Enhancement of the photodynamic activity of tri-cationic porphyrins towards proliferating keratinocytes by conjugation to poly-S-lysine
João
Nuno Silva
a,
Josiane
Haigle
b,
João P. C.
Tomé
c,
Maria G. P. M. S.
Neves
c,
Augusto C.
Tomé
c,
Jean-Claude
Mazière
def,
Cécile
Mazière
def,
René
Santus
bg,
José A. S.
Cavaleiro
c,
Paulo
Filipe
a and
Patrice
Morlière
*def
aHospital de Santa Maria, Clinica Universitária de Dermatologia, 1600, Lisbon, Portugal
bMuséum National d'Histoire Naturelle, RDDM, Photobiologie, F-75231, Paris, France
cUniversidade de Aveiro, Departamento de Química, Campus Universitário de Santiago, 3810-193, Aveiro, Portugal
dINSERM, ERI12, Laboratoire de Biochimie, CHU Amiens-Hôpital Nord, place Victor Pauchet, 80054 Amiens Cedex 1, France. E-mail: morliere.patrice@chu-amiens.fr
eCHU Amiens, Laboratoire de Biochimie, F-80054 Amiens, France
fUniversité de Picardie Jules Verne, Faculté de Médecine et de Pharmacie, EA 2087, F-80054, France
gINSERM, U697, F-75475, Paris, France
First published on 25th November 2005
Abstract
A fast uptake of the tri-cationic 5-(4-carboxyphenyl)-10,15,20-tris(4-methylpyridinium-4-yl)porphyrin tri-iodide (P-H), independent of the presence or absence of proteins in the culture medium, occurs during incubation of NCTC 2544 human keratinocytes with this porphyrin. By contrast, the uptake of the poly-S-lysine conjugate (P-(Lys)n) is faster in serum-free medium than in the supplemented medium suggesting that P-(Lys)n interacts with serum proteins. The P-(Lys)n uptake is almost an order of magnitude greater than that of P-H in serum-free or supplemented culture medium. With histidine as a specific probe of type II photodynamic reactions, the relative photosensitizing effectiveness of the conjugate is only one fourth that of P-H. Nevertheless, the photocytotoxicity of the conjugate is strongly enhanced as compared to that of P-H as a result of its larger uptake. Thus, the doses achieving 50% of photocytotoxicity after incubation with 5 µM of the conjugate and its parent cationic porphyrin are about 20 min and 1 h, respectively. Similarly, the initial rate of the cell lipid peroxidation induced by photosensitization with P-(Lys)n is about 8 times higher than that obtained with P-H. Fluorescence microscopy reveals that P-H is more diffusely located in the cytoplasm than P-(Lys)n which seems to accumulate in lysosome-like structures. Little if any staining of the nucleus is observed with both photosensitizers.
Introduction
Photodynamic therapy (PDT) is an interesting alternative treatment in several domains of oncology and ophthalmology.1–4 In dermatological oncology, PDT is currently used in the treatment of basal and squamous cell carcinoma or of pre-cancerous states such as solar keratoses. Presently authorized photoactive substances are tetrapyrrole derivatives such as the historical reference photosensitizer, Photofrin™, a mixture of photosensitizing porphyrin oligomers1 or more recently, meso-tetrakis(m-hydroxyphenyl)chlorine (Foscan®) used in palliative treatments of head and neck cancers. In dermatology, the precursor of the powerful photosensitizer, protoporphyrin IX, e.g. δ-aminolevulinic acid (ALA) or its methyl esters are currently topically used as a pro-drug to treat cutaneous unvascularized cancers.5 With the exception of Photofrin™, all these photosensitizers are rather insoluble in water. Their use implies the tedious preparation of special formulations for their systemic or topical administration. As a result, workers in the field of PDT have carried out the synthesis of water-soluble derivatives of porphyrins and chlorins.6 Thus, the water-soluble mono-L-aspartylchlorin e6 undergoes advanced clinical trials in Japan to treat lung cancer.7 Along these lines, poly-S-lysine-porphyrin and chlorin conjugates have also been synthesized. Their water solubility has been further improved by conjugating the polypeptide side-chain to cationic meso-tetra-substituted porphyrins.8–10 These derivatives are effective in the photoinactivation of both Gram (+) and Gram (−) bacteria.8 In view of other potential medical applications of poly-S-lysine conjugates, especially in dermatological oncology, effective PDT activity towards rapidly proliferating cells cannot be anticipated since the structure of bacterial walls is quite different from that of mammalian cell membranes.11 As a consequence the response of mammalian cells to PDT with the cationic porphyrin and its polylysine conjugate has been investigated. With the immortalized NCTC 2544 human skin keratinocytes as model of proliferating cutaneous epithelial cells, we have compared the uptake, the intracellular localization and the phototoxicity of a tri-cationic porphyrin, with those of the poly-S-lysine conjugate.Materials and method
Chemicals, culture media and routine spectroscopic equipment
Reagents for cell culture, minimum essential medium with Earle's salts (EMEM), Hanks' buffered saline solution containing 20 mM Hepes (HBSS), Dulbecco's phosphate-buffered saline (PBS), all without phenol red, fetal calf serum (FCS), trypsin and antibiotics were purchased from Gibco (Cergy-Pontoise, France). The Folin reagent, 1,1′,3,3′-tetraethoxypropane and L-histidine (His) were supplied by Sigma Chemical Co. (St Louis, MO, USA). Neutral Red (NR) was a Fluka (Saint-Quentin Fallavier, France) product whereas 1-butanol, Triton X100 (TX100), sodium dodecyl sulfate (SDS) and Hepes were supplied by Merck (Darmstadt, Germany). LysoTracker Green was obtained from Molecular Probes (Eugene, OR, USA). The syntheses of the tri-cationic 5-(4-carboxyphenyl)-10,15,20-tris(4-methylpyridinium)porphyrin (P-H) and of the poly-S-lysine conjugate (P-(Lys)n) have been described by Tomé et al.8 Since P-(Lys)n has no defined molecular weight (depending on the Lys chain polymerization and the degree of porphyrin linked) its concentration was determined assuming the same molar absorbance in the Soret band for the parent porphyrin (P-H) and the conjugate P-(Lys)n in 1% aqueous SDS. Taking into account the weighed material and the spectrophotometrically determined concentration it is found that the degree of polymerization n is about 35. Stock solutions of the porphyrins (500 µM) were prepared in water–DMSO mixture (1 : 1 v/v) and stored at 0–4 °C. The optical absorption spectrophotometry was carried out with an UVIKON 943 spectrophotometer provided by Kontron Instruments (Montigny les Bretonneux, France). An SLM AMINCO-BOWMAN (series 2) (Bioritech, Chamarande, France) equipped with correction of excitation and emission spectra was used for fluorescence measurements. Gel exclusion chromatography was performed with Econo-Pac® 10 DG columns (Bio-Rad France, Marnes la Coquette, France) packed with Bio-Gel® P-6DG. Solutions of P-H and P-(Lys)n at 1 µM in EMEM with or without 10% FCS were used for loading the column and elution was carried out with PBS.Cell culture and treatment
The NCTC 2544 immortalized human skin keratinocyte cell line was purchased from ICN Flow (Fontenay sous Bois, France). Cultures were propagated in EMEM supplemented with 10% FCS, 100 U mL−1 penicillin and 100 µg mL−1 streptomycin without other additives (weekly passages, 1 : 10 splitting ratio). Cells from trypsinized confluent monolayers were seeded at about 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ,000 cells per 35 mm diameter plastic Petri dish containing 2.5 mL of EMEM supplemented with FCS. They were grown for 4 days at 60–80% of confluency. Cells were incubated for various times with 1 mL of medium (HBSS or 10% FCS-supplemented EMEM) containing the desired concentration of photosensitizer. After incubation, cells were thoroughly washed twice with 4 mL of HBSS before further use (photosensitizer uptake, photosensitized lipid peroxidation, photocytotoxicity or fluorescence microscopy). Irradiations were carried out with 1 ml of HBSS, devoid of photosensitizer, covering the cell monolayers. Sham-irradiated cells, used as controls, consisted in cells kept in the dark for the same duration and under the same environmental conditions as the irradiated cells.
,000 cells per 35 mm diameter plastic Petri dish containing 2.5 mL of EMEM supplemented with FCS. They were grown for 4 days at 60–80% of confluency. Cells were incubated for various times with 1 mL of medium (HBSS or 10% FCS-supplemented EMEM) containing the desired concentration of photosensitizer. After incubation, cells were thoroughly washed twice with 4 mL of HBSS before further use (photosensitizer uptake, photosensitized lipid peroxidation, photocytotoxicity or fluorescence microscopy). Irradiations were carried out with 1 ml of HBSS, devoid of photosensitizer, covering the cell monolayers. Sham-irradiated cells, used as controls, consisted in cells kept in the dark for the same duration and under the same environmental conditions as the irradiated cells.
Cellular uptake of photosensitizers
Immediately after washing, cells were mechanically scraped in 1 mL of water. After collection, 100 µL of an 11% SDS solution in 10 mM, pH 7.0 phosphate buffer were added to the cell suspension. Fifty µL of this solution were saved for protein determination using the method of Lowry et al. with the Folin reagent.12 The rest was used for fluorometric measurement of the photosensitizer concentration, using standard photosensitizer solutions for calibration with excitation and emission wavelengths respectively set at 427 and 659 nm. The photosensitizer concentration was normalized to the protein content and the data are the mean ± standard deviation (SD) of at least three independent experiments, each performed in triplicates.Irradiation
Irradiation of cell monolayers with broad band red light was carried out with a custom-built table consisting of two 300 watt tungsten-halogen lamps whose light was filtered with Balzers Y54 and calflex 3000 optical filters. The lamps were placed below the 30 × 25 cm table thermostated at 37 °C. Under these conditions, most of the light arises from wavelengths in the range 500–750 nm as determined with a CS 1000A Minolta spectroradiometer. Absolute calibration of the light fluence rate was carried out using the photodynamic degradation of 50 µM His (see below) by 20 µM hematoporphyrin in air saturated 50 mM pH 7.3 phosphate buffer as actinometer. The quantum yield of His degradation was first measured under irradiation with 365 nm monochromatic light delivered by a filtered high pressure mercury lamp as described in ref. 13. Then, the photodynamic His degradation was performed with the red light. The relative absorbed light is equal to S∫(∫Iλ(1−10−Absλ)dλ)dt, where S is the surface of the Petri dish, Absλ the optical density at the wavelength λ and Iλ the relative light intensity at the wavelength λ, obtained from the radiometer. It is thus possible to calculate the absolute spectral distribution assuming that the His degradation quantum yield in the red range is similar to that at 365 nm. The integration over the wavelength range yields a light intensity of 5.0 × 1016 photon s−1 cm−2 corresponding to 15.2 mW cm−2. For the sake of comparison with other reports, it may be estimated that this intensity is equivalent to monochromatic light intensities Ieq of 4.9 × 1016 photon s−1 cm−2 (∼14.9 mW cm−2) and 4.7 × 1016 photon s−1 cm−2 (∼14.6 mW cm−2) for P-(Lys)n and P-H respectively, at the maximum of the first visible absorption band, namely at 648 nm for P-(Lys)n (ε = 4000 mol l−1 cm−1) and at 639 nm for P-H (ε = 1600 mol l−1 cm−1). These estimates have been calculated by the relationship: ∫Iλελdλ = Ieqε, where Iλ is the incident light intensity, ελ the molar absorbances in PBS at wavelength λ. This calculation is valid for weakly absorbing solutions.Neutral Red uptake assay
As detailed earlier,14 the photocytotoxic effect was determined by the NR uptake assay performed according to Lasarow et al.15 except challenged cells were washed and further incubated at 37 °C for 3 h with the supplemented culture medium before loading with NR. This 3 h lag was chosen to allow the initial damage to propagate but was short enough to avoid important proliferation of undamaged cells (population doubling time is about 1 day) which may alter data. The NR uptake is a widely used assay for evaluating the photocytotoxicity of exogenous drugs or hazardous compounds. It has been validated by the European Union for testing phototoxic chemicals and for the classification and labelling of hazardous chemical (EU Commission Directive 2000/33/EC).16 As reported earlier,17 in the case of NCTC 2544 keratinocytes the cell injury measured with the NR uptake closely parallels that obtained by the well-known Trypan Blue exclusion test. Data are presented as the percentage of NR uptake with respect to that of a control consisting of sham-irradiated untreated cells. The data are the mean ± SD of at least three independent experiments, each performed in triplicates.Thiobarbituric acid reactive substances (TBARS) assay
Immediately after irradiation or sham irradiation, cell supernatants (0.9 mL) were collected for TBARS assay used as an index of lipid peroxidation. TBARS were fluorometrically assayed in terms of malondialdehyde (MDA) equivalent as earlier described18 using 1,1′,3,3′-tetraethoxypropane for calibration. Cells were washed twice with PBS, scraped and collected for protein determination. The TBARS values were normalized to the cell protein content. Data are the mean ± SD of triplicates.Photosensitized His degradation
Dioxygen-saturated solutions containing 5 µM P-H or P-(Lys)n and 500 µM His in PBS were irradiated with increasing light doses at 365 nm. Histidine was monitored by HPLC using a Whatman Partisil 10/25 SCX cation exchange column and 50 mM NH4H2PO4 whose pH was adjusted to 2.3 by addition of phosphoric acid as mobile phase, as earlier described.13 Irradiations, in a 1 × 1 cm cuvette (2.5 mL), were performed with an OSRAM HBO 200 W high pressure mercury lamp whose 365 nm Hg emission line was isolated as detailed before.13 Chemical actinometry based on the photoreduction of ferrioxalate by the UV radiation was carried out according to Parker.19Fluorescence microscopy
An inverted fluorescence microscope (Eclipse TE 3000 DV Nikon) equipped with a CoolSNAPHQ™ detector cooled at −30 °C (Princeton Instruments; Division of Roper Scientific, Evry, France) was used for cell microfluorometry. This system is controlled by the Metaview/Metamorph software, which is also used for image analyses. The fluorescence was recorded with a CCD camera (1392 × 1040 pixels, 8980 × 6700 µm) through a ×100 oil immersion Phaco 3 Nikon objective. A 2 × 2 binning was used yielding 696 × 520 images corresponding to a field of about 110 × 80 µm. The exposure time was 2 s. Background fluorescence obtained from cell-free areas was subtracted from all acquired images. Fluorescence was collected through a filter block (UV2A Nikon) consisting in a dichroic mirror at 400 nm, an excitation filter in the range 330–380 nm. The emission filter of the filter block was removed and the red fluorescence of the porphyrin derivatives was collected through a 645AF75 filter (∼610–700 nm) from Omega Optical (Brattleboro, VT, USA). In the case of double labeling with LysoTracker Green, the green fluorescence was collected through a 535AF45 filter (∼510–565 nm) from Omega Optical. We checked that no porphyrin fluorescence was collected through the green emission filter and that negligible fluorescence of the LysoTracker Green was recorded through the red emission filter. The displayed images are scaled as 256 gray levels between the minimum and maximum fluorescence.Results and discussion
P-H and P-(Lys)n uptake and retention
NCTC 2544 keratinocytes were incubated for increasing times with increasing concentrations of the porphyrin derivatives (Fig. 1) in FCS-supplemented EMEM. As shown in Fig. 2A, an extremely fast uptake of P-H is observed; a plateau being reached less than half an hour after the beginning of the incubation. For P-(Lys)n, the initial uptake rate is faster, but it takes longer to reach the plateau, i.e. about 2–3 h after the beginning of the incubation. Once the plateau is reached, no major changes occur at longer incubation times (up to 18 h, data not shown). The most striking difference between P-H and P-(Lys)n resides in the cell porphyrin contents (Fig. 2A). There is about an order of magnitude between the uptake of P-(Lys)n and that of P-H. | ||
| Fig. 1 Chemical structures of P-H and P-(Lys)n. | ||
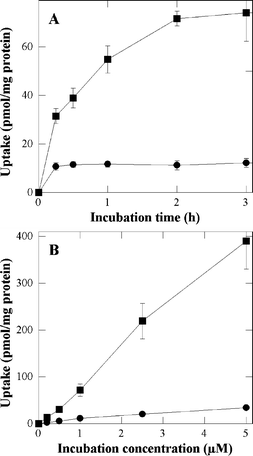 | ||
| Fig. 2 Uptake of P-H (●) and P-(Lys)n (■) as a function of the incubation time (A) and incubation concentration (B). In A, cells were incubated with 1 µM of porphyrin in FCS-supplemented EMEM. In B cells were incubated for 3 h in FCS-supplemented EMEM. Data are the mean of at least three independent experiments performed in triplicates. | ||
The uptake was also measured as a function of the porphyrin concentration after a 3 h incubation in FCS-supplemented EMEM, e.g. at times for which maximum uptakes have been observed above. As shown in Fig. 2B, the uptake is practically proportional to the incubation concentration. This figure also confirms the larger uptake of the polylysine derivative. It is noteworthy that enhanced uptake associated with the conjugation of polylysine to tetrapyrrolic photosensitizers was already reported by Hamblin et al., in the case of chlorin e6.20
A significant difference was noted in the clearance of P-H and P-(Lys)n as shown by chase experiments performed after loading cells for 18 h with 5 µM of P-H or P-(Lys)n in FCS-supplemented EMEM followed by washing. About 65% of P-H and only about 10% of P-(Lys)n were released during the first hour of incubation with porphyrin-free FCS-supplemented EMEM. In both cases, no further loss was observed up to 6 h of incubation with the porphyrin-free medium.
Photocytotoxicity of P-H and P-(Lys)n
As detailed in the experimental section, Photocytotoxicity was estimated by the NR uptake assay. Prior to irradiation, cells were incubated with 5 µM P-H or P-(Lys)n for 3 h in FCS-supplemented EMEM in order to reach maximum uptakes. Then cells were irradiated with increasing light doses (Fig. 3A). Control experiments were carried out with cells incubated with up to 0.5% DMSO in the incubation medium but in the absence of porphyrins and then, sham-irradiated. Control experiments were also performed with cells incubated with porphyrins but sham-irradiated (up to 60 min). No alteration of the NR uptake was observed with both types of control as compared to native cells. It could also be determined that neither P-H nor P-(Lys)n exhibited any cytotoxic effect in the dark for incubation concentrations up to 5 µM (data not shown). No photocytotoxicity was observed with up to 5 µM P-H and irradiation times up to 30 min (Fig. 3B). Fig. 3A also shows that the irradiation time must be increased to 60 min to observe a noticeable photocytotoxic effect with 5 µM P-H. On the other hand, after incubation with 5 µM P-(Lys)n, a marked photocytotoxic effect occurs at much shorter irradiation times (Fig. 3A). Thus, the light doses corresponding to a 50% loss of NR uptake are 20–25 min and 50–60 min for P-(Lys)n and P-H, respectively. The much greater photocytotoxicity of P-(Lys)n is also evidenced by Fig. 3B, showing that incubation with 2.5 µM of the conjugate is sufficient to produce a 50% loss of NR uptake after 30 min of irradiation.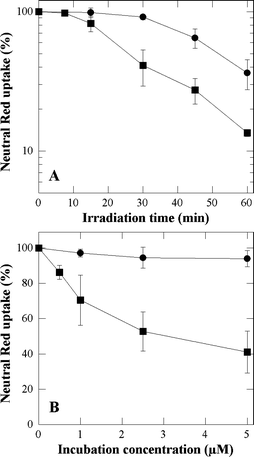 | ||
| Fig. 3 Cytotoxic effects photosensitized by P-H (●) and P-(Lys)n (■) as a function of the irradiation time (A) and of the incubation concentration (B). In A, prior to irradiation, cells were incubated for 3 h with 5 µM of porphyrin in FCS-supplemented EMEM. In B cells were incubated for 3 h with the porphyrin in FCS-supplemented EMEM, and then irradiated for 30 min. A Neutral Red uptake of 100% corresponds to sham-irradiated untreated cells. Data are the mean of at least three independent experiments performed in triplicates. | ||
The greater photocytotoxic effect of P-(Lys)n as compared to that of P-H may be the consequence of three paramaters; e.g. a larger concentration of P-(Lys)n in the cells or a stronger molar absorbance or an intrinsic greater photosensitizing effectiveness. To assess the relative intrinsic photosensitizing efficiency of these two porphyrins, the quantum yield of the photosensitized degradation of His which occurs via a type II photodynamic mechanism was determined upon monochromatic irradiation as detailed in the experimental section.21 The His degradation quantum yield was found to be 0.086 and 0.020 using, respectively, P-H and P-(Lys)n as photosensitizers, illustrating that the intrinsic photosensitizing ability is in favor of the parent compound. Using polychromatic light, the ratio of absorbable light (equal concentration) is equal to ∫IλεP-(Lys)nλdλ/∫IλεP-Hλdλ, where Iλ is the incident intensity, εP-(Lys)nλ and εP-Hλ the molar absorbances at wavelength λ of P-(Lys)n and P-H, respectively. These molar absorbances are unknown in the involved cell micro-environments. However a reasonable estimate can be obtained by using those determined in PBS, a polar solvent and in 0.3% Triton X100 non-polar micelles. As compared to the parent compounds, the visible absorption bands exhibit a hyperchromic effect and the two last visible absorption bands of P-(Lys)n undergo a few nanometers bathochromic shift. Combined with the increase of the spectral output of the lamp in the concerned wavelength range, this yields ratios in favor of P-(Lys)n equal to about 2.6 and 2.8 in PBS and 0.3% Triton X100 respectively. They are not large enough to explain alone the greater efficiency of the polylysine derivative. In view of the lower photodynamic effectiveness of P-(Lys)n it may be suggested that its larger uptake or its better retention than those of P-H strongly contributes to its greater photocytotoxicity. The enhancement of the phototoxic response due the conjugation to polylysine was also reported by Hamblin et al., in the case of related chlorin e6 conjugates.20
P-H and P-(Lys)n photo-induced oxidative stress
The photo-induced lipid peroxidation was used as an overall index of the intensity of the photo-induced oxidative stress induced in keratinocytes by the studied porphyrins. Lipid peroxidation was assayed by measuring TBARS formation. No TBARS formation was detected in the photosensitized cells in agreement with previous studies dealing with photosensitization by endogenous22 or exogenous photosensitizers.23 Then, we focused our attention on the TBARS released in the supernatant. No significant TBARS release can be observed with all the above described controls. As shown in Fig. 4, the ability of P-H and P-(Lys)n to trigger lipid peroxidation parallels their photocytotoxic effects. Furthermore it can be seen that P-(Lys)n produces much more TBARS than P-H. However it must be pointed out that this does not demonstrate that membrane damage are responsible for the observed cytotoxic effects. It simply suggests that the photocytotoxic effect of the studied derivatives is correlated to their overall ability to trigger an oxidative stress, whatever the targets.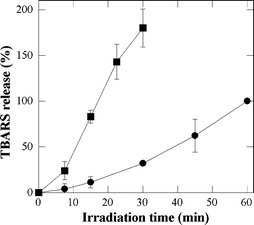 | ||
| Fig. 4 P-H- and P-(Lys)n-photosensitized lipid peroxidation in NCTC 2544 keratinocytes. TBARS were measured in the supernatant of cells incubated with 5 µM P-H (●) or P-(Lys)n (■) for 3 h in FCS-supplemented EMEM prior to irradiation. The percentage of TBARS released by P-(Lys)n-photosensitization was expressed taking as 100% the TBARS value measured with P-H after 60 min of irradiation. Data are the mean of at least three independent experiments performed in triplicates. | ||
Microfluorometry of P-H and P-(Lys)n
One of the advantages of PDT is that the currently used and potentially usable porphyrin derivatives do not accumulate in the cell nucleus.24 Thus in contrast to the psoralens used in the PUVA therapy of skin disorders25 direct damage to DNA that may lead to mutations in surviving cells are less likely. However, secondary dark reactions in the cells after porphyrin photosensitization have been shown to indirectly lead to DNA alterations.26 On the other hand it is known that because of their charge, cationic compounds accumulate in the nucleus.27 For all the above reasons it is interesting to study the role of the conjugation on the intracellular localization of P-H and P-(Lys)n before further developing this series of molecules. The two studied porphyrins fluoresce in the red with maximum emission near 650 nm. Fluorescence microscopy can therefore be used for unraveling the sites of localization of P-H and P-(Lys)n in the NCTC 2544 keratinocytes. Fig. 5 shows the phase contrast and fluorescence images obtained for control cells (untreated cells) and cells treated overnight with P-H and P-(Lys)n in FCS-supplemented culture medium. These images obtained after an overnight incubation (18 h) were similar to those obtained after a 3 h incubation (data not shown). Fluorescence images were obtained with an excitation in the near UV range (330–380 nm) and an emission in the visible range (610–700 nm). As expected28 the auto-fluorescence arises from the whole cytoplasm with larger intensities in the peri-nuclear area and the mitochondrial network (Fig. 5A and 5D). However, the auto-fluorescence level is low as compared to that obtained with treated cells (see quantified levels in Fig. 5 legend). Upon incubation with P-H, a diffuse fluorescence is observed in the whole cytoplasm as well as more intense localized fluorescence spots suggesting cytoplasmic and plasma membrane localization in addition to the accumulation in lysosomes or lysosome-like structures (Fig. 5B and 5E). In agreement with the uptake data, the fluorescence recorded after 18 h of incubation with P-(Lys)n is much larger than that obtained with P-H (Fig. 5C and 5F). Moreover, the P-(Lys)n fluorescence is more specifically localized in the lysosome-like structures. Lysosomal localization of P-(Lys)n was confirmed by double labeling with the lysosome specific probe, LysoTracker Green whose fluorescence emission is centered around 510 nm (see experimental section and legend of Fig. 6 for details). LysoTracker Green is a lysosomotropic weak base staining low pH (about 5–6) cell compartments. As shown in Fig. 6, the red fluorescence image (Fig. 6B) and the green fluorescence image (Fig. 6C) closely resemble. However, these two fluorescence images do not perfectly superimpose, suggesting that in addition to lysosomes, P-(Lys)n also localizes in non-lysosomal structures, most probably early endocytotic or pinocytic vesicles. These early endocytotic vesicles have a pH higher than that of lysosomes and are therefore poorly or not labeled by LysoTracker Green. In relation to this observation, Hamblin et al. showed that the conjugation of chlorin e6 to polylysine (positively charged at neutral pH) leads to a more punctuate intracellular fluorescence of the photosensitizer.20 It should be noted that the specific accumulation of these two porphyrins in the mitochondria of NCTC 2544 keratinocytes is unlikely in view of data obtained with the specific mitochondrial probe rhodamine 123, which show a mitochondrial network extending all over the cell (data not shown, see also ref. 29). Finally, it should be noted that the low fluorescence level of P-(Lys)n and in a lesser extent of P-H in the nuclear area, strongly suggests that there is little, if any, photosensitizer in the nucleus, despite the positive charges of these molecules. However, the same re-localization as that observed during or after the irradiation with other porphyrin derivatives26 could occur that may confer a carcinogenic potential on these molecules.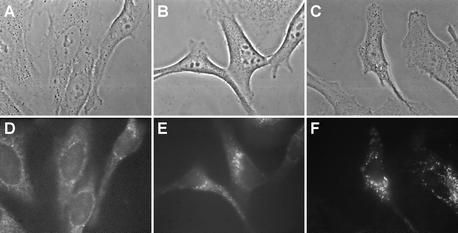 | ||
| Fig. 5 Phase contrast images (A, B, C) and fluorescence micrographs (D, E, F) obtained in NCTC 2544 keratinocytes incubated for 18 h in EMEM + 10% FCS with no additives (A, D), with 5 µM P-H (B, E) or with 5 µM P-(Lys)n (C, F). Fluorescence images were obtained with an Nikon × 100 oil immersion objective. Exposure time: 2 s. Signal attenuation: 1/16. Dichroic mirror: 400 nm. Excitation: 330–380 nm. Emission: 610–700 nm. 256 Grey levels between: −30–120 (D), −140–650 (E) and −150–2500 (F). | ||
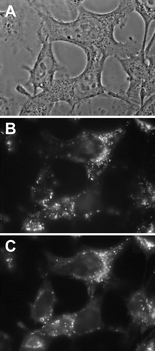 | ||
| Fig. 6 Phase contrast images (A) and fluorescence micrographs (B, C) obtained with 2544 keratinocytes incubated for 17 h with 5 µM P-(Lys)n in EMEM + 10% FCS and then for 1 h with 5 µM P-(Lys)n and 125 nM LysoTracker Green in EMEM + 10% FCS. Fluorescence images were obtained with a Nikon × 100 oil immersion objective. Exposure time: 2 s. Signal attenuation: 1/16. Dichroic mirror: 400 nm. Excitation: 330–380 nm. Emission: 610–700 nm (B) and 510–565 nm (C). 256 Grey levels between: −190–2900 (B) and −130–3100 (C). | ||
Effect of the presence of proteins in the incubation medium
The experiments described above were also performed after incubating the cells with porphyrins in serum-free medium. Though incubation in FCS-supplemented medium is closer to the in vivo situation, this study may unravel a role of serum proteins in the uptake process. As shown in Fig. 7A, an extremely fast uptake is observed for both P-H and P-(Lys)n. Whereas the kinetics of the P-H uptake is independent of the presence or absence of proteins, the P-(Lys)n uptake is faster in the absence of proteins than in their presence suggesting that the P-(Lys)n derivative interacts with serum proteins. This interaction, most probably due to the polylysine chain, may explain the lower plateau value observed in the presence of proteins (compare Fig. 7A to Fig. 2A). In this regard, solutions of the derivatives (1 µM) in culture medium with or without proteins (10% FCS) were submitted to gel exclusion chromatography. In contrast to P-H, retained at the top of the column either in the presence or in the absence of proteins, part of P-(Lys)n (about 30%) eluted with the proteins (data not shown). In the absence of proteins, P-(Lys)n eluted at longer time, strongly suggesting that it interacts with serum proteins. The higher rate of uptake of the strongly positively charged P-H probably results from the negative charge of the outer cell plasma membrane.11 As observed with the serum supplemented medium, the P-(Lys)n uptake is an order of magnitude greater than that of P-H. (Fig. 7A) and is practically proportional to the P-(Lys)n concentration after 1 h incubation with concentrations up to 5 µM (data not shown).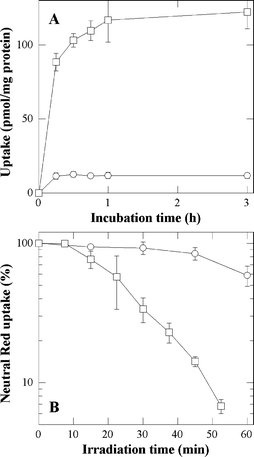 | ||
| Fig. 7 Uptake of P-H (○) and P-(Lys)n (□) as a function of the incubation time (A) and cytotoxic effects photosensitized by P-H (○) and P-(Lys)n (□) as a function of the irradiation time (B). In A, cells were incubated with 1 µM of porphyrin in HBSS. In B, cells were incubated with 5 µM of porphyrin in HBSS prior to irradiation. A Neutral Red uptake of 100% corresponds to sham-irradiated untreated cells. Data are the mean of at least three independent experiments performed in triplicates. | ||
The effect of proteins on the photocytotoxicity of P-H and P-(Lys)n was assessed by incubating the cells for 1 h in HBSS with 5 µM P-H or P-(Lys)n. Data reported in Fig. 7B can be compared to those obtained in the presence of FCS (see Fig. 3). First, it should be noted that P-H or P-(Lys)n are not cytotoxic in the dark. The phototoxicity data obtained with P-H or P-(Lys)n under these incubation conditions are similar to those obtained in the presence of serum proteins. A weak photocytotoxicity can only be observed with P-H at high incubation concentration (5 µM) and long irradiation time (60 min). The photocytotoxicity of P-(Lys)n is slightly higher in the absence than in the presence of proteins consistent with a slightly greater uptake in the former case. In conclusion, as observed for the uptake, the presence of serum proteins in the incubation medium does not significantly alter the phototoxicity.
Lipid peroxidation was also assayed after incubation in the absence of serum proteins. Data (not shown) demonstrate the lack of influence of serum proteins. A similar conclusion can be drawn regarding the intracellular localization since similar patterns are observed by microfluorometry, after incubation in HBSS.
Concluding remarks
In view of the low photodynamic effectiveness of P-(Lys)n it may be suggested that its larger uptake or its better retention than those of P-H associated with a larger absorbable light are mainly responsible for its greater photocytotoxicity. The cell microfluorescence study suggests that, in addition to the uptake, the intracellular localization could contribute to the marked differences in the cell photosensitizing effectiveness. Interestingly, the most effective polylysine derivative does not accumulate in the nucleus. However, it is obvious that a better understanding of the strong effect of the polylysine side chain on the photocytotoxic potential of the tri-cationic porphyrins requires further investigations on the relationship(s) between their cell localization and the molecular mechanisms underlying their phototoxic effect.Acknowledgements
This work was supported in part by grants from the Gulbenkian Foundation, the INSERM/GRICES exchange agreement and by the Franco-Portuguese “Pessoa” exchange Program (07958NF) and “Fundação para a Ciência e a Tecnologia” (Aveiro Organic Chemistry Research Unit). Epifluorescence microscopy experiments were performed with the equipment of the “Centre de Microscopie du Muséum”. The authors wish to thank Dr M. Gèze and M. Dellinger for their advice.References
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, Photodynamic therapy, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS.
- R. K. Pandey and G. Zheng, Porphyrins as photosensitizers in photodynamic therapy, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guillard, Academic Press, San Diego, 2000, vol. 6, pp. 157–230 Search PubMed.
- R. Bonnett, Chemical Aspects of Photodynamic Therapy, Gordon and Breach Science Publishers, London, 2000 Search PubMed.
- U. Schmidt-Erfurth and T. Hasan, Mechanism of action of photodynamic therapy with verteporfin for the treatment of age-related macular degeneration, Surv. Ophtalmol., 2000, 45, 195–214 Search PubMed.
- A. M. Soler, T. Warloe, A. Berner and K. E. Giercksky, A follow-up study of the recurrence and cosmesis in completely responding superficial and nodular basal cell carcinomas treated with methyl 5-aminolaevulinate based photodynamic therapy alone and with prior curettage, Br. J. Dermatol, 2001, 145, 467–471 CrossRef CAS.
- X. Chen, L. Hui, D. A. Foster and C. M. Drain, Efficient synthesis and photodynamic activity of porphyrin-saccharide conjugates: targeting and incapacitating cancer cells, Biochemistry, 2004, 43, 10918–10929 CrossRef CAS.
- H. Kato, K. Furukawa, M. Sato, T. Okunaka, Y. Kusunoki, M. Kawahara, M. Fukuoka, T. Miyazawa, T. Yana, K. Matsui, T. Shiraishi and H. Horinouchi, Phase II clinical study of photodynamic therapy using mono-L-aspartyl chlorin e6 and diode laser for early superficial squamous cell carcinoma of the lung, Lung Cancer, 2003, 42, 103–111 CrossRef.
- J. P. C. Tomé, M. G. P. M. S. Neves, A. C. Tomé, J. A. S. Cavaleiro, M. Soncin, M. Magaraggia, S. Ferro and G. Jori, Synthesis and anti-bacterial activity of new poly-S-lysine-porphyrin conjugates, J. Med. Chem., 2004, 47, 6649–6652 CrossRef CAS.
- M. R. Hamblin and T. Hasan, Photodynamic Therapy: a new antimicrobial approach to infectious disease?, Photochem. Photobiol. Sci., 2004, 3, 436–450 RSC.
- F. Gad, T. Zahra, K. P. Francis, T. Hasan and M. R. Hamblin, Targeted photodynamic therapy of established soft-tissue infections in mice, Photochem. Photobiol. Sci., 2004, 3, 451–458 RSC.
- E. D. Korn, Cell membranes: structure and synthesis, Ann. Rev. Biochem., 1969, 38, 263–288 CrossRef CAS.
- O. M. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, Protein measurement with the Folin phenol reagent, J. Biol. Chem., 1955, 193, 265–275.
- P. Morlière, F. Bosca, M. A. Miranda, J. V. Castell and R. Santus, Primary photochemical processes of the phototoxic neuroleptic cyamemazine: a study by laser flash photolysis and steady state irradiation, Photochem. Photobiol., 2004, 80, 535–541 CrossRef CAS.
- P. Morlière, S. Salmon, M. Aubailly, A. Risler and R. Santus, Sensitization of skin fibroblasts to UVA by excess of iron, Biochim. Biophys. Acta, 1997, 1334, 283–290 CrossRef CAS.
- R. M. Lasarow, R. V. Isseroff and E. C. Gomez, Quantitative in vitro assessment of phototoxicity by a fibroblast-neutral red assay, J. Invest. Dermatol., 1992, 98, 725–759 CrossRef.
- Commission Directive 2000/33/EC, Official Journal of the European Communities 8.6.2000, pp. L136/90–L136/107.
- P. Filipe, J. N. Silva, J. Haigle, J. P. Freitas, A. Fernandes, R. Santus and P. Morlière, Contrasting action of flavonoids on phototoxic effects induced in human skin fibroblasts by UVA alone or UVA plus cyamemazine, a phototoxic neuroleptic, Photochem. Photobiol. Sci., 2005, 4, 420–428 RSC.
- P. Morlière, A. Moysan, R. Santus, G. Huppe, J.-C. Mazière and L. Dubertret, UVA-induced lipid peroxidation in cultured human skin fibroblasts, Biochim. Biophys. Acta, 1991, 1084, 261–268 CAS.
- C. A. Parker, Photoluminescence of solutions, Elsevier, Amsterdam, 1968, pp. 208–216 Search PubMed.
- M. R. Hamblin, J. L. Miller, I. Rizvi and B. Ortel, Degree of substituiton of chlorin e6 on poly-L-lysine chains affects cellular uptake, localization and phototoxicity towards macrophages and cancer cells, J. X-Ray Sci. Technol., 2002, 10, 139–152 Search PubMed.
- E. Kohen, R. Santus and J. G. Hirschberg, Photobiology, Academic Press, San Diego, 1995, pp. 23–95 Search PubMed.
- P. Morlière, A. Moysan, R. Santus, G. Hüppe, J.-C. Mazière and L. Dubertret, UVA-induced lipid peroxidation in cultured human fibroblasts, Biochim. Biophys. Acta, 1991, 1084, 261–268 CAS.
- P. Morlière, J. Haigle, P. Filipe, J. Silva and R. Santus, An insight into the mechanisms of the phototoxic response induced by cyamemazine: a study with cultured fibroblasts and keratinocytes, Photochem. Photobiol., 2004, 79, 163–171 CrossRef CAS.
- K. Berg, Mechanisms of cell damage in photodynamic therapy, in The fundamental bases of phototherapyed, A. Young, H. Honigsmann and G. Jori, OEMF, Milan, 1996, pp. 181–207 Search PubMed.
- F. Dall'Acqua, S. Marciani Mano, F. Zambon and G. Rodighiero, Kinetic analysis of the photoreaction (365 nm) between psoralen and DNA, Photochem. Photobiol., 1979, 29, 489–495 CrossRef CAS.
- G. Ouédraogo and R. W. Redmond, Secondary reactive species extend the range of photosensitized effects in cells: DNA damage produced via initial membrane sensitization, Photochem. Photobiol., 2003, 77, 192–203 CrossRef CAS.
- R. P. Haugland, Handbook of Fluorescent Probes and Research Chemicals, Molecular Probes Inc., Eugene, 9th edn, 2002, pp. 273–283 Search PubMed.
- M. Dellinger, M. Gèze, R. Santus, E. Kohen, C. Kohen, J. G. Hirschberg and M. Monti, Imaging of cells by autofluorescence: a new tool in the probing of biopharmaceutical effects at the intracellular level, Biotechnol. Appl. Biochem., 1998, 28, 25–32 CAS.
- M. Gèze, J. M. Gaullier, M. Bazin and R. Santus, Sub-cellular localization of second generation PDT photosensitizers studied by microspectrofluorometry, in Analytical use of fluorescent probes in oncology, ed. E. Kohen and J. G. Hirschberg, Plenum Press, New York, 1996, pp. 113–121 Search PubMed.
| This journal is © The Royal Society of Chemistry and Owner Societies 2006 |