μFN and then the ST-μFN) is filled with 37 °C growth medium and placed in the incubator for 1 h.
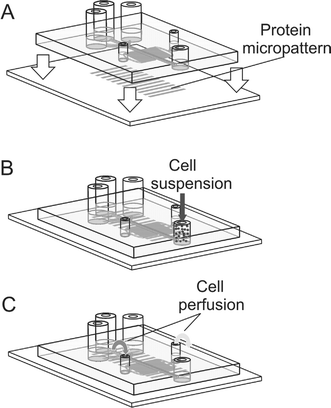 |
| | Fig. 2 Schematics of the device assembly (A), cell seeding (B) and cell perfusion (C). | |
Micropatterning of C2C12 cells.
At this point, the floor of the main channel features a micropattern of cell-adhesive lines surrounded by a non-adhesive background. The LT-μFN's inlets (Fig. 1C, ports “1” and “2”) are capped and the cell suspension is flown into the ST-μFN's main channel through the outlet. When the cells are attached (typically 15–30 min after seeding), the ST-μFN is capped and continuous perfusion flow is started via the LT-μFN and maintained for days until multinucleated myotubes are formed.
Differentiation of C2C12 cells.
C2C12 cells are myogenic, i.e. they are capable of undergoing differentiation in vitro
(fusion and production of muscle-specific proteins) upon withdrawal of growth factors from the culture medium. The reaching of the differentiated stage can be detected straightforwardly because it entails a dramatic change in cell morphology: cells fuse to form multinucleated tubular assemblies (myotubes) displaying pronounced borders in phase contrast (absence of single-cell borders, see Fig. 3C); fluorescence staining (not shown) can be used to reveal, in addition, aligned “strings” of nuclei and an uninterrupted actin cytoskeleton as further evidence of cell fusion. The cells were perfused via the LT-μFN with complete growth medium for 1 day; then, the medium was changed to differentiating medium (DM) to promote fusion.25 Typically within 4–5 days cells fused into long, well-defined myotubes often spanning the entire channel and stayed confined to the adhesive tracks (Fig. 3). In control, non-micropatterned (microfluidic and non-microfluidic) cultures the myotubes formed within the same time frame.
 |
| | Fig. 3 Micropatterned C2C12 cells in microfluidic culture under continuous perfusion. (A–B) Phase-contrast images taken 3 days after seeding at 4×
(A) and 10× magnification (B), showing confluent single myoblasts attached to adhesive tracks; white arrows indicate the direction of the medium flow; (C) Phase-contrast image taken 7 days after seeding, showing myotubes (black arrows) resulting from myoblast fusion. | |
Cells under heterogeneous flows
The exposure of cells to heterogeneous flows12,13 is of paramount interest to (a) performing assays on a subcellular scale (e.g. not the whole cell needs to be stained) and (b) generating complex concentration profiles (e.g. focal delivery or gradient generation) that more closely resemble in vivo paradigms. To generate heterogeneous flow over cells, we capped the LT-μFN's inlet and outlet and used the three inlets of the ST-μFN that converge into the ST-μFN main channel. The stimulatory / labeling molecule of interest is flowed through the center convergent channel together with Allura food dye (for flow visualization), and the two side channels are flowed with plain cell culture medium. The three ST-μFN microchannels that converge into the main channel are designed such that the center channel has 5 times more resistance (it is 5 times narrower) than the side channels, therefore for the same inlet pressure the flow rate through the channel is expected to be 5 times smaller, thus resulting in “focusing” of the center stream. Thus, assuming that most of the pressure drop occurs at the segment that spans from the inlet to the junction, a first-approximation estimate of the ratio of stream widths would be 5:1:5, i.e. the center stream occupying 1/(5+1+5)
=
∼9% fraction of the main channel width right after the junction point (where broadening by diffusion is minimal). The fraction of the channel occupied by the dye does not correspond exactly to the fraction of the flow rate contributed by that channel; rather, it is underestimated by the fact that, as the three channels merge into the main channels, the center channel “loses” both side walls but the side channels lose only the inner wall, hence the side streams are effectively “slowed down” by the outer wall at the junction point while the center channel is not; in other words, the center channel is more effective at transferring momentum to the main multi-stream channel than the side channels are. This second-approximation effect is small because the flow is mainly constricted by the roof and floor, not just the side walls. To illustrate this, an example of focused stream is shown in Fig. 1D; in the image, the central stream occupies ∼14%, i.e. 9% was indeed an underestimate. (Part of the deviation may be attributable to differences in the tubing, e.g. length and/or presence of small bubbles; or connectors that also contribute in flow resistance upstream of the junction point.)
Fig. 4 illustrates the partial labelling of cells in the microchannels using a focused central stream carrying Allura dye (for flow visualization) and CellTracker Blue (for live cell labeling). The images in Fig. 4 show the results of CellTracker labeling of cells in orthogonal (Fig. 4A), parallel (Fig. 4B), or oblique (Fig. 4C) orientation with respect to the flow. Interestingly, in the oblique configuration (Fig. 4C), as well as with seedings of cells at random (i.e. without surface micropatterning, which results in myotube cultures featuring many different orientations, not shown), the topography of the cellular structures induces homogenization of the heterogeneous flow. This observation is consistent with the results by Stroock et al.,26,27 who studied three-dimensional flows over rough surfaces in closed channels and showed that shallow grooves in the floor of the microchannels introduce pressure gradients that generate transverse flow components resulting in the mixing between adjacent streams.26,27 This type of mixing is not observed when cells (or grooves27) are positioned perpendicular (Fig. 4A) or parallel (Fig. 4B) to the flow.
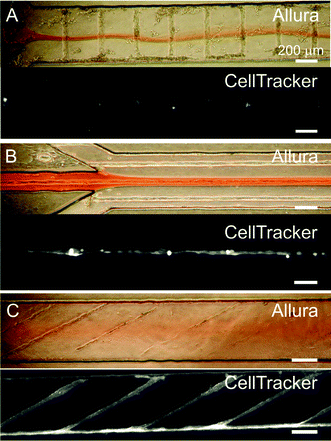 |
| | Fig. 4 Heterogeneous flows over cellular micropatterns. (A, B, and C, upper color images). Phase-contrast images showing both the cells and the heterogeneous flow pattern with a central stream carrying a red food dye (“Allura”). (A, B, and C, lower grayscale images). Fluorescence microscopy images of the cells after exposure to a live cytoplasmic dye (“CellTracker”) that was confined to the central stream. The cells' topography disturbs the flow pattern dramatically in A and in C (see text). | |
Selective surface receptor labeling
We used microfluidic patterning to selectively tag muscle cell surface receptors/ligand-gated channels, which are nicotinic acetylcholine receptors (AChRs). During skeletal muscle development, the expression of AChRs is amplified just before the fusion of myoblasts28 and the AChRs are shuffled to eventually become localized to the site of muscle-nerve contact. Soon after myotube fusion, clusters of AChRs are formed, which can be used as an early muscle differentiation marker. Snake (krait) toxin α-bungarotoxin (BTX) binds with high affinity to the a-subunit of the AChR complex. BTX coupled to a fluorescent dye (BTX*) allows to visualize AChRs. Fig. 5 shows the live labeling of AChRs (with BTX*) confined to the central zones of the myotubes. This subcellular labeling results in a sub-population of tagged receptors that could in principle be followed throughout their life span at the plasma membrane surface until subsequent internalization.
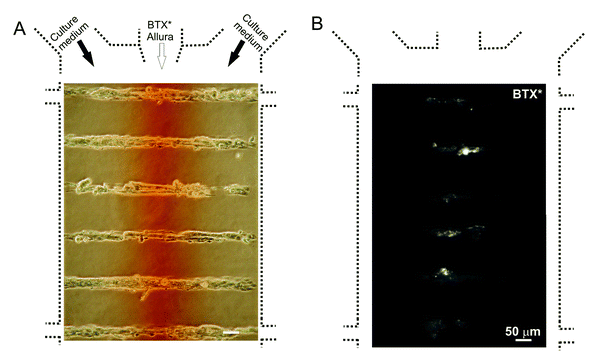 |
| | Fig. 5 Focal exposure of myotubes to fluorescently labeled α-bungarotoxin (BTX*), which tags AChRs and AChR clusters in the stimulated area. (A) Phase-contrast image showing both the myotubes and the heterogeneous flow pattern. Red dye (Allura) was added for visualization of the central stream. (B) Fluorescence microscopy image showing myotubes after exposure to BTX*. | |
Conclusions
We have demonstrated that our microfluidic system enables long-term (2 weeks) cultures of muscle cells and supports muscle cell differentiation. Cells can be reliably positioned in the microfluidic device using surface modification techniques. The process of cell fusion can be geometrically controlled to result in well-defined isolated single myotubes. A microfluidic perfusion network design that utilizes segments of high flow resistance to limit the flow rate allows for obtaining reliable low flow rates using moderate head pressures. Long-term perfusion at such low flow rates did not interfere with cell differentiation and was not observed to affect cell morphology or growth. With this setup, the target cells can be focally labeled by a fluorescent tag and/or selectively exposed to biologically-relevant molecules to study spatiotemporally-confined extracellular stimuli.
Acknowledgements
We thank Dr Stanley Froehner, Dr Marv Adams, Thomas M. Keenan and T. Fettah Kosar for their insightful comments. This work was supported by NIH grant R01 EB001474-02. A.T. is thankful to the Center for Nanotechnology (U.W.) for a Graduate Fellowship and to the Washington Technology Center for the use of their facilities. X.F. is funded in part by an NIH Oral Biology Training Grant fellowship.
References
- T. H. Park and M. L. Shuler, Biotechnol. Prog., 2003, 19(2), 243–253 CrossRef CAS.
- G. M. Walker, H. C. Zeringue and D. J. Beebe, Lab Chip, 2004, 4(2), 91–97 RSC.
- K. Viravaidya, A. Sin and M. L. Shuler, Biotechnol. Prog., 2004, 20(1), 316–323 CrossRef CAS.
- A. Sin, K. C. Chin, M. F. Jamil, J. Kostov, G. Rao and M. L. Shuler, Biotechnol. Prog., 2004, 20(1), 338–345 CrossRef CAS.
- I. K. Glasgow, H. C. Zeringue, D. J. Beebe, S.-J. Choi, J. T. Lyman, N. G. Chan and M. B. Wheeler, IEEE Trans Biomed Eng, 2001, 48(5) Search PubMed.
- D. Beebe, M. Wheeler, H. Zeringue, E. Walters and S. Raty, Theriogenology, 2002, 57, 125–135 CrossRef CAS.
- E. M. Walters, S. G. Clark, D. J. Beebe and M. B. Wheeler, Methods Mol. Biol., 2004, 254, 375–82 Search PubMed.
- S. Raty, E. Walters, J. Davis, H. Zeringue, D. Beebe, S. L. Rodriguez-Zas and M. B. Wheeler, Lab Chip, 2004, 4, 186–190 RSC.
- G. M. Walker, M. S. Ozers and D. J. Beebe, Biomed. Microdevices, 2002, 4(3), 161–166 CrossRef CAS.
- S. Takayama, J. C. McDonald, E. Ostuni, M. N. Liang, P. J. A. Kenis, R. F. Ismagilov and G. M. Whitesides, Proc. Natl. Acad. Sci. USA, 1999, 96(10), 5545 CrossRef CAS.
- B. S. Cho, T. G. Schuster, X. Zhu, D. Chang, G. D. Smith and S. Takayama, Anal. Chem., 2003, 75(7), 1671–1675 CrossRef CAS.
- S. Takayama, E. Ostuni, P. LeDuc, K. Naruse, D. E. Ingber and G. M. Whitesides, Nature, 2001, 411(6841), 1016–1016 CrossRef CAS.
- S. Takayama, E. Ostuni, P. LeDuc, K. Naruse, D. E. Ingber and G. M. Whitesides, Chem. Biol, 2003, 10(2), 123–30 CrossRef CAS.
- A. Sawano, S. Takayama, M. Matsuda and A. Miyawaki, Dev. Cell, 2002, 3, 245–257 Search PubMed.
- H. Kaji, M. Nishizawa and T. Matsue, Lab Chip, 2003, 3, 208–211 RSC.
- A. Tourovskaia, T. Barber, B. T. Wickes, D. Hirdes, B. Grin, D. G. Castner, K. E. Healy and A. Folch, Langmuir, 2003, 19, 4754–4764 CrossRef CAS.
- J. P. Bearinger, D. G. Castner, S. L. Golledge, A. Rezania, S. Hubchak and K. E. Healy, Langmuir, 1997, 13(19), 5175 CrossRef CAS.
- C. H. Thomas, J. B. Lhoest, D. G. Castner, C. D. McFarland and K. E. Healy, J. Biomech. Eng., 1999, 121(1), 40 CAS.
- Y. N. Xia and G. M. Whitesides, Angew. Chem. Int. Ed. Engl., 1998, 37(5), 551 CrossRef CAS.
- J. P. Brody, P. Yager, R. E. Goldstein and R. H. Austin, Biophys. J., 1996, 71(6), 3430–3441 CrossRef CAS.
-
R. B. Bird, W. E. Stewart and E. N. Lightfoot, Transport Phenomena, John Wiley and Sons, New York, 1960, p. 780 Search PubMed.
-
J. G. Aunins and H. J. HenzlerAeration in Cell Culture Bioreactors, ed. H. J. Rehm and G. Reed,, VCH, Weinheim, 2nd edn., 1993, vol. 3 Search PubMed.
- J. T. Keane, D. Ryan and P. P. Gray, Biotechnol. Bioeng., 2002, 81(6951), 211–220 CrossRef CAS.
- R. Nerem, R. Alexander, D. Chappell, R. Medford, S. Varner and W. Taylor, Am. J. Med. Sci., 1998, 316(6951), 109–114 Search PubMed.
- C. Neville, N. Rosenthal, M. McGrew, N. Bogdanova and S. Hauschka, Methods Cell Biol., 1997, 52, 85–116 Search PubMed.
- A. D. Stroock, S. K. W. Dertinger, A. Ajdari, I. Mezic, H. A. Stone and G. M. Whitesides, Science, 2002, 295(5555), 647–651 CrossRef CAS.
- A. D. Stroock, S. K. Dertinger, G. M. Whitesides and A. Ajdari, Anal. Chem., 2002, 74(20), 5306–5312 CrossRef CAS.
- R. M. Krause, M. Hamann, C. R. Bader, J. H. Liu, A. Baroffio and L. Bernheim, J. Physiol., 1995, 489, 779–90 CAS.
|
| This journal is © The Royal Society of Chemistry 2005 |
Click here to see how this site uses Cookies. View our privacy policy here.