Ambient spark generation to synthesize carbon-encapsulated metal nanoparticles in continuous aerosol manner
Jeong Hoon
Byeon
a,
Jae Hong
Park
b,
Ki Young
Yoon
b and
Jungho
Hwang
*bc
aLCD Division, Samsung Electronics Co., Ltd., Yongin 446-711, Republic of Korea
bSchool of Mechanical Engineering, Yonsei University, Seoul 120-749, Republic of Korea. E-mail: hwangjh@yonsei.ac.kr; Fax: +82 2 312 2821; Tel: +82 2 2123 2821
cYonsei Center for Clean Technology, Yonsei University, Seoul 120-749, Republic of Korea
First published on 3rd November 2009
Abstract
We report the use of spark generation in an inert gas atmosphere to synthesize carbon-encapsulated metal nanoparticles (CEMNs) in a continuous aerosol manner using a metal (nickel, cobalt, iron)–graphite carbon electrode configuration without the use of a vacuum. The spark-generated particles consisted of CEMNs and carbonaceous aggregated debris. The outer layer of the CEMNs showed parallel fringes (ordered graphitic nanostructures) while the debris consisted of disordered nanostructures. Electron and X-ray diffraction showed that both metal and graphite in the CEMNs were the pure elements except for iron–carbon, which contained a carbide phase. Based on the order of the activation energies for carbon diffusion into a metal: iron–carbon (10.5–16.5 kcal mol−1) < cobalt–carbon (34.7 kcal mol−1) ∼ nickel–carbon (33.0–34.8 kcal mol−1), it was concluded that carbide particles form more easily from elemental iron than nickel or cobalt. The metal-to-carbon mass fractions of the spark-generated particles from nickel (anode)–carbon (cathode), cobalt–carbon, and iron–carbon spark configurations were 18.7, 28.3, and 11.2%, respectively, while the mass fractions for the configurations of metal (cathode)–carbon (anode) were 6.4, 9.1, and 4.3%, respectively. Similarly, the yield of CEMNs from the metal (anode)–carbon (cathode) electrodes was higher (54, 61, and 53%) than that of metal (cathode)–carbon (anode) electrodes (18, 30, and 18%).
1. Introduction
Nanocrystalline metal particles are prone to rapid environmental degradation on account of their high surface-area-to-volume ratio and reactivity. This limits their practical applications, even making scientific evaluations of the nanocrystalline properties difficult.1–4 The encapsulation of nanocrystalline metal particles with a chemically stable species, such as graphite, has been a recent breakthrough in this regard.1,2,5–9 Carbon-encapsulated metal nanoparticles (CEMNs) may have applications in ferrofluids, sensor devices, hydrogen storage, xerography, micro-machinery, and recording media, as well as a variety of biomedical applications.2,10–18 Among the many types of CEMNs with various core materials, those with iron group metals (nickel, cobalt, and iron) are of particular interest, not only because of their ferromagnetic properties, but also because these metals have a unique catalyzing ability to transform carbon into graphite.5,11,19–26The arc-synthesis of CEMNs, which involves the evaporation of a metal-inserted graphite anode in an inert atmosphere, requires high power, exceeding 1 kW, and expensive vacuum systems to generate the plasma.2–7,9,10,12–14,19,23,24,27,28 Moreover, the arc method produces unwanted byproducts due to the harsh synthesis conditions arising from the high energy generated during the process. Synthesis technologies of CEMNs including the arc method are not mature, and require further research and optimization.18 Spark generation has been used to generate monometallic and bimetallic particles of a variety conducting materials with particle sizes ranging from several nanometers to ∼100 nm in the aerosol state at ambient temperatures and pressures.29,30 These metallic aerosol nanoparticles have been used as initiators for the electroless deposition of metals.31–35 This paper reports the feasibility of spark generation in an inert gas atmosphere to synthesize CEMNs in a continuous aerosol manner using a metal (nickel, cobalt, iron)–graphite carbon electrode configuration without the use of a vacuum. The morphology and structure of the synthesized particles were examined by transmission electron microscopy (TEM), selected area electron diffraction (SAED), energy dispersive X-ray spectroscopy (EDX), X-ray diffraction (XRD), and Raman spectroscopy.
2. Experimental
In the spark generation apparatus, a spark was generated between a metal (nickel, cobalt, or iron) and a graphite carbon rod (each; 3 mm diameter, 100 mm length, Nilaco, Japan) in a chamber under a pure nitrogen (<10−4 impurities) atmosphere at standard temperature and pressure (STP). The following conditions were used to generate the spark: a current of 2 mA; a voltage drop of 2.4 kV with a frequency of 667 Hz between the electrodes, which was kept stable by continuously translating the carbon rod to the metal rod to maintain a constant distance of 1 mm. When a spark was generated between the metal and carbon rods inside a reactor, the gas temperature inside the spark channel was increased beyond a critical value, which was sufficient to sublimate parts of the electrodes.29 The duration of each spark was very short (∼1.5 ms) and the vapors cooled rapidly downstream of the spark. This formed resulted in supersaturation and particle formation through nucleation/condensation. A nitrogen gasflow (0.5 L min−1) carried the spark-generated particles as they exited the spark chamber. The chamber was cleaned periodically with compressed dry particle-free air to eliminate any residual particles.The non-purified spark-generated particles were analyzed using a variety of methods. A TEM (JEM-3010, JEOL, Japan) was operated at 300 kV with an EDX attachment (Oxford). SAED patterns of the particles were used to determine their crystalline phase. The interlayer spacing of the particles was calculated using digital micrograph software and TEM. The EDX patterns of the selected local areas were used to determine the elemental composition of the particles. The phases of the particles were determined by XRD (Rigaku D/max-2100, Japan) using Cu Kα radiation (λ = 154.178 pm) at a step size of 0.08° 2θ. Raman spectroscopy (T64000, HORIBA Jobin Yvon, France) was used to determine the level of graphitization in the particles. The laser excitation wavelength was 514.5 nm. The particles were sampled on a porous carbon-coated copper grid located on a polyamide membrane filter (for TEM, SAED, and EDX) or on a glass plate (for XRD and Raman). The sampling location was 20 cm downstream of the spark generator. The size distribution of the spark-generated aerosol particles was measured using a scanning mobility particle sizer (SMPS, TSI 3936, US) system, which consisted of an electrostatic classifier (TSI 3081), a condensation particle counter (TSI 3025), and an aerosol charge neutralizer (2U500, NRD, US). The SMPS system was operated at a sample flow of 0.3 L min−1, a sheath flow of 3 L min−1, and a scan time of 180 s (measurement range between 13.8 and 723 nm).
3. Results and discussion
Fig. 1a shows a TEM image of the spark-generated particles containing core (black)–shell (gray) particles with small aggregated debris from the nickel (anode) and carbon (cathode) electrodes. The inset in Fig. 1a shows the local EDX data, indicating the presence of nickel and carbon. The other peak at approximately 8 keV corresponds to the copper grid used to support the sample. The XRD profile (Fig. 1b) indicated that there is a relatively broad peak at 2θ = 26.1°, which is close to 26.4° 2θ for graphite with a d-spacing of 0.341 nm. This is slightly larger than the 0.336 nm (d002) for graphite (JCPDS No. 75-1621). The broad diffraction peak indicates that the graphite also has a disordered structure. The large intershell distance suggests a decrease in the intershell interactions compared to perfect graphite. The two subsidiary peaks at 44.3 and 54.3° 2θ correspond to the (110) and (004) crystal planes of graphite, respectively. Another peak at 44.5° (with d-value is 0.2022 nm, which is almost consistent with the d111 (0.2034 nm) of nickel; peak range and the d-values of four measurements were 44.36–44.61° and 0.2062–0.2017 nm, respectively), 51.8°, and 76.4° 2θ were respectively assigned to the (111), (200), and (220) crystal planes of face-centered cubic (fcc) nickel metal with a lattice parameter of 0.352 nm (JCPDS No. 04-0850). The Raman spectrum (Fig. 1c) shows that the G-band (stretching vibrations in the basal plane of crystalline graphite) was close to 1582 cm−1, with a similar shape to that reported elsewhere, indicating that the structure of the sample was well oriented, and had a chemical structure similar to that of graphite.1,4,12,16 It is possible that the D-band (1343 cm−1, indicating the level of defects in the graphitic material) might have originated from certain disordered or defective parts of the core–shell particles (corners and edges) and debris around the core–shell particles.2,10,19,20,28 A TEM image (Fig. 1d) revealed the core to be darker than that of the shell, which means that the particle has a nickel core with a graphitic shell (inset of Fig. 1d). Sizes of the core–shell particles ranged from 10 to 390 nm in diameter (mode diameter = 86 nm) according to TEM (four hundred particles in low-magnification TEM images were selected), and are shown in another inset of Fig. 1d. In Fig. 1e, the diffraction pattern is a superposition of the two sets of patterns: one is a set of annular rings originating from graphitic carbon (diffraction from a two-dimensional lattice), and the spots are from a single nickel crystal (arranged in a rectangular lattice). The diffraction rings, which correspond to a layer distance of approximately 0.34, 0.18, and 0.17 nm, were assigned to the (002), (102), and (004) planes of hexagonal graphite, respectively. On the other hand, layer distances of approximately 0.11, 0.08, and 0.06 nm were assigned to the (311), (420), and (440) planes of fcc nickel crystalline, respectively. Fig. 1f shows a high-magnification TEM image of a CEMN. The parallel fringes in the shell (inset of Fig. 1f) show that they are ordered graphitic nanostructures. These contiguous carbon fringes around a nickel core are evidence of complete encapsulation by a graphitic shell. In other words, the above results suggest that using the ambient nickel–carbon spark configuration it is possible to synthesize CEMNs in a continuous manner. The number of graphitic shells and the nickel-to-carbon mass fraction of the CEMNs were detected using TEM and EDX, respectively, by single-particle analysis. The results are shown in Fig. 1g as a function of the CEMN diameter or nickel core diameter. Both the number of graphitic shells and the nickel-to-carbon mass fraction increased with increasing nickel core diameter, as observed in other materials.2,9,10,15Fig. 1h shows that the debris particles in Fig. 1a were aggregates. The inset in Fig. 1h revealed the debris to be disordered graphitic nanostructures. Further study on a suitable purification method is now in progress to synthesize pure CEMNs with narrow-distributed sizes.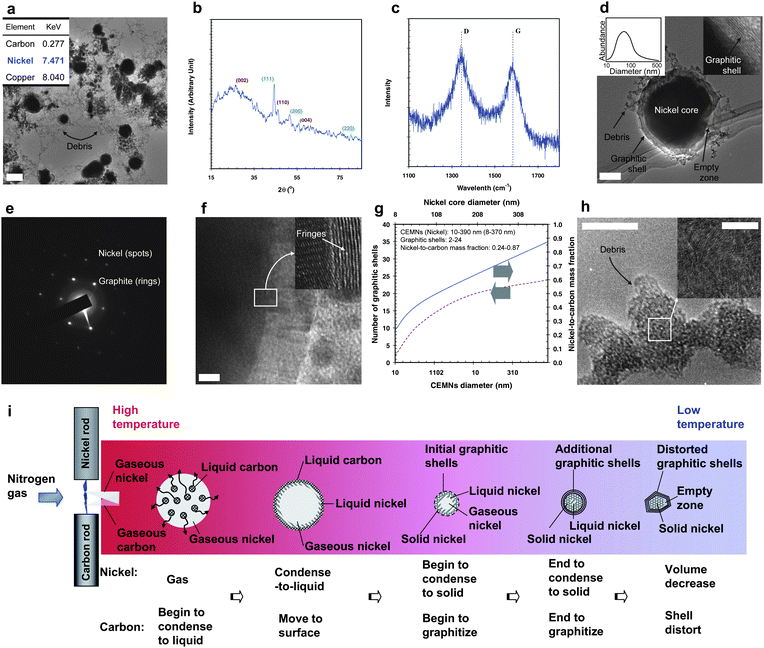 | ||
| Fig. 1 Spark-generated nickel–carbon particles and their characterization: (a) TEM image with the EDX spectrum of the spark-generated particles. Scale bar, 200 nm. (b) XRD pattern of the spark-generated particles. (c) Raman spectrum of the spark-generated particles. (d) TEM image of a single core–shell shaped particle with the size distribution. Scale bar, 20 nm. (e) SAED pattern of the particle in d. (f) High-magnification TEM image of the particle. Scale bar, 5 nm. (g) Number of graphitic shells and the nickel-to-carbon mass fraction for CEMNs. (h) High-magnification TEM images of the debris. Scale bars, 10 nm and 5 nm (inset). (i) Formation mechanism of the CEMNs. | ||
Previous studies have reported that CEMNs are formed as a result of the catalytic effect of a metal on carbon.1,3,13,20,22 In this system (Fig. 1i), when a spark channel was formed between the nickel and carbon electrodes, parts of the electrodes were gasified due to the mechanism of the spark.29,36 The condensation temperature of carbon is higher than that of nickel. Therefore, the condensation of gaseous carbon to liquid progresses more rapidly than that of nickel when the gases are swept out from the spark channel by the nitrogen gas flow. During this process, liquid carbon was pushed from the gaseous nickel toward the interface due to the temperature gradient. Simultaneously, the gaseous nickel near the interface began to condense into a liquid during carbon migration and some of the liquid carbon was prevented from being releasing away from the interface due to its high affinity for nickel.37,38 Moreover, some of the released carbon might be directly incorporated into the nickel surface—where carbon atoms join to the growing graphitic shell without dissolving into nickel.25,39 The catalytic effect of nickel and the high temperature in the spark zone caused the graphitization of carbon.39 Further cooling by the nitrogen gas flow caused the liquid nickel to solidify. Eventually, graphitization ceased when the leading solid nickel surface was encapsulated by graphitic shells, which prevented further graphitization. During graphitization, the remaining liquid nickel solidified completely resulting in a decrease in volume. Hence, an empty zone was formed between the graphitic shells and the solid nickel core. According to previous studies, an empty zone was also formed by a sufficiently high reaction temperature.9,40 When the amount of nickel vapor was insufficient in this regime, the unreacted liquid carbon escaped from the interface and became solid carbon debris (Fig. 1h) in the nitrogen gas flow.
Fig. 2a shows a TEM image of the spark-generated cobalt–carbon particles, which is similar to the nickel–carbon spark configuration case in Fig. 1. The inset in Fig. 2a shows the local EDX data revealing the presence of cobalt and carbon. The XRD (Fig. 2b) profile indicated that cores of the particles in Fig. 2a were in the form of elemental cobalt. The Raman peaks (inset of Fig. 2b) at 1571 and 1339 cm−1 demonstrate the particle sample to have an IG:ID ratio of 1.06. The high-magnification TEM images (Fig. 2c) showed that the CEMNs have a similar morphology to the nickel–carbon case. One reason for this similarity might be the comparable activation energy (33.0–34.8 kcal mol−1 for nickel–carbon vs. 34.7 kcal mol−1 for cobalt–carbon) for carbon diffusion into a metal.2,5,10,19,41
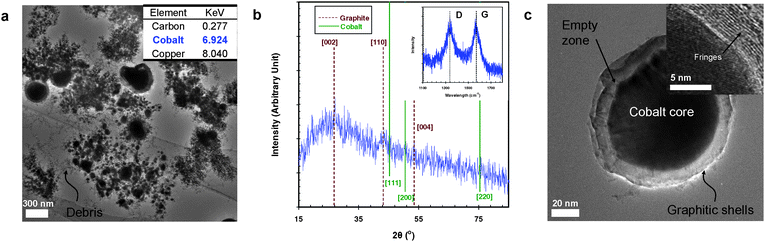 | ||
| Fig. 2 Spark-generated cobalt–carbon particles and their characterization: (a) TEM image and local EDX information (inset) of the spark-generated particles. (b) XRD pattern and Raman spectrum (inset) of the spark-generated particles. (c) TEM image of the CEMN and its high-magnification image (inset). | ||
Fig. 3a shows a TEM image of the spark-generated iron–carbon particles, which is similar to the nickel– and cobalt– carbon cases in Fig. 1 and 2, respectively. The EDX data in the inset of Fig. 3a indicates the presence of iron and carbon. The XRD profile (Fig. 3b) shows the presence of peaks associated with carbide and α-iron phases. The carbide phases, showed diffraction peaks at 37.6, 40.8, 42.8, 43.6, 44.1, 44.9, 45.7, 49.1, 54.3, 58.2, 78.4, and 86.0° 2θ. The other peaks appearing at 44.7, 65.0, and 82.3° 2θ were assigned to the (110), (200), and (211) reflections of α-iron, respectively. From single-particle TEM-SAED analyses (not shown), iron carbide was also detected in some spots of the core of a particle. Based on the order of the activation energies for carbon diffusion into a metal: iron–carbon (10.5–16.5 kcal mol−1) < cobalt–carbon ∼ nickel–carbon, it can be concluded that carbide particles form more easily from elemental iron than from nickel or cobalt.24,42,43 The Raman peaks (inset of Fig. 3b) at 1592 and 1347 cm−1 demonstrate the spark-generated particle sample to have an IG:ID ratio of 0.93. The morphology (Fig. 3c) of the CEMNs in the high-magnification TEM images is also similar to the nickel–carbon and cobalt–carbon cases. Numerous carbon nanotubes (CNTs) (Fig. 3d) were occasionally observed in the spark-generated particle samples, which had mean internal and external diameters of 3.5 and 9.2 nm, respectively. The activation energies for CNT growth are iron–carbon (16.1 kcal mol−1) < cobalt–carbon (33.0–33.3 kcal mol−1) ∼ nickel–carbon (34.7 kcal mol−1).42 Therefore, the lowest activation energy, iron–carbon, might explain why nanotubes were only observed in the iron–carbon case.
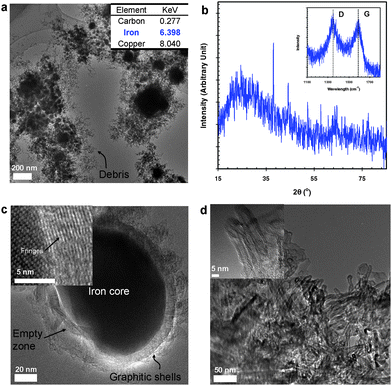 | ||
| Fig. 3 Spark-generated iron–carbon particles and their characterization: (a) TEM image and local EDX information (inset) of the spark-generated particles. (b) XRD and Raman spectra (inset) of the spark-generated particles. (c) TEM image of a CEMN and its high-magnification TEM image (inset). (d) TEM images of the spark-generated multiwall carbon nanotubes (MWCNTs). | ||
The electrical mobility equivalent diameter of all particles was measured by SMPS and ranged from 30 and 600 nm. Table 1 shows the geometric mean diameter (GMD), geometric standard deviation (GSD), and total number concentration (TNC) for the carbon–carbon (denoted as Carbon), nickel–carbon, cobalt–carbon, and iron–carbon spark configurations. The GMDs of the metal–carbon sparks were larger than carbon–carbon spark while the corresponding GSDs and TNCs were smaller. These differences between metal–carbon and carbon–carbon sparks might have originated from the mechanism (Fig. 1i) for their formation.
| Carbon | Nickel–Carbon | Cobalt–Carbon | Iron–Carbon | |
|---|---|---|---|---|
| GMD/nm | 119 | 142 | 150 | 125 |
| GSD/dimensionless | 1.72 | 1.57 | 1.58 | 1.60 |
| TNC/×107 particles cm−3 | 1.64 | 1.47 | 1.56 | 1.31 |
The effect of switching the electrode polarity of the spark on the metal-to-carbon mass fraction was investigated. The mass fraction was obtained using inductively coupled plasma atomic emission spectroscopy (ICP-AES, Elan 6000, Perkin-Elmer, US). Table 2 shows the results and corresponding yields of CEMNs. The yield was determined by the area fraction of CEMNs-to-all particles in the TEM image. For the metal (anode)–carbon (cathode) sparks, the metal-to-carbon mass fractions for nickel, cobalt and iron were 0.19, 0.28, and 0.11, respectively, with a CEMN yield of 0.54 (nickel), 0.61 (cobalt) and 0.53 (iron). For the metal (cathode)-carbon (anode) sparks, the metal-to-carbon mass fractions were 0.06, 0.09 and 0.04 for nickel, cobalt, and iron, respectively, with a CEMN yield of 0.18 (nickel), 0.30 (cobalt) and 0.18 (iron). Therefore, a higher yield was associated with a higher metal-to-carbon mass fraction. These results are in agreement with previous studies, which used a modified arc system to generate a high metal-to-carbon mass fraction, leading to a significant improvement in CEMN production.2,10,11,13,44 One study reported that the encapsulation of all metal particles during CEMN production required only a slightly higher carbon fraction than metal.7 If insufficient metal is present, the unreacted carbon would form debris and co-exist with the CEMNs in the aerosol state as they are forced way from the spark channel by the gas flow (refer Fig. 1h and 1i). The results in Table 2 show that higher yield was obtained for the metal (anode)–carbon (cathode) sparks. This might be caused by a higher temperature channel formed near the anode and thus higher evaporation and subsequent nucleation/condensation near the anode.29
| Sample | Metal-to-carbon mass fraction | Yield of CEMNs | ||
|---|---|---|---|---|
| Metal (anode)–Carbon (cathode) | Metal (cathode)–Carbon (anode) | Metal (anode)–Carbon (cathode) | Metal (cathode)–Carbon (anode) | |
| Nickel–Carbon | 0.19 | 0.06 | 0.54 | 0.18 |
| Cobalt–Carbon | 0.28 | 0.09 | 0.61 | 0.30 |
| Iron–Carbon | 0.11 | 0.04 | 0.53 | 0.18 |
4. Conclusions
CEMNs were synthesized under an inert gas atmosphere using a metal (nickel, cobalt, iron)–carbon spark configuration in a continuous manner without a vacuum or special operating conditions. Electron and X-ray diffraction showed that both metal and graphite in the sample were pure elements except for the iron–carbon case, which contained a carbide phase. Based on the order of activation energies for carbon diffusion into a metal, it was concluded that carbide particles form more easily from elemental iron than from nickel or cobalt. The Raman G- and D-bands demonstrated the particles to have IG:ID ratios of 0.81, 1.06, and 0.93 for nickel–carbon, cobalt–carbon, and iron–carbon, respectively. The corresponding metal-to-carbon mass fraction for the spark electrodes [metal (anode)–carbon (cathode)] were 18.7, 28.3, and 11.2%, while the mass fraction for the configuration of the metal (cathode)–carbon (anode) spark electrodes were 6.4, 9.1, and 4.3%. Likewise, the yields of CEMNs from the metal (anode)–carbon (cathode) electrodes were higher (54, 61, and 53%) than those of the metal (cathode)–carbon (anode) electrodes (18, 30, and 18%). This method for preparing CEMNs is simple and continuous compared with conventional methodologies and can be applied to the production of nanocrystalline encapsulates.Acknowledgements
This work was supported by a National Research Foundation (NRF) grant funded by the Korea government (MEST) (No. 20090073212).References
- W. Xiaomin, X. Bingshe, J. Husheng, L. Xuguang and I. Hideki, J. Phys. Chem. Solids, 2006, 67, 871 CrossRef.
- J.-H. Hwang, V. P. Draid, M. H. Teng, J. J. Host, B. R. Elliott, D. L. Johnson and T. O. Mason, J. Mater. Res., 1997, 12, 1076 CrossRef CAS.
- J. J. Host, J. A. Block, K. Parvin, V. P. Dravid, J. L. Alpers, T. Sezen and R. LaDuca, J. Appl. Phys., 1998, 83, 793 CrossRef CAS.
- G. Cui, Y.-S. Hu, L. Zhi, D. Wu, I. Lieberwirth, J. Maier and K. Müllen, Small, 2007, 3, 2066 CrossRef CAS.
- Y. Saito, T. Yoshikawa, M. Okuda, N. Fujimoto, S. Yamamuro, K. Wakoh, K. Sumiyama, K. Suzuki, A. Kasuya and Y. Nishina, J. Appl. Phys., 1994, 75, 134 CrossRef CAS.
- J. Jiao and S. Seraphin, J. Appl. Phys., 1998, 83, 2442 CrossRef CAS.
- M.-H. Teng, S.-W. Tsai, C.-I. Hsiao and Y.-D. Chen, J. Alloys Compd., 2007, 434–435, 678 CrossRef CAS.
- T. Hayashi, S. Hirono, M. Tomita and S. Umemura, Nature, 1996, 381, 772 CrossRef.
- J. Jiao and S. Seraphin, J. Phys. Chem. Solids, 2000, 61, 1055 CrossRef CAS.
- J. J. Host, M. H. Teng, B. R. Elliott, J. H. Hwang, T. O. Mason, D. L. Johnson and V. P. Dravid, J. Mater. Res., 1997, 12, 1268 CAS.
- A. A. Setlur, J. Y. Dai, J. M. Lauerhaas, P. L. Washington and R. P. H. Chang, J. Mater. Res., 1998, 13, 2139 CrossRef CAS.
- E. Flahaut, F. Agnoli, J. Sloan, C. O'Connor and M. L. H. Green, Chem. Mater., 2002, 14, 2553 CrossRef CAS.
- J. J. Host, V. P. Dravid and M. H. Teng, J. Mater. Res., 1998, 13, 2547 CrossRef CAS.
- K. H. Ang, I. Alexandrou, N. D. Mathur, G. A. Amaratunga and S. Haq, Nanotechnology, 2004, 15, 520 CrossRef CAS.
- M. Choi, I. S. Altman, Y.-J. Kim, P. V. Phkhitsa, S. Lee, G.-S. Park, T. Jeong and J.-B. Yoo, Adv. Mater., 2004, 16, 1721 CrossRef CAS.
- G.-X. Zhu, X.-W. Wei and S. Jiang, J. Mater. Chem., 2007, 17, 2301 RSC.
- C. Saiyasombat, N. Petchsang, I. M. Tang and J. H. Hodak, Nanotechnology, 2008, 19, 085705 CrossRef.
- J. Borysiuk, A. Grabias, J. Szczytko, M. Bystrzejewski, A. Twardowski and H. Lange, Carbon, 2008, 46, 1693 CrossRef CAS.
- Y. Saito, T. Yoshikawa, M. Okuda and N. Fujimoto, Chem. Phys. Lett., 1993, 212, 379 CrossRef CAS.
- Y. Saito, T. Yoshikawa, M. Okuda, N. Fujimoto, K. Sumiyama, K. Suzuki, A. Kasuya and Y. Nishina, J. Phys. Chem. Solids, 1849, 54 Search PubMed.
- Z. Liu, L. Ci, N. Y. Jin-Phillipp and M. Rühle, J. Mater. Chem., 2007, 17, 4619 RSC.
- R. B. Little, J. Cluster Sci., 2003, 14, 135 CrossRef CAS.
- S. Tomita, M. Hikita, M. Fujii, S. Hayashi and K. Yamamoto, Chem. Phys. Lett., 2000, 316, 361 CrossRef CAS.
- J. N. Wang, L. Zhang, F. Yu and Z. M. Sheng, J. Phys. Chem. B, 2007, 111, 2119 CrossRef CAS.
- F. Ding, A. Rosén, E. E. B. Campbell, L. K. L. Falk and K. Bolton, J. Phys. Chem. B, 2006, 110, 7666 CrossRef CAS.
- V. P. Dravid, J. J. Host, M. H. Teng, B. Elliott, J. H. Hwang, D. L. Johnson, T. O. Mason and J. R. Weertman, Nature, 1995, 374, 602 CrossRef CAS.
- R. Sergiienko, E. Shibata, Z. Akase, H. Suwa, D. Shindo and T. Nakamura, J. Mater. Res., 2006, 21, 2524 CrossRef CAS.
- N. Sano, H. Wang, I. Alexandrou, M. Chhowalla, K. B. K. Teo and K. Limura, J. Appl. Phys., 2002, 92, 2783 CrossRef CAS.
- J. H. Byeon, J. H. Park and J. Hwang, J. Aerosol Sci., 2008, 39, 888 CrossRef CAS.
- J. H. Byeon, J. H. Park, K. Y. Yoon, B. J. Ko, J. H. Ji and J. Hwang, Carbon, 2006, 44, 2106 CrossRef CAS.
- J. H. Byeon, B. J. Ko and J. Hwang, J. Phys. Chem. C, 2008, 112, 3627 CrossRef CAS.
- J. H. Byeon, J. H. Park, K. Y. Yoon and J. Hwang, Langmuir, 2008, 24, 5949 CrossRef CAS.
- J. H. Byeon, K. Y. Yoon, Y. K. Jung and J. Hwang, Electrochem. Commun., 2008, 10, 1272 CrossRef CAS.
- J. H. Byeon and J. Hwang, ACS Appl. Mater. Interfaces, 2009, 1, 261 Search PubMed.
- J. H. Byeon, R. H. Lee and J. Hwang, J. Phys. D: Appl. Phys., 2009, 42, 055303 CrossRef.
- Z. Shen and M. Nygren, J. Mater. Chem., 2001, 11, 204 RSC.
- W. L. Holstein, J. Catal., 1995, 152, 42 CrossRef CAS.
- R. T. K. Baker, Carbon, 1989, 27, 315 CrossRef CAS.
- J. Qiu, Y. Li, Y. Wang, Y. An, Z. Zhao, Y. Zhou and W. Li, Fuel Process. Technol., 2004, 86, 267 CrossRef CAS.
- Z.-J. Liu, Z.-Y. Yuan, W. Zhou, Z. Xu and L.-M. Peng, Chem. Vap. Deposition, 2001, 7, 248 CrossRef CAS.
- Y. Saito, Carbon, 1995, 33, 979 CrossRef CAS.
- Z. H. Wang, C. J. Choi, B. K. Kim, J. C. Kim and Z. D. Zhang, Carbon, 2003, 41, 1751 CrossRef CAS.
- N. M. Rodriguez, J. Mater. Res., 1993, 8, 3233 CrossRef CAS.
- B. R. Elliott, J. J. Host, V. P. Dravid, M. H. Teng and J.-H. Hwang, J. Mater. Res., 1997, 12, 3328 CAS.
| This journal is © The Royal Society of Chemistry 2009 |